 Xiangyu Tian1
Xiangyu Tian1 Luye Shi1Jia Guo1Liuyang Fu1,2Pei Du2Bingyan Huang2Yue Wu1Xinyou Zhang2*Zhenlong Wang1*
Luye Shi1Jia Guo1Liuyang Fu1,2Pei Du2Bingyan Huang2Yue Wu1Xinyou Zhang2*Zhenlong Wang1*- 1School of Life Sciences, Zhengzhou University, Zhengzhou, China
- 2Key Laboratory of Oil Crops in Huang-Huai-Hai Plains, Ministry of Agriculture and Rural Affairs, Henan Provincial Key Laboratory for Oil Crops Improvement, Henan Institute of Crop Molecular Breeding, Henan Academy of Agricultural Sciences, Zhengzhou, China
Peanuts (Arachis hypogaea L.) offer numerous healthy benefits, and the production of peanuts has a prominent role in global food security. As a result, it is in the interest of society to improve the productivity and quality of peanuts with transgenic means. However, the lack of a robust phylogeny of cultivated and wild peanut species has limited the utilization of genetic resources in peanut molecular breeding. In this study, a total of 33 complete peanut plastomes were sequenced, analyzed and used for phylogenetic analyses. Our results suggest that sect. Arachis can be subdivided into two lineages. All the cultivated species are contained in Lineage I with AABB and AA are the two predominant genome types present, while species in Lineage II possess diverse genome types, including BB, KK, GG, etc. Phylogenetic studies also indicate that all allotetraploid cultivated peanut species have been derived from a possible maternal hybridization event with one of the diploid Arachis duranensis accessions being a potential AA sub-genome ancestor. In addition, Arachis monticola, a tetraploid wild species, is placed in the same group with all the cultivated peanuts, and it may represent a transitional species, which has been through the recent hybridization event. This research could facilitate a better understanding of the taxonomic status of various Arachis species/accessions and the evolutionary relationship among them, and assists in the correct and efficient use of germplasm resources in breeding efforts to improve peanuts for the benefit of human beings.
Introduction
The genus Arachis consists of approximately 81 species, which represent nine sections and 16 genome types, and are mainly distributed in the tropics and subtropics of South America (Stalker, 2017). Among these, peanut or groundnut (Arachis hypogaea L.) is a world-famous legume crop and cultivated by more than one hundred countries in the tropical and subtropical regions (Singh and Moss, 1982; Varshney et al., 2009; Pandey et al., 2020). Peanut was domesticated about 3,500–9400 years ago in South America (Bertioli et al., 2019; Chen et al., 2019; Zhuang et al., 2019). It is known as the “longevity fruit,” “poor man’s almonds” because it is an excellent source of good fats and proteins (∼80% of seed content). Peanut has also become one of the most important contributors to human health and food security (Konate et al., 2020). In addition to cultivated peanuts, some wild species including Arachis glabrata, Arachis pintoi, Arachis stenosperma, and Arachis villosulicarpa, etc. are also used as food and medicine (Stalker, 2017). More importantly, some wild Arachis species possess many agronomic traits, such as disease and pest resistances (Subrahmanyam et al., 2001; Tallury et al., 2014), which are important in crop improvement, but these traits are not present in cultivated species (Upadhyaya et al., 2011). Although progress has been made through conventional breeding, yet the confusing species barrier between cultivated peanuts and wild species makes the utilization of genetic resources very difficult. The lack of a robust phylogeny of the Arachis genus has impeded the advances in basic biological research and molecular breeding of the cultivated peanuts.
Allotetraploidy, which are evident in soybean, Brassica, wheat, cotton, and peanut via whole chromosomal genome (Gill et al., 2009; Feldman et al., 2012; Paterson et al., 2012; Chalhoub et al., 2014; Bertioli et al., 2019; Zhuang et al., 2019), plays a critical role in the evolving history of most domesticated crop species. However, how allotetraploids species (e.g., cultivated peanut) have evolved from their diploid parents remains largely unknown (Bertioli et al., 2020; Zhuang et al., 2020). The lack of information is caused by two possible reasons: (1) morphological and molecular phylogenetic studies are not efficient in distinguishing taxonomic species for some horticulture features may have resulted from domestication. (2) Genetic diversity introduced by multiple parental inheritance makes it difficult to detect homology among sequences. According to a few previous studies, cultivated peanuts are allotetraploid (AABB genome type) and derived from two diploids wild species by a recent hybridization event (Bertioli et al., 2019; Zhuang et al., 2019). Many studies suggest that A. duranensis Krapov. & W.C.Greg. (AA) and Arachis ipaensis Krapov. & W.C.Greg. (BB) are the progenitor species, which provide valuable genetic resources to A. hypogaea (Kochert et al., 1996; Koppolu et al., 2010; Bertioli et al., 2011, 2016). However, some other studies support that cultivated peanuts may have been derived from more than two progenitor species, including Arachis diogoi Hoehne (AA), Arachis correntina (Burkart) Krapov. & W.C.Greg. (AA), Arachis cardenasii Krapov. & W.C.Greg. (AA), A. batizocoi Krapov. & W.C.Greg. (KK), A. trinitensis Krapov. & W.C.Greg. (FF), and A. williamsii Krapov. & W.C.Greg. (BB) (Stalker et al., 1991; Singh et al., 1994; Leal-Bertioli et al., 2014; Wang et al., 2019; Zhuang et al., 2019). The origination and evolution of the cultivated peanut species remains elusive, and it is extremely difficult to demarcate the boundary of some peanut species due to gene introgression, ancestral polymorphism and various speciation rates in different species (Moretzsohn et al., 2013; Bertioli et al., 2019).
