 Yakupjan Haxim1,2,3†
Yakupjan Haxim1,2,3† Gulnaz Kahar1,4†
Gulnaz Kahar1,4† Xuechun Zhang1,5Yu Si4
Xuechun Zhang1,5Yu Si4 Abdul Waheed1,2Xiaojie Liu1,2,3Xuejing Wen1,2,3
Abdul Waheed1,2Xiaojie Liu1,2,3Xuejing Wen1,2,3 Xiaoshuang Li1,2,3
Xiaoshuang Li1,2,3 Daoyuan Zhang1,2,3*
Daoyuan Zhang1,2,3*- 1State Key Laboratory of Desert and Oasis Ecology, Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Ürümqi, China
- 2Xinjiang Key Laboratory of Conservation and Utilization of Plant Gene Resources, Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Ürümqi, China
- 3Turpan Eremophytes Botanical Garden, Chinese Academy of Sciences, Turpan, China
- 4University of Chinese Academy of Sciences, College of Resources and Environment, Beijing, China
- 5School of Life Sciences, Xinjiang Normal University, Ürümqi, China
Chitinases are responsible for catalyzing the hydrolysis of chitin and contribute to plant defense against fungal pathogens by degrading fungal chitin. In this study, genome-wide identification of the chitinase gene family of wild apple (Malus sieversii) and domesticated apple (Malus domestica) was conducted, and the expression profile was analyzed in response to Valsa mali infection. A total of 36 and 47 chitinase genes belonging to the glycosyl hydrolase 18 (GH18) and 19 (GH19) families were identified in the genomes of M. sieversii and M. domestica, respectively. These genes were classified into five classes based on their phylogenetic relationships and conserved catalytic domains. The genes were randomly distributed on the chromosomes and exhibited expansion by tandem and segmental duplication. Eight of the 36 MsChi genes and 17 of the 47 MdChi genes were differentially expressed in response to V. mali inoculation. In particular, MsChi35 and its ortholog MdChi41, a class IV chitinase, were constitutively expressed at high levels in M. sieversii and domesticated apple, respectively, and may play a crucial role in the defense response against V. mali. These results improve knowledge of the chitinase gene family in apple species and provide a foundation for further studies of fungal disease prevention in apple.
Introduction
Malus sieversii is the primary progenitor of many cultivars of domesticated apple (M. domestica) and is mainly distributed in Central Asia and Xinjiang, China (Volk et al., 2013; Duan et al., 2017). A previous study of the phylogenetic relationships of cultivated apple and its ancestor revealed that M. sieversii from Xinjiang, China, was most closely related to the M. domestica cultivar ‘Golden Delicious’ (Zhou and Li, 2000). Malus sieversii is an important species in natural forest of the Tianshan Mountains in Xinjiang, and features high genetic diversity and disease resistance, thus providing a valuable genetic resource for molecular breeding of cultivated apple (Chen et al., 2007; Zhang et al., 2007; Ballester et al., 2017). The fruit of M. sieversii contain higher phenolic and flavonoid contents compared with those of cultivated apple, and potentially could be used to breed cultivars that produce fruit with red flesh and high flavonoid content (Zhang et al., 2008).
Recent studies have reported that the wild M. sieversii population in the Tianshan Mountains has experienced a dramatic decline partly as a result of Valsa canker (Bozorov et al., 2019; Liu et al., 2020). Valsa canker is caused by the necrotrophic pathogen Valsa mali, which is a major threat to apple production in China, Japan, Korea, and Central Asia, where it leads to severe yield losses (Lee et al., 2006; Suzaki, 2008; Yin et al., 2015; Wang et al., 2016). Fungicide applications are not always effective to control fungal mycelia within the host xylem (Yin et al., 2016). The mining of candidate genes and development of disease-resistant cultivars are effective and practical approaches to control the disease. The transcriptomic changes in response to V. mali infection have been investigated previously in M. sieversii (Liu et al., 2021) and M. domestica (Yin et al., 2016). The plant–pathogen interaction, plant hormone signal transduction, flavonoid biosynthesis, and phenylpropanoid biosynthesis pathways of M. sieversii (Haxim et al., 2021; Liu et al., 2021), and chitin signaling pathway of M. domestica (Yin et al., 2016) were significantly enriched in response to infection by V. mali. Exploration of the disease-resistance genes in apple responsive to V. mali infection is important for disease prevention in domesticated and wild apple species, especially M. sieversii.
Chitinases (EC 3.2.1.14) are glycosyl hydrolases that breakdown glycosidic bonds in chitin, a major structural component of fungal cell walls, and play important roles in plant defense responses (Grover Plant Chitinases, 2012; Hamid et al., 2013). Plant chitinase hydrolyzes the β-1,4-glycosidic bonds of chitin into chitin oligosaccharides during fungal infection and these oligosaccharides activate the immune responses of the host plant (Shibuya and Minami, 2001; Kaku et al., 2006; Hamid et al., 2013). To date, more than 175 different families of glycosyl hydrolases have been identified based on sequence similarity (http://www.cazy.org). On the basis of catalytic domains and amino acid sequence similarity, chitinases have been classified into the glycosyl hydrolase 18 (GH18) and 19 (GH19) families (Henrissat and Bairoch, 1993; Hamid et al., 2013). In addition, to reflect phylogenetic relationships, plant chitinases have been categorized into five classes (Class I to V), of which the GH18 family comprises classes III and V, and the GH19 family contains classes I, II, and IV (Grover Plant Chitinases, 2012). Chitinases are involved in the response to diverse abiotic stresses, such as heat, cold, salt, wounding, drought, heavy metal toxicity, ozone, and ultraviolet light, as well as a variety of plant phytohormones, including jasmonic acid, salicylic acid, ethylene, cytokinin, and indole acetic acid (Kasprzewska, 2003; Su et al., 2014). Members of the chitinase family from plant species are able to inhibit fungal growth in vitro (Li et al., 2019) and in vivo (Xiao et al., 2007), and therefore are potential candidates with which to enhance plant resistance to fungal pathogens (Kumar et al., 2018).
The availability of plant genomic sequence data allows genome-wide mining of the chitinase gene family. Recently, the chitinase gene family has been identified in the genomes of several tree species, such as Populus trichocarpa (Zhang et al., 2022), Eucalyptus grandis (Tobias et al., 2017), and Hevea brasiliensis (Misra, 2015). However, to date, the chitinase genes of apple species have not been systematically analyzed. The availability of whole-genome sequences for apple species, including M. sieversii and M. domestica, enables genome-wide identification of chitinase genes. In this study, the chitinase family members in the genomes of M. sieversii and M. domestica were surveyed and their phylogenetic relationships, gene structure, and gene duplication events were analyzed. In addition, the expression patterns of the chitinase genes in response to V. mali infection were analyzed. The present results extend knowledge of chitinases in apple species and might provide effective gene resources for improvement of apple resistance to V. mali.
Materials and methods
Plant material and pathogen infection
Malus sieversii (Ledeb.) M.Roem. seeds were purchased from the Nature and Wildlife Conservation Station of Xinyuan County, Xinjiang, China. Malus domestica ‘Golden Delicious’ seeds were purchased from the Liqun Nursery Co. Ltd. (Shandong, China). The seeds were germinated in a petri dish after removal of the husks. Three-week-old seedlings were planted in pots containing a mixture of soil and vermiculite and were grown at 24°C under a 16 h/8 h (light/dark) photoperiod. The V. mali isolate EGI1 (Liu et al., 2020) was grown on potato dextrose agar (PDA) medium (Rishui BioTechnologies, Qingdao, China) at 25°C for 3 days. Six-month-old plants were infected with V. mali as described previously (Liu et al., 2020; Haxim et al., 2021). Briefly, mycelial plugs (diameter: 5 mm) were excised from 3-day-old cultures of isolate EGI1 on PDA medium. Detached leaves of the host plant were wounded with a fabric pattern wheel and then inoculated with the mycelial plugs. The inoculated leaves were placed in petri dishes sealed with parafilm and were incubated in the dark at 25°C.
RNA isolation and real-time quantitative PCR analysis
Total RNA was isolated from M. sieversii leaves collected at different time points (0, 1, 2, and 5 days post-inoculation [dpi]) after inoculation with V. mali using the E.Z.N.A.® Plant RNA Kit (No. R6827, Omega Bio-tek, Norcross, GA, USA) in accordance with the manufacturer’s instructions. The first-strand cDNA was synthesized from 1 µg total RNA using the PrimeScript RT reagent Kit with gDNA Eraser (No. RR047Q, Takara, Dalian, China). To validate the expression level of the chitinase genes, gene-specific primers (Table S1) were designed using DNAMAN version 9.0 software (Lynnon BioSoft, Vaudreuil, Quebec, Canada) and synthesized by Sangon Biotech (Shanghai, China). The transcripts of the target genes were detected using the TB Green Premix Ex Taq II Kit (No. RR820A, Takara) with the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The thermal profile for qRT-PCR was as follows: preheating at 95°C for 30 s, then 40 cycles of 95°C for 5 s and 58°C for 30 s. A melting curve analysis was conducted to confirm the amplification specificity. The relative transcript abundance of the chitinase genes was analyzed with the 2−ΔΔCt method (Livak and Schmittgen, 2001). The EF-1α (Elongation factor 1-α) gene has been evaluated for internal reference in apple species (Zhu et al., 2019) and served as reference in our previous work (Liu et al., 2021; Haxim et al., 2021; Liu et al., 2021) because of its stable expression in our previous work under V.mali infection. Therefore, the EF-1α gene was also used as an internal reference for the qPCR quantification in the present study. Each sample comprised three biological replicates and each biological replicate was analyzed with three technical replicates. Statistical analysis of the data was performed with analysis of variance using SPSS 18 software (SPSS, Chicago, IL, USA).
RNA-sequencing data analysis
Transcriptome data for M. sieversii (NCBI BioProject: PRJNA687214) (Liu et al., 2021) and M. domestica (NCBI SRA accession: SRP034726) (Yin et al., 2016) in response to V. mali infection were used to analyze differentially expressed genes. The fragments per kilobase of transcript per million fragments mapped (FPKM) value was calculated for each gene. Differential expression analysis of three biological replicates per condition was performed using the ‘DESeq’ R package (1.18.0). The P-values were adjusted using the Benjamini–Hochberg approach to control the false discovery rate. Transcripts with an adjusted P-value < 0.05 and log2 fold change > 1 were considered to be differentially expressed. The log2 fold-change values were used for heatmap generation.
Genome-wide identification of chitinase genes
To identify potential chitinase genes in the two apple species, the M. sieversii genome (version JAHTLV010000000) and M. domestica genome (Daccord et al., 2017) (GDDH13 version 1.1) were retrieved from the NCBI (https://www.ncbi.nlm.nih.gov/) and GDR (https://www.rosaceae.org/) databases, respectively. A hidden Markov model seed profile of Glyco_hydro_18 (PF00704) and Glyco_hydro_19 (PF00182) was downloaded from the Pfam database (Mistry et al., 2021) and chitinase genes in the genomes were identified using TBtools software (Chen et al., 2020). The SMART (Letunic et al., 2021) database was used to confirm the presence of chitinase domains with a cut-off E-value < 0.0001.
