
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 17 November 2022
Sec. Plant Development and EvoDevo
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1025497
This article is part of the Research Topic Somatic Embryogenesis: 60 Years of Research Applied to Plant Cloning to Unravel Plant Totipotency, Volume II View all 12 articles
 Huangying Shu1,2,3
Huangying Shu1,2,3 Yu Zhang1Chengyao He1
Yu Zhang1Chengyao He1 Muhammad Ahsan Altaf1Yuanyuan Hao1Daolong Liao1,4Lin Li1Caichao Li1Huizhen Fu1,2Shanhan Cheng1,2
Muhammad Ahsan Altaf1Yuanyuan Hao1Daolong Liao1,4Lin Li1Caichao Li1Huizhen Fu1,2Shanhan Cheng1,2 Guopeng Zhu1,2
Guopeng Zhu1,2 Zhiwei Wang1,2,3*
Zhiwei Wang1,2,3*Regeneration is extremely important to pepper genetic development; however, the molecular mechanisms of how the callus reactivates cell proliferation and promotes cell reprogramming remain elusive in pepper. In the present study, C. baccatum (HNUCB81 and HNUCB226) and C. chinense (HNUCC22 and HNUCC16) were analyzed to reveal callus initiation by in vitro regeneration, histology, and transcriptome. We successfully established an efficient in vitro regeneration system of two cultivars to monitor the callus induction of differential genotypes, and the regenerated plants were obtained. Compared to C. chinense, there was a higher callus induction rate in C. baccatum. The phenotype of C. baccatum changed significantly and formed vascular tissue faster than C. chinense. The KEGG enrichment analysis found that plant hormone transduction and starch and sucrose metabolism pathways were significantly enriched. In addition, we identified that the WOX7 gene was significantly up-regulated in HNUCB81 and HNUCB226 than that in HNUCC22 and HNUCC16, which may be a potential function in callus formation. These results provided a promising strategy to improve the regeneration and transformation of pepper plants.
A genetic transformation is an important tool for functional genome research and an effective technique for crop breeding (Altpeter et al., 2016). The method of plant transformation depends on plant tissue culture in vitro, which is an initial step of plant transformation. However, Capsicum spp. is a recalcitrant plant that has some obstacles in tissue culture and organ differentiation in vitro (Kothari et al., 2010), which is a major bottleneck in pepper transformation. Therefore, establishing an efficient regeneration system is extremely vital for pepper genetic development.
Plants have a complex physiological and molecular process for dealing with regeneration (Ikeuchi et al., 2013). Many regulated genes have been extensively characterized, which play an essential role in the regeneration of plants. For example, previous studies revealed that phytohormones play a key role in callus induction, and auxin-related genes are regarded as an important regulator in regeneration, including Gretchen Hagen 3 (GH3), small auxin upregulated RNA (SAUR), and Auxin/Indole-3-Acetic Acid (AUX/IAA) (Méndez-Hernández et al., 2019). The transcription factor (TFs) WUSCHEL (WUS) was strongly expressed in some callus. It was found that overexpression of WUS generated callus and somatic embryos in Arabidopsis. Moreover, WUS interactions with CLV3 establish a feedback loop between the stem cells (Zuo et al., 2002; Iwase et al., 2011). Interestingly, multiple TFs have been postulated involving somatic callus competence, including LATERAL ORGAN BOUNDARIES DOMAIN (LBD) (Fan et al., 2012), WOUND INDUCED DEDIFFERENTIATION1 (WIND1), bud regeneration enhancer 1 (ESR1) (Iwase et al., 2017), Baby Boom (BBM) (Heidmann et al., 2011; Chen et al., 2022), and WUSCHEL RELATED HOMEOBOX (WOX) (Ikeuchi et al., 2021). In addition, studies have shown that DNA methylation and histone might be both associated with gene expression, and this mechanism could control cell differentiation and dedifferentiation (Ikeuchi et al., 2015).
Pepper (Capsicum spp.) is one of the most important crops and is widely applied for seasoning, pharmaceuticals, and cosmetics (Kothari et al., 2010). Pepper is cultivated worldwide because of its superior adaptability (Qin et al., 2014). The research on the pepper reference genome has accelerated the identification of genes related to important biological processes (Kim et al., 2017). A large number of studies have been widely performed to optimize the protocol of callus regeneration and transformation of various pepper species (Lee et al., 2004; Sanatombi and Sharma, 2008; Sanatombi and Sharma, 2012). However, as pepper is highly recalcitrant for regeneration and transformation, the underlying mechanism remains unclear. Therefore, it is necessary to reveal the key regulatory network of pepper regeneration.
This study constructed the regeneration system of C. baccatum and C. chinense and obtained the regenerated plants by in vitro regeneration. To find key regulated genes to the phase change of cotyledon petiole explants and callus initiation in pepper, we sought to perform histologically, and transcriptome analysis of four inbred lines with different organogenesis rates. In the process of in vitro regeneration, we found a phase transition phenomenon of pepper explants. Besides, we identified some enrichment pathways involved in organogenesis. Several candidate genes were described related to auxin transport, metabolism, and cell development, which have been previously identified to be associated with regeneration. Our findings provide an important molecular framework that supplies in-depth insight into the genetics of pepper regeneration.
6-Benzylaminopurine (6-BA) and 3-Indoleacetic acid (IAA) were applied to detect the optimum concentration of exogenous hormones for somatic callus induction. Cotyledons with petioles of 15-20 days seedlings were used as explants. In media B6 (MS+ 13.32µM 6-BA+ 2.85µM IAA), the induction rates of HNUCB81 and HNUCB226 were the highest (72.58% and 66.22%), and the best induction media for HNUCC22 and HNUCC16 was B4 (MS+ 13.32µM 6-BA+ 0.57µM IAA), and the induction rates were 48% and 37%, respectively. Compared with C. chinense in the B1 media, C. baccatum induction rate was slightly higher (57.89% and 52.86%) (Figure 1A and Supplementary Table S1). Furthermore, we induced adventitious buds by supplementing cytokinin TDZ alone, but the induction effect was poor.
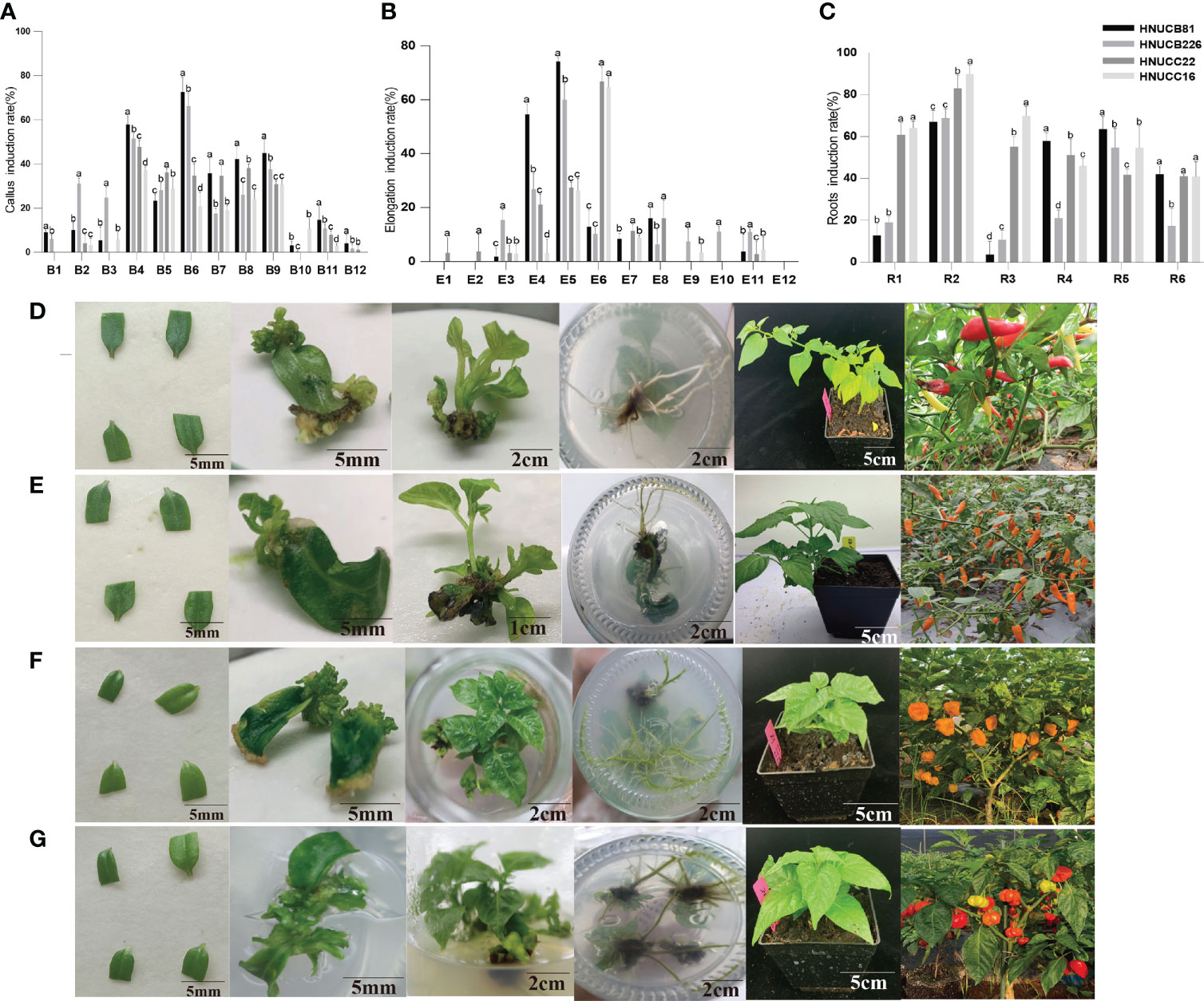
Figure 1 Construction of in vitro regeneration system. (A) callus induction rate, (B) elongation induction rate, and (C) roots induction rate of the four inbred lines. Abscissa indicated the media name. The regeneration induction growth status of pepper of (D) C. baccatum (HNUCB81), (E) C. baccatum (HNUCB226), (F) C. chinense (HNUCC16), and (G) C. chinense (HNUCC22), respectively.