The previous classification has put cultivated peanuts into two groups, subsp. hypogaea and subsp. fastigiata, based on some morphological and physiological characteristics, such as the presence of flower on main stem, time of maturation, the presence of seed dormancy, etc. (Gibbons et al., 1972; Krapovickas et al., 2007; Belamkar et al., 2011). According to some early classification work, which studied the growth habit, leaflet surface, branching pattern and pod traits of various peanuts (Ferguson et al., 2004; Krapovickas et al., 2007), subsp. hypogaea contain two botanical varieties, var. hypogaea, var. hirsute, while four varieties (var. fastigiata, var. peruviana, var. vulgaris, and var. aequatoriana) are present in subsp. fastigiata. However, classification based on morphological and physiological characteristics is not consistently supported by works done at the molecular level when employing different methods or using different genetic markers (He and Prakash, 2001; Gimenes et al., 2002; Moretzsohn et al., 2004; Koppolu et al., 2010). A molecular analysis using the AFLP approach shows that var. aequatoriana and var. peruviana are closely related to subsp. hypogaea (He and Prakash, 2001). Furthermore, a study carried out with SSRs markers put var. peruviana into subsp. hypogaea. More interestingly, var. hypogaea and var. hirsute, which are originally placed in subsp. hypogaea, are not even closely related according to Ferguson et al. (2004). The conventional classification of cultivated peanuts is supported by one recent study, which looked at high-quality SNPs in the peanut nuclear genomes (Zheng et al., 2018). However, the taxonomic boundaries among some botanical varieties cannot be clearly delimited in this study. Var. hypogaea and var. hirsute could not be distinguished due to difficulties in putting different accessions of the same variety into one cluster. A close evolutionary relationship was inferred between var. hirsute and var. vulgaris when using the plastomics approach (Wang et al., 2018, 2019). This study also supports a close relationship between var. hypogaea and var. fastigiata, which is different from what we would expect based on the previous classification. It seems that nuclear genomic sequence data is not sufficient or reliable in interpreting evolutionary relationship among allotetraploid species. Due to the lack of consistency, a study carried out with a different type of sequence data (i.e., plastomic data) or employing various analytic methods would be appropriate when trying to reconstruct the phylogeny of cultivated peanuts.
Plastomics provide a powerful tool in phylogenetic studies involving particular evolutionary events, such as interspecific hybridization, allopolyploidization, rapid evolution, etc. (Moore et al., 2007). In contrast to nuclear genomes, plastomes are maternally inherited. The evolutionary rate of plastomes is low, and there is no recombination during chloroplast division (Daniell et al., 2016). Therefore, plastomes are good resources for studying maternal evolutionary dynamics (Tonti-Filippini et al., 2017). Chloroplast genomes are highly conserved in angiosperms, which share a quadripartite structure containing a large single copy (LSC; 80–90 kb) and a small single copy (SSC; 16–27 kb) separated by two inverted repeats (IR; 20–28 kb) (Daniell et al., 2016). In green plants, plastomes typically range from 120 to 218 kb in size (Wicke et al., 2011), and such a variety in size is mainly caused by IR contraction and expansion (Choi et al., 2020; Henriquez et al., 2020). To take an extreme example, the IR region is completely lost in Erodium L’Herit. and some papilionoid legumes (Blazier et al., 2016; Lee et al., 2021). Angiosperm plastomes generally encode 110–130 genes, which include approximately 80 protein coding genes, 30 transfer RNA genes, and four ribosomal RNA genes (Daniell et al., 2016). Even though the loss of genes (Song et al., 2017; Alqahtani and Jansen, 2021) or introns (Jansen et al., 2007), and pseudogenization (Abdullah et al., 2021a; Li et al., 2021) have been reported in the plastomes of diverse plant species, plastomics still provide a reliable tool in phylogenetic studies, and plastid genomes have been largely used to reconstruct the phylogeny of many crop and horticulture species in recent years (Li et al., 2017; Xue et al., 2019; Guo et al., 2020; Hassoubah et al., 2020; Moner et al., 2020; Tyagi et al., 2020). However, there are only a limited number of peanut plastomes that have been sequenced and analyzed to date, including that of A. hypogaea and a few other related wild species (Prabhudas et al., 2016; Yin et al., 2017; Wang et al., 2018, 2019, 2021). This is insufficient in gaining a full picture of what has happened in the evolutionary history of cultivated peanuts and some wild species, and the relationship between cultivated peanuts and their potential wild maternal progenitor species is still unclear.
In this study, we assembled 33 Arachis plastomes including both cultivated and wild peanut species. Through comparative analysis with other peanut plastomes, which are currently available at NCBI, we aim to provide insights into species delimitation of Arachis and to identify the potential maternal genome progenitor species of cultivated peanuts. This work will serve as a foundation for the utilization of peanut genetic resources and the development of high-quality peanut varieties through molecular breeding.
Materials and Methods
Plant Sampling
In this study, Fresh young leave samples of 33 peanut accessions (24 species) representing 11 different genome types were collected from Henan Academy of Agricultural Sciences, Zhengzhou, China (HNAAS) and used for further analysis (Table 1). These include five botanical varieties of A. hypogaea, var. hypogae (Lainongzao), var. hirsute (Bajisitanhuapi), var. fastigiate (PI493938), var. peruviana (NcAc17090), var. vulgaris (Yiya). Samples were stored immediately in a −80°C freezer prior to DNA extraction. All the voucher specimens were deposited to the Herbarium of Zhengzhou University (Supplementary Table 1).
