 Qingqing Yang
Qingqing Yang Cunquan Yuan
Cunquan Yuan Qixiang Zhang
Qixiang Zhang- Beijing Key Laboratory of Ornamental Plants Germplasm Innovation and Molecular Breeding, National Engineering Research Center for Floriculture, Beijing Laboratory of Urban and Rural Ecological Environment, Engineering Research Center of Landscape Environment of Ministry of Education, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of Ministry of Education, School of Landscape Architecture, Beijing Forestry University, Beijing, China
Transcription factors encoded by the three-amino-acid-loop-extension (TALE) gene family play a key role in regulating plant growth and development, and are involved in plant hormone regulatory pathways and responses to various environmental stresses. Researchers are currently studying TALE genes in different species, but Prunus mumeTALE genes have not yet been studied. Therefore, based on the P. mume genome, we found a total of 23 TALE gene family members, which were distributed on eight chromosomes. TALE genes contained the characteristic domains of this family, and could be divided into KNOTTED-like homeobox (KNOX) subfamily and BEL1-like homeobox (BELL) subfamily. They can form heterodimers with each other. Fragment duplication and tandem duplication events were the main reasons for the expansion of P. mumeTALE gene family members and the TALE genes were selected by different degrees of purification. The inter-species collinearity analysis showed that the relationship between P. mume and other four Prunus species was consistent with the distance of origin. Eleven members of P. mumeTALE genes were specifically highly expressed in stem, mainly at the early stage of stem development. The cis-element analysis showed that the promoter of P. mumeTALE genes contained a variety of hormone and abiotic stress response elements, and four TALE genes responded to two kinds of abiotic stresses and four kinds of hormones at the early stage of stem development. In conclusion, this study lays a foundation to explore the role of TALE gene family in P. mume growth and development.
Introduction
Transcription factors (TFs) encoded by homeobox genes contain many members and play a key role in regulating plant growth and development (Chan et al., 1998). Vollbrecht et al. (1991) discovered a homeobox gene, maize Knotted-1 (KN1), for the first time in the plant kingdom. The kn1 mutations caused by insertion or tandem duplication of transposable elements significantly affected leaf development (Vollbrecht et al., 1991). The homeobox domain present in homeobox genes consists of 60 amino acids, forming three helical structures. The first helix and the second helix form a ring, while the second helix and the third helix form helix-turn-helix (Billeter et al., 1993). The predecessors classified Homeobox genes according to the sequence features of the homeobox domain. Originally, Homeobox genes were divided into seven categories, including Arabidopsis thaliana homeobox 8 (ATHB8), BEL1-like homeobox (BELL/BLH), GL2, homeobox from A. thaliana 1 (HAT1), homeobox from A. thaliana 2 (HAT2), KNOTTED-like homeobox (KNOX/KNAT), and Zea mays homeobox (ZM-HOX; Bharathan et al., 1997). Later, Mukherjee et al. classified them into 14 categories, including BELL/BLH, DDT, homeodomain-leucine zipper I to IV (HD-ZIP I to IV), KNOX/KNAT, luminidependens (LD), nodulin homeobox genes (NDX), plant homeodomain (PHD), PINTOX, plant zinc finger (PLINC), SAWADEE, and wuschel homeobox (WOX; Mukherjee et al., 2009). In 2017, Jin et al. reduced the homeobox genes into five categories in plant TF database PlantTFDB 4.0, including HD-ZIP, TALE, WOX, HB-PHD, and HB-others (Jin et al., 2017). Among them, TALE includes the two types of gene subfamilies BELL and KNOX, mentioned above, because the homeobox domain of these two types of genes contains three additional amino acids, which can form an additional loop connection to construct three-amino-acid-loop-extension (TALE) homeobox domain (Hamant and Pautot, 2010).
KNOTTED-like homeobox-like genes play key regulatory roles in plant meristem and leaf development. Such gene-encoded proteins typically contain four domains, including homeobox, KNOX1, KNOX2, and ELK. At first, KNOX genes were divided into two categories according to their expression and function, namely class I and II. Compared with class I, the gene expression range of class II is wider (Hake et al., 2004; Gao et al., 2015). Interestingly, later studies found that KNOX A. thaliana MEINOX protein (KNATM) only contained the KNOX domain, which was classified as a new group (class III) by phylogenetic analysis. KNATM is only found in eudicots, and some species have multiple paralogs (Magnani and Hake, 2008). Arabidopsis thaliana KNOX class I consists of four members: shoot meristemless (STM), KNAT1/BP, KNAT2, and KNAT6, which are mainly expressed in meristem and stem, and their ectopic expression in leaves can significantly affect leaf phenotype. STM is a meristem marker gene and also the first KNOX gene expressed during embryonic development (Scofield and Murray, 2006; Shani et al., 2009; Hay and Tsiantis, 2010). It affects the development and maintenance of SAM, and the formation of AM during postembryonic development (Shi et al., 2016). KNAT1/BP and KNAT6 have redundant functions with STM in maintaining SAM activity and organ separation. There is partial duplication of chromosome segments between KNAT2 and KNAT6, and single gene mutation does not significantly affect shoot development (Bharathan et al., 2002; Belles-Boix et al., 2006). KNAT1/BP can restrict the expression of KNAT6 and KNAT2 during the reproductive stage, allowing normal inflorescence development (Ragni et al., 2008). Arabidopsis thaliana KNOX class II contains four members: KNAT3, KNAT4, KNAT5, and KNAT7. Studies have shown that KNAT7 and its homologous genes affect the formation of lignin, thereby regulating the development of secondary cell wall (SCW). KNAT3 can form heterodimers with KNAT7, thus jointly promoting the formation of SCW (Li et al., 2012; Ma et al., 2019; Wang et al., 2019b). In addition, KNAT3, KNAT4, and KNAT5 play regulatory roles in A. thaliana root development with functional redundancy (Truernit and Haseloff, 2007). Medicago truncatula KNAT3/4/5-like genes regulate the development of symbiotic nodules (Di Giacomo et al., 2017). KNATM is expressed in the proximal domain of organ primordia and at the border of mature organs. Although it does not have a homeobox domain, it can still function as a transcriptional regulator. In addition, KNATM can interact with KNAT1/BP through the acidic coiled-coil domain (Magnani and Hake, 2008).
BEL1-like homeobox-like genes encoded proteins contain two conserved domains, namely homeobox and POX. Arabidopsis thaliana BELL genes has 12 members, including BEL1, A. thaliana homeobox 1 (ATH1), BLH1-10, and there is no clear classification system. In addition, BELL proteins frequently interact with KNOX proteins to perform their functions (Byrne et al., 2003). For example, KNAT1/BP can form heterodimers with PENNYWISE (PNY)/BLH9 to regulate early developmental events in inflorescence meristem (Smith and Hake, 2003). ATH1 can interact with STM to activate the expression of STM, so that STM forms a self-activating loop, thereby maintaining the activity of SAM (Cao et al., 2020). SAWTOOTH1 (BLH2/SAW1) and SAWTOOTH2 (BLH4/SAW2) can interact with KNAT1/BP, KNAT2, KNAT5, and STM, and regulate leaf edge development to a certain extent by inhibiting the expression of one or more KNOX genes (Kumar et al., 2007). BLH6 can interact with KNAT7, and this interaction enhances the inhibition ability of BLH6 and KNAT7. In addition, BLH6 and KNAT7 can directly inhibit the expression of REVOLUTA (REV) to regulate the formation of SCW (Liu et al., 2014). Recently, researchers have studied the interaction between TALE proteins in tomato and identified 75 pairs of KNOX-BLH interactions. It was found that the interaction between KNOX class I member SlKN5 and BELL proteins (SlBLH5 and SlBLH7) played an important regulatory role in the process of fruit differentiation (Ezura et al., 2022).
Previous studies have found that the TALE gene family is involved in plant hormone regulatory pathways (Tsuda and Hake, 2015; Niu and Fu, 2022). The exogenous hormone application showed that the expression of KNOX gene family members of Caucasian clover could respond to the changes of 6-BA, IAA, and KT signals (Zhang et al., 2022b). Ectopic expression of the maize KNOX-like gene KN1 in leaves enhances auxin signaling (Bolduc et al., 2012), and KN1 also can activate ga2ox1 expression by binding to its intron, which in turn negatively regulates gibberellin (GA) accumulation, thereby maintaining meristematic cell identity (Bolduc and Hake, 2009). Overexpression of tomato KNOX-like gene Tkn4 increases seedling sensitivity to GA and auxin, and increases the expression of genes related to GA and auxin synthesis (Yan et al., 2019). Ectopic expression of auxin synthesis gene in A. thaliana leaf axils can downregulate the expression of STM (Wang et al., 2015) and A. thaliana KNOX gene can rapidly activate CK biosynthetic gene expression, which is important for maintaining normal SAM development (Jasinski et al., 2005; Yanai et al., 2005). Infection of A. thaliana leaves by Rhodococcus fascians can cause local CK responses and reduce GA signaling, which may provide a suitable environment for the expression of KNOX-like genes, resulting in the formation of leaf edge serrations (Depuydt et al., 2008). Arabidopsis thaliana BLH1 can form a heterodimer with KNAT3 to activate the expression of abscisic acid (ABA) response gene ABSCISIC ACID-INSENSITIVE 3 (ABI3), which promotes plant response to ABA during seed germination and seedling stages (Kim et al., 2013). MdKNOX19, a member of the apple KNOX subfamily, is an ABA-responsive gene. Overexpression of MdKNOX19 increases the ABA sensitivity of apple callus, and can directly activate the expression of MdABI5, a key gene in the ABA signaling pathway (Jia et al., 2021). The rice KNOX-like gene homeobox1 (OSH1) can activate brassinosteroid (BR) degradation-related genes to inhibit the BR pathway, and the loss of OSH1 function leads to the increase of BR level (Tsuda et al., 2014). Litchi LcKNAT1 can inhibit the expression of ethylene biosynthesis genes by directly binding to their promoters, thereby inhibiting fruit abscission (Zhao et al., 2020).
Furthermore, TALE gene family members are not only involved in hormonal regulatory pathways but also in response to a variety of abiotic stresses (Hao et al., 2021). The promoters of TALE gene family members in soybean contained cis-elements that responded to various stresses, and the expression level of GmTALE genes could change in response to salt stress and drought stress (Wang et al., 2021). Overexpression of the KNOX-like gene TaKNOX11-A of Triticum aestivum in A. thaliana can increase the content of proline and reduce the content of malondialdehyde, thereby enhancing the salt tolerance and drought resistance of the plant (Han et al., 2022). Cotton BELL-like gene GhBLH5-A05 plays an important role in combating drought stress. Silencing GhBLH5-A05 by virus induced gene silencing (VIGS) technology reduced the drought resistance of cotton, and overexpression of GhBLH5-A05 could increase the expression level of drought-responsive genes, thereby increasing the drought resistance of cotton (Zhang, 2021). In addition, GhBLH5-A05 can enhance its own function through protein interaction with KNOX-like gene GhKNAT6-A03 (Zhang et al., 2021b). Glycine max H1 Sbh1 (GmSBH1), a member of the soybean KNOX subfamily, has a key regulatory role in response to high temperature and humidity (HTH) stress. At the same time, GmSBH1 protein can form a heterodimer with GmBLH4 to participate in the response to HTH stress during seed development (Shu et al., 2015; Tao et al., 2018). Populus alba × P. glandulosa KNOX subfamily member PagKNAT2/6b can be significantly induced by drought treatment, and can mediate drought response by down-regulating PagGA20ox1 gene of GA pathway (Song et al., 2021). Members of the pear KNOX subfamily can respond to drought stress treatment, specifically, the expression of PbKNOX7/13 is increased under drought stress, while the expression of PbKNOX5/16 is inhibited under drought stress (Liu et al., 2022).
Prunus mume belongs to genus Prunus of Rosaceae and its TALE gene family has not yet been studied, but the research on the KNOX genes of other Prunus plants has been reported. Misexpression of peach KNOPE1 (belonging to KNOX class I) may be involved in leaf hyperplasia caused by leaf curl disease (Testone et al., 2008). In addition, KNOPE1 is expressed in cortex and procambium at the primary growth stage of stems and prevents the lignification of primary stems by inhibiting lignin-related genes (Testone et al., 2012). Subsequent studies found that KNOPE1 could inhibit the expression of PpGA3ox1 to affect gibberellin(GA) homeostasis, thereby regulating peel differentiation (Testone et al., 2015). Peach KNOPE3 (belonging to KNOX class II) may be involved in the regulation of sugar transport processes (Testone et al., 2009). The members of the KNOX subfamily of pear have an important regulatory effect on the growth and development of pear. Specifically, they are highly expressed in robust hybrid progeny and weakly expressed in dwarf hybrid progeny (Liu et al., 2022). In addition, PbKNOX in pear can inhibit the level of lignin, thereby regulating the formation of stone cells in fruit (Cheng et al., 2019). The transformation of maize KNOX1 into plum can significantly improve the regeneration efficiency of plum explants (Srinivasan et al., 2011).
In this study, we used P. mume as experimental material to identify members of the TALE gene family based on genome-wide data, and performed gene and protein structure analysis, phylogenetic analysis, chromosome location analysis, collinearity analysis, promoter element prediction, and protein interaction prediction. At the same time, the response of TALE genes to hormones and abiotic stresses was explored during the early development of stem, thus laying a foundation for the functional study of the P. mume TALE gene family.
Materials and methods
Identification and gene structure analysis of TALE gene family members in Prunus mume
We obtained the genome information of P. mume (Zhang et al., 2012) on NCBI,1 and used Pfam (http://pfam.xfam.org; Mistry et al., 2021) to download the seed sequences of POX, KNOX1, KNOX2, and ELK domains. The genes containing at least one of the above domains (required E-value>10–5) were obtained from the P. mume genome with the help of hmmer 3.0 (Johnson et al., 2010) and defined as members of the P. mume TALE gene family. We used the GSDS website (http://gsds.gao-lab.org/index.php; Hu et al., 2015) to draw the intron-exon structure map of the TALE gene family members.
Basic information of TALE protein sequences in Prunus mume
The isoelectric point (pI), molecular weight (MW), overall average hydropathic coefficient (GRAVY), and instability coefficient of the TALE gene family members were analyzed using the EXPASY website (https://web.expasy.org/protparam; Wilkins et al., 1999). Transmembrane domain analysis was performed using the TMHMM website.2 Subcellular localization prediction was performed using the WoLF PSORT website (https://wolfpsort.hgc.jp; Horton et al., 2007). Protein secondary structure prediction was performed using the SOPMA website (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html; Geourjon and Deleage, 1995). Protein phosphorylation sites were predicted using the NetPhos website (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1; Blom et al., 2004).
Phylogenetic analysis and conserved motif analysis of TALE protein sequences in Prunus mume
In order to elucidate the phylogenetic relationship of TALE genes in P. mume, the TALE gene family in the genome of two other Prunus species (P. armeniaca, P. persista) and the model plant A. thaliana was analyzed together. The sequences of the A. thaliana TALE gene family were derived from UniProt (https://www.uniprot.org/blast; Bateman et al., 2021). The genomes of P. armeniaca and P. persista were downloaded from NCBI (see footnote 1). MEGA7.0 [neighbor-joining method (NJ), 1,000 Bootstrap repeats] were used to construct the TALE gene family evolution tree of P. mume, P. armeniaca, P. persista, and A. thaliana, so as to predict the homologous genes of the A. thaliana TALE gene family in three kinds of Prunus species.
Motif prediction was performed on the amino acid sequences of the TALE gene family of P. mume using the MEME website (https://meme-suite.org/meme/tools/meme; Bailey et al., 2015), using the default parameters, except that the number of motifs was set as 6, and the motif structure map was mapped using TBtools (Chen et al., 2020). Pfam’s batch sequence search tool (http://pfam.xfam.org/search#tabview=tab1; Mistry et al., 2021) was used to align the amino acid sequences of the TALE gene family of P. mume with known domains, and TBtools were used to map and beautify the analysis results (Chen et al., 2020), followed by analysis of the corresponding relationship between the motif prediction results and the structural domain alignment results.
