
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 23 September 2022
Sec. Functional and Applied Plant Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1001583
This article is part of the Research TopicResolving the Complexity of Plant Genomes and Transcriptomes with Long Reads, Volume IIView all 7 articles
 Biao Han1†
Biao Han1† Longxin Wang2†
Longxin Wang2† Yang Xian1
Yang Xian1 Xiao-Man Xie1Wen-Qing Li1
Xiao-Man Xie1Wen-Qing Li1 Ye Zhao3Ren-Gang Zhang4
Ye Zhao3Ren-Gang Zhang4 Xiaochun Qin2
Xiaochun Qin2 De-Zhu Li5*
De-Zhu Li5* Kai-Hua Jia6*
Kai-Hua Jia6*Quercus variabilis (Fagaceae) is an ecologically and economically important deciduous broadleaved tree species native to and widespread in East Asia. It is a valuable woody species and an indicator of local forest health, and occupies a dominant position in forest ecosystems in East Asia. However, genomic resources from Q. variabilis are still lacking. Here, we present a high-quality Q. variabilis genome generated by PacBio HiFi and Hi-C sequencing. The assembled genome size is 787 Mb, with a contig N50 of 26.04 Mb and scaffold N50 of 64.86 Mb, comprising 12 pseudo-chromosomes. The repetitive sequences constitute 67.6% of the genome, of which the majority are long terminal repeats, accounting for 46.62% of the genome. We used ab initio, RNA sequence-based and homology-based predictions to identify protein-coding genes. A total of 32,466 protein-coding genes were identified, of which 95.11% could be functionally annotated. Evolutionary analysis showed that Q. variabilis was more closely related to Q. suber than to Q. lobata or Q. robur. We found no evidence for species-specific whole genome duplications in Quercus after the species had diverged. This study provides the first genome assembly and the first gene annotation data for Q. variabilis. These resources will inform the design of further breeding strategies, and will be valuable in the study of genome editing and comparative genomics in oak species.
Quercus L. (oak) is an ecologically and economically important genus of deciduous and evergreen forest ecosystems throughout the Northern Hemisphere. The genus comprises approximately 450 species (Cavender-Bares, 2016, 2019; Plomion et al., 2016), and Quercus species not only play pivotal roles in ecosystem functioning (e.g., biodiversity maintenance, water and soil conservation and carbon sequestration), but also provide raw materials for timber, starch, tannin, cork and medicinal resources. Due to the economic value of these trees, their presence in many common habitats, and their dominant positions in many ecosystems and landscapes across the Northern Hemisphere (Chai et al., 2016), Quercus species have been the focus of many genetic, ecological and evolutionary studies (Eaton et al., 2015; Gugger et al., 2021; Fu et al., 2022). However, classification of oak trees is challenging, because of the large inter-and intraspecific morphological variation, and because of the conflicting phylogenies derived from analysis of plastid and low-copy nuclear markers (Manos et al., 1999; Simeone et al., 2013, 2016; Hubert et al., 2014; Vitelli et al., 2017; Zhang et al., 2020). With the accumulation of molecular and morphological evidence, eight Quercus sections, corresponding to clades, have been accepted: the Old World sections Cyclobalanopsis, Cerris and Ilex, and the New World sections Quercus, Lobatae, Virentes, Protobalanus and Ponticae (Gil-Pelegrín et al., 2017; Hipp et al., 2020).
The Chinese cork oak, Quercus variabilis (Q. variabilis) belongs to the East Asian Cerris lineage in subgenus Cerris (Hipp et al., 2020). It is an important tree species in warm-temperate deciduous broadleaved woodland, and it is native to and widespread in East Asia, including China, the Korean Peninsula, Japan, Laos and Thailand (Fujiwara and Harada, 2015). Q. variabilis is characterized by its thick corky bark, which is peeled to make the corks used as bottle stoppers in the wine industry (Pereira, 2011), and Q. variabilis is also a valuable timber species. Furthermore, Q. variabilis, together two other East Asian oak species (Q. acutissima and Q. chenii), is proposed as an indicator species for local forest health, due to its importance in the local ecology (Zilliox and Gosselin, 2014; Chen et al., 2020b; Asbeck et al., 2021).
Previous studies investigating Q. variabilis have mainly focused on its morphological characteristics (Du et al., 2021; Sun et al., 2021), its responses and adaptations to climate change (Gao et al., 2020; Xia et al., 2022), or its adaptive evolution and introgression, as assessed using whole genome resequencing (Fu et al., 2022). However, to date, no nuclear genomic resources are available for Q. variabilis. Here, we present the first chromosome-scale high-quality genome assembly of Q. variabilis, generated using a combination of Pacific Biosciences high-fidelity (PacBio HiFi) and Hi-C technologies. We performed structural gene annotation, identified repetitive sequences, and also conducted a comparative genomics study with the genomes of a further 13 plants. This study will provide important resources for the further investigation of genetic diversity in Q. variabilis and will improve the resolution of the oak phylogeny.
Quercus variabilis samples were collected from an ancient tree (more than 400 years old) growing in Culai Mountain National Forest Park, Shandong Province, China.
Fresh leaves were collected and immediately frozen in liquid nitrogen for transport back to the lab. The genomic DNA was then isolated using a Plant DNeasy Mini kit (Qiagen China, Shanghai, China) according to the manufacturer’s instructions. The quality and quantity of the DNA were determined using agarose gel electrophoresis and with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). A library of short-insert-size genomic DNA fragments of length 300–400 bp was constructed according to the manufacturer’s instructions, and was sequenced on a DNBSEQ platform (Beijing Genomics Institute, Shenzhen, China) for 150 bp pair end sequencing. For long-read sequencing, a 20 kb high-fidelity (HiFi) library was constructed following the manufacturer’s protocol1 on the PacBio Sequel II platform (Pacific Biosciences of California, Inc.). To increase continuity of the genome, a Hi-C library was constructed and sequenced on the DNBSEQ platform (BGI, Shenzhen, China).
For RNA sequencing, fresh leaves, young twigs, fruits and seeds were sampled and immediately frozen in liquid nitrogen. RNA was extracted using TRIzol reagent (Invitrogen), the genomic DNA was eliminated using DNase and the samples were then mixed for RNA sequencing. We used agarose gel electrophoresis, a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and an Agilent Bioanalyzer 2,100 (Agilent Technologies, Santa Clara, CA, United States) to evaluate the quality of the RNA. High-quality RNA was then used to build a cDNA library following the manufacturer’s instructions. Paired-end sequencing was performed on the DNBSEQ platform (BG I, Shenzhen, China), generating 150-bp paired-end reads.
SOAPnuke V1.6.5 (Chen et al., 2018b) was employed to filter out PCR duplications, low-quality reads (≥ 10% of nucleotides with a quality score ≤ 20 or the proportion of N is greater than 1%) and adapter sequences, with the following parameters: -n 0.01-l 20-q 0.1-i -Q 2-G -M 2-A 0.5 –d. Next, the software Jellyfish v2.1.4 (Marçais and Kingsford, 2011) was used to count k-mers of length 17–31. GenomeScope 2.0 (Ranallo-Benavidez et al., 2020) was then applied for the estimation of genome size and other features such as heterozygosity and repetition rate. Smudgeplot (Ranallo-Benavidez et al., 2020) was used for the estimation of ploidy.