Phylogenetic relationships, gene organization, and conserved motif analysis
To study evolutionary relationships, the full-length amino acid sequences of chitinase proteins from M. domestica, M. sieversii, and Arabidopsis thaliana were aligned using Clustal X2 (http://www.clustal.org/). A phylogenetic tree was generated using MEGA-X software (https://www.megasoftware.net/) with the maximum likelihood method and topological support was assessed by means of a bootstrap analysis with 1000 replicates. The exon–intron organization of the chitinase genes was visualized using TBtools (Chen et al., 2020). Conserved motifs and domains were identified with the MEME Suite (Bailey et al., 2006) and SMART database (Letunic et al., 2021), respectively, and were visualized using TBtools (Chen et al., 2020). The domain signatures were identified using the PROSITE database (Sigrist et al., 2013). The SignalP 5.0 online server (Almagro Armenteros et al., 2019) was used to identify signal peptides in the chitinase proteins of M. sieversii and M. domestica.
Chromosomal location and gene duplication analysis
The chromosomal location of the chitinase genes was visualized with the MG2C online tool (http://mg2c.iask.in/mg2c_v2.1/) based on genomic annotation data. Gene duplication events and synteny relationships of the chitinase genes were analyzed using the multiple collinearity scan toolkit MCScanX (Wang et al., 2012) and were visualized with TBtools (Chen et al., 2020). For tandem duplication events, two or more chitinase genes separated by five or fewer genes within an interval of 100 kb on the same chromosome were considered to be tandem duplicated genes (Wang et al., 2010; Zhao et al., 2018). Substitution rates of synonymous (Ks) and non-synonymous (Ka) chitinase genes were calculated with KaKs_Calculator 3.0 (Zhang, 2022). The evolutionary duplication time (T) was calculated with the formula T = Ks/2λ × 10−6 million years ago (Mya), where λ = 1.5 × 10−8 in apple (Zhang et al., 2018).
Prediction of cis-acting elements in promoter regions
The promoter sequence (the 2.0 kb DNA sequence upstream of the start codon) of each chitinase gene was retrieved from the M. sieversii genome (version JAHTLV010000000) and M. × domestica genome (GDDH13 version 1.1). The cis-acting elements in each gene was predicted using the PlantCARE online tools (Lescot, 2002).
Gene co-expression network analysis for M. sieversii
Gene co-expression network analysis was performed using the weighted gene co-expression network analysis (WGCNA) tools from the BMKCloud platform (https://international.biocloud.net/zh/software/tools/list). Normalized gene expression data (FPKM > 1) in the 12 samples from M. sieversii were used as the input, and the expression level of the chitinase genes was used as a trait. The WGCNA modules of eigengenes correlated with chitinase gene expression profiles were identified with the default settings using a dynamic tree cut-off algorithm (minimum cluster size 30 and merging threshold function 0.3). Module membership (MM) was calculated based on Pearson correlation analysis between the expression level and the module eigengenes (ME; representative of the gene expression profile in each module). Gene significance (GS) was calculated as the correlation between the trait data and MEs.
Transient expression assay
For the transient expression, the full length of MsChi35 gene was cloned into pBI121 plant expression vector by using an In-fusion HD cloning kit (TAKRA, Code No.639648). The pBI121-MsChi35 was transiently expressed in detached M.sieverssi leaves by agroinfiltration method as described (Liu et al., 2021). After the 3 days of agroinfiltration, these leaves were wounded by a fabric pattern wheel and inoculated with V.mali isolate EGI1 as described (2.1) (Liu et al., 2020; Liu et al., 2021). Leaves inoculated with sterile ddH2O were used as controls. After a 4-days of inoculation of the V. mali, the lesion areas were measured by ImageJ software and lesion ratios (%) was calculated as proportion of lesion area in the whole leaf. Each sample contained ten leaves with three biological replicates. Statistical analysis of the data was performed with analysis of variance using SPSS 18 software (SPSS, Chicago, IL, USA).
Results
Genome-wide identification of chitinase gene family in two apple species
The whole-genome sequences of M. sieversii and M. × domestica were used for genome-wide exploration and phylogenetic analysis of the chitinase gene family. We used the Pfam database (Mistry et al., 2021) and the Glyco_hydro_18 (PF00704) and Glyco_hydro_19 (PF00182) domains to search for chitinases in the two apple genomes. The SMART (Letunic et al., 2021) database was used to verify the predicted genes. We accurately identified 36 putative chitinase genes in M. sieversii, of which 25 genes were members of the GH18 family and 11 genes belonged to the GH19 family (Table S2). In the M. × domestica genome, 47 chitinase genes were identified (Table S2), of which 33 genes were identified as members of the GH18 family and 14 genes belonged to the GH19 subfamily. For M. sieversii, the chitinase genes were annotated as MsChi1 to MsChi36, whereas for M. × domestica the genes were annotated MdChi1 to MdChi47, where the genes were numbered sequentially based on their chromosomal location in the genome.
The length of the 36 predicted MsChi proteins ranged from 101 (MsChi36) to 517 (MsChi1) amino acid residues, of which only MsChi1 was longer than 500 amino acids. The predicted molecular weight (MW) and isoelectric point (PI) ranged from 10.97 kDa (MsChi36) to 57.42 kDa (MsChi1) and from 4.34 (MsChi7) to 9.64 (MsChi23), respectively. Of the 36 MsChi proteins, 25 were predicted to contain a signal peptide in the C-terminus.
Chromosomal location and phylogenetic analyses
To accurately locate the chromosomal position of the genes, a chromosomal distribution map was constructed based on the start–end position of each chitinase gene. The 36 MsChi genes were mapped to 11 chromosomes, whereas the 47 MdChi genes were distributed on 13 chromosomes (Figures 1A, B).
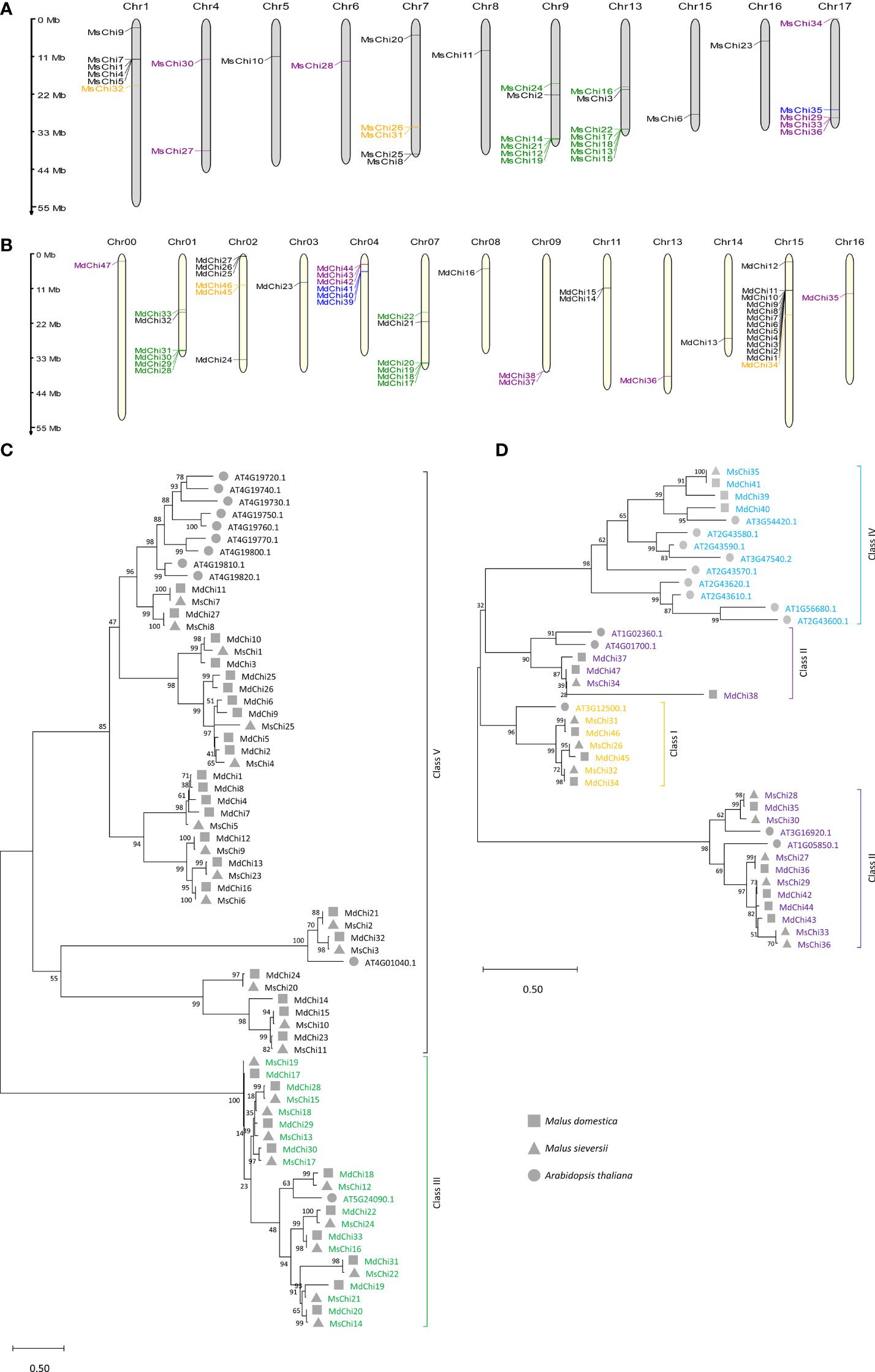
Figure 1 Chromosomal locations and phylogenetic relationship of MsChis and MdChis. Distribution of chitinase genes on M.sieversii (A) and M.domestica (B) chromosomes. MG2C online tools were used (http://mg2c.iask.in/mg2c_v2.1/) to depict chromosomal location, and genes were marked with short lines. The tandem duplicated gene pairs are marked by red braces. The major classes are represented as follows: orange=Class I, purple=Class II, green=Class III, blue= Class IV and black=Class V. Scale bar represents Mb. The full-length of amino acid sequences of putative chitinase protein from two glycosyl hydrolase families: GH18 (C) Class III (green) and V (black), and GH19 (D) Class IV (sky blue), Class II (purple), Class I (orange) in M.domestica, M.sieversii and A.thaliana were aligned using ClustalW method in MEGAX. An unrooted phylogenetic tree was built using the Maximum Likelihood method with 1000 replicates. The roman numerals (I–V) representing each gene cluster and genes from each species were labelled with different shapes. The numbers at the nodes represent statistical frequency.