Chromosomal location and collinearity analysis of TALE gene family members in Prunus mume
The MG2C website3 was used to analyze the chromosomal location of the P. mume TALE gene family. The intra-species collinearity analysis of the P. mume TALE gene family was performed using MCScanX (Wang et al., 2012, 2013), and the non-synonymous substitution (Ka) and synonymous substitution (Ks) values of the collinear pair were calculated with the help of the PAL2NAL website (http://www.bork.embl.de/pal2nal; Suyama et al., 2006). The evolution time of collinear pairs was predicted using the formula T = Ks/2λ × 106 Mya (λ = 1.5 × 10−8 for dicots; Wang et al., 2019a). We selected four other Prunus species, including P. armeniaca, P. persica, P. avium, and P. dulcis, and used MCScanX (Wang et al., 2012, 2013) to analyze the inter-species collinearity between P. mume and the above four species. The genomes of the other four plants (Ezhova, 2003; Alioto et al., 2020; Pinosio et al., 2020; Zhang et al., 2021a) were downloaded from NCBI (see footnote 1) and the Ensembl Plants website.4
Promoter analysis and protein interaction prediction of TALE gene family members in Prunus mume
The 2000 bp promoter sequence upstream of ATG of the TALE gene family of P. mume was extracted by TBtools (Chen et al., 2020), and the cis-acting elements on the promoter sequence were analyzed by the PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html; Rombauts et al., 1999). The STRING website (https://cn.string-db.org; Szklarczyk et al., 2021) was used to predict the protein interactions between members of the TALE gene family, and Cytoscape was used to map protein interaction networks (Shannon et al., 2003).
Analysis of expression patterns of the TALE gene family members in Prunus mume
Tissue-specific expression analysis of TALE gene family members in P. mume: The expression profiles of TALE genes in five different tissues (root, stem, leaf, flower bud, and fruit) were mapped using previous transcriptome data (GEO No. GSE40162), and P. mume wild species in Tongmai town, Tibet, China (accession No. BJFU1210120008) was used in this previous study (Zhang et al., 2012). TBtools was used to draw the heatmap with parameters set to row scale and scale size by area (Chen et al., 2020).
Expression profile analysis of P. mume TALE genes at different stem developmental stages: The plant material P. mume cv. “Jiangmei” used in this study was planted on the campus of Beijing Forestry University. To explore the effect of the TALE gene family on stem development, annual stems (that is, 1-year-old branches) with the same growth length (about 20 cm) were obtained from the same tree in May. The three developmental stages of the stem were divided as follows: the first developmental stage included the apical bud and the first to third stem nodes counting down from the apical bud of each annual stem (leaves removed), the second developmental stage included the fourth to sixth stem nodes of each annual stem (leaves removed), and the third developmental stage included the seventh to ninth stem nodes of each annual stem (leaves removed). Samples for each developmental stage were obtained in three replicates. All samples were snap-frozen in liquid nitrogen and stored in a −80°C freezer.
Expression profile analysis of P. mume TALE genes under hormone or abiotic stress treatments: In order to explore the response of the TALE gene family to exogenous hormones and abiotic stresses in early stem development, annual stems with the same growth length (20 cm) were obtained from the same tree in May, and the annual stems were treated with four hormones and two stresses, then the first developmental stage samples of the treated annual stems were used for the fluorescence quantitative assay. Various treatments were carried out by inserting annual stems into MS liquid medium containing different hormones or stress treatment-related substances. Each liter of MS liquid medium contains 4.43 g MS powder, 25 g sucrose and 7 g agar. Hormone treatments included 1 mmol/L 6-BA, 1 mmol/L NAA, 1 mmol/L GA3, and 1 mmol/L ABA. Abiotic stress included 300 mmol/L NaCl and 300 mmol/L mannitol. The treatment time included 1, 3, 6, and 12 h, and the untreated (that is, 0 h) first developmental stage samples of the annual stems were the blank control. The test was carried out in an incubator, and the setting conditions were 16 h (25°C) during the day, 8 h (18°C) at night, and 80% humidity. Each treatment experiment was repeated three times. All samples were snap-frozen in liquid nitrogen and stored in a −80°C freezer.
RNA extraction and qRT-PCR
RNA was extracted from samples by RNA Extraction Kit (Takara, Dalian, China), the residual DNA in each sample was removed by DNase I, and the first strand of cDNA was synthesized by PrimeScript RT reagent kit (Takara, Dalian, China). Primer design was completed by NCBI primer tool and the primers were synthesized by Beijing Qingke Biotechnology Co., Ltd. (Supplementary Table S1). qRT-PCR was performed with SYBR premix ex Taq II Kit (Takara, Dalian, China). The total reaction system was 20 μl: SYBR Mix 10 μl, 10-fold diluted cDNA template 1 μl, 0.4 μl of forward and reverse primers (10 μmol/L) respectively, and H2O 8.2 μl. The reaction procedure is as follows: pre denaturation at 95°C for 3 min; denaturation at 95°C for 10 s, renaturation at 60°C for 1 min, 40 cycles; fluorescence collection at 60°C. Actin was used as the internal reference gene in the experiment (Xu et al., 2015). Three technical repeats were set up and the gene expression level was calculated by 2–ΔΔCt (Schmittgen and Livak, 2008). Microsoft Office Excel was used for data analysis and histogram plotting.
Results
Identification and gene structure analysis of TALE gene family members in Prunus mume
In order to obtain members of the TALE gene family in P. mume, we used the seed sequences of POX, KNOX1, KNOX2, and ELK domains downloaded from Pfam to align and analyze the P. mume genome, and the genes containing at least one of the above domains were defined as members of the TALE gene family. Finally, 23 P. mume TALE genes were obtained, and according to the order number of the genes in the genome, they were named PmTALE1–PmTALE23 in order from small to large.
Analysis of the basic information of the P. mume TALE gene family showed that a total of 12 members of the family were located on the sense strand, and the other 11 members were located on the antisense strand. The gene length of PmTALE19 (837 bp) and the length of its coding region (327 bp) were the shortest. The length of PmTALE15 (6,905 bp) and the coding region of PmTALE1 (2,445 bp) were the longest. All members of the P. mume TALE gene family except PmTALE15 and PmTALE21 genes contain 4–5 exons and 3–4 introns, while PmTALE15 contains 10 exons and nine introns and PmTALE21 contains only two exons and one intron (Table 1). Gene structure analysis showed that compared with other members, PmTALE1, PmTALE11, PmTALE13, and PmTALE23 all contained a longer intron with lengths of 2,766, 3,869, 2,995, and 4,094 bp respectively, while PmTALE18 contained the shortest exon with a length of only 24 bp (Figure 1).
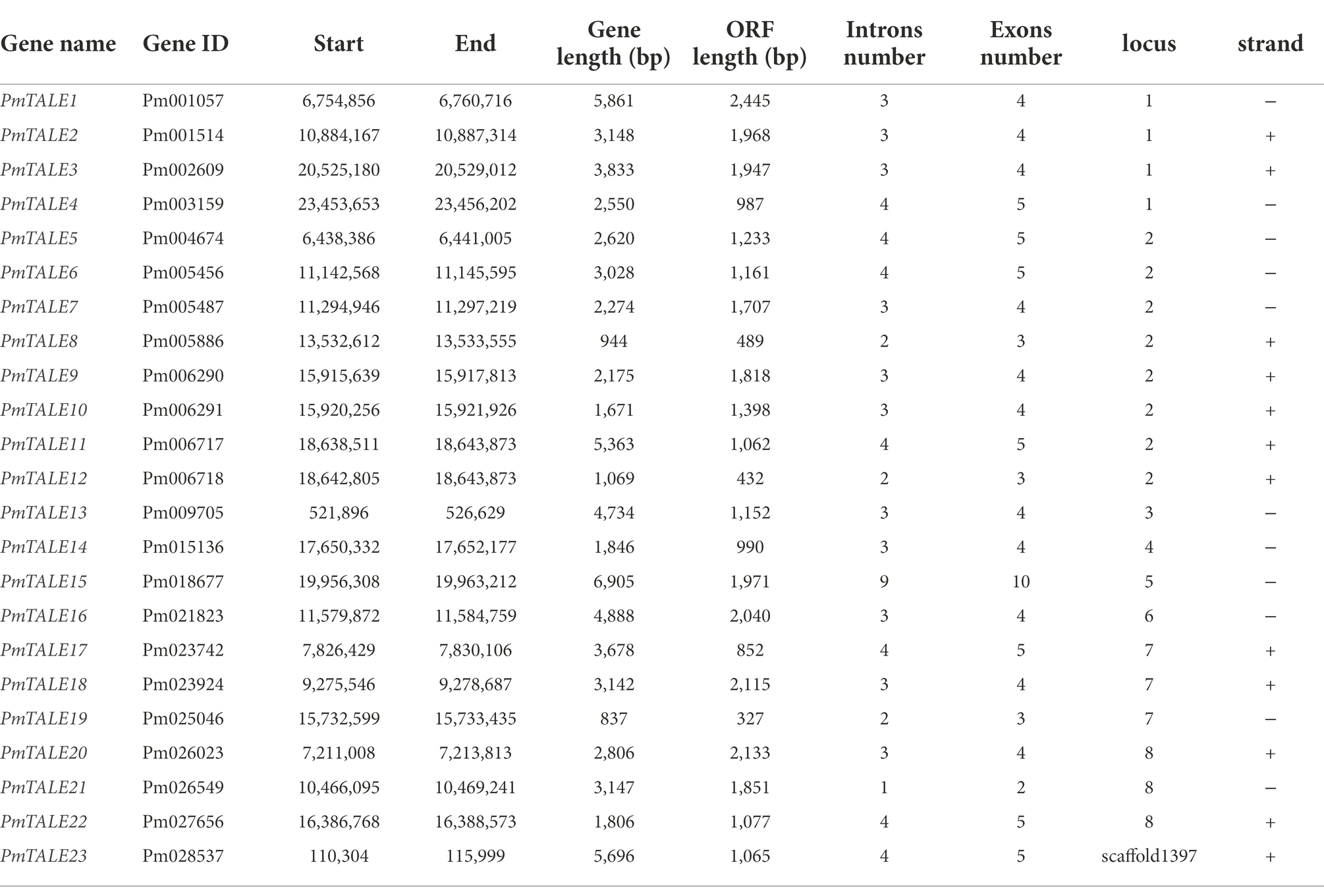
Table 1. Basic information of Prunus mume TALE gene family members.
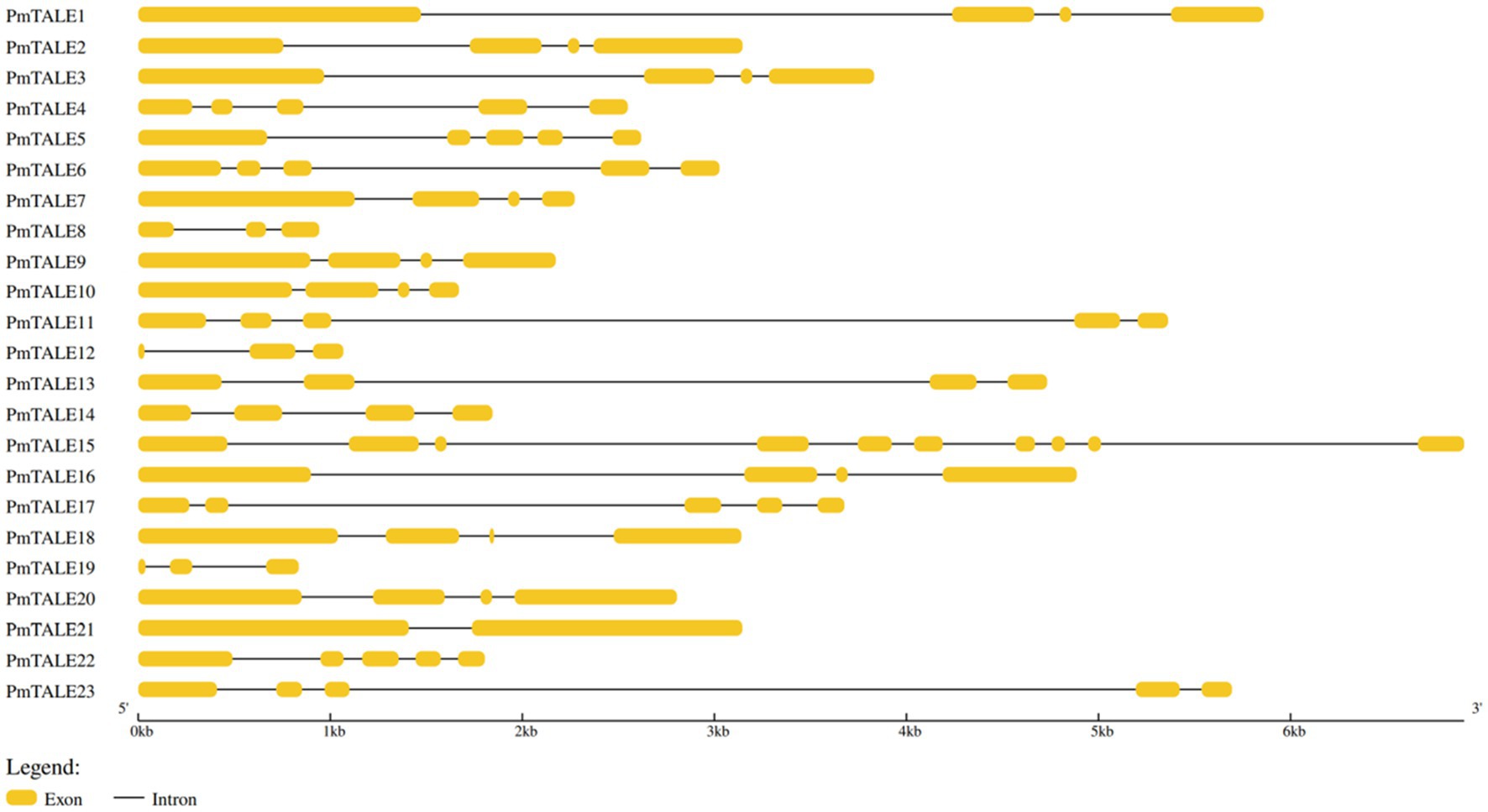
Figure 1. Schematic diagram of the exon-intron structure of the three-amino-acid-loop-extension (TALE) gene family members in Prunus mume.
Basic information analysis of TALE protein sequences in Prunus mume
Analysis of the basic information of the P. mume TALE gene family proteins showed that the number of amino acids ranged from 108 to 814. Among them, PmTALE19 protein sequence has the shortest length and the smallest molecular weight, which is 12042.49 Da. PmTALE1 protein sequence has the longest length and the largest molecular weight, which is 89285.16 Da. The isoelectric points of PmTALE1 and PmTALE2 proteins are 7.17 and 7.21 respectively, showing near neutral. The isoelectric point of PmTALE15 protein is 9.52, showing its alkaline nature. The protein isoelectric points of other 20 gene family members are all less than 7, showing their acidic nature. The GRAVY of all gene family proteins was negative, and the instability coefficients were all greater than 40, indicating that all proteins were soluble and the protein structures were unstable. The prediction results of the transmembrane domain showed that there was no transmembrane helical region in all protein amino acid sequences, so they were all distributed outside the membrane. Subcellular localization prediction results showed that all proteins were localized in the nucleus, suggesting their function as transcription factors (Supplementary Table S2). The secondary structures of all proteins were similar, with a high proportion of α-helix and random coil, a small proportion of β-sheet and extended linkage (Figure 2A). In addition, the prediction results of protein phosphorylation sites show that there are various phosphorylation sites on the amino acid sequence of all members, among which serine residues (S) account for the highest proportion, while threonine residues (T) and lysine residues (Y) account for a small proportion, indicating that the biological function of P. mume TALE gene family members may be regulated by kinases (Figure 2B).