We further found that the application of 6-BA + IAA with differential GA3 led to an increased elongation rate. The best elongation induction rates of HNUCC22 and HNUCC16 were 66.78% and 64.61% in E6 media (MS + 13.32µM 6-BA + 5.71µM IAA + 11.55µM GA3), respectively. While HNUCB81 and HNUCB 226 could obtain higher elongation induction rates (74.27% and 60%) in media E5 (13.32µM 6-BA + 5.71µM IAA + 5.78µM GA3). Besides, a high concentration of 6-BA resulted in a low elongation induction rate (Figure 1B and Supplementary Table S2).
The rooting induction rate of supplemented IAA was the optimum among the four inbred lines. In media R2 (1/2MS + 1.14µM IAA), the highest rooting induction rate of the four inbred lines was obtained, which was over 60%. In addition, adding a certain amount of NAA can also induce a good rooting induction rate in C. chinense. On the contrary, it could obtain a quite low induction rate in C. baccatum (Figure 1C and Supplementary Table S4). Therefore, we constructed the regeneration systems of two cultivars (Figures 1D–G). Further studies were carried out through the comparison of different genotypes.
Compared with elongation and root induction rate, C. chinense callus differentiating rate was lower than C. baccatum. We investigated the phenotypic differences between in vitro plantlets at an early stage. For C. baccatum genotypes HNUCB81 and HNUCB226, a few adventitious-like buds could be observed at the cotyledon petiole tip at the S2 stage. Some plantlet-like structures were growing at the S3 stage. HNUCC22 and HNUCC16 produced only some callus and did not format adventitious during the three stages (Figure 2A).
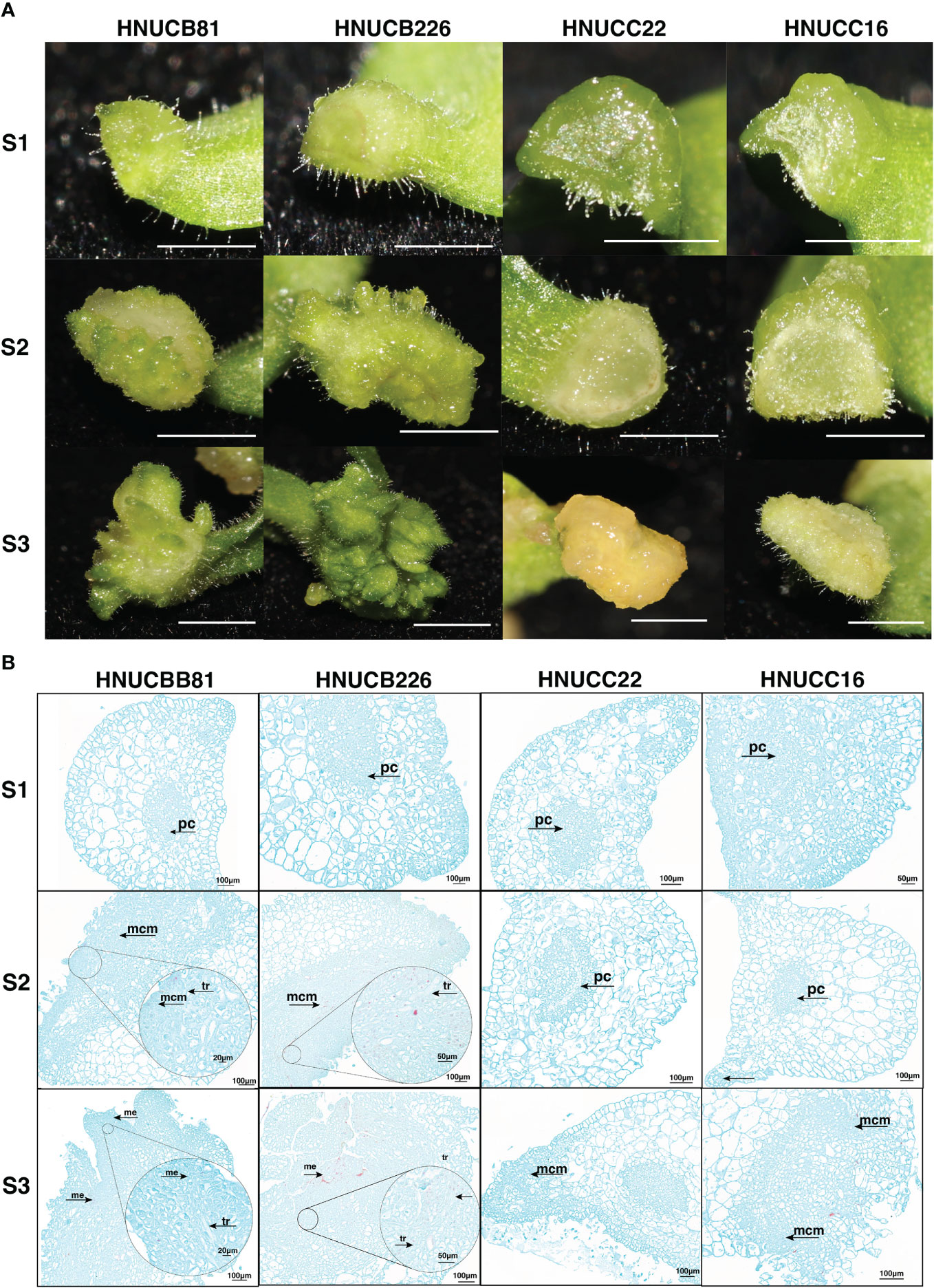
Figure 2 Phenotypic of pepper callus at three development stages. (A) The growth status of the embryonic callus of four genotypes, Bar=2mm; (B) The anatomical morphology of callus at different stages. (tr: vessels, pc. Parenchyma cells, mcm: Meristematic cell mass, and me: Meristemoid.).
To gain insight into the morphological variation of four lines, the histological analysis examined the organogenesis of pepper regeneration. It was found that the differentiation of parenchyma cells near the epidermis was dedifferentiated and restored the ability to divide under the stimulation of phytohormones from C. baccatum. Compared with C. chinense, the cells of HNUCB81 and HNUCB226 were arranged more densely during the S2 and S3 stages. However, loosely packed with large cellular spaces of HNUCC22 and HNUCC16. Furthermore, we found that the bud primordia produced by pepper tissue culture originated from several layers of parenchyma cells with the callus (Figure 2B).
We sequenced the transcriptome of the embryonic callus at three different stages to find out how these regulated genes affect pepper shoot regeneration in the early stages of growth. A total of 36 libraries were established by the transcriptome sequencing of different callus levels of four genotypes and by evaluating the repeatability of the data (Supplementary Figure S1). About 151.89 million clean reads were generated. The average GC content was 42.21%, and the Q30 was ~92.56%. Interestingly, the proportion of 92.68% to 84.03% sequencing reads mapped to the C. chinense reference genome, and the remaining reads ranging from 82.25% to 94.07% were uniquely mapped to the four genotypes (Supplementary Table S4).
C. baccatum and C. chinense demonstrated the greatest differences in S2 and S3; however, they suggested a similar expression profile in the S1 (Figure 3A). Four cluster profiles were indicated, including 7, 17442, 9322, and 1027, respectively (Figure 3B). Moreover, the Venn diagram showed that the common DEGs were higher than the specific DEGs among four genotypes (Supplementary Figure S2). Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. Figures 3C-F shows several pathways that play a role in callus regeneration, such as phenylpropanoid biosynthesis, flavonoid biosynthesis, starch and sucrose metabolism.
