 Jing Sun†
Jing Sun† Wenzhong Song
Wenzhong Song Tiegang Lu
Tiegang Lu Zhiguo Zhang
Zhiguo Zhang- Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China
Lesion mimic mutants have become an effective material for understanding plant-microbe interactions and the immune mechanism in plants. Although many mechanisms responsible for the lesion mimic phenotype have been clarified in plants, the mechanism by which lesion mimic is regulated by posttranslational modification remained largely elusive, especially in rice. In this study, a mutant with the lesion mimic phenotype was obtained and named lmp1-1. Physiological measurements and quantitative real-time PCR analysis showed that the defense response was activated in the mutants. Transcriptome analysis showed that the phenylalanine ammonia lyase (PAL) pathway was activated in the mutant, causing the accumulation of salicylic acid (SA). The results of mapping based cloning showed that OsLMP1 encodes a deubiquitinase. OsLMP1 can cleave ubiquitination precursors. Furthermore, OsLMP1 epigenetically modifies SA synthetic pathway genes by deubiquitinating H2B and regulates the immune response in rice. In summary, this study deepens our understanding of the function of OsLMP1 in the plant immune response and provides further insight into the relationship between plants and pathogenic microorganisms.
Introduction
Rice, one of the main food crops in the world, supports half of the world’s population. Pathogenic microorganism invasion often decreases grain weight and the seed setting rate, which seriously affects the yield and quality of crops. Improving the disease resistance of crops itself is one of the most environmentally friendly, economic and effective ways to defend against diseases and is of great significance for maintaining stable and high crop yields (Vejan et al., 2016). Lesion mimic is a phenomenon in which local cell necrosis occurs spontaneously in plants without invasion by external pathogens. Most lesion mimic mutants show increased disease resistance to at least one microorganism; thus, they are good materials with which to study the mechanism of the plant defense response (Wu et al., 2008).
As an increasing number of disease resistance genes have been cloned and their functions elucidated, the mechanism responsible for the lesion mimic phenotype can be summarized by the following four observations. First, the resistance genes nucleotide binding site/leucine rich repeats (NBS-LRRs) were found to lead to the disorder of signaling pathways in the defense response, triggering the irreversible death of cells and eventually the occurrence of lesion mimic in plants. Mutations in rice necrotic leaf sheath1 (NLS1) (Tang et al., 2011), maize resistance protein (RP1) (Sun et al., 2001), and Arabidopsis Toll/interleukin-1 receptor (TIR)-NBS (TN13) (Cai et al., 2021) activate the expression of defense genes and thus cause high levels of salicylic acid (SA) accumulation and leaf necrosis spots. Second, blockade of the synthesis of some intermediate metabolic products or the accumulation of these products directly or indirectly leads to the lesion mimic phenotype. Due to mutations in rice lesion initiation 1 (rlIN1) (Sun et al., 2011) and Arabidopsis mosaic death1 (MOD1) (Mou et al., 2000), cell death occurs. Third, activation of the programmed cell death (PCD) pathway in plants results in the lesion mimic phenotype. Arabidopsis lesions simulating disease resistance 1 (LSD1) (Kaminaka et al., 2006), which encodes a zinc finger protein, promotes the production of excessive hydrogen peroxide in mutants and negatively regulates the PCD pathway. Fourth, the occurrence of lesion mimic is accompanied by changes in plant hormones, such as SA (Mosher et al., 2010; Singh et al., 2018), jasmonic acid (Feng et al., 2020), ethylene (Bouchez et al., 2007) and abscisic acid (ABA) (Mosher et al., 2010); Among these hormones, increasing the SA content significantly improves disease resistance (Singh et al., 2018). Most of the abovementioned regulatory proteins are disease-resistant NBS-LRRs, metabolic enzymes and transcription factors and drive lesion mimic at the transcriptional level and rarely at the posttranscriptional and translational levels.
Ubiquitination and deubiquitination are a kind of posttranslational modification. Deubiquitinases are some of the most abundant proteases in the ubiquitin system (Hu, 2012). Based on their amino acid sequences, deubiquitinases can be divided into cysteine proteases and metalloproteinases. Cysteine proteases include four subfamilies: ubiquitin-specific proteases (UBPs), ubiquitin C-terminal hydrolase (UCH), ovarian tumor-related proteases (OTUs), Machado Joseph domain-containing proteases (MJDs), and the Jab1/MPN/Mov34 (JAMM) family (Amerik and Hochstrasser, 2004). Deubiquitination is involved in the regulation of not only important life activities in animals and yeast (Clague et al., 2013) but also plant growth and development (Katsiarimpa et al., 2014). Upon the mutation of osubp6 in Oryza sativa var. japonica cv. Dongjin, the seedlings grew slowly but then showed normal growth at a later stage (Moon et al., 2009). Seed development and heading date were affected in an atubp26 mutant (Luo et al., 2008). Further study showed that the AtUBP26 mutation caused the accumulation of ubiquitin H2B, which indirectly influenced the levels of H3K27me3 and H3K36me3 at the flowering locus C (FLC) site, and the transcription of FLC was inhibited in the atubp26 mutant (Schmitz et al., 2009). H2B-Ub stimulates Dot1L activity and the contacts mediated by Dot1L and the H4 tail induce a conformational change in the globular core of histone H3 that reorients K79 from an inaccessible position, thus enabling this side chain to insert into the active site in a position primed for catalysis (Worden et al., 2019). AtUBP27 is located in mitochondria, and its mutation does not influence mitochondrial morphology. However, mitochondrial morphology did change when AtUBP27 was overexpressed (Pan et al., 2014). In addition, UBP/UCH family members participate in multiple signaling pathways. UBP6 participates in the Ca2+ signaling pathway (Moon et al., 2005). UBP24 deubiquitination modulated the responses to ABA and salt stress (Zhao et al., 2016). Overexpression of UCH1 in Arabidopsis restored the auxin insensitivity phenotype caused by the mutation of axr1-3 and axr2-1, indicating that UCH1 may be involved in the auxin signaling pathway (Yang et al., 2007). In conclusion, deubiquitination not only regulates the growth and development of plants but also plays a very important role in mediating photomorphogenesis, hormone signal transduction, and abiotic stress responses. These diverse functions highlight the importance of deubiquitination in biological processes; however, the immune response induced by deubiquitination remained largely elusive, especially in rice.
In the past decade, the studies had been established a role for epigenetic mechanisms in plant–pathogen interactions. Kong et al. (2017) found that the cytoplasmic effector PsAvh23 produced by the soybean pathogen Phytophthora sojae plays as a regulatory factor of histone acetyltransferase (HAT) in plants. PsAvh23 interfered with the association of ADA2, subunit of the HAT complex SAGA and disrupts the catalytic activities of ADA2 module. Thus, PsAvh23 regulated ADA2 module by suppressing H3K9 acetylation and improved plant susceptibility. PsAvh52 as an early-induced RxLR effector from the soybean root rot pathogen, P. sojae, interacted with GmTAP1 (Li et al., 2018). During early infection, PsAvh52 caused GmTAP1 to relocate into the nucleus and made histones H2A and H3 acetylation, thereby promoting susceptibility to P. sojae. In the absence of pathogen, GmTAP1 remained confined to the cytoplasm and did not modify plant susceptibility. Recent, PICI1, encoding a deubiquitinase, plays as an immunity hub for PTI and ETI in rice (Zhai et al., 2021). PICI1 is targeted for degradation by blast fungal effectors, including AvrPi9, to dampen PTI. Nucleotide-binding domain, leucine-rich-repeat-containing receptors (NLRs) in the plant immune system, such as PigmR, protect PICI1 from effector-mediated degradation to reboot the methionine-ethylene cascade. Although generational defense priming has only been in a few cases using model plants, the fine regulation between pathogenic microorganisms and plants needs to be further explored.
To study the immune response mechanism in rice, a lesion mimic mutant was obtained and named lmp1-1. In this study, OsLMP1 was cloned by a map-based cloning strategy. A series of assays verified that OsLMP1, which encodes a deubiquitinase, epigenetic modifies H2B and regulates the plant immune response. This study broadens the function of deubiquitination in rice and deepens our understanding of the role of epigenetic modification in plant disease resistance.
Materials and Methods
Plant Materials and Growth Conditions
The lmp1-1, wild-type (Nipponbare), and OsLMP1 complementation lines and OsLMP1 knockout lines were planted in an experimental field under normal growth conditions at Langfang station in summer (22°C, 16 h light/8 h dark) and in Sanya in winter (22°C, 16 h light/8 h dark). The lmp1-1 and wild-type lines were crossed to determine the inheritance law in the F2 progeny.
Map-Based Cloning of OsLMP1
A genetic mapping population was constructed using lmp1-1 as the recipient and the Indica variety Dular as the donor with wide compatibility. Among the F2 progeny, segregated plants with the lesion mimic phenotype were selected for coarse and fine mapping. SNP and indel molecular markers were designed based on whole-genome differences between Dular and Nipponbare. The candidate genes within the positioning interval were predicted, amplified and sequenced.
Complementation Experiment
A complementation experiment was constructed as follows. First, the full coding sequence of OsLMP1 was amplified and ligated into the pCambia2300 vector. The coding sequence was driven by maize ubiquitin1. The fused complementation vector (OsLMP1-pCambia2300) was transferred to the Agrobacterium tumefaciens AGL1 strain. NPTII was selected as the selectable marker. Calli of the lmp1-1 strain were infected by the strain containing OsLMP1-pCambia2300. The positive plants were identified by PCR amplification and sequencing (Hiei et al., 1994).
Creation of the OsLMP1 Knockout Lines
The CRISPR/Cas9 method was used to knock out OsLMP1 with sgRNA designed to target OsLMP1. A single sgRNA was designed and inserted in the BGK03 vector containing Cas9, which was introduced into Agrobacterium strain AGL01 and transformed into Nipponbare calli. In total, twenty independent lines transfected with the sgRNA were obtained. Sequence alignment showed that OsLMP1 deletion lines had been obtained.
qRT-PCR Analysis
Total RNA was extracted from the lmp1-1, wild-type and complementation lines at the later tiller stage using TRIzol RNA reagent. Total RNA (1.5 μg) from each sample was reverse-transcribed with an oligo(dT) primer and Ace enzyme (Toyobo, Japan) according to standard procedure. The cDNA solutions were diluted at least 10-fold. The amplification procedure was as follows: an initial 94°C denaturation step for 5 min, followed by 25–30 cycles of 94°C for 30 s, 56°C for 20 s, and 72°C for 20 s and a final 72°C extension step for 10 min. OsActin1 was selected as a control. Pathogenic genes were selected for further analysis. The expression pattern of OsLMP1 was determined in root, leaf, panicle, leaf sheath, and stem tissues at the seedling and flowering stages. Relative expression was calculated using the ΔΔCT method (Livak and Schmittgen, 2001). Wild-type leaves at the later tiller stage were treated with rice bacterial blight (PXO99). Tissues 2 cm from the incision were sampled every 4 h. The untreated wild type (mock) was selected as control.
DAB Staining
The lmp1-1 and wild-type leaves at the flowering stage were stained with DAB dye (Jubany-Mari et al., 2009), decolorized using ethanol, and then photographed.
Measurement of the H2O2 Content
The H2O2 content was measured based on the protocol used by Zhang et al. (2013). Briefly, 2 g of fresh leaves from the lmp1-1 and wild-type lines at the flowering stage were collected, mixed with acetone at a ratio of 1:1 and ground with a small amount of quartz sand to form a homogenate. The sample was centrifuged at 12,000 × g for 5 min. One milliliter of the supernatant was obtained and mixed with 5 ml of Ti(SO4)2 and 0.2 ml of 19% ammonia to make a precipitate. The precipitate was washed with acetone at least 3–5 times until the green pigment was removed. The resulting pellet was dissolved in 5 ml of 2 M H2SO4. The H2O2 content was calculated according to a standard curve made with H2O2 ranging from 0 to 10 mM.
