 Wendi Ma1
Wendi Ma1 Vijai Bhadauria
Vijai Bhadauria- 1Department of Plant Pathology, College of Plant Protection, China Agricultural University, Beijing, China
- 2Ministry of Agriculture and Rural Affairs, Key Laboratory for Crop Pest Monitoring and Green Control, China Agricultural University, Beijing, China
- 3College of Agronomy, Henan Agricultural University, Zhengzhou, China
Didymella leaf blight (DLB) caused by Didymella glomerata is a new fungal disease of maize (Zea mays), first detected in 2021 in Panjin, Liaoning province of China. Here we report the reference genome assembly of D. glomerata to unravel how the fungal pathogen controls its virulence on maize at the molecular level. A maize-infecting strain Pj-2 of the pathogen was sequenced on the Illumina NovaSeq 6000 and PacBio Sequel II platforms at a 575-fold genomic coverage. The 33.17 Mb gapless genome assembly comprises 32 scaffolds with L/N50 of 11/1.36 Mb, four of which represent full-length chromosomes. The Pj-2 genome is predicted to contain 10,334 protein-coding genes, of which 211, 12 and 134 encode effector candidates, secondary metabolite backbone-forming enzymes and CAZymes, respectively. Some of these genes are potentially implicated in niche adaptation and expansion, such as colonizing new hosts like maize. Phylogenomic analysis of eight strains of six Didymella spp., including three sequenced strains of D. glomerata, reveals that the maize (Pj-2)- and Chrysanthemum (CBS 528.66)-infecting strains of D. glomerata are genetically similar (sharing 92.37% genome with 98.89% identity), whereas Pj-2 shows truncated collinearity with extensive chromosomal rearrangements with the Malus-infecting strain M27-16 of D. glomerata (sharing only 55.01% genome with 88.20% identity). Pj-2 and CBS 528.66 carry four major reciprocal translocations in their genomes, which may enable them to colonize the different hosts. Furthermore, germplasm screening against Pj-2 led to the identification of three sources of DLB resistance in maize, including a tropical inbred line CML496. DLB resistance in the line is attributed to the accumulation of ROS H2O2 in the apoplastic space of the infected cells, which likely restricts the fungal growth and proliferation.
Introduction
Didymella (syn. Peyronellaea or Phoma) glomerata (Corda) Q. Chen & L. Cai is an ascomycete fungal pathogen, which causes Didymella leaf blight (DLB). The pathogen has a broad host range and is known to infect both monocots and dicots, including although not limited to flax (Linum usitatissimum) (Reeder and Vanterpool, 1953), oilseed rape (Brassica napus) (Taber and Vanterpool, 1963), bread wheat (Triticum aestivum), durum wheat (Triticum turgidum) and triticale (Triticale hexaploide) (Hosford, 1975), pea (Pisum sativum) (Tran et al., 2014), sunflower (Helianthus annuus) (Roustaee et al., 2000), grapevine (Vitis vinifera) (Khani, 2014), peach (Prunus persica) (Thomidis et al., 2011), kiwi (Actinidia chinensis) (Pan et al., 2018), pistachio (Pistacia vera) (Moral et al., 2018), Chinese cornel dogwood (Cornus officinalis) (Huang et al., 2018) and apple (Malus spp.).
During a disease survey in the 2021 growing season, we observed gray leaf blight symptoms on maize in Panjin, Liaoning, a northeastern province of China. The symptoms differed from those caused by D. zeae-maydis, the only Didymella species infectious on maize and the causal agent of yellow leaf blight. Culture characteristics and molecular phylogeny based on multi-locus DNA barcodes (ITS, Actin and β-tubulin) showed that the single spore culture isolate Pj-2 retrieved from the infected leaf tissues was morphologically and genetically identical with the D. glomerata isolate D/034 from soybean (Glycine max) (Irinyi et al., 2009). This was the first report of D. glomerata causing DLB on maize (Ma et al., 2022).
D. glomerata is likely a heterothallic fungal pathogen as it harbors the mating-type gene scaffold1.t522 (MAT1-2), whose protein product contains the characteristic high mobility group box domain. The pathogen likely deploys a subcuticular intramural necrotrophic infection strategy, whereby the pathogen colonizes maize leaves, resulting in DLB. The infection cycle starts when fungal spores (conidia) land on the leaf surface and germinate to form a germ tube. However, unlike hemibiotrophic fungal pathogens, the germ tube does not differentiate into appressoria (specialized structures dedicated to mechanized penetration of the host surface) but instead directly perforates the host cuticle, presumably via an infection peg. The pathogen can also gain access to host tissues through natural openings, such as stomata. Following successful penetration, the invasive hyphae emanating from the infection pegs rapidly colonize the host parietal layers and eventually kill and macerate the host cells. Black lesions appear on the maize leaves five to seven days after infection. Single-celled ellipsoidal conidia are produced in glabrous pycnidia developing in these lesions and are dispersed by splashing raindrops and wind to infect healthy neighboring plants.
Thus far, studies have revealed that D. glomerata produces phytotoxins zinniol (Sugawara and Strobel, 1986) and 5-dihydroxymethylfuran (Cimmino et al., 2021), which contribute to the virulence of the pathogen on sunflowers and grapevine, respectively. However, molecular and genetic factors regulating the D. glomerata virulence on various hosts, let alone maize, are still lacking. Only seven species in the genus Didymella have been sequenced, and the genomes thereof are available in the public domain. Of the sequenced Didymella species, two are pathogenic to humans (D. keratinophila [the causal agent finger-hand lesions in humans; GenBank accession GCA_011058865.1] and D. heteroderae [GenBank accession GCA_011058895.1]), whereas five are pathogenic to plants (D. pinodes [the causal agent of Ascochyta blight of pea; GenBank GCA_004151525.1], D. lethalis [pea root rot pathogen; GenBank accession GCA_004335245.1], D. arachidicola [the causal agent of web blotch of peanut; GenBank accession GCA_016630955.1], D. exigua [the causal agent of Didymella wilt and necrosis of yellow starthistle; GenBank accession GCA_010094145.1] and D. rabiei [the causal agent of Ascochyta blight of chickpea; GenBank accession: GCA_004011695.1]). More recently, fragmented genome assemblies of two D. glomerata strains CBS 528.66 (accession GCA_022559905.1; host: Chrysanthemum spp.) and M27-16 (accession GCA_022225945.1; host: Malus spp.) were deposited to GenBank.
In this study, we sequenced, assembled and annotated the genome of D. glomerata strain Pj-2, which infects maize and causes DLB. Therefore, the high-quality gapless assembly reported in the study represents the first sequenced genome of the maize-infecting D. glomerata strain. Furthermore, we also identified three genetic sources of DLB resistance in maize germplasm, including an elite tropical inbred line CML496, for potential introgression of resistance alleles into elite cultivars lacking resistance through intraspecific hybridization.
Results and discussion
High-quality gapless genome assembly of maize-infecting D. glomerata
We sequenced the D. glomerata strain Pj-2 genome on the second-generation (Illumina NovaSeq 6000) and third-generation (PacBio Sequel II) sequencing platforms, which yielded a de novo reference genome assembly of 33.17 Mb with a 575-fold genomic coverage (Table 1). Thirty-two contiguous super-sequences (called supercontigs or scaffolds) were assembled from 2,054,785 clean SMRT reads (average read length = 8,300 bp; summed read length = 17,055,541,969 bp) originating from the sequencing of a long DNA fragment (insert size = 10 Kb) library on PacBio Sequel II. The scaffolds were confirmed and corrected by 250-bp 11,853,984 high-quality paired-end reads (summed length = 2,876,946,471 bp; average phred quality score on a sliding window of four nucleotides ≥30) generated from the sequencing of a short paired-end library (insert size = 500 bp) on NovaSeq 6000. As a result, a virtually gapless high-quality genome assembly was generated and consisted of 32 scaffolds with only a single gap of 100 bp in scaffold14 (coordinate: 515,915–516,014 bp). The analysis of K-mer frequency at the optimal length of 17 bp within 250-bp 11,853,984 paired-end reads revealed that the genome of Pj-2 is estimated to be 34.90 Mb in size. The genome size is comparable to the recently sequenced genome assemblies of the Chrysanthemum-infecting strain CBS 528.66 (33.98 Mb) and the apple-infecting strain M27-16 (35.52 Mb) of D. glomerata.
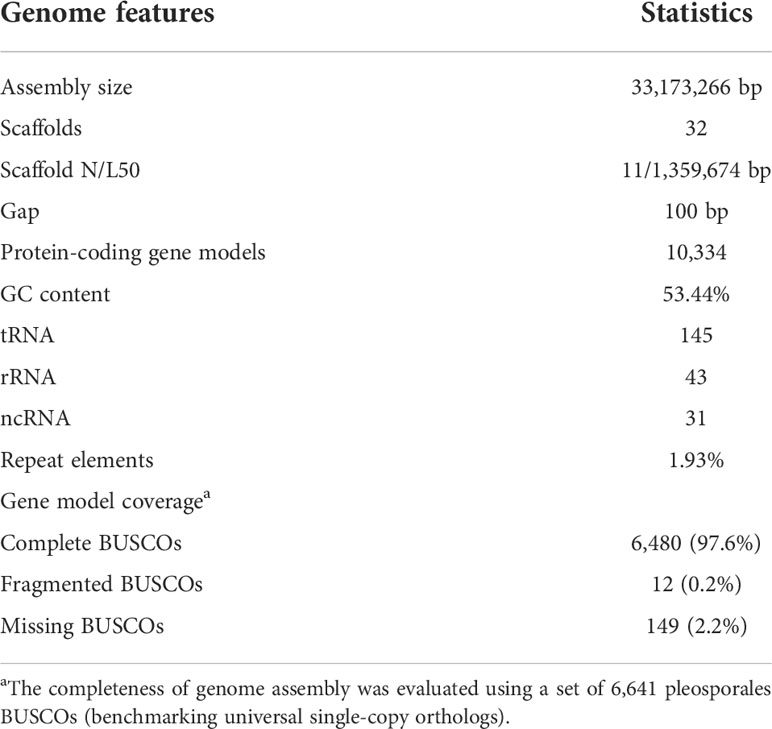
Table 1 The Didymella glomerata Pj-2 genome assembly features.