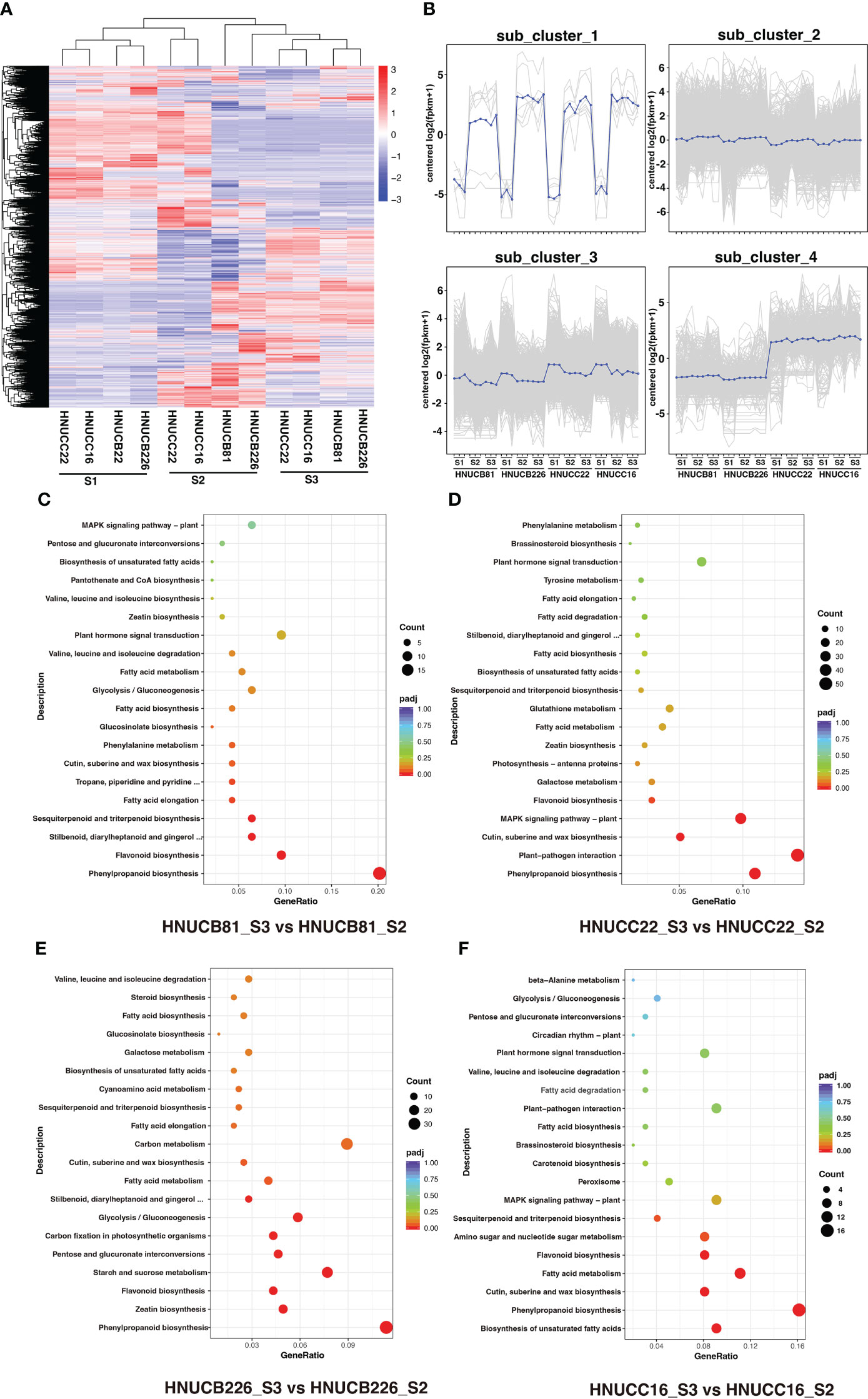
Figure 3 DEGs between four inbred lines at differential stages. (A) Heatmap and cluster analysis expression level of DEGs. (B) Magnified regions of 4 subclusters in all DEGs, blue line indicate consensus of each subcluster. (C-F) The top 20 KEGG pathway terms for the DEGs.
Distributed to twenty-three modules to reveal the correlation between the genes at different stages based on the matrix set as soft-thresholding power =10 (Figure 4A). The modules were presented in a hierarchical clustering dendrogram, in which different colors indicate different modules; each branch represents an individual gene (Figure 4B). After filtering. lightcyan, lightyellow, and midnightblue modules were identified, which showed high-association specificity in S2 or S3 (Figures 4C, D).
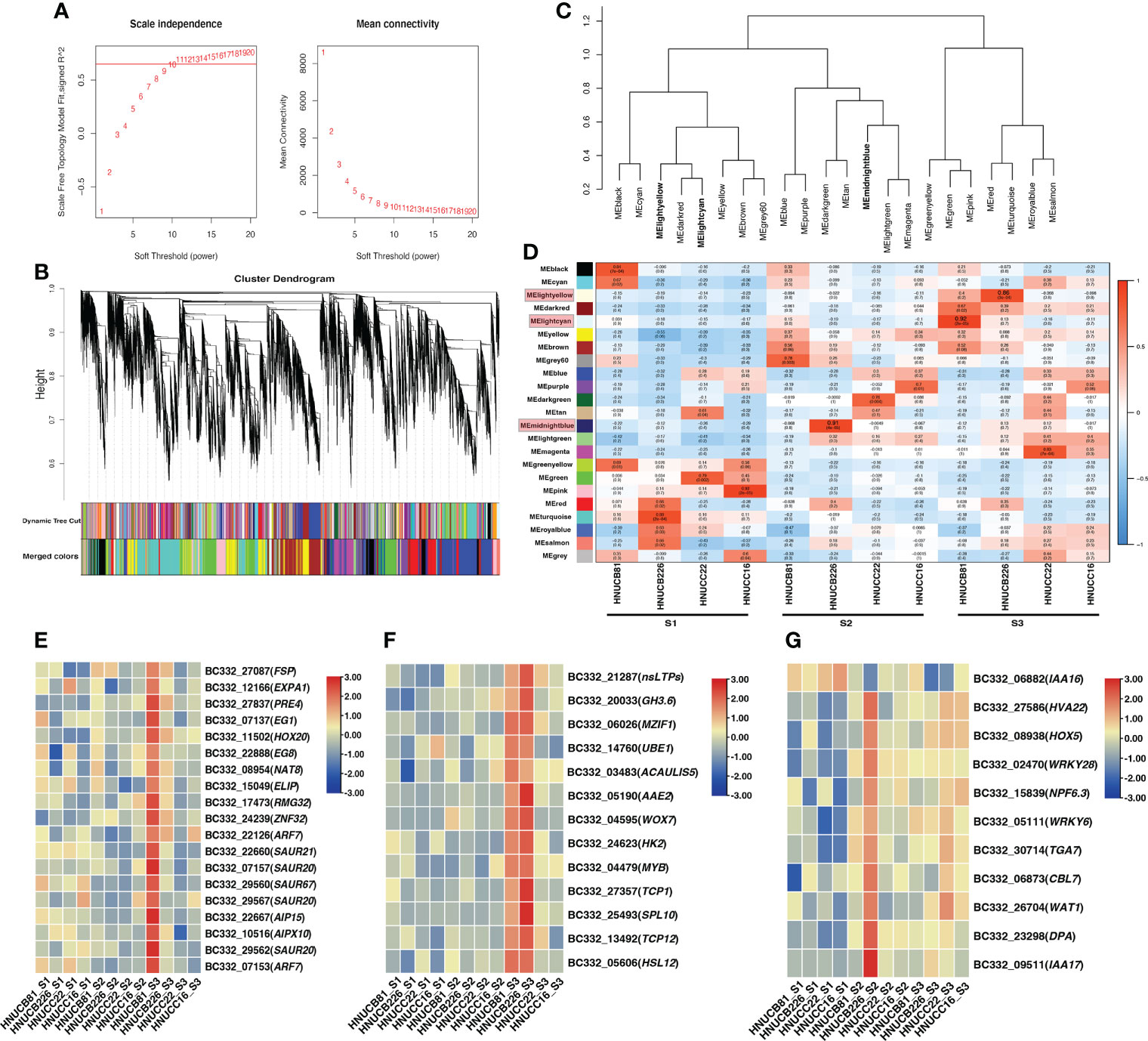
Figure 4 Weight-gene co-expression network analysis of pepper callus regeneration. (A) Soft-threshold of pepper callus regeneration. (B) Hierarchical clustering presents 23 modules having co-expressed genes. (C) Hierarchical cluster dendrogram of eigengenes. (D) Heatmap presents the relationship between the module and developmental stages. Red indicated a positive correlation and blue indicates a negative correlation. The heat map indicates gene expression of DEGs in the lightcyan module (E), lightyellow module (F), and midnightblue module (G). The scale shows FPKM values.
Most of the selected somatic regeneration-related genes were significantly up-regulated with a higher organogenesis rate at S2 and S3 (Figures 4E-G). HOX20 was specifically expressed in HNUCB81 but not expressed in the S1 stage. WOX7 was mainly expressed in HNUCB81 and HNUCB226 at S3 but not in S1. AAE2 and SPL10 were specifically expressed in C. baccatum at the S3, and the highest expression was found in HNUCB226 (Figure 4F). IAA16 was specifically at the S1 but had lower expression at the S3 (Figure 4G). A total of 200, 107, and 230 genes were included in three modules, respectively (Supplementary Table S5). The 26 genes that are called “hub genes” were shown in Figure 5 and Table 1 as part of the study.