Subcellular Localization
The full OsLMP1 coding sequence without its termination codon was amplified using PCR and ligated with PAN580 to generate 35S:OsLMP1:GFP. The fusion construct (35S:OsLMP1:GFP) and pMcherry (35S:OsMADS3:RFP, as a control) were transiently transformed into rice protoplasts extracted from 14-day-old seedlings by the PEG transformation method (Zhang et al., 2011). The pMcherry construct (35S:OsMADS3:RFP) has been described by Gao et al. (2013). After 16 h at 28°C in the dark, the transformed protoplasts were observed using confocal laser scanning microscopy (Leica TCS SP2) (Chen et al., 2006). Then observed with a 20× mirror and GFP fluorescence images were taken with a 488-nm argon laser and a 505–530-nm bandpass emission filter. Analyzed with ZEN 2009 Light Edition software, Scale bar = 5 μm.
GUS Staining
The 2K promoter of OsLMP1 was amplified and ligated with the pCambia1391Z vector. The fusion vector (OsLMP1-1391Z) was transferred to the A. tumefaciens AGL1 strain. The strain was transformed to Nipponbare calli. The positive plants were stained, and tissue expression patterns were determined using the GUS staining method (Jefferson et al., 1987).
Transcriptome Analysis
Total RNA was extracted from both the wild-type and lmp1-1 lines at the later tiller stage (about 2 months growth period) using TRIzol reagent, and the RNA was sent to Novogene for transcriptome analysis. Each sample was assayed in triplicate. The transcriptome data were deposited at NCBI1.
Deubiquitinase Activity Assay
A deubiquitinase activity assay was performed as described by Shi et al. (2019). The ubiquitinated OsUBQ1 (LOC_Os03g13170) and polyubiquitinated OsUBQ10 (LOC_Os02g06640) proteins from rice were selected as substrates (Moon et al., 2009). Recombinant GST-OsLMP1, GST-OsLMP1-m (in which an “A” base was inserted as position 2,604, generating a truncated protein), OsUBP10, and OsUBP1 plasmids were constructed and co-transformed into Escherichia coli Transetta (DE3). Expression of the proteins was induced by IPTG at 23°C for 8 h. Total proteins were extracted using a Millipore ProteoPrep kit and then boiled at 95°C for 10 min. After electrophoresis, the proteins were transferred to a PVDF membrane and then incubated with primary antibodies. The antibodies used in this study were anti-GST (CWBio) and anti-ubiquitin (Abcam). Signals were detected using a Pierce ECL Plus Western Blotting Detection Kit and visualized with an imaging system.
Western Blot Analysis
Total histones were extracted from the leaf tissues of the wild-type, lmp1-1, crisp-m1, and complementation lines using a Millipore ProteoPrep kit at the later tiller stage. Total histones were boiled at 95°C for 10 min before immunoblotting. After electrophoresis, the proteins were transferred to a PVDF membrane and then incubated with the primary antibodies. Anti-H2B (1:1000) was purchased from Abcam (ab1790), and anti-H2B-ubi (1:1000) (Lys 120) was purchased from Active Motif (39623). Anti-H3 (ab1791) (1:2000), anti-H3K4me2 (07-030) (1:2000), anti-H3K36me2 (07-369) (1:2000), anti-H3K4me3 (07-73) (1:2,000), anti-H3K9me2 (05-1354) (1:2000), anti-H2A (ABE327) (1:1000), and anti-monoubiquitinated H2A (clone E6C5) (1:1000) were purchased from Abcam or Millipore. Signals were detected using a Pierce ECL Plus Western Blotting Detection Kit and visualized with an imaging system.
ChIP-qPCR Analysis
ChIP was conducted as follows (Clontech, cat# 640166). First, cell nuclei were isolated from wild type seedlings, respectively, at the 5th leaf stage cross-linked with formaldehyde, and sonicated to shear the chromatin to fragment with an average size of 0.2–1.0 kb. The samples were incubated with protein A agarose beads (40 μl; 16–157, Millipore). Then, the resulting mixture was incubated at 4°C overnight with anti-H2B-ubi (Lys 120) combined with protein A agarose beads. The product was washed successively with high-salt NaCl, LiCl, and TE buffers. The washed samples were digested by proteinase K. The DNA fragments were cleaned up using a PCR DNA purification kit (Qiagen). The resulting DNA was analyzed by qRT-PCR using designed primers (Supplementary Table 6). The control was selected using protein A agarose beads incubated with chromatin samples in the absence of anti-H2B-ubi (Lys 120) antibody. The experiment was repeated three times. OsActin1 was used as negative control.
Blast and Bacterial Blight Resistance Experiments
Bacterial blight strain PXO99, C1, C2, and C6 were used to infect the lmp1-1 plants and wild type, respectively (Wang et al., 2014). Magnaporthe oryzae (M. oryzae) strain Dao72 was used to infect the lmp1-1 plants and wild type (Wang et al., 2017a). The infected results were evaluated 1 weeks after inoculation.
Measurement of the Salicylic Acid Content
Flag leaves (0.3 g) at the flowering stage (about 3 month period) from the wild-type and lmp1-1 were prepared. SA was extracted and quantified as described (Chen and Jastreboff, 1995).
Statistical Analyses
The experiments were repeated at least three times. The mean standard deviations are shown in the figures. Significant differences based on Student’s t-test are marked with asterisks (*P < 0.05, **P < 0.01).
Primer and Gene Sequences
The primers were designed according to Primer3 Input (version 0.4.0)2. The primers used are shown in Supplementary Table 6.
Results
Identification of the lmp1-1 Mutant
Previously, we constructed a rice T-DNA insertion population and obtained ∼100 lesion mimic mutants at different developmental stages (Wan et al., 2009). To study the mechanism of the immune response in rice, a lesion mimic phenotype mutant, named lmp1-1, was selected for further study. The lmp1-1 plants began to exhibit necrotic spots on their leaves at the early tiller stage and showed a lesion mimic phenotype at the late tiller stage (Figure 1A). At the flowering stage, the leaves of lmp1-1 plants became completely brown (Figures 1B,C).
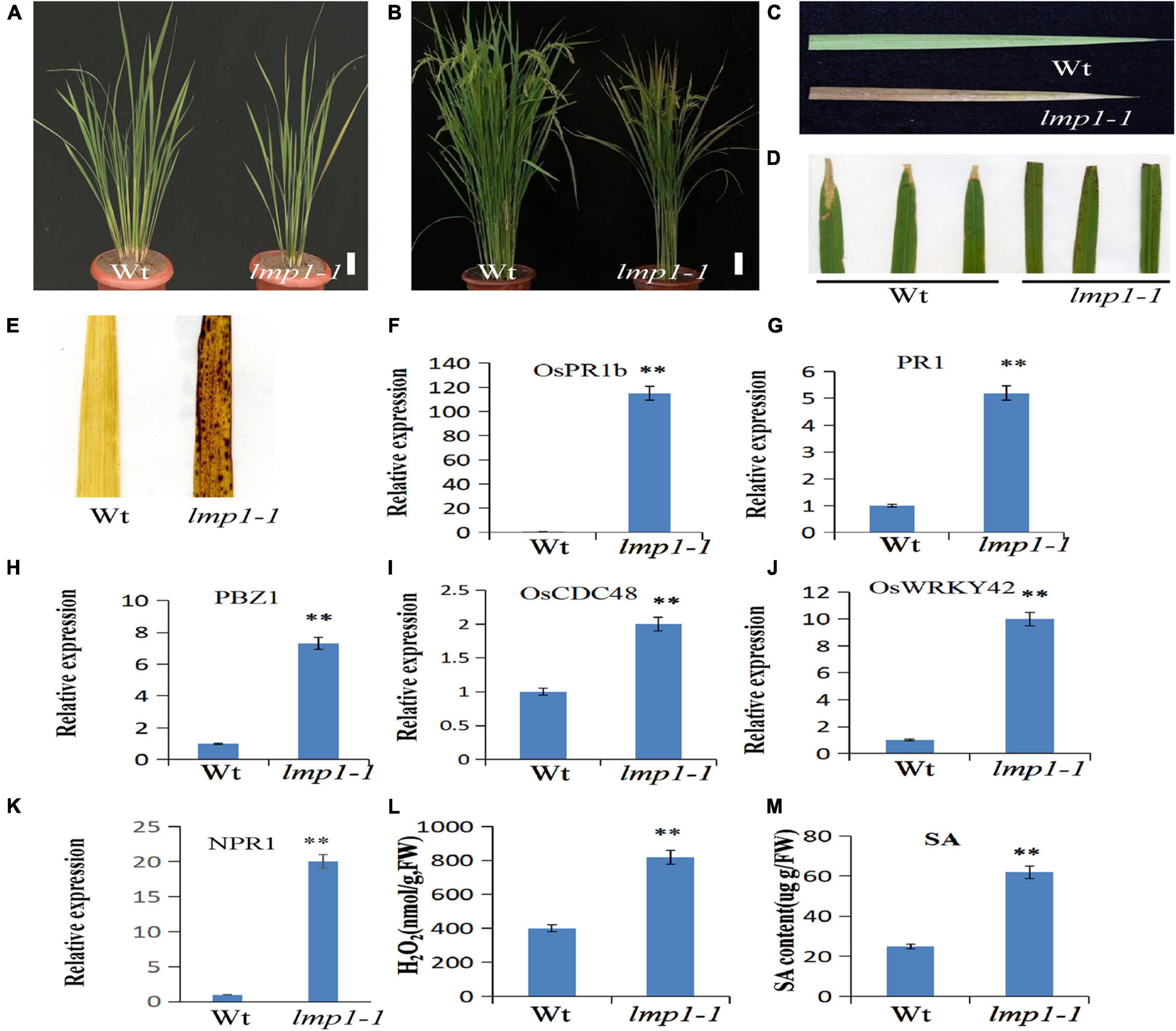
Figure 1. Morphological characterization of the lmp1-1 and wild-type lines. (A) Phenotype comparison of field-grown wild-type (wt) and lmp1-1 lines at the later tiller stage (bar = 5 cm). (B) Phenotype comparison of field-grown wt and lmp1-1 lines at the flowering stage (bar = 5 cm). (C) Leaves collected from wt and lmp1-1 plants shown in panel (A), which also shows the lesion mimic phenotype in lmp1-1. (D) Enhanced resistance of lmp1-1 to rice bacterial blight (PXO99). (E) Detection of accumulated H2O2 in wt and lmp1-1 leaves at the flowering stage using DAB staining. (F–K) The expression levels of pathogenic genes in wt and lmp1-1 plants at the later tiller stage were detected using qRT-PCR (n = 3). (L) The H2O2 content of wt and lmp1-1 plants at the flowering stage was measured (n = 3). (M) The SA content of wt and lmp1-1 plants at the flowering stage was measured (n = 3). The significant differences were marked with asterisks based on student’s t-test (**P < 0.01).
A cross test between lmp1-1 and wild-type plants showed that the segregation ratio in the F2 progeny of wild-type (156) and mutant-like (50) plants reached 3:1 (x2 = 0.81, x20.05,1), indicating that lmp1-1 is controlled by a single recessive nuclear locus.