The scaffold N/L50 of the D. glomerata Pj-2 genome is 11/1,359,674 bp, which is far superior than the other two genome assemblies of D. glomerata strains CBS 528.66 (177 scaffolds; N/L50 = 7/635,919 bp) and M27-16 (946 scaffolds; N/L50 = 26/469,072 bp). In addition, the Pj-2 genome assembly is either comparable or better than that of other sequenced Didymella species: D. keratinophila 9M1 (577 scaffolds; N/L50 = 76/141,571 bp), D. heteroderae 28M1 (620 scaffolds; N/L50 = 38/283,873 bp), D. pinodes WTN-11-157 (1,593 scaffolds; N/L50 = 91/106,671 bp), D. lethalis GRM-16-623 (512 scaffolds; N/L50 = 50/196,696), D. arachidicola YY187 (25 scaffolds; N/L50 = 8/2,070,310 bp) (Li et al., 2021); D. exigua CBS 183.55 (176 scaffolds; N/L50 = 64/145,421 bp), D. rabiei ArMe14 (34 scaffolds; N/L50 = 9/1,812,190) (Shah et al., 2020) and D. segeticola GZSQ-4 (23 scaffolds; N/L50 = 6/2,254,797) (Ren et al., 2019).
Tandem telomeric repeats (CCCTAA)n or (TTAGGG)n were present in at least one end of the 23 scaffolds of the D. glomerata Pj-2 genome assembly. Of these 23 scaffolds, four (scaffold1 [1.90 Mb], scaffold14 [1.09 Mb], scaffold18 [1.05 Mb] and scaffold19 [1.03 Mb]) contain the telomeric repeats at both ends and hence represent full-length chromosomes. Transposable elements (TEs) comprise only 1.93% of the D. glomerata genome assembly (0.99% [328,869 bp] of retroelements, 0.90% [297,166 bp] of DNA transposons and 0.02% [7,437 bp] of unclassified TEs) and are scattered in the low GC content regions of the genome (Table S1). The GC content in the Pj-2 genome is 53.44%, which is similar to the CBS 528.66 (53.10%) and M27-16 (53.30%) genomes and other sequenced Didymella spp. A total of 6,480 (97.6%) out of 6,641 pleosporales (the order within Dothidiomycetes) single-copy genes were mapped on the D. glomerata Pj-2 genome, suggesting a near-complete genome assembly with regard to the genic content thereof. Overall, the assembled PacBio reads corrected by the Illumina reads produced a high-quality (32 scaffolds with 575-fold genome coverage), nearly gapless (single gap of 100 bp) genome assembly of the maize-infecting strain Pj-2 of D. glomerata.
Gene families implicated in the virulence/pathogenicity of D. glomerata on maize
The D. glomerata Pj-2 genome is predicted to contain 10,334 protein-coding genes, whose number is markedly lower than that of the Chrysanthemum-infecting strain CBS 528.66 (11,200) and the Malus-infecting strain M27-16 (11,650). Similarly, the predicted gene content of Pj-2 encodes is markedly lower than that of other Didymella spp., e.g., D. keratinophila 9M1 (11,880), D. heteroderae 28M1 (11,640), D. exigua CBS 183.55 (12,356) and D. rabiei ArMe14 (11,257), except D. pinodes WTN-11-157 (10,505). The clustering of 90,823 proteins coded by the eight strains based on their sequence similarity (≥90% over 50 aligned aa) showed that the strains carry 1,027 core orthologous proteins. Pj-2 shares 2,734 more orthologous proteins with CBS 528.66 (7,660) than M27-16 (4,927), suggesting that the maize-infecting D. glomerata strains are phylogenetically closely related to the Chrysanthemum-infecting D. glomerata strain. Pj-2 shares 5,444 orthologous proteins with 9M1, 4,771 with 28M1, 3,847 with CBS 183.55 and 1,958 with ArMe14 (Table S2).
Only a subset of genes in the genomes encodes virulence/pathogenicity-related factors, including although not limited to effectors, carbohydrate-active enzymes (CAZymes) and secondary metabolism enzymes. Effectors are the pathogen-secreted small proteins and are an example of the extended phenotype as they function in hosts instead of pathogens, whose genomes carry the effector-coding genes. Pathogens deliver effectors into plants to subdue the innate immune system of plants, thereby enabling colonization and proliferation (Stergiopoulos and De Wit, 2009). Only 1.88% of the Pj-2 genes encode effector candidates (211), whose number is higher than that of the Chrysanthemum-infecting strain CBS 528.66 (176) and the Malus-infecting strain M27-16 (196) even though the latter two strains of D. glomerata have a larger gene content than the former (Tables S3-S5). Similarly, the pea-infecting D. pinodes WTN-11-157 genome carried a reduced number of effector candidate genes (180; Table S6). However, effector content in other four strains were slightly larger than that in Pj-2, e.g., 9M1 (231; Table S7), 28M1 (240; Table S8), CBS 183.55 (240; Table S9) and ArMe14 (223; Table S10). The majority of these effectors lack any known functional domain, e.g., 195 Pj-2 effectors show homology to hypothetical and uncharacterized proteins. Some of these effectors function in the host cell apoplast, which is replete with cysteine proteases, whereas some effectors are translocated into host cells wherein they subvert host cell immunity, viz., effector-triggered immunity (ETI). EffectorP (Sperschneider and Dodds, 2022) predicted that 111 encoded by the Pj-2 genome are likely translocated into the host cell apoplast. Seventy-four of the 111 apoplastic effector candidates possess ≥2% cysteine residues; cysteine residues in some of these effectors form disulfide bonds, which may provide stability to them in the hostile protease-rich milieu in the host cell apoplast (Figure 1A). The distribution of apoplastic effector candidates in the eight strains followed a similar pattern as effector candidates, i.e., Pj-2 carries a larger repertoire of apoplastic effector candidates than that in CBS 528.66 (71), M27-16 (97) and WTN-11-157 (88). A total of 58 effector candidates are likely transported into the host cell cytoplasm; notably, the number of these cytoplasmic effector candidates is lower than that in CBS 528.66 (64), M27-16 (60) and WTN-11-157 (59) (Figure 1A). Effectors in some plant pathogens, such as Leptosphaeria maculans (the causal agent of blackleg disease of oilseed crop), Fusarium spp., Magnaporthe oryzae (the causal agent of rice blast disease) and Ustilago maydis (the causal agent of corn smut), are localized in chromosomal regions enriched with transposable elements, including telomeres, or clustered in AT-rich blocks of the genomes (Kämper et al., 2006; Farman, 2007; Ma et al., 2010; Rouxel et al., 2011). Such a genomic environment drives rapid diversification of genes, including those coding for effectors, thereby enabling pathogens to evade the innate immune system of host cultivars. However, the effector candidates in Pj-2 are randomly distributed across the genome, with no evidence for enrichment of particular scaffolds, including full-length chromosomes (scaffold1, scaffold14, scaffold18 and scaffold19), or clustering in the proximity of genomic regions replete with repetitive elements, e.g., telomeres. The relatively larger effector repertoire of Pj-2, especially cytoplasmic effectors, may be an adaptation of D. glomerata to colonize new host maize under field conditions, which we reported recently (Ma et al., 2022).
