
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 November 2022
Sec. Plant Development and EvoDevo
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1024515
This article is part of the Research Topic Recent Advances in Flower and Fruit Development in Perennial plants View all 22 articles
 Yuanhui Yu1,2†
Yuanhui Yu1,2† Miaomiao Yang1,2†
Miaomiao Yang1,2† Xinya Liu1,2
Xinya Liu1,2 Yan Xia1,2
Yan Xia1,2 Ruoqian Hu1,2
Ruoqian Hu1,2 Qingqing Xia1,2
Qingqing Xia1,2 Danlong Jing1,2*
Danlong Jing1,2* Qigao Guo1,2*
Qigao Guo1,2*The WUSCHEL (WUS)-related homeobox (WOX) gene family plays a crucial role in stem cell maintenance, apical meristem formation, embryonic development, and various other developmental processes. However, the identification and function of WOX genes have not been reported in perennial loquat. In this study, 18 EjWOX genes were identified in the loquat genome. Chromosomal localization analysis showed that 18 EjWOX genes were located on 12 of 17 chromosomes. Gene structure analysis showed that all EjWOX genes contain introns, of which 11 EjWOX genes contain untranslated regions. There are 8 pairs of segmental duplication genes and 0 pairs of tandem duplication genes in the loquat WOX family, suggesting that segmental duplications might be the main reason for the expansion of the loquat WOX family. A WOX transcription factor gene named EjWUSa was isolated from loquat. The EjWUSa protein was localized in the nucleus. Protein interactions between EjWUSa with EjWUSa and EjSTM were verified. Compared with wild-type Arabidopsis thaliana, the 35S::EjWUSa transgenic Arabidopsis showed early flowering. Our study provides an important basis for further research on the function of EjWOX genes and facilitates the molecular breeding of loquat early-flowering varieties.
Loquat is a subtropical evergreen fruit tree of the Rosaceae family (Cao et al., 2013; Zhang et al., 2016). Compared with many fruit trees, loquat has unique characteristics of flowering in winter and fruit ripening in late spring or early summer (Jiang et al., 2019a). Previous studies have found that EjTFL1, EjFRI, EjFT, EjGI, EjCO, EjFD, EjSOC1, EjLEY, EjSVP, EjAP1, and EjSPL played important roles in regulating loquat flowering (Chen et al., 2020; Esumi et al., 2005; Jiang et al., 2019a, 2019b, 2019c; Reig et al., 2017; Liu et al., 2013; Zhang et al., 2019a; 2016b). However, studies of transcription factors regulating loquat flowering were still limited.
Plant flowering is regulated by both external environmental factors and internal genetic factors. At present, the understanding of angiosperm flowering relies mainly on the studies of flowering regulation in the model plant Arabidopsis. In Arabidopsis, flower bud differentiation is mainly regulated by the photoperiod pathway, vernalization pathway, gibberellin (GA) pathway, autonomous flowering pathway, heat-sensing pathway, and age pathway. In this regulatory network, about 180 genes interact to regulate Arabidopsis flowering (Bergonzi et al., 2013; Zhou et al., 2013). Compared with Arabidopsis, the studies of flowering regulation in woody plants are relatively lacking and need further research.
The WOX family is a class of plant-specific transcription factors (Feng et al., 2021). Its members possess 60-65 amino acid residues with the helix-loop-helix-turn-helix domain (referred to as homeodomain) and specifically bind DNA by the homeodomain to activate or depress the expression of the target gene in plants (Ikeda et al., 2009; Shafique Khan et al., 2021). WOX genes were divided into three separate clades, modern/WUS clade (WC), intermediate clade (IC), and ancient clade (AC) according to the time of their appearance during plant evolution (Alvarez et al., 2018). Based on phylogenetic analysis, WOX genes were further divided into nine subgroups (Zhang et al., 2010). In addition to the homeodomain, some WOX proteins contain three other functional domains: the acidic region (Rich in glutamic acid and aspartic acid), the WUS-box (T-L-X-L-F-P-X-X, X is an uncertain amino acid), and the EAR-like motif (X-L-X-L-X-L, X is an uncertain amino acid). The WUS-box is critical for regulating stem cell identity and floral meristem size. The acidic region is the only activation domain of the WUS proteins. In addition to the WUS-box, the EAR-like motif is also a repression domain (Ikeda et al., 2009). The WUS gene is the earliest gene identified in the WOX gene family (Xu, 2021). The negative feedback loop between WUS and CLAVATA3 (CLV3) underlies the maintenance of stem cell homeostasis in the shoot apical meristem (SAM) (Yadav, 2012; Xiao et al., 2018; Lopes et al., 2021). Previous studies have demonstrated that WUS protein regulates the expression of CLV3 gene in the organizing center and central zone by forming homodimers with itself and heterodimers with SHOOT MERISTEMLESS (STM), respectively. In turn, CLV3 forms a signaling complex with CLAVATA1 (CLV1) and CLAVATA2 (CLV2) to regulate the expression of the WUS gene in the organizing center (Daum et al., 2014; Perales et al., 2016; Zhou et al., 2018; Su et al., 2020). Previous studies have demonstrated that the WOX genes regulated plant flowering and development (Tvorogova et al., 2021). However, genome-wide identification and functional analysis of the WOX genes in loquat have not been reported.
In this study, we systematically identified the WOX family in the loquat genome and analyzed the chromosomal localization, gene structure, conserved motifs, and basic characteristics of the loquat WOX family. A WOX transcription factor gene named EjWUSa was isolated from the triploid loquat ‘Wuhezaoyu’. The subcellular localization of EjWUSa was observed in Nicotiana benthamiana leaves. Meanwhile, the interactions between EjWUSa with EjWUSa and EjSTM were verified. Finally, we overexpressed EjWUSa in wild-type Arabidopsis for functional analysis.
Triploid loquat ‘Wuhezaoyu’ was cultivated in the orchard of loquat resources belonging to the College of Horticulture and Landscape Architecture, Southwest University. In our previous study, the development of loquat buds was divided into nine stages (Jing et al., 2020), and from July to November 2021, the loquat flower buds at 9 stages were collected from the 12 years of triploid loquat ‘Wuhezaoyu’. After removing the superficial fluff, the loquat flower buds were immediately frozen in liquid nitrogen and stored in an ultra-low temperature refrigerator at −80°C until use.
Wild-type Arabidopsis was used for stable transformation of the EjWUSa gene. Tobacco was used in transient expression assays. Both Arabidopsis and tobacco were grown under long-day conditions (16 h light/8 h dark) at 22°C in a controlled environment room. Arabidopsis leaves for qRT-PCR were immediately frozen in liquid nitrogen and stored in an ultra-low temperature refrigerator at −80°C until use.
The Hidden Markov Model (HMM) of the Homeobox (HOX) superfamily (PF00046) was obtained at the Pfam website (http://pfam.xfam.org/). The WOX protein sequences in the model plant Arabidopsis were obtained from the TAIR database (https://www.arabidopsis.org/). The WOX family is one of six families in the HOX superfamily (Feng et al., 2021). The HMM of HOX superfamily was used as a query to search for candidate EjHOX genes in the loquat genome (The data presented in the study are deposited in NGDC repository accession No. GWHBOTF00000000). A total of 134 candidate EjHOX genes were identified in loquat. The conserved domain of candidate EjHOX genes was analyzed in NCBI (http://www.ncbi.nlm.nih.gov). As shown in Figure S1, 18 EjHOX genes and 15 AtWOX genes were in the same branch. Therefore, 18 EjWOX genes were finally identified in loquat and named according to their homology with the AtWOX genes (Figure S1; Table S1).
The apple genome file (V 3.0) was downloaded from the apple information resource (GDR, https://www.rosaceae.org) (Wang, 2021). Similar to the EjHOX genes, 130 candidate MdHOX genes were identified in the apple genome. The conserved domain of candidate MdHOX genes was analyzed in NCBI. As shown in Figure S2, 17 MdHOX genes and 15 AtWOX genes were in the same branch. Therefore, 17 MdWOX genes were finally identified in apple and named according to their homology with the AtWOX genes (Figure S2; Table S2).
The WOX protein sequences in the model plant tomato were obtained from the article published by Li et al. (2018). A phylogenetic tree including WOX proteins in loquat, apple, Arabidopsis, and tomato was established by MEGA software (v 11.0) with the Neighbor-joining method based on the following parameters: pairwise deletion and bootstrap analysis with 1000 replicates (Munir et al., 2016). The phylogenetic tree was imported into the iTOL website (https://itol.embl.de/) for further beautification. Multiple sequence alignment of WOX proteins in loquat were analyzed using DNAMAN software (v 9.0) with the default settings. The ExPASY database (https://web.expasy.org/protparam/) was used to forecast the characteristics of EjWOX proteins including the coding sequence length, theoretical isoelectric point (PI), molecular weight, and amino acid length (Wilkins et al., 1999).
The MEME suite (http://meme-suite.org/tools/meme) was used to identify the conserved motifs (Bailey et al., 2009). Chromosomal localization, gene structure, conserved motif, and synteny analysis of the EjWOX genes were visualized using TBtools software (v 1.098763) (Chen et al., 2020).
Genes and promoter sequences were obtained in the loquat genome. Gene was amplified from the cDNA of the triploid loquat ‘Wuhezaoyu’ flower buds by PCR using Phanta Max Super-Fidelity DNA polymerase (Vazyme, China). The PCR product was ligated with the pMD19-T vector. Then, the cloned product was sequenced. Finally, multiple sequence alignment was performed using DNAMAN software. Based on the same method, the promoter was isolated from the DNA of ‘Wuhezaoyu’. The specific primers were listed in Table S3. Then, the putative cis-acing elements on the promoter region were found in the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Wilkins et al., 2005). The result was visualized using TBtools software.
The total RNA was extracted by EASYspin Plus plant RNA extraction kit (Aidlab, China), and the cDNA was synthesized using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Japan). qRT-PCR was performed using NovoStart® SYBR qPCR SuperMix plus (Novoprotein, China). The loquat EjActin gene and Arabidopsis AtActin gene were used as internal controls, with the special primers in Table S3. Three biological replicates were applied and data were analyzed with the 2−ΔΔCt method (Livak and Schmittgen, 2001).