The PacBio SMRT-Analysis2 was used as quality control to eliminate adaptors and low-quality short reads, producing a total of 51.7 G bases (~ 72 × coverage) of PacBio HiFi data. The initial assemblies were then performed in HiFi-asm v0.15.2 (Cheng et al., 2021). To acquire high-quality, chromosome-level assemblies, Hi-C reads were compared to the contigs assembled above using Juicer (Durand et al., 2016b). Unique mapped reads with map-quality scores > 40 were subsequently used for Hi-C association chromosome assembly using the 3D-DNA pipeline (Dudchenko et al., 2017). Scaffolds were then manually checked and refined with Juicebox (Durand et al., 2016a) and visualized in Hicplotter (Akdemir and Chin, 2015). A BUSCO analysis was conducted to determine gene/genome completeness using BUSCO v4 (Simão et al., 2015) together with the embryophyta_odb10 database with 1,614 plant single-copy orthologues. BWA (Li, 2013) was used to map short reads of DNBSEQ data against the assembly. SAMtools (Danecek et al., 2021) was then employed to create a pile-up file summary of the aligned reads, and the results were imported to BCFtools (Danecek et al., 2021) for SNP and INDEL calling. The heterozygosity was then calculated as the proportion of the heterozygous sites to the total sites.
Several different methods were employed to annotate the repetitive sequences. First, Tandem Repeats Finder v4.09 (Price et al., 2005) was used for the identification of tandem repeats. Then, RepeatProteinMask v4.07 and RepeatMasker v4.07 (Chen, 2004) were used with their default parameters against RepBase v21.12 (Bao et al., 2015) to identify known repeats in a homology-based approach. Thirdly, RepeatMasker (Bedell et al., 2000) identified repeat elements with a de novo library, built in RepeatModeler (Abrusán et al., 2009) and LTR_FINDER v1.06 (Zhao and Hao, 2007).
To annotate the protein-coding genes, we combined RNA-based, homology-based and de novo methods. For the RNA-based method, we generated 63.27 million raw reads (9.49 Gb) with DNBSEQ sequencing (Supplementary Table S1). After quality control and filtering by fastp (Chen et al., 2018a), 9.48 Gb clean data were retained and aligned to scaffolds using hisat2 v2.2.1 (Kim et al., 2019). Reference genome-guided transcriptome assemblies were then constructed with StringTie v2.2.0 (Pertea et al., 2015). For homology-based predictions, the Q. variabilis genome was aligned against the Arabidopsis thaliana, Q. lobata, Q. robur and Q. suber genomes using TBLASTN v2.2.26 (Mount, 2007) with an E-value cutoff of 1e–5. Finally, GeneWise v2.4.1 (Birney et al., 2004) was employed for structural inspection of these alignments. For ab initio gene prediction, MAKER v3.01.03 (Holt and Yandell, 2011) was used to compute annotation edit distance (AED) for each protein-coding gene, based on transcript assembly from the transcriptome data, as well as from homologous annotations of the four genomes. Augustus v3.4.0 (Stanke et al., 2008; Keller et al., 2011) and SNAP (Johnson et al., 2008) were then employed for ab initio gene prediction using model training, based on coding sequences of 1,200 genes with structural integrity selected based on AED. Finally, the predictions obtained using these methods were combined using EVM v1.1.1 (Haas et al., 2008). The predicted genes were functionally annotated using seven public biological databases: NR, TrEMBL (Boeckmann et al., 2003), SwissProt (Boeckmann et al., 2003), KEGG (Kanehisa and Goto, 2000), InterPro (Zdobnov and Apweiler, 2001), KOG (Koonin et al., 2004), and GO (Consortium, 2004). Blast v2.2.26 was used for homolog searches with an E-value cutoff of 1e-5, and InterproScan v5.55 (Jones et al., 2014) was used for protein function prediction based on the conserved protein domains.
Homology-based non-coding RNA (ncRNA) was identified using Infernal v1.14 (Nawrocki and Eddy, 2013) by mapping plant small nuclear RNA (snRNA) and microRNA (miRNA) genes from the Rfam database (Kalvari et al., 2018). Transfer RNAs (tRNAs) were detected with tRNAscan-SE v1.3.1 (Lowe and Chan, 2016). BLASTN was used for the identification of ribosomal RNAs (rRNAs) by alignment with known plant rRNA sequences (Vitales et al., 2017).
OrthoFinder v2.5.41 (Emms and Kelly, 2019) was used to identify homologous gene families among the assembled genomes of Q. variabilis and 13 further representative flowering plant species (Amborella trichopoda, Arabidopsis thaliana, Castanea crenata, Castanena mollissima, Eucalyptus grandis, Juglans regia, Oryza sativa, Prunus persica, Q. lobata, Q. robur, Q. suber, Vitis vinifera and Xanthoceras sorbifolia). GO enrichment analysis was conducted using ClusterProfiler with an adjusted p value cutoff of 0.05 (Wu et al., 2021). We performed collinearity analysis of homologous gene pairs between Q. lobata, Q. robur, and Q. variabilis using MCScanX (Wang et al., 2012).
For phylogenetic analysis and estimation of species divergence time, MUSCLE (Edgar, 2004) was applied to align the amino acid sequences of single-copy orthologous genes. The concatenated amino acid sequences were further used for construction of the phylogenetic tree in IQ-TREE2 (Minh et al., 2020). MCMCTREE of PAML (Yang, 2007) was used to estimate phylogenetic dating using a BRMC method (Sanderson, 2003) with the soft fossil calibrations obtained from the TimeTree website3: split of A. trichopoda from O. sativa, 173–199 million years ago (MYA); split of X. sorbifolia from A. thaliana, 96–104 MYA; split of A. thaliana from J. regia, 107–135 MYA; split of V. vinifera from A. thaliana, 89–113 MYA. Gene families were filtered out if more than 200 genes were present in one species but only 2 or fewer in the other species. The remaining gene families were used to infer the expansions and contractions of protein family in CAFÉ v3.0 (Han et al., 2013).
Searches for putative paralogous genes were conducted for Q. variabilis and P. persica against each other using BLASTP (E-value ≤ 1e–5). Syntenic blocks were then identified using MCScanX (Wang et al., 2012) with parameters of –a –e 1e–5–s 5. Synonymous substitutions per synonymous site (Ks) values were calculated with codeml in the PAML package (Yang, 2007). For interspecific orthologues, the protein sequences of the homologous genes in Q. variabilis, Q. robur, and Q. lobata were aligned in BLASTP (E-value ≤1e-5), and the results were sorted according to their bit-scores and E-values to obtain reciprocal optimal gene pairs. Then codeml was used to calculate the Ks values of reciprocal optimal gene pairs. Finally, the Ks distributions of intraspecific paralogs and interspecific orthologues were evaluated to infer whole genome duplication (WGD) events and divergence time in the species genome.