The qRT-PCR analysis at the later tiller stage showed that the expression levels of pathogenesis-related genes, such as OsWRKY42, OsPR1b, and PBZ1, were significantly increased in the mutants (Figures 1F–K). An infection experiment at the flowering stage using four bacterial blight strains and one M. oryzae strain showed that the lmp1-1 t plants were more resistant to the bacterial blight strain and M. oryzae strain than the wild-type plants, similar to most of the lesion mimic mutants (Figure 1D and Supplementary Figure 1).
Disease Resistance in the lmp1-1 Plants Is Associated With H2O2 Accumulation
Lesion mimic is often accompanied by the accumulation of reactive oxygen species, H2O2 (Tang et al., 2011). H2O2 in leaves can be stained with the dye 3,3′–diaminobenzidine (DAB) (Jubany-Mari et al., 2009). A DAB staining assay in mature leaves at the flowering stage showed that the leaves of lmp1-1 plants were more deeply stained than those of the wild-type plants, indicating that more H2O2 had accumulated in the mutants (Figure 1E). Measurement of the H2O2 content in the leaves also verified this result (Figure 1L). Thus, we speculated that H2O2 accumulation in the mutants activated the defense response and caused the lesion mimic phenotype.
Map-Based Cloning of OsLMP1
The lmp1-1 mutant did not cosegregate with the hygromycin gene, which indicated that the mutation may have originated from somatic variation induced by tissue culture. To clone OsLMP1, we first constructed an F2 mapping population (lmp1-1xDular). Using map-based cloning, OsLMP1 was roughly mapped to the region between Indel1-1 and Indel1-2 on chromosome 9 using 50 F2 mutant-like plants (Figure 2A). Fine positioning using 950 plants showed that OsLMP1 was localized within 110 kilobase pairs (kbp) (Figure 2B). This region contains eight open reading frames. All the genes were sequenced, and a mutation in LOC_Os09g32740 was identified in the lmp1-1 mutant (Figure 2C). An insertion of one base (“A”) at position 2,604 of the second exon in lmp1-1 eventually led to early translation termination and an additional amino acids (Figures 2D,E).
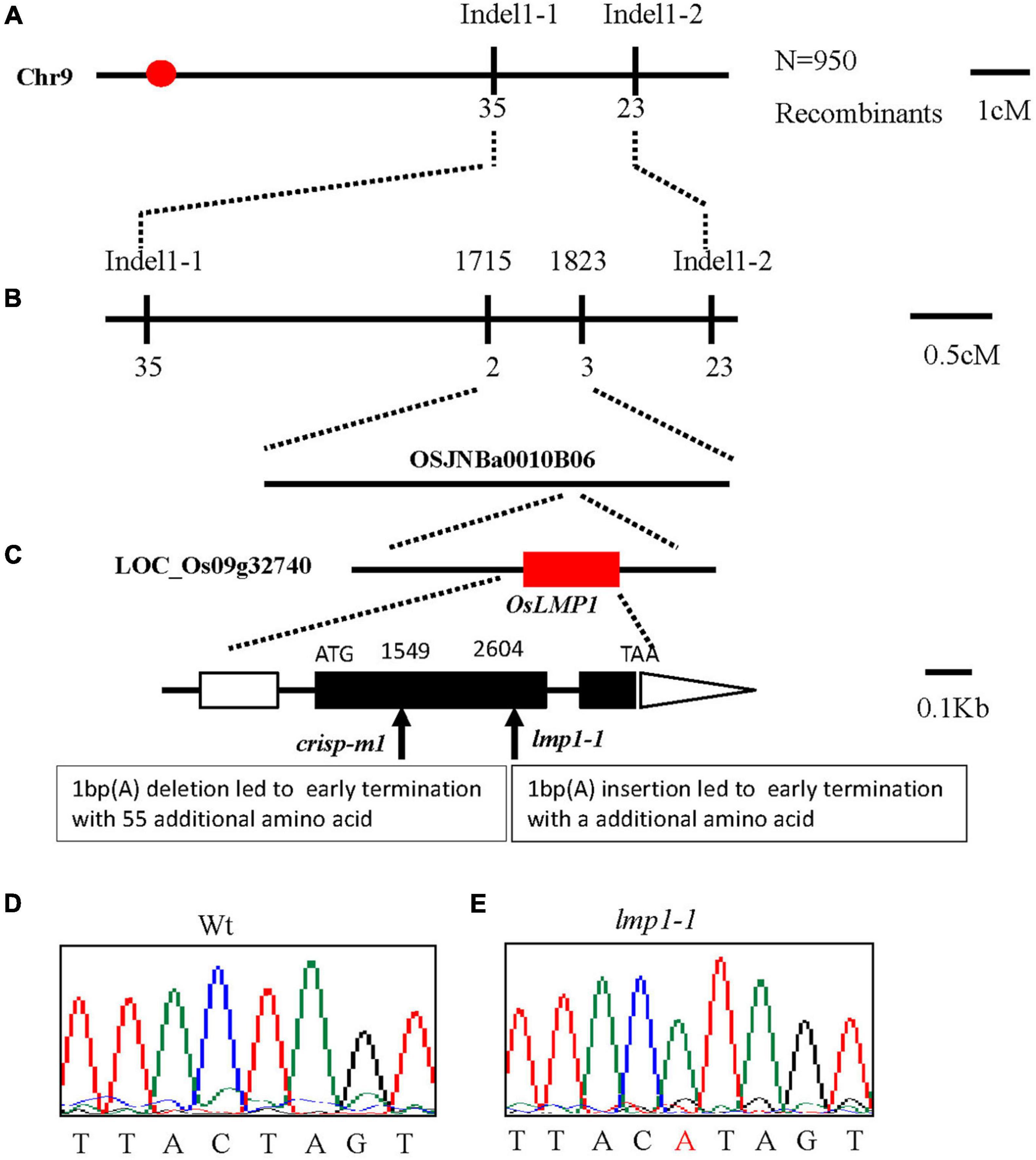
Figure 2. Map-based cloning of OsLMP1. (A) The OsLMP1 locus was mapped to a 3-cM interval on chromosome 9L between indel markers Indel1-1 and Indel1-2. (B) The location of the OsLMP1 locus was narrowed down to a 110-kb region between indel markers 1,715 and 1,823 in the BAC clone OSJNBa0010B06 using 950 F2 homozygous mutant plants. (C) Eight open reading frames (ORFs) were predicted in the mapped region. Structure of OsLMP1. ATG, TAA: start and stop codons, respectively. Boxes: exons. Lines between black boxes: introns. One single nucleotide insertion results in a frame shift mutation in lmp1-1. The deletion site in crisp-m1 is labeled. The black box denotes the coding sequence from ATG to TAA. (D,E) Comparison of the mutant site of LOC_Os09g32740 between the wild-type (D) and lmp1-1 mutant (E). The inserted base is labeled in red in (E).
A complementation vector was constructed by fusing the LOC_Os09g32740 coding sequence with pCambia2300. The A. tumefaciens strain containing pCambia2300-OsLMP1 was transformed into the lmp1-1 calli. The regenerated lines were observed with the naked eye and detected by PCR amplification (Figure 3A). Ten positive lines showed restoration of the normal leaf color (Figure 3B). The expression level of OsLMP1 in the complementation lines was restored to the expression level in the wild-type plants (Supplementary Figure 2). Measurement of the H2O2 contents in the leaves of the complementation lines (CP1–CP3) showed restoration to the wild-type levels (Figure 3C).
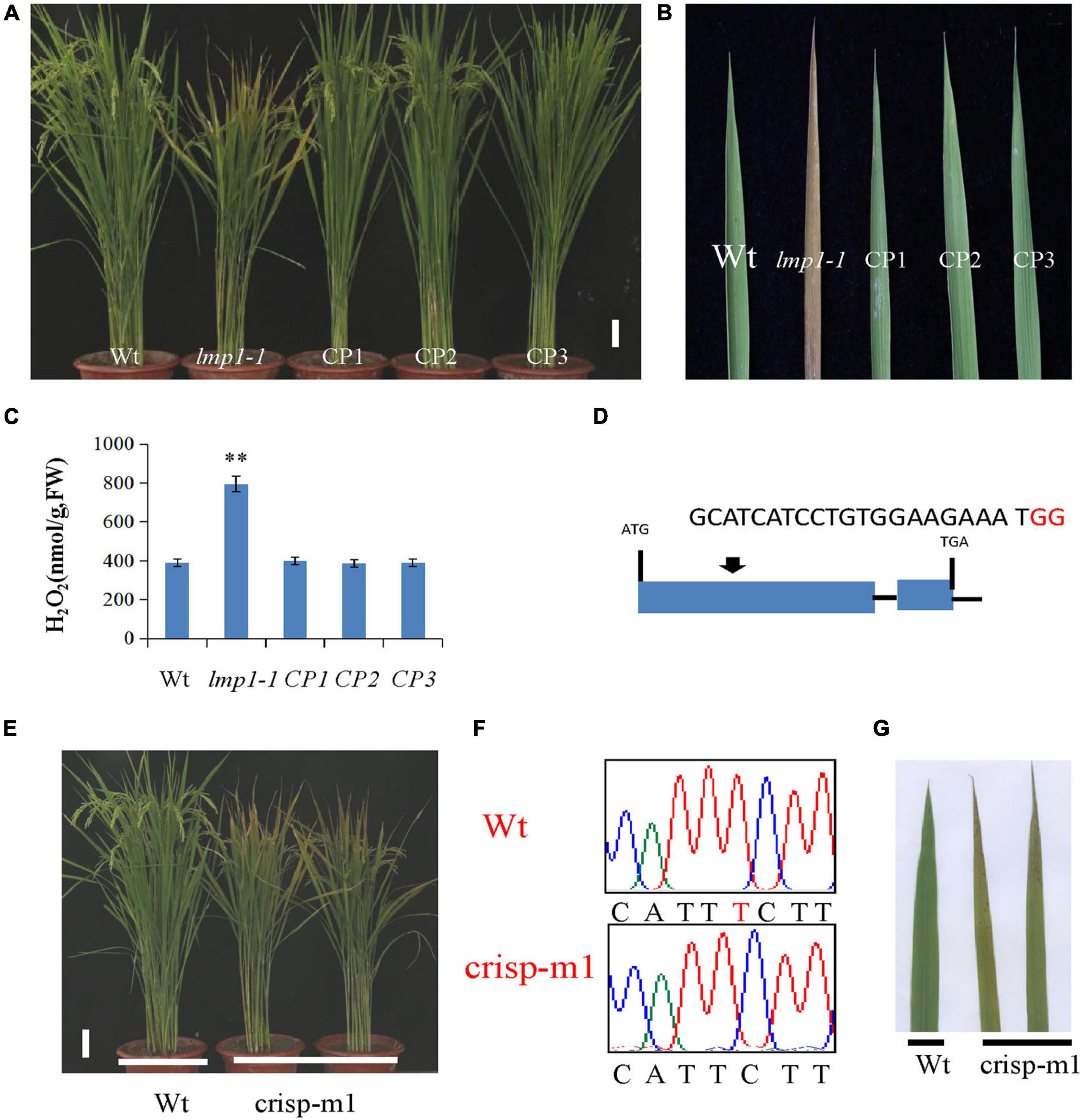
Figure 3. Verification of OsLMP1 by complementation assay and gene editing technology. (A) A complementation assay was conducted using full-length OsLMP1 cDNA. Phenotype comparison of field-grown wt, lmp1-1, and complementation lines (CP1–3) at the flowering stage (bar = 5 cm). (B) Leaves from the wt, lmp1-1, and complementation (CP1–3) lines collected from the plants shown in panel (A) show that complementation restored the lesion mimic phenotype at the flowering stage. (C) The H2O2 content of the wt, lmp1-1, and complementation (CP1–3) lines was measured at the flowering stage (n = 3). The significant differences were marked with asterisks based on student’s t-test (**P < 0.01). (D) Schematic map of the genomic region of OsLMP1 and the sgRNA target site; the arrow shows the sgRNA target site on the OsLMP1 genomic sequence, and the PAM motif (NGG) is shown in red. Blue boxes indicate OsLMP1 exons, and black lines indicate introns. (E) Phenotype comparison of the wt and crisp-m1 homozygous mutants at the flowering stage (bar = 5 cm). (F) The deleted base (1,549) is labeled in red in the wt. (G) Comparison of the wt and crisp-m1 leaves shows that crisp-m1 has the lesion mimic phenotype at the flowering stage.