The coding sequence (without stop codon) of EjWUSa was cloned into the modified pCAMBIA1300 vector (Jing et al., 2020) with the special primers in Table S3. The constructed fusion vector or empty vector was transformed into Agrobacterium strain GV3101, respectively, and then tobacco leaves were used for transient expression. An Olympus (BX35) fluorescence fluorescence signals.
The coding sequence (without stop codon) of EjWUSa was constructed into the pXY104 vector and the special primers with restriction sites (Sal I and BamH I) were listed in Table S3. The coding sequence of EjWUSa or EjSTM was constructed into the pXY106 vector and the special primers with restriction sites (Sal I and BamH I) were listed in Table S3. The constructed fusion vectors or empty vectors were transformed into Agrobacterium strain GV3101, respectively, and then tobacco leaves were used for transient expression. An Olympus (BX35) fluorescence microscope (Tokyo, Japan) was used to observe the fluorescence signals. Three independent leaves were observed. The method was described by Liu et al. (2021).
The coding sequence of EjWUSa was constructed into the pCAMBIA-CLuc vector and the special primers with restriction sites (Sal I and BamH I) were listed in Table S3. The coding sequence (without stop codon) of EjWUSa or EjSTM was constructed into the pCAMBIA-NLuc vector and the special primers with restriction sites (Sal I and BamH I) were listed in Table S3. The constructed fusion vectors or empty vectors were transformed into Agrobacterium strain GV3101, respectively, and then tobacco leaves were used for transient expression. After 2 days of dark treatment, 1 mmol of fluorescein (Promega, USA) was applied to the injection site of tobacco leaves and dark treated for six minutes. The signals were then captured with a CCD imaging instrument (Alliance, UK). Three independent leaves were observed. The method was described by Liu et al. (2021).
The coding sequence of EjWUSa was cloned into the pFGC5941 vector with the special primers in Table S3. The constructed fusion vector was transformed into Agrobacterium strain GV3101, and then wild-type Arabidopsis (WT) were used for stable expression assay. The seeds of 35S::EjWUSa transgenic Arabidopsis and WT were planted in the soil after treatment at 4°C for 24 h. Basta was used to screen 35S::EjWUSa transgenic Arabidopsis. DNA was extracted from 35S::EjWUSa transgenic Arabidopsis and WT leaves and then PCR was performed. RNA was extracted from 35S::EjWUSa transgenic Arabidopsis and WT and then expression analysis was performed using special primers in Table S3. Finally, we obtained the T3 homozygous 35S::EjWUSa transgenic lines. We counted the bolting time and the flowering time of 35S::EjWUSa transgenic Arabidopsis and WT, respectively. Meanwhile, we counted the number of rosette leaves of 35S::EjWUSa transgenic Arabidopsis and WT when the flowering shoot was 1 cm, respectively. All data were analyzed for significance by SPSS 26.0 software with One‐way ANOVAs analysis.
The HMM of HOX superfamily was used as a query to search for candidate EjHOX genes in the loquat genome. A total of 134 candidate EjHOX genes were identified in the loquat genome, and the conserved domain of candidate EjHOX genes was analyzed in NCBI. The result showed 18 EjWOX genes were finally identified in the loquat (Figure S1). Phylogenetic analysis showed that the WOX proteins in loquat, apple, Arabidopsis, and tomato were similar (Figure 1). The EjWOX genes were classified into three well-supported clades (Figure 1). The WUS clade had the largest number of members, 11 in total, and the intermediate clade and ancient clade contained 4 and 3 members, respectively (Figure 1). The EjWOX genes were divided into nine subgroups based on phylogenetic analysis (Figure 1). Multiple sequence alignment showed that 13 amino acid residues are strictly conserved in the homeodomain of the EjWOX proteins, including Q and L in helix1, G in loop, P and L in helix2, G in turn, and N, V, W, F, Q, N, and R in helix3 (Figure 2A). An extra amino acid residue in the black box was observed in the homeodomain of the EjWOX proteins within the WUS subgroup (Figure 2A). The amino acid residue might be essential for their biological function. The WUS-box was found only in EjWOX proteins within the WUS clade (Figure 2B). The EAR-like motif was found only in EjWOX proteins within the WUS subgroup and WOX5 subgroup (Figure 2C). The acidic region was found only in EjWOX proteins within the WUS subgroup (Figure 2D).
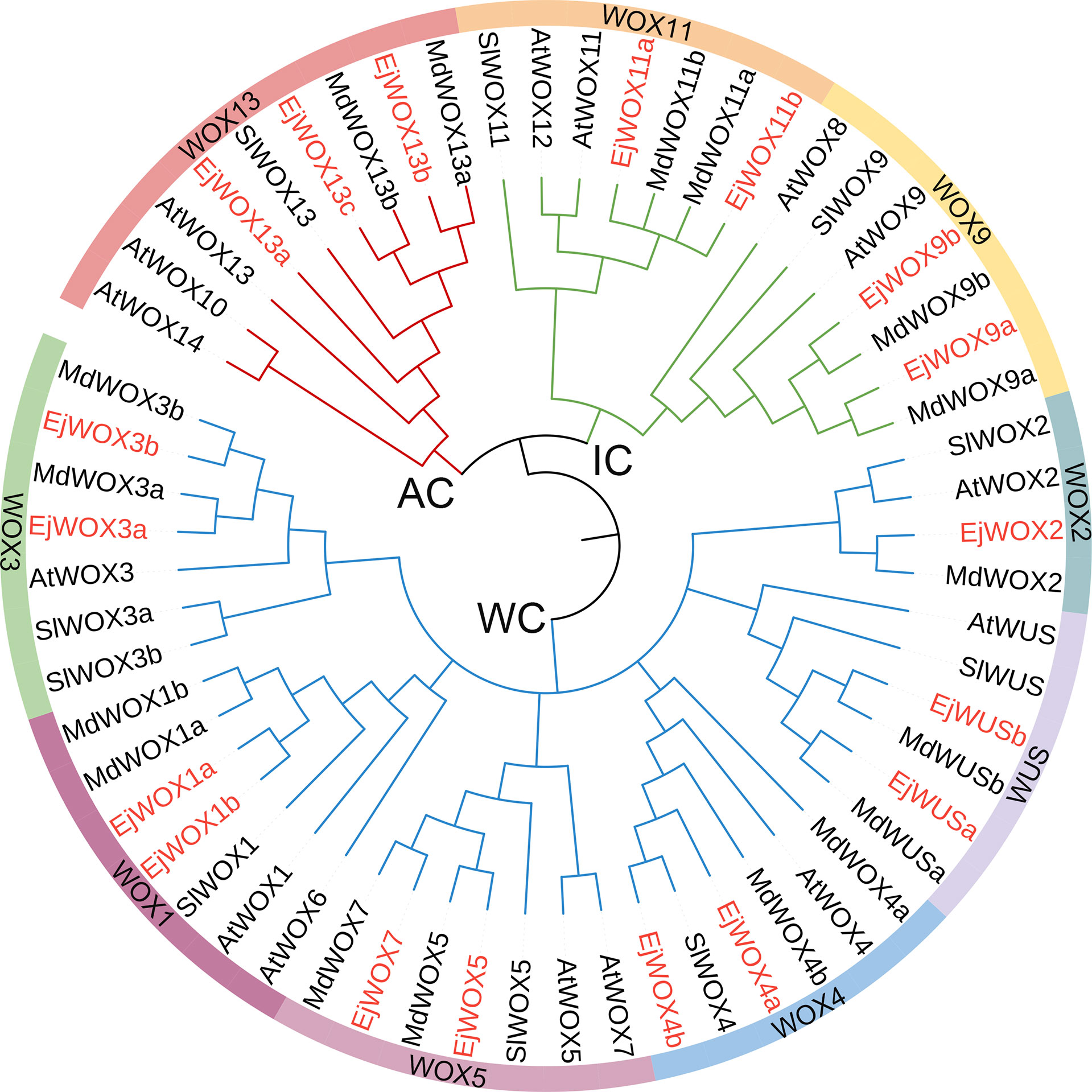
Figure 1 The phylogenetic tree including WOX proteins from Eriobotrya japonica, Malus domestica, Solanum lycopersicum, and Arabidopsis thaliana. Nine subgroups of the WOX family are represented by different colors. WC, WUS clade. IC, intermediate clade. AC, ancient clade. The WOX proteins in loquat are marked with red.