We sequenced the Q. variabilis genome using a combination of PacBio and Hi-C technologies, and obtained a high-quality diploid reference genome (Smudgeplot, Supplementary Figure S1). A 20 kb DNA library was constructed and sequenced on a PacBio Sequel II platform, generating 51.70 Gb HiFi reads, approximately 72 × the estimated genome size (713.93 Mb; Supplementary Figure S2; Supplementary Table S1). Then, initial genome sequences spanning 796.30 Mb (327 contigs, N50 of 26.04 Mb; Supplementary Table S2) were constructed, slightly larger than the total genome size as estimated at 713.93 Mb using the 21-mer peak and distribution from DNBSEQ data (Supplementary Figure S2; Supplementary Table S1). This is perhaps due to chimerism caused by the relatively high heterozygosity (estimated to be 2.15%; Supplementary Figure S2). The contig N50 of Q. variabilis is significantly higher than that of other published congeneric species, e.g., Q. acutissima (1.44 Mb; Fu et al., 2022), Q. mongolica (2.64 Mb; Ai et al., 2020), Q. robur (0.07 Mb; Plomion et al., 2018), Q. lobata (1.9 Mb; Sork et al., 2022) and Q. suber (0.08 Mb; Ramos et al., 2018; Table 1). We next used 3D-DNA derived from the Hi-C data (Supplementary Table S1) to generate 12 pseudo-chromosomes (787.15 Mb, Supplementary Table S2), with lengths ranging from 39.05 to 97.21 Mb (Figure 1A; Supplementary Table S3). Interestingly, the number of pseudo-chromosomes of the assembled haploid is the same as that of other Quercus genomes (Q. acutissima, Q. mongolica, Q. robur, Q. lobata, and Q. suber). The chromosomal genome of Q. variabilis was characterized by 245 scaffolds, with a scaffold N50 of 64.86 Mb which is similar to that of Q. mongolica (66.7 Mb), slightly smaller than that of Q. lobata (75 Mb), but ~ 22-fold, ~ 50-fold and ~ 130-fold larger than those of Q. acutissima, Q. robur, and Q. suber, respectively (Table 1). We calculated the heterozygosity based on the 10,014,769 heterozygous sites (including SNPs and INDELs), and found that the heterozygosity of this genome was 1.26%, which was slightly lower than estimated (2.15%) due to underestimation (considering only SNPs and INDELs). We further evaluated the completeness of the genome assembly using the BUSCO.v4 plant datasets, and identified 1,587 (98.3%) of the 1,614 plant single-copy orthologues, with 1,526 (94.5%) presented as single-copy (Supplementary Table S4), a value superior to that of Q. lobata (95%), Q. robur (91%), Q. suber (95%), Q. acutissima (91%) and Q. mongolica (92.71%), indicating that our genome assembly is of high quality and nearly complete.
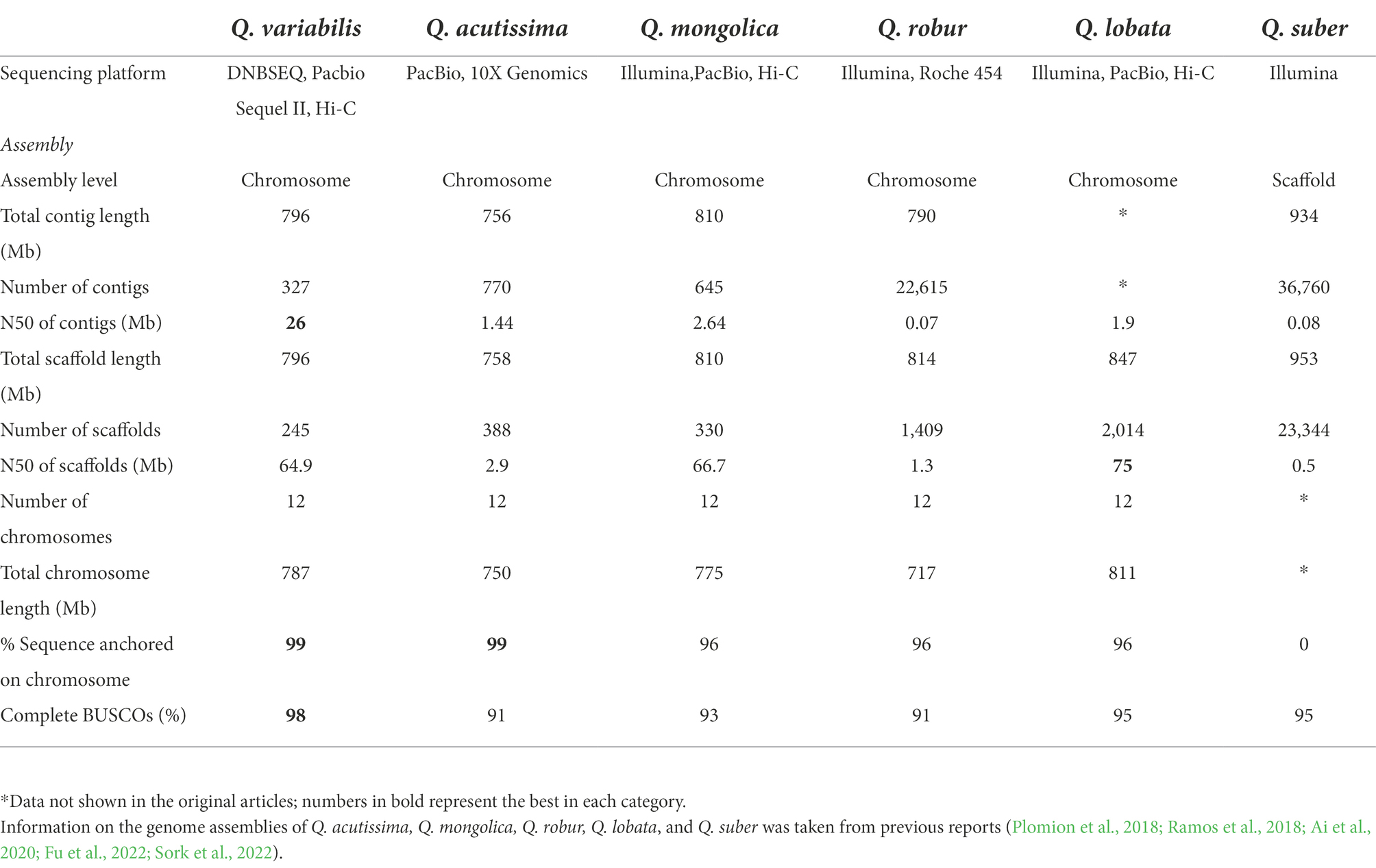
Table 1. The statistics for genome assembly of six Quercus species.
