 Xiangyu Qi1Huadi Wang1,2Shuangshuang Chen1Jing Feng1Huijie Chen1Ziyi Qin1,3
Xiangyu Qi1Huadi Wang1,2Shuangshuang Chen1Jing Feng1Huijie Chen1Ziyi Qin1,3 Ikram Blilou4
Ikram Blilou4 Yanming Deng1,2,3*
Yanming Deng1,2,3*- 1Jiangsu Key Laboratory for Horticultural Crop Genetic Improvement, Institute of Leisure Agriculture, Jiangsu Academy of Agricultural Sciences, Nanjing, China
- 2School of Life Sciences, Jiangsu University, Zhenjiang, China
- 3College of Horticulture, Nanjing Agricultural University, Nanjing, China
- 4Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Jasmine [Jasminum sambac (L.) Aiton] is a commercially important cultivated plant species known for its fragrant flowers used in the perfume industry, medicine and cosmetics. In the present study, we obtained a draft genome for the J. sambac cultivar ‘Danbanmoli’ (JSDB, a single-petal phenotype). We showed that the final genome of J. sambac was 520.80 Mb in size (contig N50 = 145.43 kb; scaffold N50 = 145.53 kb) and comprised 35,363 genes. Our analyses revealed that the J. sambac genome has undergone only an ancient whole-genome duplication (WGD) event. We estimated that the lineage that has given rise to J. sambac diverged from the lineage leading to Osmanthus fragrans and Olea europaea approximately 31.1 million years ago (Mya). On the basis of a combination of genomic and transcriptomic analyses, we identified 92 transcription factors (TFs) and 206 genes related to heat stress response. Base on a combination of genomic, transcriptomic and metabolomic analyses, a range of aroma compounds and genes involved in the benzenoid/phenylpropanoid and terpenoid biosynthesis pathways were identified. In the newly assembled J. sambac genome, we identified a total of 122 MYB, 122 bHLH and 69 WRKY genes. Our assembled J. sambac JSDB genome provides fundamental knowledge to study the molecular mechanism of heat stress tolerance, and improve jasmine flowers and dissect its fragrance.
Introduction
Jasmine [Jasminum sambac (L.) Aiton] is a diploid (2n = 2x = 26) evergreen ornamental species belonging to the family Oleaceae. It is one of the most important commercial flower plant species in many countries, and be used extensively in bouquets, ornamental displays, tea, cosmetics and perfumery (Cai et al., 2007; Wikee et al., 2011). In China, jasmine cultivation dates back more than 2,000 years on account of its usage in traditional Chinese medicine and its high value in scenting the famous ‘jasmine tea’ (Deng et al., 2016). Jasmine plants generally exhibit single-, double-, or multi-petal phenotypes (Deng et al., 2017). Among which the J. sambac cultivar ‘Danbanmoli’ (JSDB, a single-petal phenotype) is one of the main cultivars cropped in China, for its flowers are considered to be the most fragrant (Deng et al., 2017). The perfumed flowers contain an essential oil known as the “attar of jasmine”, which is rich in low molecular weight aroma compounds, and the most prominent of which are benzenoids, phenylpropanoids and terpenoids (Bera et al., 2017).
In plants, benzenoids/phenylpropanoids are generated via the aromatic acid phenylalanine produced from phenylpyruvate and arogenate pathways, which diverge from the shikimate pathway (Maeda and Dudareva, 2012; Qian et al., 2019). Monoterpenes/diterpenes and sesquiterpenes are derived from the 2-C-methylerythritol-4-phosphate (MEP) and mevalonate (MVA) pathways, respectively (Vranová et al., 2013). Studies have identified numerous genes involved in the phenylpropanoid, MEP and MVA pathways (Achnine et al., 2004; Degenhardt et al., 2009; Olofsson et al., 2011). Furthermore, transcription factors (TFs), including MYB, bHLH and WRKY families are also involved in terpenoids synthesis (Shang et al., 2020). However, the molecular mechanism of aroma compounds biosynthesis in jasmine is still not very clear and needs further exploration.
As global warming progresses, heat stress is becoming a threat to the environment as well as plant populations (Parmesan and Yohe, 2003). Heat stress compromises plant growth by causing reactive oxygen species generation, protein denaturation and membrane destabilization (Mittler et al., 2012). Under heat stress, heat shock transcription factors (HSFs) are rapidly activated and enhance the expression of many genes that encode heat shock proteins (HSPs) (Ohama et al., 2016). HSFs are core regulators of the heat stress response. HSPs protect cellar components by preventing protein denaturation and aggregation. J. sambac is one of the evergreen species that bloom in the hot summer, during which the temperature rises above 38°C. Therefore, it is important to elucidate the molecular mechanism involved in the heat stress tolerance of jasmine, and this will be helpful to understand the plant’s adaptability to high temperature condition.
Genomic resources are essential for molecular and evolutionary studies and are becoming increasingly attainable. With the ongoing rapid developments in sequencing technology, an increasing number of genomes are being sequenced and released (Chen et al., 2018a; Chen et al., 2019). To date, several species in the Oleoideae subfamily of the Oleaceae, including Fraxinus exceisior (Sollars et al., 2017), Olea europaea (Unver et al., 2017), Osmanthus fragrans (Yang et al., 2018), and Forsythia suspensa (Li et al., 2020) have been sequenced and reported. Jasmine cultivars are characterized by differing floral phenotypes ranging from single- to multi-petal flowers. These differences not only have a notable influence on floral morphology, but also influence floral fragrance, because petals contribute to producing aroma components. Although a genome of J. sambac cultivar ‘Trifoliatum’ was reported recently (Xu et al., 2021), there is currently a lack of genomic resource for single-petal jasmine, which is essential for linking an understanding of flower fragrance biosynthetic genes with aroma scent emission during flowering.
In this study, we obtained a draft genome of J. sambac single-petal cultivar JSDB based on PacBio and Illumina sequencing. On the basis of a combination of genomic and transcriptomic analyses, we identified TFs and genes related to its heat stress response. Base on a combination of genomic, transcriptomic, and metabolomic analyses, we identified a range of aroma compounds and genes involved in the benzenoid/phenylpropanoid and terpenoid biosynthesis pathways. Our data provide new insights into the molecular mechanisms underlying heat stress tolerance and aroma scent emission of J. sambac.
Materials and methods
Plant materials, library construction and sequencing
The J. sambac JSDB was maintained in the Preservation Centre of the Jasmine Germplasm Resources, Jiangsu Academy of Agricultural Sciences, Nanjing, China (latitude: 32°05′N, longitude: 118°08′E; 68 m above sea level) (Figure S1). Tender young leaves of individual plant were collected from J. sambac. The genomic DNA was extracted using the cetyltrimethylammonium bromide (CTAB) method (Li et al., 2013). A total of four PacBio 20-kb libraries were generated using an SMRTbell Template Prep Kit (PacBio) and were sequenced on the PacBio Sequel platform (Table S1). For Illumina library construction, the genomic DNA was fragmented and size-fractionated, then subjected to library construction and sequenced on the Illumina HiSeq 2000 system with paired-end 150-bp reads (Table S1). All the above sequencing was performed by Shanghai Personal Biotechnology Company Limited (Shanghai, China).
Estimation of J. sambac genome size
Fresh leaves were collected from the same J. sambac plant used for sequencing. The genome size was determined based on flow cytometry (Doležel et al., 2007), with three parallel experiments being carried out for each sample. The Solanum lycopersicum L. ‘Stupicke polni tyckove rane’ was used as an internal standard. The sequencing data were analyzed through FACSTM 1.0.0.650 software, and the statistical analysis was conducted using SPSS 17.0.
The J. sambac genome size was also estimated by using a k-mer (k = 17) analysis-based approach with quality-filtered Illumina paired-end short reads. Jellyfish software (version 2.1.4) was applied for counting k-mers in the DNA samples (Marcais and Kingsford, 2011). GCE software (version 1.0) was used for estimating genome size (Liu et al., 2013). Finally, the heterozygosity of JSDB was determined (Kajitani et al., 2014).
Genome assembly and quality assessment
Raw Illumina reads were processed to collapse duplicated read pairs into unique read pairs. Duplicated read pairs were defined as those having identical bases at positions 14 to 90 in both left and right reads. Then, the resulting reads were processed to remove adaptor and low-quality sequences using AdapterRemoval (version 2.1.7) (Schubert et al., 2016). Reads shorter than 50 bp at either end was further discarded. Finally, sequencing errors in paired-end reads were corrected using SOAPec (parameter ‘-kmer-len 17’) (Luo et al., 2012).
For J. sambac genome assembly, we used DBG2OLC (Ye et al., 2016). The high-quality cleaned paired-end reads were initially assembled into contigs using Platanus (version 1.8.8 parameters ‘-k 32 -s 10 -c 2 -a 10.0 -u 0.1 -d 0.5’), and thereafter connected to scaffolds with DBG2LOC using all paired-end reads. Following assembly, the third-generation sequencing raw data were used to correct the scaffolds using Arrow (version 2.2.2) in two rounds, and the high-quality next-generation sequencing data were then used to correct scaffolds in a further two rounds using Pilon (version 1.22) with paired-end reads. To evaluate the accuracy and completeness of the genome assemblies, BUSCO (version 3.0.2) was performed using the embryophyta_odb10 plant database (Simão et al., 2015).