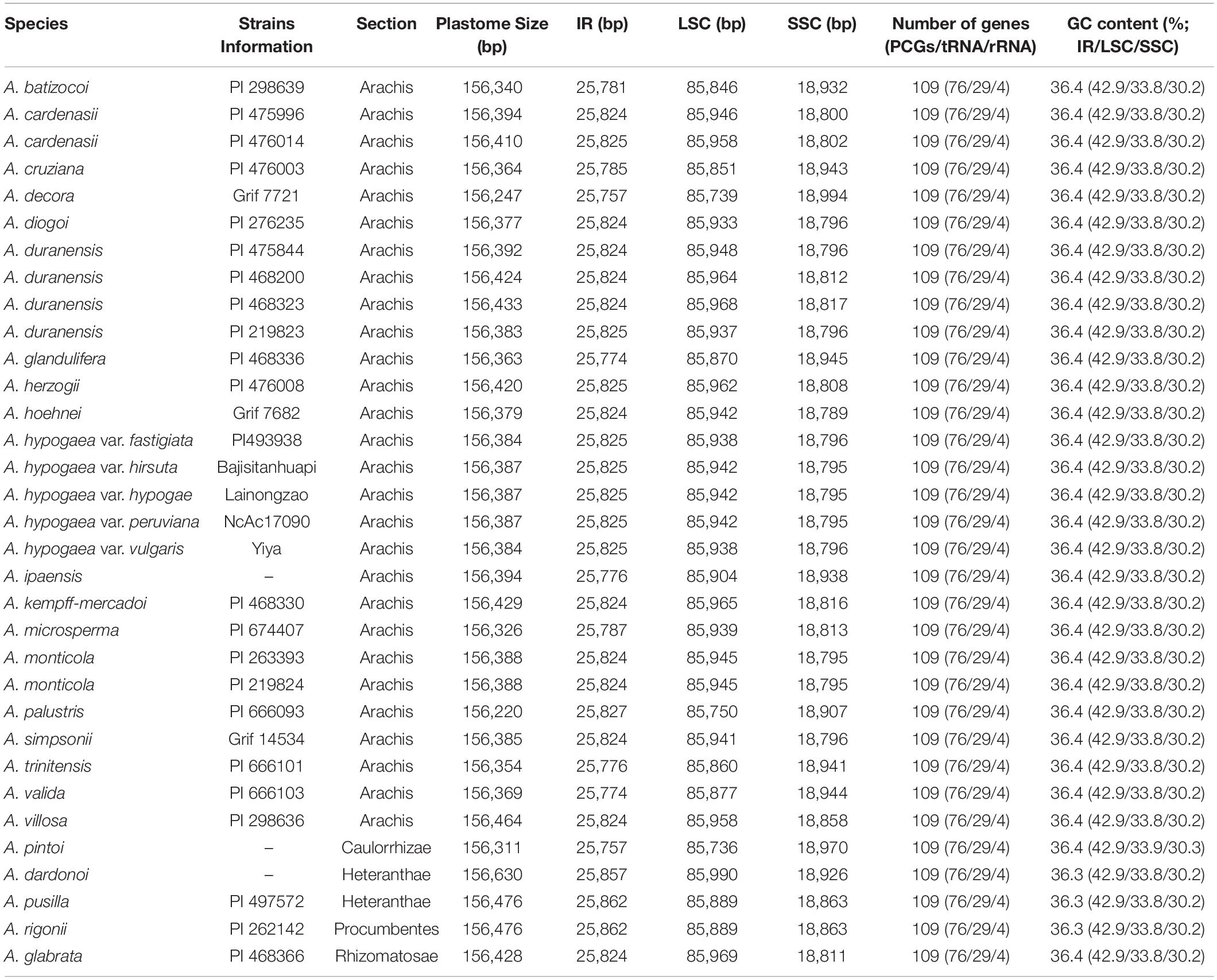
Table 1. Complete plastome features of the 33 Arachis accessions.
Genomic DNA Extraction and Sequencing
Total genomic DNA of the 33 samples were extracted with the Tiangen Plant Genomic DNA Kit (Tiangen Inc., China) following the protocol provided by the manufacturer. DNA purity was assessed using the Qubit 2.0 (Invitrogen Inc., United States) and a NanoDrop machine (Thermo Scientific Inc., United States). DNA libraries were constructed using the Illumina Paired-End DNA library Kit and sequenced with a NovaSeq 6000 platform (Illumina Inc., United States) with a paired-end read length of 150 bp (NovoGene Inc., China). Upon completion, more than 6.0 GB raw reads were retrieved for each sample. The GetOrganelle toolkit was used for de novo assembling of the complete plastid genomes (Jin et al., 2020). The published plastomic sequences of Arachis (Supplementary Table 1) from GenBank were used as the seed file (“embplant_pt”) for the assembling process, as well as a template to estimate the possible circular sequence pattern.
Plastome Annotation and Comparison
The Plastid Genome Annotator (PGA) software (Qu et al., 2019) was employed in the annotation of the selected peanut plastomes using A. hypogaea (accession no. MT712165) as a reference. The accuracy of annotation was evaluated with GeSeq (Tillich et al., 2017), HMMER (Wheeler and Eddy, 2013), and tRNAscan-SE (Lowe and Eddy, 1997) programs implemented in the CHLOROBOX web toolbox1 with a default setting. Chloroplot was used to visualize the plastid genomes as a physical map (Zheng et al., 2020). MISA-web (Beier et al., 2017) was used to identify simple sequence repeats (SSRs) with the following criteria: 10, 5, 4, 3, 3, and 3 repeat units are for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. In addition, forward, palindrome, reverse, and complement repeated elements were identified using REPuter (Kurtz et al., 2001) with a minimal length of 30 bp, an identity value of more than 90% and a Hamming distance of 3. The comparison among whole chloroplast genomes in genus Arachis species were using data from 33 new sequenced plastomes, and published plastomes of five cultivated peanuts (Prabhudas et al., 2016; Wang et al., 2018), 12 wild peanuts (Wang et al., 2019) which downloaded from the NCBI database (Supplementary Table 1). Nucleotide diversity (Pi) of the plastomic sequences of Arachis species were obtained in this study and the published sequences were calculated using a sliding window method with a window length of 600 bp and a step size of 200 bp by DnaSP (Rozas et al., 2017).
Phylogenetic Analysis
To reconstruct the phylogeny of peanut species and to identify the potential maternal progenitor species, the complete plastomes of 53 species (Supplementary Table 1) were retrieved from various databases and used to make a multiple sequence alignment with MAFFT under a default setting (Katoh and Standley, 2013). Among these species, Dalbergia hupeana Hance from the Tribe Dalbergieae was defined as outgroup. Phylogenetic trees were constructed with the 53 sequences using both the Maximum likelihood (ML) and the Bayesian inference method (BI), which are implemented in IQ-TREE (Nguyen et al., 2014) and MrBayes (Ronquist et al., 2012), respectively. The best fit nucleotide substitution models, TVM + F + R3 for ML analysis and GTR + F + I + G4 for BI analysis, were selected using the ModelFinder (Kalyaanamoorthy et al., 2017) according to the AIC criterion. In the ML analysis, 50,000 bootstrap replicates were carried out with the SH-aLRT branch test. The BI analysis was performed with two independent Markov Chain Monte Carlo chains with 2,000,000 generations, and it was considered to be stationary when the average standard deviation of split frequencies fell below 0.01. The first 25% of trees were discarded as burn-ins, and the remaining trees were used to construct a consensus tree.