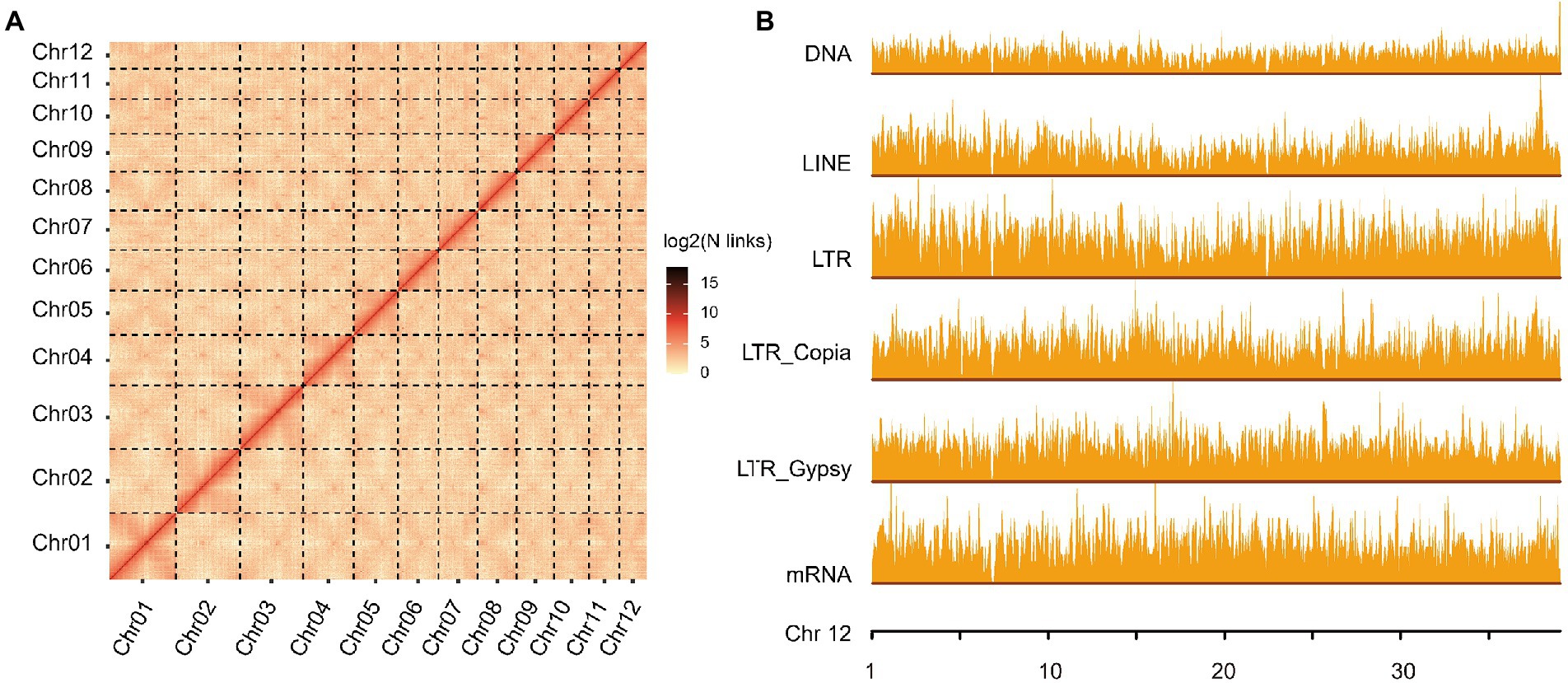
Figure 1. Overview of Quercus variabilis genome assembly and annotation. (A) Genome-wide chromatin interaction analysis of the Q. variabilis genome based on Hi-C data. (B) The distribution of gene and repeat sequences (DNA, LINE, LTR_Copia, LTR_Gypsy, and LTR_other) across Chr12. The height of the bars represents the distribution density of these categories and the window size was set to 50 kb.
The total length of the repetitive sequences in the Q. variabilis genome was 538.34 Mb, covering 67.6% of the assembled genome (Supplementary Table S5). This proportion was higher than that observed in the Q. mongolica genome (435.34 Mb, ~53.75% of the genome) identified using the same process (Ai et al., 2020), and also higher than those in Q. lobata (54%; Sork et al., 2022), Q. suber (51%; Ramos et al., 2018), and E. grandis (55%; Myburg et al., 2014), which have been calculated using other processes. TEs accounted for 61.09% of the Q. variabilis genome (Supplementary Table S6). Long terminal repeat retrotransposons (LTR-RT), which often contribute to variations in genome size (Feschotte et al., 2002; Du et al., 2010), were identified as being the most abundant repeats (46.63%), followed by long interspersed nuclear elements (LINE; 8.14%) and DNA elements (17.88%; Figure 1B; Supplementary Figures S3–S13; Supplementary Table S6).
Gene models for the Q. variabilis genome were obtained using a comprehensive approach including ab initio, RNA sequence-based and homology-based predictions (Supplementary Table S7). In total, we predicted 32,466 protein-coding genes with an average gene length of 5,272.04 bp, an average coding-sequence length of 1,139.49 bp, an average exon length of 226.50 bp and an average exon number per gene of 5.03 (Supplementary Table S7). Functional annotation using the NR, Swissprot, KEGG, KOG, TrEMBL, Interpro and GO databases allowed 30,878 (95.11%) of the total 32,466 genes to be assigned putative functions (Supplementary Table S8). Of these, 52.32% (16,985) of the total genes could be functionally annotated through NR, InterPro, KEGG, SwissProt and KOG simultaneously (Figure 2A). We also predicted 12,220 rRNA, 942 tRNA, 157 miRNA, and 1,148 snRNA genes in the Q. variabilis genome (Supplementary Table S9).