In addition, an allelic mutant named crisp-m1 was created by CRISPR-Cas9 technology with the deletion of a single base (“T”) at position 1549 in the second exon, which led to early termination and an additional 55 amino acids (Figures 2C, 3D,F). The crisp-m1 plants also showed the lesion mimic phenotype at the flowering stage (Figures 3E,G), which was consistent with the observations of lmp1-1 plants. Overall, the complementation experiment and the allelic line verified that OsLMP1 is responsible for the lesion mimic phenotype.
Bioinformatics Prediction and Expression Pattern of OsLMP1
BLASTP sequence analysis with the NCBI database showed that OsLMP1 encodes a ubiquitin-specific protease containing a zinc finger motif at its N-terminus from amino acids (aa) 132 to 190 and a C-terminal hydrolase catalytic C19 domain from aa 226 to 1,051. Peptidase C19 contains ubiquitin hydrolases, which are intracellular peptidases that remove ubiquitin molecules from polyubiquitinated peptides by the cleavage of isopeptide bonds. The ubiquitin/proteasome system is responsible for most protein turnover in mammalian cells, and with over 50 members, the C19 family is one of the largest families of peptidases in the human genome (Piao et al., 2015). We performed cluster analysis of UCHs in Arabidopsis and rice. OsLMP1 shows low homology with other UCH family genes in rice and is most closely related to AtUBP1 and AtUBP2, with a sequence homology of 23.59% and 26.23% in Arabidopsis, respectively (Supplementary Figure 3). Similar conserved domain architecture (CDART) analysis with the NCBI database showed that the conserved domain of OsLMP1 exhibits the highest homology with the ubiquitin-specific protease UBP8 (Saccharomyces cerevisiae) (Supplementary Figure 4).
OsLMP1 encodes a deubiquitinase composed of 1,055 amino acids with a molecular weight of 116 kDa. To assess the localization pattern of OsLMP1, a fusion construct (35S:OsLMP1:GFP) and pMcherry (35S:OsMADS3:RFP, as a control) were transiently transformed into rice protoplasts by PEG transformation method. After 16 h of incubation at 28°C in the dark, the transformed protoplasts were observed using confocal laser scanning microscopy. GFP fluorescence in the image almost merged with the pMcherry fluorescence. Thus, the localization assay showed that OsLMP1 is mainly localized in the nucleus (Figures 4A–D).
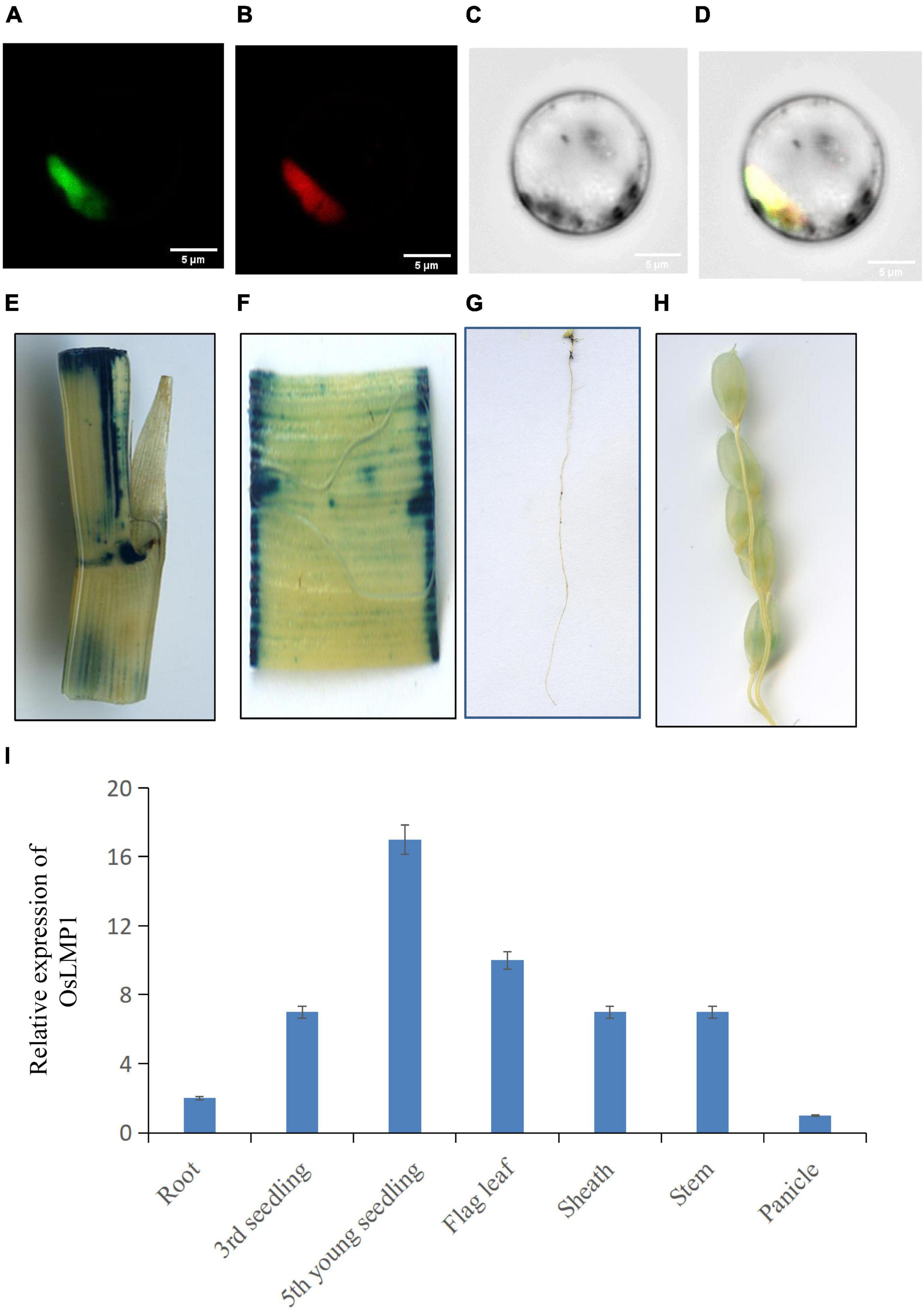
Figure 4. Subcellular location and the expression pattern of OsLMP1. (A–D) The 35S:OsLMP1:GFP vector was transformed into rice protoplasts, and the green fluorescent signal was visualized using confocal microscopy. OsMADS3, which localizes to the nuclei, was selected as the marker. The results revealed colocalization of the GFP signal with OsMADS3 fluorescence, indicating that OsLMP1 is mainly targeted to the rice nucleus. Bar: 100 μm. (E–H) Transgenic plants containing the OsLMP1-1391Z vector were stained using GUS dye at the flowering stage. (E) Stem; (F) leaf blade; (G) root; (H) panicle. The results showed that the stem, leaf blade and root were more deeply stained, whereas the panicle was weakly stained. (I) qRT-PCR analysis of OsLMP1 in the root, seedling, flag leaf, stem, sheath, and panicle tissues of the wt. The results showed that OsLMP1 is mainly expressed in young leaf at the early tiller stage.
The 2K promoter of OsLMP1 was amplified and fused with pCambia1391Z. The fused recombinant construct (OsLMP1-1391Z) was transformed into Nipponbare calli. A GUS staining experiment in the positive lines showed that OsLMP1 is preferably expressed in the leaf, stem and root and weakly expressed in the panicle (Figures 4E–H). The qRT-PCR results showed that OsLMP1 is most highly expressed in the young seedlings (at the tiller stage); moderately expressed in flag leaves, leaf blades; and weakly expressed in the roots and panicles, which is generally consistent with the GUS staining results (Figure 4I).
Transcriptome Analysis
To determine the regulatory pathways that OsLMP1 may be involved in, transcriptome analysis was conducted using wild-type and lmp1-1 mutant plants at the later tiller stage. A total of 2,737 genes were upregulated, and 1,064 genes were downregulated in lmp1-1 (at least three repeats, p ≤ 0.01) (Figure 5A and Supplementary Table 1). Some disease resistance-responsive family proteins, such as WRKYs and PRs, were activated in the lmp1-1 plants, which was consistent with the qRT-PCR results. We then performed Gene Ontology (GO) analysis to classify and identify the functions of the differentially expressed genes (DEGs) in the lmp1-1 plants (Figure 5C). A total of 64 GO terms were divided on the up-regulated datasets (at least three repeats, p ≤ 0.01). Among the GO terms 20 GO terms belong to biosynthetic process 42 terms belong to molecular function, and 2 terms belong to Component process (Supplementary Table 2). The “molecular function,” “biosynthetic process,” and “catalytic activity” terms were the terms most enriched in the DEGs, with p-values of 1.97E-10, 0.003708, and 6.78E-19, respectively (upregulated) (Supplementary Figure 7). A total of 38 GO terms were enriched in the downregulated DEGs, among which “catalytic activity,” “ion binding,” and “transferase activity” were the most enriched in the downregulated DEGs, with p-values of 5.01E-05, 0.0042861, and 6.72E-08, respectively (downregulated) (Supplementary Figure 8). Among the GO terms enriched in the downregulated and upregulated DEGs, the “catalytic activity” term was among the top three, indicating that OsLMP1 may regulate plant disease resistance through its catalytic activity. The biological pathways enriched in the lmp1-1 mutant plants were also explored, and we performed KEGG enrichment analysis of the DEGs between the lmp1-1 mutant and wild-type plants. The DEGs were mainly enriched in the “plant–pathogen interaction,” “phenylpropanoid biosynthesis,” and “biosynthesis of secondary metabolites” pathways (Figure 5B). In-depth analysis suggested that the upregulated DEGs were significantly enriched in seven pathways (P ≤ 0.01), among which (Supplementary Table 3 and Supplementary Figure 5) the “plant–pathogen interaction,” “alpha-linolenic acid metabolism,” “diterpenoid biosynthesis,” “phenylpropanoid biosynthesis,” “biosynthesis of secondary metabolites,” “biosynthesis of unsaturated fatty acids,” and “phenylalanine, tyrosine and tryptophan biosynthesis” pathways were the most enriched and prominent (upregulated). In contrast, the downregulated DEGs were enriched in six predominant pathways, among which the “diterpenoid biosynthesis,” “carbon fixation in photosynthetic organisms,” “biosynthesis of secondary metabolites,” “cyanoamino acid metabolism,” “carotenoid biosynthesis,” and “photosynthesis” pathways were the most significantly enriched pathways, with p-values of 0.00021, 0.00333, 0.00472, 0.01487, 0.01559, and 0.02610, respectively (downregulated) (Supplementary Table 3 and Supplementary Figure 6). These results indicated that OsLMP1-mediated disease resistance is likely associated with phenylalanine metabolic pathway in up-regulated database sets and the photosynthesis in down-regulated database sets.