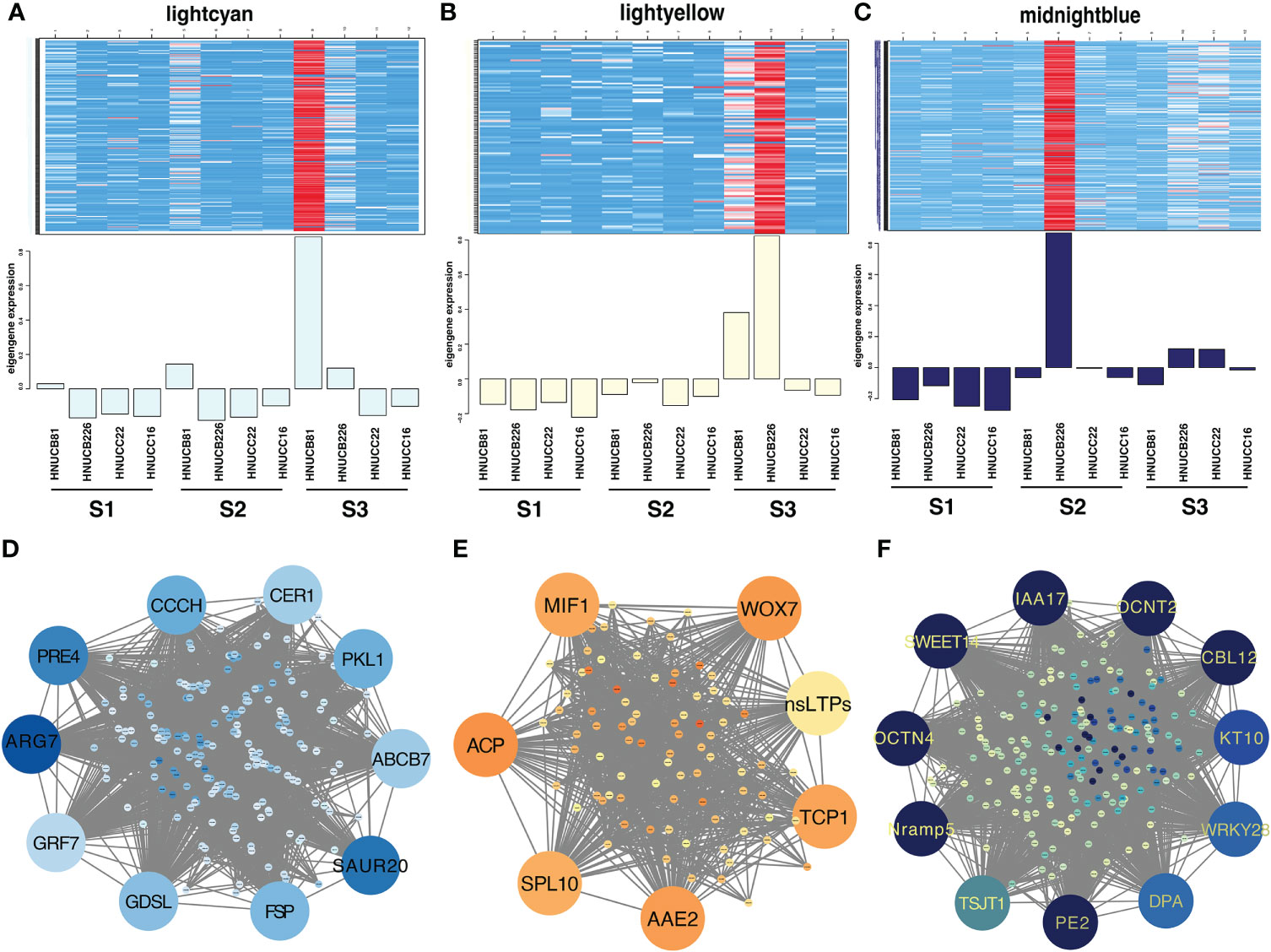
Figure 5 Gene network and candidate genes. Heat map showing the relative FPKM of each gene from lightcyan module (r2 = 0.92, p =2e-05) (A), lightyellow module (r2 = 0.89, p =3e-04) (B), and midnightblue module (r2 = 0.91, p =4e-05) (C). The correlation network of the hub gene of lightcyan module (D), lightyellow module (E), and midnightblue module (F).
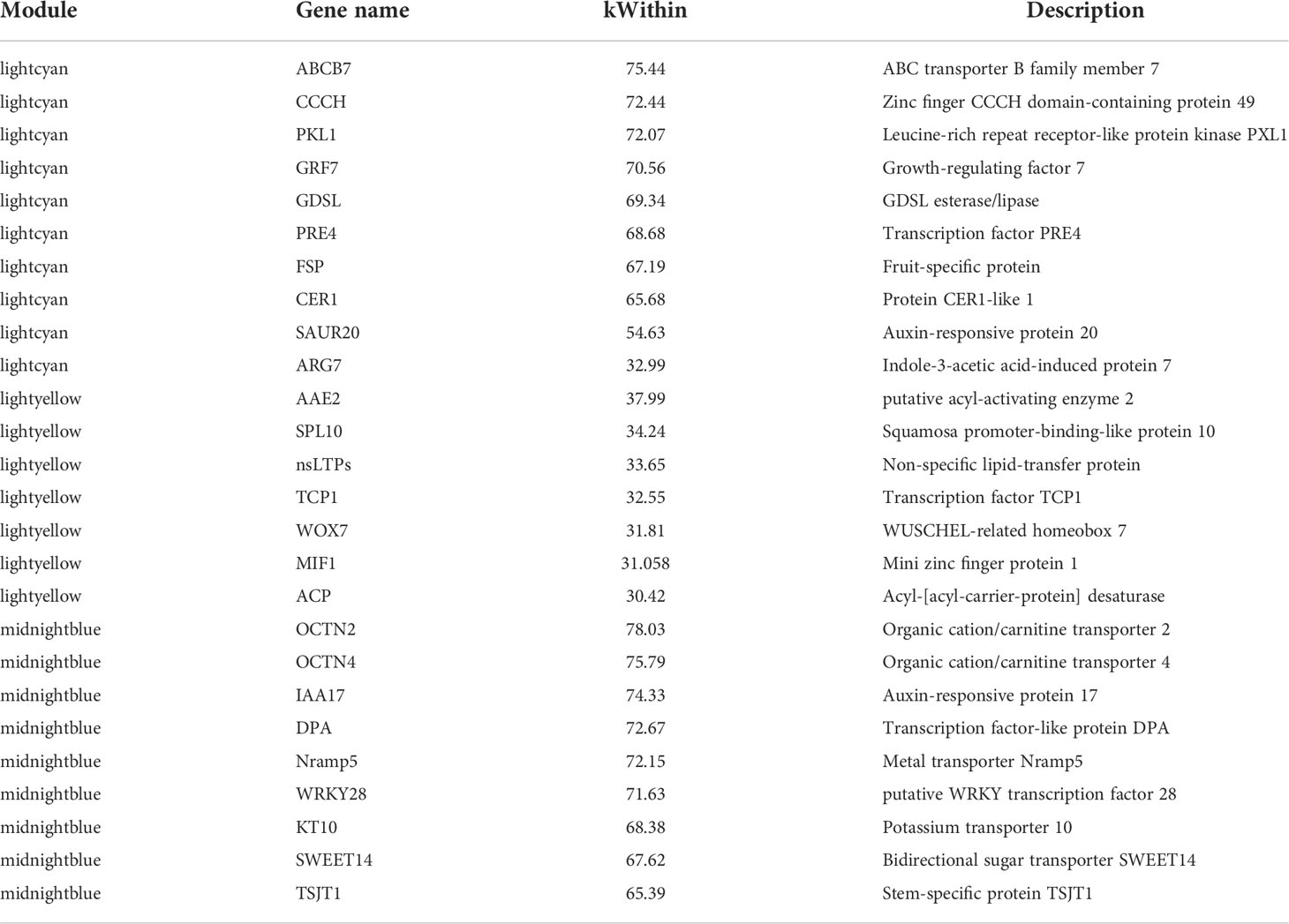
Table 1 Information of hub genes.
We conducted KEGG on three modules. Interestingly, plant hormone signal transduction and starch and sucrose metabolism were significantly enriched (Supplementary Table S6). In the plant hormone signal transduction pathways, five DEGs were up-regulated in HNUCB81_S3 but down-regulated in HNUCC22_S3 and HNUCC16_S3 of the lightcyan module, which all belong to the auxin-inducible TF family (Figure 6). A total of four and two DEGs were involved in the starch and sucrose metabolism pathway between the lightcyan and lightyellow module, and all of the genes were up-regulated in C. baccatum at the S3. However, the results of the midnightblue module are inconsistent with the other two modules, suggesting that these pathways may not have been activated at the S2 (Figure 7). Furthermore, Gene Ontology (GO) describes the molecular and biological functions of the genes involved in regeneration. DEGs were distributed in the molecular function (MF), cellular component (CC), and biological process (BP) categories, respectively (Supplementary Figure S3 and Supplementary Table S7).