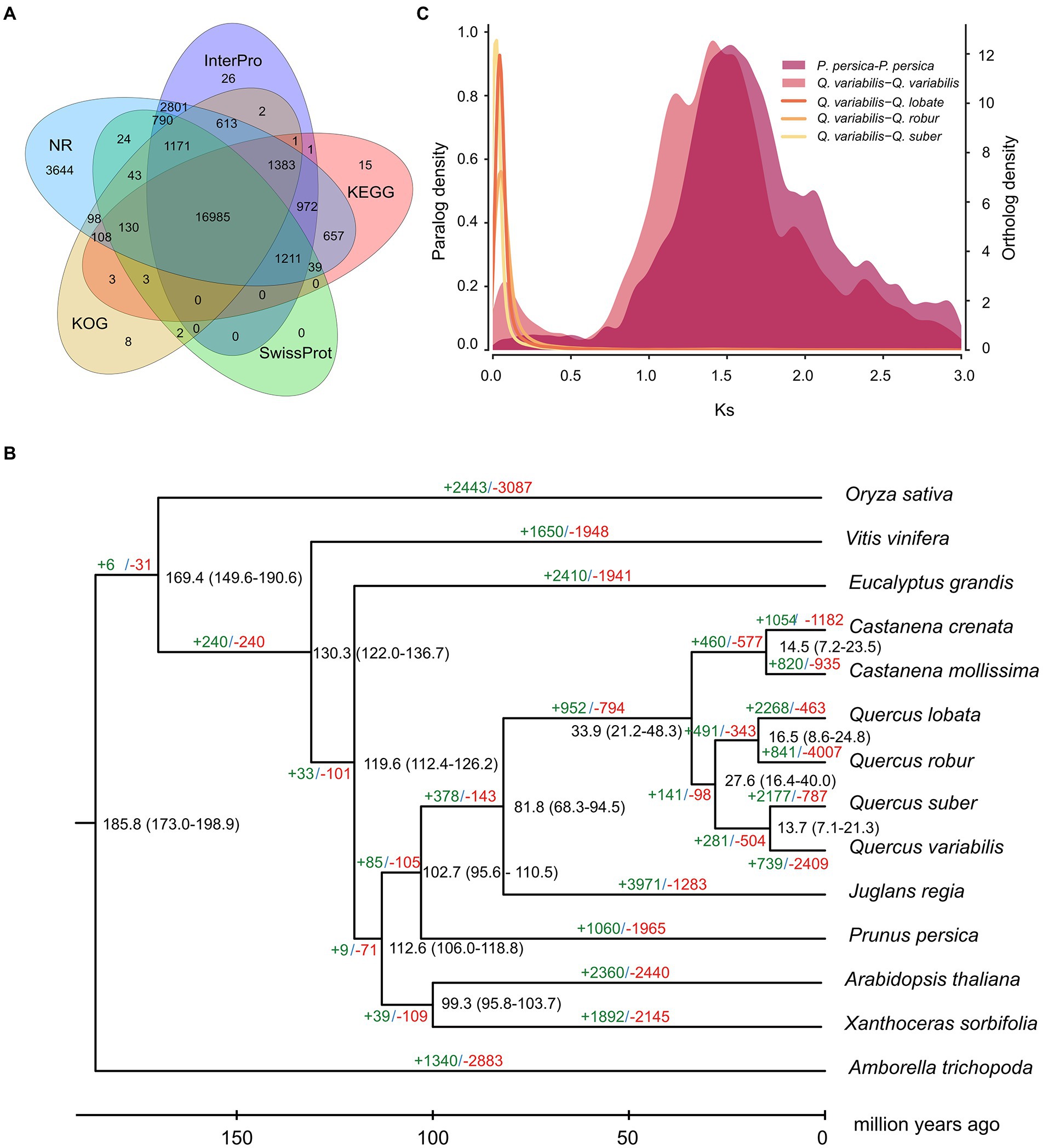
Figure 2. Genome functional annotation and evolution of Q. variabilis. (A) Venn diagram showing shared and unique gene functional annotations among InterPro, KEGG, SwissProt, KOG and NR databases. (B) Phylogenetic tree, divergence time and gene family expansion and contraction among 14 plant species. (C) Ks distribution of Q. variabilis-Q. lobata, Q. variabilis-Q. robur, Q. variabilis-Q. suber, Q. variabilis-Q. variabilis and P. persica-P. persica based on orthologous and paralogous gene pairs.
Orthologous gene families were identified using the proteomes of Q. variabilis predicted in our project and those of 13 other flowering plant species, including the three congeneric species (Q. lobata, Q. robur and Q. suber; Supplementary Table S10). In total, the 29,808 Q. variabilis genes (91.81% of the total) clustered into 14,930 gene families, of which 6,245 gene families (including 11,113 Q. variabilis genes) were shared among all the 14 plant species. We also found that 964/4,854, 747/3,644, 1,821/12,372, 37/95, 891/6,186, 206/1,814, 2,486/12,784, 438/1,633, 340/891, 129/370, 4,311/11,160, 326/2,693, 814/3,015, 286/938 gene families/genes appeared to be unique to A. trichopoda, A. thaliana, C. crenata, C. mollissima, E. grandis, J. regia, O. sativa, P. persica, Q. lobata, Q. robur, Q. suber, Q. variabilis, V. vinifera, and X. sorbifolia, respectively. The gene families unique to Q. variabilis were mainly enriched in “glycine catabolic process,” “serine family amino acid catabolic process,” “organic acid catabolic process,” “oxaloacetate metabolic process,” “tricarboxylic acid cycle” and “nuclear chromosome segregation” (Figure 3; Supplementary File S1).
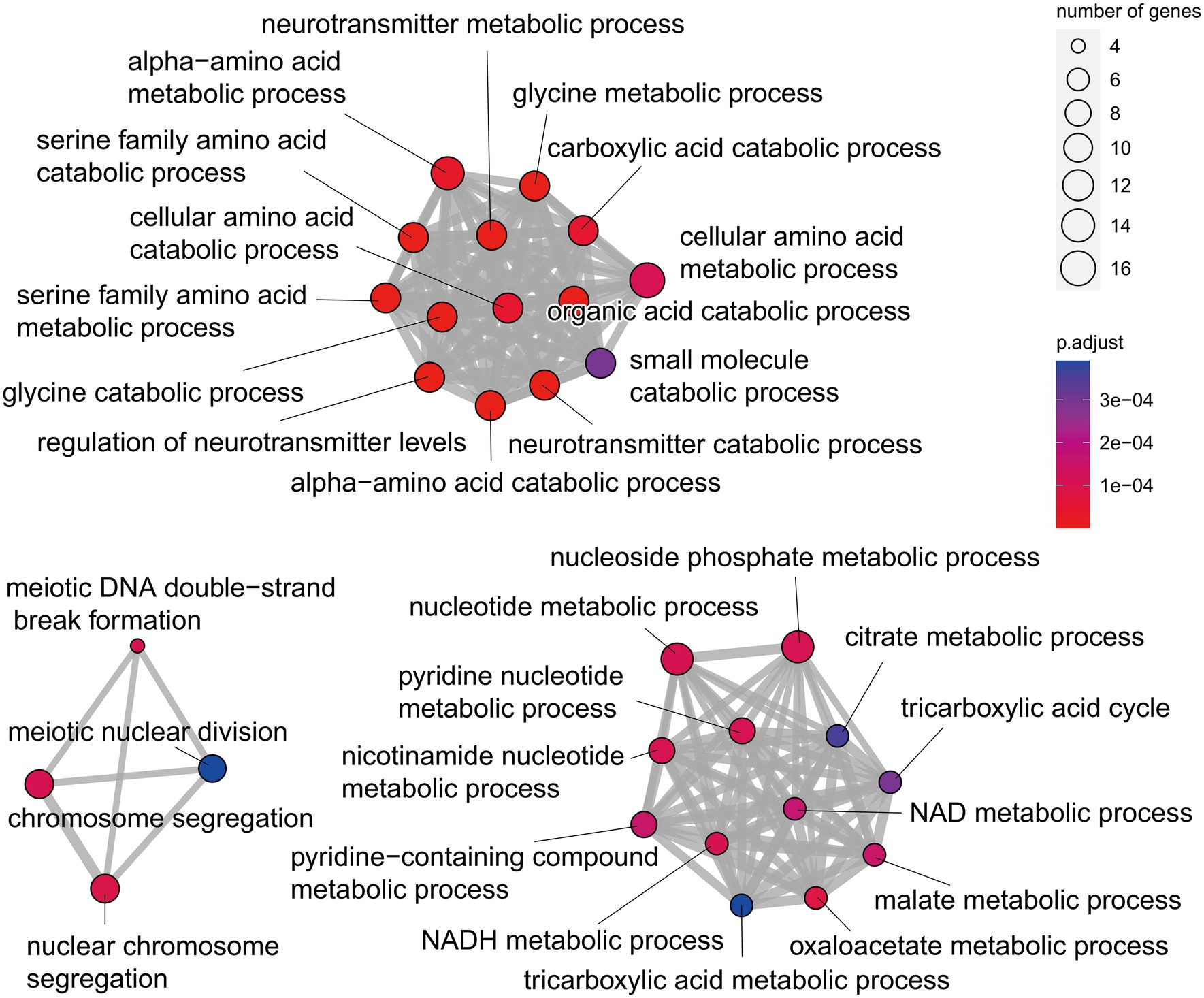
Figure 3. Gene ontology (GO) functional enrichment analysis of the unique genes of Q. variabilis.