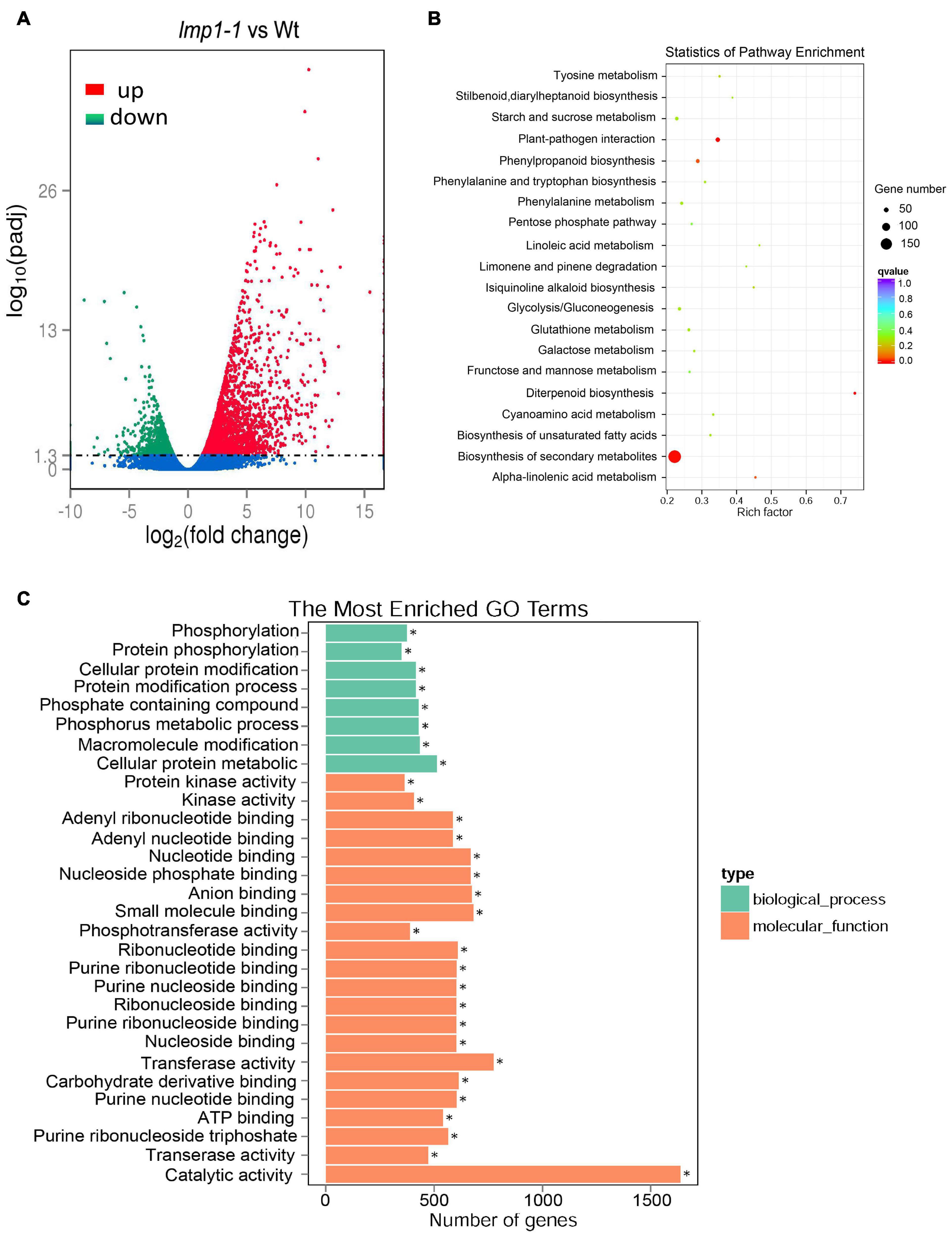
Figure 5. Transcriptome analysis and comparison of the lmp1-1 and wt lines. (A) The X-axis indicates the multiple of the difference after log2 conversion, and the Y-axis indicates the significance value after –log10 conversion for the lmp1-1 and wt lines. Points represent the percentage of the number of DEGs (ratios of the number of DEGs to the total number of detected genes enriched in the same GO terms) identified by paired transcriptome analysis. The numbers of upregulated DEGs are on the right of the backslashes, and the numbers of downregulated DEGs are on the left. vs., versus. (B) KEGG enrichment analysis of the lmp1-1 and wt liens. The X-axis indicates the enrichment ratio (the ratio of the number of genes annotated to an entry in the selected gene set to the total number of genes annotated to the entry in the species). The Y-axis indicates the KEGG pathway, and the size of the bubble indicates the number of genes. The color represents the enriched Q-value; the darker the color is, the smaller the Q-value is. (C) GO annotation of the DEGs from the leaves of the lmp1-1 and wt lines at the later tiller stage. The top 30 GO terms with P < 0.05 are shown here, and more information about the GO annotations is shown in Supplementary Table 2.
OsLMP1 Encodes a Functional Deubiquitinase Enzyme
Since OsLMP1 contains a C-terminal hydrolase catalytic domain, we determined whether OsLMP1 has deubiquitinase enzyme activity. Vectors for GST-OsLMP1, GST-OsLMP1-m (in which an “A” base was inserted at bp 2,604, generating a truncated protein) and GST (as a control) were constructed and expressed in E. coli. Western blot experiments using an antibody against GST showed that purified GST-OsLMP1 showed the expected molecular weight of 142 kDa and that GST-OsLMP1-m showed a molecular weight of 94 kDa, as expected for truncation (Figure 6A). Then, the GST-OsLMP1 and GST-OsLMP1-m vectors were coexpressed with the substrate His-UBQ10 in E. coli, respectively. Western blot assays were conducted using an anti-ubiquitin antibody. The results showed that GST-OsLMP1 could cleave ubiquitin molecules from the substrate His-UBQ10, but the ubiquitin precursor His-OsUBQ10 was not cleaved by GST-OsLMP1-m (Figure 6B). Similarly, the substrate His-OsUBQ1 was completely cleaved by GST-OsLMP1, but only a small amount was cleaved by GST-OsLMP1-m (Figure 6C). The above results indicated that OsLMP1 has functional deubiquitinase activity, and the mutant OsLMP1 could not cleave ubiquitin molecules or had weak ubiquitin cleavage activity.
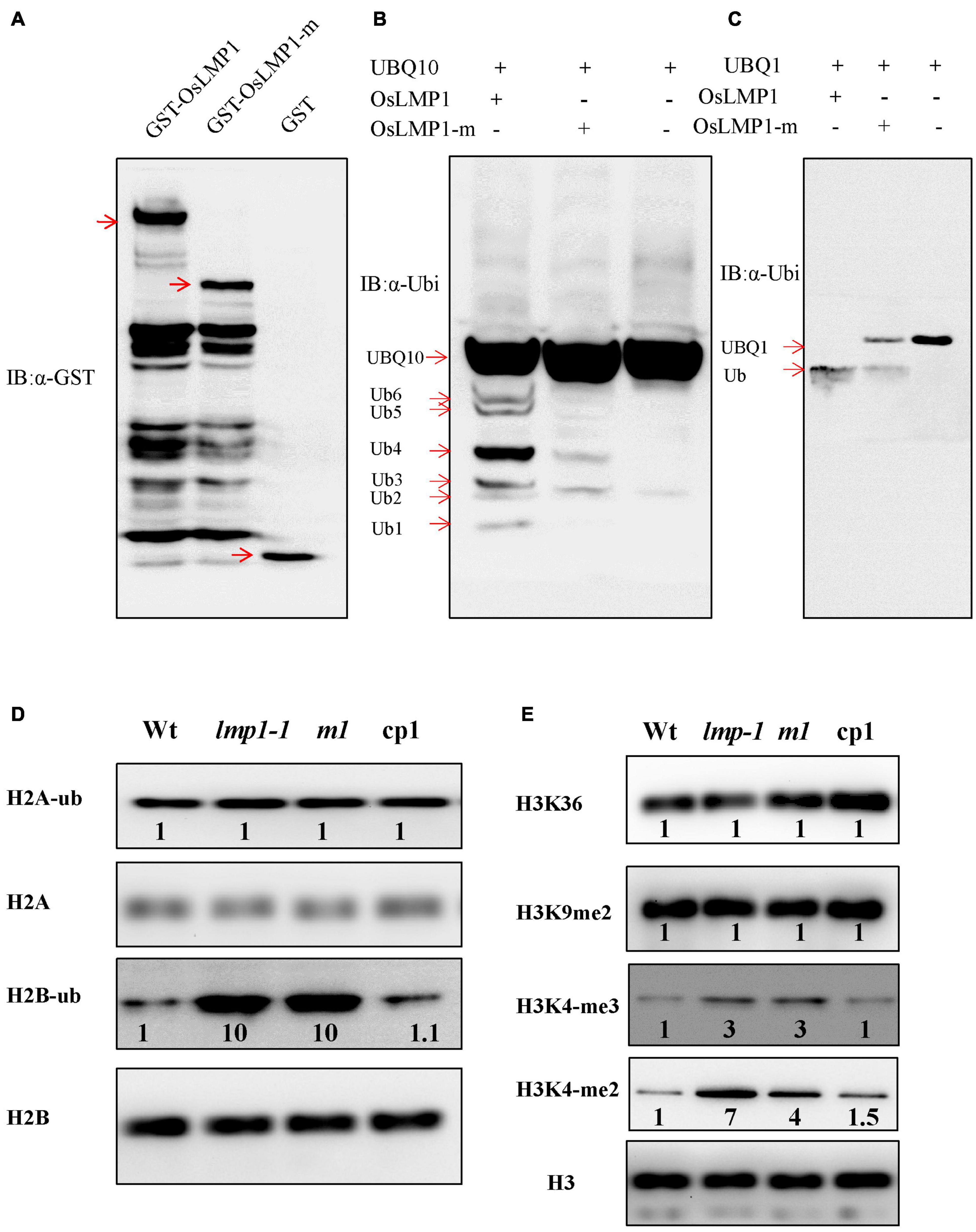
Figure 6. OsLMP1 is an active deubiquitinase. (A) The fused proteins GST-OsLMP1, GST-OsLMP1-m, and GST were purified in E. coli. Western blotting was conducted using an anti-GST antibody. Red arrows denote the expected bands. (B) The fused proteins GST-OsLMP1, GST-OsLMP1-m, and GST were coexpressed with His-UBQ10 in E. coli. Western blotting was conducted using an anti-ubiquitin (α-ubi) antibody. Immunoblot analysis showed that GST-OsLMP1 could cleave His-UBQ10, but GST-OsLMP1-m did not cleave His-UBQ10. Red arrows denoted the ubiquitination precursor bands. (C) The fused proteins GST-OsLMP1, GST-OsLMP1-m, and GST were coexpressed with His-UBQ1 in E. coli. Western blotting was conducted using an anti-ubiquitin antibody (α-ubi). Immunoblot analysis showed that GST-OsLMP1 could cleave His-UBQ1, but GST-OsLMP1-m did not cleave His-UBQ1. Red arrows denoted the ubiquitination precursor bands. (D) Western blot analysis of purified rice histone proteins at the later tiller stage with an anti-H2A/H2B antibody and anti-ubiquitin antibody. The levels of H2B-ub were higher in lmp1-1 and crisp-m1 plants than in wt plants. Band intensities were quantified using ImageJ analysis of the Western blots. The fold change shown above the blot is relative to wt controls normalized by antibody against H2A or H2B in each lane (n = 3). (E) Western blot analysis of purified rice histone proteins at the later tiller stage was conducted with an anti-H3 antibody or specific antibodies. The results show that the levels of H3K4-me2 and H3K4-me3 were higher in lmp1-1 and crisp-m1 plants than in wt plants. Band intensities were quantified using ImageJ analysis of the Western blots. The fold change shown above the blot is relative to wt controls normalized by anti-H3 antibody in each lane (n = 3).