Repetitive elements and non-coding RNA annotation
In order to search for transposable elements in the assembled J. sambac genome, an integrated strategy based on de novo prediction and a homology-based method was adopted. For de novo prediction, we identified repetitive elements using RepeatModeler (version 1.0.4; http://www.repeatmasker.org/RepeatModeler/), RECON (version 1.0.8; http://selab.janelia.org/recon.html) and RepeatScout (version1.0.5; http://repeatscout.bioprojects.org/), with default parameters. Homology-based repetitive elements were identified by comparison with consensus sequences in the Repbase library (version 20150807) using RepeatMasker (version 4.0.5; http://www.repeatmasker.Org/) (Kapitonov and Jurka, 2008).
We searched for LTR-RTs in the genome using LTR_finder with parameters ‘-D 5000 -d 100 -L 20000 -l 1000 -p 20 -M 0.3’ (Xu and Wang, 2007) and LTRharvest with parameters ‘-v -mintsd 4 -maxtsd 6’ (Ellinghaus et al., 2008). Then, the identified LTR-RT candidates were filtered using LTRdigest (Steinbiss et al., 2009) with default parameters ‘-trnas -hmms’. The insert time (T) of intact LTRs was estimated using the formula T = K/2r, where K is the number of nucleotide substitutions per site between each pair of LTRs and r refers to the general nucleotide substitution rate, which was set to 1.3 × 10-8 per site per year (Ma et al., 2020).
We predicted tRNAs using tRNAscan-SE (version 1.3.1) (Lowe and Eddy, 1997). rRNAs were predicted using RNAmmer (version 1.2) (Lagesen et al., 2007), and other ncRNAs were predicted using the Perl program Rfam-scan.pl (version 1.0.4) by inner calling using Infernal (version 1.1.1) (Nawrocki and Eddy, 2013).
Gene prediction and annotation
Based on the repeat-masked genome, we combined evidence obtained from three source (ab initio gene prediction, homolog searching and UniGene-based prediction) to predict non-redundant protein-encoding gene models. For ab initio gene prediction, Augustus (version 3.0.3) (Stanke and Morgenstern, 2005), SNAP (version 2006-07-28) (Korf, 2004), and GlimmHMM (version 3.0.4) (Majoros et al., 2004) were used to annotate genes, whereas, for the homolog-based prediction, we mapped the J. sambac genome against the published protein sequences of A. thaliana, Erythranthe guttata, O. europaea, Sesamum indicum and Vitis vinifera using Exonerate (version2.2.0, http://www.animalgenome.org/bioinfo/resources/manuals/exonerate/exonerate.man.html). To accurately identify alignments, we used GeneWise (version 2.4.1) to filter the initially aligned coding sequences (Birney et al., 2004), and for the UniGene-based prediction, Trinity (version r20140717) was used to assemble the RNA-seq data (Haas et al., 2013). Thereafter, we applied PASA software (version r20140417) to improve the gene structure (Haas et al., 2008). All three prediction methods were then integrated by EvidenceModeler (version r2012-06-25) (Haas et al., 2008). Finally, we used PASA software (Haas et al., 2008) to obtain annotation information for the 5′ and 3′ UTRs of genes, as well as variations in alternative splicing.
We performed functional annotation of the genes based on BLASTP (E-value< e-6) searches against the NCBI NR, SwissProt and eggNOG (version 4) databases (Shang et al., 2020). We determined the motifs and domains of genes using InterProScan (version 5.28) (Jones et al., 2014), whereas we determined the Gene Ontology (GO) classification of genes using InterPro or Pfam entry, and obtained KO and Pathway annotations of protein-coding genes using KAAS (Moriya et al., 2007) and the KEGG database.
Genome comparison and phylogenomic analysis
To identify orthologous genes among 15 plant genomes, the complete genome sequences of 14 other plants (A. thaliana, Begonia fuchsioides, Beta vulgaris, Betula pendula, Cercis canadensis, Durio zibethinus, Helianthus annuus, Nelumbo nucifera, Oryza sativa, Prunus mume, Solanum pennellii, V. vinifera, O. europaea and O. fragrans) from the appropriate websites were retrieved (Table S2). We used OrthoFinder (version 2.2.6) pipeline to identify gene families (Emms and Kelly, 2019), and then single-copy orthologs genes were used for MUSCLE alignment. We constructed phylogenomic tree using RAxML (version 8.2.12) (Stamatakis, 2014). We estimated divergence times among the 15 examined plant species using the program Mcmctree (version 4.0) in the PAML package (version 4.8) (Yang, 2007), with three corrected divergence times point [A. thaliana-O. sativa (148–173 Mya), A. thaliana-V. vinifera (105–115 Mya) and A. thaliana-D. zibethinus (81–94 Mya) obtained from the TimeTree website (http://www.timetree.org/)] being used to adjust the divergence times. Expansion and contraction events in gene families were computationally identified using cafe` (version 3.0) software (De Bie et al., 2006).
Whole-genome duplication analysis
We used four-fold synonymous third-codon transversion (4DTv) to estimate whole-genome duplication (WGD) events. Initially, the respective paralogs of J. sambac, V. vinifera, O. europaea and A. thaliana, and the respective orthologs of J. sambac and V. vinifera, J. sambac and O. europaea and J. sambac and A. thaliana were identified. Then, we identified the conserved paralogs and orthologs based on BLASTP (E-value< e-5) searches, and calculated WGD events based on their 4DTv values.
Aroma compounds analysis
Fresh flowers at three different stages of development (S1, young floral bud stage; S2, mature floral bud stage; S3, initial opening flower stage), defined by flower size, were picked from the same plants at the time of samples collection for transcriptome study (Figure S2). To identify aroma compounds and determine the quantity of floral scent, headspace solid-phase microextraction (HS-SPME) combined with gas chromatography-mass spectrometry (GC-MS) was used (Yang et al., 2018). The spectra obtained for volatile compounds were auto-matched based on comparisons with those in the NIST08 mass spectral library and those reported in specialized literature. The quantities of the volatile compounds were based on the normalization of peak areas.
Transcriptome libraries preparation and sequencing
To obtain information that can be used to assist gene annotation, we collected mixed tissues (leaves, stems, roots, buds and flowers at the aforementioned three developmental stages) from the J. sambac plant used for genome sequencing. We also collected flowers at the three stages from three plants at the time of sample collection for identifying aroma compounds (each with three biological replicates) (Figure S2). We isolated total RNA using Trizol Reagent (Invitrogen Life Technologies, USA). The concentration, quality and integrity of which were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). We generated sequencing libraries using a TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA), and these were subsequently sequenced with the HiSeq 2000 platform using paired-end sequencing with 150-bp reads (Table S1). We mapped the RNA-seq reads of each sample to the reference genome of J. sambac.
Identification and analysis of heat stress response and aroma compound biosynthesis genes
Using heat stress response related genes from A. thaliana (Ohama et al., 2017), terpene biosynthesis genes from A. thaliana (Vranová et al., 2013) and phenylpropanoid biosynthesis genes from Petunia hybrida (Maeda and Dudareva, 2012) as baits, the corresponding genes in J. sambac were identified based on genome annotation and local blast searches against J. sambac genome using a filtered parameter (E-value < 10−6, identity ≥ 40% and coverage ≥ 30%). TPS genes in the J. sambac genome were identified as previously reported (Song et al., 2018). Multiple sequence alignment of the amino acid sequences of TPS genes, Hsfs, MYB TFs, bHLH TFs, and WRKY TFs were performed using the default parameters of MUSCLE followed by maximum likelihood phylogenetic analyses performed using RAxML (version 8.2.12) (Stamatakis, 2014). The 3000-bp upstream region of TPS genes was defined as the promoter fragment, and the cis-acting regulatory elements within these promoter sequences were identified using PlantCARE (Lescot et al., 2002) and PLACE (Higo et al., 1999). Based on the expression levels of genes at the three different floral developmental stages, a heat map was illustrated using TBtools (Chen et al., 2018b).
Results
Determination of genome size and heterozygosity
Given genome size varies between species and cultivars, using flow cytometry, we sought to determine the J. sambac genome size using fresh leaves from the same individual used for genome sequencing (Figures S3A), and accordingly established that the size of the single-petal J. sambac genome was 583 Mb (Table S3).
The J. sambac genome size and the rate of heterozygosity were also estimated from raw short-reads sequenced using the k-mer based method. A total of 54.67 Gb short paired-end reads (2 × 150 bp) was obtained by Illumina sequencing, of which 52.36 Gb remained after filtering out low-quality reads (Tables S1, S4). The 17-mer frequency of short reads with the main peak appeared at a depth of 84 (Figure S3B). On the basis of these data, the genome size was estimated to be 555.45 Mb, and the rate heterozygosity was 0.84% (Table S5).
Sequencing and assembly of the J. sambac genome
For the J. sambac genome, we generated 54.7 Gb (~98×) Illumina and 15.9 Gb (~28×) PacBio reads (Tables S4, S6). The integrated work-flow of the genome assembly was shown in Figure S4. The assembly yielded a draft genome of 521 Mb, representing ~94% (521 Mb/555 Mb) of the estimated genome size, with contig N50 and scaffold N50 length values of 145.43 and 145.53 kb, respectively (Table 1) and 95.0% BUSCO completion (Table S7).