Phylogenetic analysis was conducted based on the 483 single-copy gene families derived from Q. variabilis and 13 further flowering plant species (Supplementary Table S10). We found that within this subclade, Q. variabilis is more closely related to Q. suber than to Q. lobata or Q. robur (Figure 2B). The divergence between Q. variabilis and Q. suber occurred at approximately 13.7 (7.1–21.3) MYA, while Q. lobata and Q. robur, which belong to a different subclade, diverged from the Q. variabilis-Q. suber subclade ~27.6 (16.4–40.0) MYA (Figure 2B).
To investigate potential WGD events in the evolutionary history of Q. variabilis, we studied the distribution of the Ks between homologous gene pairs derived from Q. variabilis, Q. lobata, Q. robur, Q. suber, and P. persica. One peak was found based on the paralogous gene pairs in Q. variabilis and P. persica (~ 1.5 Ks units), indicating a shared ancient WGD event (γ) for these two species (Murat et al., 2015; Figure 2C). The divergence between Q. variabilis and three congeneric species (Q. lobata, Q. robur and Q. suber; 0.02–0.05 Ks units) occurred later than the WGD event (Figure 2C). Further investigation of the genomic collinearity between Q. variabilis and Q. lobata, Q. robur showed a clear one-to-one syntenic relationship, and the overall gene synteny was largely conserved (Figure 4), suggesting that no large amounts of chromosome fusion or species-specific WGD events occurred after species divergence (Ai et al., 2020).
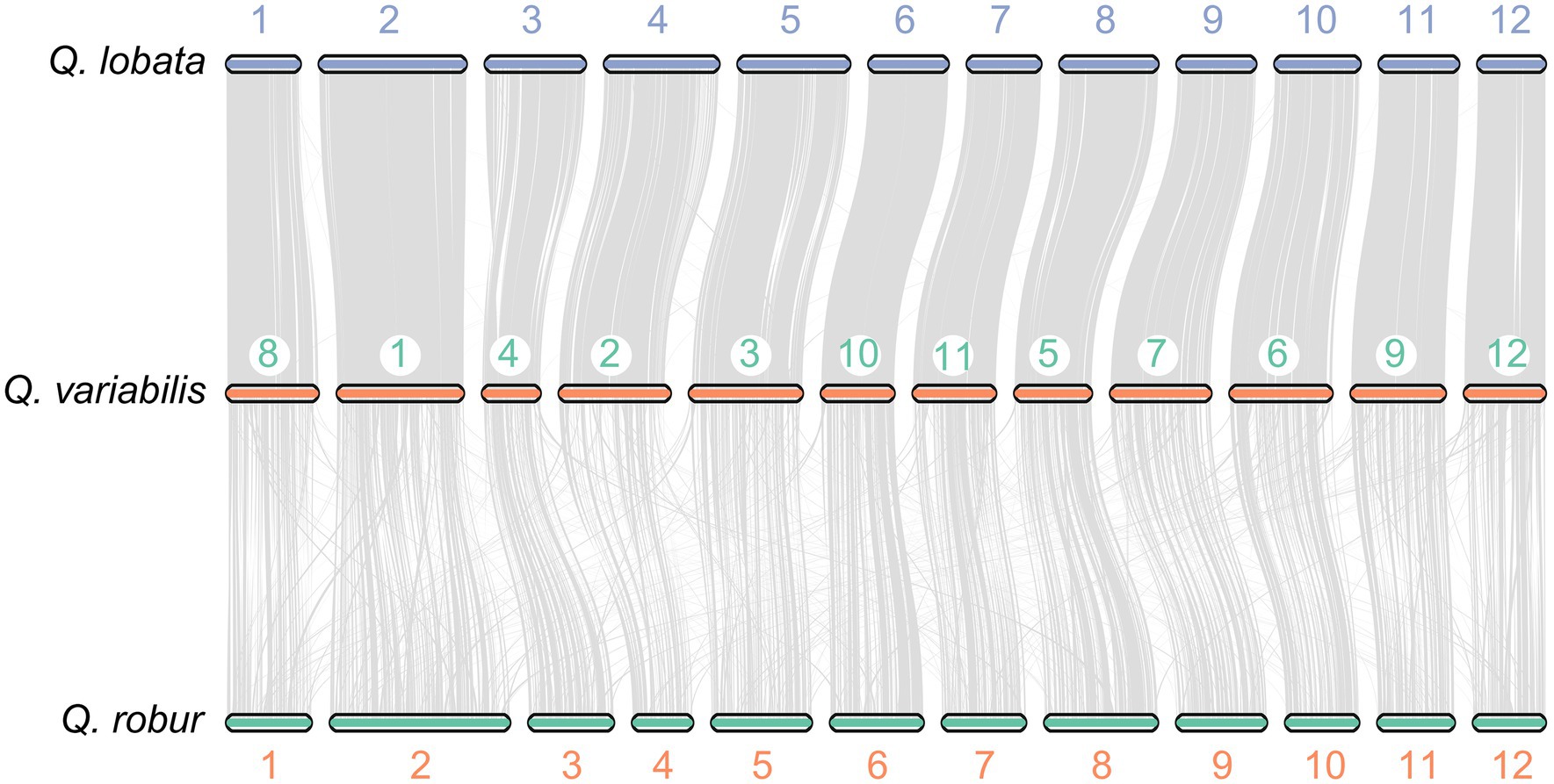
Figure 4. Synteny blocks identified between Q. variabilis and Q. lobata, and Q. variabilis and Q. robur.
We analyzed gene family expansion and contraction based on the gene families in the 14 studied flowering plant genomes (Supplementary Table S10) using OrthoFinder. The number of expanded/contracted gene families in Q. variabilis compared with its common ancestor were 739/2,409, while in Q. suber, which is genomically the most similar to Q. variabilis, these numbers were 2,177/787 (Figure 2B). A significant number of expanded genes in Q. variabilis were enriched in “monoterpene metabolic process,” “terpene biosynthetic process,” “intrachromosomal DNA recombination,” “hydrocarbon biosynthetic process” and “oxaloacetate metabolic process” (Supplementary Figure S14; Supplementary File S2), while contracted gene families were enriched in “glutathione metabolic process,” “isoflavonoid biosynthetic process,” “toxin catabolic process,” “programmed cell death induced by symbiont” and “regulation of response to red or far red light” (Supplementary Figure S15; Supplementary File S3).