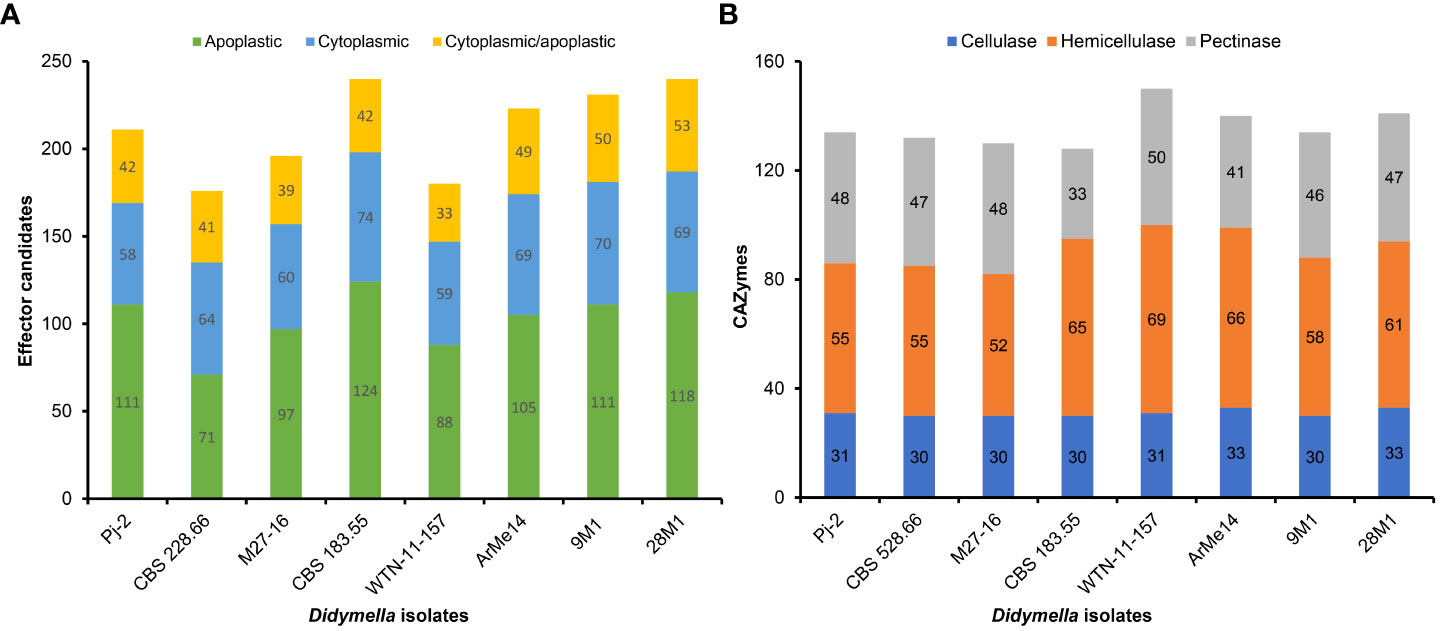
Figure 1 Distribution of effector candidates (A) and carbohydrate-active enzymes (CAZymes; B) in eight strains representing six Didymella spp. (Didymella glomerata Pj-2, CBS 528.66 and M27-16, D. pinodes WTN-11-157, D. keratinophila 9M11, D. heteroderae 28M1, D. exigua CBS 183.55 and Ascochyta [Didymella] rabiei ArMe14).
Pj-2 is equipped with 134 carbohydrate-active enzymes (CAZymes; Figure 1B and Table S11) that likely degrade the host cell wall, composed mainly of cellulose, hemicellulose and pectin, to colonize the host tissues and drive nutrition from the macerated tissues (van den Brink and de Vries, 2011). The other two D. glomerata strains possess a slightly lower number of CAZymes, i.e., CBS 528.66 (132; Figure 1B and Table S12) and M27-16 (130; Figure 1B and Table S13). The number of CAZymes in other Didymella spp. (Figure 1B and Tables S1-S17) except CBS 183.55 (128; Figure 1B and Table S18) is higher than that in Pj-2. Notably, the number of hemicellulases (55) is higher than cellulases (31) and pectinases (48) in Pj-2, which is presumably associated with a higher amount of hemicellulose in the maize cell wall.
In fungal pathogens, secondary metabolism genes are generally localized in biosynthetic gene clusters (BGCs), and each BGC contains a backbone gene and a few decorating genes, and encodes a specific secondary metabolite (Collemare et al., 2008). Secondary metabolites are indispensable for fungal virulence on hosts (Soanes et al., 2012). The Pj-2 genome contains 12 BGCs, similar to CBS 528.66 (12) but lower than M27-16 (16) (Tables 2, S19-S26). Likewise, the number of backbone genes encoding polyketide synthase (PKS) and nonribosomal peptide synthase (NRPS) is slightly higher in M27-16 (6 PKS and 7 NRPS-like) than Pj-2 (4 PKS and 4 NRPS-like) and CBS 528.66 (5 PKS and 4 NRPS-like) (Tables 2, S19-S26). Therefore, the Malus-infecting D. glomerata strain M27-16 may have more capacity to produce secondary metabolites than that of the maize- and Chrysanthemum-infecting D. glomerata strains.
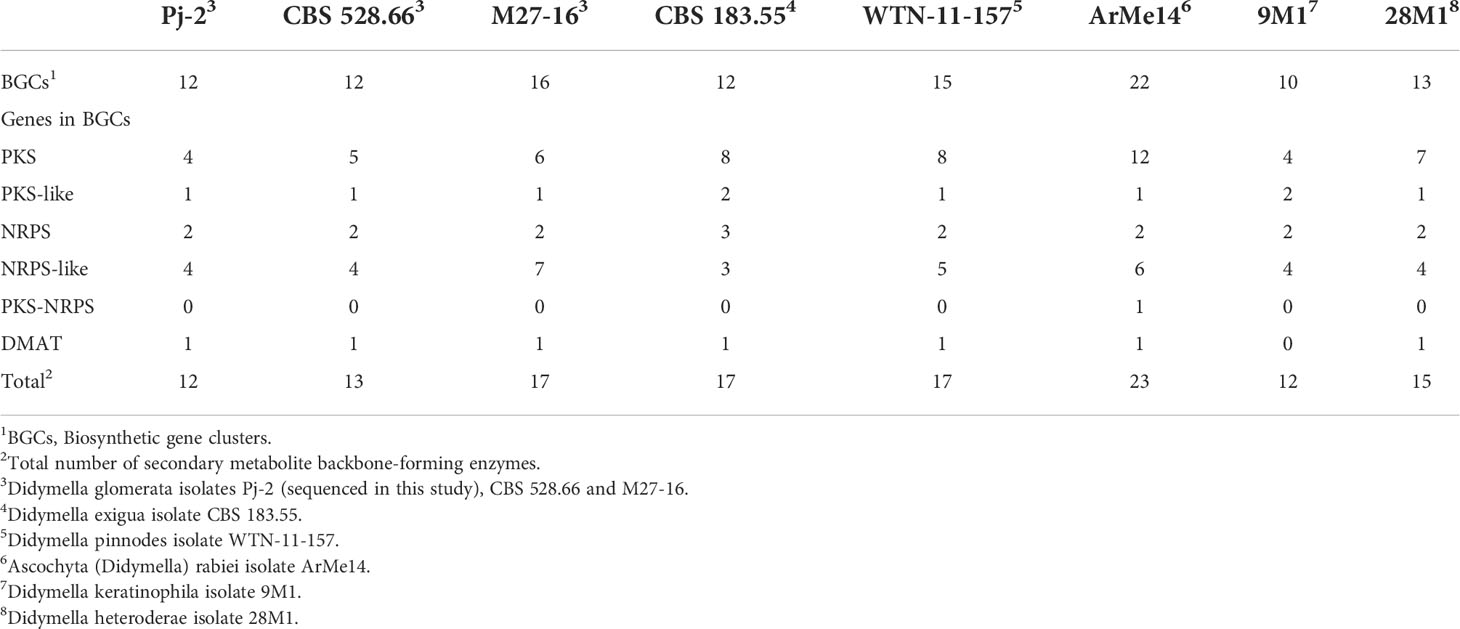
Table 2 Biosynthetic gene clusters (BGCs) and secondary metabolite backbone-forming enzymes within BCGs in Didymella spp.
Phylogenomics reveals intra- and inter-specific genetic diversity among Didymella spp.
The D. glomerata strain Pj-2 genome (33.17 Mb) is similar in size to the Chrysanthemum-infecting strain CBS 528.66 (33.98 Mb), apple-infecting strain M27-16 (35.52 Mb), as well as other Didymella spp., e.g., D. pinodes WTN-11-157 (33.77 Mb), and D. exigua CBS 183.55 (34.39 Mb). However, the other four sequenced Didymella spp. have slightly larger genomes, e.g., D. rabiei ArMe14 (40.92 Mb), D. keratinophila 9M1 (36.22 Mb) and D. heteroderae 28M1 (37.07 Mb).
To gauge genetic diversity among Didymella spp., we performed genome similarity and synteny analyses among eight strains, representing six species. An eight-strain whole-genome alignment was contrived using the following parameters: 50-bp seeds and extension thereof with HOXD scoring matrix (Chiaromonte et al., 2002) and ≥100-bp alignment block. Pairwise distance matrices between the genomes were calculated from the alignment blocks of size ≥100 bp and were expressed as alignment percentage (AP) and average nucleotide identity (ANI) (Figure 2B). The D. glomerata strain Pj-2 shares 92.37% (i.e., AP) of its genome with CBS 528.66, followed by 57.71% with 9M1, 55.01% with M27-16, 45.04% with WTN-11-157, 43.14% with 28M1, 23.31% with CBS 183.55 and 5.19% with ArMe14. The shared genomic regions showed ≥85% ANI; Pj-2 displayed 98.89% nucleotide similarity with CBS 528.66, followed by 88.79% with 9M1, 88.20% with M27-16, 87.01% with 28M1, 86.99% with WTN-11-1, 85.11% with CBS 183.55 and 84.56% with ArMe14. A neighbor-joining tree constructed from the pairwise distance matrices revealed that a clade comprising the three D. glomerata strains Pj-2, CBS 528.66 and M27-16, and D. keratinophila 9M1. These strains share 55.01% to 92.37% genomes (AP), with ANI values ranging from 88.20% to 98.89%. Within this clade, Pj-2 and CBS 528.66 formed a subclade, which is in agreement with high AP (92.37%) and ANI (98.89%) between their genomes (Figure 2B). Synteny analysis showed that the two D. glomerata strains possess a similar genomic architecture. However, a few structural variations (chromosomal rearrangements) exist between their genomes (Figure 2B). To further investigate these genomic rearrangements, whole-genome collinearity analysis was performed between the Pj-2 and the genomes of seven strains (CBS 528.66, M27-16, WTN-11-15, CBS 183.55, ArMe14, 9M1 and 28M1) (Figure 2). The Pj-2 genome is collinear with the CBS 528.66 genome, whereas it shows fragmented collinearity with the other genomes (due to low and extensive structural variations), including M27-16, which precluded further investigation (Figures 2, S1).