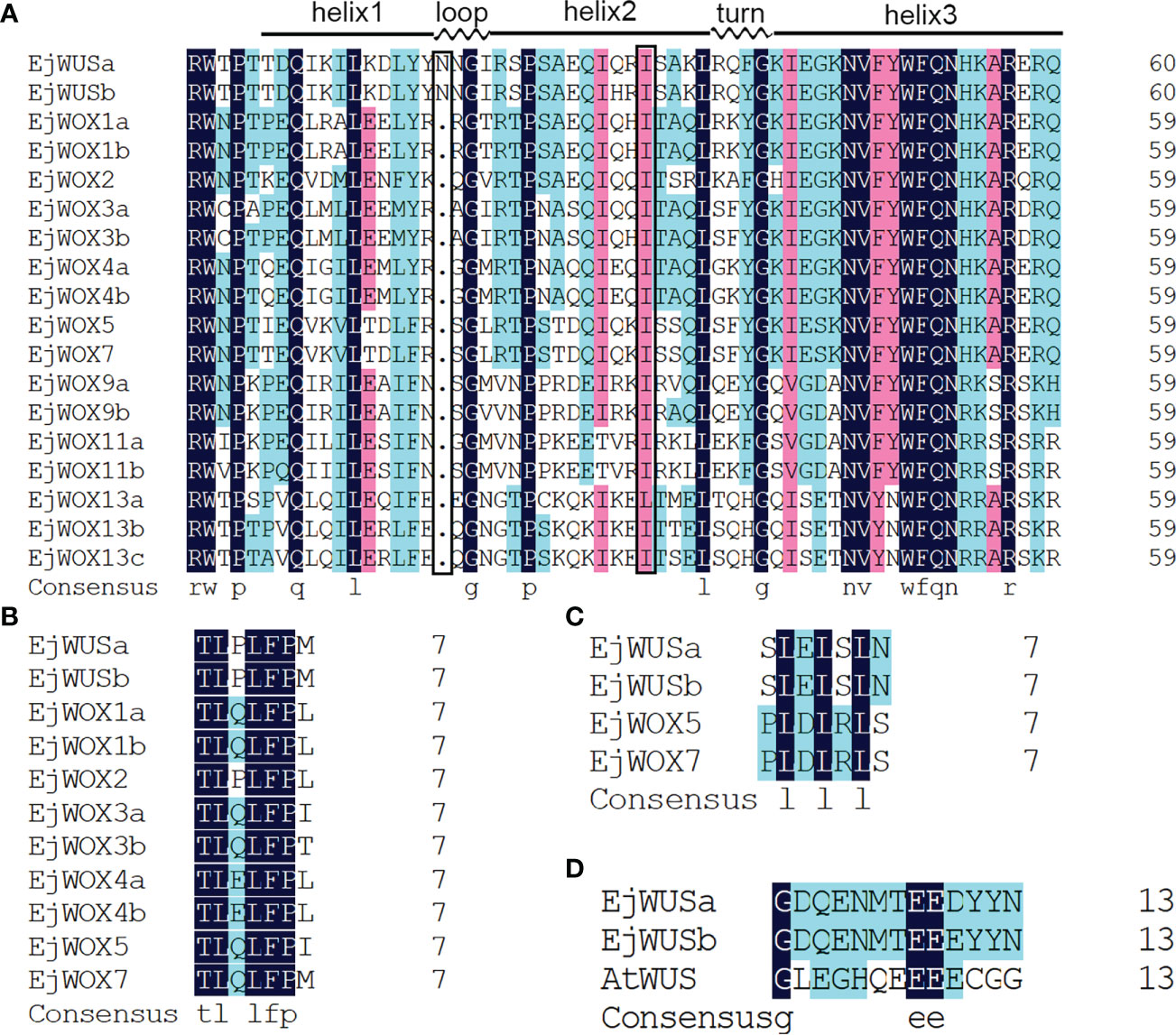
Figure 2 Multiple sequence alignment of WOX proteins in loquat. (A) Multiple sequence alignment of the homeodomain. An extra amino acid residue in the black box was observed in the homeodomain of the EjWOX proteins within the WUS subgroup. (B) Multiple sequence alignment of the WUS-box. (C) Multiple sequence alignment of the EAR-like motif. (D) Multiple sequence alignment of the acidic region. The identical residues are in black boxes.
We analyzed the localization of 18 EjWOX genes on 17 loquat chromosomes. The 18 EjWOX genes were unevenly distributed on 12 of the 17 chromosomes (Figure 3A). No EjWOX genes were distributed on chromosomes 7, 8, 11, 12, and 13 (Figure 3A). Chromosomes 1, 2, 4, 10, 15, 16, and 17 had only one EjWOX gene (Figure 3A). Chromosomes 5, 6, 9, and 14 had two EjWOX genes (Figure 3A). Moreover, chromosome 3 had the highest number of EjWOX genes, with three EjWOX genes (Figure 3A). A total of 8 pairs of segmental duplication genes and 0 pairs of tandem duplication genes were identified in the loquat WOX family (Figure 3B). The length of EjWOX proteins ranged from 176 aa (EjWOX7) to 409 aa (EjWOX9a) with an average of 286.94 aa (Table 1). Their molecular weights ranged from 20.16 KDa (EjWOX7) to 45.14 KDa (EjWOX9a), with an average of 32.25 KDa (Table 1). In addition, these proteins might be mainly composed of basic amino acids with isoelectric points ranging from 4.71 (EjWOX13a) to 9.26 (EjWOX4b), with an average of 7.48 (Table 1).
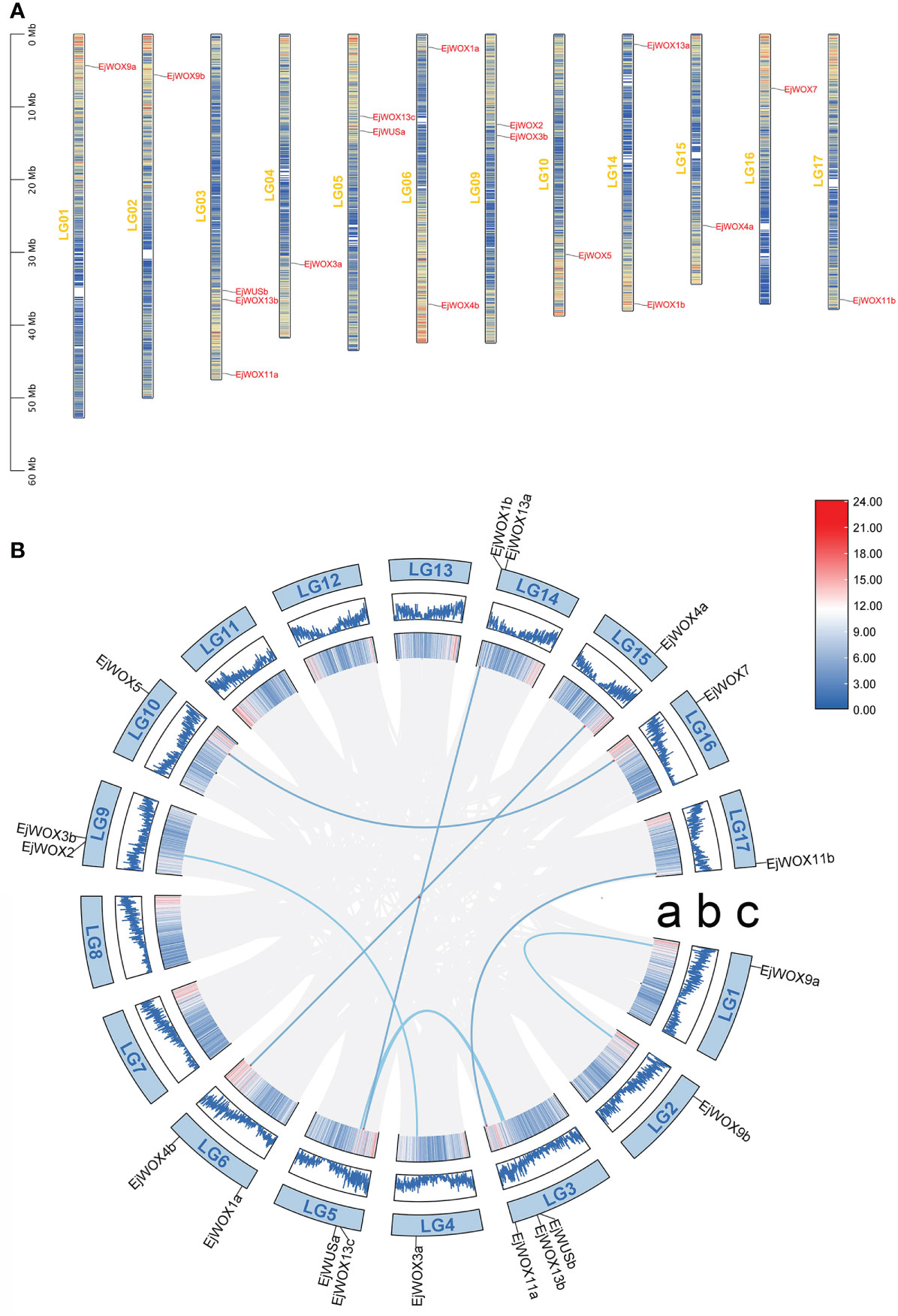
Figure 3 Chromosomal localization and synteny analysis of the EjWOX genes. (A) The chromosomal localization of the EjWOX genes. The chromosome numbers are marked on the left of each chromosome. Gene names are marked to the right of each chromosome. The black line with the scale on the left represents the length of the chromosomes. The colored lines within these columns represent gene density. (B) The synteny analysis of the EjWOX genes. Syntenic gene pairs are connected by blue lines. a: The colored lines indicate gene density. b: The width of the blue lines represents the gene density. c: LG01-LG17 represent 17 loquat chromosomes.
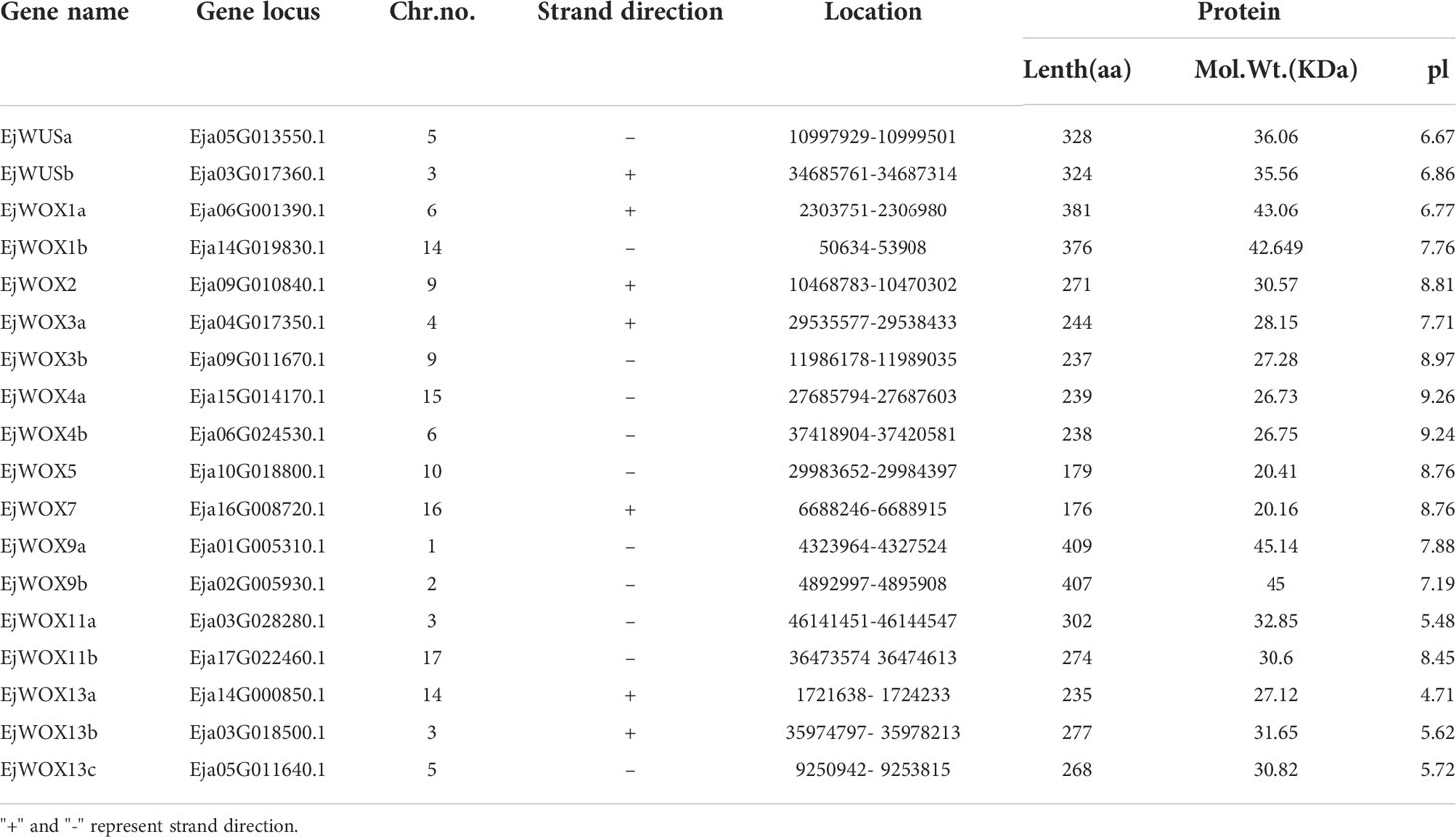
Table 1 The characteristics of EjWOX genes in loquat.