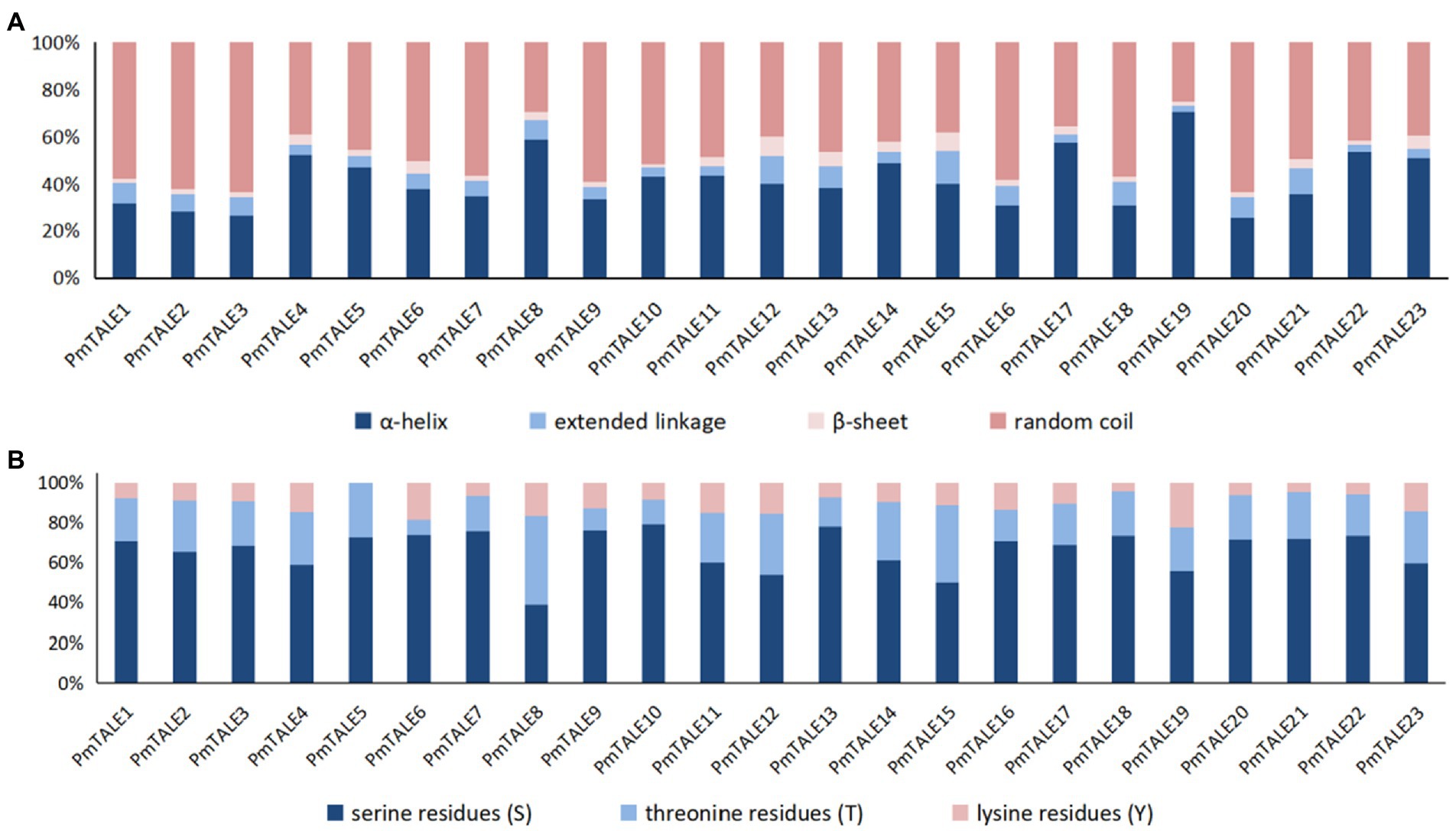
Figure 2. Analysis of protein secondary structure and phosphorylation site of Prunus mume TALE gene family members, (A) reflects the proportion of the four types of secondary structures of the TALE proteins; (B) reflects the proportion of the three types of phosphorylation sites of the TALE proteins. S, serine residues; T, threonine residues; and Y, lysine residues.
Phylogenetic analysis and conserved motif analysis of TALE protein sequence in Prunus mume
In order to predict the phylogenetic relationship of TALE gene family members in P. mume, the phylogenetic tree was constructed from the protein sequences of TALE gene family members in P. mume, two other Prunus species (P. armeniaca and P. persica) and A. thaliana. The results showed that the TALE gene families of these four species can all be divided into two subfamilies (Figure 3A), and the number of members of the two subfamilies is not much different (Figure 3B), indicating that the overall evolutionary relationships within the TALE gene families of these three Prunus species are all similar to A. thaliana. According to the clustering results of P. mume and A. thaliana, PmTALE1-3, PmTALE7, PmTALE9-10, PmTALE15-16, PmTALE18, and PmTALE20-21, a total of 11 members clustered with the A. thaliana BELL subfamily, and the remaining 12 members clustered with the A. thaliana KNOX subfamily, therefore, the 11 TALE gene family members clustered with the A. thaliana BELL subfamily were named as the P. mume BELL subfamily, and the remaining 12 TALE gene family members were named as the P.mume KNOX subfamily (Figure 3).
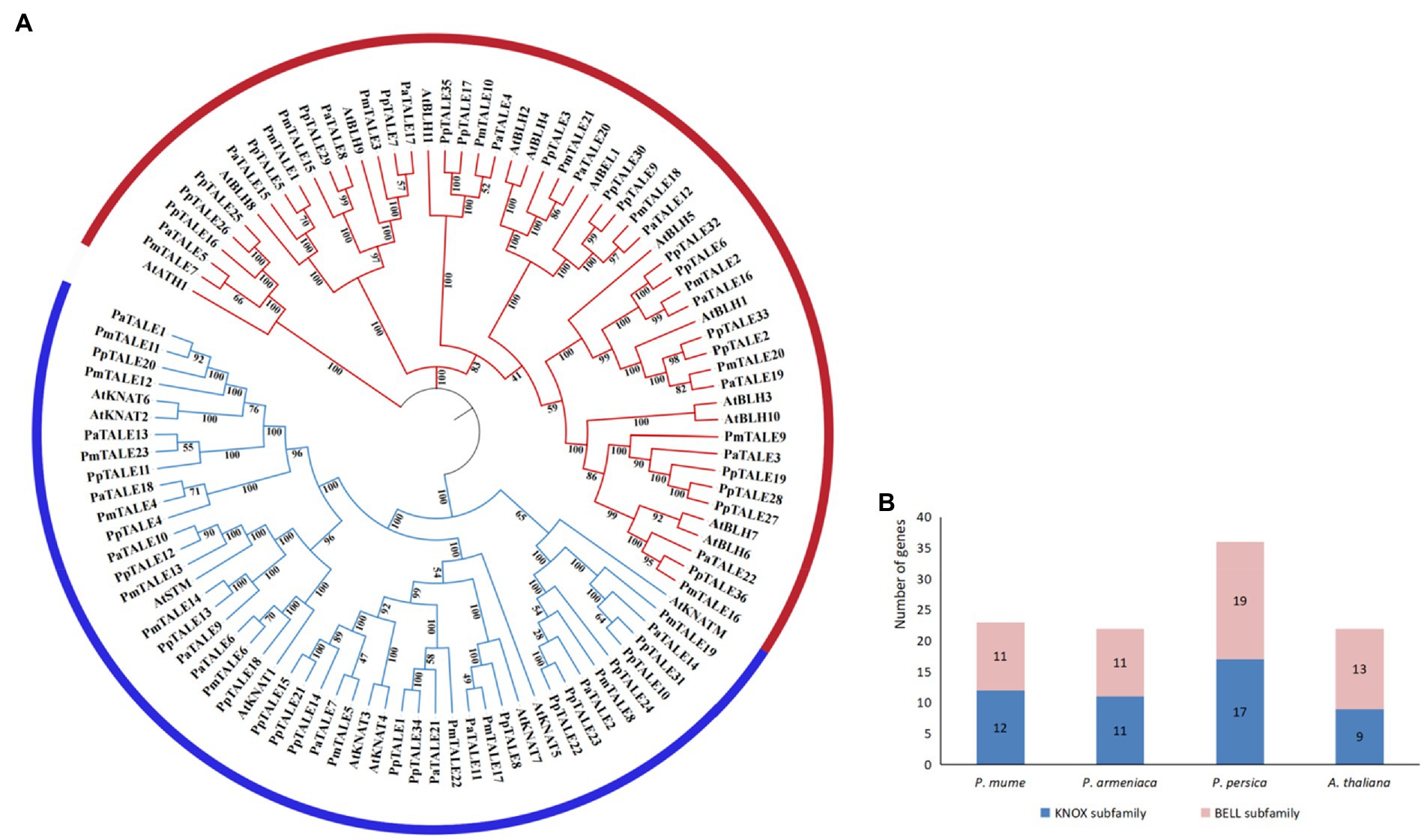
Figure 3. Phylogenetic analysis of TALE gene family protein sequences in Prunus mume and other plant species, (A) shows phylogenetic tree of TALE gene family protein sequences in P. mume, Prunus armeniaca, Prunus persica, and Arabidopsis thaliana. PmTALE represents the P. mume TALE protein, PaTALE represents the P. armeniaca TALE protein, PpTALE represents the P. persica TALE protein, and the rest represent the A. thaliana TALE proteins. On the periphery of the figure, the blue semi-circle represents the KNOTTED-like homeobox (KNOX) subfamily, and the red semi-circle represents the BEL1-like homeobox (BELL) subfamily; (B) shows the gene numbers of subfamily in P. mume, P. armeniaca, P. persica, and A. thaliana.
We used MEME website to predict amino acid sequences of P. mume TALE gene family members and a total of six kinds of motifs were found (Figure 4A). Analysis of the motif contained in all the members of the TALE gene family found that 19 TALE gene family members except PmTALE8, PmTALE18, PmTALE19, and PmTALE21 contained motif1, and 21 TALE gene family members except PmTALE8 and PmTALE19 contained motif3. Analysis of the motif contained in the BELL subfamily found that all the members contained motif2 and motif6. Analysis of the motif contained in the KNOX subfamily found that, except for PmTALE12 and PmTALE19, the rest of the members contained motif4 and motif5, while PmTALE19 only contained motif5, and PmTALE12 contained neither motif4 nor motif5 (Figure 4B).
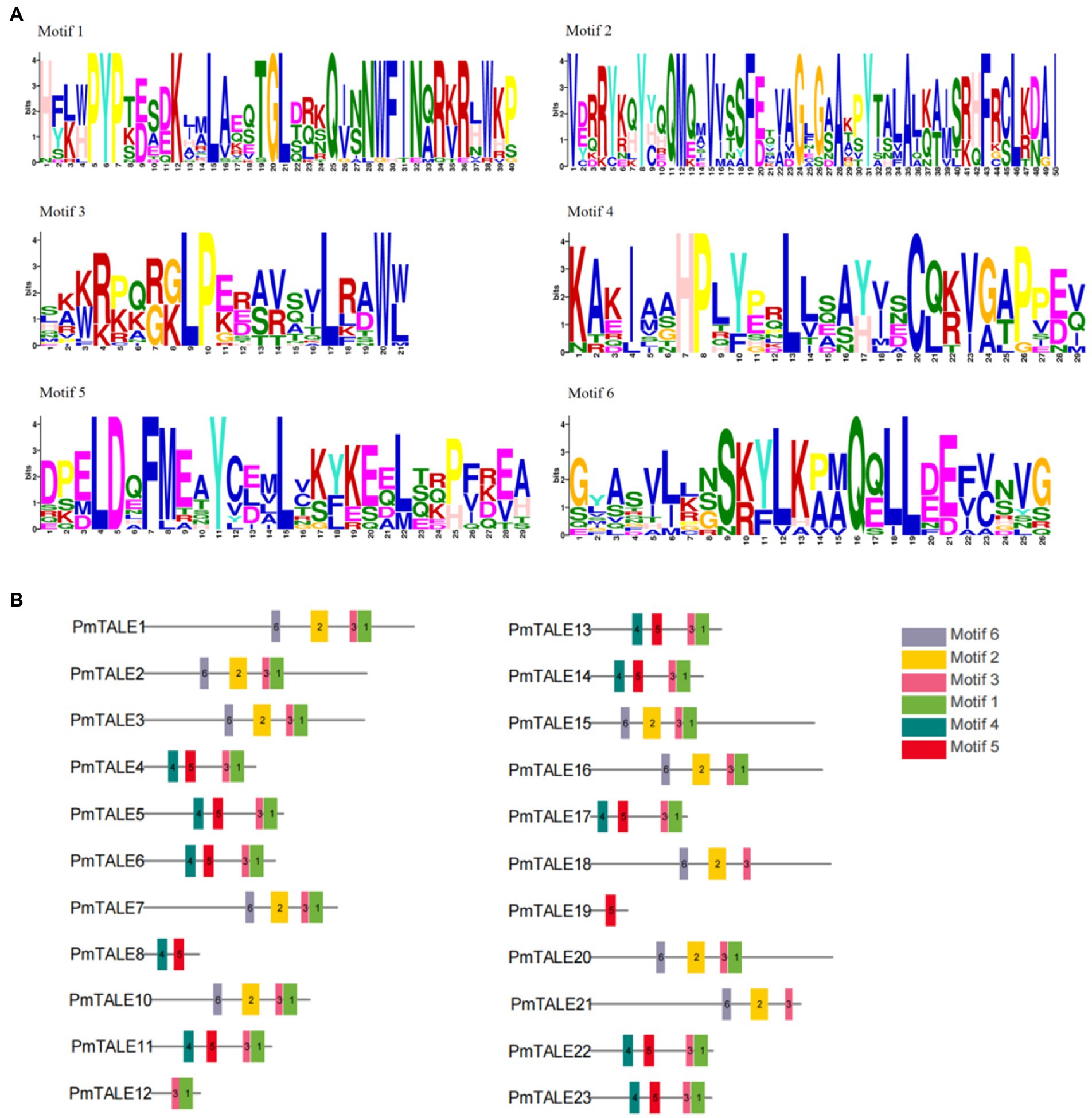
Figure 4. Motif analysis of the protein sequence of the Prunus mume TALE gene family, (A) shows the sequences of the six kinds of motifs; (B) reflects the distribution of the six kinds of motifs on the P. mume TALE protein sequences.
We used Pfam to align the amino acid sequences of P. mume TALE gene family members with known domains, and it was found that the family contained seven domains. Analysis of the domains contained in all members of TALE gene family showed that 19 TALE gene family members except PmTALE8, PmTALE18, PmTALE19, and PmTALE21 contained Homeodomain or Homeobox_KN domain. Analysis of the domains contained in the BELL subfamily found that all members of this subfamily contained POX domains. Analysis of the domains contained in the KNOX subfamily found that, except for PmTALE8, PmTALE12, and PmTALE19, the rest of the members contained KNOX1 and KNOX2 domains, while PmTALE8 and PmTALE19 only contained KNOX2 domains, and PmTALE12 contained neither KNOX1 nor KNOX2 domain. Furthermore, with the exception of PmTALE8, PmTALE17 and PmTALE19, the remaining members of this subfamily contained ELK domain (Figure 5).