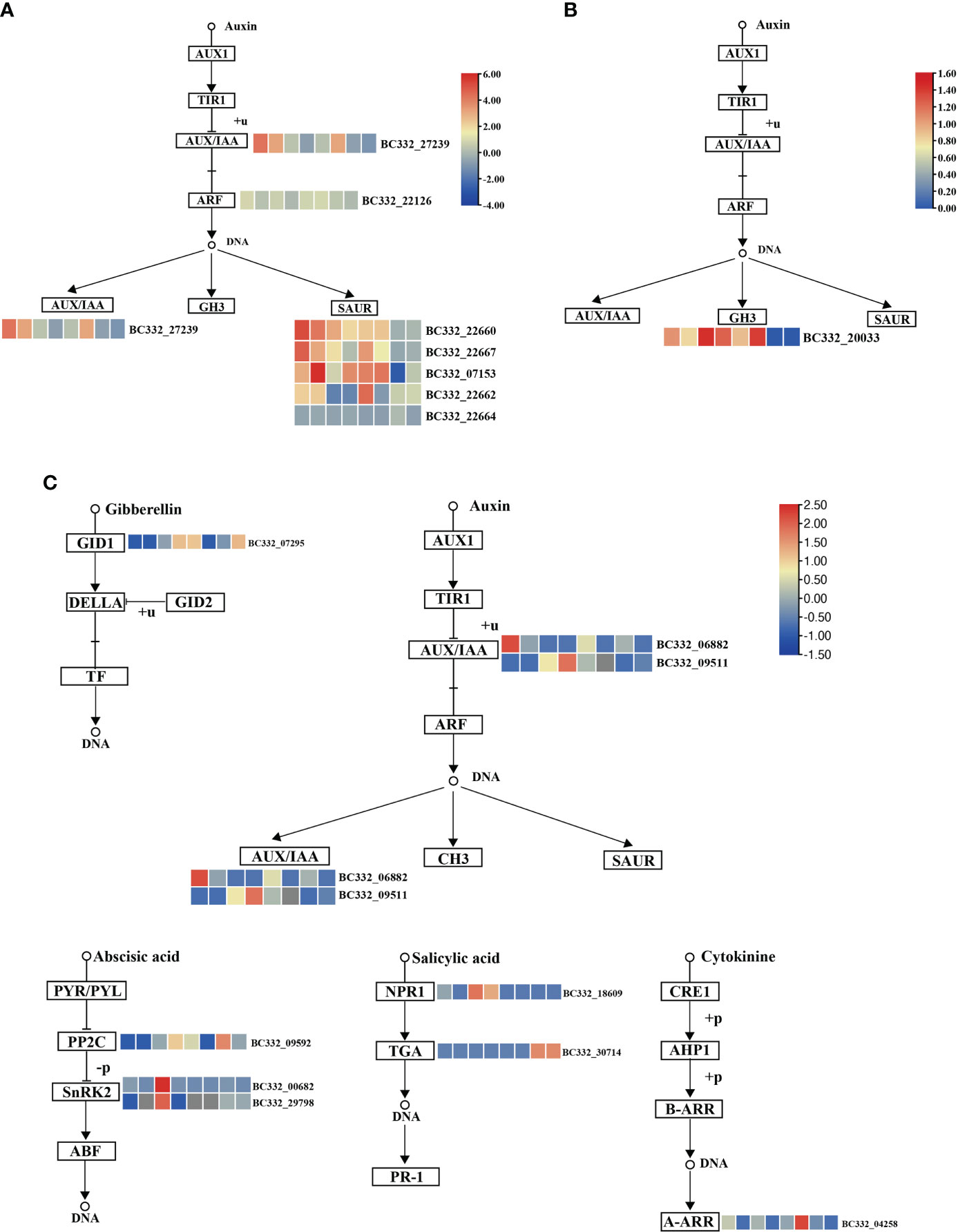
Figure 6 Pepper regeneration candidate DEGs in plant hormone signal transduction pathway. (A) lightcyan module, (B) lightyellow module, and (C) midnightblue module. The columns in the heatmap were displayed as HNUCB81_S3 vs HNUCC22_S3, HNUCB81_ S3 vs HNUCC16_ S3, HNUCB226_S3 vs HNUCC22_S3, HNUCB226_S3 vs HNUCC16_S3, HNUCB81_S3 vs HNUCB81_S2, HNUCB226_S3 vs HNUCB226_S2, HNUCC22_S3 vs HNUCC22_S2, and HNUCC16_S3 vs HNUCC16_ S2 of pepper regeneration from left to right, respectively.
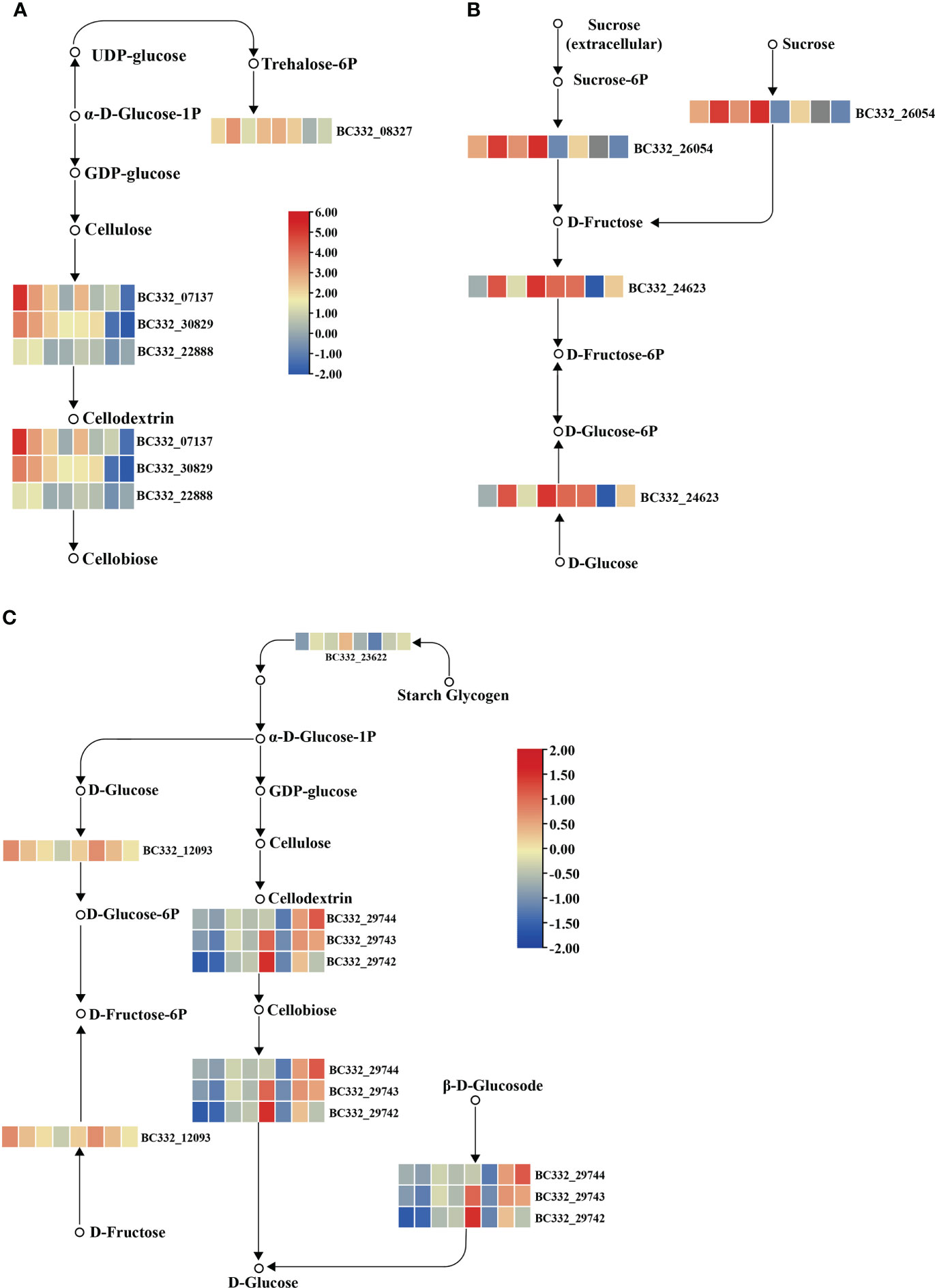
Figure 7 Pepper regeneration candidate DEGs in starch and sucrose metabolism pathway. (A) lightcyan module, (B) lightyellow module, and (C) midnightblue module. The columns in the heatmap were displayed as HNUCB81_S3 vs HNUCC22_S3, HNUCB81_S3 vs HNUCC16_S3, HNUCB226_ S3 vs HNUCC22_S3, HNUCB226_S3 vs HNUCC16_S3, HNUCB81_S3 vs HNUCB81_S2, HNUCB226_S3 vs HNUCB226_S2, HNUCC22_S3 vs HNUCC22_S2, and HNUCC16_S3 vs HNUCC16_ S2 of pepper regeneration from left to right, respectively.
Previous studies have indicated that TFs play a key role in callus formation by regulating cell differentiation and proliferation (Ikeuchi et al., 2019). We further analyzed the TFs between four inbred lines; 737, 156, 116, and 610 TFs were identified in HNUCC22, HNUCC16, HNUCB81, and HNUCB226 between S2 and S3, respectively. Pkinase, p450, NB-ARC, WRKY, LRRNT_2, HLH, and MYB TFs account for a large proportion (Supplementary Table S8).
To validate the transcriptome sequencing data, 9 DEGs involved in enrichment pathways were selected for qRT-PCR. The results suggested that the expression patterns of qRT-PCR were the same as those of RNA-seq data, revealing that the RNA-seq data is reliable (Figure 8).