To explore the evolutionary relationships of the chitinase gene family, an unrooted phylogenetic tree for the 36, 47, and 25 chitinase proteins from M. sieversii, M. × domestica, and A. thaliana, respectively, was constructed using the maximum likelihood method. The GH18 subfamily was grouped into three lineages, whereas the GH19 subfamily was grouped into four branches. Based on the classification of A. thaliana chitinases (Passarinho and Vries, 2002), the MsChi and MdChi genes were further classified into five classes (Figures 1C, D). The number of members in each class differed between M. sieversii and M. × domestica (Table S3), especially in Class V. Interestingly, Class IV chitinases comprised only one member in M. sieversii, but three members in M. × domestica.
Gene duplication and synteny analysis
In the course of evolution, gene duplication plays a crucial role in the expansion of a gene family (Cannon et al., 2004; Yanai, 2022). To further study the expansion mechanism of the chitinase genes, we employed the basic local alignment search tool for proteins (BLASTP) and MCScanX tools to identify duplication events in the chitinase gene family of the two apple species. Segmentally duplicated genes in the M. sieversii and M. × domestica genomes were identified by collinearity analysis (Figures 2A, B). In detail, 14 pairs of MsChi genes were segmentally duplicated among 10 chromosomes, whereas 12 MdChi genes were segmentally duplicated among nine chromosomes (Table S4). In addition, 16 and 22 chitinase genes were tandemly duplicated in M. sieversii and M. × domestica, respectively (Table S4). These results demonstrated that tandem duplication was a major driving force in expansion of the chitinase gene family in M. sieversii and M. × domestica.
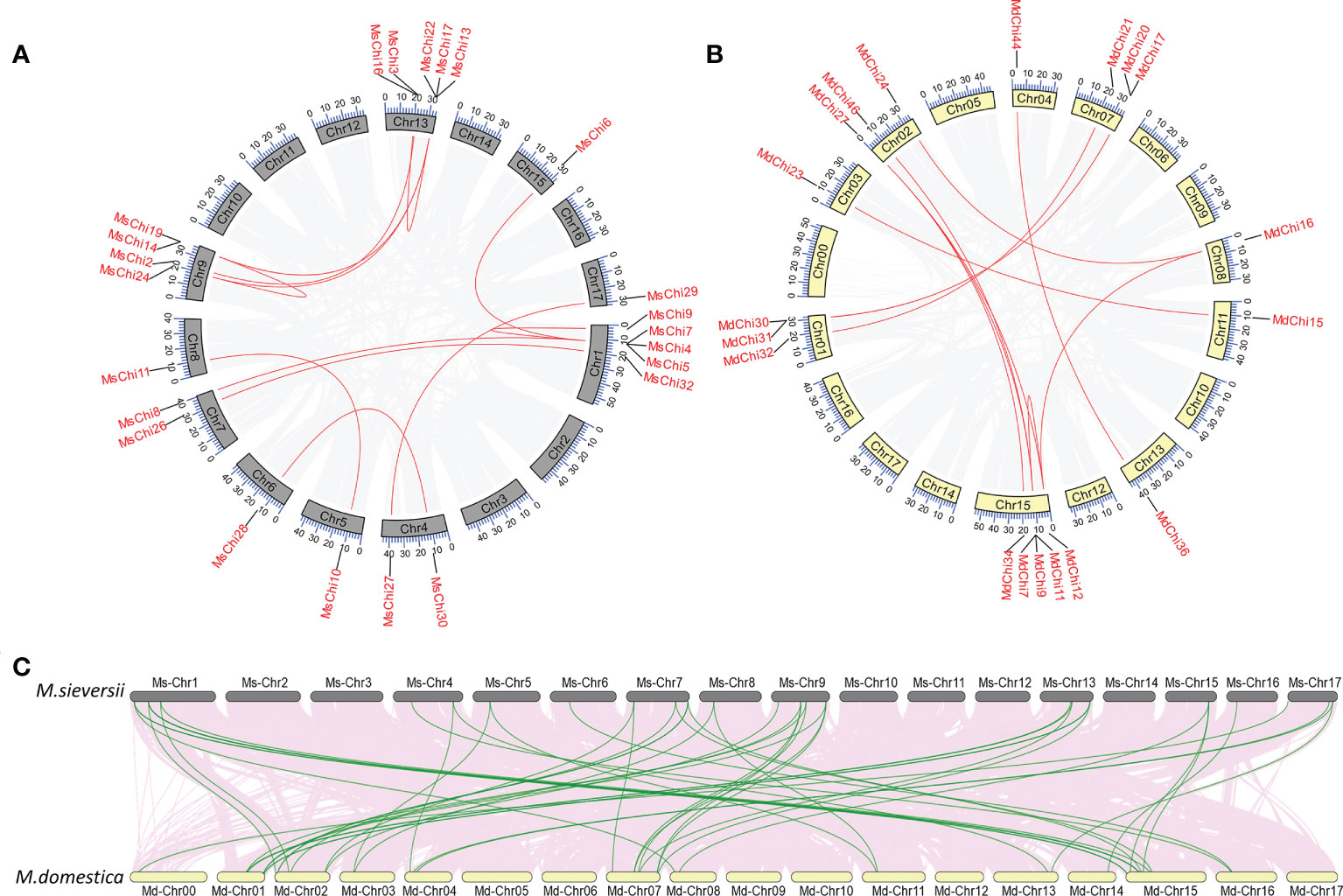
Figure 2 Collinearity analysis of MsChis and MdChis. Distribution of segmental duplication of chitinase genes on M.sieversii (A) and M.domestica (B) chromosomes. Chromosomes are represented by gray or yellow boxes. Segmental duplication genes are connected with red lines. The gray lines indicate all synteny blocks within the genome, and the chromosome numbers were indicated at the inside of the box. (C) Synteny analysis of chitinase genes in M.sieversii and M.domestica. Gray lines in the background indicate the collinear blocks within M.sieversii and M.domestica genomes, while the red lines highlight the syntenic chitinase gene pairs.
To further explore the evolutionary history of the chitinase family, we calculated the Ka and Ks substitution rates for duplicated chitinase gene pairs (Table S4). Almost half of the segmental or tandem duplicated MsChi genes were subjected to positive selection (Ka/Ks > 1), whereas the remainder had undergone purifying selection (Ka/Ks < 1). The segmental or tandem duplication of MsChi genes was estimated to have occurred from 3.62 to 128.71 Mya. Among MdChi genes, purifying selection was dominant for segmental duplication, whereas positive selection accounted for the highest proportion of tandem duplication events. Duplication of MdChi genes was estimated to have occurred between 1.93 and 138.52 Mya.
To understand the orthologous relationships within the M. sieversii and M. × domestica, we further analyzed the synteny of chitinase genes. A tightly conserved collinearity relationship and strong orthologs of chitinase genes were observed between the syntenic regions among the M. sieversii and M. × domestica genomes (Figure 2C). Thirty orthologous chitinase gene pairs were identified between M. sieversii and M. × domestica (Table S5).
Gene structure and conservative motif analyses
To determine structural features of the chitinase genes from the two apple species, we analyzed the exon–intron distribution of the genes. Most GH18 chitinase genes contained one or two exons, except for the genes of Class V, which contained seven or eight exons (Figure 3B). In contrast, three exons were most common for GH19 chitinase genes (Figure 3E). Interestingly, three genes (MsChi15, MsChi21, and MdChi29) in Class III (Figure 3B) and only one gene (MsChi33) in Class II contained relatively long introns (Figure 3E). Most of the closely related genes in the two apple species were similar in length and number of exons/introns.
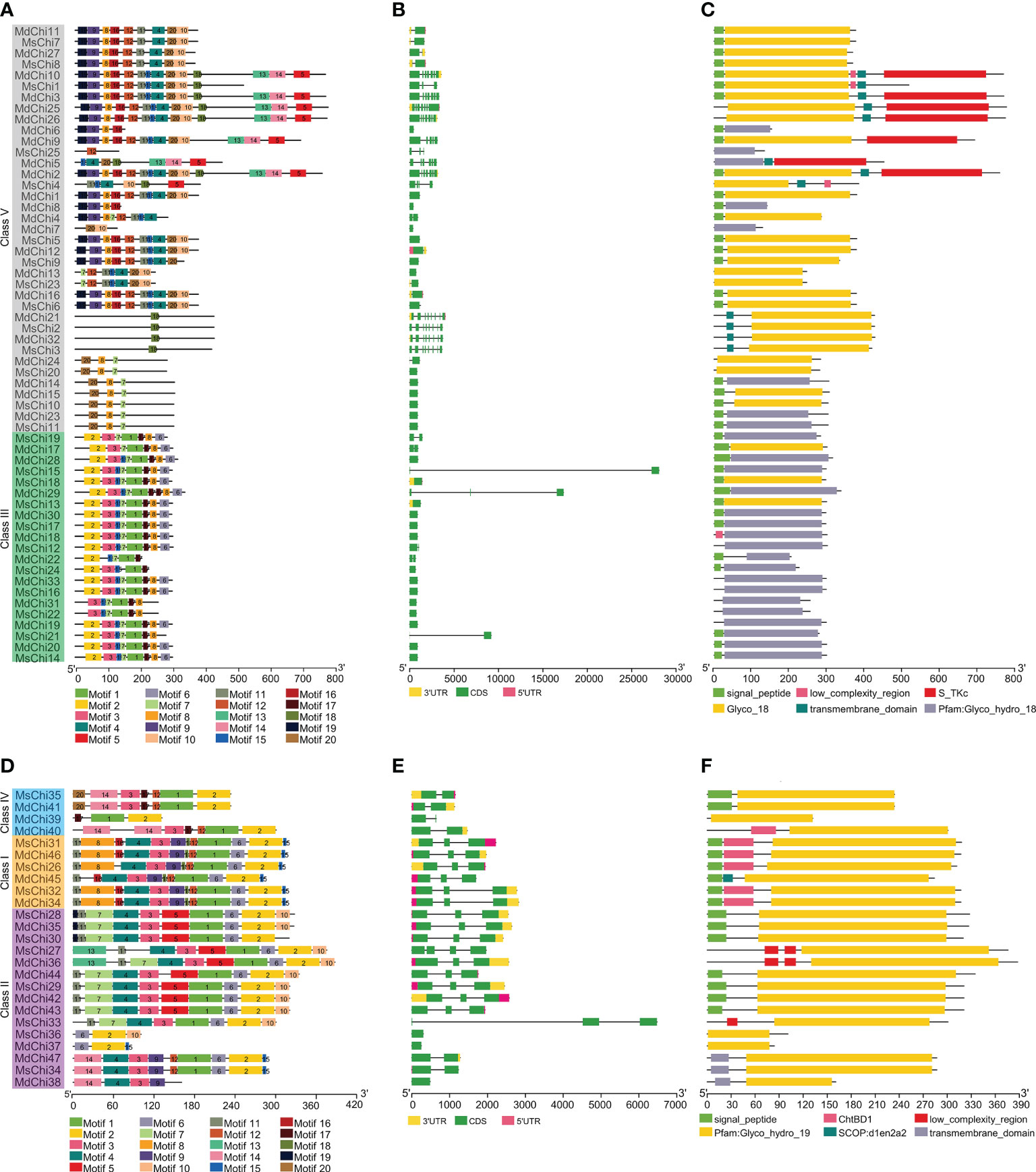
Figure 3 Structural analysis MsChis and MdChis. The conserved motifs in GH18 chitinase subfamily (A) and GH19 chitinase (D) proteins from M.domestica and M.sieversii were identified by MEME (Bailey et al., 2006). Schematic representation of exon-intron structure of GH18 (B) and GH19 (E) chitinase subfamilies in M.domestica and M.sieversii. Active domains of GH18 (C) and GH19 (F) chitinase proteins from M.sieversii and M.domestica were identified by SMART (Letunic et al., 2021) database. Motifs, domains and exon-intron structures were visualized by TBtools software (Chen et al., 2020). Each single signature is indicated by a colored box on bottoms of the figure and presented proportionally.