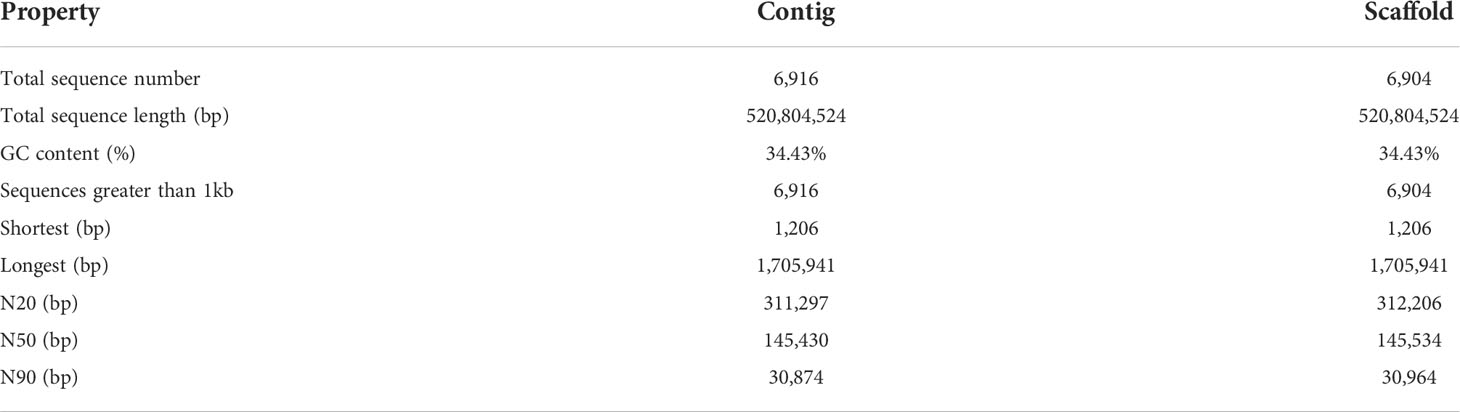
Table 1 Statistics of J. sambac cultivar JSDB genome assembly.
Annotation of the genome
On the basis of a combination of de novo annotation and homolog-based approaches, 49.01% of the J. sambac assembled genome was identified as being repetitive sequences (Table 2). It was found that most of these replicated sequences were transposable elements (TEs) (comprising 48.64% of the genome) (Table 2). Among the major types of TEs identified, long terminal repeat retrotransposons (LTR-RTs) comprised the largest proportion (accounting for 20.56% of the genome). In the genome, 11.23% of LTRs were Gypsy elements and 9.20% were Copia elements (Table 2). Unclassified repeats ranked the second most abundant, accounting for 20.24% of the genome (Table 2). In addition to the LTRs and unclassified repeats, 6.14% of the genome were annotated as DNA transposons, and 1.70% as long interspersed nuclear elements (LINEs), with the remaining repeats being assigned to other elements (Table 2). Moreover, 3,394 complete LTR-RTs were identified in the genome, and the estimated time of LTR-RT burst was approximately 0.2 million years ago (Mya) (Figure S5). Non-coding RNA (ncRNA) genes were also annotated, and accordingly we identified 261 miRNAs, 630 tRNAs, 122 rRNAs and 781 snRNAs, respectively (Table S8).
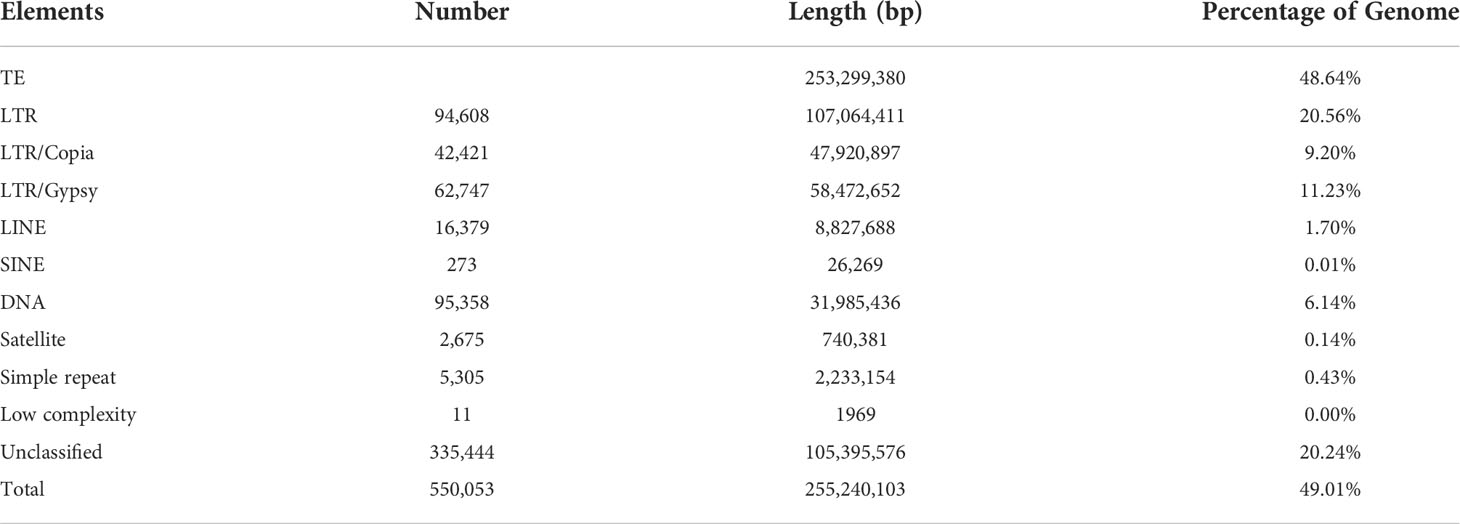
Table 2 Statistics of repetitive element contents in the J. sambac cultivar JSDB genome.
For the purposes of identifying protein-coding genes, a combination of homolog-based prediction, ab initio prediction and transcriptome-assisted prediction were used, which enabled us to predict final set of 35,363 protein-coding genes, with an average transcript length of 3,323.1 bp, an average coding sequence length of 1,025.3 bp, and an average of 4.8 exons per gene (Table S9). Among the annotated genes, 29,921 (84.6%) of the genes were functionally classified based on reference to five databases (Table S10).
Genome evolution
To investigate the evolutionary position and distinct traits of single-petal jasmine, a comparative analysis of the genome of J. sambac and 14 other plant species was performed (Table S2). On the basis of the proteomic databases, we identified 61 gene families unique to J. sambac, comprising 519 genes (Figure S6, Table S11).
To evaluate the phylogenetic relationships between J. sambac and other plant species, a phylogenomic tree based on 352 single-copy genes was constructed, and thereby we estimated the divergence times. It revealed that J. sambac was closely related to the fragrant tree (O. fragrans) and the European olive (O. europaea) (Figure 1C). The lineage giving rise to J. sambac was found to diverge from that leading to O. fragrans and O. europaea at ~31.1 Mya, and the lineage that gave rise to the Oleaceae species diverged from the lineage giving rise to S. pennellii at ~65.8 Mya (Figure 1C). Moreover, 1,541 gene families were found to have undergone an expansion, whereas 5,124 gene families have undergone contractions (Figure 1C).
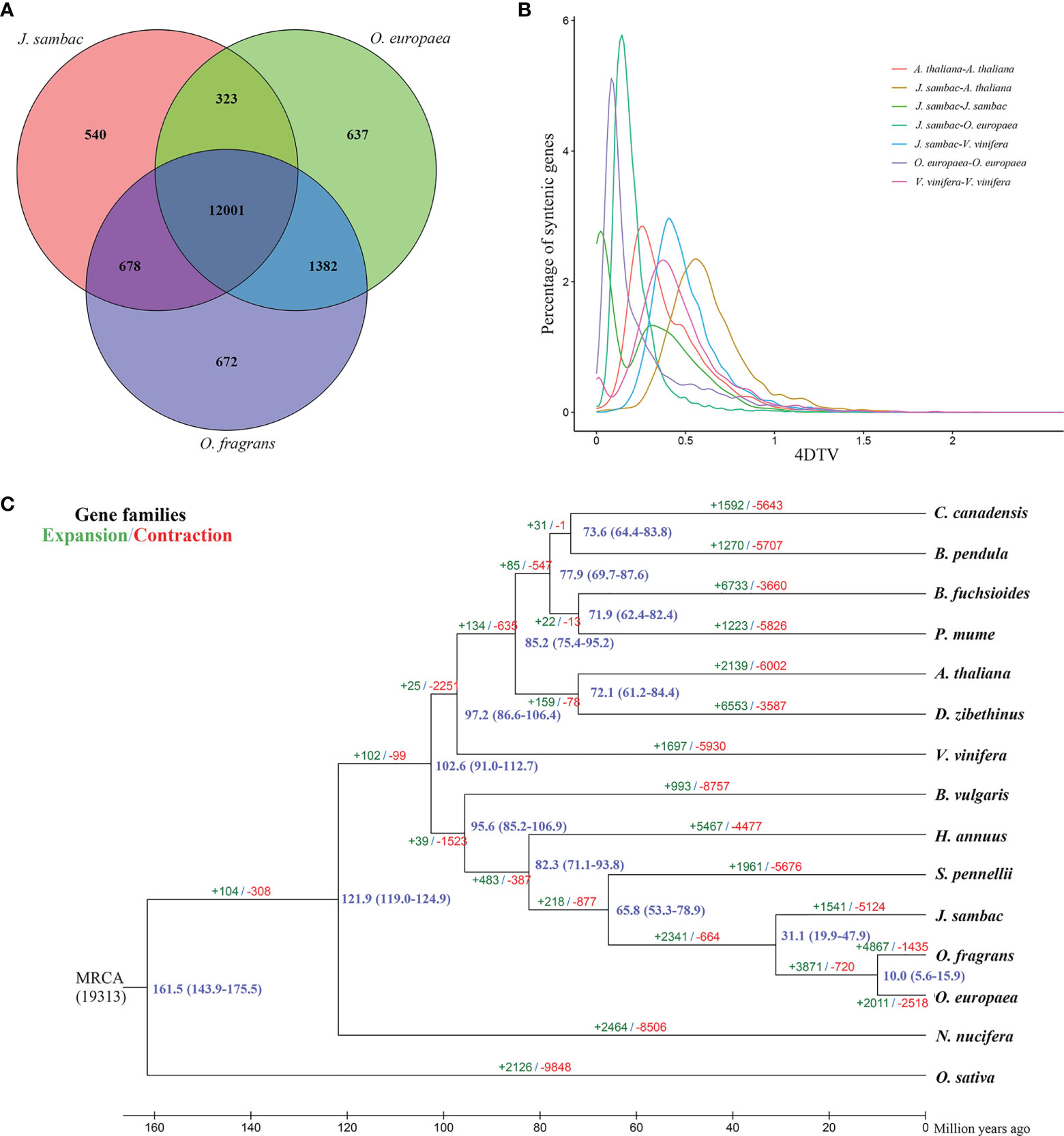
Figure 1 Evolution and comparative analysis of the J. sambac cultivar JSDB genome. (A) Venn diagram of J. sambac, O. europaea and O. fragrans. Each number in the diagram was the gene family number within a group. (B) Fourfold degenerate distributions for J. sambac, O. europaea, A. thaliana, and V. vinifera. (C) Phylogenomics relationships, divergence times and gene family expansion and contraction of 15 plant species. The blue numbers on the nodes were divergence time to present (in Mya). The green and red numbers above or under each branch denoted the expanded and contracted gene families after the diversification from the most recent common ancestor (MRCA), respectively.