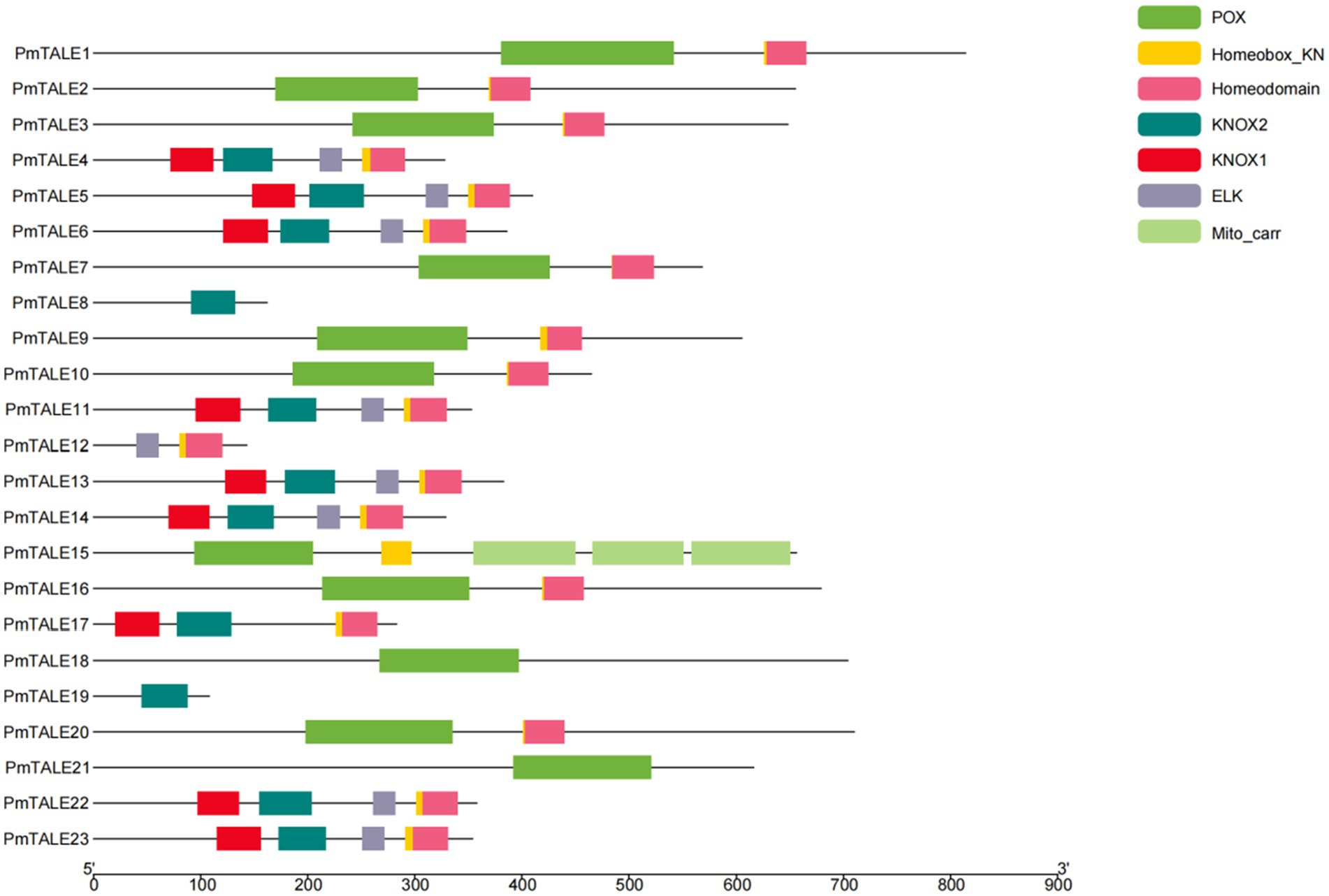
Figure 5. Analysis of the protein sequence domain of the Prunus mume TALE gene family.
Finally, we compared the motif prediction results and the structural domain alignment results, and found that the two results had a corresponding relationship. Motif1 and motif3 roughly correspond to Homeodomain and Homeobox_KN domains, motif2 and motif6 roughly correspond to POX domains, motif4 roughly corresponds to KNOX1, and motif5 roughly corresponds to KNOX2. It is worth mentioning that PmTALE8 and PmTALE19 clustered with the A. thaliana KNATM in the phylogenetic tree (Figure 3). The protein length of the A. thaliana KNATM is very short, and does not contain the Homeodomain domain of the Homeobox gene family, and only contains the KNOX1 and KNOX2 domains, but it can still play biological functions in the process of plant growth and development (Magnani and Hake, 2008). The domain characteristics of PmTALE8 and PmTALE19 were similar to KNATM, indicating that the predicted protein structure was consistent with the phylogenetic tree clustering results.
Chromosome location and collinearity analysis of Prunus mume TALE gene family
Chromosome location results showed that except for PmTALE23 gene, 22 members of the TALE gene family in P. mume were all located on chromosomes and were distributed on eight chromosomes. Specifically, chromosome 2 contained eight TALE genes. Chromosome 1 contains four TALE genes, chromosomes 7 and 8 contain three TALE genes, and chromosomes 3–6 contain only one TALE gene (Figure 6).
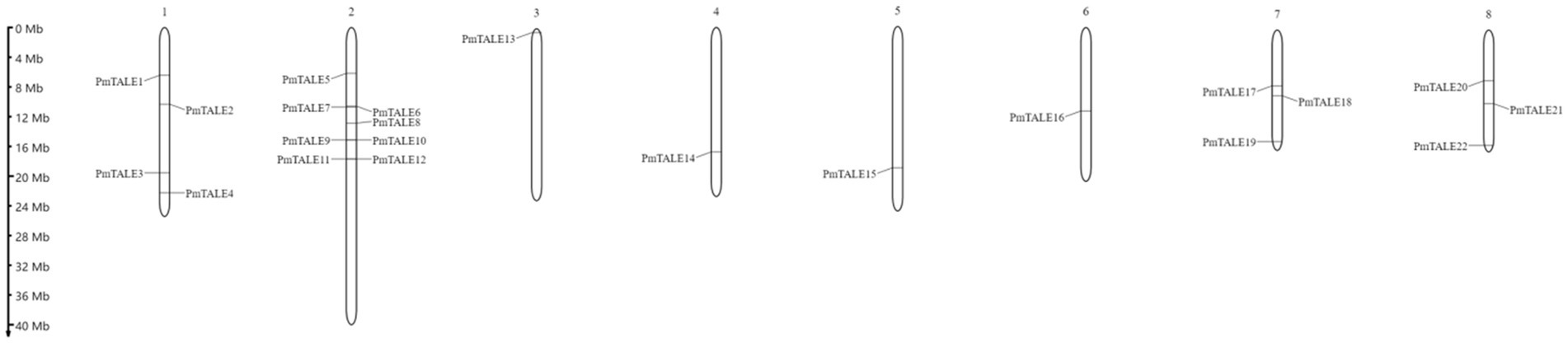
Figure 6. Chromosomal location of the Prunus mume TALE gene family.
Intra-species collinearity analysis was performed on 22 members of the TALE gene family located on chromosomes and the results showed that a total of 13 members participated in the formation of seven collinear pairs, as follows: PmTALE2 and PmTALE20, PmTALE3 and PmTALE15, PmTALE5 and PmTALE22, PmTALE9 and PmTALE16, PmTALE13 and PmTALE14, PmTALE9 and PmTALE10, and PmTALE11 and PmTALE12. Among them, the first five pairs were genome-wide replication (WGD) or fragment replication, and the last two pairs were tandem replication. It indicated that fragment duplication and tandem duplication events were the main reasons for the expansion of members of the TALE gene family in P. mume (Figure 7).
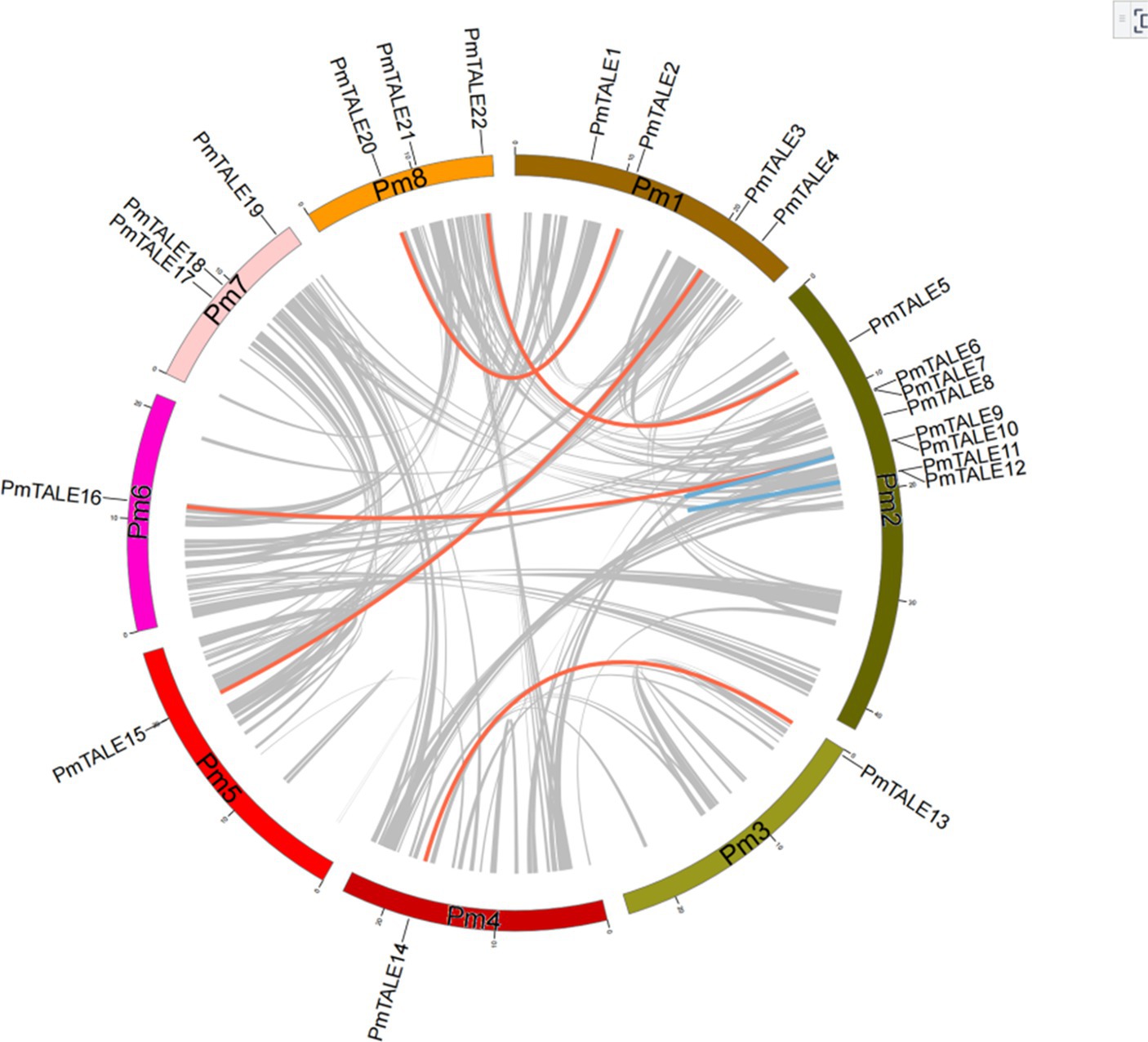
Figure 7. Intra species collinearity analysis of Prunus mume TALE gene family. Red lines indicate gene pairs that form genome-wide replication (WGD) or segmental duplications, blue lines indicate gene pairs that form tandem duplications, and grey lines represent other collinear gene pairs of non-TALE gene family members within the genome.
The Ka to Ks ratio was calculated for collinear pairs with WGD or fragment duplication and tandem duplication to predict the adaptive evolution of gene CDS regions. The results showed that the ratios of Ka to Ks for all collinear pairs ranged from 0.02 to 0.67, which were all less than 1, indicating that these genes were selected by different degrees of purification. In addition, the evolutionary time of the five collinear pairs with WGD or fragment duplication was relatively close, which was estimated to be 89.2–95.7 MY. However, the evolutionary time of the two collinear pairs with tandem duplication was far apart, specifically, PmTALE9 and PmTALE10 evolved as early as about 1485.3 MY, while PmTALE11 and PmTALE12 evolved at about 5.5 MY (Table 2).
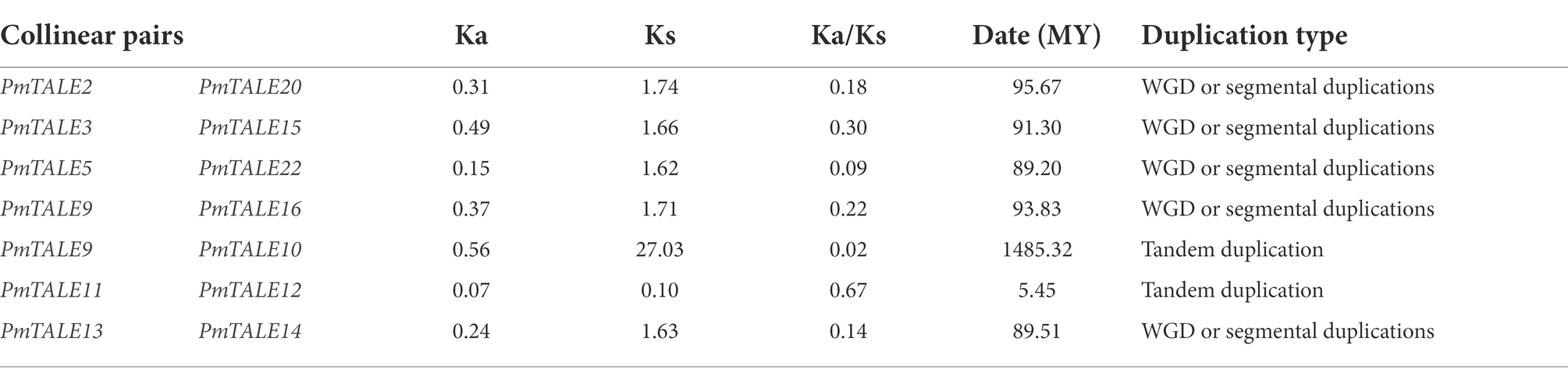
Table 2. Ka/Ks value calculation and evolution time prediction of collinear pairs within the Prunus mume TALE gene family.
In order to preliminarily explore the evolutionary relationship of TALE gene family between P. mume and other Prunus species, we performed the inter-species collinearity analysis of the 22 members of this family located on chromosomes. The other four Prunus species selected for comparative analysis were P. armeniaca, P. persica, P. avium, and P. dulcis. China is the main origin of P. armeniaca, while P. persica has been cultivated in China as early as 2,000 BC. Different from the first two kinds of plants, the latter two kinds of plants are mainly distributed in foreign countries. Among them, P. avium is a cherry native to Europe, western Turkey, northwestern Africa, and western Asia. P. dulcis is native to Israel, west Jordan, Lebanon, south Turkey, Turkmenistan, and Uzbekistan.
The results of inter-species collinearity analysis showed that genes with collinear relationship with members of the P. mume TALE gene family could be found in other four Prunus species. Specifically, a total of 24 genes on the P. armeniaca chromosome formed 32 collinear gene pairs with P. mume TALE genes. These 24 genes were distributed on eight chromosomes of P. armeniaca. Among them, chromosome 1 contained the largest number of genes, a total of 7, while chromosomes 3, 4, and 8 contained only one gene. A total of 22 genes on the P. persica chromosome formed 31 collinear gene pairs with the P. mume TALE genes. These 22 genes were distributed on eight chromosomes of P. persica. Among them, chromosome 1 contained the largest number of genes, a total of 6, while chromosomes 3, 4, and 8 contained only one gene. Overall, the results of the collinearity analysis of TALE gene family between P. mume with P. armeniaca are similar to that between P. mume with P. persica. On the chromosome of P. dulcis, only five genes formed five collinear gene pairs with members of the P. mume TALE gene family, and these five genes were distributed on chromosomes 4, 5, and 7. On the P. avium chromosome, there were only two genes that formed three collinear gene pairs with the P. mume TALE genes, and these two genes were distributed on chromosomes 1 and 4. Overall, compared with P. armeniaca and P. persica, the number of collinear gene pairs of TALE genes formed between the P. mume with P. dulcis or P. avium was very small. To some extent, this indicates that P. mume is closely related to P. armeniaca and P. persisca, but far away from P. dulcis and P. avium. The origins of P. mume, P. armeniaca and P. persica are mainly distributed in China, while the origins of P. dulcis and P. avium are mainly distributed in foreign countries, indicating that the distance of origin of these five Prunus species is consistent with the distance of collinear relationship (Figure 8).