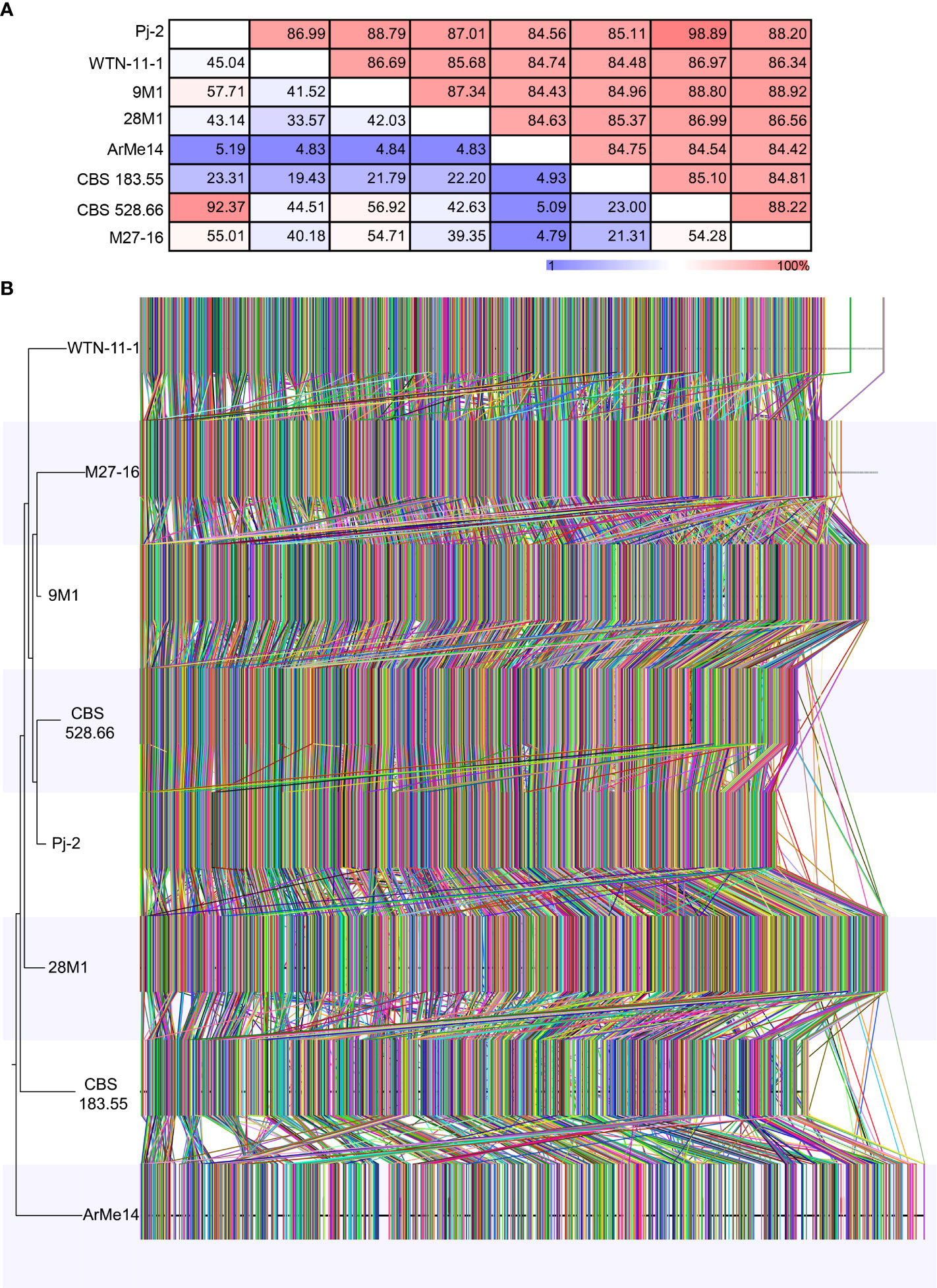
Figure 2 Phylogenomics of Didymella spp. The whole genomes of eight Didymella spp. strains are aligned using CLC Genomic Workbench with 50 bp seeds: D. glomerata Pj-2 (33.17 Mb; 32 scaffolds), CBS 528.66 (33.98 Mb; 177 scaffolds) and M27-16 (35.52 Mb; 946 scaffolds), D. keratinophila 9M1 (36.22 Mb; 577 scaffolds), D. heteroderae 28M1 (37.07 Mb; 620 scaffolds), D. pinodes WTN-11-157 (33.77 Mb; 1,593 scaffolds), D. exigua CBS 183.55 (34.39 Mb; 176 scaffolds) and D. rabiei ArMe14 (40.92 Mb; 34 scaffolds). The seeds were extended using a HOXD scoring matrix until the local alignment score dropped. Pairwise distance matrices between the aligned genomes are calculated from the alignment blocks of size ≥100 bp and are expressed as alignment percentage (AP; upper) and average nucleotide identity (ANI; lower) (A). Genome-wide synteny analysis among the eight strains (B). Each colored link represents a syntenic block (≥1,000 bp) that is shared among genomes. A neighbor-joining tree is also shown to the left of the whole-genome alignment.
Although the dot plot shows a high degree of collinearity between the two D. glomerata strains Pj-2 and CBS 528.6; however, there exist four major reciprocal translocations, viz. Pj-2 scaffold14/CBS 528.66 scaffold12 and Pj-2 scaffold18 (274 kb), Pj-2 scaffold22/CBS 528.66 scaffold6 and Pj-2 scaffold3 (166 kb), Pj-2 scaffold26/CBS 528.66 scaffold17 and Pj-2 scaffold13 (131 kb translocation), and Pj-2 scaffold26/CBS 528.66 scaffold17 and Pj-2 scaffold8 (217 kb translocation) (Figure 3A).
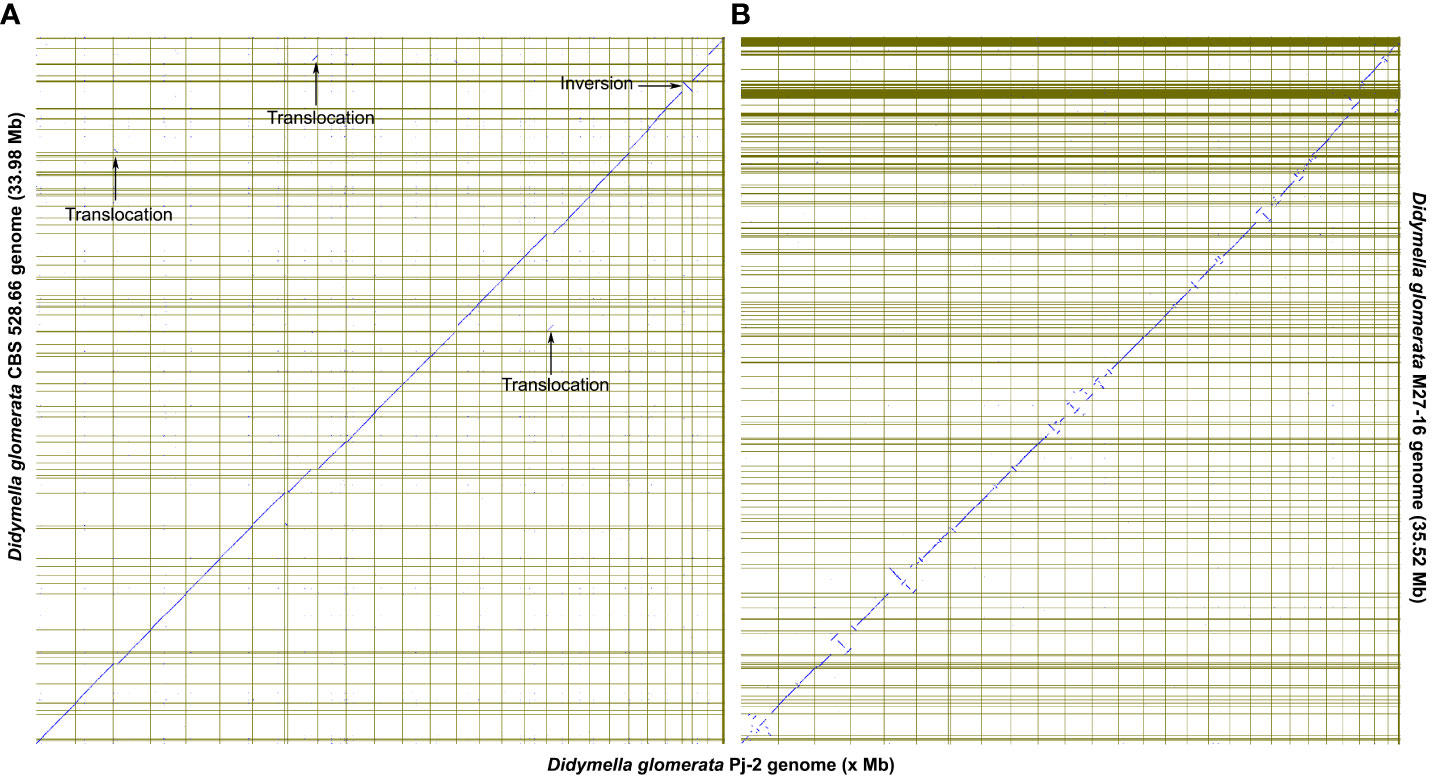
Figure 3 The dot plots show the collinearity of the D. glomerata Pj-2 genome (X-axis) with that of CBS 528.66 (Y-axis; A) and M27-16 (Y-axis; B). Four translocations are conspicuously present between Pj-2 and CBS 528.66.