OsLMP1 Deubiquitinates H2B
Similar CDART analysis showed that the conserved domains of OsLMP1 show the highest homology with the ubiquitin-specific protease UBP8 from S. cerevisiae (Supplementary Figure 3). In S. cerevisiae, Ubp8, Sgf11, Sus1, and Sgf73 form a large subcomplex known as the deubiquitination (DUB) module (Morgan et al., 2016). The catalytic domain of Ubp8 contacts H2B, leading to H2B deubiquitination at multiple stages of nucleosome disassembly and reassembly during transcription (Morgan et al., 2016). Thus, we speculated that OsLMP1 may also deubiquitinate histone H2A or H2B. Monoclonal antibodies specific for ubiquitinated histone H2A or H2B were used to detect the levels of H2B-ub and H2A-ub in the wild-type, lmp1-1, crisp-m1 and complementation lines at the later tiller stage. The results showed that the amount of H2A-ub in the mutants was comparable to that in the wild-type lines, but the amount of H2B-ub was significantly greater (by ∼10 times) in the lmp1-1 and crisp-m1 lines compared to the wild-type line (Figure 6D). The amount of H2B-ub in the complementation lines was restored to the level in the wild-type line. The above experimental results indicated that OsLMP1 may function by deubiquitinating H2B.
H2B ubiquitination is often a prerequisite for H3K4 and H3K9 methylation, and the two processes are closely coupled (Sridhar et al., 2007). Western blot assays at the later tiller stage were used to detect the H3 methylation status in the wild-type, lmp1-1, crisp-m1 and complementation lines with an anti-methylated H3 antibody (H3K4me2, H3K4me3, H3K9me2, and H3K36me2). The band intensities were quantified using ImageJ analysis of western blots. The fold change shown above the blot is relative to wt controls normalized by antibody against H2A or H2B in each lane. The imaging results showed that the levels of H3K4me2 and H3K4me3 methylation were significantly increased by at least 3 times in the lmp1-1 and crisp-m1 lines compared with the wild-type line, whereas H3K9me2 and H3K36me2 methylation levels in the lmp1-1 and crisp-m1 lines were comparable to those in the wild-type line (Figure 6E). The H3K4me2 and H3K4me3 methylation levels in the complementation lines were restored to the level in the wild-type line. Dot1L and Set1 build a ‘histone crosstalk’ bridge between ubiquitination of histone H2B on K120 and histone H3 (Worden et al., 2019). Our experiment supported that methylation of histone H3K4 was associated with active transcription, which was closely related to H2B deubiquitination.
OsLMP1 Is Involved in the Deubiquitination of OsPAL6 and OsPAL7 Chromatin
Gene expression is often coupled with increased H2B monoubiquitination (Batta et al., 2011). Thus, OsLMP1 may deubiquitylate H2B at the chromatin level. Many SA synthesis genes were activated in the lmp1-1 line (Supplementary Table 5). The expression levels of those SA synthesis genes were verified by qRT-PCR (Supplementary Figure 9), and the results were essentially consistent with the transcriptome data. In addition, the SA content in the lmp1-1 line at the heading stage was measured. The results showed that the SA content was significantly higher in the lmp1-1 line than in the wild-type line (Figure 1M). Because SA accumulated in the mutant plants, we speculated that the SA synthesis pathway genes may had undergone ubiquitination modification and thus the immune responses were activated in mutants. We examined this hypothesis using chromatin immunoprecipitation qPCR (ChIP -qPCR). Transcriptome analysis showed that OsLMP1 mediates immune responses associated with the phenylalanine metabolic pathway (Supplementary Table 5). SA synthesis pathway genes (OsPAL1, OsPAL6, OsPAL7, and OsICS1) were selected for ChIP -qPCR analysis. ChIP-qPCR assays at the 5th leaf stage revealed substantial levels of hyperubiquitinated H2B in OsPAL6 and OsPAL7 chromatin (Figures 7A,B), but not in OsPAL1 or OsICS1 chromatin in wild type (Figures 7C,D). The hyperubiquitinated region was located near the translation initiation codon in OsPAL6 and OsPAL7. The degree of ubiquitination ranged from ∼5 to 8 times higher than ubiquitination of the wild-type OsPAL6 and OsPAL7 chromatin (Figures 7A,B). The effect on OsPAL7 monoubiquitination was more significant than that on OsPAL6 monoubiquitination. In all, examination of H2B-ub throughout the length of OsPALs chromatin by chromatin immunoprecipitation (ChIP) analysis revealed enrichment of this histone modification within the promoter of the genes in wild type (Figures 7A,B).
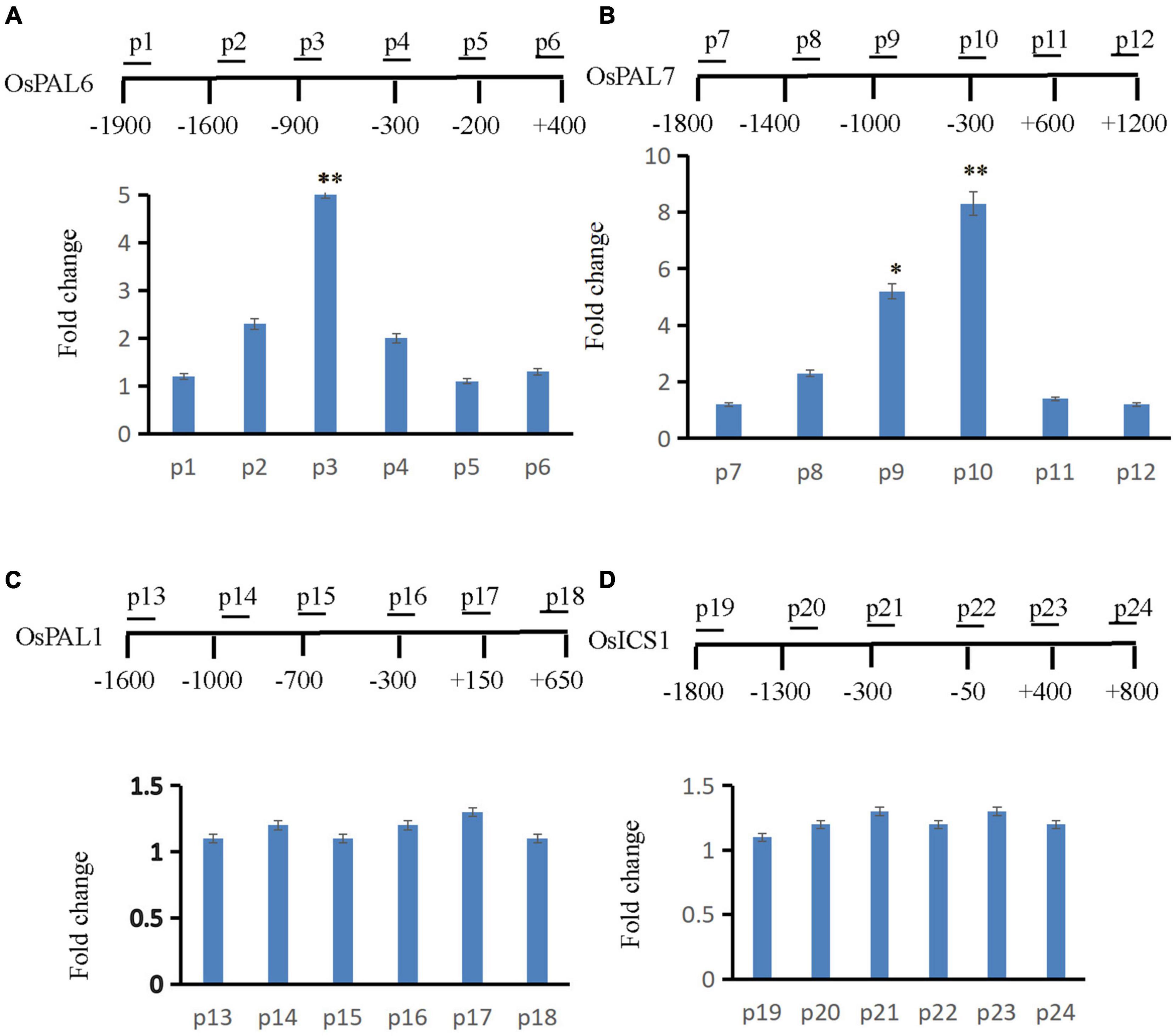
Figure 7. ChIP assay to analyze SA synthesis pathway genes. The supernatant was immunoprecipitated with an anti-H2B-Lys 120 antibody, and the resulting DNA of wt was eluted and subjected to quantitative PCR to assess genes involved in the SA synthesis pathway (A: OsPAL6, B: OsPAL7, C: OsPAL1, and D: OsICS1). The immunoprecipitated genomic fragments were quantified using real-time quantitative PCR and normalized to OsActin1 as an internal control.
Discussion
OsLMP1 Regulates the Immune Response in Rice
Lesion mimic mutants are ideal materials for the study of disease resistance. At present, at least 31 genes have been cloned in rice (Supplementary Table 4), and most act at the transcriptional level. Epigenetic modification is a posttranscriptional regulatory mechanism and plays an important role in the rice immune response. The JMJ705 protein, which contains the Jumonji C domain, is involved in the defense response in rice as it modifies histone H3 lysine 27 trimethylation (Li et al., 2013). PigmR confers broad spectrum resistance and is subjected to tight epigenetic regulation (Deng et al., 2017). In this study, we cloned OsLMP1 by map-based cloning and found that it encodes a deubiquitinase enzyme. The functions of UBP family members in plants have rarely been reported for the following reasons. First, many UCH family members exhibit functional redundancy in plants. For example, AtUBP3 and AtUBP4 are essential for pollen development and transmission in Arabidopsis (Doelling et al., 2007). GIGANTEA recruits the deubiquitylases AtUBP12 and AtUBP13 to regulate accumulation of the ZTL photoreceptor complex (Lee et al., 2019). Thus, it is difficult to identify the function in a single gene. However, sequence analysis showed that the rice genome contains no proteins with an amino acid similarity with OsLMP1 of more than 20%, suggesting that rice contains no redundant genes; thus, a single mutation in OsLMP1 causes a lesion mimic phenotype.
A series of assays confirmed that OsLMP1 regulates the plant immune response through the following findings. First, in the lmp1-1 mutant, the expression of pathogenesis-related genes, such as OsWRKY42, OsPR1, and PBZ1, was greatly increased compared to that in the wild-type line, which indicates that the defense response in the lmp1-1 mutant is significantly activated. Second, DAB staining and H2O2 measurements experiments pointed out that H2O2 contents were greatly increased in the mutant leaves, which indicated that PCD was activated and thus resulted in resistance to bacterial blight. Third, the amount of H2B-ub was significantly increased by ∼10 times in the lmp1-1 mutant. OsLMP1 functions by deubiquitinating H2B. Fourth, the SA content was higher in the lmp1-1 mutant than in the wild-type line. A ChIP-qPCR assay indicated that OsLMP1 was more enriched in the promoters of OsPAL6 and OsPAL7 than other OsPALs family members. The above four lines of evidence verified that OsLMP1 regulates the immune response by histone ubiquitination.