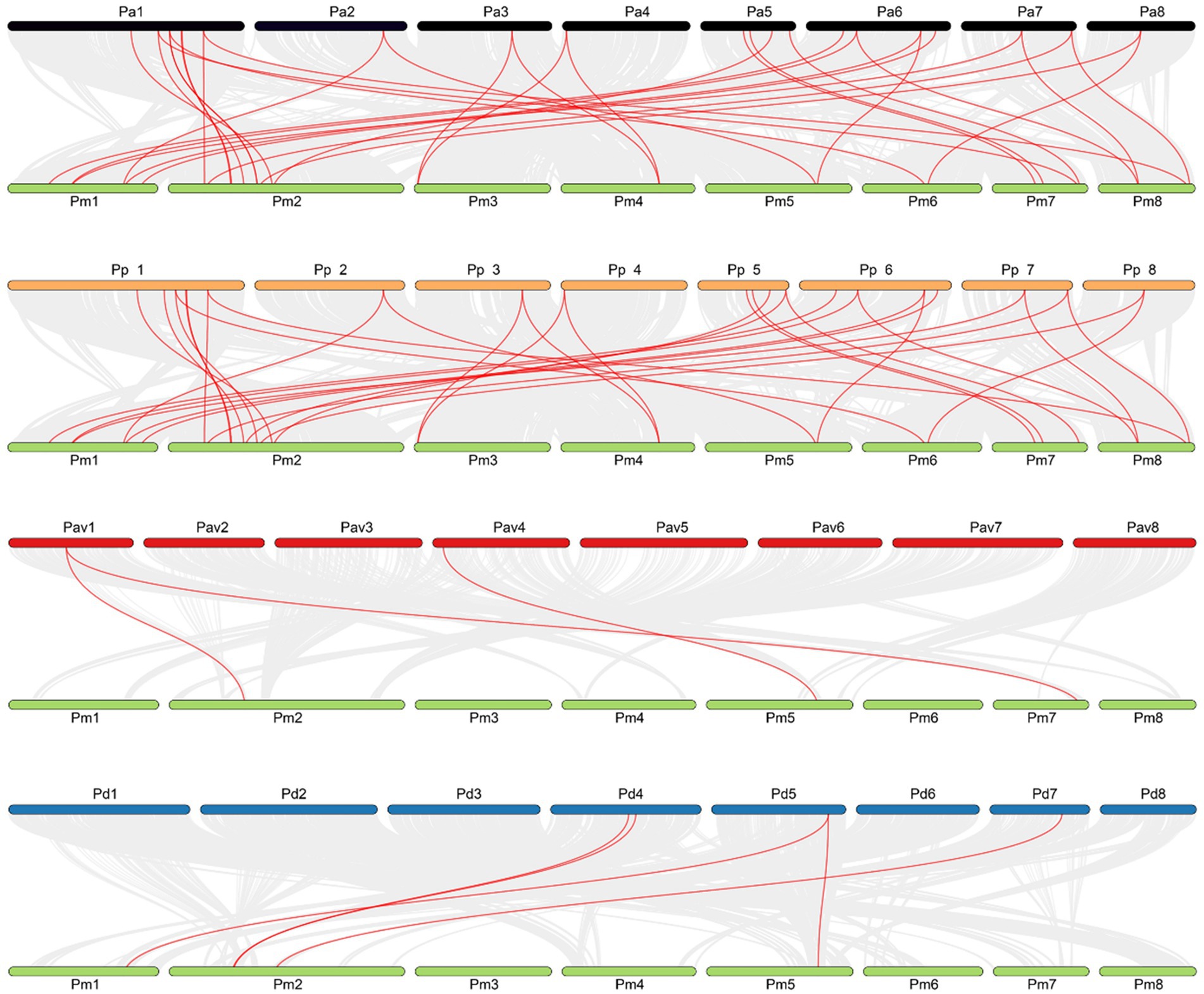
Figure 8. Collinear analysis of TALE gene family between Prunus mume and four kinds of Prunus species. The red lines represent gene pairs with a collinear relationship, grey lines represent other collinear gene pairs of non-TALE gene family members across genomes; Green represents P. mume chromosomes, black represents P. armeniaca chromosomes, orange represents P. persica chromosomes, red represents P. avium chromosomes, and blue represents P. dulcis chromosomes.
Prediction of protein interactions and promoter elements of Prunus mume TALE gene family members
We used STRING website to predict the protein interactions among members of P. mume TALE gene family, and the results showed that there were many interactions among members of this family. First, there are four types of protein interactions within the KNOX subfamily, specifically, STM (PmTALE13, PmTALE14) and KNAT1 (PmTALE6), STM (PmTALE13, PmTALE14) and KNAT7 (PmTALE17), KNAT6 (PmTALE4, PmTALE11, PmTALE12, and PmTALE23) and KNAT7 (PmTALE17), KNAT7 (PmTALE17) and KNAT1 (PmTALE6). In addition, there are also eight kinds of protein interactions within the BELL subfamily, including ATH1 (PmTALE7) and BLH1 (PmTALE2, PmTALE20), BLH1 (PmTALE2, PmTALE20) and BEL1 (PmTALE18), BLH1 (PmTALE2, PmTALE20) and BLH2 (PmTALE21), BLH1 (PmTALE2, PmTALE20) and BLH3 (PmTALE9), BLH2 (PmTALE21) and BEL1 (PmTALE18), BLH3 (PmTALE9) and BLH7 (PmTALE16), BLH3 (PmTALE9) and RPL (PmTALE3, PmTALE15), and BEL1 (PmTALE18) and BLH3 (PmTALE9). Moreover, there are also interactions between the KNOX subfamily and the BELL subfamily, and the number is large, with a total of 29 types. For example, STM (PmTALE13, PmTALE14) and ATH1 (PmTALE7), KNAT3 (PmTALE5, PmTALE22) and RPL (PmTALE3, PmTALE15), and so on. In summary, the members of the P. mume TALE gene family may form dimers or multimers through protein interactions to perform transcriptional regulatory functions (Figure 9).
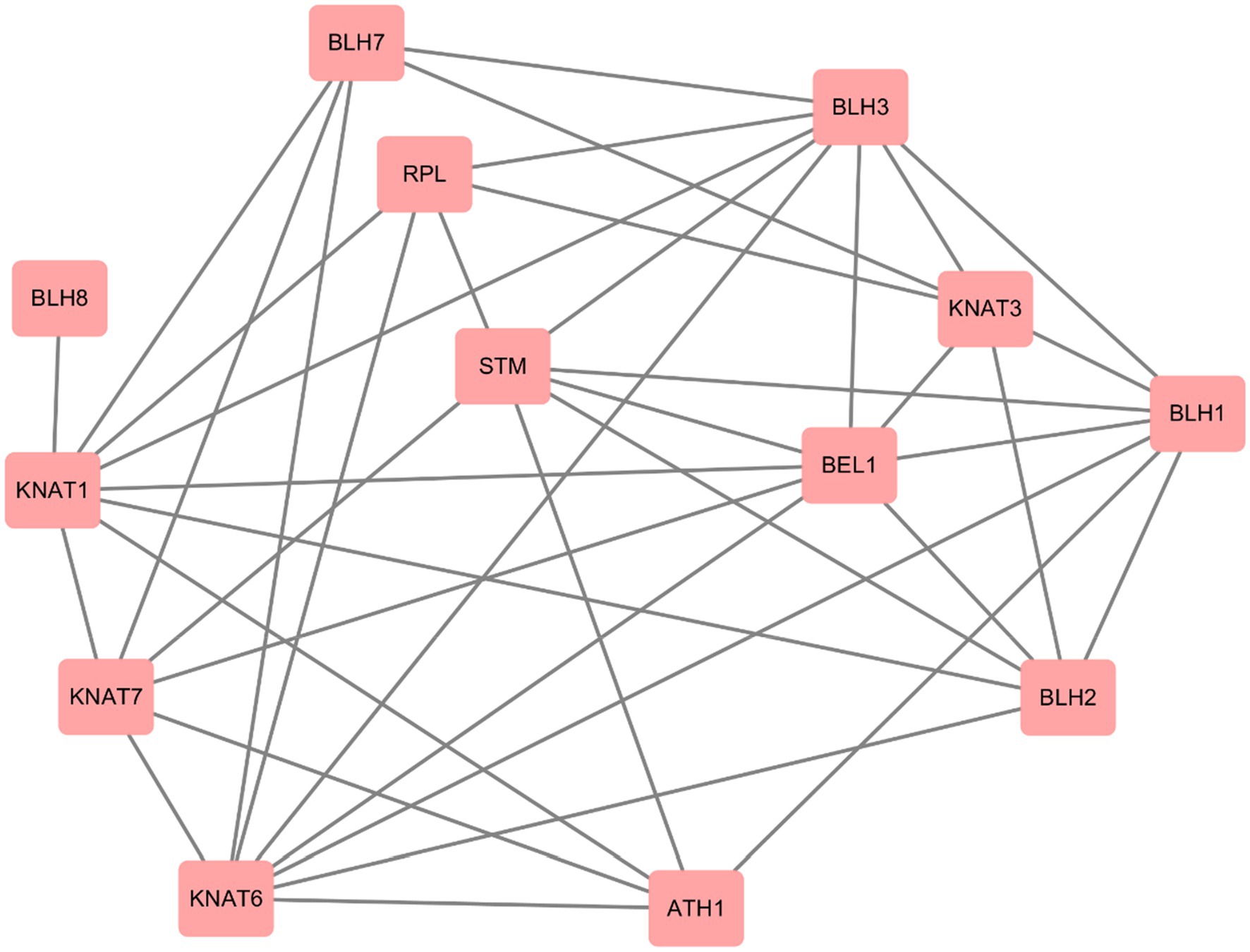
Figure 9. Prediction of protein interactions within Prunus mume TALE gene family. STM: PmTALE13, PmTALE14; KNAT1: PmTALE6; KNAT3: PmTALE5, PmTALE22; KNAT6: PmTALE4, PmTALE11, PmTALE12, PmTALE23; KNAT7: PmTALE17; RPL: PmTALE3, PmTALE15; ATH1: PmTALE7; BEL1: PmTALE18; BLH1: PmTALE2, PmTALE20; BLH2: PmTALE21; BLH3: PmTALE9; BLH7: PmTALE16; BLH8: PmTALE1.
Promoter analysis results showed that there were 22 light-responsive elements in the promoters of the P. mume TALE gene family members. Among them, the PmTALE12 promoter contained the most types of light-responsive elements, with a total of 11 types, while the PmTALE11 and PmTALE18 promoters contained the least types of light-responsive elements, three types. The PmTALE23 promoter contained the largest number of light-responsive elements, with a total of 18, and the PmTALE18 promoter contained the least number of light-responsive elements, only 6 (Figure 10A). In addition, there were 10 types of hormone response elements in the promoters of the P. mume TALE gene family members, including three kinds of auxin response elements, three kinds of gibberellin response elements, two kinds of methyl jasmonate response elements, one kind of abscisic acid response element, and one kind of salicylic acid response element. Among them, the PmTALE22 promoter contained the largest number of hormone response elements, with a total of 20, and the PmTALE19 promoter contained the least number of hormone response elements, only one (Figure 10B). Moreover, the promoters of the P. mume TALE gene family members also contained regulatory elements that respond to abiotic stress and plant development. Specifically, abiotic stress includes low temperature, drought, hypoxia induction, trauma response, etc., and plant development includes palisade mesophyll cell differentiation, meristem development, endosperm development, cell cycle, circadian rhythm, etc. (Figure 10C). In conclusion, the P. mume TALE gene family may be regulated by light signals and various hormonal signals, participate in the development of P. mume and respond to various abiotic stresses.
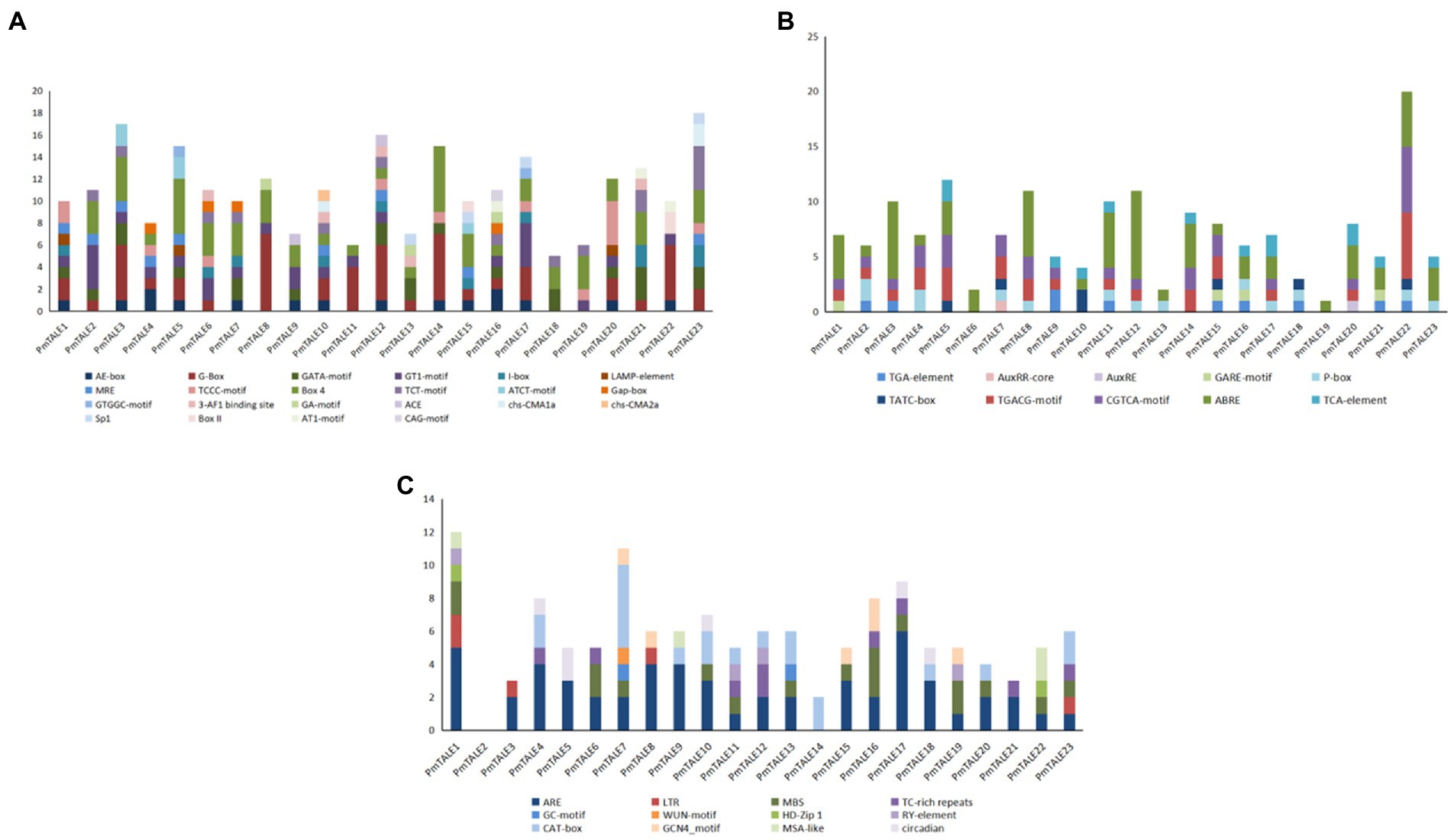
Figure 10. Cis element analysis of promoter of Prunus mume TALE gene family members. (A) shows the types and numbers of light-responsive elements contained in the TALE gene promoter; (B) shows the types and numbers of hormone response elements contained in the TALE gene promoter; and (C) shows the types and numbers of other elements contained in the TALE gene promoter. The ordinate represents the number of elements.
Expression profile of Prunus mume TALE gene family members in different tissues and different stem developmental stages
The expression profiles of P. mume TALE gene family members in five different tissues showed significant differences in the expression distribution of members. The first 11 members were highly expressed in the stem (Figure 11A). Among these 11 members, PmTALE6 has the highest expression, while PmTALE19 has the lowest expression in stem (Figure 11B). In addition, PmTALE4, PmTALE15, and PmTALE23 were highly expressed in flower buds, PmTALE21 and PmTALE8 were highly expressed in leaves, PmTALE16 and PmTALE17 were significantly expressed in roots, and PmTALE2, PmTALE20, and PmTALE22 showed high expression in fruits (Figure 11A).