Pj-2 scaffold14 (size = 1094.13 kb; full-length chromosome) and CBS 528.66 scaffold12 (size = 763.58 kb) are collinear with a high degree of similarity in the genomic region located between 282 kb and 764 kb, whereas a 274-kb genomic region of CBS 528.66 scaffold12 (8 to 282 kb) shows alignment with a high degree of homology with a similar size genomic region located at the 5’-end of Pj-2 scaffold18 (size = 1051.93 kb; full-length chromosome). This is called the reciprocal translocation between Pj-2 scaffold14/CBS 528.66 scaffold12 and Pj-2 scaffold18. The 274 kb translocation region contains 92 genes in Pj-2 and 100 genes in CBS 528.66, 78 of which are homologous. This translocation encodes three apoplastic effector candidates with unknown functions. The Pj-2 effector candidates scaffold18.g41 (141 aa) and scaffold18.g46 (238 aa) are 100% identical to CBS 528.66 scaffold12.g4610 and scaffold12.g4605, respectively. The third effector candidate scaffold18.g5 (269 aa) is aligned with 100% identity from 6 aa to 265 aa of the 526 aa CBS 528.66 protein scaffold12.g4649 (526 aa); likewise, the third CBS 528.66 effector scaffold12.g4639 (186 aa) is matched with 97% identity from 1 aa to 184 aa of the 675 aa Pj-2 protein scaffold18.g16. Therefore, these proteins represent the truncated version of larger proteins and thus are unlikely effector candidates. In addition to effector candidates, the translocation codes for three CAZymes. The Pj-2 cellulase scaffold18.g13 (394 aa) and the CBS 528.66 protein scaffold12.g4641 (439 aa) are homologous proteins, showing 98% identity over the first 327 aligned aa, whereas the C-termini thereof are highly variable and former lacks 45 aa in this region. The Pj-2 hemicellulase scaffold18.g6 (837 aa) is homologous to the CBS 528.66 hemicellulase scaffold12.g4648 (837 aa), exhibiting 99% identity over the entire length of proteins. The Pj-2 pectinase scaffold18.g16 (675 aa) and the CBS 528.66 scaffold12.g4639 (186 aa) are homologous proteins with 99% identity over the first 184 aligned aa; however, scaffold12.g4639 was predicted as an effector candidate due to its length ≤300. Therefore, scaffold12.g4639 is a truncated pectinase rather than an effector candidate. The translocation does not carry any secondary metabolite backbone enzyme.
Pj-2 scaffold22 (size = 915.48 kb) and CBS 528.66 scaffold6 (size = 1123.25 kb) are collinear with a high degree of similarity in the genomic region located between the 7–911 kb (Pj-2 scaffold22) and 174–911 Kb (CBS 528.66 scaffold6); however, a 166-kb genomic region of CBS 528.66 scaffold12 (8–174 kb), which Pj-2 scaffold22 lacks, aligns with a high degree of homology with a similar size to the genomic region (25–191 kb) located at the 5’-end of Pj-2 scaffold3 (size = 1813.70 kb). This is the reciprocal translocation between Pj-2 scaffold22/CBS 528.66 scaffold6 and Pj-2 scaffold3. The 166 kb translocation contains 54 genes in Pj-2 and 60 genes in CBS 528.66, 53 of which are homologous. This translocation encodes two apoplastic and/or cytoplasmic effector candidates with unknown functional domains and one CAZyme hemicellulase; however, it does not code for any secondary metabolite backbone enzyme. The Pj-2 effector candidates scaffold3.g27 (226 aa; apoplastic/cytoplasmic localization) and scaffold3.g51 (300 aa; apoplastic localization) are 100% identical to the CBS 528.66 effector candidates sacffold6.g2757 and scaffold6.g2777, respectively. The Pj-2 hemicellulase scaffold3.g49 (338 aa) is homologous to the CBS 528.66 hemicellulase scaffold6.g2776 (1120 aa), displaying 99% over the 334-aligned aa, whereas the former lacks the remaining aa. Therefore, scaffold3.g49 is a truncated protein.
Pj-2 scaffold26 (size = 703.34 kb) and CBS 528.66 scaffold17 (size = 703.44 kb) are collinear with a high degree of similarity in the genomic region located between 1–277 kb (Pj-2 scaffold22) and 434–703 Kb (CBS 528.66 scaffold6). The remaining genomic region of Pj-2 scaffold26, spanning from 277 kb to 703 kb, is collinear with CBS 528.66 scaffold29 (size = 481.49 kb). However, a majority of the 430-kb genomic region located at the 5’-end of CBS 528.66 scaffold17 (15–430 kb), which Pj-2 scaffold26 lacks, matches with a high degree of homology with two genomic regions located at distinct Pj-2 scaffolds, i.e., scaffold13 (1,133–1,264 kb) and scaffold8 (1,222–1,439 kb). Therefore, there are two reciprocal translocations: Pj-2 scaffold26/CBS 528.66 scaffold17 and Pj-2 scaffold13 (131 kb translocation) and Pj-2 scaffold26/CBS 528.66 scaffold17 and Pj-2 scaffold8 (217–250 kb translocation). The 131-kb translocation contains 46 genes in Pj-2 and 55 genes in CBS 528.66, 45 of which are homologous genes. The translocation codes for one apoplastic and/or cytoplasmic effector candidate with an unknown functional domain; however, it does not encode
CAZymes and secondary metabolite backbone enzymes. The Pj-2 effector candidate scaffold13.g401(239 aa) is homologous to CBS 528.66 Scaffold17.g5676, showing 100% identity. The 217–250 kb translocation contains 77 genes in Pj-2 and 89 genes in CBS 528.66 (146-396 kb; scaffold17 of CBS 528.66), 77 of which are homologous genes. The translocation codes for two apoplastic and/or cytoplasmic effector candidates with an unknown function; however, it does not encode CAZymes and secondary metabolite backbone enzymes. The 136 aa effector candidates scaffold8.g410 (Pj-2) and scaffold17.g5766 (CBS 528.66) are homologous proteins, exhibiting 95% identity over their entire length; seven aa differ between the two effector candidates. Both are predicted to be localized in the apoplast and/or cytoplasm. However, the second effector candidate (294 aa; scaffold8.g423 in Pj-2 and scaffold17.g5781 in CBS 528.66) does not show any polymorphism between the two strains and is likely localized in the cytoplasm.
A spurious inversion is evident in the collinearity plot between Pj-2 and CBS 528.66. Pj-2 scaffold27 is flipped with respect to the 3’-end of CBS 528.66 scaffold4; the 3’-end of CBS 528.66 scaffold4 is aligned with Pj-2 scaffold 24. Therefore, we conclude that Pj-2 scaffold24 and scaffold27 are part of the same contiguous sequence.
The structural variations in the genomes are the major drivers of genetic diversity within and between species of the same genus, which enable the species or strains within the same species to colonize distinct ecological niches. For example, the maize- and Chrysanthemum-infecting strains of D. glomerata are genetically similar, sharing 92.37% genome with 98.89% sequence similarity. However, the Malus-infecting strain of D. glomerata M27-16 is highly diverse from the other two strains, Pj-2 (sharing only 55.01% genome with 88.20% sequence similarity) and CBS 528.66 (sharing 54.28% genome with 88.22% sequence similarity) (Figures 2A, 3B). The four translocations between Pj-2 and CBS 528.66 may be responsible for the two strains colonizing different hosts, Z. mays and Chrysanthemum spp. However, it is unlikely that CAZymes and secondary metabolism genes located the translocations. We found only one effector candidate showing polymorphism between the two strains, Pj-2 (scaffold8.g410) and CBS 528 (scaffold17.g5766). It would be interesting to investigate whether the targeted deletion of these two effectors in their respective strains contributes to virulence on their hosts.
Germplasm screening to identify the genetic sources of DLB resistance in maize
Deployment of resistant cultivars in the field offers an environmentally friendly way to control crop diseases and thus reduces the overreliance on pesticides, thereby increasing farm crop cash receipts. However, developing a resistant cultivar requires an intraspecific hybridization through the wide/top cross between a susceptible cultivar (elite cultivar) and a donor parent (inbred line, landrace or wild relative) that carries alleles conferring genetic resistance to the disease of interest. Identifying such genetic sources of disease resistance is the first key step in cultivar development. With the above objective and given that DLB is a new disease of maize, we conducted germplasm screening to identify the genetic source of DLB resistance in maize. Thirty maize lines representing inbred lines, cultivars and landraces were screened for reactions to the D. glomerata isolate Pj-2 in a growth chamber through spray-inoculation following an augmented randomized block design (Table S27). Three of these lines expressed complete resistance (immune; no lesion) to D. glomerata, followed by respectively sixteen and eleven lines exhibiting moderate resistance (few confined scattered lesions) and susceptible (coalesced lesions) reactions (Figure 4 and Table S27). The resistant lines were CML496, Jitiaojiao and Denglonghong; CML496 is an elite tropical inbred line, developed by CIMMYT, whereas the other two are local cultivars. CML496 is highly resistant to Puccinia polysora, the causal agent of southern corn rust (SCR) and carries a major QTL RppCML496 for resistance to SCR (Lv et al., 2021). Among the moderately resistant lines were elite inbred lines, such as Ye478 (lesion area/2500 mm2: 12.7 ± 19.2 mm2), Zheng58 (16.7 mm2), Chang7-2 (39.7 mm2), CML470 (51.0 mm2), PH4CV (32.6 mm2), PH6WC (6.5 mm2) and B73 (66.6 mm2) (Figure S2 and Table S27). Ye478, Zheng58 and Chang7-2 are elite lines, heavily used as parental lines in hybrid breeding programs in China because of their high general combining ability and superior agronomic performance (Zheng et al., 2009). All three lines display moderate resistance to Bipolaris maydis (the causal agent of southern corn leaf blight) and Puccinia sorghi (common rust pathogen), whereas they are susceptible to Curvularia lunata (the causal agent of Curvularia leaf spot) and P. polysora. In addition, Zheng58 and Chang7-2 are moderately resistant to Exserohilum turcicum (northern corn leaf blight pathogen), and the latter line also manifests a similar resistance response to Cercospora zeae-maydis (gray leaf spot pathogen) (Wang et al., 2014). CML470, a CIMMYT inbred line, carries a dominant gene RppC, which controls resistance to P. polysora (Sun et al., 2020). PH4CV and PH6WC are the elite inbred lines developed by Pioneer HiBred International; however, their reactions to fungal pathogens are largely unexplored. B73 is susceptible to B. maydis, C. lunata, C. zeae-maydis and P. polysora, whereas the line shows moderate resistance to E. turcicum and complete resistance to P. sorghi.