Results
Characterization of the Peanut Plastomes
The size of the studied Arachis plastomes ranges from 156,220 bp (Arachis palustris) to 156,630 bp (Arachis dardonoi) in length (Table 1 and Figure 1), while the Arachis species ranging from 156,220 bp (A. palustris) to 156,878 bp (A. hypogaea var. hirsute AHL) in whole chloroplast genome length. Moreover, the cultivated peanut plastomes ranges from 156,354 to 156,878 bp in length, with A. hypogaea var. hirsute AHL being the largest. There is a 10–100 bp difference in length when comparing our sequencing data with the published data of a few species including A. batizocoi, A. cardenasii, A. duranensis, A. ipaensis, Arachis villosa, and A. monticola PI 219824. All the sequenced plastomes share a G + C content of 36.4% except for A. dardonoi, A. pusilla, and A. rigonii, which share a G + C content of 36.3%. All peanut plastomes contain a large single-copy (LSC), a small single-copy (SSC), and two inverted repeats (IRa/IRb). The LSC regions range from 85,736 bp (A. pintoi) to 85,990 bp (A. dardonoi) in length, with the G + C contents falling between 33.8 and 33.9%. The SSC regions vary from 18,789 bp (Arachis hoehnei) to 18,994 bp (Arachis decora) in length and the G + C content falls between 30.2 and 30.3%. A. pintoi has the smallest IRs, which is 25,757 bp in length, while a maximum IR length of 25,862 bp was observed in both A. pusilla and A. rigonii. These regions have a G + C content of 42.9%, which is significantly higher than that of the LSCs and SSCs. All plastomes included in our studies contain 109 unique genes, encoding 76 protein genes, 29 tRNAs, and 4 rRNAs (Table 1 and Figure 1), which is comparable with some well-studied Arachis species. Based on their annotated functions, these genes can be classified into four categories (Table 2), namely self-replication genes, photosynthesis related genes, other genes, and unknown function genes.
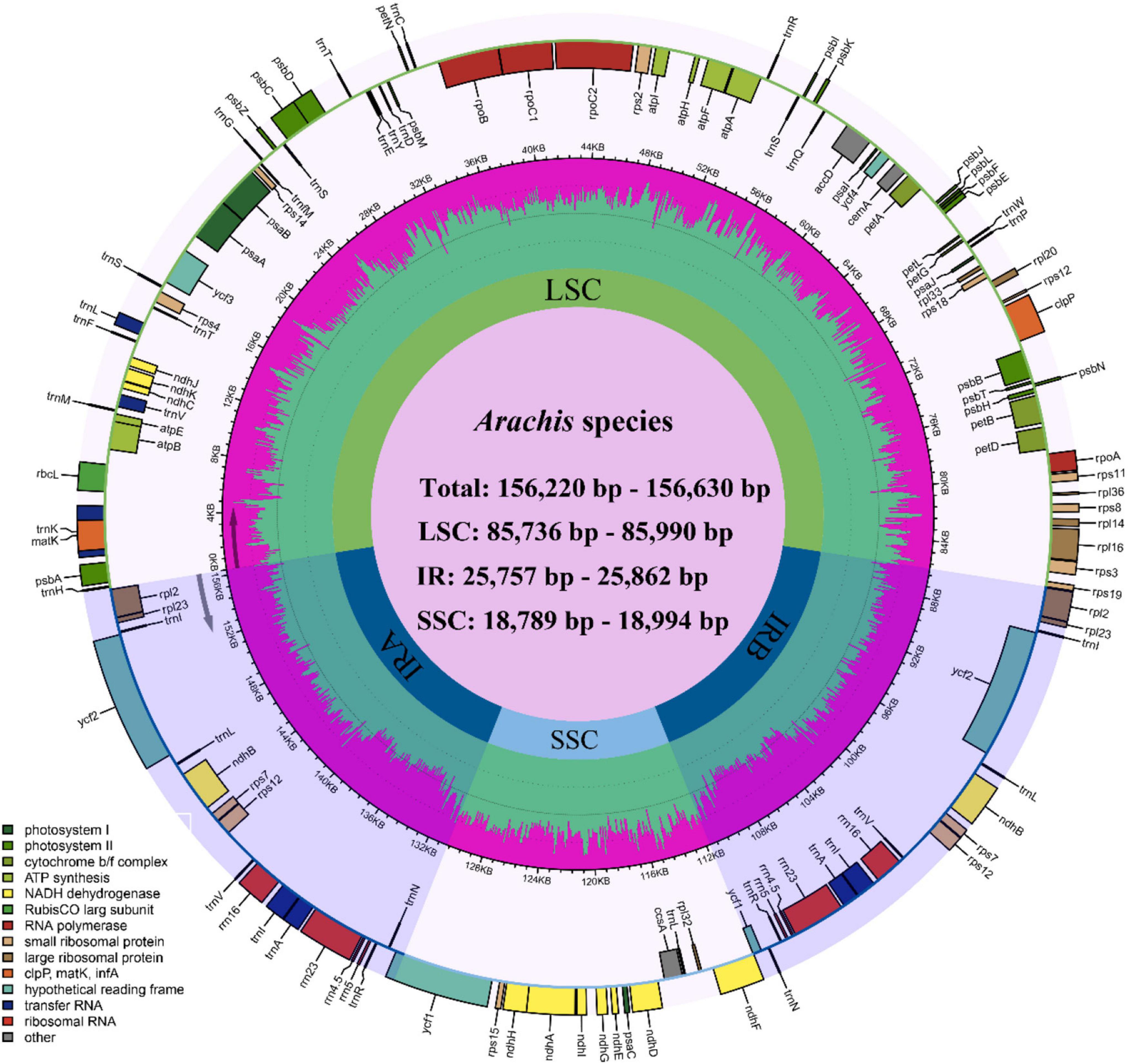
Figure 1. Circular plastome map of Arachis. Genes of different functional groups are color coded. The green in the inner circle corresponds to the GC content, while the pink corresponds to the AT content.