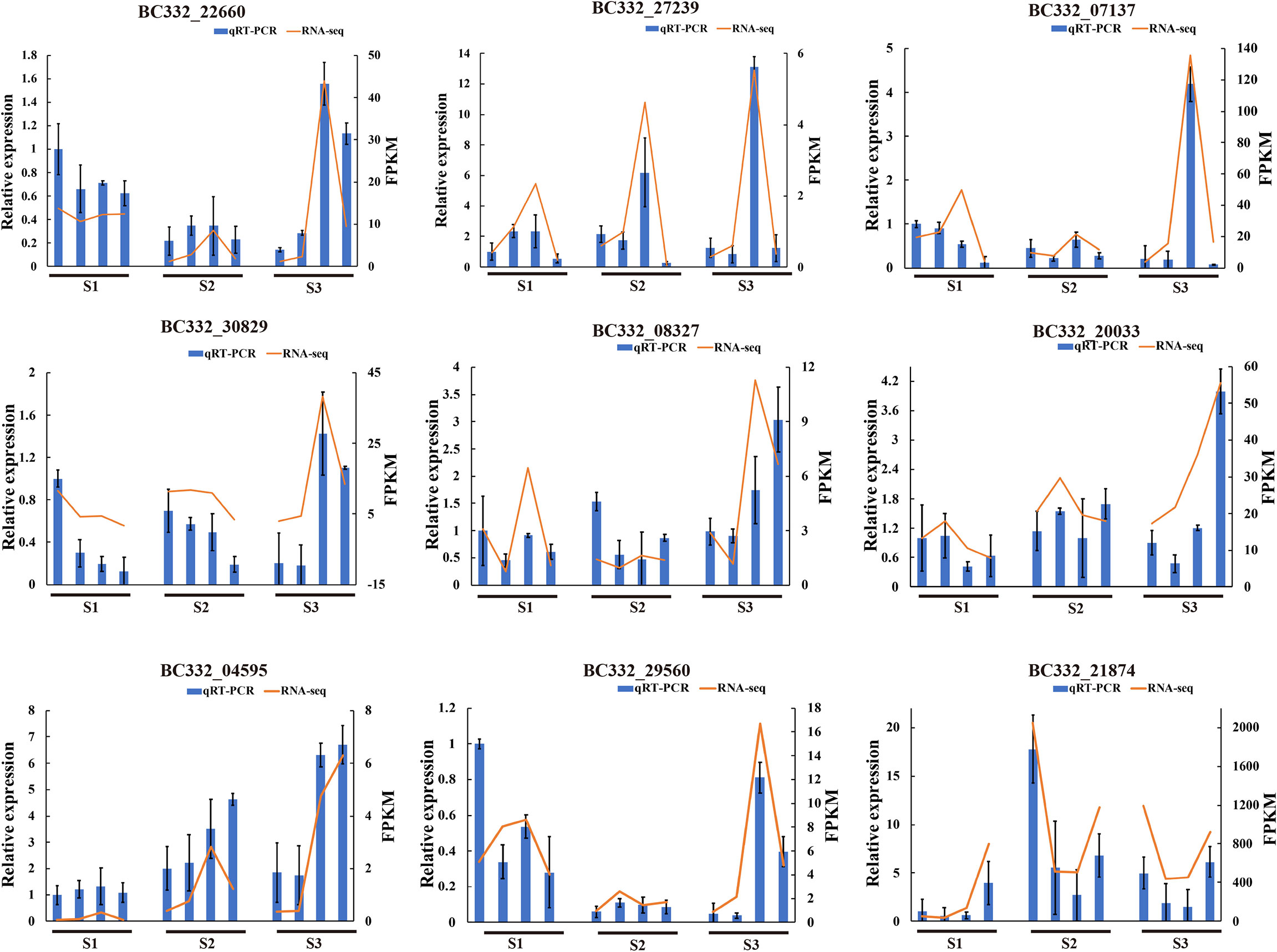
Figure 8 Correlation between qRT-PCR and RNA-seq based on differentially expressed genes of enrichment pathways at three differentiation stages. HNUCC22, HNUCC16, HNUCB81, and HNUCC226 are represented from left to right at three stages, respectively.
Pepper is a highly genotype-dependent, and recalcitrant plant, which leads to the development of in vitro regeneration system being relatively slow. Traditional breeding methods are being gradually updated. Plant biotechnology, including tissue culture and genetic engineering, is gradually becoming an important breeding method to promote the improvement of pepper breeding. It is well known that several genes are involved in explant regeneration, including WOX, BBM, LEC, and WUS (Zhao et al., 2022). Our results indicated that phytohormones play an important role in callus formation by regulating the expression of regeneration-related genes. In addition, WGCNA analysis identified many genes linked to plant hormone regulation, which has a positive correlation with cell differentiation. This study illustrated how pepper conveys a diverse range of signals to regulate the early regeneration process in pepper.
Auxin and cytokinin regulate cell division, callus differentiation, and somatic embryogenesis (Pinto et al., 2011). It was reported that the percentage of bud-forming explants was the highest under the induction of 3.4 µM TDZ (Santanabuzzy et al., 2005). Venkataiah reported that the highest induction rate was obtained for C. baccatum when supplemented with 2,4-D and Kinetin (Venkataiah et al., 2016). This study constructed the in vitro regeneration systems of four inbred lines of pepper. C. chinense and C. baccatum somatic callus were produced in the presence of 6-BA and IAA. However, there are distinct differences between the two cultivated species. Our results showed that supplementation with GA3 markedly promoted elongation, but the higher levels of 6-BA inhibited the induction of elongation. Furthermore, we observed that the application of IAA significantly increased the induction of roots in C. chinense. In the induction media of 1.07µM IAA, the ability of pepper to induce rooting was the most significant. These results laid the foundation for genetic transformation and molecular-assisted breeding of pepper.
Previous studies have reported that plant hormones regulate many aspects of plant growth and development (Su et al., 2011). Auxin, ethylene, and abscisic acid (ABA) hormones are considered to play a positive role in organogenesis and regeneration (Méndez-Hernández et al., 2019). The auxin biosynthesis-related gene AtSAUR36 has been linked to plant cell expansion (Hou et al., 2013). A recent study revealed that ZmSAUR15 has an adverse effect on the embryogenic callus (ECs) formation of maize (Wang Y. et al., 2021). Our study demonstrated that the plant hormone transduction and biosynthesis genes were involved in the process of pepper regeneration potential. We found that most of the auxin-related genes were up-regulated in C. baccatum but down-regulated in C. chinense, indicating that auxin transduction was the key factor of callus regeneration. Moreover, we performed qRT-PCR, showing that the expression of GH3.6 (BC332_20033) and SAUR67 (BC332_29560) increased dramatically at S3, suggesting that these auxin-related genes could enhance pepper’s to promote plant cell division and differentiation. Furthermore, previous studies revealed that IAA might interact with different ARFs, resulting in transcriptional inhibition or activation (Su et al., 2014). These results show that the callus induction process is based on the synergistic effect of phytohormones.
Sucrose is the main carbohydrate supplement in the culture media. It plays an important role as an energy source and osmosis in the process of organogenesis (Verma and Dougall, 1977; Cui et al., 2019). Moreover, the correlation between starch metabolism and shoot regeneration has been reported in rice (Lee and Huang, 2013; Lee and Huang, 2014) and sorghum (Zhou et al., 2021). Previous studies indicated that SWEET13 and SWEET14 proteins were linked to sugar transport and involved in gibberellin-mediated physiological processes at the seedling stage of Arabidopsis (Kanno et al., 2016). In the present study, SWEET14 was identified as a hub gene in the network. It may be related to the sucrose metabolic pathway. Furthermore, the majority of genes regulated by starch and sucrose were up-regulated in HNUCB81 and HNUCB226 compared with HNUCC22 and HNUCC16. The results revealed that C. baccatum expression was significantly higher than C. chinense during S2 and S3. The findings suggest that these genes may regulate the metabolism of starch and sucrose and play a key role in the process of regeneration.
WOX family genes are important in a variety of processes, ranging from callus formation to organ development (Kim et al., 2018; Wang C. et al., 2021; Willoughby and Nimchuk, 2021). It was reported that WOX2 and WOX8/9 were involved in differentiation and somatic embryogenesis (Palovaara et al., 2010; Zhao et al., 2022). Based on the activity of WIND1, WOX13 was rapidly induced, which is a key regulator of callus formation (Ikeuchi et al., 2021). In addition, previous studies found that WOX7 inhibited lateral root initiation and primordium growth (Kong et al., 2016). In contrast, we have shown rapid increases in WOX7 expression in C. baccatum lines between S2 and S3 but it is down-regulated in C. chinense lines. Interestingly, WOX7 was a significant expression within the WGCNA module, consistent with RT-qPCR. It was also found that WOX genes were up-regulated in the high-regeneration lines of maize (Zhang et al., 2019). These results demonstrated that WOX could reflect a positive function in regeneration.
Several hub genes have been identified via network analysis that may be key regulating factors in regeneration. Previous studies have indicated that some WRKY genes induce callus of papaya and Panax ginseng (Jamaluddin et al., 2017; Yang et al., 2020). Moreover, WRKY genes may upregulate callus formation in bread wheat (Chu et al., 2017). In our study, WRKY TFs were up-regulated between HNUCB226_S3 vs HNUCB226_S2, but there was no significant expression in C. chinense. These results revealed that WRKY TFs play an important role in the process of regeneration.