Figure 4 Maize germplasm screening to identify the genetic sources of resistance to Didymella leaf blight caused by Didymella glomerata. Fourth leaves of thirty lines (cultivars, inbred lines and landraces) at the V3 stage were spray-inoculated with conidial suspension (1 x 107 conidia/ml in 0.025%) until runoff. Three of the lines expressed complete resistance (R; no lesion) to D. glomerata, followed by respectively sixteen and twelve lines exhibiting moderate resistance (MR; few confined scattered lesions) and susceptible (S; coalesced lesions) reactions. An augmented randomized complete block design was used to evaluate the maize germplasm for DLB resistance. The leaves were photographed five days post-inoculation.
Furthermore, we selected the most susceptible inbred line (P178) and the most resistant inbred line (CML496) for histochemical analysis. P178 is an inbred line, which is resistant to P. polysora SCR (six QTL for SCR resistance) (Tian et al., 2018) and Exserohilum turcicum (Wang et al., 2019). However, P178 exhibited extensive leaf blight following the inoculation with the D. glomerata strain Pj-2; over 50% of the inoculated leaf area showed blight (lesion area/2500 mm2: 1278.6 ± 38.5 mm2) while the CML496 leaves remained healthy (Figures 4, S2 and Table S27). DAB-staining of the D. glomerata-infected leaf tissues revealed the accumulation of H2O2, a reactive oxygen species, in the apoplastic space of the infected issues of CML496, not in P718 (Figure 5). The rapid and transient burst of ROS (called oxidative burst) at the infection site is the hallmark of the pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and ETI, and is ensued upon successful recognition of pathogens by host cells (Torres, 2010). The CML496 infected cells likely successfully perceived D. glomerata, which in turn activated defense responses, such as oxidative burst that arrested fungal growth and proliferation (Figures 5A, C). The pathogen apparently requires successful penetration of the host epidermal cell cuticle, which may trigger its perception in the CML496 epidermal cells, leading to DLB resistance (Figure 5C). However, D. glomerata likely dodges surveillance (PTI and ETI) in the P178 epidermal cells, facilitating fungal growth and proliferation (Figures 5B, D). Altogether, the DLB of maize is likely a gene-for-gene disease in which the resistant maize genotypes (e.g., CML496, Jitiaojiao and Denglonghong) and C. graminicola recognize each other by corresponding pairs of the nucleotide-binding leucine-rich repeat receptors and effectors.
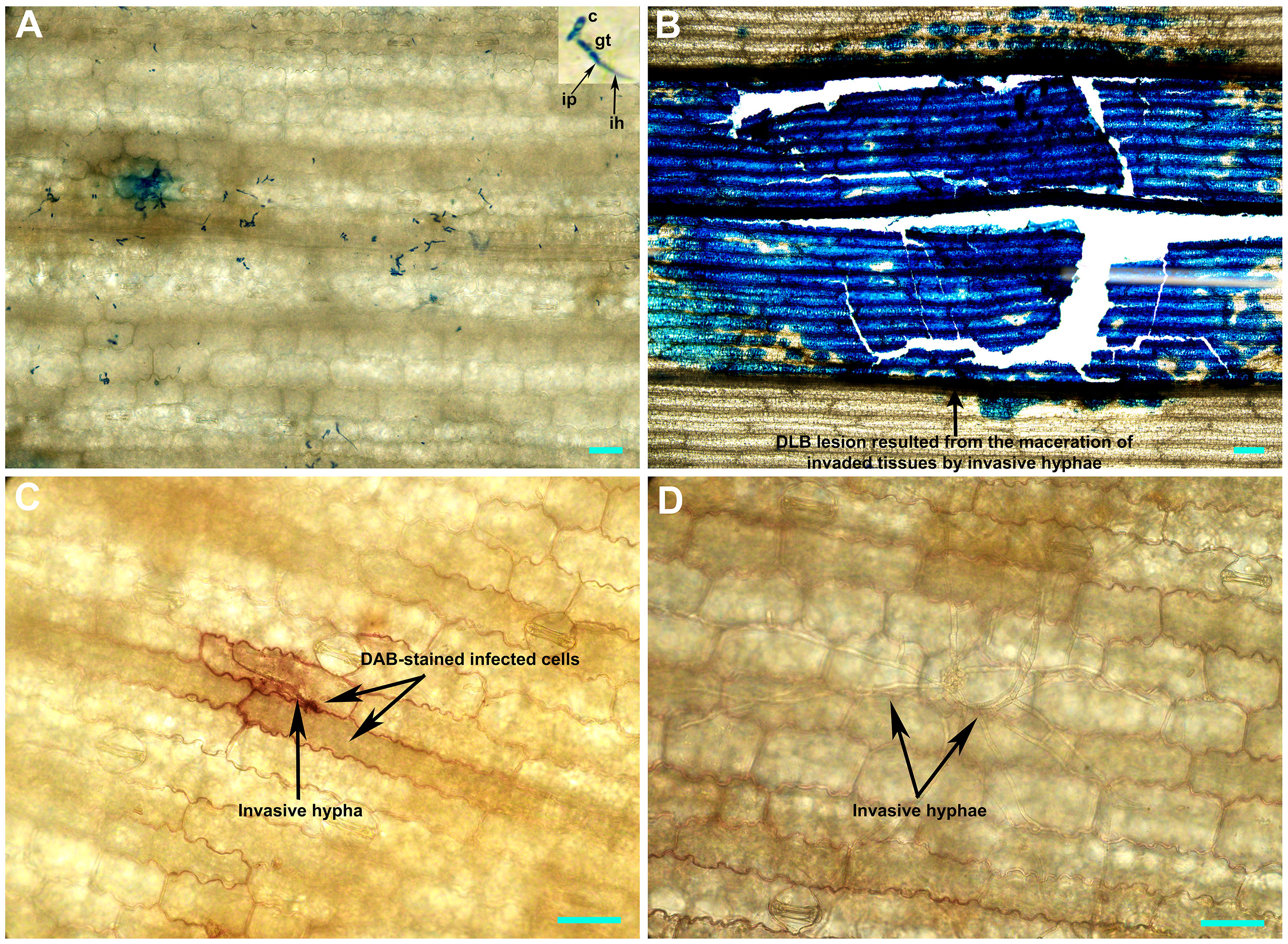
Figure 5 Histochemical staining of the Didymella glomerata strain Pj-2-infected tissues of susceptible (P178) and resistant (CML496) inbred lines of maize. The top image panel shows the acidic aniline blue-stained CML496 (A) and P718 (B) leaf tissues, and the bottom image panel illustrates the DAB-stained CML496 (C) and P178 (D) leaf tissues five days post-inoculation. The D. glomerata conidia germinate to form germ tubes, which directly breach the host cell cuticle, presumably via infection pegs. This invasion is restricted to the first infected epidermal cells (A) and leads to the accumulation of H2O2 (a reactive oxygen species; c) in CML496, whereas the invasion in P178 leads to the maceration of infected tissues (B) by invasive hyphae (D). c, conidia; gt, germ tube; ip, infection peg; ih, invasive hypha; Bars = 20 µm.
Concluding remarks
DLB is a new disease of maize, which we recently identified in Panjin, Liaoning, a northeastern province of China (Ma et al., 2022). In this study, we presented a nearly gapless genome assembly of the D. glomerata strain Pj-2, which comprises 32 scaffolds, including four full-length scaffolds (representing chromosomes) carrying telomeres at both ends. Phylogenomic analysis shows that the maize- and Chrysanthemum-infecting strains of D. glomerata are genetically similar and share 93.37% genome with 98.89% sequence similarity. However, they carry four major reciprocal translocation regions in their genomes, which may allow their adaptation to different hosts. Furthermore, germplasm screening against DLB identified three maize inbred lines (e.g., CML496, Jitiaojiao and Denglonghong) expressing complete resistance to the D. glomerata strain Pj-2. Therefore, these lines can be used as a donor parent to transfer genetic resistance into other elite cultivars lacking DLB resistance through intraspecific hybridization introgression. Further research, however, is required to identify genes in CML496 conferring resistance to DLB. In addition, field research should be undertaken to assess the economic impact of this new maize disease, both in terms of yield loss and down-gradation of grain quality.