Quercus variabilis, the Chinese cork oak, is an ecologically and economically valuable deciduous broadleaved tree species native to and widespread in East Asia (Fujiwara and Harada, 2015). Here, we present a chromosome-scale high-quality de novo genome assembly for Q. variabilis using a combination of PacBio Sequel II and Hi-C sequencing data. This Q. variabilis genome is 796.30 Mb, of which approximately 98.85% (787.15 Mb, Supplementary Table S2) can be anchored to 12 chromosomes. The quality of the Q. variabilis genome assembly was higher than that of several other published Quercus genomes, including those of Q. acutissima (Fu et al., 2022), Q. mongolica (Ai et al., 2020), Q. robur (Plomion et al., 2018), Q. lobata (Sork et al., 2022) and Q. suber (Ramos et al., 2018), although the Q. lobata genome had a slightly longer scaffold N50 than did that of Q. variabilis. It is worth noting that 98.3% of the plant single-copy orthologs was detected in the assembly genome, which is a higher percentage than detected in Q. lobata (95%), Q. robur (91%), Q. suber (95%), Q. acutissima (91%) or Q. mongolica (92.71%; Table 1). Altogether, the assembly of Q. variabilis is relatively accurate and complete. This is the first reference genome for Q. variabilis and will lay the foundation for understanding the evolution of this species and will provide important resources for the further investigation of genetic diversity in Q. variabilis and related species.
The raw sequencing data presented in the study are deposited in the NCBI SRA, BioProject No. PRJNA849150. The whole-genome sequencing data reported in the study are deposited into CNGB Sequence Archive (CNSA) (Guo et al., 2020) of China National GeneBank DataBase (CNGBdb) (Chen et al., 2020a), accession number CNP0003390, and is publicly accessible at https://db.cngb.org/.
BH and D-ZL conceived and designed the study. K-HJ, YX, X-MX, W-QL, YZ, and R-GZ analyzed the data. LW wrote the manuscript. BH, D-ZL, K-HJ, and XQ edited and improved the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Project funded by the Postdoctoral Science Foundation “Research and development of key technologies and equipment of germplasm bank” (BSHCX202101), the Postdoctoral Station Recruitment Subsidy of Shandong Province “Collection, preservation, evaluation and utilization of Quercus acutissima and Q. variabilis Germplasm Resources” (BSHCX202102), and the Agricultural Science and Technology Innovation Project of SAAS (CXGC2022E13).
We appreciate the facilitation provided by National Wild Plant Germplasm Resource Center.
R-GZ was employed by Ori (Shandong) Gene Science and Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1001583/full#supplementary-material
Abrusán, G., Grundmann, N., DeMester, L., and Makalowski, W. (2009). TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330. doi: 10.1093/bioinformatics/btp084
Ai, W., Liu, Y., Mei, M., Zhang, X., Tan, E., Liu, H., et al. (2020). A chromosome-scale genome assembly of the Mongolian oak (Quercus mongolica). Mol. Ecol. Resour. 22, 2396–2410. doi: 10.1111/1755-0998.13616
Akdemir, K. C., and Chin, L. (2015). HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 1–8. doi: 10.1186/s13059-015-0767-1
Asbeck, T., Großmann, J., Paillet, Y., Winiger, N., and Bauhus, J. (2021). The use of tree-related microhabitats as forest biodiversity indicators and to guide integrated forest management. Curr. For. Rep. 7, 59–68. doi: 10.1007/s40725-020-00132-5
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 1–6. doi: 10.1186/s13100-015-0041-9
Bedell, J. A., Korf, I., and Gish, W. (2000). MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041. doi: 10.1093/bioinformatics/16.11.1040
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.-C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Cavender-Bares, J. (2016). Diversity, distribution and ecosystem services of the north American oaks. International Oaks 27, 37–48.
Cavender-Bares, J. (2019). Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytol. 221, 669–692. doi: 10.1111/nph.15450
Chai, Z., Sun, C., Wang, D., and Liu, W. (2016). Interspecific associations of dominant tree populations in a virgin old-growth oak forest in the Qinling Mountains. China Bot. Stud. 57, 1–13. doi: 10.1186/s40529-016-0139-5
Chen, N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10. 11–14.10. 14. doi: 10.1002/0471250953.bi0410s05
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018b). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7:gix120. doi: 10.1093/gigascience/gix120
Chen, Y., Shao, Y., Xi, J., Yuan, Z., Ye, Y., and Wang, T. (2020b). Community preferences of woody plant species in a heterogeneous temperate forest, China. Front. Ecol. Evol. 8:165. doi: 10.3389/fevo.2020.00165
Chen, F. Z., You, L. J., Yang, F., Wang, L. N., Guo, X. Q., Gao, F., et al. (2020a). CNGBdb: China national genebank database. Hereditas 42, 799–809. doi: 10.16288/j.yczz.20-080
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018a). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Consortium, G. O. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, 258D–2261D. doi: 10.1093/nar/gkh036
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10:giab008. doi: 10.1093/gigascience/giab008
Du, J., Tian, Z., Hans, C. S., Laten, H. M., Cannon, S. B., Jackson, S. A., et al. (2010). Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 63, 584–598. doi: 10.1111/j.1365-313X.2010.04263.x
Du, B., Zhu, Y., Kang, H., and Liu, C. (2021). Spatial variations in stomatal traits and their coordination with leaf traits in Quercus variabilis across eastern Asia. Sci. Total Environ. 789:147757. doi: 10.1016/j.scitotenv.2021.147757
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016a). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. doi: 10.1016/j.cels.2015.07.012
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016b). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Eaton, D. A., Hipp, A. L., González-Rodríguez, A., and Cavender-Bares, J. (2015). Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69, 2587–2601. doi: 10.1111/evo.12758
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Feschotte, C., Jiang, N., and Wessler, S. R. (2002). Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 3, 329–341. doi: 10.1038/nrg793
Fu, R., Zhu, Y., Liu, Y., Feng, Y., Lu, R.-S., Li, Y., et al. (2022). Genome-wide analyses of introgression between two sympatric Asian oak species. Nat. Ecol. Evol. 6, 924–935. doi: 10.1038/s41559-022-01754-7
Fujiwara, K., and Harada, A. (2015). “Character of warm-temperate Quercus forests in Asia” in Warm-temperate Deciduous Forests Around the Northern Hemisphere. eds. E. O. Box and K. Fujiwara (Berlin: Springer), 27–80.
Gao, W. Q., Lei, X. D., Fu, L. Y., Duan, G. S., Zhou, M. L., and Cao, J. (2020). Radial growth response of two oaks to climate at their disparate distribution limits in semiarid areas, Beijing, China. Ecosphere 11:e03062. doi: 10.1002/ecs2.3062
Gil-Pelegrín, E., Peguero-Pina, J. J., and Sancho-Knapik, D. (2017). “Oaks physiological ecology” in Exploring the Functional Diversity of Genus Quercus L.. eds. E. Gil-Pelegrín, J. J. Peguero-Pina, and D. Sancho-Knapik (Berlin: Springer), 13–38.