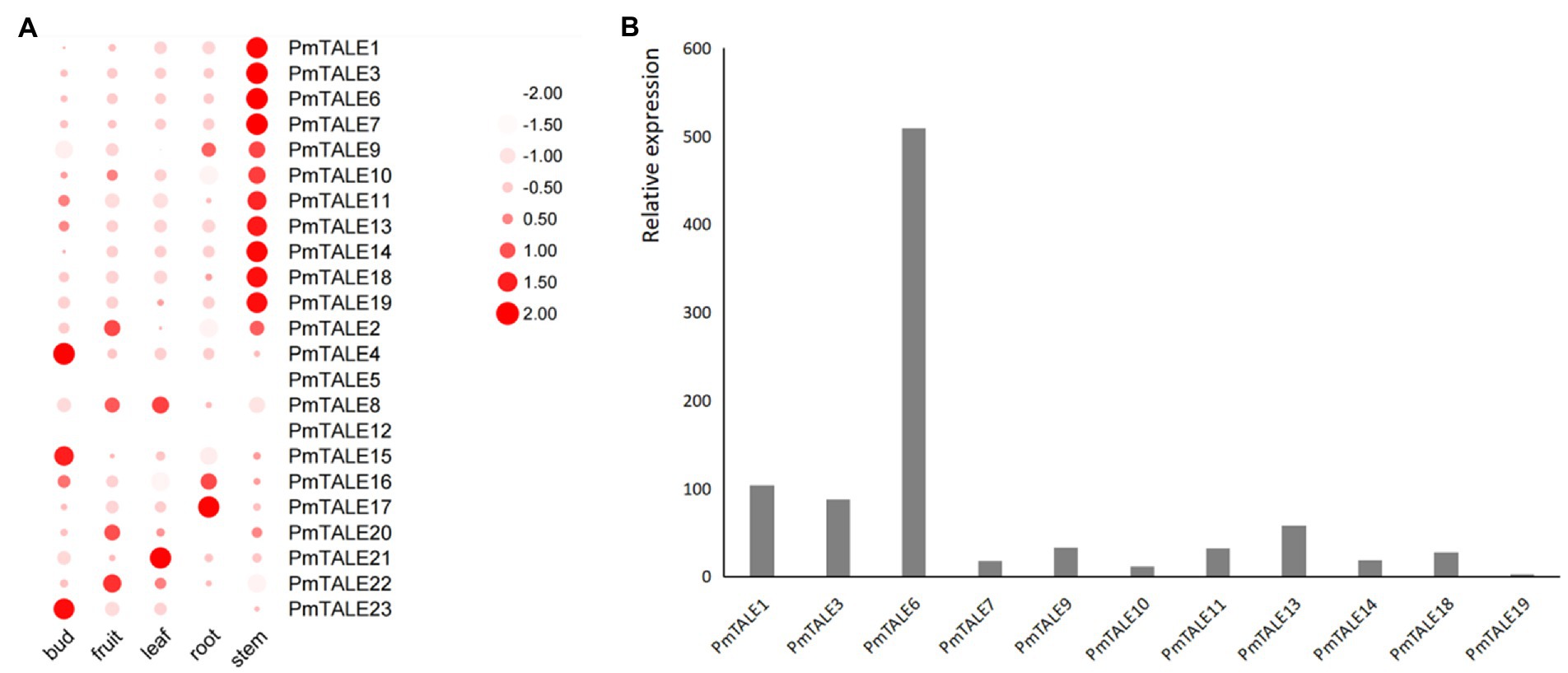
Figure 11. Expression profiling of the Prunus mume TALE gene family based on RNA sequencing results. (A) The heatmap parameters are set to: row scale and scale size by area; (B) Relative expression levels of some TALE genes in stem. The abscissa represents the 11 TALE genes that are specifically highly expressed in the stem, and the ordinate represents the relative expression of genes in the stem.
Previous reports have shown that TALE genes are involved in stem development of plants (Testone et al., 2012; Zhao et al., 2019; Que et al., 2022). In this study, in order to preliminarily explore the effect of TALE genes on P. mume stem development, we selected 11 members highly expressed in the P. mume stem to perform further expression pattern analysis in three different developmental stages of stem. According to the level of expression and the significance of the difference, the expression patterns can be divided into four types (Figure 12). The first type included PmTALE7 and PmTALE19, and there was no significant difference in the expression levels among the three developmental stages. The second type only included PmTALE6, and the expression level in the third stage was significantly lower than that in the first and second stages, but there was no significant difference between the first and second stages. The third type included PmTALE1, PmTALE3, and PmTALE13, and the expression levels in the first stage were significantly higher than that in the second and third stages, but there was no significant difference in the expression levels between the second and third stages. The fourth type included PmTALE9, PmTALE10, PmTALE11, PmTALE14, and PmTALE18, and the expression levels decreased gradually from the first stage to the third stage, and there were significant differences between each stage. Overall, all nine members except PmTALE7 and PmTALE19 were highly expressed in the first stage of stem development. Therefore, it is speculated that the highly expressed TALE genes in stem may be involved in regulating the early development of P. mume stem.
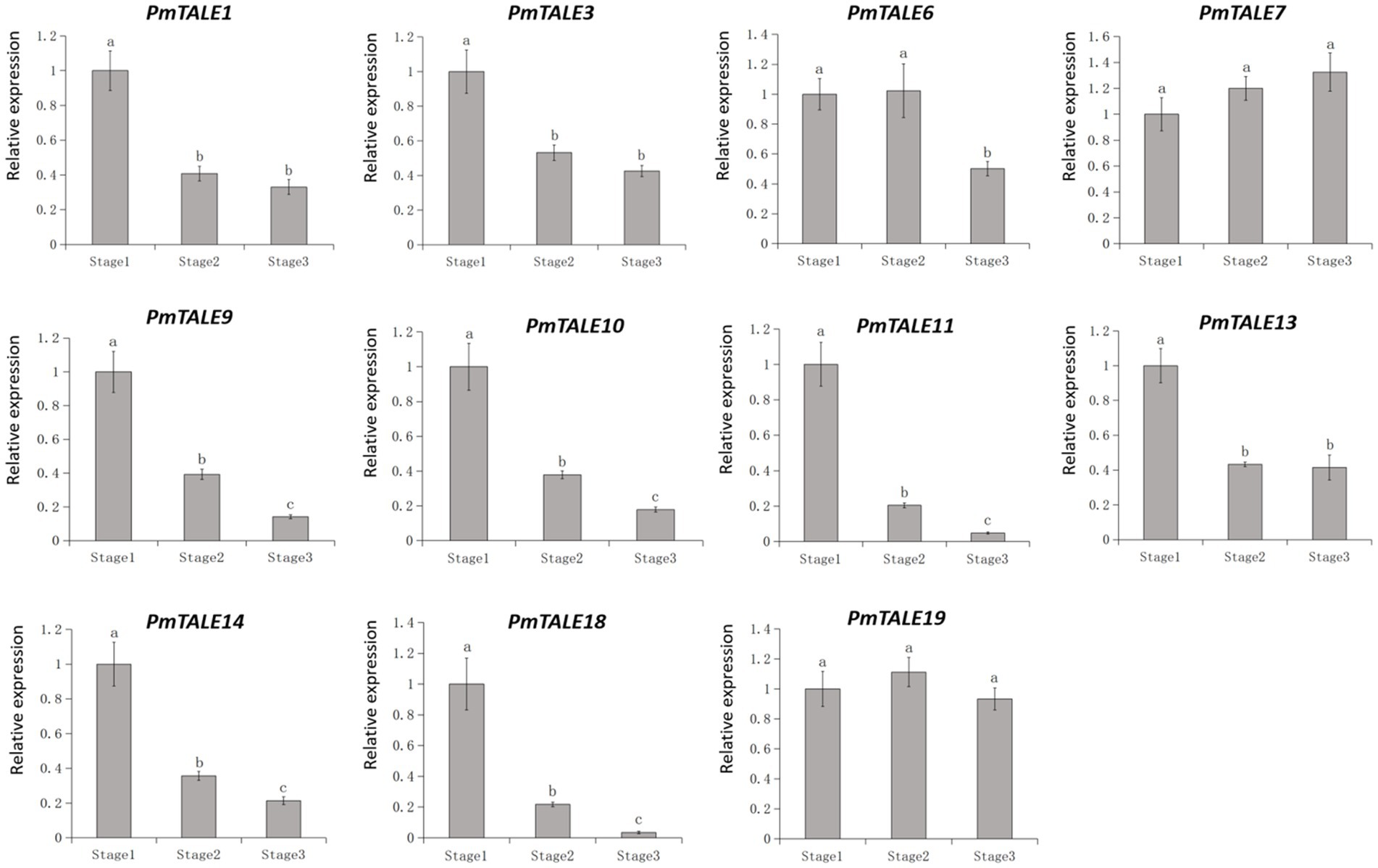
Figure 12. The expression profile of Prunus mume TALE gene family at different developmental stages of the stem. The abscissa represents the three stages of Prunus mume stem development, and the ordinate represents the relative expression level.
Expression profile of Prunus mume TALE gene family members under different hormone treatments and different abiotic stress conditions
Based on the above studies, nine of the 11 highly expressed TALE genes in P. mume stem showed high expression in the early stage of stem development. Among these nine genes, PmTALE1, PmTALE3, PmTALE6, and PmTALE13 had the highest expression levels (Figure 11B). Therefore, we selected these genes for further study to explore their responses against four kinds of hormone treatments and two kinds of abiotic stress treatments during the early development of P. mume stem. 6-BA treatment upregulated the expression of these four TALE genes, PmTALE3 was significantly upregulated at 1 h, PmTALE1 and PmTALE13 were significantly upregulated at 3 h, and PmTALE6 was significantly upregulated at 6 h. NAA treatment downregulated the expression of these four TALE genes, PmTALE1 and PmTALE6 were significantly down-regulated at 1 h, PmTALE3 was significantly downregulated at 3 h, and PmTALE13 was significantly downregulated at 6 h. GA3 treatment also downregulated the expression of these four TALE genes, and all of them were significantly downregulated at 1 h. The expression of PmTALE1, PmTALE3, and PmTALE6 were all upregulated under ABA treatment, while the expression of PmTALE13 was downregulated, and their expression levels at 1 h were significantly different from those at 0 h. NaCl treatment downregulated the expression of these four genes, PmTALE1, PmTALE6, and PmTALE13 were significantly downregulated at 1 h, and PmTALE3 was significantly downregulated at 3 h. After mannitol treatment, the expression of four genes was significantly down-regulated at 1 h. In conclusion, in the early stage of P. mume stem development, four TALE genes responded to all hormone treatments, specifically, 6-BA promoted the expression, NAA and GA3 inhibited the expression, while the effects of ABA on TALE genes were not consistent. In addition, the four TALE genes were all downregulated under salt stress and drought stress, which may be due to the poor resistance of the P. mume young stem to external stress in the early developmental stage or the downregulated expression of these genes may help to improve the resistance of P. mume (Figure 13).
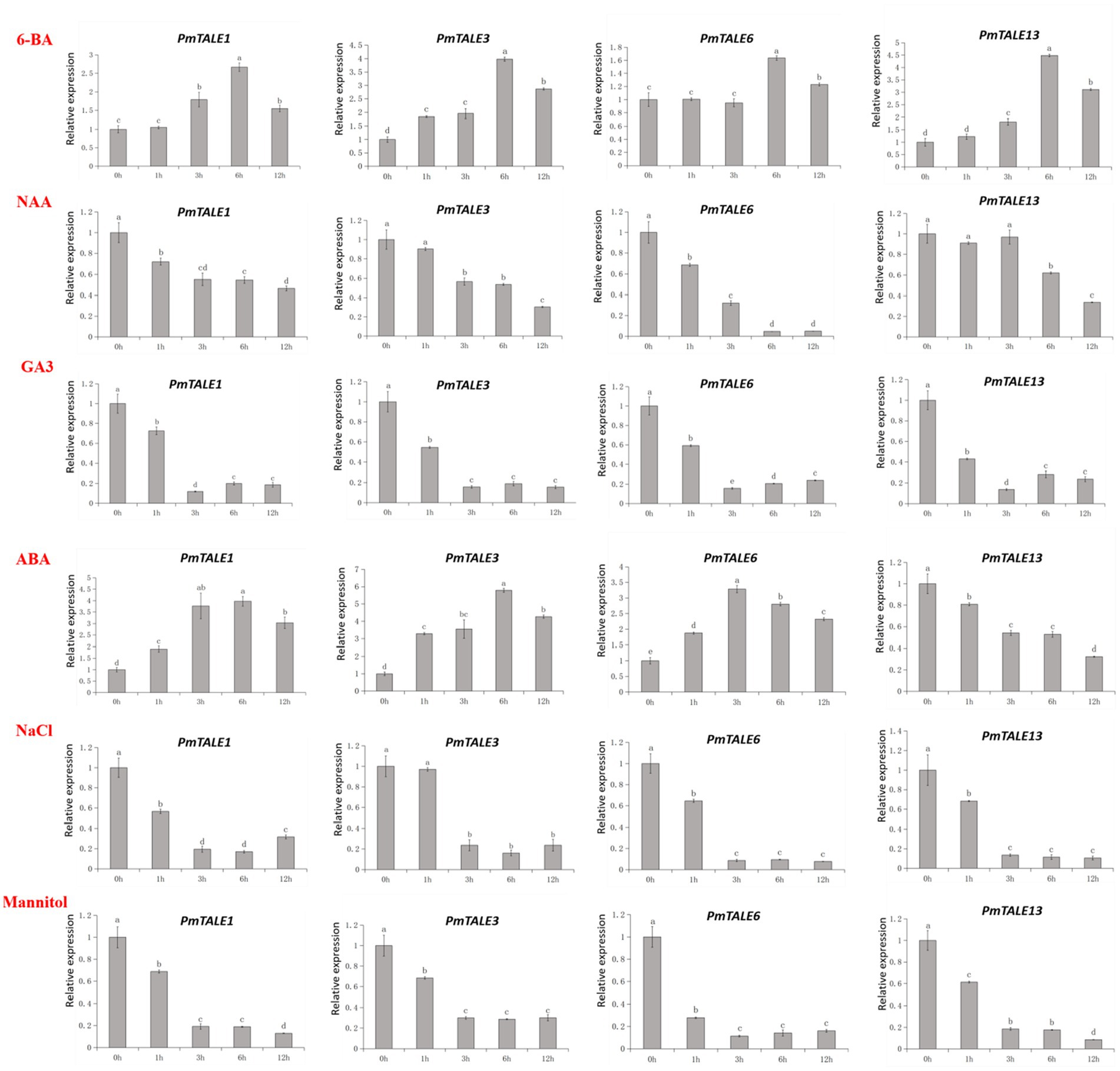
Figure 13. Expression profiling of Prunus mume TALE genes under different hormone treatments and different abiotic stress conditions. The abscissa represents the treatment time of hormone or abiotic stress, and the ordinate represents the relative expression level.
Discussion
As early as 3,000 years ago, P. mume has been domesticated as an ornamental plant in China, and the fruit is edible. In addition, P. mume is also one of the earliest Prunus plants to have their genome sequenced (Zhang et al., 2012). In this study, a total of 23 TALE genes are identified based on the P. mume genome, which is close to the number of TALE genes (22) in A. thaliana and P. armeniaca. However, the number of TALE genes (36) in P. persica, which is also a Prunus plant, is relatively large. There are also differences in the number of TALE genes found by researchers in other species. For example, Punica granatum contains only 17 members (Wang et al., 2020), radish, walnut, and poplar contain 33, 30, and 35 members, respectively (Zhao et al., 2019, 2022; Guo et al., 2022), and G. hirsutum contains as many as 89 members (Razzaq et al., 2020). The phylogenetic tree results showed that, similar to A. thaliana, the TALE proteins of P. mume and two other Prunus species (P. armeniaca and P. persica) could all be divided into two subfamilies, KNOX and TELL subfamilies, respectively, and the number of members of the two subfamilies is not much different. In addition, the KNOX subfamilies of the above three Prunus species all had KNATM branch. It is worth mentioning that this branch is only present in eudicots, for example, Theobroma cacao, Populus trichocarpa, and Gossypium hirsutum all contain KNATM like proteins (Zhang et al., 2021b), while no member of KNATM like proteins has been identified in the KNOX proteins of orchids (Zhang et al., 2022a). The domain analysis of P. mume TALE proteins showed that almost all members contained Homebox_KN domain, all members of BELL subfamily contained POX domain, and most members of KNOX subfamily contained KNOX1, KNOX2 and ELK domain. Previous studies have shown that the Homebox_KN domain is located at the C-terminal of the protein and is related to the function of DNA binding, while ELK domain can be used as a nuclear localization signal and is related to transcriptional inhibition (Kerstetter et al., 1994; Scofield and Murray, 2006).