The conserved motifs of the loquat WOX proteins were analyzed using MEME program and five different motifs were obtained (Figure 4A). All EjWOX proteins contain motif 1 and motif 2 (Figure 4A). Sequence analysis showed that motif 1 and motif 2 make up the homeodomain. All EjWOX proteins within the WUS clade contain motif 4 (Figure 4A). Sequence analysis showed that motif 4 is the WUS-box. All EjWOX proteins within the ancient clade contain motif 3 (Figure 4A). Except for EjWOX11b, all EjWOX proteins within the intermediate clade contain motif 5 (Figure 4A). EjWOX proteins within the same clade contained similar motifs, indicating that they might undertake similar biological functions (Figure 4A). We further analyzed the exon-intron structure of the EjWOX genes. All EjWOX genes contain introns (Figure 4B). The intron number of EjWOX genes varied from one to three (Figure 4B). Among the 18 EjWOX genes, there were 11 genes containing untranslated regions (Figure 4B). Only 7 EjWOX genes did not contain untranslated regions, i.e., EjWUSa, EjWUSb, EjWOX2, EjWOX5, EjWOX7, EjWOX11a, and EjWOX11b (Figure 4B). The number and length of exons, introns, and untranslated regions (UTRs) were conserved in EjWOX genes within the same subgroup (Figure 4B).
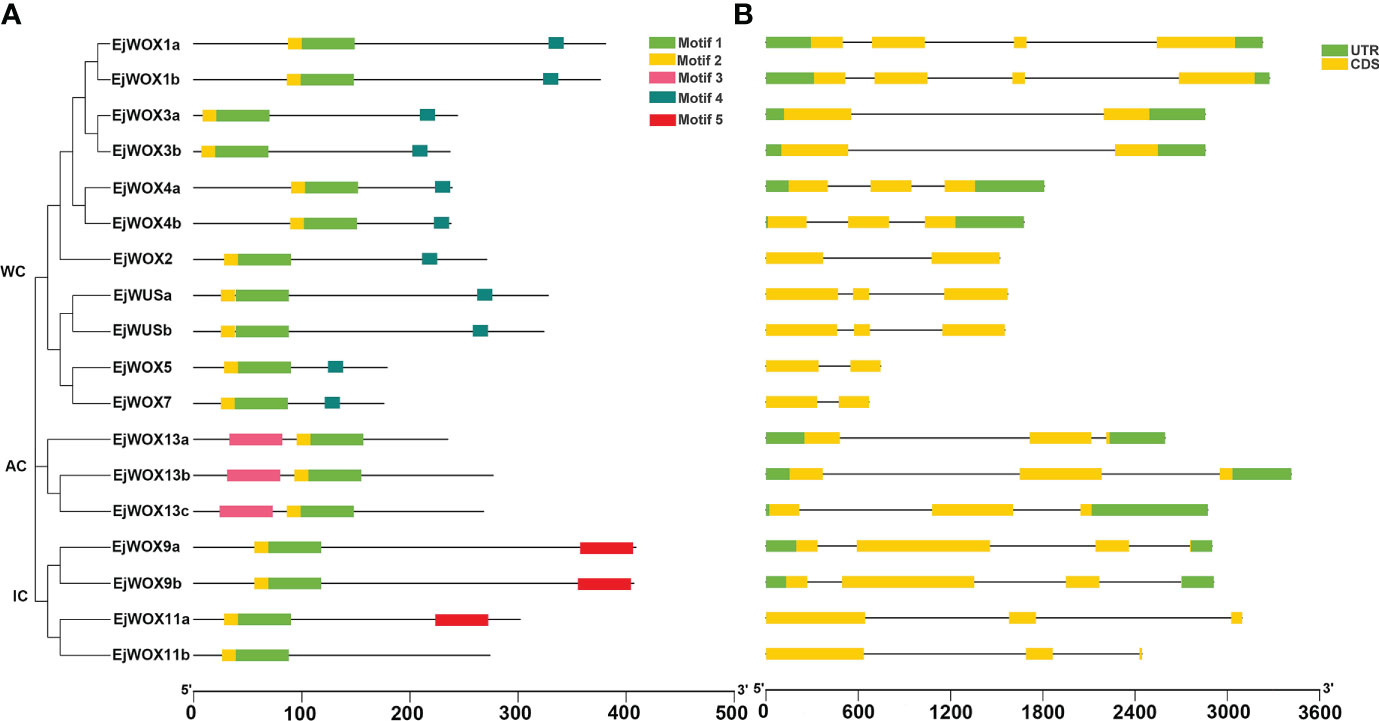
Figure 4 Conserved motif and gene structure analysis of loquat WOX genes. (A) Conserved motifs in EjWOX proteins are represented by colored boxes. (B) UTRs, exons, and introns are represented by green squares, yellow squares, and gray lines, respectively. Black lines indicate length. WC, WUS clade. IC, intermediate clade. AC, ancient clade.
From July to November 2021, the loquat flower buds at 9 stages were collected from the 12-year-old triploid loquat ‘Wuhezaoyu’. The RNA degradation of loquat flower buds at the petal fall was serious, so the loquat flower buds at the petal fall were not suitable for qRT-PCR (Figure 5A, B). Transcriptome analysis of loquat flowers showed that WUS might be a floral meristem identity gene (Jing et al., 2020). We analyzed the expression levels of EjWUSa and EjWUSb in loquat flowers at different stages. In the early stages of flower development, the expression level of EjWUSa was significantly higher than that of EjWUSb (Figure 5C), suggesting that EjWUSa might play an important role in the transition from the vegetative apex to reproductive apex. Therefore, we further cloned EjWUSa from loquat and then investigated its role in loquat flowering.
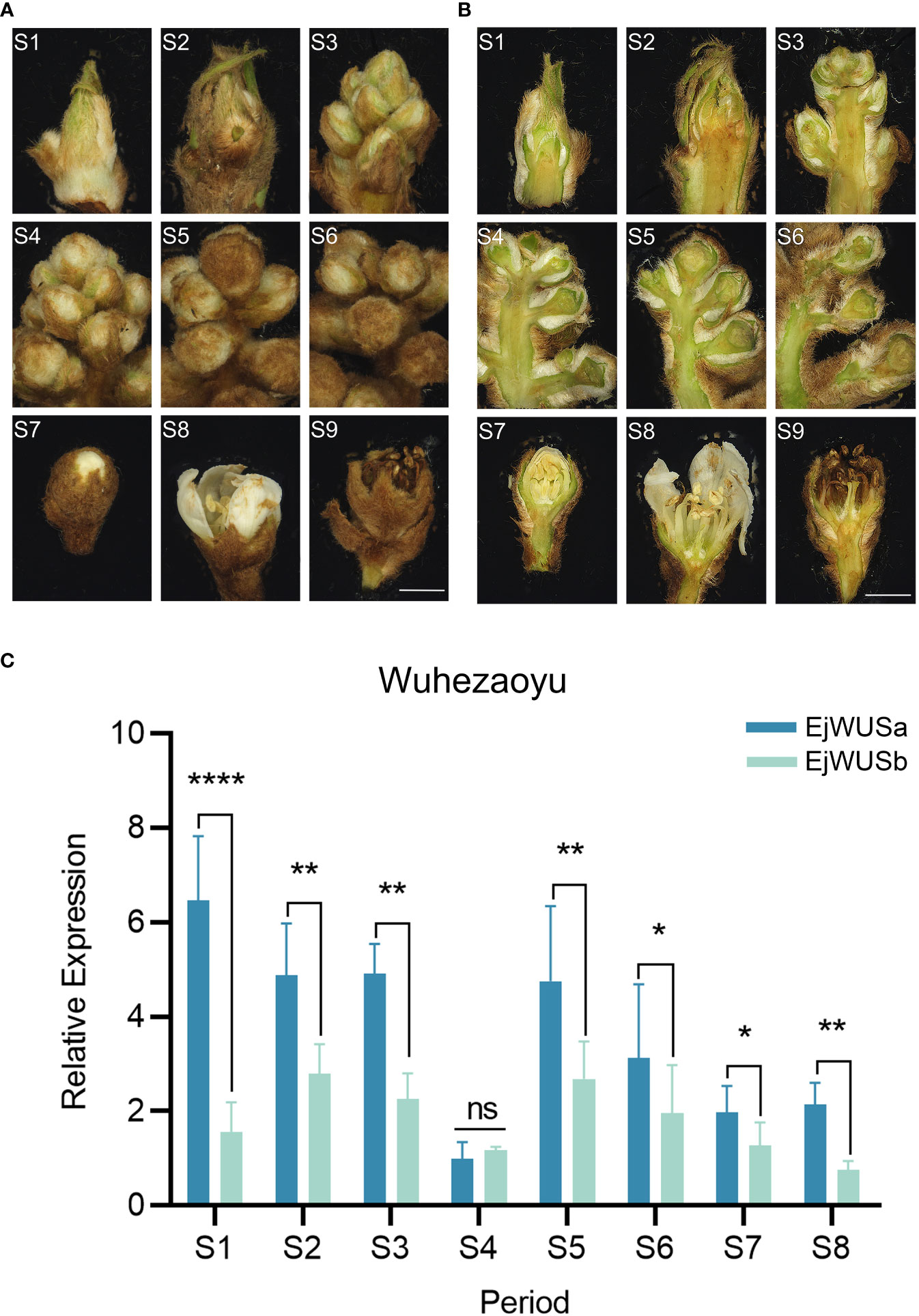
Figure 5 Inflorescence morphology and morphological anatomy of the triploid loquat ‘Wuhezaoyu’ and the temporal expression patterns of EjWUSa and EjWUSb in the triploid loquat ‘Wuhezaoyu’ flower buds. (A, B) Inflorescence morphology and morphological anatomy of the triploid loquat ‘Wuhezaoyu’. S1: Vegetative apex; S2: Floral meristem initiation and flower bud differentiation; S3: Rapid differentiation of floral buds; S4: Panicle elongation; S5: Floral bud elongation with visible floral buds; S6: Elongation of branches in a panicle; S7: White corollas of floral buds; S8: Floral anthesis and full bloom; S9: Petal fall. Bars = 2000µm. (C) The temporal expression patterns of EjWUSa and EjWUSb in the triploid loquat ‘Wuhezaoyu’ flower buds. Ejactin as an internal control. Error bars indicate Standard Error (SE) from three biological replicates. Asterisks or n.s. indicate significant differences between EjWUSa and EjWUSb, *p<0.05, **p<0,01, **** P < 0.0001, ns, no significant difference, by One-way ANOVAs.