To further study the architecture of chitinase proteins, the conserved motifs were predicted with the MEME Suite. A total of 20 distinct motifs were identified (Figures 3A, D). The number and location of motifs of certain closely related genes in the two apple species were highly conserved, providing further evidence to support the phylogenetic and functional relationships. Furthermore, several class-specific motifs were detected in the GH18 family. For instance, motifs1, 2, and 17 were restricted to Class III, wheras motif18 was the only motif in some members of Class V (Figure 3A). Motif2 was common in each class of GH19, except in MdChi38. In addition, motif17 and motif16 were unique to Class IV and Class I, respectively (Figure 3D).
Conserved domains and active site analysis of chitinase genes
To examine the similarity and diversity of domain architecture of chitinase proteins from the two apple species, the localization of the catalytic domain within the protein was analyzed by searching for featured domains using the SMART database. Glycosyl hydrolase family 18 (GH18) domains were present in classes III and V chitinases, and Glycosyl hydrolase family 19 (GH19) domains were detected in classes I, II, and IV. To locate the catalytic domain and active site in each chitinase protein, we generated a multiple sequence alignment and conducted a motif-based sequence analysis. Analysis of the amino acid sequences combined with the domain signature revealed that Class III chitinases possessed the GH18 catalytic domain with the CHITINASE_18 active site signature (PS01095, [LIVMFY]-[DN]-G-[LIVMF]-[DN]-[LIVMF]-[DN]-x-E) except for MdChi30 and MdChi17 (Figure 4A). Among Class V chitinases, 10 members had the CHITINASE_18 active site signature (Figure 4B). The Class I chitinases contained the GH19 catalytic domain with the CHITINASE_19_2 signature (PS00774; [LIVM]-[GSA]-F-x-[STAG](2)-[LIVMFY]-W-[FY]-W-[LIVM]) and CHITINASE_19_1 signature (PS00773; C-x(4,5)-F-Y-[ST]-x(3)-[FY]-[LIVMF]-x-A-x(3)-[YF]-x(2)-F-[GSA]) (Figure 5A). In addition, Class I chitinases had the chitin-binding domain with the CHIT_BIND_I_1 signature (PS00026; C-x(4,5)-C-C-S-x(2)-G-x-C-G-x(3,4)-[FYW]-C) except for MdChi45. Class IV chitinases contained the GH19 catalytic domain with the CHITINASE_19_1 and CHITINASE_19_2 signatures. Only MdChi40 had the chitin-binding domain with the CHIT_BIND_I_1 signature (Figure 5B). Among Class II chitinases, the CHITINASE_19_1 signature was detected in only three members (MdChi38, MdChi47, and MsChi34), and no CHITINASE_19_2 signature was detected in any members of this class (Figure 5C).
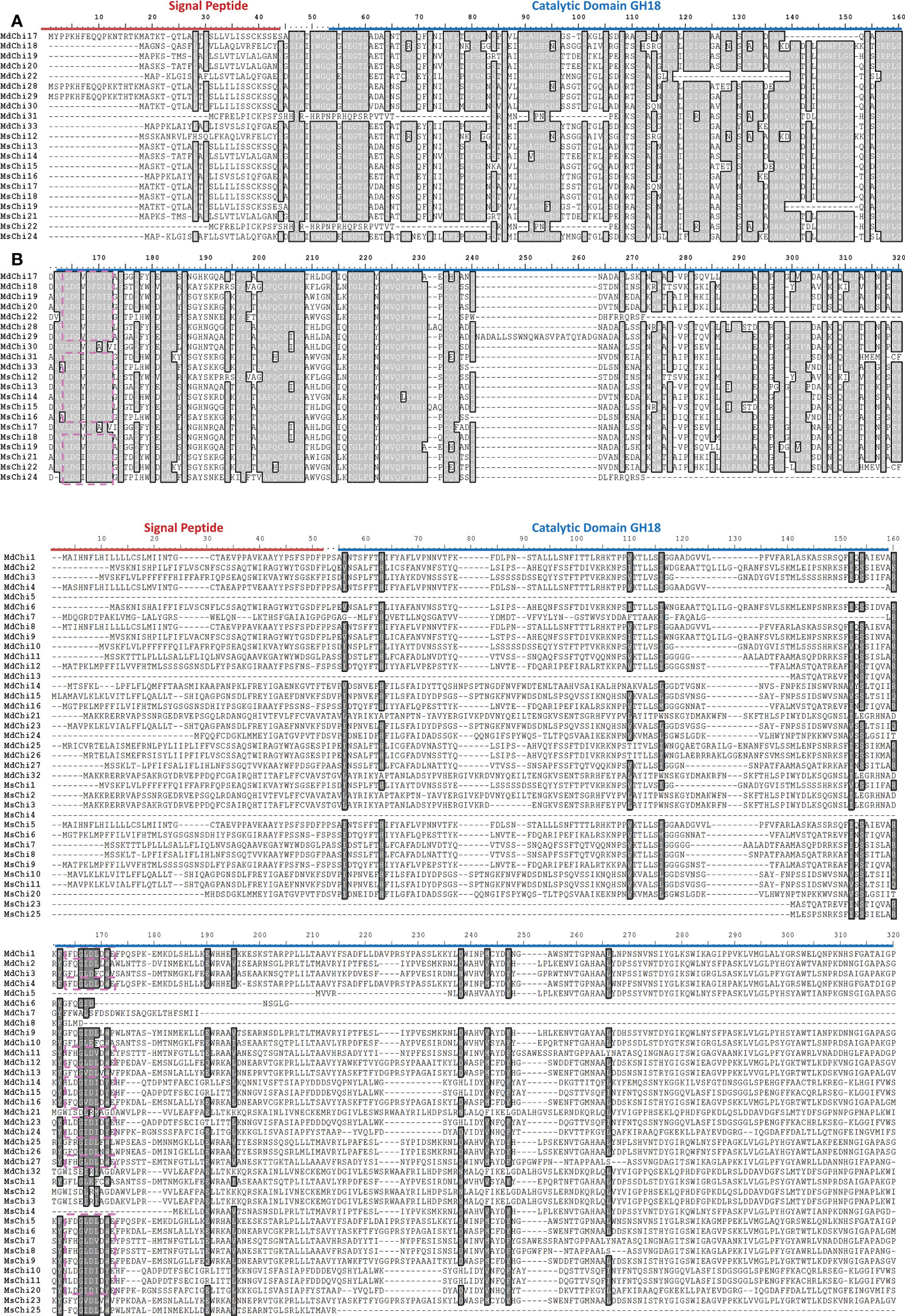
Figure 4 Multiple alignments of GH18 chitinase subfamily Class III (A) and Class V (B) of MsChis and MdChis. Amino acid sequences were aligned using ClustalW and visualized using BioEdit. Shaded amino acid sequences are 75–100% homologous. Red line indicates signal peptide sequence and blue line for GH18 catalytic domain. The amino acids in the purple dash line box represent residues essential for catalytic activity, Chitinase_18 signature (PS01095). The partial amnio acid sequences were presented in (B).
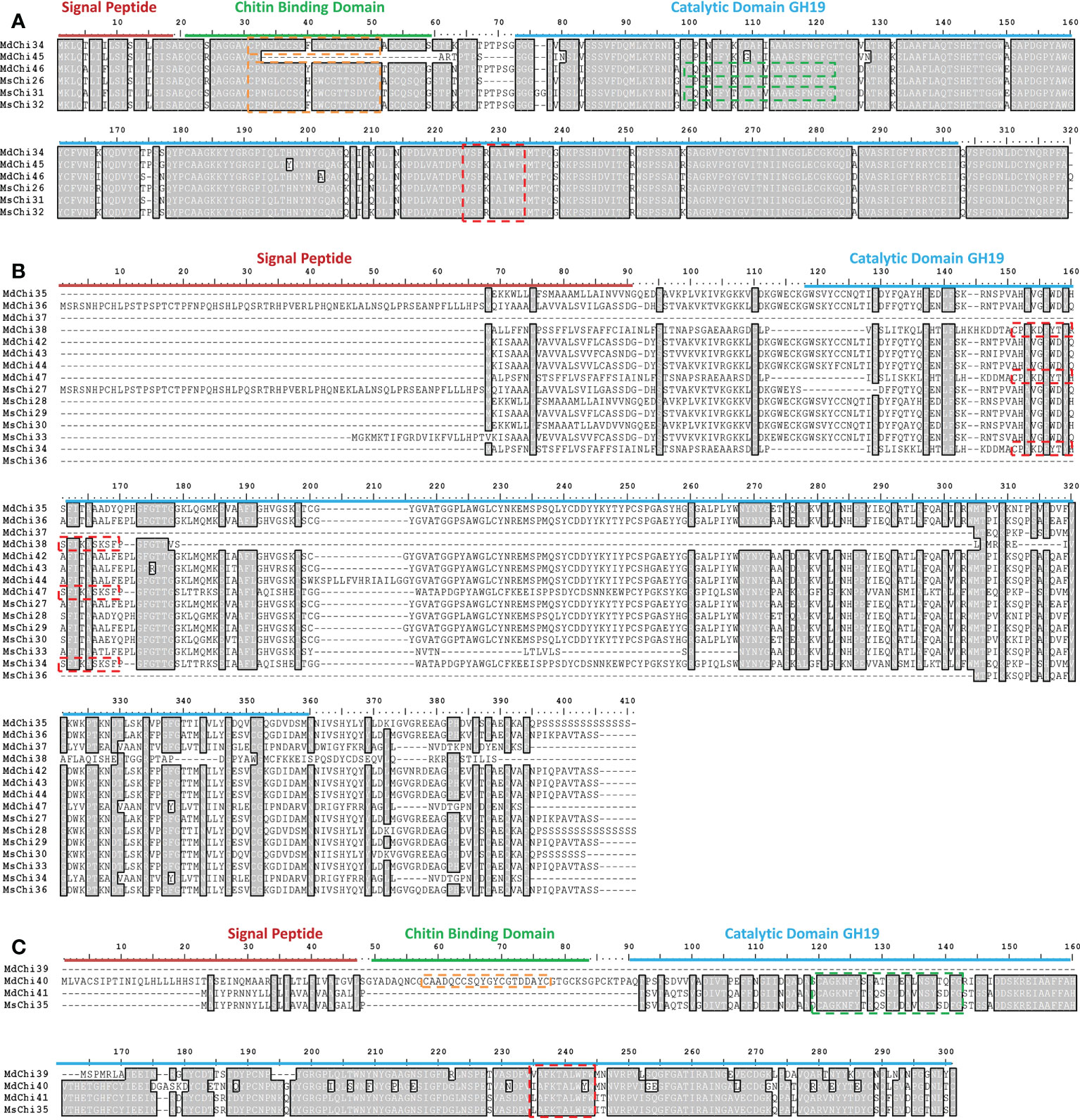
Figure 5 Multiple alignments of GH19 chitinase subfamily Class I (A), Class II (B) and Class IV (C) of MsChis and MdChis. Amino acid sequences were aligned using Clustal W and visualized by using BioEdit. Shaded amino acid sequences are 75–100% homologous. Red line indicates signal sequence and blue line for GH19 catalytic domain. The amino acids in the green dash line box represents CHITINASE_19 _1 signature (PS00773); red dash line box represents CHITINASE_19_2 signature (PS00774); orange dash line box represents CHIT_BIND_I_1 signature (PS00026).