OsLMP1 Mediates Histone Ubiquitination in Rice
The ubiquitination and deubiquitination of histone H2B, like other histone modifications, regulate chromosome structure and gene transcription (Hu, 2012). Catalytic histone ubiquitination and deubiquitination enzymes have been successfully identified and cloned in yeast, animals and plants. Usp22 regulates histone H2B monoubiquitination and exhibits both oncogenic and tumor-suppressor roles in cancer (Jeusset and McManus, 2017). The deubiquitinase OTLD1 targets histone H2B to regulate seed size in Arabidopsis (Keren and Citovsky, 2016). However, only a few deubiquitinases have been identified in rice. OTUB1, due to its deubiquitinase activity, defines a new plant type associated with higher grain yield (Wang et al., 2017b). OsUBP15, a deubiquitinase, plays an important role in regulating grain width and size (Shi et al., 2019). Recent research has shown that the posttranslational regulation of H2B participates in non-host resistance and pathogen defense in plants (Ramirez-Prado et al., 2018). For example, AtHUB1 and AtHUB2, which encode two RING E3 ubiquitin ligase enzymes, catalyze H2B monoubiquitination. AtHUB1 and AtHUB2 are essential to fungal pathogens in Arabidopsis (Dhawan et al., 2009). Furthermore, reduced tomato H2B monoubiquitination by SiHUB1 and SiHUB2 knock down enhanced plant sensitivity to Botrytis cinerea (Zhang et al., 2015). In this study, we tested whether OsLMP1 mediates histone ubiquitination in rice. In E. coli assays, GST-OsLMP1 could cleave the ubiquitination precursor (Figure 6). Purified GST-OsLMP1 appeared as not only the targeted band but also many unknown bands at small molecular weights (Figure 6A). We speculated that the OsLMP1 protein may undergo unknown modifications, or the observation could have been due to incomplete expression of the full-length reading frame of OsLMP1. In this experiment, we found that the OsLMP1-m lanes contained some bands that were present in the wild-type OsLMP1 sample but not in the negative control (Figure 6B). We speculated that mutant OsLMP1 has weak deubiquitination activity, especially that in the OsLMP1 lane, with OsU BQ1 as a substrate (Figure 6C).
The ChIP-qPCR showed that OsLMP1 may be more enriched in the promoters of OsPAL6 and OsPAL7 but not those of OsPAL genes. He reported that planthopper resistance was regulated by SA from the PAL pathway in rice (He et al., 2020). In the study, the level of SA is significantly reduced in the OsPAL6 co-suppressed or OsPAL RNAi plants, but increased in the plants overexpressing OsPAL8. This study provide additional support for the notion that the PAL pathway is an important route of SA biosynthesis in rice (He et al., 2020). Higher SA content in lpm1-1 was perhaps a combined action of OsPAL6 and OsPAL7. The expression levels of OsPAL2 and OsPAL4 were slightly increased in the mutants, which may have been due to feedback regulation of SA signaling. Isochorismate synthase (ICS) and phenylalanine ammonia lyase (PAL) are two important genes in the plant SA synthesis pathway. OsPALs, and not OsICS1, may be primarily responsible for SA synthesis in rice (He et al., 2020). However, AtICS1 is mainly responsible for SA synthesis in Arabidopsis. Furthermore, the temporal and spatial expression patterns and mechanisms of PALs and ICSs in Arabidopsis and rice need to be further studied. In addition, we tried to verify the interaction of OsLMP1 with H2B variants, but the results did not verify their interaction. Thus, OsLMP1 and H2B may be connected through a new regulatory link.
A Suggested Model of OsLMP1 Regulation
We suggest a model (Supplementary Figure 11) in which the OsLMP1 complex maintains deubiquitinase activity under normal growth conditions in the wild-type, which maintains the level of histone H2B ubiquitination at the background level or lower at different developmental stages, thus inhibiting the expression of disease resistance-related genes. In the lmp1-1 mutant, the deubiquitination activity of the OsLMP1 complex is decreased, and the levels of histone H2B ubiquitination and methylation are increased, which activates the expression of disease resistance-related genes. Under conditions of pathogen infection, unknown factors may reduce OsLMP1 complex activity (Supplementary Figure 10), increasing H2B ubiquitination levels, thus activating the expression of disease resistance-related genes and conferring resistance in plants (Figure 7).
In summary, this study preliminarily reveals that OsLMP1 regulates plant disease resistance by epigenetically modifying histone H2B. OsLMP1 loss of function led to high levels of histone H2B ubiquitination and histone H3-K4me2/3 methylation, directly activated SA synthesis genes (OsPAL6 and OsPAL7) and improved plant disease resistance. This pathway may be conserved in animals and plants. However, whether other UCH family members also participate in H2B deubiquitination requires further study. Moreover, this study also provides important insight suggesting that other UCH family members in plants induced by pathogenic bacteria are also related to plant disease resistance. In addition, because the deubiquitination and ubiquitination processes are coupled, we would expect to find ubiquitination-related proteins in rice related to disease resistance.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/ and https://www.ncbi.nlm.nih.gov/sra/PRJNA694229.
Author Contributions
JS and YC performed the map-based cloning of OsLMP1. WS made the expression analysis and subcellular localization experiments. YW and WS performed western blotting and ChIP experiments. YC conducted the transgenic experiments. ZZ and TL designed the research and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (31771884), the Agricultural Science and Technology Innovation Program (CAAS-ZDXT2019003), and National Key Research and Development Program of China (2020YFA0907603).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.814465/full#supplementary-material
Supplementary Figure 1 | Enhanced resistance in the lmp1-1 mutant to Magnaporthe oryzae and Xanthomonas oryzae pv. oryzae isolates.
Supplementary Figure 2 | The expression level of OsLMP1 in the complementation lines (CP1–CP3) was restored to the expression level in the wild-type line.
Supplementary Figure 3 | Phylogenetic tree showing the predicted relationships between OsLMP1 and homologous proteins in rice and Arabidopsis.
Supplementary Figure 4 | Similar conserved domain architecture (CDART) analysis with NCBI showed that the conserved domain in OsLMP1 exhibits the highest homology with the ubiquitin-specific protease UBP8 (Saccharomyces cerevisiae).
Supplementary Figure 5 | Up-regulated genes KEGG enrichment analysis of the lmp1-1 and wt lines.
Supplementary Figure 6 | Down-regulated genes KEGG enrichment analysis of the lmp1-1 and wt lines.
Supplementary Figure 7 | GO annotation of the up-regulated DEGs from the leaves of the lmp1-1 and wt lines at the later tiller stage.
Supplementary Figure 8 | GO annotation of the down-regulated DEGs from the leaves of the lmp1-1 and wt lines at the later tiller stage.
Supplementary Figure 9 | The expression levels of SA synthesis genes, including OsPAL1-7 and OsICS1, were verified by qRT-PCR (A–H).
Supplementary Figure 10 | Time course of OsLMP1 expression in the later tiller stage in the seedlings of wt plants treated with rice bacterial blight (PXO99) using the leaf cutting method.
Supplementary Figure 11 | A model of the role of OsLMP1 in the defense response.
Supplementary Table 1 | Differential analysis of the lmp1-1 vs. wt lines.
Supplementary Table 2 | Results of GO enrichment analysis of DEGs in the lmp1-1 line vs. the wt line.
Supplementary Table 3 | Results of KEGG analysis of the lmp1-1 line vs. the wt line.
Supplementary Table 4 | Cloned lesion mimic genes in rice.
Supplementary Table 5 | Fold change in the expression of SA-related genes in the lmp1-1 line vs. the wt line.
Supplementary Table 6 | Primer sequences used.
Footnotes
References
Amerik, A. Y., and Hochstrasser, M. (2004). Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207. doi: 10.1016/j.bbamcr.2004.10.003
Batta, K., Zhang, Z., Yen, K., Goffman, D. B., and Pugh, B. F. (2011). Genome-wide function of H2B ubiquitylation in promoter and genic regions. Genes Dev. 25, 2254–2265. doi: 10.1101/gad.177238.111
Bouchez, O., Huard, C., Lorrain, S., Roby, D., and Balague, C. (2007). Ethylene is one of the key elements for cell death and defense response control in the Arabidopsis lesion mimic mutant vad1. Plant Physiol. 145, 465–477. doi: 10.1104/pp.107.106302
Cai, H., Wang, W., Rui, L., Han, L., Luo, M., Liu, N., et al. (2021). The TIR-NBS protein TN13 associates with the CC-NBS-LRR resistance protein RPS5 and contributes to RPS5-triggered immunity in Arabidopsis. Plant J. 107, 775–786. doi: 10.1111/tpj.15345
Chen, G. D., and Jastreboff, P. J. (1995). Salicylate-induced abnormal activity in the inferior colliculus of rats. Hear Res. 82, 158–178. doi: 10.1016/0378-5955(94)00174-o
Chen, W., Liu, P., Zhang, A., Ren, J., and Xu, L. X. (2006). Quantification of quantum dots in HUVECs by confocal laser scanning microscopy. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006, 1478–1481. doi: 10.1109/IEMBS.2006.260265
Clague, M. J., Barsukov, I., Coulson, J. M., Liu, H., Rigden, D. J., and Urbe, S. (2013). Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315. doi: 10.1152/physrev.00002.2013
Deng, Y., Zhai, K., Xie, Z., Yang, D., Zhu, X., Liu, J., et al. (2017). Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355, 962–965. doi: 10.1126/science.aai8898
Dhawan, R., Luo, H., Foerster, A. M., Abuqamar, S., Du, H. N., Briggs, S. D., et al. (2009). Histone monoubiquitination1 interacts with a subunit of the mediator complex and regulates defense against necrotrophic fungal pathogens in Arabidopsis. Plant Cell 21, 1000–1019. doi: 10.1105/tpc.108.062364
Doelling, J. H., Phillips, A. R., Soyler-Ogretim, G., Wise, J., Chandler, J., Callis, J., et al. (2007). The ubiquitin-specific protease subfamily UBP3/UBP4 is essential for pollen development and transmission in Arabidopsis. Plant Physiol. 145, 801–813. doi: 10.1104/pp.106.095323
Feng, X., Zhang, L., Wei, X., Zhou, Y., Dai, Y., and Zhu, Z. (2020). OsJAZ13 negatively regulates jasmonate signaling and activates hypersensitive cell death response in Rice. Int. J. Mol. Sci. 21:4379. doi: 10.3390/ijms21124379
Gao, H., Zheng, X. M., Fei, G., Chen, J., Jin, M., Ren, Y., et al. (2013). Ehd4 encodes a novel and Oryza-genus-specific regulator of photoperiodic flowering in rice. PLoS Genet. 9:e1003281. doi: 10.1371/journal.pgen.1003281
He, J., Liu, Y., Yuan, D., Duan, M., Liu, Y., Shen, Z., et al. (2020). An R2R3 MYB transcription factor confers brown planthopper resistance by regulating the phenylalanine ammonia-lyase pathway in rice. Proc. Natl. Acad. Sci. U. S. A. 117, 271–277. doi: 10.1073/pnas.1902771116
Hiei, Y., Ohta, S., Komari, T., and Kumashiro, T. (1994). Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 6, 271–282.
Hu, H. Y. (2012). Editorial: protein ubiquitination and deubiquitination. Curr. Protein Pept. Sci. 13:413. doi: 10.2174/138920312802430626
Jefferson, R. A., Kavanagh, T. A., and Bevan, M. W. (1987). GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907.