The EjWUSa was isolated from the cDNA of triploid loquat ‘Wuhezaoyu’ flowers (Figure S3), which contains three exons and two introns, but no untranslated region (Figure 4B). The coding sequence of EjWUSa is 987 bp, encoding 328 amino acids (Table 1). The EjWUSa contains four conserved domains from the N-terminal to the C-terminal, followed by the homeodomain, the acidic region, the WUS-box, and the EAR-like motif (Figure 2). The EjWUSa promoter was cloned from the DNA of the triploid loquat ‘Wuhezaoyu’ leaves (Figure S3; Table S4). We further analyzed the binding elements on the EjWUSa promoter (Figure S4). The result showed that the EjWUSa promoter contains one CAT-box related to meristem expression (Figure S4) (Lin et al., 2022), suggesting that the EjWUSa might be involved in forming the shoot apical meristem. In addition, the EjWUSa promoter also contains two Box4s and two TCCC-motifs (Figure S4) (Chen and Qiu, 2020), indicating that the expression of EjWUSa might be regulated by light signals.
The coding sequence (without stop codon) of EjWUSa was cloned into the pCAMBIA1300 vector to generate a fusion protein in tobacco cell. The green fluorescent protein (GFP) in the control group was localized in the nucleus and cell membrane, while the EjWUSa-GFP fusion protein in the experimental group was localized in the nucleus (Figure 6). It showed that the EjWUSa protein was localized in the nucleus, consistent with the characteristics of transcription factors.
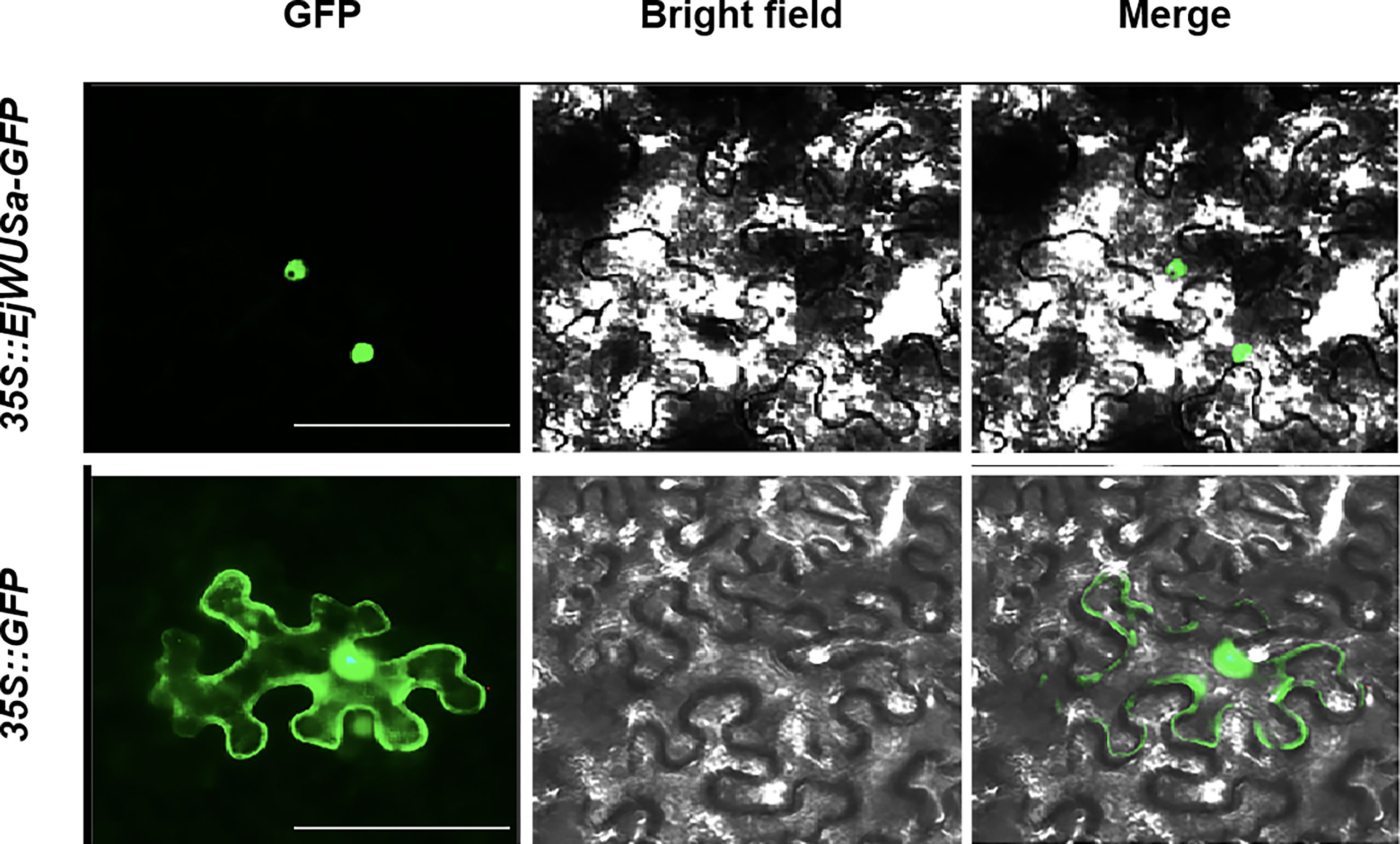
Figure 6 Subcellular localization of EjWUSa in tobacco leaves. GFP, GFP fluorescence channel; Merged, merged image of GFP and bright field. Bars = 50 µm.
To investigate whether the protein interactions between WUS and WUS and STM are conserved across species, we isolated the EjSTM from the cDNA of the triploid loquat ‘Wuhezaoyu’ flower buds (Figure S3; Table S5). The BiFC assay and LCI assay were used to verify whether EjWUSa can form a dimer with EjSTM or EjWUSa in tobacco cells. In the experimental groups, strong signals were observed in tobacco cells, while no signals were observed in the control groups (Figure 7). This suggested that EjWUSa formed dimers with EjWUSa or EjSTM, respectively, which provided the basis for the formation of a complete YFP or LUC.