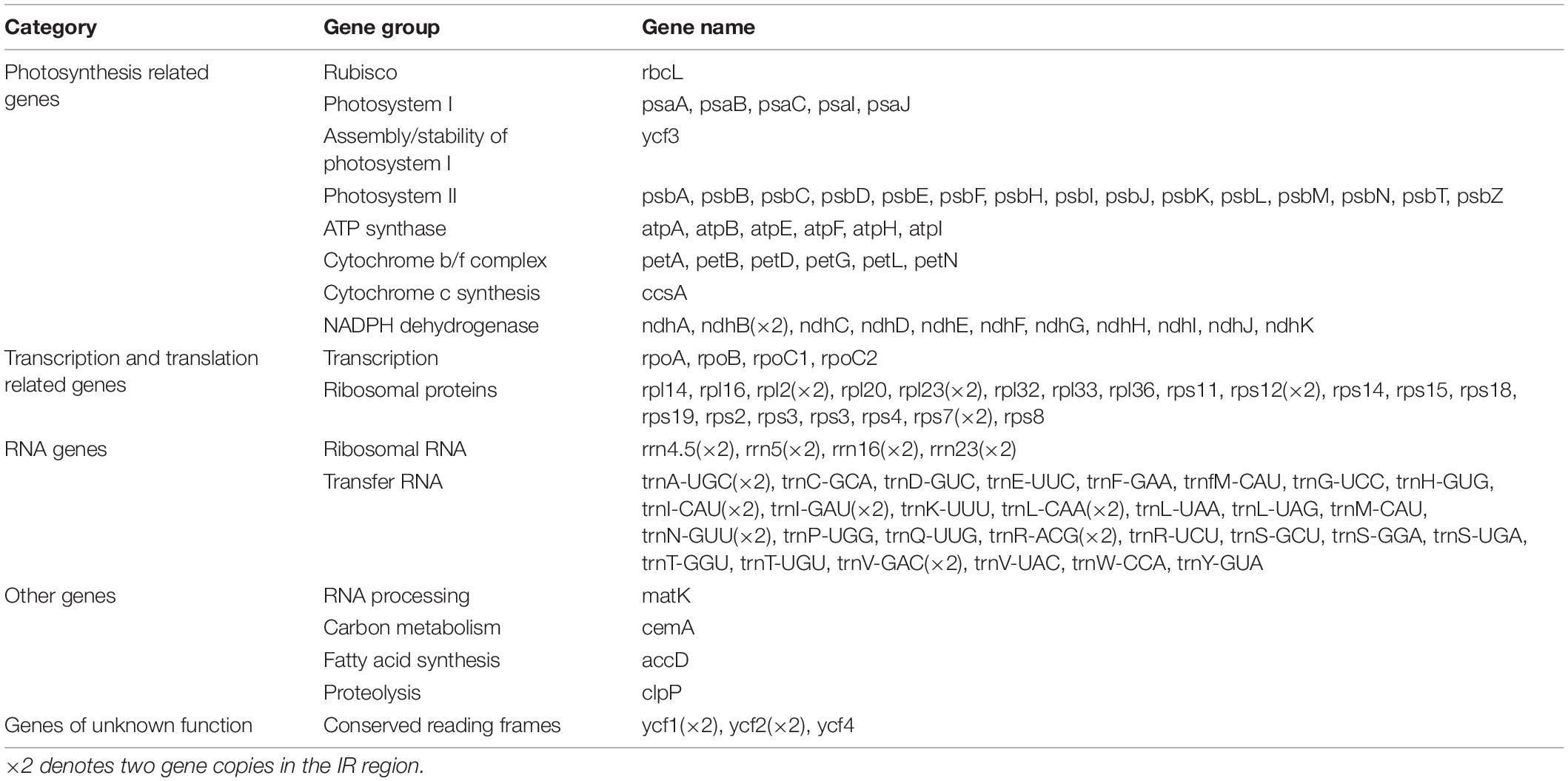
Table 2. List of the annotated genes in the plastomes of the selected Arachis accessions.
Comparative Plastomic Analysis
Analysis of the 33 new sequenced plastomes revealed 1,593 tandem repeats with complement, forward, reverse, and palindromic elements (>30 bp). The number of repeats present in each plastome varies considerably, ranging from 38 in A. decora to 50 in most other species (Figure 2A). In average, 17 forward, 27 palindromic, 2 complement, and 3 reverse repeats were estimated in each plastome. Among the species, in which repeats were identified, A. dardonoi lacks complement repeats, while A. pintoi, A. pusilla, and A. rigonii do not have complement or reverse repeats. Most repeats among Arachis species plastomes are present in the intergenic spacer regions. With the MISA analysis, 60 universal SSR loci were detected in the plastomes of A. pusilla and A. rigonii while 83 was in A. dardonoi (Figure 2B). Based on the SSR analysis, 40–57 of the identified SSRs are mononucleotidic, 14–20 are dinucleotidic, 1–4 are trinucleotidic, and 5–9 are tetranucleotidic (Supplementary Table 2). Among these SSRs, most of the identified mononucleotidic SSRs are composed of A/T, and the dinucleotidic ones contain AT/TA. Moreover, the pentanucleotidic SSRs in A. dardonoi and A. ipaensis have a typical sequence of AATAG/CTATT or TATAA/TTATA, and the hexanucleotidic SSRs in A. cardenasii, A. dardonoi, A. duranensis, A. glabrata, Arachis herzogii, and Arachis microsperma contain either AATGGA/TCCATT or ATAGCA/TGCTAT (Figure 2B).
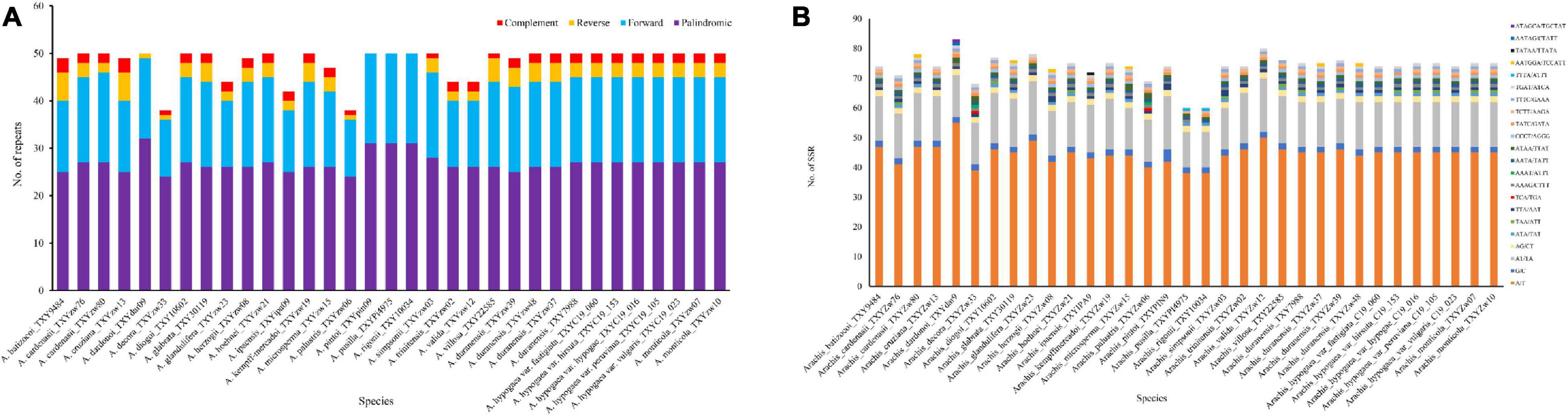
Figure 2. Analyses of repeated sequences in the plastomes of the 33 Arachis accessions. (A) Numbers of repeats and their types; (B) number of identified SSRs motifs and their types.