In order to further analyze the evolutionary relationship within the TALE gene family of P. mume, we conducted an intra-species collinearity analysis and found a total of seven pairs of collinearity, of which four pairs existed in the BELL subfamily, and the other three pairs existed in the KNOX subfamily. The seven collinear pairs included four fragment duplications and two tandem duplication events, leading to the expansion of the TALE gene family in P. mume. In addition, the Ka/Ks values of the collinear pairs were all less than 1, indicating that these gene pairs were all selected by different degrees of purification. Studies have shown that evolution caused by gene duplication events is prevalent in angiosperms. For example, there is obvious expansion phenomenon in woody bamboo TALE gene family, which is caused by many duplication events (Que et al., 2022). Whole-genome duplication events occurred in G. raimondii and G. arboretum (Huang et al., 2020), and there were 34 gene pairs with fragment duplication in the TALE genes of G. hirsutum obtained by crossing the above two species, and also selected by purification (Zhang et al., 2021b). In addition, interspecies collinearity analysis can be used to study the evolution of gene families among species. For example, Que. et al. found that the TALE genes of Moso Bamboo and the TALE genes of rice have a high collinearity relationship (Que et al., 2022). In this study, we used the inter-species collinearity analysis to study the collinearity of TALE genes between P. mume and four other Prunus species. The origin of P. mume, P. armeniaca, and P. persica are mainly distributed in China, and the results show that their collinearity is also closer. At the same time, the phylogenetic tree analysis mentioned above also showed that the overall evolutionary pattern of TALE gene family of P. armeniaca and P. persica is similar to P. mume. While the origin of P. dulcis and P. avium are mainly distributed abroad, and the analysis results show that the collinear relationship between P. mume and these two species of Prunus is relatively far. Therefore, it can be seen that the distance of the collinearity is consistent with the distance of the origin.
The Homebox_KN domain and ELK domain of the TALE gene family are both related to transcriptional regulation, and as a transcription factor, this family plays an important role in regulating plant growth and development. In recent years, there have been many reports on the function of the TALE genes. For example, Wang et al. predicted that the TALE genes of pomegranate may have regulatory effects on SAM, flower, and ovule development (Wang et al., 2020). The expression levels of TALE genes in walnut were different at different stages of flower bud development (Guo et al., 2022). Downregulated expression of cotton TALE gene family member GhSTM3 affects flowering time (Zhang et al., 2021b). During the rapid growth period of Phyllostachys edulis, members of the TALE gene family may affect the formation of secondary cell walls in internodes by regulating the synthesis of xylan (Que et al., 2022). Nearly half of (17) TALE genes in poplar are highly expressed in stem, which may play a key regulatory role in wood formation (Zhao et al., 2019). Similar to the results of poplar, in this study, we also found nearly half of (11) TALE genes that were specifically highly expressed in P. mume stem based on the expression profiles of different tissues, indicating that P. mume TALE genes may have an important regulatory role in stem development. In addition, although the expression levels of these genes were different in different developmental stages of stem, they were mainly highly expressed in the early developmental stage of stem, further indicating that TALE genes may mainly play a role in the early developmental process of stem. Similarly, it has been previously reported that KNOPE1, a member of the peach TALE gene family, is also involved in early stem development and is expressed in cortex and procambium, preventing lignification of primary stem by inhibiting lignin-related genes (Testone et al., 2012). However, considering that some TALE genes of P. mume are not specifically highly expressed in stem, but their expression levels are not low in stem, they may also have important functions in stem development and are worthy of further research in the future. In addition, TALE proteins often form dimers to function, for example, yeast two-hybrid experiments found that different poplar TALE proteins can form heterodimers (Zhao et al., 2019). Similarly, this study also predicted that heterodimers may be formed within the P. mume KNOX and BELL subfamilies and between the two subfamilies. Therefore, the TALE proteins of P. mume may affect the early development of stem through the formation of heterodimers.
The effects of transcription factors on growth and development are usually closely related to hormonal pathways, and previous studies have found that the function of the TALE genes is related to the hormone pathway of plants. For example, the promoter region of the orchid KNOX genes was enriched with MeJA and ABA-responsive cis-elements (Zhang et al., 2022a), the promoter sequence of pomegranate TALE genes contained auxin and gibberellin-responsive cis-elements (Wang et al., 2020), and the expression of the KNOX genes of Caucasian clover could respond to changes in 6-BA, IAA, and KT signals from external application (Zhang et al., 2022b). Infection of A. thaliana leaves by R. fascians can cause local CK responses and reduce GA signaling, which may provide a suitable environment for the expression of KNOX genes, resulting in the formation of leaf edge serration (Depuydt et al., 2008). MdKNOX19, a member of the apple KNOX subfamily, is an ABA-responsive gene and overexpression of MdKNOX19 increases the ABA sensitivity of apple callus (Jia et al., 2021). In this study, the 2000 bp promoter of the P. mume TALE genes was analyzed, and a variety of hormone response elements were also found, including auxin, gibberellin, methyl jasmonate, abscisic acid and salicylic acid. At the same time, the exogenous hormone application experiment was used to explore the response of the top four TALE genes in P. mume stem expression (PmTALE1, PmTALE3, PmTALE6, and PmTALE13) to four kinds of hormones during the early stage of stem development, and it was found that the expression of these four genes could all respond to the changes of external hormones. The 6-BA upregulated their expression, NAA and GA3 downregulated their expression, and ABA had inconsistent effects on them, showing the complexity of hormone action, and further indicating that hormones may affect early development of P. mume stem by regulating the expression of TALE genes.
There is a correlation between hormone response and plant resistance to abiotic stress. For example, ethylene-related gene expression models suggest that ethylene may indirectly participate in the induction of dormancy, thereby enhancing cold/freezing tolerance of P. mume (Li et al., 2021). Populus alba × P. glandulosa KNOX subfamily member PagKNAT2/6b can mediate drought response by down-regulating PagGA20ox1 gene of GA pathway (Song et al., 2021). Similarly, the expression of TALE genes is not only regulated by hormones, but also affected by abiotic stress (Tsuda and Hake, 2015; Niu and Fu, 2022). The promoters of TALE gene family members in soybean contained cis-elements that responded to various stresses, and the expression level of GmTALE gene could change in response to salt stress and drought stress (Wang et al., 2021). Eleven members of poplar TALE genes respond to salt stress (Zhao et al., 2019). A variety of TALE genes in cotton are upregulated under various abiotic stresses, and may play a role in coping with stressful environments (Razzaq et al., 2020). A later study found that the silencing of GhKNOX10 and GhKNOX14 in cotton reduced the tolerance of seedlings to salt stress, while the silencing of GhKNOX2 enhanced the salt tolerance of cotton seedlings (Zhang et al., 2021b). Members of the pear KNOX subfamily can respond to drought stress treatment, specifically, the expression of PbKNOX7/13 is increased under drought stress, while the expression of PbKNOX5/16 is inhibited under drought stress (Liu et al., 2022). In this study, the promoter analysis of TALE genes of P. mume revealed a variety of abiotic stress elements, including low temperature, drought, hypoxia induction, and trauma response. Then, the responses of the top four TALE genes in P. mume stem expression (PmTALE1, PmTALE3, PmTALE6, and PmTALE13) to drought stress and salt stress during the early stage of stem development were investigated. The results showed that these four genes were all downregulated under stress conditions, which may be due to the poor resistance of the P. mume young stem to external stress in the early developmental stage. Alternatively, the downregulated expression of these genes may help to improve the resistance of P. mume, which requires further research in the future.
Conclusion
We identified a total of 23 TALE genes in P. mume. Phylogenetic tree showed that TALE proteins were divided into KONX subfamily and BELL subfamily. The results of protein interaction prediction showed that a variety of heterodimers could be formed between TALE proteins. Intra species collinearity analysis showed that fragment replication and tandem replication events were the main reasons for the expansion of the TALE gene family members and the collinearity analysis between species showed that the collinearity of TALE genes between P. mume and the other four prunus species was consistent with the distance of origin. Eleven members of the P. mume TALE genes were specifically highly expressed in the stem, mainly in the early stage of stem development. Cis element analysis showed that the promoter of P. mume TALE genes contained a variety of hormones and abiotic stress response elements, and TALE genes could respond to hormone or abiotic stress treatments during the early stage of stem development.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author contributions
QY, CY, and QZ conceived and designed the experiments. QY performed the experiments and analyzed the data. TC carried out some of the experiments. JW supervised the conduct of the experiment. QY prepared the original draft. CY and QZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Key R & D Program of China (2019YFD1001500) and Special Fund for Beijing Common Construction Project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1006360/full#supplementary-material
Footnotes
1. ^https://www.ncbi.nlm.nih.gov
2. ^https://services.healthtech.dtu.dk/service.php?TMHMM-2.0
References
Alioto, T., Alexiou, K. G., Bardil, A., Barteri, F., Castanera, R., Cruz, F., et al. (2020). Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 101, 455–472. doi: 10.1111/tpj.14538
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Bateman, A., Martin, M. J., Orchard, S., Magrane, M., Agivetova, R., Ahmad, S., et al. (2021). Uni Prot: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Belles-Boix, E., Hamant, O., Witiak, S. M., Morin, H., Traas, J., and Pautot, V. (2006). KNAT6: an Arabidopsis homeobox gene involved in meristem activity and organ separation. Plant Cell 18, 1900–1907. doi: 10.1105/tpc.106.041988
Bharathan, G., Goliber, T. E., Moore, C., Kessler, S., Pham, T., and Sinha, N. R. (2002). Homologies in leaf form inferred from KNOXI gene expression during development. Science 296, 1858–1860. doi: 10.1126/science.1070343
Bharathan, G., Janssen, B. J., Kellogg, E. A., and Sinha, N. (1997). Did homeodomain proteins duplicate before the origin of angiosperms, fungi, and metazoa? Proc. Natl. Acad. Sci. U. S. A. 94, 13749–13753. doi: 10.1073/pnas.94.25.13749
Billeter, M., Qian, Y. Q., Otting, G., Muller, M., Gehring, W., and Wuthrich, K. (1993). Determination of the nuclear magnetic resonance solution structure of an Antennapedia homeodomain-DNA complex. J. Mol. Biol. 234, 1084–1097. doi: 10.1006/jmbi.1993.1661
Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S., and Brunak, S. (2004). Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649. doi: 10.1002/pmic.200300771
Bolduc, N., and Hake, S. (2009). The maize transcription factor KNOTTED1 directly regulates the gibberellin catabolism gene ga 2ox1. Plant Cell 21, 1647–1658. doi: 10.1105/tpc.109.068221
Bolduc, N., Yilmaz, A., Mejia-Guerra, M. K., Morohashi, K., O’Connor, D., Grotewold, E., et al. (2012). Unraveling the KNOTTED1 regulatory network in maize meristems. Genes Dev. 26, 1685–1690. doi: 10.1101/gad.193433.112
Byrne, M. E., Groover, A. T., Fontana, J. R., and Martienssen, R. A. (2003). Phyllotactic pattern and stem cell fate are determined by the Arabidopsis homeobox gene BELLRINGER. Development 130, 3941–3950. doi: 10.1242/dev.00620
Cao, X. W., Wang, J., Xiong, Y. Y., Yang, H. B., Yang, M. L., Ye, P., et al. (2020). A self-activation loop maintains meristematic cell fate for branching. Curr. Biol. 30:1893. doi: 10.1016/j.cub.2020.03.031
Chan, R. L., Gago, G. M., Palena, C. M., and Gonzalez, D. H. (1998). Homeoboxes in plant development. Biochim. Biophys. Acta 1442, 1–19. doi: 10.1016/S0167-4781(98)00119-5
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, X., Li, M. L., Abdullah, M., Li, G. H., Zhang, J. Y., Manzoor, M. A., et al. (2019). In Silico genome-wide analysis of the pear (Pyrus bretschneideri) KNOX family and the functional characterization of PbKNOX1, an Arabidopsis BREVIPEDICELLUS Orthologue gene, involved in Cell Wall and lignin biosynthesis. Front. Genet. 10:632. doi: 10.3389/fgene.2019.00632
Depuydt, S., Dolezal, K., Van Lijsebettens, M., Moritz, T., Holsters, M., and Vereecke, D. (2008). Modulation of the hormone setting by Rhodococcus fascians results in ectopic KNOX activation in Arabidopsis. Plant Physiol. 146, 1267–1281. doi: 10.1104/pp.107.113969
Di Giacomo, E., Laffont, C., Sciarra, F., Iannelli, M. A., Frugier, F., and Frugis, G. (2017). KNAT3/4/5-like class 2 KNOX transcription factors are involved in Medicago truncatula symbiotic nodule organ development. New Phytol. 213, 822–837. doi: 10.1111/nph.14146
Ezhova, T. A. (2003). Genetic control of totipotency of plant cells in vitro. Ontogenez 34, 245–252. doi: 10.1023/A:1024940130511
Ezura, K., Nakamura, A., and Mitsuda, N. (2022). Genome-wide characterization of the TALE homeodomain family and the KNOX-BLH interaction network in tomato. Plant Mol. Biol. 109, 799–821. doi: 10.1007/s11103-022-01277-6
Gao, J., Yang, X., Zhao, W., Lang, T. G., and Samuelsson, T. (2015). Evolution, diversification, and expression of KNOX proteins in plants. Front. Plant Sci. 6:882. doi: 10.3389/fpls.2015.00882
Geourjon, C., and Deleage, G. (1995). SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684. doi: 10.1093/bioinformatics/11.6.681
Guo, C. H., Quan, S. W., Zhang, Z. R., Kang, C., Liu, J. M., and Niu, J. X. (2022). Genome-wide identification, characterization and expression profile of TALE gene family in (Juglans regia L.). Sci. Hortic. 297:110945. doi: 10.1016/j.scienta.2022.110945
Hake, S., Smith, H. M. S., Holtan, H., Magnani, E., Mele, G., and Ramirez, J. (2004). The role of Knox genes in plant development. Annu. Rev. Cell Dev. Biol. 20, 125–151. doi: 10.1146/annurev.cellbio.20.031803.093824
Hamant, O., and Pautot, V. (2010). Plant development: A TALE story. C. R. Biol. 333, 371–381. doi: 10.1016/j.crvi.2010.01.015