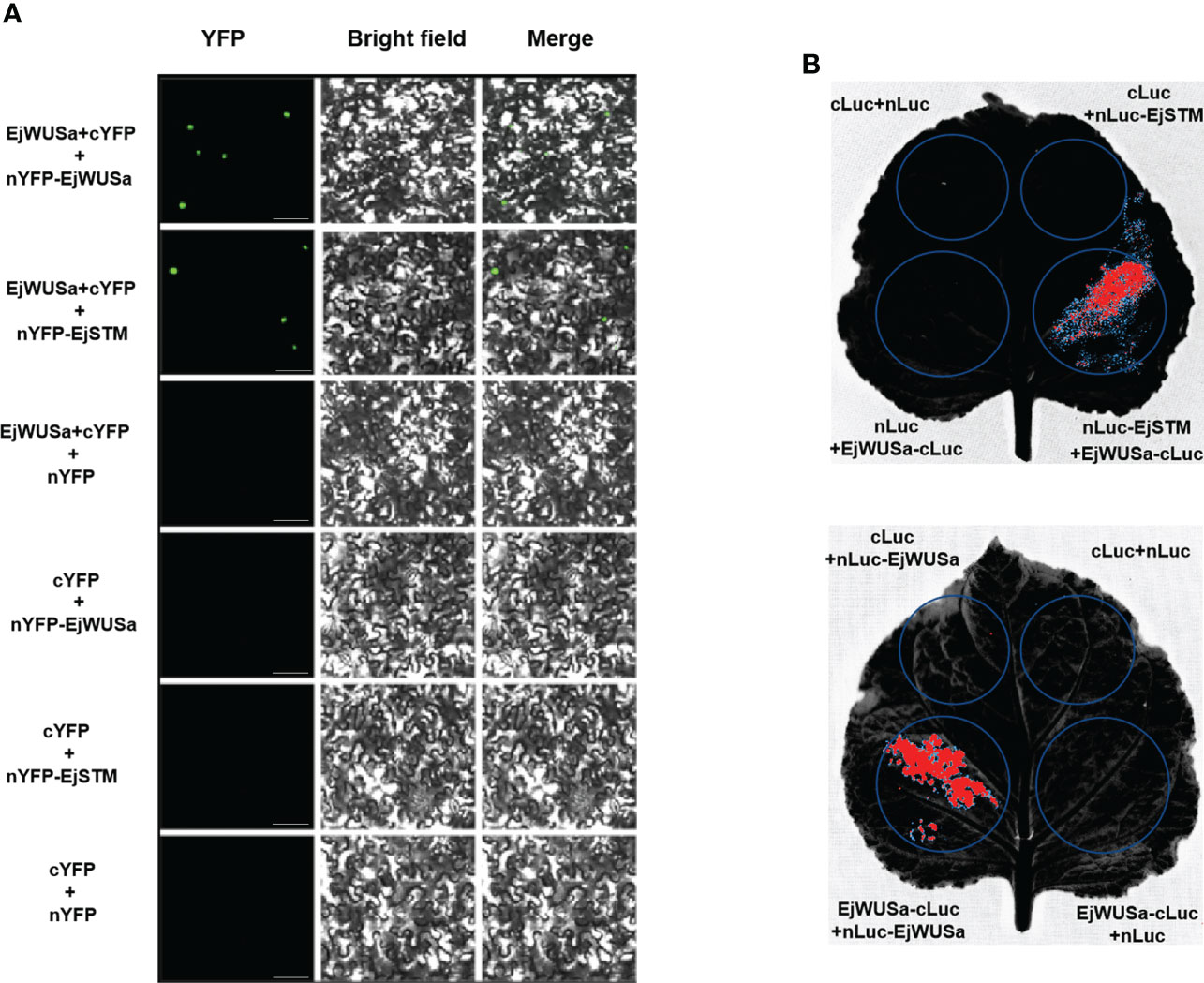
Figure 7 Protein interactions between EjWUSa with EjWUSa and EjSTM. (A) Protein interactions between EjWUSa with EjWUSa and EjSTM were demonstrated in the BIFC assay. The scale bar is 20 μm. (B) Protein interactions between EjWUSa with EjWUSa and EjSTM were demonstrated in the LCI assay.
To investigate the function of EjWUSa, an overexpression vector containing the coding sequence of the EjWUSa gene was constructed and transformed into wild-type Arabidopsis (WT). After Basta screening and PCR identification, we obtained T3 homozygous 35S::EjWUSa transgenic lines (Figure S5). The expression level of EjWUSa in 35S::EjWUSa transgenic lines was higher than in WT (Figure 8D). Under the same growing conditions, WT had about 12 rosette leaves, whereas the 35S::EjWUSa transgenic lines had only seven to nine rosette leaves (Figures 8B, C). All 35S::EjWUSa transgenic lines had approximately 10 days early bolting and 9 days early flowering compared to WT (Figures 8A, B). In conclusion, all 35S::EjWUSa transgenic lines showed an early flowering phenotype compared to WT. Compared to WT, the 35S::EjWUSa transgenic lines had no significant difference in morphological characteristics such as flower organs, leaf shape, siliques, stems, and leaves. From the above results, EjWUSa has the function of promoting Arabidopsis flowering.
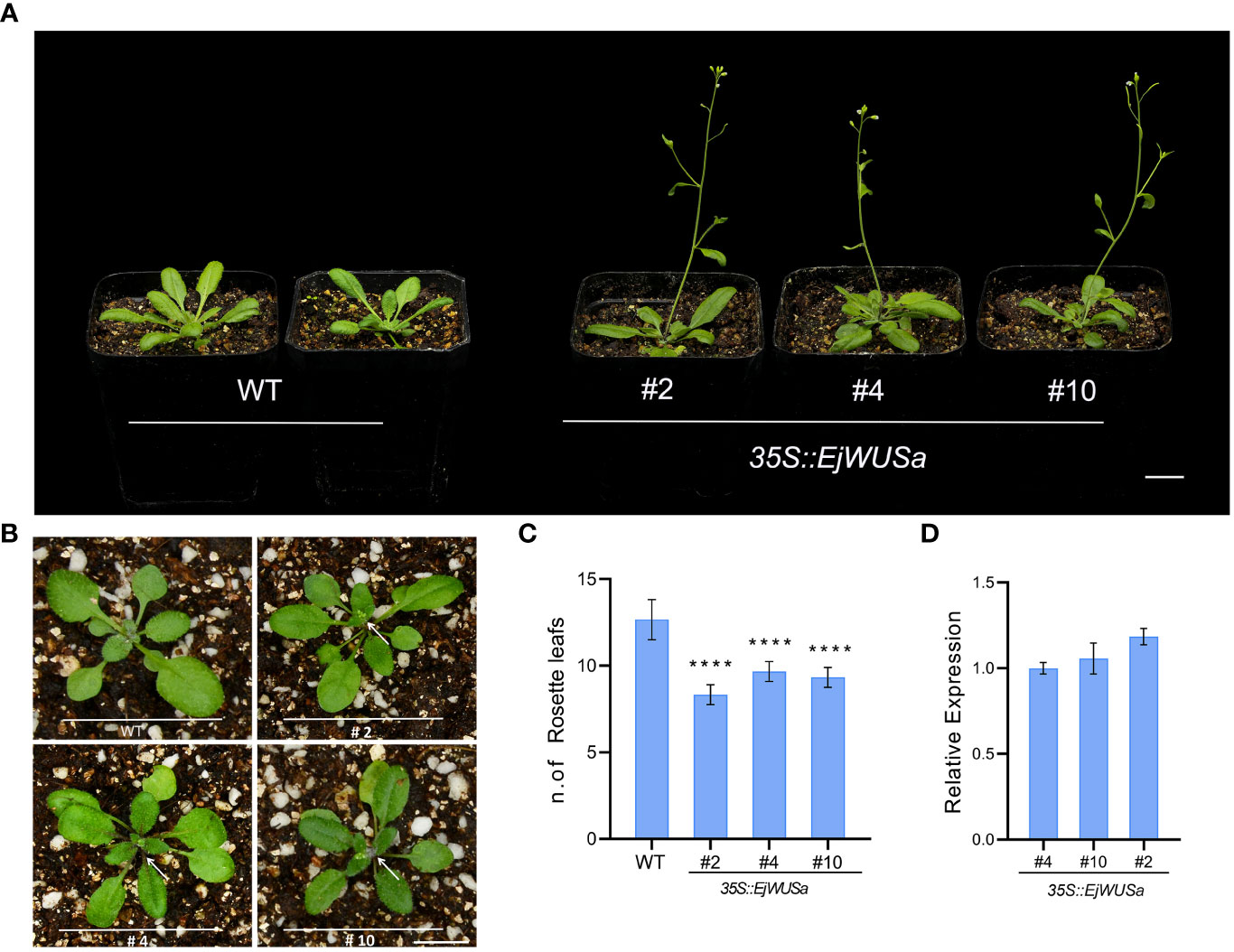
Figure 8 Overexpression of EjWUSa promotes Arabidopsis flowering. (A) 35S::EjWUSa transgenic lines showed early flowering compared to WT. Bars = 1cm. (B) 35S::EjWUSa transgenic lines showed early bolting compared to WT. Bars = 1cm. (C) The number of rosette leaves of 35S::EjWUSa transgenic lines and WT while blooming. (D) The expression level of EjWUSa in 35S::EjWUSa transgenic lines and WT. The leaves of 35S::EjWUSa transgenic Arabidopsis and WT were collected after flowering and used for qRT-PCR analysis. Error bars indicate SE from three biological replicates. Asterisks indicate significant differences between 35S::EjWUSa transgenic lines and WT, ****p < 0.0001, by One-way ANOVAs.
As a category of transcription factors regulating stem cell fate, WOX proteins are involved in many physiological processes related to plant growth and development (van der Graaff et al., 2009; Gambino et al., 2011; Lin et al., 2012; Kanchan and Sembi, 2020). Due to advances in omics technologies, genome-wide identification of the WOX gene family has been accomplished in several species, including Arabidopsis, sorghum, maize, tobacco, potato, walnut, and sweet orange. These plants contain 15, 11, 21, 10, 8, 14, and 8 WOX genes, respectively (2020; Vandenbussche et al., 2009; Zhang et al., 2010; Li et al., 2018; Shafique Khan et al., 2021). However, genome-wide identification and function of the WOX genes have not been reported in loquat. In the present study, we performed the genome-wide identification of the WOX gene family in loquat. Meanwhile, we further investigated the role of EjWUSa in loquat flowering.
A total of 18 EjWOX genes were identified in the loquat genome (Table S1). Like the AtWOX genes, the EjWOX genes can be divided into three clades or nine subgroups. Nevertheless, the homologs of Arabidopsis AtWOX6, AtWOX10, and AtWOX14 could not be found in loquat (Figure 1). The substitutability of WOX genes might make their loss possible (Feng et al., 2021). Gene duplication is the main reason for gene family expansion (Feng et al., 2021). In our study, 8 pairs of segmental duplication genes and 0 pairs of tandem duplication genes were identified in the loquat WOX family (Figure 3B), suggesting that segmental duplications might be the main reason for the expansion of the loquat WOX family. The homeodomain is conserved in the WOX gene family among different species and maintains the functional integrity of WOX genes (Zhang et al., 2010; Kanchan and Sembi, 2020). As shown in Figure 2A, the 11th amino acid residue (In the black box) in helix 2 of the homeodomain is not strictly conserved in loquat WOX proteins, which differs from that of other species, such as apple (Figure S6), walnut, tomato, potato, rice, sorghum, maize, Arabidopsis, and poplar (Zhang et al., 2010; Li et al., 2018; Chang et al., 2019). We found a fragment of the EjWUS proteins consisting of 10 amino acids residues, rich in glutamate residues and aspartate residues, and with the same number of acidic residues as the AtWUS protein (Figure 2D). It should be considered as an acidic region. Therefore, the acidic region is not a conserved domain specific to the AtWUS protein. However, the acidic region was not detectable in WUS proteins in rice, sorghum, or maize (Zhang et al., 2010). The EjWOX genes within the ancient clade were more conserved in the number and length of exons, introns, and UTRs compared to those within the intermediate and modern clades, consistent with previous studies (Figure 4B) (Deveaux et al., 2008).