A total number of 3,416 polymorphic sites (Pi: 0.227%) were detected in the 52 cultivated and wild peanut plastomes (Supplementary Table 1), including 1,670 singleton variable sites and 1,746 parsimony informative sites. The alignment of seventeen peanut complex (see discussion) sharing high sequence similarity revels 54 singleton variable sites and 20 parsimony informative sites, which are also highly conserved across all the analyzed plastomes. Pi values among different plastomes were computed by a sliding window method with a window length of 600 bp and a step size of 200 bp (Figure 3). In addition, six hotspot regions with high Pi values were identified among various cultivated peanut accessions and other wild species, which include two protein-coding genes (rpoC2 and ycf1) and four intergenic spacer regions (trnS-UGA-psbC, atpA-trnR-UCU, psbE-petL, and rpl32-trnL-UAG). These regions could be potentially used as DNA markers in phylogenetic studies of different Arachis species.
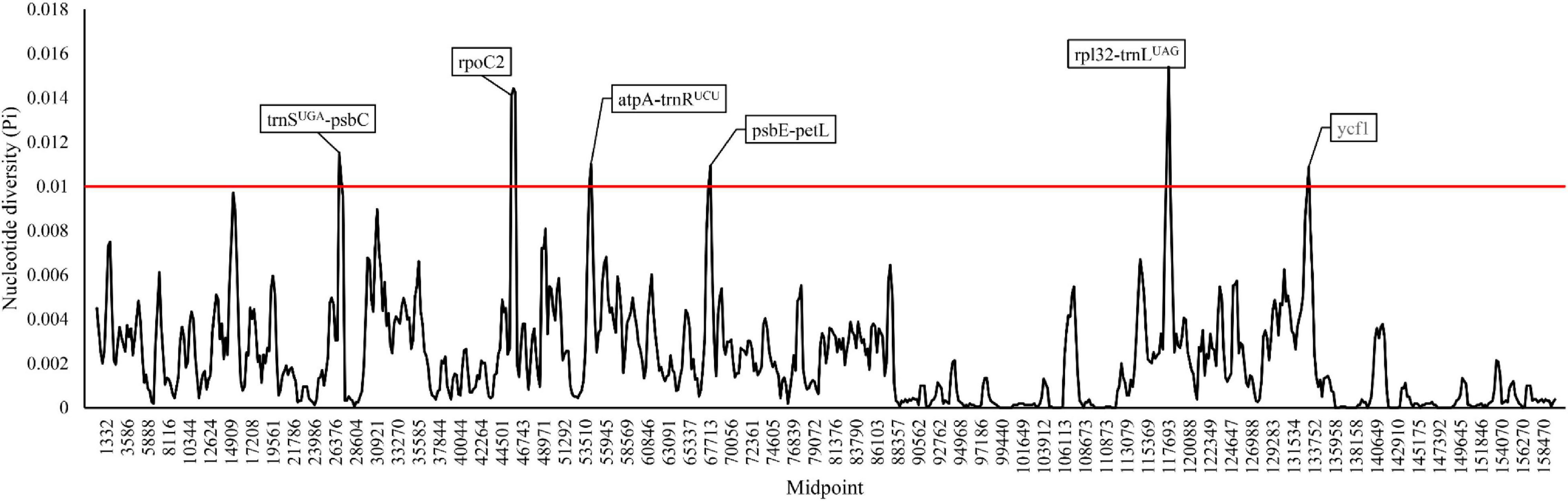
Figure 3. Sliding window analysis of the whole plastomes of the 52 Arachis accessions.
Phylogeny of Arachis Based on Whole Plastomes
In our study, Arachis is recovered as monophyletic, which is well supported by both ML and BI analyses (Figures 4A,B). The basal position of A. dardonoi (HH) from section Heteranthae is strongly supported by both methods. Within section Arachis, two major lineages, Lineage I and II, were clearly defined (Figure 4). Another species A. pusilla (HH) from section Heteranthae is grouped into one clade with A. rigonii (PR) from section Procumbentes. However, ML and BI analysis did not provide consistent result in terms of the taxonomic statuses of A. duranensis, A. monticola, and cultivated peanut (Figures 4A,B).