Prediction of potential cis-acting elements in chitinase genes
To explore the transcriptional modulation of the chitinase genes, we predicted the presence of cis-acting elements in the promoter region (within 2000 bp upstream of the coding region) of the genes in the two apple genomes. The number of cis-acting elements involved in response to drought, low temperature, light, and phytohormones were predicted (Figures S1, S2). Among these cis-acting elements, the W-box (TTGACC) (Rushton and Somssich, 1998), P-box (CCTTTTG) (Lois et al., 1989), L-box (ATCCCACCTAC) (Lois et al., 1989), and S-box (AGCCACC) (Kirsch et al., 2000) are reported to respond to fungal pathogen infection. The most frequent cis-acting elements in all promoters were the TATA-box, CAAT-box, and AT-TATA box.
Expression of chitinase genes in response to V. mali inoculation
To investigate expression of the chitinase genes of M. sieversii and M. domestica in response to V. mali attack, we analyzed the transcriptome of M. sieversii and M. domestica leaves after inoculation with V. mali. A total of 36 MsChi transcripts from M. sieversii were subjected to further differential expression analysis through comparisons of fold-change expression. Five M. sieversii GH18 genes (MsChi1, MsChi7, MsChi9, MsChi18, and MsChi19) and one M. domestica GH19 gene (MsChi35) were up-regulated, whereas a single M. sieversii GH18 gene (MsChi26) was down-regulated significantly in response to V. mali infection (Figure 6A). For M. domestica, we analyzed previous transcriptome data in response to V. mali infection (NCBI SRA accession ID : SRP034726) (Yin et al., 2016). Thirteen genes were up-regulated, whereas four genes were down-regulated (Figure 6C). To validate the expression pattern of each transcript, we quantified the transcript abundance of each MsChi and MdChi gene by qRT-PCR at different time points after inoculation. The qRT-PCR results for the MsChi transcripts were consistent with the RNA-sequencing data (Figures 6B, D).
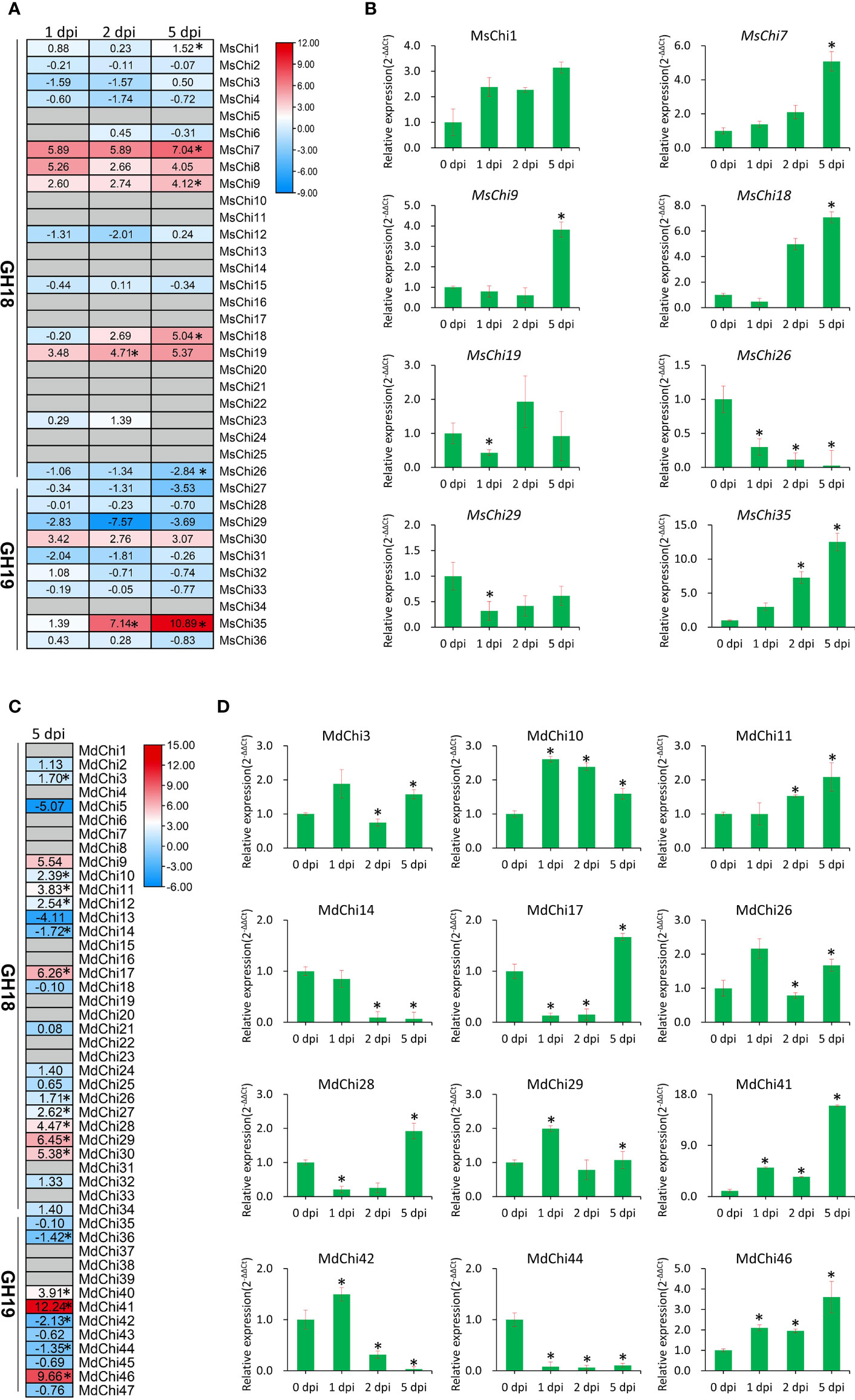
Figure 6 Expression profile of MsChis and MdChis during V.mali infection. RNA-seq data analysis of chitinase genes in M.sieversii (A) or M.domestica (C) in response to V.mali. The heatmap generated by using TBtools (Chen et al., 2020) software based on the log2fold change values of each chitinase genes. The colors represent expression levels, red colors represent high expression and blue colors represent low expression. ‘*’ represented significantly (P<0.05) different expression. Verification of the expression of chitinase genes in M.sieversii (B) or M.domestica (D) under V.mali infection by qRT-PCR. EF1-α was used as an internal reference gene. The relative expression data were analyzed by one-way ANOVA method. ‘*’ represented significantly different (P< 0.05, n = 3), and error bars indicate mean ± SE.
Co-expression network of MsChi genes
To further predict the transcription factors associated with chitinase gene expression, we preformed WGCNA based on the FPKM values from the RNA-sequencing data. After filtering (FPKM > 1), 8780 genes were subjected to further analysis and 20 co-expressed gene modules were identified. A module with a correlation coefficient (r) > 0.80 and p < 0.05 was defined as a MsChi-specific module. In this manner, 10 MsChi-specific modules were identified (Figure 7B). Notably, the MEblack module with 1010 genes was positively correlated with six up-regulated MsChi genes (MsChi1, MsChi7, MsChi9, MsChi18, MsChi19, and MsChi35) in response to V. mali infection. Calculation of MM and GS for the 1010 genes in the MEblack module revealed that 55.6% of the genes (562/1010) had high MM (>0.8) and GS (>0.8) scores, implying that they had relatively high connectivity (r = 0.83, p < 1e−200) within the module (Figure 7C). Twenty-nine genes were identified as transcription factors in the MEblack module of which seven were WRKY transcription factors (Figure 7D). These results inferred that WRKY transcription factors may function as upstream positive regulators of a majority of the MsChi genes in response to V. mali infection.