A comparison of the genomes of J. sambac, O. europaea and O. fragrans revealed that 12,001 gene families were common to these Oleaceae species, whereas 540 gene families were identified as being unique to J. sambac genome (Figure 1A). We further applied 4DTv analysis to investigate J. sambac WGD events, and the analysis indicated that J. sambac has undergone only an ancient WGD event (Figure 1B).
Genes involved in heat stress
In order to reveal the heat stress tolerance mechanism of J. sambac, we identified genes related to heat stress response, and accordingly 92 transcription factors (TFs) and 206 genes were identified (Tables S12–S14). We found a set of genes had expanded in both J. sambac and H. annuus compared with A. thaliana, including dehydration-responsive element-binding protein 2 (DREB2), NAC (NAM, ATAF and CUC), squamosa-promoter binding-like (SPLs), heat shock proteins (HSP), nuclear factor Y subunit A2 (NF-YA2), nuclear factor Y subunit B3 (NF-YB3), cyclin-dependent kinase A1 (CDKA1), calmodulin-binding protein kinase 3 (CBK3), heat-intolerant 4 (HIT4), decrease in DNA methylation 1 (DDM1) and cyclic nucleotide-gated channel (CNGCs) (Tables S12, S13). Furthermore, three HsfA1s that act as the master regulators in the heat response were identified in the 17 Hsf genes (Figure 2A; Table S14). HsfA1s and DREB2A displayed the same expression pattern, and both of them had high expression levels at the S1 stage. Meanwhile, the abundance of their transcript decreased with the flower development (Figures 2B; Figure S7).
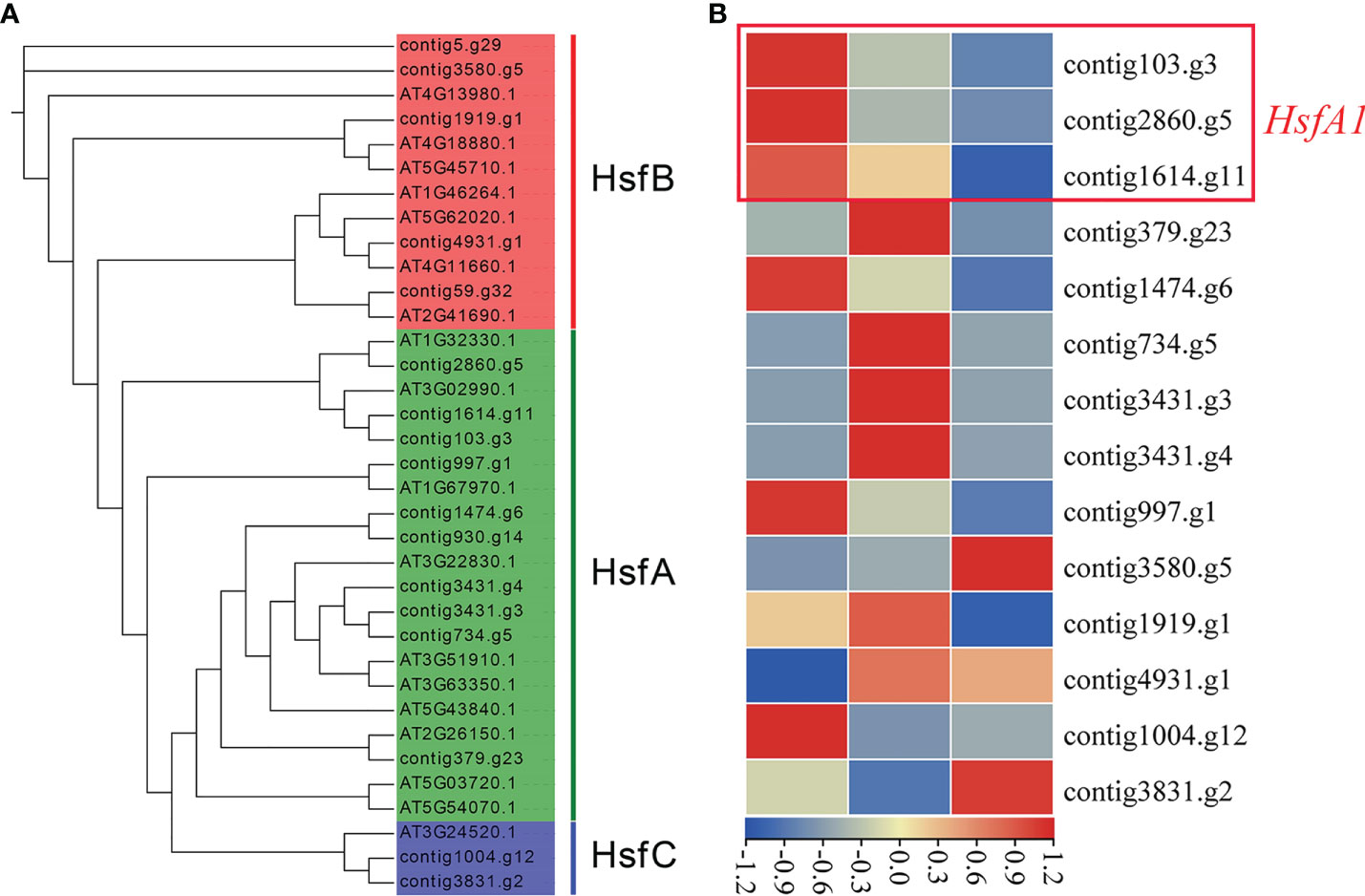
Figure 2 Phylogenetic analysis of Hsf transcriptional factors identified in the J. sambac cultivar JSDB and A. thaliana genomes (A) and the expression of Hsfs in the three floral developmental stages of J. sambac cultivar JSDB (B). S1, young floral bud stage; S2, mature floral bud stage; S3, initial opening flower stage.
Genes involved in benzenoid/phenylpropanoid biosynthesis
To establish a direct linkage between biosynthetic genes and flower fragrance development, a metabolomic approach was used to determine the aroma compounds synthesized at three flower developmental stages (Figure S2). On the basis of HS-SPME/GC-MS combined analysis, we accordingly identified over 50 aroma compounds (Figure S8; Table S15).
The results indicated that benzenoids/phenylpropanoids were the predominant components of floral volatile organic compounds (VOCs) (Table S15). We identified 16 genes involved in the shikimate pathway, and 13 genes in phenylpyruvate and arogenate pathways (Figure 3A; Table S16). Our findings indicate that WGD and tandem duplication events played prominent roles in the genes involved in the shikimate pathway, phenylpyruvate and arogenate pathways and benzenoid/phenylpropanoid pathway, which resulted in the high rate of paralog formation. To investigate the biological processes associated with these aroma compounds, we generated RNA-seq data from flowers at the aforementioned developmental stages (Table S17) and focused on those genes involved in benzenoid/phenylpropanoid synthesis pathway (Figure 3B).