Methods
Fungal and plant material
The D. glomerata strain Pj-2 was routinely maintained on PDA plates under a 12 h photoperiod at 25°C. Maize germplasm involving 30 lines representing cultivars, inbred lines and landraces (Table S27) was evaluated for reactions to Pj-2.
Genome sequencing, size, assembly and completeness
Conidia (ca. 107) were collected by swamping the five-day-old six-cm PDA culture plates of Pj-2 with five ml of distilled deionized H2O. One ml of conidial suspension was incubated in a one-liter flask containing 200 ml of complete medium for four days in an orbital shaker set at 25°C and 120 rpm. Mycelia balls were collected by filtering the liquid culture through a one-layered Miracloth. The ENZA Fungal DNA kit (Omega Bio-Tek, Norcross, USA) was used to isolate genomic DNA (gDNA) from the mycelia.
Covaris E220 (Covaris, Brighton, UK) was used to fragment one µg of high-quality gDNA, and end-repaired fragments were used to construct a short insert (500 bp) paired-end library using TruSeq DNA LT Kit (Illumina, San Diego, USA). The library was assessed for the insert size and quality using BioAnalyzer 2100 using Agilent 1000 DNA Chip (Agilent Technologies, Palo Alto, USA) and sequenced on NovaSeq 6000 (Illumina, San Diego, USA), generating 2 x 250 bp reads. AdapterRemoval v2 (Schubert et al., 2016) was used to clip adapters and low-quality bases from the paired-end reads. SOAPec v2.0 (Luo et al., 2012) was used to correct sequencing errors based on the K-mer (17) frequency in the paired-end reads. Finally, Jellyfish v2.0 (Marçais and Kingsford, 2011) was used to count K-mer (17) within the high-quality paired-end reads to estimate the D. glomerata Pj-2 genome size.
g-TUBE (Covaris, Brighton, UK) was used to shear five µg of high-quality gDNA, and BluePippin (Sage Science, Beverly, USA) was utilized to size-select ~10 kb gDNA fragments. Finally, the end-repaired large fragments were used to construct a long insert (10 kb) library using SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, Menlo Park, USA). The resulting library was assessed for insert size and quality on BioAnalyzer 2100 and sequenced on PacBio Sequel II (Pacific Biosciences, Menlo Park, USA) using Continuous Long Read mode. The polymerase reads generated on PacBio Sequel II were checked for adapter contamination, quality and size using SMRT analysis v2.3.0; the subreads (quality >0.8 and size >1,000 bp) were extracted from the adapter-free polymerase reads. Unicycler (Wick et al., 2017) was employed to contrive a hybrid genome assembly using high-quality 250-bp paired-end reads (NovaSeq 6000) and 1,000–10,000 bp subreads (PacBio Sequel II).
Phylogenomics
The whole genomes of eight strains of six Didymella spp. (D. glomerata Pj-2, CBS 528.66 and M27-16, D. keratinophila 9M1, D. heteroderae 28M1, D. pinodes WTN-11-157, D. exigua CBS 183.55 and D. rabiei ArMe14) were aligned using the Whole Genome Alignment tool plugged in CLC Genomics Workbench (Qiagen, Aarhus, Denmark) with the following parameters: 50-bp seed and minimum alignment block size ≥100 bp. AP and ANI were calculated from the whole-genome alignment using pairwise distance matrices.
Genome annotation
The following genomes were masked for repetitive DNA elements using RepeatMasker v4.0.6 (Smit et al., 1996) and RepeatModeler v2.1 (Smit and Hubley, 2015): the three D. glomerata strains (Pj-2, CBS 528.66 and M27-16), D. exigua CBS 183.55, D. pinodes WTN-11-157, D. rabiei ArMe14, D. keratinophila 9M1 and D. heteroderae 28M1. The masked genome of Pj-2 was assessed for its completeness using BUSCO v5.1.3 (Simão et al., 2015) with a set of 6,641 benchmarking universal single-copy orthologs (BUSCOs) in Pleosporales.
Augustus v3.03 (Stanke and Morgenstern, 2005), glimmerHMM v3.0.1 (Majoros et al., 2004) and GeneMark-ES v4.35 (Ter-Hovhannisyan et al., 2008) were used to predict gene models ab initio in the masked genomes. Protein models from the closely related Didymella species (D. keratinophila, D. heteroderae, D. exigua and D. rabiei) were mapped onto the D. glomerata genome using Exonerate v2.2.0 (http://www.ebi.ac.uk). The ab initio predictions and the Exonerate alignments were combined using EVidenceModeler v.r2012-06-25 (Haas et al., 2008) to obtain final gene models. CAZymes and secondary metabolism genes were predicted as described previously (Bhadauria et al., 2019). Genes that encode protein equal to or less than 300 aa in length, carrying N-terminal signal peptide (predicted using SignalP v5.0), lacking transmembrane domain (TMHMM v2.0) and GPI anchor addition site (PredGPI) were considered as effector candidate genes. In addition, EffectorP v3.0 was employed to discern their potential localization in the host cells, i.e., apoplast, cytoplasm and apoplast/cytoplasm. We also screened the effector candidates for possible N- and O-glycosylation sites using NetNGlyc v1.0 and NetOGlyc v4.0, respectively, as such sites allow their tethering to plasma membrane.
Germplasm screening and statistical analysis
Thirty maize lines were individually grown in pots (6 x 10 cm) containing peat moss (soilless medium) and perlite in a 2:1 ratio in a growth chamber following an augmented randomized block design. Plants were spray-inoculated at the V3 growth stage with the Pj-2 conidia (1 x 107 conidia/ml) following the method described previously (Vargas et al., 2012). The fourth leaves of the inoculated plants were photographed six days post-inoculation (dpi), and the lesion area (mm2) was measured using ImageJ (https://imagej.net/). A linear mixed-effects model using the R lm() function was used to calculate least-squares (LS) means of lesion areas on the maize lines (a fixed effect) while considering blocks (biological replications) as a random effect.
Histochemical analysis of the Pj-2 infected maize tissues
Acidic aniline blue and 3, 3-diaminobenzidine (DAB) staining of Pj-2 infected leaf tissues of the inbred lines P178 and CML496 were performed by methods described by Bhadauria et al. (2010) and Thordal-Christensen et al. (1997), respectively.
Data availability statement
The D. glomerata Pj-2 (BioSample: SAMN23554018) genome (32 scaffolds) has been deposited to GenBank/ENA/DDBJ under the accession number JAJOHN000000000 (BioProject: PRJNA785361).
Author contributions
VB conceived and performed research. Y-LP, VB, JD, WZ, JY and WM analyzed data. JD provided maize germplasm and VB, Y-LP, JD, WZ and JY wrote the paper. All authors contributed to the article and approved the submitted version.
Acknowledgments
The research was supported by grants from the National Natural Science Foundation of China (Grant No. 32172363) and the Chinese Universities Scientific Fund (Grant No. 10092004). Technical support for the incumbent study was provided by Hongjie Yuan at the Communication University of China, Beijing, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1022819/full#supplementary-material
Supplementary Figure 1 | Genome collinearity of D. glomerata Pj-2 with Didymella keratinophila 9M1, Didymella heteroderae, Didymella pinnodes WTN-11-157, Didymella exigua CBS 183.55 and the Didymella (Ascochyta) rabiei ArMe14.
Supplementary Figure 2 | Reactions of the Didymella glomerata strain Pj-2 on 30 maize lines representing cultivars, inbred lines and landraces five days post-inoculation. Lesion areas were measured using ImageJ (https://imagej.net/).
References
Bhadauria, V., MacLachlan, R., Pozniak, C., Cohen-Skalie, A., Li, L., Halliday, J., et al. (2019). Genetic map-guided genome assembly reveals a virulence-governing minichromosome in the lentil anthracnose pathogen Colletotrichum lentis. New Phytol. 221 (1), 431–445. doi: 10.1111/nph.15369
Bhadauria, V., Miraz, P., Kennedy, R., Banniza, S., Wei, Y. (2010). Dual trypan-aniline blue fluorescence staining methods to study fungal-plant interactions. Biotech Histochem. 85 (2), 99–105. doi: 10.3109/10520290903132196
Chiaromonte, F., Yap, V. B., Miller, W. (2002). Scoring pairwise genomic sequence alignments. PSB. 7, 115–126. doi: 10.1142/9789812799623_0012
Cimmino, A., Bahmani, Z., Masi, M., Abdollahzadeh, J., Amini, J., Tuzi, A., et al. (2021). Phytotoxins produced by Didymella glomerata and truncatella angustata, associated with grapevine trunk diseases (GTDs) in Iran. Nat. Prod. Res. 36 (17), 1478–6419. doi: 10.1080/14786419.2021.1979544
Collemare, J., Pianfetti, M., Houlle, A. E., Morin, D., Camborde, L., Gagey, M. J., et al. (2008). Magnaporthe grisea avirulence gene ACE1 belongs to an infection-specific gene cluster involved in secondary metabolism. New Phytol. 179, 196–208. doi: 10.1111/j.1469-8137.2008.02459.x
Farman, M. L. (2007). Telomeres in the rice blast fungus Magnaporthe oryzae: the world of the end as we know it. FEMS Microbiol. 273, 125–132. doi: 10.1111/j.1574-6968.2007.00812.x
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9 (1), R7. doi: 10.1186/gb-2008-9-1-r7
Hosford, R. (1975). Phoma glomerata, a new pathogen of wheat and triticales. Phytopathology 65, 1236–1239. doi: 10.1094/Phyto-65-1236
Huang, S. L., Wang, L., Wang, T., Jiao, Z. J., Pang, F. H., Tao, A. L., et al. (2018). First report of didymella leaf blight on cornus officinalis caused by Didymella glomerata in China. Plant Dis. 102 (5), 1031. doi: 10.1094/PDIS-07-17-0933-PDN
Irinyi, L., Kövics, G. J., Sándor, E. (2009). Taxonomical re-evaluation of phoma-like soybean pathogenic fungi. Mycol. Res. 113, 249–260. doi: 10.1016/j.mycres.2008.11.003
Kämper, J., Kahmann, R., Bölker, M., Ma, L., Brefort, T., Saville, B. J., et al. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444 (7115), 97–101. doi: 10.1038/nature05248
Khani, M. (2014). Aspects of epidemiology of phoma koolunga (Ascochyta blight of field pea) (Adelaide: University of Adelaide).