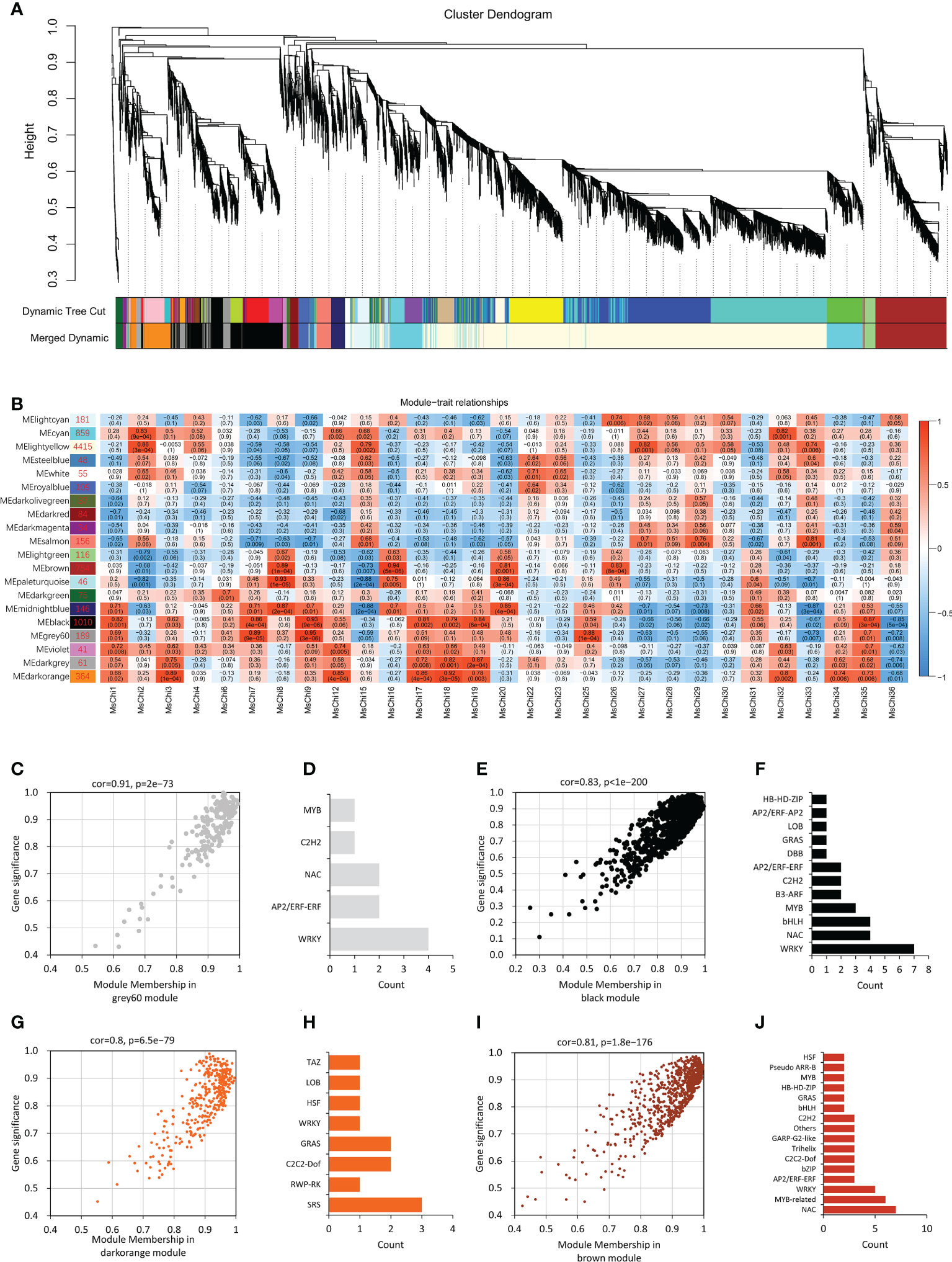
Figure 7 Weighted Gene Co-Expression Network Analysis (WGCNA) of MsChi genes. (A) Gene dendrogram obtained using dynamic tree cut-off. Each colored row in the bottom represents a module which contains group of highly connected genes. (B) Association of module eigengenes (MEs) with expression profiles of selected chitinase genes. Corresponding P-values of module-chitinase gene expression correlations are indicated in parenthesis. The panel on the left side shows the 20 modules and number of genes in each module. The color scale on the right side shows module-trait correlation from −1 (blue) to 1 (red). Scatter plot of the correlation of module membership vs. gene significance in grey60 (C), black (E), darkorange (G) and brown modules (I). Statistics of transcription factors in grey60 (D), black (F), orange (H) and brown modules (J).
Discussion
Apple is among the most commonly consumed fruits worldwide. Fungal diseases, such as Valsa canker, are a major threat to apple production. Fungal resistance proteins, such as chitinases, are essential to prevent canker diseases in apple. Chitinases are synthesized in diverse organisms, including bacteria, fungi, plants, insects, animals, and humans (Hamid et al., 2013). The chitinase gene family has been identified in numerous plant species, such as vegetable (Bartholomew et al., [[NoYear]]; Cao and Tan, 2019; Mir et al., 2020), crop (Su et al., 2015; Xu et al., 2016), and tree species (Tobias et al., 2017; Zhang et al., 2022). Reliable genomic sequence information and annotations are available for a number of Malus species (Daccord et al., 2017). However, genome-wide identification of the chitinase gene family and its expression patterns under fungal infection have not been reported previously for apple species. In the present study, 36 and 47 putative chitinase genes were identified in the genomes of the wild apple M. sieversii and domesticated apple M. domestica, respectively. Based on their phylogenetic relationships and functional domains, the chitinases were classified into five classes (I–V).
It is noteworthy that the chitinase genes were not evenly distributed on chromosomes. The chitinase genes of wild apple were mainly concentrated on chromosomes 1, 7, 9, 13, and 17, whereas those of domesticated apple were concentrated on chromosomes 1, 2, 4, 7, and 15 (Figures 1A, B). In some instances, the chitinase genes clustered on individual chromosomes were members of the same class. For instance, MdChi2, MdChi5, MdChi9, and MdChi10 belonged to class V and were clustered on chromosome 15 (Table S4). The distribution pattern of chitinases in wild and domesticated apple showed similarity with the genome of other tree species, such as E. grandis (Tobias et al., 2017).
Human activity, including agricultural breeding and genetic engineering, have driven rapid evolutionary changes in domesticated plants and animals (Palumbi, 2001; Turcotte et al., 2017). We identified a greater number of chitinase genes in domesticated apple compared with wild apple, reflecting that domestication drives evolution. The structural diversity of chitinase genes may provide an improved understanding of the evolutionary patterns of chitinase genes. The average exon number of each class of chitinase genes has remained almost unchanged in wild and domesticated apple, and Class V chitinase genes have more exons compare with the other classes (Figure S3). For instance, the exon number of 28 MdChi genes preserved the number in ancestral orthologs (Table S7), implying that the gene structure has remained conserved during evolution. With regard to intron number, stress-responsive genes generally contain relatively fewer introns (Jeffares et al., 2008). For example, the cotton Class IV chitinase gene GrChi28 has a single intron and is strongly up-regulated in response to Verticillium dahliae infection (Xu et al., 2016). Similarly, MsChi35 (Class IV) and MdChi41 (Class IV) were strongly up-regulated (Figure 6) after V. mali inoculation and each contained a single intron (Figure 3D). In accordance with this pattern, all significantly up- or down-regulated chitinase genes in wild or domesticated apple contained two or fewer introns (Figure 3).
Gene duplication is a major driving force of expansion of a gene family. The expansion of chitinase genes in wild and domesticated apple resulted from segmental duplication and tandem duplication. Duplicated genes likely experience functional specification (Prince and Pickett, 2002). In wild apple, 27% of chitinase genes originated from tandem duplication, whereas 36% were segmentally duplicated (Table S4). These duplicated chitinase genes maintained a close phylogenetic relationship but structural variation was evident. For instance, MsChi25 lost the signal peptide sequence compared with its paralog MsChi8. The genes MsChi1, MsChi4, and MsChi7 formed a tandemly arrayed gene cluster, but only MsChi7 possessed the CHITINASE_18 active site signature (PS01095) in the GH18 catalytic domain (Figure 4B) and was strongly up-regulated under infection by V. mali. In addition, MsChi13, MsChi15, MsChi17 and MsChi18 were tandemly arrayed on chromosome 13, but exhibited various expression patterns in response to V. mali infection. Similarly, duplicated chitinase genes in domesticated apple showed high structural variety despite their phylogenetic affinity. For example, MdChi40 and MdChi41 were tandemly duplicated gene pairs under purifying selection, but MdChi40 gained a single chitin-binding domain at the N-terminus (Figure 5C). Interestingly, MdChi41 was up-regulated in response to V. mali infection and its expression level was ten-times higher than that of MdChi40. We hypothesize that the gain of the chitin-binding region of MdChi40 may affect its enzymatic activity and lead to functional diversification. Although the present results infer that duplicated genes may acquire a new structure and function, additional evidence is required to confirm the functional differences between duplicated chitinase genes.
Chitinases vary in molecular structure, substrate specificity, and catalytic mechanism (Hamid et al., 2013). In the current study, structural variation of the chitinase proteins was detected. Similar motif composition was observed in classes I and III, suggesting that these classes show functional similarity (Figure 3). The variation in motif composition was not obvious between classes II and IV, but very distinct in Class V chitinases (Figure 3). In addition, a majority of the chitinase proteins in this study included a signal peptide, which was absent in several chitinase proteins. Plant chitinase proteins mature by trimming of the N-terminal signal peptide (Taira et al., 2009) and confer enhanced plant resistance to fungal pathogens (Singh et al., 2015). Chitinase genes that were significantly up-regulated in response to V. mali infection all carried the signal peptide sequence, indicating that they are secreted into the apoplast and may be involved in plant–pathogen interaction.
To explore the mechanism by which the chitinase genes were transcriptionally regulated, we analyzed the promoter regions of the chitinase genes. We detected a number of pathogen-responsive cis-acting elements, such as the W-box. Some of the chitinase genes, such as MsChi35 or its ortholog MdChi41, were observed to have two W-box cis-elements in the promoter region (Figures S1, S2) and were strongly up-regulated in response to V. mali infection (Figure 6). These results indicated that expression of MsChi35 and MdChi41 was likely regulated via the W-box cis-acting element in the promoter region. A gene co-expression network revealed that MsChi35 was highly co-expressed with seven WRKY transcription factors under V. mali infection (Figure 7). Further analysis showed that six of the seven WRKY transcription factors were significantly differentially expressed at 5 dpi (Figure S4). WRKY transcription factors are among the largest families of transcriptional regulators in plants and bind to the W-box cis-acting element in the promoter of the target gene (Zhu et al., 2017). In Lilium regale, LrWRKY2 can activate expression of LrCHI2, which encodes a chitinase, through binding to the W-box cis-element in the promoter of LrCHI2 to enhance host resistance to root rot caused by Fusarium oxysporum (Li et al., 2021). The tobacco Class I chitinase gene NtCHN48 has two W-boxes in the promoter region that are recognized by NtWRKY1 and NtWRKY4, respectively (Yamamoto et al., 2004). In a previous study, we demonstrated that MsWRKY16, one of the seven WRKY transcription factors, enhances resistance to V. mali (Liu et al., 2021). Based on these results, it is speculated that WRKY transcription factors bind to the W-box cis-element in the promoter region of MsChi35 or MdChi41 to positively regulate their expression in response to V. mali infection. The genes MsChi35 and MdChi41 are orthologs belonging to Class IV chitinase. The two genes were highly conserved in their composition and arrangement of exons–introns (Figure 3E) or motifs (Figure 3D) and shared a high level of homology (100%) in amino acid sequence and enzymatic domain (Figure 5C). The MsChi35 and MdChi41 were the most highly expressed chitinase genes in response to V. mali infection. Moreover, the transient over expression of MsChi35 enhance M.sieverssi resistance to V. mali infection (Figure S5). Therefore, these results suggested that MsChi35 and MdChi41 were the dominant fungal resistance chitinase genes in the two apple species.