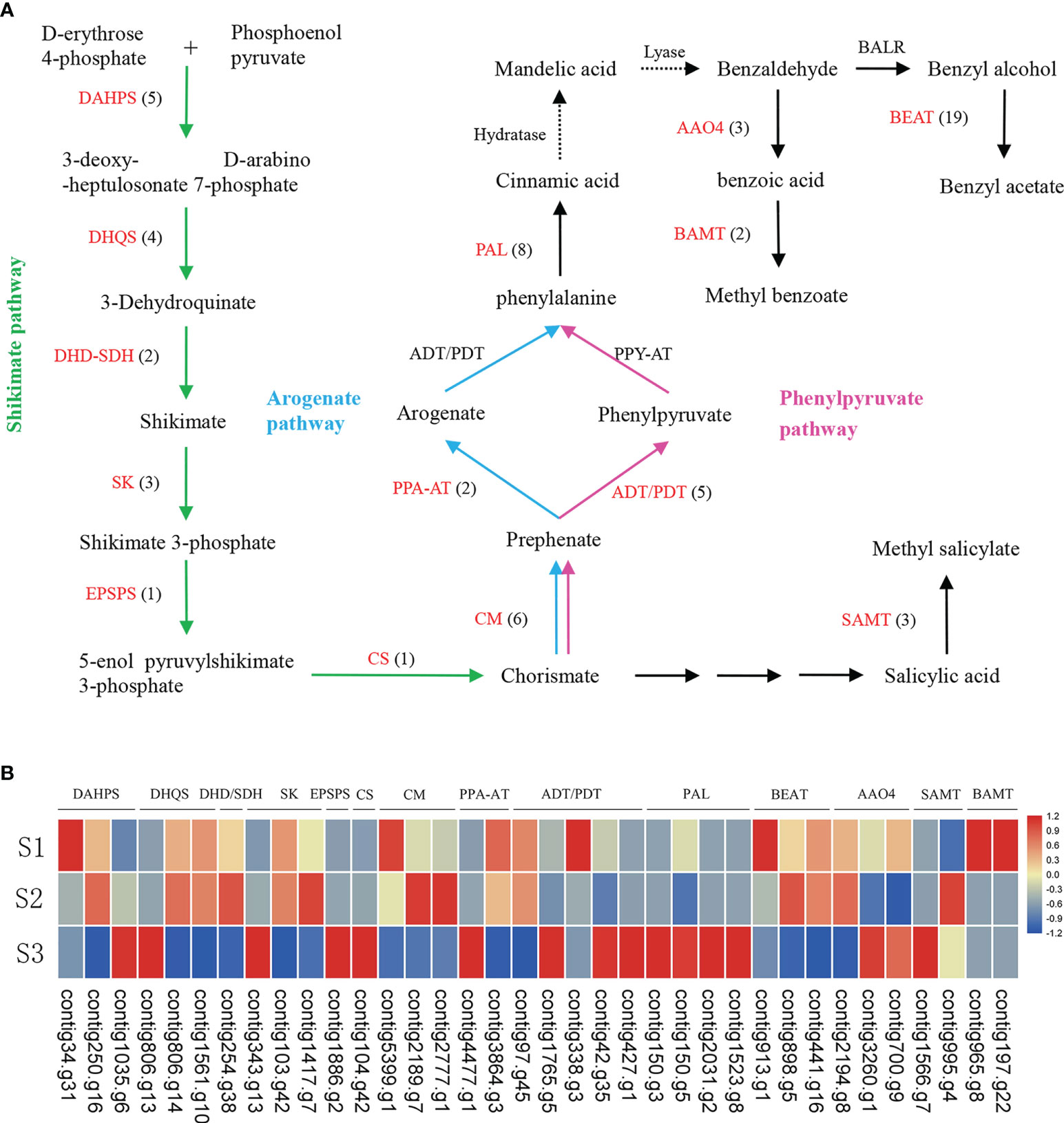
Figure 3 The biosynthesis pathways of benzenoid/phenylpropanoid (A) and expression of key genes involved in benzenoid/phenylpropanoid biosynthesis in J. sambac cultivar JSDB (B). Abbreviations for enzymes in each catalytic step were shown in red letters. The black numbers in parentheses represent the gene number in J. sambac genome. The gradient color for each gene represented the gene expression levels at three floral developmental stages (S1, young floral bud stage; S2, mature floral bud stage; S3, initial opening flower stage). These genes were expressed in at least one of the three developmental stages. DAHPS, 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase; DHQS, 3-dehydroquinate synthase; DHD/SDH, dehydroquinate dehydratase/shikimate dehydrogenase; SK, shikimate kinase; EPSPS, 3-phosphoshikimate 1-carboxyvinyltransferase; CS, chorismate synthase; CM, chorismate mutase; PPA-AT, prephenate aminotransferases; ADT/PDT, arogenate dehydratase/prephenate dehydratase; PAL, phenylalanine ammonialyase; BEAT, acetyl-CoA: benzylalcohol acetyltransferase; AAO4, arabidopsis aldehyde oxidase; BAMT, benzoic acid carboxyl methyltransferase; SAMT, salicylic acid methyltransferases.
Our analyses revealed benzyl acetate to be one the most prominent compounds among the floral VOCs in single-petal phenotype of J. sambac, the emission of which was detected at all three assessed developmental stages (Figure S8, Table S15). Benzyl acetate is synthesized from benzyl alcohol catalyzed by benzyl alcohol acetyltransferase (BEAT) via an acetyl-CoA-dependent reaction. A total of 19 BEAT genes were detected in the present J. sambac genome (Figure 3A), two genes have been generated as a consequence of a WGD event, and 10 are derived from tandem duplication (Table S16). Transcriptome analysis revealed that contig913.g1 showed a high expression level at the S1 developmental stage, but contig898.g5 were highly expressed at the S2 stage (Figure 3B). Methyl salicylate was also identified as a major component of the floral VOCs in J. sambac, and it was found to be present at both S2 and S3 stages (Figure S8, Table S15). Furthermore, we identified three salicylic acid methyltransferases (SAMTs) (Figure 3A), with contig995.g4 and contig1566.g7 being highly expressed at the S2 and S3 stages, respectively (Figure 3B). Methyl benzoate was similarly identified as a major component of jasmine floral VOCs, which was found to accumulate at stage S3 (Figure S8, Table S15). In addition, we identified two benzoic acid methyltransferases (BAMTs) (Figure 3A), and each of them were highly expressed at the S1 stage (Figure 3B).
Genes involved in terpenoid biosynthesis
We found terpenoids as the second major class of compounds among the VOCs produced by J. sambac JSDB (Table S15), so genes involved in MEP and MVA pathways were identified (Figure 4). The results showed that genes involved in both pathways, including HDR and HMGR, were generated by a WGD event (Table S18). Transcriptome analysis revealed that DXS (contig1719.g3), DXR, MCT, CMK, MCS, HDS, GGPPS (contig461.g8, contig69.g4 and contig3659.g1) and HMGR (contig549.g16, contig624.g16 and contig1594.g1) were highly expressed at the S1 stage (Figure 4), whereas HDR, GGPPS (contig3396.g1 and contig4356.g2), ACAT, MVK, PMK and IDI showed high expression at the S2 stage (Figure 4). At the same time, DXS (contig189.g8 and contig254.g35), HMGS, HMGR (contig2431.g2 and contig137.g29), MDC and FPPS tended to be more prominently expressed during the S3 stage (Figure 4).
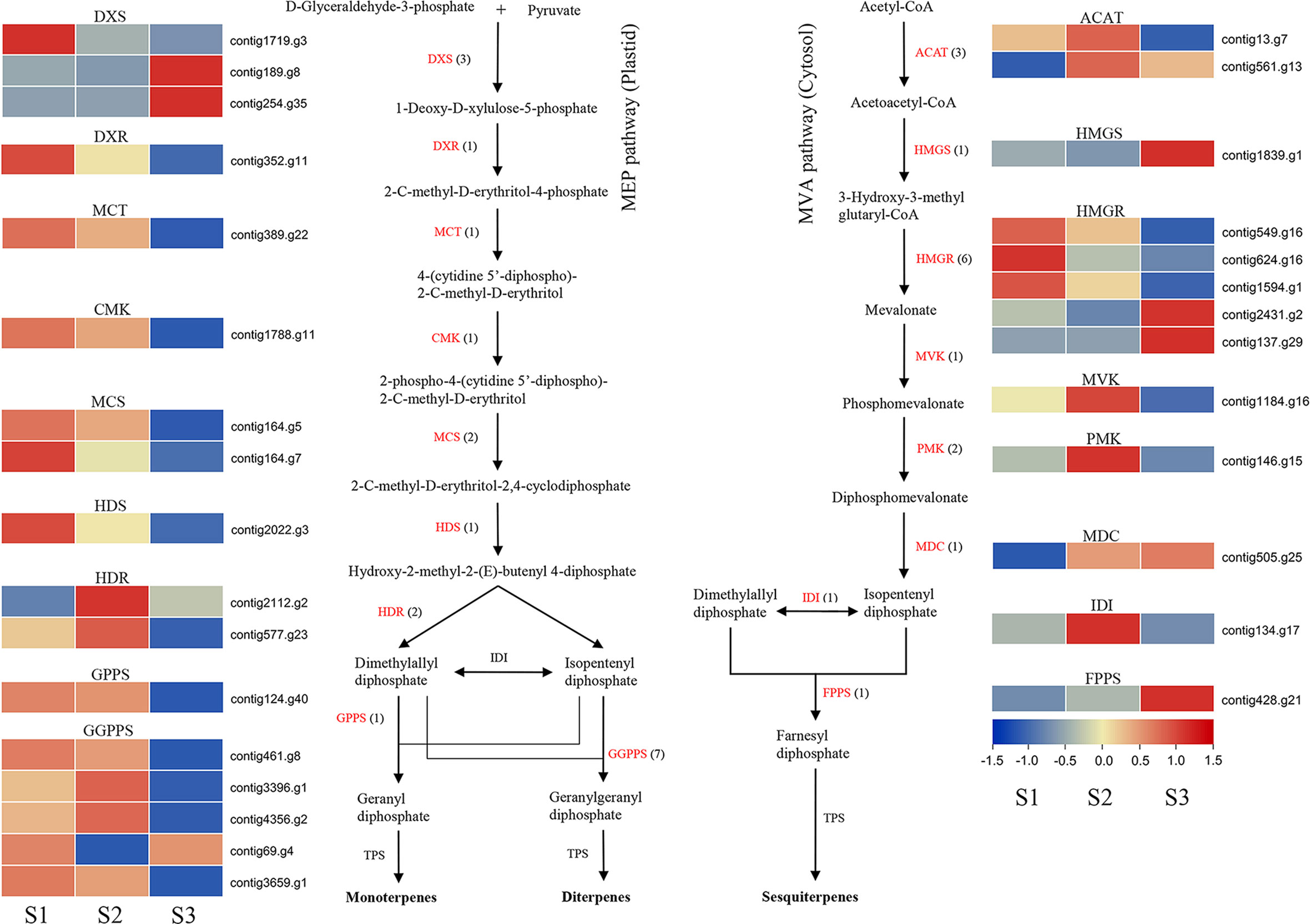
Figure 4 Expression profiles of genes encoding enzymes involved in terpene biosynthesis in J. sambac cultivar JSDB. Abbreviations for enzymes in each catalytic step were shown in red letters. The black numbers in parentheses represent the gene number in J. sambac genome. The gradient color for each gene represented the gene expression levels at three floral developmental stages (S1, young floral bud stage; S2, mature floral bud stage; S3, initial opening flower stage). DXS, 1-deoxy-D-xylulose 5-phosphate synthase; DXR, 1-deoxy-D-xylulose 5-phosphate reductoisomerase; MCT, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CMK, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; MCS, 2-C-methyl-D-erythritol-2,4-cyclodiphosphate synthase; HDS, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase; HDR, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase; GPPS, geranyl diphosphate synthase; GGPPS, geranylgeranyl diphosphate synthase; ACAT, acetyl-CoA acetyltransferase; HMGS, hydroxymethylglutaryl-CoA synthase; HMGR, hydroxymethylglutaryl-CoA reductase; MVK, mevalonate kinase; PMK, phosphomevalonate kinase; MDC, diphospho-MVA decarboxylase; IDI, isopentenyl diphosphate isomerase; FPPS, farnesyl diphosphate synthase.