Li, S., Xue, X., Gao, M., Wang, N., Cui, X., Sang, S., et al. (2021). Genome resource for peanut web blotch causal agent Peyronellaea arachidicola strain YY187. Plant Dis. 105 (4), 1177–1178. doi: 10.1094/PDIS-04-20-0898-A
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1 (1), 18. doi: 10.1186/2047-217X-1-18
Lv, M., Deng, C., Li, X., Zhao, X., Li, H., Li, Z., et al. (2021). Identification and fine-mapping of RppCML496, a major QTL for resistance to Puccinia polysora in maize. Plant Genome 14, e20062. doi: 10.1002/tpg2.20062
Ma, W., Yang, J., Gao, X., Han, T., Liu, J., Ding, J., et al. (2022). First report of didymella glomerata causing didymella leaf blight on maize in China. Plant Dis. 106 (9), 2522. doi: 10.1094/PDIS-02-22-0282-PDN
Ma, L. J., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in. Fusarium Nat. 464 (7287), 367–373. doi: 10.1038/nature08850
Majoros, W. H., Pertea, M., Salzberg, S. L. (2004). TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20 (16), 2878–2879. doi: 10.1093/bioinformatics/bth315
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 (6), 764–770. doi: 10.1093/bioinformatics/btr011
Moral, J., Lichtemberg, P. S. F., Papagelis, A., Sherman, J., Michailides, T. J. (2018). Didymella glomerata causing leaf blight on pistachio. Eur. J. Plant Pathol. 151, 1095–1099. doi: 10.1007/s10658-018-1422-y
Pan, H., Chen, M. Y., Deng, L., Wang, Z. P., Li, L., Zhong, C. H. (2018). First report of Didymella glomerata causing black spot disease of kiwifruit in China. Plant Dis. 102 (12), 2654. doi: 10.1094/PDIS-04-18-0583-PDN
Reeder, E. T., Vanterpool, T. C. (1953). Phoma spp. on flax in Saskatchewan. Proc. Can. Phytopathol. Soc 21, 16.
Ren, Y., Li, D., Zhao, X., Wang, Y., Bao, X., Wang, X., et al. (2019). Whole genome sequences of the tea leaf spot pathogen Didymella segeticola. Phytopathology 109, 1676–1678. doi: 10.1094/PHYTO-02-19-0050-A
Roustaee, A., Costes, S., Dechamp-Guillaume, G., Barrault, G. (2000). Phenotypic variability of Leptosphaeria lindquistii (anamorph: Phoma macdonaldii), a fungal pathogen of sunflower. Plant Pathol. 49, 227–234. doi: 10.1046/j.1365-3059.2000.00451.x
Rouxel, T., Grandaubert, J., Hane, J., Hoede, C., van de Wouw, A. P., Couloux, A., et al. (2011). Effector diversification within compartments of the leptosphaeria maculans genome affected by repeat-induced point mutations. Nat. Commun. 2, 202. doi: 10.1038/ncomms1189
Shah, R. M., Williams, A. H., Hane, J. K., Lawrence, J. A., Farfan-Caceres, L. M., Debler, J. W., et al. (2020). Reference genome assembly for Australian Ascochyta rabiei isolate ArME14. G3 (Bethesda Md.) 10 (7), 2131–2140. doi: 10.1534/g3.120.401265
Simão, D., Pinto, C., Piersanti, S., Weston, A., Peddie, C. J., Bastos, A. E., et al. (2015). Modeling human neural functionality in vitro: three-dimensional culture for dopaminergic differentiation. Tissue Eng Part A 21 (3-4), 654–668. doi: 10.1089/ten.tea.2014.0079
Smit, A., Hubley, R. (2015) Repeat modeler open-1.0. Available at: http://repeatmasker.org.
Smit, B., McNabb, D., Smithers, J. (1996). Agricultural adaptation to climatic variation. Clim. Change 33, 7–29. doi: 10.1007/BF00140511
Soanes, D. M., Chakrabarti, A., Paszkiewicz, K. H., Dawe, A. L., Talbot, N. J. (2012). Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PloS Pathog. 8, e1002514. doi: 10.1371/journal.ppat.1002514
Sperschneider, J., Dodds, P. N. (2022). EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant Microbe Interact. 35 (2), 146–156. doi: 10.1094/MPMI-08-21-0201-R
Stanke, M., Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33 (W), W465–W467. doi: 10.1093/nar/gki458
Stergiopoulos, I., De Wit, P. J. G. M. (2009). Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263. doi: 10.1146/annurev.phyto.112408.132637
Sugawara, F., Strobel, G. (1986). Zinniol, a phytotoxin, is produced by Phoma macdonaldii. Plant Sci. 43 (1), 19–23. doi: 10.1016/0168-9452(86)90102-0
Sun, Q., Yu, S., Guo, Z. (2020). Calmodulin-like (CML) gene family in Medicago truncatula: Genome-wide identification, characterization and expression analysis. Int. J. Mol. Sci. 21 (19), 7142. doi: 10.3390/ijms21197142
Taber, R. A., Vanterpool, T. C. (1963). Alternaria species on rape in Western Canada. Proc. Can. Phytopathol.l Soc 30, 10–21.
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18 (12), 1979–1990. doi: 10.1101/gr.081612.108
Thomidis, T., Michailides, T. J., Exadaktylou, E. (2011). Phoma glomerata (Corda) wollenw. & hochapfel a new threat causing cankers on shoots of peach trees in Greece. Eur. J. Plant Pathol. 131, 171–178. doi: 10.1007/s10658-011-9796-0
Thordal-Christensen, H., Zhang, Z., Wei, Y., Collinge, D. B. (1997). Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley–powdery mildew interaction. Plant J. 11 (6), 1187–1194. doi: 10.1046/j.1365-313X.1997.11061187.x
Tian, Z., Ai, T., Deng, C., You, S., Shi, Q., Ma, Y., et al. (2018). QTL mapping of resistance to northern corn leaf blight in maize inbred line P178 and effect analysis. J. Henan Agric. Sci. 47 (2), 73–76.
Torres, M. A. (2010). ROS in biotic interactions. Physiol. Plant 138 (4), 414–429. doi: 10.1111/j.1399-3054.2009.01326.x
Tran, H. S., You, M. P., Lanoiselet, V., Khan, T. N., Barbetti, M. J. (2014). First report of Phoma glomerata associated with the ascochyta blight complex on field pea (Pisum sativum) in Australia. Plant Dis. 98, 427. doi: 10.1094/PDIS-08-13-0809-PDN
van den Brink, J., de Vries, R. P. (2011). Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 91, 1477–1492. doi: 10.1007/s00253-011-3473-2
Vargas, W. A., Sanz Martín, J. M., Rech, G. E., Rivera, L. P., Benito, E. P., Díaz-Mínguez, J. M., et al. (2012). Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotrichum graminicola in maize. Plant Physiol. 158 (3), 1342–1358. doi: 10.1104/pp.111.190397
Wang, X., Zhang, Y., Xu, X., Li, H., Wu, X., Zhang, S., et al. (2014). Evaluation of maize inbred lines currently used in Chinese breeding programs for resistance to six foliar diseases. Crop J. 2 (4), 213–222. doi: 10.1016/j.cj.2014.04.004
Wang, S., Chen, Z., Tian, L., Ding, Y., Zhang, J., Zhou, J., et al. (2019). Comparative proteomics combined with analyses of transgenic plants reveal ZmREM1.3 mediates maize resistance to southern corn rust. Plant Biotechnol. J. 17 (11), 2153–2168. doi: 10.1111/pbi.13129
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13 (6), e1005595. doi: 10.1371/journal.pcbi.1005595
Keywords: emerging maize diseases, fungal genomes, effectors, CAZymes, secondary, metabolism genes, genetic source of Didymella leaf blight resistance
Citation: Ma W, Yang J, Ding J, Zhao W, Peng Y-L and Bhadauria V (2022) Gapless reference genome assembly of Didymella glomerata, a new fungal pathogen of maize causing Didymella leaf blight. Front. Plant Sci. 13:1022819. doi: 10.3389/fpls.2022.1022819
Received: 19 August 2022; Accepted: 30 September 2022;
Published: 26 October 2022.
Edited by:
Cheng Liu, Shandong Academy of Agricultural Sciences, ChinaReviewed by:
Yunsheng Wang, Hunan Agricultural University, ChinaYanping Tian, Shandong Agricultural University, China
Copyright © 2022 Ma, Yang, Ding, Zhao, Peng and Bhadauria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vijai Bhadauria, dmlqYWkuYmhhZGF1cmlhQGNhdS5lZHUuY24=