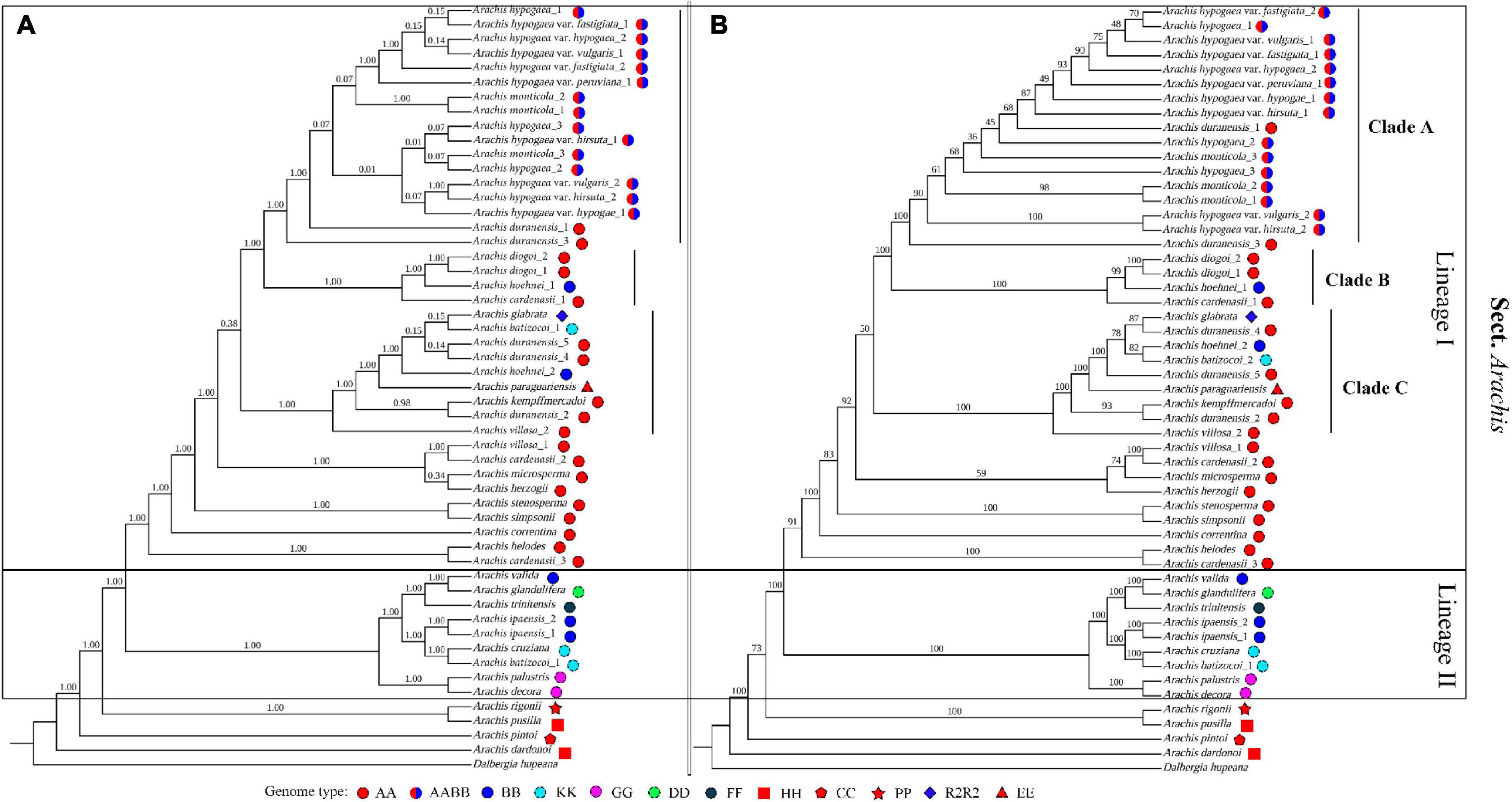
Figure 4. Phylogenetic trees constructed with 52 Arachis plastomic sequences using the Bayesian inferred (A) and Maximum likelihood (B) methods. Numbers above the branch represent the confidence level.
Species with a genome type of AA are mainly distributed in Lineage I, which are further divided into three clades. Based on the BI analysis, the newly sequenced peanut varieties, var. hypogaea and var. hirsuta, are clustered into one clade, while var. fastigiata, var. vulgaris, and var. peruviana are placed in another clade with relatively low bootstrap values (Figure 4A). In the ML analysis, the six varieties are placed in one big clade, and it is impossible to draw a clear boundary between subsp. hypogaea and subsp. fastigiated (Figure 4B). Var. vulgaris (Yiya vs. AHZ) and var. hirsute (Bajisitanhuapi vs. AHL) are placed in two separate clades in this study. The cultivated peanuts and two wild species, A. monticola and A. duranensis (PI219823 and PI 475844), are grouped together as the “peanut complex” clade (Clade A), members of which demonstrate a diversity in morphological features, and the boundary between Clade A and other clades is not well defined or supported by the phylogenetic analyses (Figure 5). Both the ML and BI analyses support that A. duranensis (AA) is the wild diploid progenitor of all cultivated peanuts. In Lineage I, Clades B and C are not monophyletic, which contain species with various genome types, such as A. hoehnei (BB), A. glabrata (R2) from section Rhizomatosae, A. batizocoi (KK), and Arachis paraguariensis (EE) from section Erectoides (Figure 4).

Figure 5. Morphological differences among selected Arachis species (three accessions of A. duranensis, two accessions of A. cardenasii and five cultivated peanuts).
The phylogenetic structure of Lineage II is strongly supported by both ML and BI analyses. It contains nine Arachis species/accessions with diverse genome types. For example, the two accessions of A. ipaensis and Arachis valida contain BB genome type. A. decora and A. palustris (2n = 18), share a genome type of GG and are placed together with high confidence scores. A. valida shows a sisterhood relationship with Arachis trinitensis (FF) and Arachis glandulifera (DD), while A. ipaensis, another possible diploid progenitor of cultivated peanuts, is grouped together with A. batizocoi (AA) and Arachis cruziana (KK).
Discussion
Arachis Plastomes Are Highly Conserved
All Arachis plastomes share a typical quadripartite structure, consisting of one LSC region and one SSC regions separated by a pair of IRs. The same structure has also been reported in other angiosperms (Xu et al., 2015; Daniell et al., 2016; Tonti-Filippini et al., 2017). All the Arachis plastomes covered in this study are highly conserved in genome size and structure, G + C content, and gene number, which are also comparable to the plastomes of previously published Arachis species (Prabhudas et al., 2016; Yin et al., 2017; Wang et al., 2018, 2019). Plastomes of angiosperms tend to vary in size, the size of a typical Arachis plastome is approximately 156 kb (Supplementary Table 1), similar with the plastomes length of soybean (Glycine) in 152 kb, but more than the length of wheat (Tribe Triticeae), which varies from 133 to 137 kb (Middleton et al., 2014), rice (Oryza) of 135 kb in size (Asaf et al., 2017), and less than buckwheat (Fagopyrum) of 159 kb in total length (Wang et al., 2017). Genome size change was suggested to be linked variation of intergenic region, InDel events and oligonucleotide/microsatellites repeats within the related species, while gene loss, expansion/contraction of an IR region among seed plants (Xu et al., 2015; Zheng et al., 2017).