In the MVA and MEP pathways, terpene synthases (TPSs) are responsible for the final catalytic reaction in the generation of terpenoid compounds. On the basis of the assembled genomes of J. sambac JSDB, we identified 31 TPSs. Phylogenetic analysis revealed that these TPS genes were clustered into five discrete groups, namely TPS-a (6), TPS-b (12), TPS-c (6), TPS-e/f (4) and TPS-g (3) (Figure 5A). Linalool was identified as the major component of monoterpenes in the aroma compounds of J. sambac JSDB, and was found to be accumulated at the S3 stage (Figure S8 and Table S15). We identified two linalool synthase genes in the present genome, and each of them was highly expressed at the S1 stage (Figure 5B). β-Ocimene was another major monoterpene in J. sambac JSDB and presented in the S3 stage, and it was synthesized by contig203.g8 (Figure 5B and Table S15). The contig203.g8 showed a high expression level at the S3 stage, and its expression pattern was consistent with β-Ocimene emission (Figure 5B; Table S15). Caryophyllene was found to be the major sesquiterpene in J. sambac JSDB, and the emission of which was detected at stages S2 and S3 (Figure S8 and Table S15). Furthermore, we identified three caryophyllene synthase genes, among which, contig2155.g2 and contig1096.g3 were characterized by high expression levels at the S2 stage, while contig445.g1 was highly expressed at the S3 stage (Figure 5B).
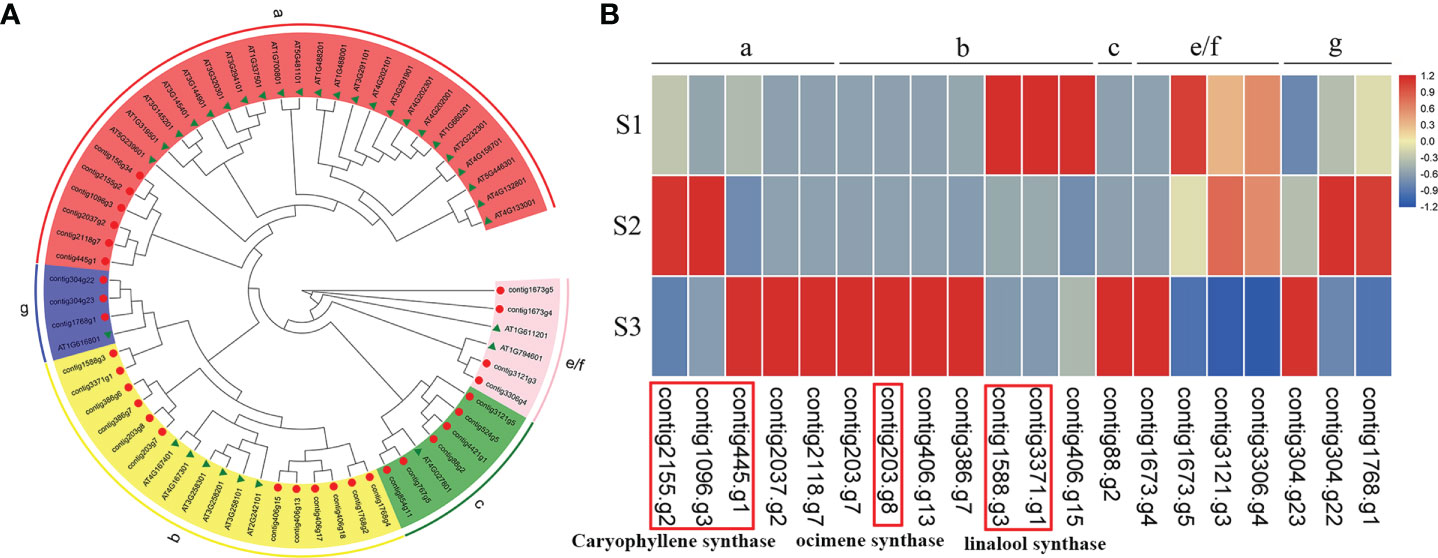
Figure 5 The phylogenetic tree and expression of TPS genes in J. sambac cultivar JSDB. (A) The maximum likelihood phylogenetic tree of TPS protein identified in J. sambac. J. sambac (red circle) and A. thaliana (green triangle) were shown in the tree with corresponding gene ID, respectively. (B) The expression of TPSs at three different floral developmental stages (S1, young floral bud stage; S2, mature floral bud stage; S3, initial opening flower stage). Note the genes were expressed in at least one stage.
In addition to TPS genes, we identified a group TFs involved in the biosynthesis of terpenes (Shang et al., 2020). Screening of the 3000-bp regions upstream of all TPS genes in J. sambac revealed that defense and stress responsive elements, including MYB-, bHLH-, and WRKY-binding elements, were significantly enriched (Table S19). In the newly assembled J. sambac genome, we identified a total of 122 MYB, 122 bHLH, and 69 WRKY genes (Figures S9A, S10, S12A). R2R3-MYB subgroup 4, 6, and 7 genes have been shown to regulate phenylpropanoid metabolism in different species (Dubos et al., 2010). In the present J. sambac genome, we identified eight genes from the subgroups 4, 6, and 7 (Figure S9A), and transcriptional analysis revealed that among these genes, the subgroup 4 genes were highly expressed at the S3 stage, whereas the expression of subgroup 6 and 7 genes was more pronounced at stage S1 (Figure S9B). Similarly, most of the bHLH and WRKY TFs were highly expressed at the S1 stage (Figures S11, S12B).
Discussion
J. sambac is one of the most popularly cultivated ornamental plant species in many countries. It has been cultivated for over 2,000 years in China because of its attractive flower scent, its usage in traditional Chinese medicine, and high value in the famous ‘jasmine tea’. In the study on J. sambac cultivar ‘Trifoliatum’ genome assembly, a set of Nanopore long reads (49.00 Gb, ~96×), Illumina paired-end short reads (55.97 Gb, ~110×) and Hi-C data (46.5 Gb, ~91×) were generated (Xu et al., 2021). In another study, they generated 15.35 Gb of HiFi PacBio (single-petal jasmine) and 12.11 Gb of PacBio Sequel (double-petal jasmine) with an estimated coverage depth of over 30-folds for both genomes (Wang et al., 2022). Here, a draft genome of J. sambac cultivar JSDB comprising 520.80 Mb was assembled based on Pacbio and Illumination sequencing technologies. Furthermore, combining genomic and transcriptomic analyses, we gained deep insight into heat stress tolerance and aroma compound biosynthesis in the single-petal flowers of J. sambac.
TEs play a significant role in genome expansion and evolution, which could lead to an increase in genome size (Bennetzen and Wang, 2014). Based on transposition mechanism, TEs can be broadly divided into two major classes, namely, class I (retrotransposons) and class II (DNA transposons) (Levin and Moran, 2011). Of these, retrotransposons, especially the LTR-RT class, are the most abundant in plant genomes (Wicker et al., 2007). In this study, numerous repetitive elements in the genome (49.01%) were detected, among which, LTR retrotransposons (accounting for 20.56% of the repetitive elements) were the most abundant (Table 2). High content of repetitive elements is a common characteristic among the genomes of plant species. For example, in the Lonicera japonica genome, transposable elements occupy 58.2% of the genome, of which 45.6% being LTRs (Pu et al., 2020). Similarly, 53.27% of the Isatis indigotica genome is repetitive sequences, with LTRs constituting 30.09% (Kang et al., 2020). However, the repetitive elements in the genome of J. sambac cultivar JSDB was higher than that of the J. sambac cultivar ‘Trifoliatum’ (Xu et al., 2021). In this study, we detected a recent burst of LTR-RTs in the genome (Figure S5), a phenomenon that has also been identified in the genomes of Lonicera japonica, Chrysanthemum nankingense and Chimonanthus praecox (Song et al., 2018; Pu et al., 2020; Shang et al., 2020). Consequently, these findings indicate that LTR-RTs may make an important contribution to an increase in the genome size of J. sambac.