Han, Y. X., Zhang, L. L., Yan, L. Y., Xiong, X. X., Wang, W. J., Zhang, X. H., et al. (2022). Genome-wide analysis of TALE superfamily in Triticum aestivum reveals TaKNOX11-a is involved in abiotic stress response. BMC Genomics 23:89. doi: 10.1186/s12864-022-08324-y
Hao, S. H., Wang, Y. R., Yan, Y. X., Liu, Y. H., Wang, J. Y., and Chen, S. (2021). A review on plant responses to salt stress and their mechanisms of salt resistance. Horticulturae 7:665439. doi: 10.3390/horticulturae7060132
Hay, A., and Tsiantis, M. (2010). KNOX genes: versatile regulators of plant development and diversity. Development 137, 3153–3165. doi: 10.1242/dev.030049
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Hu, B., Jin, J. P., Guo, A. Y., Zhang, H., Luo, J. C., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Huang, G., Wu, Z. G., Percy, R. G., Bai, M. Z., Li, Y., Frelichowski, J. E., et al. (2020). Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 52:516. doi: 10.1038/s41588-020-0607-4
Jasinski, S., Piazza, P., Craft, J., Hay, A., Woolley, L., Rieu, I., et al. (2005). KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 15, 1560–1565. doi: 10.1016/j.cub.2005.07.023
Jia, P., Xing, L., Zhang, C., Zhang, D., Ma, J., Zhao, C., et al. (2021). MdKNOX19, a class II knotted-like transcription factor of apple, plays roles in ABA signalling/sensitivity by targeting ABI5 during organ development. Plant Sci. 302:110701. doi: 10.1016/j.plantsci.2020.110701
Jin, J. P., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J. C., et al. (2017). Plant TFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Johnson, L. S., Eddy, S. R., and Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11:431. doi: 10.1186/1471-2105-11-431
Kerstetter, R., Vollbrecht, E., Lowe, B., Veit, B., Yamaguchi, J., and Hake, S. (1994). Sequence analysis and expression patterns divide the maize knotted 1-like homeobox genes into two classes. Plant Cell 6, 1877–1887. doi: 10.1105/tpc.6.12.1877
Kim, D., Cho, Y. H., Ryu, H., Kim, Y., Kim, T. H., and Hwang, I. (2013). BLH1 and KNAT3 modulate ABA responses during germination and early seedling development in Arabidopsis. Plant J. 75, 755–766. doi: 10.1111/tpj.12236
Kumar, R., Kushalappa, K., Godt, D., Pidkowich, M. S., Pastorelli, S., Hepworth, S. R., et al. (2007). The Arabidopsis BEL1-LIKE HOMEODOMAIN proteins SAW1 and SAW2 act redundantly to regulate KNOX expression spatially in leaf margins. Plant Cell 19, 2719–2735. doi: 10.1105/tpc.106.048769
Li, E. Y., Bhargava, A., Qiang, W. Y., Friedmann, M. C., Forneris, N., Savidge, R. A., et al. (2012). The class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 194, 102–115. doi: 10.1111/j.1469-8137.2011.04016.x
Li, P., Zheng, T. C., Zhuo, X. K., Zhang, M., Yong, X., Li, L. L., et al. (2021). Photoperiod- and temperature-mediated control of the ethylene response and winter dormancy induction in Prunus mume. Hortic. Plant J. 7, 232–242. doi: 10.1016/j.hpj.2021.03.005
Liu, Y. Y., You, S. J., Taylor-Teeples, M., Li, W. L., Schuetz, M., Brady, S. M., et al. (2014). BEL1-LIKE HOMEODOMAIN6 and KNOTTED ARABIDOPSIS THALIANA7 interact and regulate secondary cell wall formation via repression of REVOLUTA. Plant Cell 26, 4843–4861. doi: 10.1105/tpc.114.128322
Liu, J., Zhang, C., Han, J., Fang, X., Xu, H., Liang, C., et al. (2022). Genome-wide analysis of KNOX transcription factors and expression pattern of dwarf-related KNOX genes in pear. Front. Plant Sci. 13:806765. doi: 10.3389/fpls.2022.806765
Ma, Q., Wang, N. H., Hao, P. B., Sun, H. R., Wang, C. C., Ma, L., et al. (2019). Genome-wide identification and characterization of TALE superfamily genes in cotton reveals their functions in regulating secondary cell wall biosynthesis. BMC Plant Biol. 19:432. doi: 10.1186/s12870-019-2026-1
Magnani, E., and Hake, S. (2008). KNOX lost the OX: the Arabidopsis KNATM gene defines a novel class of KNOX transcriptional regulators missing the homeodomain. Plant Cell 20, 875–887. doi: 10.1105/tpc.108.058495
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Mukherjee, K., Brocchieri, L., and Burglin, T. R. (2009). A comprehensive classification and evolutionary analysis of plant Homeobox genes. Mol. Biol. Evol. 26, 2775–2794. doi: 10.1093/molbev/msp201
Niu, X. L., and Fu, D. Q. (2022). The roles of BLH transcription factors in plant development and environmental response. Int. J. Mol. Sci. 23:3731. doi: 10.3390/ijms23073731
Pinosio, S., Marroni, F., Zuccolo, A., Vitulo, N., Mariette, S., Sonnante, G., et al. (2020). A draft genome of sweet cherry (Prunus avium L.) reveals genome-wide and local effects of domestication. Plant J. 103, 1420–1432. doi: 10.1111/tpj.14809
Que, F., Liu, Q. N., Zha, R. F., Xiong, A. S., and Wei, Q. (2022). Genome-wide identification, expansion, and evolution analysis of homeobox gene family reveals TALE genes important for secondary cell wall biosynthesis in Moso bamboo (Phyllostachys edulis). Int. J. Mol. Sci. 23:4112. doi: 10.3390/ijms23084112
Ragni, L., Belles-Boix, E., Gunl, M., and Pautot, V. (2008). Interaction of KNAT6 and KNAT2 with BREVIPEDICELLUS and PENNYWISE in Arabidopsis inflorescences. Plant Cell 20, 888–900. doi: 10.1105/tpc.108.058230
Razzaq, A., Ashraf, J., Malik, W., Shaban, M., Zhang, R., Liang, C. Z., et al. (2020). In Silico analyses of TALE transcription factors revealed its potential role for organ development and abiotic stress tolerance in cotton. Int. J. Agric. Biol. 23, 1083–1094. doi: 10.17957/IJAB/15.1389
Rombauts, S., Dehais, P., Van Montagu, M., and Rouze, P. (1999). Plant CARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 27, 295–296. doi: 10.1093/nar/27.1.295
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C-T method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Scofield, S., and Murray, J. A. H. (2006). KNOX gene function in plant stem cell niches. Plant Mol. Biol. 60, 929–946. doi: 10.1007/s11103-005-4478-y
Shani, E., Burko, Y., Ben-Yaakov, L., Berger, Y., Amsellem, Z., Goldshmidt, A., et al. (2009). Stage-specific regulation of Solanum lycopersicum leaf maturation by class 1 KNOTTED1-LIKE HOMEOBOX proteins. Plant Cell 21, 3078–3092. doi: 10.1105/tpc.109.068148
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shi, B. H., Zhang, C., Tian, C. H., Wang, J., Wang, Q., Xu, T. F., et al. (2016). Two-step regulation of a Meristematic cell population acting in shoot branching in Arabidopsis. PLoS Genet. 12:e1006168. doi: 10.1371/journal.pgen.1006168
Shu, Y. J., Tao, Y., Wang, S., Huang, L. Y., Yu, X. W., Wang, Z. K., et al. (2015). GmSBH1, a homeobox transcription factor gene, relates to growth and development and involves in response to high temperature and humidity stress in soybean. Plant Cell Rep. 34, 1927–1937. doi: 10.1007/s00299-015-1840-7
Smith, H. M. S., and Hake, S. (2003). The interaction of two homeobox genes, BREVIPEDICELLUS and PENNYWISE, regulates internode patterning in the Arabidopsis inflorescence. Plant Cell 15, 1717–1727. doi: 10.1105/tpc.012856
Song, X., Zhao, Y., Wang, J., and Lu, M.-Z. (2021). The transcription factor KNAT2/6b mediates changes in plant architecture in response to drought via down-regulating GA20ox1 in Populus alba × P. glandulosa. J. Exp. Bot. 72, 5625–5637. doi: 10.1093/jxb/erab201
Srinivasan, C., Liu, Z. R., and Scorza, R. (2011). Ectopic expression of class 1 KNOX genes induce adventitious shoot regeneration and alter growth and development of tobacco (Nicotiana tabacum L) and European plum (Prunus domestica L). Plant Cell Rep. 30, 655–664. doi: 10.1007/s00299-010-0993-7
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets (vol 49, pg D605, 2021). Nucleic Acids Res. 49:10800. doi: 10.1093/nar/gkab835
Tao, Y., Chen, M., Shu, Y. J., Zhu, Y. J., Wang, S., Huang, L. Y., et al. (2018). Identification and functional characterization of a novel BEL1-LIKE homeobox transcription factor GmBLH4 in soybean. Plant Cell Tissue Organ Cult. 134, 331–344. doi: 10.1007/s11240-018-1419-4
Testone, G., Bruno, L., Condello, E., Chiappetta, A., Bruno, A., Mele, G., et al. (2008). Peach [Prunus persica (L.) Batsch] KNOPE1, a class 1 KNOX orthologue to Arabidopsis BREVIPEDICELLUS/KNAT1, is misexpressed during hyperplasia of leaf curl disease. J. Exp. Bot. 59, 389–402. doi: 10.1093/jxb/erm317
Testone, G., Condello, E., Di Giacomo, E., Nicolodi, C., Caboni, E., Rasori, A., et al. (2015). The KNOTTED-like genes of peach (Prunus persica L. Batsch) are differentially expressed during drupe growth and the class 1 KNOPE1 contributes to mesocarp development. Plant Sci. 237, 69–79. doi: 10.1016/j.plantsci.2015.05.005
Testone, G., Condello, E., Verde, I., Caboni, E., Iannelli, M. A., Bruno, L., et al. (2009). The peach (Prunus persica [L.] Batsch) homeobox gene KNOPE3, which encodes a class 2 knotted-like transcription factor, is regulated during leaf development and triggered by sugars. Mol. Gen. Genomics. 282, 47–64. doi: 10.1007/s00438-009-0445-7
Testone, G., Condello, E., Verde, I., Nicolodi, C., Caboni, E., Dettori, M. T., et al. (2012). The peach (Prunus persica L. Batsch) genome harbours 10 KNOX genes, which are differentially expressed in stem development, and the class 1 KNOPE1 regulates elongation and lignification during primary growth. J. Exp. Bot. 63, 5417–5435. doi: 10.1093/jxb/ers194
Truernit, E., and Haseloff, J. (2007). A role for KNAT class II genes in root development. Plant Signal. Behav. 2, 10–12. doi: 10.4161/psb.2.1.3604
Tsuda, K., and Hake, S. (2015). Diverse functions of KNOX transcription factors in the diploid body plan of plants. Curr. Opin. Plant Biol. 27, 91–96. doi: 10.1016/j.pbi.2015.06.015
Tsuda, K., Kurata, N., Ohyanagi, H., and Hake, S. (2014). Genome-wide study of KNOX regulatory network reveals Brassinosteroid catabolic genes important for shoot meristem function in Rice. Plant Cell 26, 3488–3500. doi: 10.1105/tpc.114.129122
Vollbrecht, E., Veit, B., Sinha, N., and Hake, S. (1991). The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 350, 241–243. doi: 10.1038/350241a0
Wang, Q., Kohlen, W., Rossmann, S., Vernoux, T., and Theres, K. (2015). Auxin depletion from the leaf axil conditions competence for axillary meristem formation in Arabidopsis and tomato (vol 26, pg 2068, 2014). Plant Cell 27:946. doi: 10.1105/tpc.15.00205
Wang, Y. P., Li, J. P., and Paterson, A. H. (2013). MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 29, 1458–1460. doi: 10.1093/bioinformatics/btt150
Wang, N. H., Ma, Q., Ma, J. J., Pei, W. F., Liu, G. Y., Cui, Y. P., et al. (2019a). A comparative genome-wide analysis of the R2R3-MYB gene family among four Gossypium species and their sequence variation and association with fiber quality traits in an Interspecific G. hirsutum x G. barbadense population. Front. Genet. 10:741. doi: 10.3389/fgene.2019.00741
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, L., Yang, X. Y., Gao, Y. Q., and Yang, S. P. (2021). Genome-wide identification and characterization of TALE superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 22:4117. doi: 10.3390/ijms22084117
Wang, S. G., Yang, H. L., Mei, J. S., Liu, X. L., Wen, Z., Zhang, L. J., et al. (2019b). Rice Homeobox protein KNAT7 integrates the pathways regulating cell expansion and wall stiffness. Plant Physiol. 181, 669–682. doi: 10.1104/pp.19.00639
Wang, Y. Y., Zhao, Y. J., Yan, M., Zhao, H. L., Zhang, X. H., and Yuan, Z. H. (2020). Genome-wide identification and expression analysis of TALE gene family in pomegranate (Punica granatum L.). Agronomy 10:829. doi: 10.3390/agronomy10060829
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J.-C., Williams, K. L., Appel, R. D., et al. (1999). “Protein identification and analysis tools in the ExPASy server” in BT—2-D Proteome Analysis Protocols. ed. A. J. Link (Totowa, NJ: Humana Press), 531–552.
Xu, Z. D., Sun, L. D., Zhou, Y. Z., Yang, W. R., Cheng, T. R., Wang, J., et al. (2015). Identification and expression analysis of the SQUAMOSA promoter-binding protein (SBP)-box gene family in Prunus mume. Mol. Gen. Genomics. 290, 1701–1715. doi: 10.1007/s00438-015-1029-3
Yan, F., Deng, W., Pang, X., Gao, Y., Chan, H., Zhang, Q., et al. (2019). Overexpression of the KNOX gene Tkn 4 affects pollen development and confers sensitivity to gibberellin and auxin in tomato. Plant Sci. 281, 61–71. doi: 10.1016/j.plantsci.2018.12.024
Yanai, O., Shani, E., Dolezal, K., Tarkowski, P., Sablowski, R., Sandberg, G., et al. (2005). Arabidopsis KNOXI proteins activate cytokinin biosynthesis. Curr. Biol. 15, 1566–1571. doi: 10.1016/j.cub.2005.07.060
Zhang, Q. X., Chen, W. B., Sun, L. D., Zhao, F. Y., Huang, B. Q., Yang, W. R., et al. (2012). The genome of Prunus mume. Nat. Commun. 3:1318. doi: 10.1038/ncomms2290
Zhang, J. B. (2021). Study on the Roles of Drought Stress-Responsive Genes GhBLH5-A05 and GhHDT4D in Cotton Response to Drought Stress. Central China Normal University. Available at: https://kns.cnki.net/KCMS/detail/detail.aspx?dbname=CDFDLAST2022&filename=1021158716.nh
Zhang, X., Jiang, J., Yang, Y., Ma, Z., Meng, L., Cui, G., et al. (2022b). Identification and responding to exogenous hormone of HB-KNOX family based on transcriptome data of Caucasian clover. Gene 828:146469. doi: 10.1016/j.gene.2022.146469
Zhang, D., Lan, S., Yin, W.-L., and Liu, Z.-J. (2022a). Genome-wide identification and expression pattern analysis of KNOX gene family in Orchidaceae. Front. Plant Sci. 13:901089. doi: 10.3389/fpls.2022.901089
Zhang, Q. P., Zhang, D. Y., Yu, K., Ji, J. J., Liu, N., Zhang, Y. P., et al. (2021a). Frequent germplasm exchanges drive the high genetic diversity of Chinese-cultivated common apricot germplasm. Hortic. Res. 8:215. doi: 10.1038/s41438-021-00650-8
Zhang, X. H., Zhao, J. J., Wu, X. Y., Hu, G. H., Fan, S. L., and Ma, Q. F. (2021b). Evolutionary relationships and divergence of KNOTTED1-like family genes involved in salt tolerance and development in cotton (Gossypium hirsutum L.). Front. Plant Sci. 12:774161. doi: 10.3389/fpls.2021.774161
Zhao, M., Li, C., Ma, X., Xia, R., Chen, J., Liu, X., et al. (2020). KNOX protein KNAT1 regulates fruitlet abscission in litchi by repressing ethylene biosynthetic genes. J. Exp. Bot. 71, 4069–4082. doi: 10.1093/jxb/eraa162
Zhao, W., Li, X., Wang, H., Jia, H., Song, J., Yang, W., et al. (2022). Identification and analysis of the TALE transcription factor family in radish. Sheng Wu Gong Cheng Xue Bao 38, 343–358. doi: 10.13345/j.cjb.210321
Zhao, K., Zhang, X. M., Cheng, Z. H., Yao, W. J., Li, R. H., Jiang, T. B., et al. (2019). Comprehensive analysis of the three-amino-acid-loop-extension gene family and its tissue-differential expression in response to salt stress in poplar. Plant Physiol. Biochem. 136, 1–12. doi: 10.1016/j.plaphy.2019.01.003
Keywords: Prunus mume, TALE transcription factors, stem development, abiotic stress, hormone response
Citation: Yang Q, Yuan C, Cong T, Wang J and Zhang Q (2022) Genome-wide identification of three-amino-acid-loop-extension gene family and their expression profile under hormone and abiotic stress treatments during stem development of Prunus mume. Front. Plant Sci. 13:1006360. doi: 10.3389/fpls.2022.1006360
Edited by:
Penghui Li, Anhui Agricultural University, ChinaReviewed by:
Wenjing Yao, Nanjing Forestry University, ChinaShengji Wang, Shanxi Agricultural University, China
Su Chen, Northeast Forestry University, China
Copyright © 2022 Yang, Yuan, Cong, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cunquan Yuan, eXVhbmN1bnF1YW5AMTYzLmNvbQ==; Qixiang Zhang, enF4YmpmdUAxMjYuY29t