The present research allows proposal of a framework for the response of chitinase family genes to V. mali infection in M. sieversii and M. domestica (Figure 8). The results provide useful insights for further functional investigation of the chitinase gene family, particularly of MsChi35 and MdChi41, in the mediation of fungal pathogen resistance in apple species.
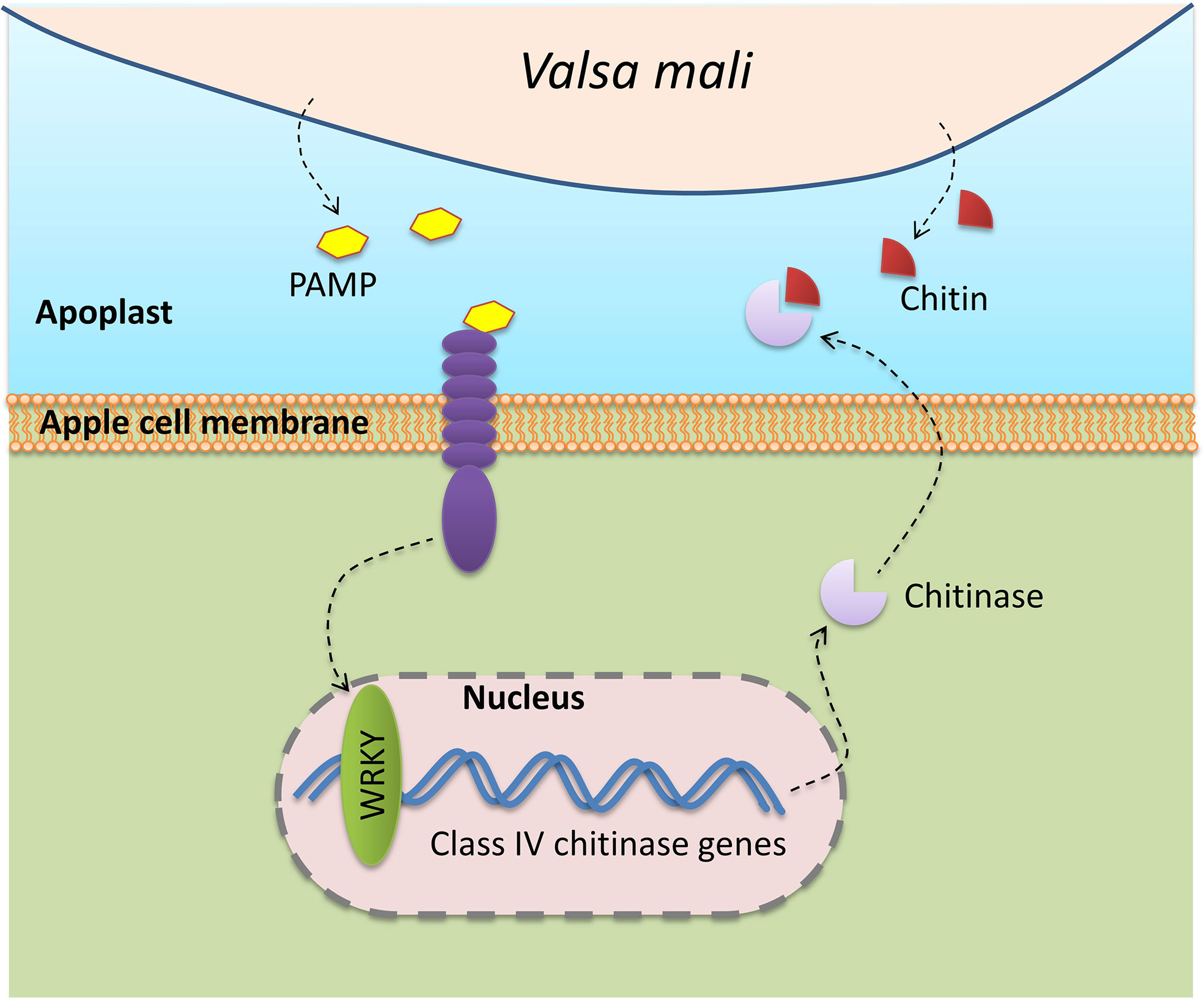
Figure 8 Schematic model illustrating the responses of apple chitinase family genes against to V.mali infection.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: The M.sieversii transcriptome datasets that used in the current article are available at the Sequence Read Archive (SRA) database of National Center for Biotechnology Information under project accession number PRJNA687214(https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA687214). The M.domestica transcriptom datasets that used in the current article are available at the Sequence Read Archive (SRA) database of National Center for Biotechnology Information under accession number SRP034726 (https://www.ncbi.nlm.nih.gov/sra/?term=SRP034726). The M.sieversii genome data are available at genome database of National Center for Biotechnology Information under accession number JAHTLV000000000.1 (https://www.ncbi.nlm.nih.gov/nuccore/JAHTLV000000000.1). The M.domestica genome data are available at https://iris.angers.inra.fr/gddh13/the-apple-genome-downloads.html.
Author contributions
The experimental design, data analyzation, manuscript organization were completed by YH and GK. XZ, XL (6th author) and XW were assistant with RNA quantification. DZ and XL (8th author) conceived the project, supervised the analysis, and critically revised the manuscript. AW was revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502030403), Biological Resources Program, Chinese Academy of Sciences (KFJ-BRP-007-008) and the Youth Innovation Promotion Association, Chinese Academy of Sciences (No. 2018478) and the China Postdoctoral Science Foundation (2021M693380).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1007936/full#supplementary-material
Supplementary Table S1 | List of the qRT-PCR primers of the MsChi and MdChi genes.
Supplementary Table S2 | Detailed information of predicted chitinase genes in M.sieversii and M.domestica.
Supplementary Table S3 | Number of different classes of chitinase from different species.
Supplementary Table S4 | Duplicated chitinase genes in M.sieversii and M.domestica.
Supplementary Table S5 | Ortholog chitinase genes in M.sieversii and M.domestica.
Supplementary Table S6 | RNA-seq expression data of seven WRKY transcription factors in M.sieversii in respons to V.mali infection.
Supplementary Table S7 | Exon number in the ortholog pairs of MsChi and MdChi genes.
References
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/S41587-019-0036-Z
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Ballester, A.-R., Norelli, J., Burchard, E., Abdelfattah, A., Levin, E., González-Candelas, L., et al. (2017). Transcriptomic response of resistant (PI613981–malus sieversii) and susceptible (“Royal gala”) genotypes of apple to blue mold (Penicillium expansum) infection. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01981
Bartholomew, E. S., Black, K., Feng, Z., Liu, W., Shan, N., Zhang, X., et al. (2019). Comprehensive analysis of the chitinase gene family in cucumber (Cucumis sativus l.): From gene identification and evolution to expression in response to fusarium oxysporum. Int. J. Mol. Sci. 20, 5309. doi: 10.3390/ijms20215309
Bozorov, T. A., Luo, Z., Li, X., Zhang, D. (2019). Agrilus mali matsumara (Coleoptera: Buprestidae), a new invasive pest of wild apple in western China: DNA barcoding and life cycle. Ecol. Evol. 9, 1160–1172. doi: 10.1002/ece3.4804
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in arabidopsis thaliana. BMC Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Cao, J., Tan, X. (2019). Comprehensive analysis of the chitinase family genes in tomato (Solanum lycopersicum) 8, 52. doi: 10.3390/plants8030052
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/J.MOLP.2020.06.009
Chen, X., Feng, T., Zhang, Y., He, T., Feng, J., Zhang, C. (2007). Genetic diversity of volatile components in xinjiang wild apple (Malus sieversii). J. Genet. Genomics 34, 171–179. doi: 10.1016/S1673-8527(07)60018-6
Daccord, N., Celton, J.-M., Linsmith, G., Becker, C., Choisne, N., Schijlen, E., et al. (2017). High-quality. novo assembly apple Genome methylome dynamics early Fruit Dev. Nat. Genet. 49, 1099–1106. doi: 10.1038/ng.3886
Duan, N., Bai, Y., Sun, H., Wang, N., Ma, Y., Li, M., et al. (2017). Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 8, 1–11. doi: 10.1038/s41467-017-00336-7
Grover Plant Chitinases (2012). Genetic diversity and physiological roles. Crit. Rev. Plant Sci. 31 (1), 57–73. doi: 10.1080/07352689.2011.616043
Hamid, R., Khan, M. A., Ahmad, M., Ahmad, M. M., Abdin, M. Z., Musarrat, J., et al. (2013). Chitinases: An update. J. Pharm. Bioallied Sci. 5, 21. doi: 10.4103/0975-7406.106559
Haxim, Y., Si, Y., Liu, X., Wen, X., Kahar, G., Ding, Y., et al. (2021). Genome-wide characterization of HSP90 gene family in malus sieversii and their potential roles in response to valsa mali infection. For. 12, 1232. doi: 10.3390/F12091232
Henrissat, B., Bairoch, A. (1993). New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 293, 781–788. doi: 10.1042/BJ2930781
Jeffares, D. C., Penkett, C. J., Bähler, J. (2008). Rapidly regulated genes are intron poor. Trends Genet. 24, 375–378. doi: 10.1016/J.TIG.2008.05.006
Kaku, H., Nishizawa, Y., Ishii-Minami, N., Akimoto-Tomiyama, C., Dohmae, N., Takio, K., et al. (2006). Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc. Natl. Acad. Sci. 103, 11086–11091. doi: 10.1073/PNAS.0508882103
Kasprzewska, A. (2003). Plant chitinases-regulation and function. Cell Mol. Biol. Lett. 8, 809–824. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12949620.