Previous chloroplast genome and genome analyses have placed J. sambac in the Oleaceae family (Qi et al., 2020; Xu et al., 2021). Here, a phylogenomic tree based on a comparison of the J. sambac genome with that of 14 other plant species was constructed (Figure 1C). We accordingly established that J. sambac is closely related to O. fragrans and O. europaea, belonging to the Oleaceae family (Unver et al., 2017; Yang et al., 2018). WGD plays a central role in plant genome evolution, as it generally leads to a sudden increase in genome size (Panchy et al., 2016; Wendel et al., 2016). Previous research showed that there are two WGDs in O. europaea (Unver et al., 2017). In this study, we revealed that the J. sambac JSDB genome has only undergone an ancient WGD event, and there was a recent WGD event in the O. europaea genome that distinguishes O. europaea from J. sambac (Figure 1B). Therefore, this is a plausible explanation for the size of O. europaea genome is larger than J. sambac genome (1.48 Gb vs 520.8 Mb). Thus, the present sequencing of the JSDB genome provides significant insights for further genomic studies on J. sambac, including phenotypic diversity and evolution.
Heat stress generally damages photosynthetic activity and reduces cell division and compromises plant growth (Hasanuzzaman et al., 2013). So it is important to elucidate the molecular mechanism involved in the heat stress response. Here, we identified 92 TFs and 206 genes related to heat stress tolerance in J. sambac JSDB, and we also found that some gene families expanded in J. sambac (Tables S12, S13). These will provide useful information for studying the complex transcriptional regulatory networks involved in heat stress response in J. sambac. As well known, heat shock transcription factors (Hsf) played a critical role in heat stress response, and HsfA1s served as ‘master regulators’ during the process (Ohama et al., 2017). In Arabidipsis and tomato, the mutation of HsfA1s genes resulted in the reduced induction of heat stress responsive genes and heat stress sensitive phenotypes (Mishra et al., 2002; Yoshida et al., 2011; Fragkostefanakis et al., 2016). DREB2A is a plant-specific TF involved in heat stress response, which is directly regulated by HsfA1s genes (Ohama et al., 2016). In the present study, both HsfA1s and DREB2A had highly expressed levels in the S1 stage, and both of them displayed a similar expression profile (Figures 2, S7). These indicated that HsfA1s and DREB2A perhaps play a vital role in jasmine’s heat stress response.
Basing on the assembled genome, and using the terpenoid biosynthesis genes in A. thaliana and phenylpropanoid biosynthesis genes in Petunia hybrida as baits, the homologs genes in J. sambac were identified, which revealed a notable duplication of particular genes in J. sambac, particularly PAL, BEAT and TPS genes (Tables S16, S19). In the Chimonanthus praecox genome, it has been established that the expansion of BEAT and TPS genes were attributed to tandem duplication (Shang et al., 2020). Therefore, it can be reasonably speculated that a tandem duplication event has influenced the evolution of PAL, BEAT and TPS genes in J. sambac JSDB.
To analyze the genes that contribute to aroma compound biosynthesis in J. sambac, we performed comparative transcriptome analysis in combination with metabolome studies. PAL enzymes play a vital role in the initial step of the phenylpropanoid pathway by deaminating L-Phe to yield trans-cinnamic acid (Achnine et al., 2004). In this study, there were four PAL genes highly expressed at the S3 stage (Figure 3B), a pattern of expression that is similar to a former report (Bera et al., 2017). We detected that methyl benzoate accumulated at the S3 stage, whereas the two identified BAMT genes were highly expressed at the S1 stage (Figure 3B, Table S15), indicating that these genes may be actively expressed at the S1 stage in preparation for the following release of methyl benzoate during the S3 stage. The production and diversification of terpenes is mainly determined by the TPS family genes and their transcription levels (Chen et al., 2011; Vranová et al., 2013). In this study, we revealed a dynamic expression of TPSs (Figure 5C), which may account for the observed diversification of the terpene profiles. Within the TPS gene family, the members of the TPS-b subfamily are involved in the synthesis of monoterpenes (Tholl, 2006; Chen et al., 2011). In the present study, we also established that two linalool synthase genes were highly expressed at the stage S1, whereas linalool was observed to be accumulated at stage S3 (Figures 5, S8, Table S15). This is consistent with the findings of previous studies that have revealed a marked increase in linalool levels coinciding with the initial opening of jasmine flowers (Yu et al., 2017), thereby indicating that these genes are highly expressed at the S1 stage preparing for the release of linalool during stage S3. However, the monoterpene β-Ocimene was only detected in the S3 stage. This is consistent with the expression pattern of the gene (TPS-b subfamily) being responsible for β-Ocimene biosynthesis (Figure 5B, Table S15).
TFs, including those in the MYB (Reddy et al., 2017), bHLH (Hong et al., 2012) and WRKY (Spyropoulou et al., 2014) families, are involved in the regulation of terpenoid biosynthesis (Shang et al., 2020). In the present study, we detected multiple MYB-, bHLH- and WRKY-binding elements in the promoter sequences of JsTPS genes (Table S19). Our results indicate that these TFs may play a key role in regulating the expression of TPS genes and will provide valuable information for further studies, in which intend to examine the co-expression patterns of TFs and aroma compound pathway genes.
Conclusion
In summary, in the present study, we generated a draft genome of a single-petal phenotype of J. sambac, a culturally and commercially important plant in the Oleaceae family. The newly assembled genome will provide a solid foundation for further research of resistance to abiotic stresses, aroma compound biosynthesis and genomic evolution. Moreover, the genome will contribute to gain a more comprehensive understanding of the molecular mechanisms underlying heat stress tolerance, flower development and its scent formation.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI, BioProject PRJNA690159 and BioSample SAMN17245799. The genome assembly and annotation files are available publicly at FigShare (https://doi.org/10.6084/m9.figshare.17030054.v1).
Author contributions
YD and XQ managed and organized the project. HW and HC collected plant samples and performed experiments. XQ and SC contributed to genome assembly, genome annotation and evolutionary analyses. XQ, HW, JF and ZQ analyzed the data. XQ, IB and YD wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant No.: 31772338), and the Basic Scientific Research Business Special Project of Jiangsu Academy of Agricultural Sciences (0090756100ZX).
Acknowledgments
We thank the staffs of the Central Laboratory at Jiangsu Academy of Agricultural Sciences for their help in HS-SPME and GC-MS analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1045194/full#supplementary-material
References
Achnine, L., Blancaflor, E. B., Rasmussen, S., Dixon, R. A. (2004). Colocalization of l-phenylalanine ammonia-lyase and cinnamate 4-hydroxylase for metabolic channeling in phenylpropanoid biosynthesis. Plant Cell 16, 3098–3109. doi: 10.1105/tpc.104.024406
Bennetzen, J. L., Wang, H. (2014). The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505. doi: 10.1146/annurev-arplant-050213-035811
Bera, P., Mukherjee, C., Mitra, A. (2017). Enzymatic production and emission of floral scent volatiles in Jasminum sambac. Plant Sci. 256, 25–38. doi: 10.1016/j.plantsci.2016.11.013
Birney, E., Clamp, M., Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Cai, H., Biswas, D. K., Shang, A. Q., Zhao, L. J., Li, W. D. (2007). Photosynthetic response to water stress and changes in metabolites in Jasminum sambac. Photosynthetica 45, 503–509. doi: 10.1007/s11099-007-0087-0
Chen, F., Dong, W., Zhang, J. W., Guo, X. Y., Chen, J. H., Wang, Z. J., et al. (2018a). The sequenced angiosperm genomes and genome databases. Front. Plant Sci. 9, 418. doi: 10.3389/fpls.2018.00418
Chen, F., Song, Y. F., Li, X. J., Chen, J. H., Mo, L., Zhang, X. T., et al. (2019). Genome sequences of horticultural plants: past, present, and future. Hortic. Res. 6, 112. doi: 10.1038/s41438-019-0195-6
Chen, F., Tholl, D., Bohlmann, J., Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Chen, C. J., Xia, R., Chen, H., He, Y. H. (2018b). TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv, 289660. doi: 10.1101/289660
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Degenhardt, J., Köllner, T. G., Gershenzon, J. (2009). Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70, 1621–1637. doi: 10.1016/j.phytochem.2009.07.030
Deng, Y. M., Jia, X. P., Liang, L. J., Gu, C. S., Sun, X. B. (2016). Morphological anatomy, sporogenesis and gametogenesis in flowering process of jasmine (Jasminum sambac aiton). Sci. Hortic. 198, 257–266. doi: 10.1016/j.scienta.2015.11.036
Deng, Y. M., Sun, X. B., Gu, C. S., Jia, X. P., Liang, L. J., Su, J. L. (2017). Identification of pre-fertilization reproductive barriers and the underlying cytological mechanism in crosses among three petal-types of Jasminum sambac and their relevance to phylogenetic relationships. PLos One 12, e0176026. doi: 10.1371/journal.pone.0176026
Doležel, J., Greilhuber, J., Suda, J. (2007). Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244. doi: 10.1038/nprot.2007.310