Jeusset, L. M., and McManus, K. J. (2017). Ubiquitin Specific Peptidase 22 Regulates histone H2B mono-ubiquitination and exhibits both oncogenic and tumor suppressor roles in cancer. Cancers (Basel) 9:167. doi: 10.3390/cancers9120167
Jubany-Mari, T., Munne-Bosch, S., Lopez-Carbonell, M., and Alegre, L. (2009). Hydrogen peroxide is involved in the acclimation of the Mediterranean shrub, Cistus albidus L., to summer drought. J. Exp. Bot. 60, 107–120. doi: 10.1093/jxb/ern274
Kaminaka, H., Nake, C., Epple, P., Dittgen, J., Schutze, K., Chaban, C., et al. (2006). bZIP10-LSD1 antagonism modulates basal defense and cell death in Arabidopsis following infection. EMBO J. 25, 4400–4411. doi: 10.1038/sj.emboj.7601312
Katsiarimpa, A., Munoz, A., Kalinowska, K., Uemura, T., Rojo, E., and Isono, E. (2014). The ESCRT-III-interacting deubiquitinating enzyme AMSH3 is essential for degradation of ubiquitinated membrane proteins in Arabidopsis thaliana. Plant Cell Physiol. 55, 727–736. doi: 10.1093/pcp/pcu019
Keren, I., and Citovsky, V. (2016). The histone deubiquitinase OTLD1 targets euchromatin to regulate plant growth. Sci. Signal 9:ra125. doi: 10.1126/scisignal.aaf6767
Kong, L., Qiu, X., Kang, J., Wang, Y., Chen, H., Huang, J., et al. (2017). A phytophthora effector manipulates host histone acetylation and reprograms defense gene expression to promote infection. Curr. Biol. 27, 981–991. doi: 10.1016/j.cub.2017.02.044
Lee, C. M., Li, M. W., Feke, A., Liu, W., Saffer, A. M., and Gendron, J. M. (2019). GIGANTEA recruits the UBP12 and UBP13 deubiquitylases to regulate accumulation of the ZTL photoreceptor complex. Nat. Commun. 10:3750. doi: 10.1038/s41467-019-11769-7
Li, H., Wang, H., Jing, M., Zhu, J., Guo, B., Wang, Y., et al. (2018). A Phytophthora effector recruits a host cytoplasmic transacetylase into nuclear speckles to enhance plant susceptibility. Elife 22:e40039. doi: 10.7554/eLife.40039
Li, T., Chen, X., Zhong, X., Zhao, Y., Liu, X., Zhou, S., et al. (2013). Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 25, 4725–4736. doi: 10.1105/tpc.113.118802
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, M., Luo, M. Z., Buzas, D., Finnegan, J., Helliwell, C., Dennis, E. S., et al. (2008). Ubiquitin-specific protease 26 is required for seed development and the repression of PHERES1 in Arabidopsis. Genetics 180, 229–236. doi: 10.1534/genetics.108.091736
Moon, B. C., Choi, M. S., Kang, Y. H., Kim, M. C., Cheong, M. S., Park, C. Y., et al. (2005). Arabidopsis ubiquitin-specific protease 6 (AtUBP6) interacts with calmodulin. FEBS Lett. 579, 3885–3890. doi: 10.1016/j.febslet.2005.05.080
Moon, Y. K., Hong, J. P., Cho, Y. C., Yang, S. J., An, G., and Kim, W. T. (2009). Structure and expression of OsUBP6, an ubiquitin-specific protease 6 homolog in rice (Oryza sativa L.). Mol. Cells 28, 463–472. doi: 10.1007/s10059-009-0138-4
Morgan, M. T., Haj-Yahya, M., Ringel, A. E., Bandi, P., Brik, A., and Wolberger, C. (2016). Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science 351, 725–728. doi: 10.1126/science.aac5681
Mosher, S., Moeder, W., Nishimura, N., Jikumaru, Y., Joo, S. H., Urquhart, W., et al. (2010). The lesion-mimic mutant cpr22 shows alterations in abscisic acid signaling and abscisic acid insensitivity in a salicylic acid-dependent manner. Plant Physiol. 152, 1901–1913. doi: 10.1104/pp.109.152603
Mou, Z., He, Y., Dai, Y., Liu, X., and Li, J. (2000). Deficiency in fatty acid synthase leads to premature cell death and dramatic alterations in plant morphology. Plant Cell 12, 405–418. doi: 10.1105/tpc.12.3.405
Pan, R., Kaur, N., and Hu, J. (2014). The Arabidopsis mitochondrial membrane-bound ubiquitin protease UBP27 contributes to mitochondrial morphogenesis. Plant J. 78, 1047–1059. doi: 10.1111/tpj.12532
Piao, J., Tashiro, A., Nishikawa, M., Aoki, Y., Moriyoshi, E., Hattori, A., et al. (2015). Expression, purification and enzymatic characterization of a recombinant human ubiquitin-specific protease 47. J. Biochem. 158, 477–484. doi: 10.1093/jb/mvv063
Ramirez-Prado, J. S., Abulfaraj, A. A., Rayapuram, N., Benhamed, M., and Hirt, H. (2018). Plant immunity: from signaling to epigenetic control of defense. Trends Plant Sci. 23, 833–844. doi: 10.1016/j.tplants.2018.06.004
Schmitz, R. J., Tamada, Y., Doyle, M. R., Zhang, X., and Amasino, R. M. (2009). Histone H2B deubiquitination is required for transcriptional activation of FLOWERING LOCUS C and for proper control of flowering in Arabidopsis. Plant Physiol. 149, 1196–1204. doi: 10.1104/pp.108.131508
Shi, C., Ren, Y., Liu, L., Wang, F., Zhang, H., Tian, P., et al. (2019). Ubiquitin specific protease 15 has an important role in regulating grain width and size in Rice. Plant Physiol. 180, 381–391. doi: 10.1104/pp.19.00065
Singh, S. K., Sung, T. Y., Chung, T. Y., Lin, S. Y., Lin, S. C., Liao, J. C., et al. (2018). ACR11 modulates levels of reactive oxygen species and salicylic acid-associated defense response in Arabidopsis. Sci. Rep. 8:11851. doi: 10.1038/s41598-018-30304-0
Sridhar, V. V., Kapoor, A., Zhang, K., Zhu, J., Zhou, T., Hasegawa, P. M., et al. (2007). Control of DNA methylation and heterochromatic silencing by histone H2B deubiquitination. Nature 447, 735–738. doi: 10.1038/nature05864
Sun, C., Liu, L., Tang, J., Lin, A., Zhang, F., Fang, J., et al. (2011). RLIN1, encoding a putative coproporphyrinogen III oxidase, is involved in lesion initiation in rice. J. Genet. Genomics 38, 29–37. doi: 10.1016/j.jcg.2010.12.001
Sun, Q., Collins, N. C., Ayliffe, M., Smith, S. M., Drake, J., Pryor, T., et al. (2001). Recombination between paralogues at the Rp1 rust resistance locus in maize. Genetics 158, 423–438. doi: 10.1093/genetics/158.1.423
Tang, J., Zhu, X., Wang, Y., Liu, L., Xu, B., Li, F., et al. (2011). Semi-dominant mutations in the CC-NB-LRR-type R gene, NLS1, lead to constitutive activation of defense responses in rice. Plant J. 66, 996–1007. doi: 10.1111/j.1365-313X.2011.04557.x
Vejan, P., Abdullah, R., Khadiran, T., Ismail, S., and Nasrulhaq, B. A. (2016). Role of plant growth promoting rhizobacteria in agricultural sustainability-A Review. Molecules 21:573. doi: 10.3390/molecules21050573
Wan, S., Wu, J., Zhang, Z., Sun, X., Lv, Y., Gao, C., et al. (2009). Activation tagging, an efficient tool for functional analysis of the rice genome. Plant Mol. Biol. 69, 69–80. doi: 10.1007/s11103-008-9406-5
Wang, C., Fan, Y., Zheng, C., Qin, T., Zhang, X., and Zhao, K. (2014). High-resolution genetic mapping of rice bacterial blight resistance gene Xa23. Mol. Genet. Genomics 289, 745–753. doi: 10.1007/s00438-014-0848-y
Wang, S., Lei, C., Wang, J., Ma, J., Tang, S., Wang, C., et al. (2017a). SPL33, encoding an eEF1A-like protein, negatively regulates cell death and defense responses in rice. J. Exp. Bot. 68, 899–913. doi: 10.1093/jxb/erx001
Wang, S., Wu, K., Qian, Q., Liu, Q., Li, Q., Pan, Y., et al. (2017b). Non-canonical regulation of SPL transcription factors by a human OTUB1-like deubiquitinase defines a new plant type rice associated with higher grain yield. Cell Res. 27, 1142–1156. doi: 10.1038/cr.2017.98
Worden, E., Hoffmann, N., Hicks, C., and Wolberge, R. (2019). Mechanism of cross-talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 176, 1490–1501. doi: 10.1016/j.cell.2019.02.002
Wu, C., Bordeos, A., Madamba, M. R., Baraoidan, M., Ramos, M., Wang, G. L., et al. (2008). Rice lesion mimic mutants with enhanced resistance to diseases. Mol. Genet. Genomics 279, 605–619. doi: 10.1007/s00438-008-0337-2
Yang, P., Smalle, J., Lee, S., Yan, N., Emborg, T. J., and Vierstra, R. D. (2007). Ubiquitin C-terminal hydrolases 1 and 2 affect shoot architecture in Arabidopsis. Plant J. 51, 441–457. doi: 10.1111/j.1365-313X.2007.03154.x
Zhai, K., Liang, D., Li, H., Jiao, F., Yan, B., Liu, J., et al. (2021). NLRs guard metabolism to coordinate pattern- and effector-triggered immunity. Nature doi: 10.1038/s41586-021-04219-2 [Epub ahead of print].
Zhang, Y., Li, D., Zhang, H., Hong, Y., Huang, L., Liu, S., et al. (2015). Tomato histone H2B monoubiquitination enzymes SlHUB1 and SlHUB2 contribute to disease resistance against Botrytis cinerea through modulating the balance between SA- and JA/ET-mediated signaling pathways. BMC Plant Biol. 15:252. doi: 10.1186/s12870-015-0614-2
Zhang, Y., Su, J., Duan, S., Ao, Y., Dai, J., Liu, J., et al. (2011). A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 7:30. doi: 10.1186/1746-4811-7-30
Zhang, Z., Zhang, Q., Wu, J., Zheng, X., Zheng, S., Sun, X., et al. (2013). Gene knockout study reveals that cytosolic ascorbate peroxidase 2 (OsAPX2) plays a critical role in growth and reproduction in rice under drought, salt and cold stresses. PLoS One 8:e57472. doi: 10.1371/journal.pone.0057472
Keywords: rice, lesion mimic, epigenetic modification, histone deubiquitination, immune response, SA
Citation: Sun J, Song W, Chang Y, Wang Y, Lu T and Zhang Z (2022) OsLMP1, Encoding a Deubiquitinase, Regulates the Immune Response in Rice. Front. Plant Sci. 12:814465. doi: 10.3389/fpls.2021.814465
Received: 13 November 2021; Accepted: 28 December 2021;
Published: 18 January 2022.
Edited by:
Juan Ignacio Vilchez Morillas, Universidade Nova de Lisboa, PortugalReviewed by:
Ana Santos, Universidade Nova de Lisboa, PortugalMeng Yuan, Huazhong Agricultural University, China
Copyright © 2022 Sun, Song, Chang, Wang, Lu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiegang Lu, bHV0aWVnYW5nQGNhYXMuY24=; Zhiguo Zhang, emhhbmd6aGlndW9AY2Fhcy5jbg==
†These authors have contributed equally to this work