Kirsch, C., Takamiya-Wik, M., Schmelzer, E., Hahlbrock, K., Somssich, I. E. (2000). A novel regulatory element involved in rapid activation of parsley ELI7 gene family members by fungal elicitor or pathogen infection. Mol. Plant Pathol. 1, 243–251. doi: 10.1046/j.1364-3703.2000.00029.x
Kumar, M., Brar, A., Yadav, M., Chawade, A., Vivekanand, V., Pareek, N. (2018). Chitinases–potential candidates for enhanced plant resistance towards fungal pathogens. Agric 8, 88. doi: 10.3390/AGRICULTURE8070088
Lee, D. H., Lee, S. W., Choi, K. H., Kim, D. A., Uhm, J. Y. (2006). Survey on the occurrence of apple diseases in Korea from 1992 to 2000. Plant Pathol. J. 22, 375–380. doi: 10.5423/PPJ.2006.22.4.375
Lescot, M. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Khedkar, S., Bork, P. (2021). SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 49, D458–D460. doi: 10.1093/nar/gkaa937
Li, S., Hai, J., Wang, Z., Deng, J., Liang, T., Su, L., et al. (2021). Lilium regale Wilson WRKY2 regulates chitinase gene expression during the response to the root rot pathogen fusarium oxysporum. Front. Plant Sci. 12. doi: 10.3389/FPLS.2021.741463/BIBTEX
Li, C., Li, X., Bai, C., Zhang, Y., Wang, Z. (2019). A chitinase with antifungal activity from naked oat (Avena chinensis) seeds. J. Food Biochem. 43, e12713. doi: 10.1111/jfbc.12713
Liu, X., Li, X., Bozorov, T. A., Ma, R., Ma, J., Zhang, Y., et al. (2020). Characterization and pathogenicity of six cytospora strains causing stem canker of wild apple in the tianshan forest, China. For. Pathol 50, e12587. doi: 10.1111/efp.12587
Liu, X., Li, X., Wen, X., Zhang, Y., Ding, Y., Zhang, Y., et al. (2021a). PacBio full-length transcriptome of wild apple (Malus sieversii) provides insights into canker disease dynamic response. BMC Genomics 22, 52. doi: 10.21203/rs.3.rs-47953/v1
Liu, X., Zhang, Y., Zhou, T., Li, X., Wen, X., Zhang, D. (2021b). Full-length transcriptome-wide characteristic and functional identification of WRKY family in malus sieversii during the valsa canker disease response. 12, 790. doi: 10.3390/F12060790
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2^-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lois, R., Dietrich, A., Hahlbrock, K., Schulz, W. (1989). A phenylalanine ammonia-lyase gene from parsley: structure, regulation and identification of elicitor and light responsive cis-acting elements. EMBO J. 8, 1641–1648. doi: 10.1002/j.1460-2075.1989.tb03554.x
Mir, Z. A., Ali, S., Shivaraj, S. M., Bhat, J. A., Singh, A., Yadav, P., et al. (2020). Genome-wide identification and characterization of chitinase gene family in brassica juncea and camelina sativa in response to alternaria brassicae. Genomics 112, 749–763. doi: 10.1016/j.ygeno.2019.05.011
Misra, B. (2015). Molecular evolution and functional divergence of chitinase gene family in hevea brasiliensis genome. The Winnower 9, e144125.54243. doi: 10.15200/WINN.144125.54243
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Palumbi, S. R. (2001). Humans as the world’s greatest evolutionary force. Sci. (80-. ). 293, 1786–1790. doi: 10.1126/SCIENCE.293.5536.1786/ASSET/9BE6BB2E-67AD-4424-96A2-61ADAA1C6349/ASSETS/GRAPHIC/SE3519735002.JPEG
Passarinho, P. A., Vries, S. (2002). Arabidopsis Chitinases: A genomic survey. Arab. B. 1, e0023. doi: 10.1199/tab.0023
Prince, V. E., Pickett, F. B. (2002). Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 3, 827–837. doi: 10.1038/nrg928
Rushton, P. J., Somssich, I. E. (1998). Transcriptional control of plant genes responsive to pathogens. Curr. Opin. Plant Biol. 1, 311–315. doi: 10.1016/1369-5266(88)80052-9
Shibuya, N., Minami, E. (2001). Oligosaccharide signalling for defence responses in plant 59, 223–233. doi: 10.1006/pmpp.2001.0364
Sigrist, C. J. A., de Castro, E., Cerutti, L., Cuche, B. A., Hulo, N., Bridge, A., et al. (2013). New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347. doi: 10.1093/nar/gks1067
Singh, H. R., Deka, M., Das, S. (2015). Enhanced resistance to blister blight in transgenic tea (Camellia sinensis [L.] o. kuntze) by overexpression of class I chitinase gene from potato (Solanum tuberosum). Funct. Integr. Genomics 15, 461–480. doi: 10.1007/S10142-015-0436-1/FIGURES/12
Su, Y., Xu, L., Fu, Z., Yang, Y., Guo, J., Wang, S., et al. (2014). ScChi, encoding an acidic class III chitinase of sugarcane, confers positive responses to biotic and abiotic stresses in sugarcane. Int. J. Mol. Sci. 15, 2738–2760. doi: 10.3390/IJMS15022738
Su, Y., Xu, L., Wang, S., Wang, Z., Yang, Y., Chen, Y., et al. (2015). Identification, phylogeny and transcript of chitinase family genes in sugarcane. Sci. Rep. 5, 1–15. doi: 10.1038/srep10708
Suzaki, K. (2008). Population structure of valsa ceratosperma, causal fungus of valsa canker, in apple and pear orchards. J. Gen. Plant Pathol. 74, 128–132. doi: 10.1007/s10327-008-0078-4
Taira, T., Hayashi, H., Tajiri, Y., Onaga, S., Uechi, G. I., Iwasaki, H., et al. (2009). A plant class V chitinase from a cycad (Cycas revoluta): Biochemical characterization, cDNA isolation, and posttranslational modification. Glycobiology 19, 1452–1461. doi: 10.1093/GLYCOB/CWP119
Tobias, P. A., Christie, N., Naidoo, S., Guest, D. I., Külheim, C. (2017). Identification of the eucalyptus grandis chitinase gene family and expression characterization under different biotic stress challenges. Tree Physiol. 37, 565–582. doi: 10.1093/treephys/tpx010
Turcotte, M. M., Araki, H., Karp, D. S., Poveda, K., Whitehead, S. R. (2017). The eco-evolutionary impacts of domestication and agricultural practices on wild species. Philos. Trans. R. Soc B Biol. Sci. 372, 20160033. doi: 10.1098/RSTB.2016.0033
Volk, G. M., Henk, A. D., Richards, C. M., Forsline, P. L., Thomas Chao, C. (2013). Malus sieversii: A diverse central Asian apple species in the USDA-ARS national plant germplasm system. HortScience 48, 1440–1444. doi: 10.21273/HORTSCI.48.12.1440
Wang, L., Guo, K., Li, Y., Tu, Y., Hu, H., Wang, B., et al. (2010). Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 10, 1–16. doi: 10.1186/1471-2229-10-282/COMMENTS
Wang, S., Hu, T., Wang, Y., Luo, Y., Michailides, T. J., Cao, K. (2016). New understanding on infection processes of valsa canker of apple in China. Eur. J. Plant Pathol. 146, 531–540. doi: 10.1007/s10658-016-0937-3
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49. doi: 10.1093/NAR/GKR1293
Xiao, Y. H., Xian-Bi, L. I., Yang, X. Y., Luo, M., Hou, L., Guo, S. H., et al. (2007). Cloning and characterization of a balsam pear class I chitinase gene (Mcchit1) and its ectopic expression enhances fungal resistance in transgenic plants. J. Agric. Chem. Soc Japan 71, 1211–1219. doi: 10.1271/bbb.60658
Xu, J., Xu, X., Tian, L., Wang, G., Zhang, X., Wang, X., et al. (2016). Discovery and identification of candidate genes from the chitinase gene family for verticillium dahliae resistance in cotton. Sci. Rep. 2016 61 6, 1–12. doi: 10.1038/srep29022
Yamamoto, S., Nakano, T., Suzuki, K., Shinshi, H. (2004). Elicitor-induced activation of transcription via W box-related cis-acting elements from a basic chitinase gene by WRKY transcription factors in tobacco. Biochim. Biophys. Acta - Gene Struct. Expr. 1679, 279–287. doi: 10.1016/j.bbaexp.2004.07.005
Yanai, I. (2022). Quantifying gene duplication. Nat. Rev. Genet. 23, 196–197. doi: 10.1038/s41576-022-00457-w
Yin, Z., Ke, X., Kang, Z., Huang, L. (2016). Apple resistance responses against valsa mali revealed by transcriptomics analyses. Physiol. Mol. Plant Pathol. 93, 85–92. doi: 10.1016/j.pmpp.2016.01.004
Yin, Z., Liu, H., Li, Z., Ke, X., Dou, D., Gao, X., et al. (2015). Genome sequence of valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 208, 1202–1216. doi: 10.1111/nph.13544
Zhang, Z. (2022). KaKs_calculator 3.0: Calculating selective pressure on coding and non-coding sequences. Genomics Proteomics Bioinf S1672-0229, 00259-X. doi: 10.1016/j.gpb.2021.12.002
Zhang, C., Chen, X., He, T., Liu, X., Feng, T., Yuan, Z. (2007). Genetic structure of malus sieversii population from xinjiang, China, revealed by SSR markers. J. Genet. Genomics 34, 947–955. doi: 10.1016/S1673-8527(07)60106-4
Zhang, X. Y., Chen, X., Peng, Y., Wang, H. B., Jun, S. (2008). Genetic Diversity of Mineral Elements, Sugar and Acid Components in Malus sieversii. Acta Hortic. Sin. 35, 277–280. Available at: https://www.ahs.ac.cn/EN/Y2008/V35/I2/277.
Zhang, Y. J., Ren, L. L., Lin, X. Y., Han, X. M., Wang, W., Yang, Z. L. (2022). Molecular evolution and functional characterization of chitinase gene family in populus trichocarpa. Gene 822, 146329. doi: 10.1016/J.GENE.2022.146329
Zhang, Z., Yuan, L., Liu, X., Chen, X., Wang, X. (2018). Evolution analysis of dof transcription factor family and their expression in response to multiple abiotic stresses in malus domestica. Gene 639, 137–148. doi: 10.1016/j.gene.2017.09.039
Zhao, P., Wang, D., Wang, R., Kong, N., Zhang, C., Yang, C., et al. (2018). Genome-wide analysis of the potato Hsp20 gene family: Identification, genomic organization and expression profiles in response to heat stress. BMC Genomics 19, 1–13. doi: 10.1186/S12864-018-4443-1/FIGURES/6
Zhou, Z. Q., Li, Y. N. (2000). The RAPD evidence for the phylogenetic relationship of the closely related species of cultivated apple. Genet. Resour. Crop Evol. 2000 474 47, 353–357. doi: 10.1023/A:1008740819941
Zhu, L., Weichen, N., Liu, S., Cai, B., Xing, H., Wang, S. (2017). Transcriptomics analysis of apple leaves in response to alternaria alternata apple pathotype infection. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00022
Keywords: apple canker, chitinase, Malus sieversii, gene family, Valsa mali
Citation: Haxim Y, Kahar G, Zhang X, Si Y, Waheed A, Liu X, Wen X, Li X and Zhang D (2022) Genome-wide characterization of the chitinase gene family in wild apple (Malus sieversii) and domesticated apple (Malus domestica) reveals its role in resistance to Valsa mali. Front. Plant Sci. 13:1007936. doi: 10.3389/fpls.2022.1007936
Received: 31 July 2022; Accepted: 03 October 2022;
Published: 07 November 2022.
Edited by:
Youxiong Que, Fujian Agriculture and Forestry University, ChinaReviewed by:
Changhai Liu, Northwest A&F University, ChinaDahui Li, Anhui Agricultural University, China
Copyright © 2022 Haxim, Kahar, Zhang, Si, Waheed, Liu, Wen, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daoyuan Zhang, emhhbmdkeUBtcy54amIuYWMuY24=
†These authors have contributed equally to this work