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., Lepiniec, L. (2010). MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. doi: 10.1016/j.tplants.2010.06.005
Ellinghaus, D., Kurtz, S., Willhoeft, U. (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinf. 9, 18. doi: 10.1186/1471-2105-9-18
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Fragkostefanakis, S., Mesihovic, A., Simm, S., Paupiere, M. J., Hu, Y. J., Paul, P., et al. (2016). HsfA2 controls the activity of developmentally and stress-regulated heat stress protection mechanisms in tomato male reproductive tissues. Plant Physiol. 170, 2461–2477. doi: 10.1104/pp.15.01913
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Hasanuzzaman, M., Nahar, K., Alam, M. M., Roychowdhury, R., Fujita, M. (2013). Physiological, biochemical, and molecular mechanisms of heat stress tolerance in plants. Int. J. Mol. Sci. 14, 9643–9684. doi: 10.3390/ijms14059643
Higo, K., Ugawa, Y., Iwamoto, M., Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Hong, G. J., Xue, X. Y., Mao, Y. B., Wang, L. J., Chen, X. Y. (2012). Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 24, 2635–2648. doi: 10.1105/tpc.112.098749
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W. Z., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kang, M. H., Wu, H. L., Yang, Q., Huang, L., Hu, Q. J., Ma, T., et al. (2020). A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine. Hortic. Res. 7, 18. doi: 10.1038/s41438-020-0240-5
Kapitonov, V. V., Jurka, J. (2008). A universal classification of eukaryotic transposable elements implemented in repbase. Nat. Rev. Genet. 9, 411–412. doi: 10.1038/nrg2165-c1
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Peer, Y. V. D., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Levin, H. L., Moran, J. V. (2011). Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615–627. doi: 10.1038/nrg3030
Li, L. F., Cushman, S. A., He, Y. X., Li, Y. (2020). Genome sequencing and population genomics modeling provide insights into the local adaptation of weeping forsythia. Hortic. Res. 7, 130. doi: 10.1038/s41438-020-00352-7
Liu, B. H., Shi, Y. J., Yuan, J. Y., Hu, X. S., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 1308, 1–47. doi: 10.48550/arXiv.1308.2012
Li, J. L., Wang, S., Yu, J., Wang, L., Zhou, S. L. (2013). A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48, 72–78. doi: 10.3724/SP.J.1259.2013.00072
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. doi: 10.1186/2047-217X-1-18
Maeda, H., Dudareva, N. (2012). The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu. Rev. Plant Biol. 63, 73–105. doi: 10.1146/annurev-arplant-042811-105439
Majoros, W. H., Pertea, M., Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Marcais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Ma, Q. Y., Sun, T. L., Li, S. S., Wen, J., Zhu, L., Yin, T. M., et al. (2020). The Acertruncatum genome provides insights into the nervonic acid biosynthesis. Plant J. 104, 662–678. doi: 10.1111/tpj.14954
Mishra, S. K., Tripp, J., Winkelhaus, S., Tschiersch, B., Theres, K., Nover, L., et al. (2002). In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 16, 1555–1567. doi: 10.1101/gad.228802
Mittler, R., Finka, A., Goloubinoff, P. (2012). How do plants feel the heat? Trends Biochem. Sci. 37, 118–125. doi: 10.1016/j.tibs.2011.11.007
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, 182–185. doi: 10.1093/nar/gkm321
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ohama, N., Kusakabe, K., Mizoi, J., Zhao, H. M., Kidokoro, S., Koizumi, S., et al. (2016). The transcriptional cascade in the heat stress response of arabidopsis is strictly regulated at the level of transcription factor expression. Plant Cell 28, 181–201. doi: 10.1105/tpc.15.00435
Ohama, N., Sato, H., Shinozaki, K., Yamaguchi-Shinozaki, K. (2017). Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22, 53–65. doi: 10.1016/j.tplants.2016.08.015
Olofsson, L., Engström, A., Lundgren, A., Brodelius, P. E. (2011). Relative expression of genes of terpene metabolism in different tissues of Artemisia annua l. BMC Plant Biol. 11, 45. doi: 10.1186/1471-2229-11-45
Panchy, N., Lehti-Shiu, M., Shiu, S. H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316. doi: 10.1104/pp.16.00523
Parmesan, C., Yohe, G. (2003). A globally coherent fingerprint of climate change impacts across natural systems. Nature 421, 37–42. doi: 10.1038/nature01286
Pu, X. D., Li, Z., Tian, Y., Gao, R. R., Hao, L. J., Hu, Y. T., et al. (2020). The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytol. 227, 930–943. doi: 10.1111/nph.16552
Qian, Y. C., Lynch, J. H., Guo, L. Y., Rhodes, D., Morgan, J. A., Dudareva, N. (2019). Completion of the cytosolic post-chorismate phenylalanine biosynthetic pathway in plants. Nat. Commun. 10, 15. doi: 10.1038/s41467-018-07969-2
Qi, X. Y., Chen, S. S., Wang, Y. J., Feng, J., Wang, H. D., Deng, Y. M. (2020). Complete chloroplast genome of Jasminum sambac l. (Oleaceae). Braz. J. Bot. 43, 855–867. doi: 10.1007/s40415-020-00638-z
Reddy, V. A., Wang, Q., Dhar, N., Kumar, N., Venkatesh, P. N., Rajan, C., et al. (2017). Spearmint R2R3-MYB transcription factor MsMYB negatively regulates monoterpene production and suppresses the expression of geranyl diphosphate synthase large subunit (MsGPPS.LSU). Plant Biotechnol. J. 15, 1105–1119. doi: 10.1111/pbi.12701
Schubert, M., Lindgreen, S., Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88. doi: 10.1186/s13104-016-1900-2
Shang, J. Z., Tian, J. P., Cheng, H. H., Yan, Q. M., Li, L., Jamal, A., et al. (2020). The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 21, 200. doi: 10.1186/s13059-020-02088-y
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Sollars, E. S. A., Harper, A. L., Kelly, L. J., Sambles, C. M., Ramirez-Gonzalez, R. H., Swarbreck, D., et al. (2017). Genome sequence and genetic diversity of European ash trees. Nature 541, 212–216. doi: 10.1038/nature20786
Song, C., Liu, Y. F., Song, A. P., Dong, G. Q., Zhao, H. B., Sun, W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant 11, 1482–1491. doi: 10.1016/j.molp.2018.10.003
Spyropoulou, E. A., Haring, M. A., Schuurink, R. C. (2014). RNA Sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genomics 15, 402. doi: 10.1186/1471-2164-15-402
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, 465–467. doi: 10.1093/nar/gki458
Steinbiss, S., Willhoeft, U., Gremme, G., Kurtz, S. (2009). Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013. doi: 10.1093/nar/gkp759
Tholl, D. (2006). Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 9, 297–304. doi: 10.1016/j.pbi.2006.03.014
Unver, T., Wu, Z. Y., Sterck, L., Turktas, M., Lohaus, R., Li, Z., et al. (2017). Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 114, E9413–E9422. doi: 10.1073/pnas.1708621114
Vranová, E., Coman, D., Gruissem, W. (2013). Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700. doi: 10.1146/annurev-arplant-050312-120116
Wang, P. J., Fang, J. P., Lin, H. Z., Yang, W. W., Yu, J. X., Hong, Y. P., et al (2022). Genomes of single- and double-petal jasmines (Jasminum sambac) provide insights into their divergence time and structural variations. Plant Biotechnol. J., 1–3. doi: 10.1111/pbi.13820
Wendel, J. F., Jackson, S. A., Meyers, B. C., Wing, R. A. (2016). Evolution of plant genome architecture. Genome Biol. 17, 37. doi: 10.1186/s13059-016-0908-1
Wicker, T., Sabot, F. O., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 10, 276. doi: 10.1038/nrg2165-c4
Wikee, S., Cai, L., Pairin, N., Mckenzie, E. H. C., Su, Y. Y., Chukeatirote, E., et al. (2011). Colletotrichum species from jasmine (Jasminum sambac). Fungal Divers. 46, 171–182. doi: 10.1007/s13225-010-0049-x
Xu, S. X., Ding, Y. L., Sun, J. T., Zhang, Z. Q., Wu, Z. Y., Yang, T. Z., et al. (2021). A high-quality genome assembly of Jasminum sambac provides insight into floral trait formation and oleaceae genome evolution. Mol. Ecol. Resour. 00, 1–16. doi: 10.1111/1755-0998.13497
Xu, Z., Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268. doi: 10.1093/nar/gkm286
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, X. L., Yue, Y. Z., Li, H. Y., Ding, W. J., Chen, G. W., Shi, T. T., et al. (2018). The chromosome-level quality genome provides insights into the evolution of the biosynthesis genes for aroma compounds of Osmanthus fragrans. Hortic. Res. 5, 72. doi: 10.1038/s41438-018-0108-0
Ye, C. X., Hill, C. M., Wu, S. G., Ruan, J., Ma, Z. S. (2016). DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 6, 31900. doi: 10.1038/srep31900
Yoshida, T., Ohama, N., Nakajima, J., Kidokoro, S., Mizoi, J., Nakashima, K., et al. (2011). Arabidopsis HsfA1 transcription factors function as the main positive regulators in heat shock-responsive gene expression. Mol. Genet. Genomics 286, 321–332. doi: 10.1007/s00438-011-0647-7
Keywords: genome evolution, heat stress, benzenoid/phenylpropanoid biosynthesis, terpenoid biosynthesis, terpene synthase
Citation: Qi X, Wang H, Chen S, Feng J, Chen H, Qin Z, Blilou I and Deng Y (2022) The genome of single-petal jasmine (Jasminum sambac) provides insights into heat stress tolerance and aroma compound biosynthesis. Front. Plant Sci. 13:1045194. doi: 10.3389/fpls.2022.1045194
Received: 15 September 2022; Accepted: 03 October 2022;
Published: 19 October 2022.
Edited by:
Tangchun Zheng, Beijing Forestry University, ChinaReviewed by:
Liangsheng Zhang, Zhejiang University, ChinaGuogui Ning, Huazhong Agricultural University, China
Copyright © 2022 Qi, Wang, Chen, Feng, Chen, Qin, Blilou and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanming Deng, ZGVuZ3ltQGphYXMuYWMuY24=