Gugger, P. F., Fitz-Gibbon, S. T., Albarrán-Lara, A., Wright, J. W., and Sork, V. L. (2021). Landscape genomics of Quercus lobata reveals genes involved in local climate adaptation at multiple spatial scales. Mol. Ecol. 30, 406–423. doi: 10.1111/mec.15731
Guo, X., Chen, F., Gao, F., Li, L., Liu, K., You, L., et al. (2020). CNSA: a data repository for archiving omics data. Database 2020 2020:baaa055. doi: 10.1093/database/baaa055
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7–R22. doi: 10.1186/gb-2008-9-1-r7
Han, M. V., Thomas, G. W., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. doi: 10.1093/molbev/mst100
Hipp, A. L., Manos, P. S., Hahn, M., Avishai, M., Bodénès, C., Cavender-Bares, J., et al. (2020). Genomic landscape of the global oak phylogeny. New Phytol. 226, 1198–1212. doi: 10.1111/nph.16162
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 1–14. doi: 10.1186/1471-2105-12-491
Hubert, F., Grimm, G. W., Jousselin, E., Berry, V., Franc, A., and Kremer, A. (2014). Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst. Biodivers. 12, 405–423. doi: 10.1080/14772000.2014.941037
Johnson, A. D., Handsaker, R. E., Pulit, S. L., Nizzari, M. M., O'Donnell, C. J., and De Bakker, P. I. (2008). SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939. doi: 10.1093/bioinformatics/btn564
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kalvari, I., Nawrocki, E. P., Argasinska, J., Quinones-Olvera, N., Finn, R. D., Bateman, A., et al. (2018). Non-coding RNA analysis using the Rfam database. Curr. Protoc. Bioinformatics 62:e51. doi: 10.1002/cpbi.51
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Koonin, E. V., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Krylov, D. M., Makarova, K. S., et al. (2004). A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, R7–R28. doi: 10.1186/gb-2004-5-2-r7
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv [Epub ahead of preprint], doi: 10.6084/M9.FIGSHARE.963153.V1
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Manos, P. S., Doyle, J. J., and Nixon, K. C. (1999). Phylogeny, biogeography, and processes of molecular differentiation in Quercus subgenus Quercus (Fagaceae). Mol. Phylogenet. Evol. 12, 333–349. doi: 10.1006/mpev.1999.0614
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mount, D. W. (2007). Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007:pdb.top17. doi: 10.1101/pdb.top17
Murat, F., Zhang, R., Guizard, S., Gavranović, H., Flores, R., Steinbach, D., et al. (2015). Karyotype and gene order evolution from reconstructed extinct ancestors highlight contrasts in genome plasticity of modern rosid crops. Genome Biol. Evol. 7, 735–749. doi: 10.1093/gbe/evv014
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature 510, 356–362. doi: 10.1038/nature13308
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Plomion, C., Aury, J. M., Amselem, J., Alaeitabar, T., Barbe, V., Belser, C., et al. (2016). Decoding the oak genome: public release of sequence data, assembly, annotation and publication strategies. Mol. Ecol. Resour. 16, 254–265. doi: 10.1111/1755-0998.12425
Plomion, C., Aury, J.-M., Amselem, J., Leroy, T., Murat, F., Duplessis, S., et al. (2018). Oak genome reveals facets of long lifespan. Nature Plants 4, 440–452. doi: 10.1038/s41477-018-0172-3
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Ramos, A. M., Usié, A., Barbosa, P., Barros, P. M., Capote, T., Chaves, I., et al. (2018). The draft genome sequence of cork oak. Scientific Data 5, 1–12. doi: 10.1038/sdata.2018.69
Ranallo-Benavidez, T. R., Jaron, K. S., and Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1–10. doi: 10.1038/s41467-020-14998-3
Sanderson, M. J. (2003). r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Simeone, M. C., Grimm, G. W., Papini, A., Vessella, F., Cardoni, S., Tordoni, E., et al. (2016). Plastome data reveal multiple geographic origins of Quercus group ilex. PeerJ 4:e1897. doi: 10.7717/peerj.1897
Simeone, M. C., Piredda, R., Papini, A., Vessella, F., and Schirone, B. (2013). Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): problems, prospects and phylogenetic implications. Bot. J. Linn. Soc. 172, 478–499. doi: 10.1111/boj.12059
Sork, V. L., Cokus, S. J., Fitz-Gibbon, S. T., Zimin, A. V., Puiu, D., Garcia, J. A., et al. (2022). High-quality genome and methylomes illustrate features underlying evolutionary success of oaks. Nat. Commun. 13, 1–15. doi: 10.1038/s41467-022-29584-y
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Sun, J., Shi, W., Wu, Y., Ji, J., Feng, J., Zhao, J., et al. (2021). Variations in acorn traits in two oak species: Quercus mongolica Fisch. Ex Ledeb. And Quercus variabilis Blume. Forests 12:1755. doi: 10.3390/f12121755
Vitales, D., D’Ambrosio, U., Galvez, F., Kovařík, A., and Garcia, S. (2017). Third release of the plant rDNA database with updated content and information on telomere composition and sequenced plant genomes. Plant Syst. Evol. 303, 1115–1121. doi: 10.1007/s00606-017-1440-9
Vitelli, M., Vessella, F., Cardoni, S., Pollegioni, P., Denk, T., Grimm, G. W., et al. (2017). Phylogeographic structuring of plastome diversity in Mediterranean oaks (Quercus group ilex, Fagaceae). Tree Genet. Genomes 13, 1–17. doi: 10.1007/s11295-016-1086-8
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovations 2:100141. doi: 10.1016/j.xinn.2021.100141
Xia, K., Daws, M. I., and Peng, L. L. (2022). Climate drives patterns of seed traits in Quercus species across China. New Phytol. 234, 1629–1638. doi: 10.1111/nph.18103
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, R.-S., Yang, J., Hu, H.-L., Xia, R.-X., Li, Y.-P., Su, J.-F., et al. (2020). A high level of chloroplast genome sequence variability in the sawtooth oak Quercus acutissima. Int. J. Biol. Macromol. 152, 340–348. doi: 10.1016/j.ijbiomac.2020.02.201
Keywords: Quercus variabilis, genome assembly, PacBio HiFi sequencing, Hi-C sequencing, comparative genomics
Citation: Han B, Wang L, Xian Y, Xie X-M, Li W-Q, Zhao Y, Zhang R-G, Qin X, Li D-Z and Jia K-H (2022) A chromosome-level genome assembly of the Chinese cork oak (Quercus variabilis). Front. Plant Sci. 13:1001583. doi: 10.3389/fpls.2022.1001583
Edited by:
Kyung Do Kim, Myongji University, South KoreaReviewed by:
Minkyu Park, University of Florida, United StatesCopyright © 2022 Han, Wang, Xian, Xie, Li, Zhao, Zhang, Qin, Li and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai-Hua Jia, a2FpaHVhamlhX3NhYXNAMTYzLmNvbQ==; De-Zhu Li, ZHpsQG1haWwua2liLmFjLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.