Furthermore, several callus-related genes, including GRF, GDSL, TSJT1, and CCCH, have been identified as being associated with dedifferentiation (Zhang et al., 2022). Our results indicated that these hub genes were highly expressed in HNUCB81 and HNUCB226, but without fluctuation in C. chinense. In addition, it is reported that the ABC transport family mediates the transport of auxin in roots to promote basipetal transport (Ko et al., 2014). Interestingly, we identified a hub gene for ABCB7 in the lightcyan module, suggesting its essential role in initial callus induction.
A comprehensive understanding of the genes associated with pepper callus development will help to understand the process of pepper regeneration. In the present study, we successfully established an efficient in vitro regeneration system in two cultivars to monitor callus induction in differential genotypes. In addition, we performed histology and transcriptome analysis of the pepper regeneration and demonstrated a high correlation between somatic callus induction and plant hormone pathway and starch and sucrose metabolism pathways. This study provides novel information for the induction of regeneration and will have great potential for transgenic plants or molecular-assisted breeding of pepper.
We kept Capsicum baccatum (HNUCB81 and HNUCB226) and Capsicum chinense (HNUCC22 and HNUCC16) on bud induction media (B1-B12), which was supplemented with 4.44 - 22.2µM (6-BA) and 0.57 - 2.85µM IAA (Supplementary Table S1). We also kept on the plant elongation media (E1-E12), which was supplemented with 13.32 – 22.2µM (6-BA) + 2.85-5.71µM IAA + 0 – 11.55µM GA3 (Supplementary Table S2), rooting media (R1-R6) supplemented with 0.57 - 2.85µM IAA or 0.54 - 2.69µM NAA mg · L−1 (Supplementary Table S3). MS media including 6 g/L agar and 30g/L sucrose (Hopebio, Qingdao) and phytohormone (Sigma, St. Louis, MO, USA). Each treatment contained at least 30 explants derived from petiole of cotyledons of sterile seedling aged 15-20 days (0.5-1cm in length). All media were adjusted to pH 5.8 and then autoclaved at 116°C for 30 minutes. The explants were inoculated on B media for 15-20 d to get the plantlets, and after elongation incubated in E media to proliferate buds, and was transplanted to R media to induct roots. All of the explants were cultured in MS media at 24 ± 2°C with 16 h light/d for differentiation at Hainan University. All experiments were repeated three times. The measurement of data was calculated with the equation: percentage of callus induction: Callus induction (%) = No. of Callus/No. of explants ×100%; Plantlets induction (%) = No. of plantlets/No. of Callus ×100%; Rooting induction (%) = No. of rooting plants/No. of plantlets ×100%. Statistical analysis was done using IBM SPSS (v26). The significance of the difference between the mean values was tested using Duncan’s multiple range test (P ≤ 0.05). The results are indicated as mean ± standard error of three duplications.
Based on the regeneration results, we collected early differentiated callus supplemented with 13.32µM 6-BA+ 2.85µM IAA at three different stages, including S1 (1-3d), S2 (4-6d), and S3 (7-9d). The callus tissues were collected in a 50% FAA solution. After fixation, the slices were dehydrated successively with xylene and ethanol. After that, the slices were dyed in Safranin O-Fast Green staining solution. Moreover, slices were placed in clean xylene and stuck tissue slices with neutral balm (Servicebio, Wuhan). Finally, the slices were observed and photographed under the microscope (Nikon Eclipse E100 and Nikon DS-U3). Triplicate biological replicates for each sample.
We selected explants with normal development (No contamination and browning) for mixed sampling at three early development stages, including S1 (1-3d), S2 (4-6d), and S3 (7-9d). Total RNA was isolated with Trizol Reagents (Thermo Fisher Scientific, Shanghai, China). Thirty-six non-directional libraries were produced using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, United States) and were sequenced on the Illumina Novaseq platform. Clean reads were obtained by filtering low-quality reads and aligned to the C. chinense reference genome (https://www.ncbi.nlm.nih.gov/genome/?term=Capsicum+chinense) using HISAT2 (v.2.0.5) (Kim et al., 2015).
The fragments per kilo-base of exon per million fragments mapped (FPKM) was to estimate the transcript expression level of all samples, which was calculated by StringTie (v1.3.3b) (Pertea et al., 2016). Significantly differential expression genes (DEGs) were identified by DESeq2 R package (v1.20.0) (Love et al., 2014) with a threshold included |log2 (Foldchange)| > 1 and p-adjust < 0.05. The Venn diagram of the DEGs was created using Jvenn (Bardou et al., 2014), and the heat map was plotted using the R package of pheatmap.
Differential expression genes enrichment analysis of Gene Ontology (GO) was applied by GOseq (v1.22) software (Young et al., 2010). The setting parameter is that the p- adjust < 0.05. Various metabolic pathways were identified based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The enrichment analysis of DEGs was calculated by KOBAS 2.0 software (Mao et al., 2005). p-adjust < 0.05 was distributed to be significantly enriched in KEGG.
WGCNA is applied to analyze gene expression patterns in different samples to obtain gene modules with similar expression patterns (Langfelder and Horvath, 2008). In this WGCNA network, the soft power was selected at 10 (R2 = 0.8), mergeCutHeight was set as > 0.75, and minModuleSize was set as > 30. WGCNA network was constructed by WGCNA in the R package and the genes from each module were visualized using Cytoscape (v3.8.0).
Total RNA was subjected to reverse transcription using Hiscript III RT SuperMix for qPCR (Vazyme Biotech, China), and qPCR was analyzed using SYBR qPCR Master Mix (Vazyme Biotech, China). The amplification program was based on the standard protocol of the Applied Biosystems QuantStudio 1 Real-Time PCR Instrument (Thermo Fisher Scientific, USA), as follows 95°C for 10 min, 40 cycles of 95°C for 15s, and 60°C for 10s, and thermal denaturation step is then performed to generate a melting curve to verify the amplification specificity. The primers were designed by primer 5 (v5.0) that were listed in Supplementary Table S9.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA790105.
For research articles: Conceptualization, ZW, HS; methodology and validation, HS; investigation, YZ, CH, MA, YH, DL, LL, CL, FH, SC, and GZ; writing—original draft preparation, HS; supervision, ZW. All authors have read and agreed to the published version of the manuscript.
This work was supported by a grant from the Project of the Administrative Bureau of Sanya Yazhou Bay Science and Technology City (HNF202210), the Major Science and Technology Plan of Hainan Province (ZDKJ2021010) and the National Key Research and Development Program of China (2018YFD1000800).