EjWUSa was shown to interact with EjWUSa and EjSTM (Figure 7), consistent with studies in Arabidopsis (Perales et al., 2016; Zhou et al., 2018). This suggested that protein interactions between WUS with WUS and STM might be conserved across species. In Arabidopsis, AtWOX proteins were essential for embryonic patterning, stem cell maintenance, and organ formation (Ji et al., 2010). AtWOX2 was involved in the formation of apical-basal axis (2008; Palovaara, 2010). The wox3/prs1 mutant Arabidopsis exhibited short stature, abnormal sepal number and morphology, and the absence of stamens and stipules (Shimizu et al., 2009). OsWOX3A, which is the rice homolog of AtWOX3, was involved in the pleiotropic effects of organ development (Cho et al., 2013). As a homolog of AtWOX9, Cymose Petunia WOX9 played an important role in the inflorescence development (Rebocho et al., 2008). AtWOX13 and AtWOX14 played important roles in the development of roots and the formation of floral organs (Deveaux et al., 2008). AtWOX5 maintained the maintenance of stem cell homeostasis in the root apical meristem (RAM) (Sarkar et al., 2007; Nardmann et al., 2009; Tian et al., 2014). In Arabidopsis shoot and floral meristems, the AtWUS gene was required for stem cell identity (Schoof et al., 2000; Nardmann et al., 2007). When the AtWUS gene was mutated, it caused an early termination of inflorescence meristem (Laux et al., 1996). Arabidopsis overexpressing the AtWUS gene showed ectopic flower bud growth and callus-like formation (Xu et al., 2005). Functional verification of the WUS gene in other plants has been reported. For example, overexpression of the GhWUS gene increased the embryogenic callus formation in Gossypium hirsutum (Xiao et al., 2018). WUS protein has anti-virus activity by repressing MTase expression (Xu, 2021). In this study, 35S::EjWUSa transgenic Arabidopsis exhibits early flowering (Figure 8). Unlike studies in Arabidopsis, we did not find the callus-like tissue on the stems of transgenic lines. Multiple sequence alignment showed that the similarity between EjWUSa and AtWUS was only 32.94%, which might lead to their functional differences (Figure S7). This also reflects the difference in genetic information between woody plants and herbs.
We reported the first genome-wide identification of the WOX family in perennial loquat. In the present study, 18 EjWOX genes were identified in the loquat genome. We further analyzed the evolutionary features and basic characteristics of the loquat WOX genes. A total of 8 pairs of segmental duplication genes and 0 pairs of tandem duplication genes were identified in the loquat WOX family, suggesting that segmental duplications might be the main reason for the expansion of the loquat WOX family. Compared with WT, 35S::EjWUSa transgenic Arabidopsis exhibits early flowering. Our study provides an important basis for further research on the function of EjWOX genes in the future and also facilitates the molecular breeding of loquat early-flowering varieties.
The original contributions presented in the study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/gwh, GWHBOTF00000000.
YY and MY performed the experiments and drafted the manuscript. XL contributed to morphology analysis of diploid loquat. YX, RH, and QX contributed to the data analysis. QG, DJ, and YX provided plant tissues, laboratory facilities, and project supervision. All authors approved the final draft of the manuscript.
This work was financially supported by The National Natural Science Foundation of China (Grant Numbers: 32102321), the National Key R&D Program of China (No. 2019YFD1000900), the Chongqing Science and Technology Commission (cstc2021jscx-gksbX0010 and cstc2021jcyj-msxmX1156), the Innovation Research Group Funds for Chongqing Universities (CXQT19005), the Chongqing Forestry Administration (YuLinKeYan2022-14), and the Characteristic Fruit Industry and Technology System Innovation Team of Chongqing Agriculture and Rural Affairs Commission: No.2020[3]01.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1024515/full#supplementary-material
Alvarez, J. M., Bueno, N., Cañas, R. A., Avila, C., Cánovas, F. M., Ordás, R. J. (2018). Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in Pinus pinaster: New insights into the gene family evolution. Plant Physiol. Biochem. 123, 304–318. doi: 10.1016/j.plaphy.2017.12.031
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37 (Web Server issue), W202–W208. doi: 10.1093/nar/gkp335
Bergonzi, S., Albani, M. C., Ver Loren van Themaat, E., Nordström, K. J. V., Wang, R., Schneeberger, K., et al. (2013). Mechanisms of age-dependent response to winter temperature in perennial flowering of Arabis alpina. Sci. (New York N.Y.) 340 (6136), 1094–1097. doi: 10.1126/science.1234116
Cao, S., Yang, Z., Zheng, Y. (2013). Sugar metabolism in relation to chilling tolerance of loquat fruit. Food Chem. 136 (1), 139–143. doi: 10.1016/j.foodchem.2012.07.113
Chang, Y., Song, X., Zhang, Q., Liu, H., Bai, Y., Lei, X., et al. (2019). Genome-wide identification of WOX gene family and expression analysis during rejuvenational rhizogenesis in walnut (Juglans regia l.). Forests 11 (1), 16. doi: 10.3390/f11010016
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, S., Qiu, G. (2020). Cloning and activity analysis of the promoter of nucleotide exchange factor gene ZjFes1 from the seagrasses Zostera japonica. Sci. Rep. 10 (1), 17291. doi: 10.1038/s41598-020-74381-6
Chen, W., Wang, P., Wang, D., Shi, M., Xia, Y., He, Q., et al. (2020). EjFRI, FRIGIDA (FRI) ortholog from Eriobotrya japonica, delays flowering in Arabidopsis. Int. J. Mol. Sci. 21 (3), E1087. doi: 10.3390/ijms21031087
Cho, S.-H., Yoo, S.-C., Zhang, H., Pandeya, D., Koh, H.-J., Hwang, J.-Y., et al. (2013). The rice narrow leaf2 and narrow leaf3 loci encode WUSCHEL-related homeobox 3A (OsWOX3A) and function in leaf, spikelet, tiller and lateral root development. New Phytol. 198 (4), 1071–1084. doi: 10.1111/nph.12231
Daum, G., Medzihradszky, A., Suzaki, T., Lohmann, J. U. (2014). A mechanistic framework for noncell autonomous stem cell induction in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 111 (40), 14619–14624. doi: 10.1073/pnas.1406446111
Deveaux, Y., Toffano-Nioche, C., Claisse, G., Thareau, V., Morin, H., Laufs, P., et al. (2008). Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evol. Biol. 8, 291. doi: 10.1186/1471-2148-8-291
Esumi, T., Tao, R., Yonemori, K. (2005). Isolation of LEAFY and TERMINAL FLOWER 1 homologues from six fruit tree species in the subfamily maloideae of the rosaceae. Sex. Plant Reprod. 17 (6), 277–287. doi: 10.1007/s00497-004-0239-3
Feng, C., Zou, S., Gao, P., Wang, Z. (2021). In silico identification, characterization expression profile of WUSCHEL-related homeobox (WOX) gene family in two species of kiwifruit. PeerJ 9, e12348. doi: 10.7717/peerj.12348
Gambino, G., Minuto, M., Boccacci, P., Perrone, I., Vallania, R., Gribaudo, I. (2011). Characterization of expression dynamics of WOX homeodomain transcription factors during somatic embryogenesis in Vitis vinifera. J. Exp. Bot. 62 (3), 1089–1101. doi: 10.1093/jxb/erq349
Ikeda, M., Mitsuda, N., Ohme-Takagi, M. (2009). Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 21 (11), 3493–3505. doi: 10.1105/tpc.109.069997
Jiang, Y., Peng, J., Wang, M., Su, W., Gan, X., Jing, Y., et al. (2019a). The role of EjSPL3, EjSPL4, EjSPL5, and EjSPL9 in regulating flowering in loquat (Eriobotrya japonica lindl.). Int. J. Mol. Sci. 21 (1), E248. doi: 10.3390/ijms21010248
Jiang, Y., Peng, J., Zhang, Z., Lin, S., Lin, S., Yang, X. (2019b). The role of EjSVPs in flower initiation in Eriobotrya japonica. Int. J. Mol. Sci. 20 (23), E5933. doi: 10.3390/ijms20235933
Jiang, Y., Peng, J., Zhu, Y., Su, W., Zhang, L., Jing, Y., et al. (2019c). The role of EjSOC1s in flower initiation in Eriobotrya japonica. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00253
Jing, D., Chen, W., Hu, R., Zhang, Y., Xia, Y., Wang, S., et al. (2020). An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in loquat. Int. J. Mol. Sci. 21 (14), E5107. doi: 10.3390/ijms21145107
Jing, D., Chen, W., Xia, Y., Shi, M., Wang, P., Wang, S., et al. (2020). Homeotic transformation from stamen to petal in Eriobotrya japonica is associated with hormone signal transduction and reduction of the transcriptional activity of EjAG. Physiol. Plant. 168 (4), 893–908. doi: 10.1111/ppl.13029
Ji, J., Shimizu, R., Sinha, N., Scanlon, M. J. (2010). Analyses of WOX4 transgenics provide further evidence for the evolution of the WOX gene family during the regulation of diverse stem cell functions. Plant Signa Behav. 5 (7), 916–920. doi: 10.4161/psb.5.7.12104
Kanchan, M., Sembi, J. (2020). Comparative transcriptomic analyses of four phalaenopsis species to identify and characterize the WUSCHEL -related homeobox (WOX ) gene family. Biotechnologia 101 (4), 309–322. doi: 10.5114/bta.2020.100423