We thank Dr. Muhammad Ali Mumtaz and Dr. Sunjeet Kumar from School of Horticulture for their suggestions on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1025497/full#supplementary-material
Altpeter, F., Springer, N. M., Bartley, L. E., et al. (2016). Advancing crop transformation in the era of genome editing. Plant Cell 28, 1510–1520. doi: 10.1105/tpc.16.00196
Bardou, P., Mariette, J., Escudié, F., et al. (2014). Jvenn: An interactive Venn diagram viewer. BMC Bioinf. 15, 293. doi: 10.1186/1471-2105-15-293
Chen, J., Tomes, S., Gleave, A. P., et al. (2022). Significant improvement of apple (Malus domestica borkh.) transgenic plant production by pre-transformation with a baby boom transcription factor. Hortic. Res. 9. doi: 10.1093/hr/uhab014
Chu, Z., Chen, J., Sun, J., et al. (2017). De novo assembly and comparative analysis of the transcriptome of embryogenic callus formation in bread wheat (Triticum aestivum l.). BMC Plant Biol. 17, 244. doi: 10.1186/s12870-017-1204-2
Cui, G., Zhang, Y., Zhang, W., et al. (2019). Response of carbon and nitrogen metabolism and secondary metabolites to drought stress and salt stress in plants. J. Plant Biol. 62, 387–399. doi: 10.1007/s12374-019-0257-1
Fan, M., Xu, C., Xu, K., et al. (2012). LATERAL ORGAN BOUNDARIES DOMAIN transcription factors direct callus formation in Arabidopsis regeneration. Cell Res. 22, 1169–1180. doi: 10.1038/cr.2012.63
Heidmann, I., de Lange, B., Lambalk, J., et al. (2011). Efficient sweet pepper transformation mediated by the BABY BOOM transcription factor. Plant Cell Rep. 30, 1107–1115. doi: 10.1007/s00299-011-1018-x
Hou, K., Wu, W., Gan, S. S. (2013). SAUR36, a small auxin up RNA gene, is involved in the promotion of leaf senescence in Arabidopsis. Plant Physiol. 161, 1002–1009. doi: 10.1104/pp.112.212787
Ikeuchi, M., Favero, D. S., Sakamoto, Y., et al. (2019). Molecular mechanisms of plant regeneration. Annu. Rev. Plant Biol. 70, 377–406. doi: 10.1146/annurev-arplant-050718-100434
Ikeuchi, M., Iwase, A., Ito, T., et al. (2021). Wound-inducible WUSCHEL RELATED HOMEOBOX 13 is required for callus growth and organ reconnection. Plant Physiol 188, 425–441. doi: 10.1093/plphys/kiab510
Ikeuchi, M., Iwase, A., Sugimoto, K. (2015). Control of plant cell differentiation by histone modification and DNA methylation. Curr. Opin. Plant Biol. 28, 60–67. doi: 10.1016/j.pbi.2015.09.004
Ikeuchi, M., Sugimoto, K., Iwase, A. (2013). Plant callus: mechanisms of induction and repression. Plant Cell 25, 3159–3173. doi: 10.1105/tpc.113.116053
Iwase, A., Harashima, H., Ikeuchi, M., et al. (2017). WIND1 promotes shoot regeneration through transcriptional activation of ENHANCER OF SHOOT REGENERATION1 in Arabidopsis. Plant Cell 29, 54–69. doi: 10.1105/tpc.16.00623
Iwase, A., Mitsuda, N., Koyama, T., et al. (2011). The AP2/ERF transcription factor WIND1 controls cell dedifferentiation in Arabidopsis. Curr. Biol. 21, 508–514. doi: 10.1016/j.cub.2011.02.020
Jamaluddin, N. D., Mohd Noor, N., Goh, H. H. (2017). Genome-wide transcriptome profiling of Carica papaya l. embryogenic callus. Physiol. Mol. Biol. Plants 23, 357–368. doi: 10.1007/s12298-017-0429-8
Kanno, Y., Oikawa, T., Chiba, Y., et al. (2016). AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 7, 13245. doi: 10.1038/ncomms13245
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, S., Park, J., Yeom, S. I., et al. (2017). New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 18, 210. doi: 10.1186/s13059-017-1341-9
Kim, J. Y., Yang, W., Forner, J., et al. (2018). Epigenetic reprogramming by histone acetyltransferase HAG1/AtGCN5 is required for pluripotency acquisition in Arabidopsis. EMBO J. 37, e98726. doi: 10.15252/embj.201798726
Ko, D., Kang, J., Kiba, T., et al. (2014). Arabidopsis ABCG14 is essential for the root-to-shoot translocation of cytokinin. Proc. Natl. Acad. Sci. U.S.A. 111, 7150–7155. doi: 10.1073/pnas.1321519111
Kong, D., Hao, Y., Cui, H. (2016). The WUSCHEL related homeobox protein WOX7 regulates the sugar response of lateral root development in Arabidopsis thaliana. Mol. Plant 9, 261–270. doi: 10.1016/j.molp.2015.11.006
Kothari, S. L., Joshi, A., Kachhwaha, S., et al. (2010). Chilli peppers–a review on tissue culture and transgenesis. Biotechnol. Adv. 28, 35–48. doi: 10.1016/j.biotechadv.2009.08.005
Langfelder, P., Horvath, S. (2008). WGCNA: An r package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Lee, S. T., Huang, W. L. (2013). Cytokinin, auxin, and abscisic acid affects sucrose metabolism conduce to de novo shoot organogenesis in rice (Oryza sativa l.) callus. Bot. Stud. 54, 5. doi: 10.1186/1999-3110-54-5
Lee, S. T., Huang, W. L. (2014). Osmotic stress stimulates shoot organogenesis in callus of rice (Oryza sativa l.) via auxin signaling and carbohydrate metabolism regulation. Plant Growth Regul. 73, 193–204. doi: 10.1007/s10725-013-9880-x
Lee, Y. H., Kim, H. S., Kim, J. Y., et al. (2004). A new selection method for pepper transformation: callus-mediated shoot formation. Plant Cell Rep. 23, 50–58. doi: 10.1007/s00299-004-0791-1
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Mao, X., Cai, T., Olyarchuk, J. G., et al. (2005). Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Méndez-Hernández, H. A., Ledezma-Rodríguez, M., Avilez-Montalvo, R. N., et al. (2019). Signaling overview of plant somatic embryogenesis. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00077
Palovaara, J., Hallberg, H., Stasolla, C., et al. (2010). Comparative expression pattern analysis of WUSCHEL-related homeobox 2 (WOX2) and WOX8/9 in developing seeds and somatic embryos of the gymnosperm Picea abies. New Phytol. 188, 122–135. doi: 10.1111/j.1469-8137.2010.03336.x
Pertea, M., Kim, D., Pertea, G. M., et al. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pinto, D., Almeida, A., Rêgo, M., et al. (2011). Somatic embryogenesis from mature zygotic embryos of commercial passionfruit (Passiflora edulis sims) genotypes. Plant Cell Tiss. Org. 107, 521–530. doi: 10.1007/s11240-011-0003-y
Qin, C., Yu, C., Shen, Y., et al. (2014). Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. U.S.A. 111, 5135–5140. doi: 10.1073/pnas.1400975111
Sanatombi, K., Sharma, G. J. (2008). In vitro plant regeneration in six cultivars of Capsicum spp. using different explants. Biol. Plantarum 52, 141–145. doi: 10.1007/s10535-008-0029-0
Sanatombi, K., Sharma, G. J. (2012). In vitro regeneration of Capsicum chinense jacq. Curr. Trends Biotechnol. Pharm. 6, 66–72.
Santanabuzzy, N., Cantoflick, A., Barahonaperez, F., et al. (2005). Regeneration of habanero pepper (Capsicum chinense jacq.) via organogenesis. HortScience 40, 1829–1831. doi: 10.21273/HORTSCI.40.6.1829
Su, L., Bassa, C., Audran, C., et al. (2014). The auxin sl-IAA17 transcriptional repressor controls fruit size via the regulation of endoreduplication-related cell expansion. Plant Cell Physiol. 55, 1969–1976. doi: 10.1093/pcp/pcu124
Su, Y. H., Liu, Y. B., Zhang, X. S. (2011). Auxin-cytokinin interaction regulates meristem development. Mol. Plant 4, 616–625. doi: 10.1093/mp/ssr007
Venkataiah, P., Bhanuprakash, P., Suman Kalyan, S., et al. (2016). Somatic embryogenesis and plant regeneration of Capsicum baccatum l. J. Genet. Eng. Biotechnol. 14, 55–60. doi: 10.1016/j.jgeb.2016.02.001
Verma, D. C., Dougall, D. K. (1977). Influence of carbohydrates on quantitative aspects of growth and embryo formation in wild carrot suspension cultures. Plant Physiol. 59, 81–85. doi: 10.1104/pp.59.1.81
Wang, Y., He, S., Long, Y., et al. (2021). Genetic variations in ZmSAUR15 contribute to the formation of immature embryo-derived embryonic calluses in maize. Plant J 109:980–991. doi: 10.1111/tpj.15609
Wang, C., Zhao, B., He, L., et al. (2021). The WOX family transcriptional regulator SlLAM1 controls compound leaf and floral organ development in Solanum lycopersicum. J. Exp. Bot. 72, 1822–1835. doi: 10.1093/jxb/eraa574
Willoughby, A. C., Nimchuk, Z. L. (2021). WOX going on: CLE peptides in plant development. Curr. Opin. Plant Biol. 63, 102056. doi: 10.1016/j.pbi.2021.102056
Yang, Y., Wang, N., Zhao, S. (2020). Functional characterization of a WRKY family gene involved in somatic embryogenesis in panax ginseng. Protoplasma 257, 449–458. doi: 10.1007/s00709-019-01455-2
Young, M. D., Wakefield, M. J., Smyth, G. K., et al. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Zhang, Y., Li, J., Li, C., et al. (2022). Gene expression programs during callus development in tissue culture of two eucalyptus species. BMC Plant Biol. 22, 1. doi: 10.1186/s12870-021-03391-x
Zhang, X., Wang, Y., Yan, Y., et al. (2019). Transcriptome sequencing analysis of maize embryonic callus during early redifferentiation. BMC Genomics 20, 159. doi: 10.1186/s12864-019-5506-7
Zhao, R., Qi, S., Cui, Y., et al. (2022). Transcriptomic and physiological analysis identifies a gene network module highly associated with brassinosteroid regulation in hybrid sweetgum tissues differing in the capability of somatic embryogenesis. Hortic. Res. 9. doi: 10.1093/hr/uhab047
Zhou, C., Wang, S., Zhou, H., et al. (2021). Transcriptome sequencing analysis of sorghum callus with various regeneration capacities. Planta 254, 33. doi: 10.1007/s00425-021-03683-4
Keywords: Capsicum chinense, Capsicum baccatum, somatic callus, in vitro regeneration transcriptome, histology, WGCNA
Citation: Shu H, Zhang Y, He C, Altaf MA, Hao Y, Liao D, Li L, Li C, Fu H, Cheng S, Zhu G and Wang Z (2022) Establishment of in vitro regeneration system and molecular analysis of early development of somatic callus in Capsicum chinense and Capsicum baccatum. Front. Plant Sci. 13:1025497. doi: 10.3389/fpls.2022.1025497
Received: 23 August 2022; Accepted: 19 October 2022;
Published: 17 November 2022.
Edited by:
Paloma Moncaleán, Neiker Tecnalia, SpainReviewed by:
Victor Aguilar-Hernández, National Council of Science and Technology (CONACYT), MexicoCopyright © 2022 Shu, Zhang, He, Altaf, Hao, Liao, Li, Li, Fu, Cheng, Zhu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiwei Wang, d2FuZ3poaXdlaUBoYWluYW51LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.