Laux, T., Mayer, K. F., Berger, J., Jürgens, G. (1996). The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Dev. (Cambridge England) 122 (1), 87–96. doi: 10.1242/dev.122.1.87
Li, X., Hamyat, M., Liu, C., Ahmad, S., Gao, X., Guo, C., et al. (2018). Identification and characterization of the WOX family genes in five solanaceae species reveal their conserved roles in peptide signaling. Genes 9 (5), E260. doi: 10.3390/genes9050260
Li, Z., Liu, D., Xia, Y., Li, Z., Jing, D., Du, J., et al. (2020). Identification of the WUSCHEL-related homeobox (WOX) gene family, and interaction and functional analysis of TaWOX9 and TaWUS in wheat. Int. J. Mol. Sci. 21 (5), E1581. doi: 10.3390/ijms21051581
Lin, H., Niu, L., McHale, N. A., Ohme-Takagi, M., Mysore, K. S., Tadege, M. (2012). Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc. Natl. Acad. Sci. U. S. A. 110 (1), 366–371. doi: 10.1073/pnas.1215376110
Lin, M., Yan, J., Ali, M. M., Wang, S., Tian, S., Chen, F., et al. (2022). Isolation and functional characterization of a green-tissue promoter in japonica rice (Oryza sativa subsp. Japonica). Biology 11 (8), 1092. doi: 10.3390/biology11081092
Liu, Y., Song, H., Liu, Z., Hu, G., Lin, S. (2013). Molecular characterization of loquat EjAP1 gene in relation to flowering. Plant Growth Regul. 70 (3), 287–296. doi: 10.1007/s10725-013-9800-0
Liu, Y., Wen, L., Shi, Y., Su, D., Lu, W., Cheng, Y., et al. (2021). Stress-responsive tomato gene SlGRAS4 function in drought stress and abscisic acid signaling. Plant Sci. 304, 110804. doi: 10.1016/j.plantsci.2020.110804
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods (San Diego Calif.) 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Lopes, F. L., Galvan-Ampudia, C., Landrein, B. (2021). WUSCHEL in the shoot apical meristem: old player, new tricks. J. Exp. Bot. 72 (5), 1527–1535. doi: 10.1093/jxb/eraa572
Munir, S., Khan, M. R. G., Song, J., Munir, S., Zhang, Y., Ye, Z., et al. (2016). Genome-wide identification, characterization and expression analysis of calmodulin-like (CML) proteins in tomato (Solanum lycopersicum). Plant Physiol. Biochem. 102, 167–179. doi: 10.1016/j.plaphy.2016.02.020
Nardmann, J., Reisewitz, P., Werr, W. (2009). Discrete shoot and root stem cell-promoting WUS/WOX5 functions are an evolutionary innovation of angiosperms. Mol. Biol. Evol. 26 (8), 1745–1755. doi: 10.1093/molbev/msp084
Nardmann, J., Zimmermann, R., Durantini, D., Kranz, E., Werr, W. (2007). WOX gene phylogeny in poaceae: A comparative approach addressing leaf and embryo development. Mol. Biol. Evol. 24 (11), 2474–2484. doi: 10.1093/molbev/msm182
Palovaara, J. (2008). Conifer WOX-related homeodomain transcription factors, developmental consideration and expression dynamic of WOX2 during Picea abies somatic embryogenesis. Plant Mol. Biol. 66 (5), 533–549. doi: 10.1007/s11103-008-9289-5
Palovaara, J. (2010). Comparative expression pattern analysis of WUSCHEL-related homeobox 2 (WOX2) and WOX8/9 in developing seeds and somatic embryos of the gymnosperm Picea abies. New Phytol. 188 (1), 122–135. doi: 10.1111/j.1469-8137.2010.03336.x
Perales, M., Rodriguez, K., Snipes, S., Yadav, R. K., Diaz-Mendoza, M., Reddy, G. V. (2016). Threshold-dependent transcriptional discrimination underlies stem cell homeostasis. Proc. Natl. Acad. Sci. U. S. A. 113 (41), E6298–E6306. doi: 10.1073/pnas.1607669113
Rebocho, A. B., Bliek, M., Kusters, E., Castel, R., Procissi, A., Roobeek, I., et al. (2008). Role of EVERGREEN in the development of the cymose petunia inflorescence. Dev. Cell 15 (3), 437–447. doi: 10.1016/j.devcel.2008.08.007
Reig, C., Gil-Muñoz, F., Vera-Sirera, F., García-Lorca, A., Martínez-Fuentes, A., Mesejo, C., et al. (2017). Bud sprouting and floral induction and expression of FT in loquat [Eriobotrya japonica (Thunb.) lindl.]. Planta 246 (5), 915–925. doi: 10.1007/s00425-017-2740-6
Sarkar, A. K., Luijten, M., Miyashima, S., Lenhard, M., Hashimoto, T., Nakajima, K., et al. (2007). Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 446 (7137), 811–814. doi: 10.1038/nature05703
Schoof, H., Lenhard, M., Haecker, A., Mayer, K. F., Jürgens, G., Laux, T. (2000). The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100 (6), 635–644. doi: 10.1016/s0092-8674(00)80700-x
Shafique Khan, F., Zeng, R.-F., Gan, Z.-M., Zhang, J.-Z., Hu, C.-G. (2021). Genome-wide identification and expression profiling of the WOX gene family in Citrus sinensis and functional analysis of a CsWUS member. Int. J. Mol. Sci. 22 (9), 4919. doi: 10.3390/ijms22094919
Shimizu, R., Ji, J., Kelsey, E., Ohtsu, K., Schnable, P. S., Scanlon, M. J. (2009). Tissue specificity and evolution of meristematic WOX3 function. Plant Physiol. 149 (2), 841–850. doi: 10.1104/pp.108.130765
Su, Y. H., Zhou, C., Li, Y. J., Yu, Y., Tang, L. P., Zhang, W. J., et al. (2020). Integration of pluripotency pathways regulates stem cell maintenance in the Arabidopsis shoot meristem. Proc. Natl. Acad. Sci. U. S. A. 117 (36), 22561–22571. doi: 10.1073/pnas.2015248117
Tian, H., Wabnik, K., Niu, T., Li, H., Yu, Q., Pollmann, S., et al. (2014). WOX5-IAA17 feedback circuit-mediated cellular auxin response is crucial for the patterning of root stem cell niches in Arabidopsis. Mol. Plant 7 (2), 277–289. doi: 10.1093/mp/sst118
Tvorogova, V. E., Krasnoperova, E. Y., Potsenkovskaia, E. A., Kudriashov, A. A., Dodueva, I. E., Lutova, L. A. (2021). [What does the WOX say? review of pegulators, targets, partners]. Mol. Biol. 55 (3), 362–391. doi: 10.31857/S0026898421030174
Vandenbussche, M., Horstman, A., Zethof, J., Koes, R., Rijpkema, A. S., Gerats, T. (2009). Differential recruitment of WOX transcription factors for lateral development and organ fusion in petunia and Arabidopsis. Plant Cell 21 (8), 2269–2283. doi: 10.1105/tpc.109.065862
van der Graaff, E., Laux, T., Rensing, S. A. (2009). The WUS homeobox-containing (WOX) protein family. Genome Biol. 10 (12), 248. doi: 10.1186/gb-2009-10-12-248
Wang, Y. (2021). A draft genome, resequencing, and metabolomes reveal the genetic background and molecular basis of the nutritional and medicinal properties of loquat (Eriobotrya japonica (Thunb.) lindl). Hortic. Res. 8 (1), 231. doi: 10.1038/s41438-021-00657-1
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., et al. (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. (Clifton N.J.) 112, 531–552. doi: 10.1385/1-59259-584-7:531
Xiao, Y., Chen, Y., Ding, Y., Wu, J., Wang, P., Yu, Y., et al. (2018). Effects of GhWUS from upland cotton (Gossypium hirsutum l.) on somatic embryogenesis and shoot regeneration. Plant Sci. 270, 157–165. doi: 10.1016/j.plantsci.2018.02.018
Xu, L. (2021). WUSCHEL: The versatile protein in the shoot apical meristem. Sci. China Life Sci. 64 (1), 177–178. doi: 10.1007/s11427-020-1870-4
Xu, Y.-Y., Wang, X.-M., Li, J., Li, J.-H., Wu, J.-S., Walker, J. C., et al. (2005). Activation of the WUS gene induces ectopic initiation of floral meristems on mature stem surface in Arabidopsis thaliana. Plant Mol. Biol. 57 (6), 773–784. doi: 10.1007/s11103-005-0952-9
Yadav, R. K. (2012). WUSCHEL protein movement and stem cell homeostasis. Plant Signaling Behav. 7 (5), 592–594. doi: 10.4161/psb.19793
Zhang, L., Jiang, Y., Zhu, Y., Su, W., Long, T., Huang, T., et al. (2019). Functional characterization of GI and CO homologs from Eriobotrya deflexa nakai forma koshunensis. Plant Cell Rep. 38 (5), 533–543. doi: 10.1007/s00299-019-02384-3
Zhang, L., Yu, H., Lin, S., Gao, Y. (2016). Molecular characterization of FT and FD homologs from Eriobotrya deflexa nakai forma koshunensis. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00008
Zhang, X., Zong, J., Liu, J., Yin, J., Zhang, D. (2010). Genome-wide analysis of WOX gene family in rice, sorghum, maize, Arabidopsis and poplar. J. Integr. Plant Biol. 52 (11), 1016–1026. doi: 10.1111/j.1744-7909.2010.00982.x
Zhou, Y., Yan, A., Han, H., Li, T., Geng, Y., Liu, X., et al. (2018). HAIRY MERISTEM with WUSCHEL confines CLAVATA3 expression to the outer apical meristem layers. Sci. (New York N.Y.) 361 (6401), 502–506. doi: 10.1126/science.aar8638
Keywords: loquat, WOX gene family, WUS transcription factor, flowering time, protein interaction
Citation: Yu Y, Yang M, Liu X, Xia Y, Hu R, Xia Q, Jing D and Guo Q (2022) Genome-wide analysis of the WOX gene family and the role of EjWUSa in regulating flowering in loquat (Eriobotrya japonica). Front. Plant Sci. 13:1024515. doi: 10.3389/fpls.2022.1024515
Received: 21 August 2022; Accepted: 19 October 2022;
Published: 03 November 2022.
Edited by:
Shunquan Lin, South China Agricultural University, ChinaReviewed by:
Tangchun Zheng, Beijing Forestry University, ChinaCopyright © 2022 Yu, Yang, Liu, Xia, Hu, Xia, Jing and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danlong Jing, amluZ2RhbmxvbmcxMTBAMTI2LmNvbQ==; Qigao Guo, cWdndW9AMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.