All the published Arachis plastomes share the same number of protein coding genes (Table 2) with only a few exceptions. Prabhudas et al. (2016) was not able to detect NADH dehydrogenase subunit 2 gene (ndhB) in A. hypogaea Co7, and orf42 and ycf68 were miss annotated in another two studies by Yin et al. (2017) and Wang et al. (2019). A closer look at the coding regions reveals that five tRNAs and 11 protein coding genes harbor at least one intron. Among these, ycf3, clpP, and rps12 (a trans-splicing gene) contain two introns (Xu et al., 2015; Liu et al., 2020). The total number of tRNA genes present in our sequenced plastomes is 29, and the same conclusion was reached in two other studies by Schwarz et al. (2015) and Wang et al. (2019). However, one extra tRNA gene was annotated in one previous study carried out by Prabhudas et al. (2016). This one extra gene is trnP-GGG, which overlaps with another tRNA gene2. According to wild Roses, trnP-GGG gene in the region of trnP-UGG gene also exists (Jeon and Kim, 2019). Former studies demonstrated a widely distributed of trnP-GGG gene present in charophyte to gymnosperm, while trnP-UGG gene in plastomes from algae to higher plants (Turmel et al., 2002; Sugiura and Sugita, 2004).
Microsatellites and oligonucleotide repeats play an important role in the identification of regions with a large number of mutations, and are helpful in the study of population genetics (Ahmed et al., 2012; Abdullah et al., 2019). A consistent result was obtained when comparing SSRs and oligonucleotide repeats across different Arachis plastomes (Yin et al., 2017; Wang et al., 2019), with A/T and AT/TA being the most common mononucleotidic SSRs and mononucleotidic SSRs, respectively. A similar pattern is also reported in plastomes of many other angiosperms (Tian et al., 2019; Mehmood et al., 2020; Abdullah et al., 2021b). The SSRs loci identified in this work could serve as potential molecular markers for understanding the population genetic structure among various Arachis species. Here, we also identified some oligonucleotide repeats, which are associated with nucleotide substitution, mutation and InDel events in the genomes (Abdullah et al., 2021b,c). Most of the oligonucleotide repeats were found in the intergenic regions, and a similar pattern is observed in the plastomes of many other vascular plants (Kuang et al., 2011; Li et al., 2017; Sigmon et al., 2017; Wang et al., 2019). Our results also showed a high abundance of complement and forward oligonucleotide repeats across different Arachis species. Oligonucleotide repeats could be used for the identification of regions with mutations and the reconstruction of accurate phylogeny of Arachis species (Mehmood et al., 2020; Abdullah et al., 2021b).
Linking Phylogeny With Genome Type
The genus Arachis consists of 81 species demonstrating a huge diversity in genome types (A, B, AB, C, D, E, EX, F, H, K, PR, R1, R2, T, and TE). Linking phylogenetic analysis with their genome type information could allow us to better understand the origination and evolution of cultivated peanuts. Based on our study, hybridization seems to play a major role in the evolution history of cultivated species (Garcia et al., 1995; Jarvis et al., 2003). However, problems within several clades are still unsolved. Our results show that the taxonomic relationship based on morphology should be revised (He et al., 2014; Vishwakarma et al., 2017). Two studies working with plastomics data (Wang et al., 2019) and microsatellite markers (Moretzsohn et al., 2013) also reached the same conclusion. In addition, one clade may contain species with various genome types, which is supported by this study and two other phylogenetic studies working with intron sequences and microsatellite markers (Moretzsohn et al., 2013). Again, it is very difficult to delimit the boundary of different Arachis species. In fact, all Arachis species look very similar morphologically, and leaf shape could probably be the only morphological trait, which could potentially be used in putting species into different taxonomic groups (Supplementary Figure 1). We speculate that recent speciation events play an important role in the evolution of Arachis. Both underground fruiting and clistogamy are thought to limit gene flows and seed dispersal in peanuts (Tan et al., 2010; Zhang et al., 2017), which should allow each species to keep its distinct identity (Yu et al., 2020). However, it is very interesting to see that the flowers and stems of Arachis plant could attract small insects, such as ants (Supplementary Figure 2). The movement of ants between different plants could cause the pollen of one species to be transferred to another species, and therefore promote gene flow between different Arachis species. In fact, genome introgression was detected among the interspecific hybrid population of peanuts (Garcia et al., 1995).
Plastomes are highly conserved and tend to have low nucleotide variations (Sigmon et al., 2017; Wang et al., 2018; Nock et al., 2019) (Figure 4). In this study, only 74 nucleotide polymorphisms were detected among different species of the cultivated peanut complex, indicating that the plastomes of cultivated peanuts are highly conserved (Wang et al., 2018). This observation could also be explained with a low nucleotide substitution rate. Peanut has only been domesticated for several thousand years, there is not enough time to accumulate many genetic variations (Bertioli et al., 2019). Although most botanical varieties examined in this study do demonstrate differences in their morphology (Figure 5), there are no distinguishable morphological features, which could be used to put different species into the two subspecies groups. For example, var. fastigiate, var. vulgaris and var. hirsute coming from two different groups all have three or more seeds in each shell (Figure 4A). The overall phylogeny obtained in this study is in agreement with the conventional classification based on studies looking at other features, including morphology (Krapovickas et al., 2007), AFLP markers (He and Prakash, 2001), simple sequence repeats (Ferguson et al., 2004), and single nucleotide polymorphisms (Zheng et al., 2018). However, violations do exist when it comes to the phylogenetic relationship of different varieties, such as, var. peruviana does not belong to subsp. fastigiata (He and Prakash, 2001; Ferguson et al., 2004). Var. hypogaea and var. hirsute should not be placed in subsp. hypogaea according to the conventional classification.
Maternal Hybridization Event in the History of Cultivated Peanuts
Our results strongly support the hypothesis that A. duranensis is the wild diploid progenitor (with a genome type of A) of cultivated peanuts (Figure 4). This result is compatible with the earlier view, which is based on multiple lines of evidence from comparative genomics, geographic distribution, phylogenetic reconstruction, etc. (Kochert et al., 1996; Seijo et al., 2004; Fávero et al., 2006; da Cunha et al., 2008; Bertioli et al., 2016; Chen et al., 2016; Wang et al., 2019). Furthermore, phylogenomic investigation using both ML and BI methods suggests that A. duranensis have diverged into two groups. A. duranensis (PI219823 and PI 475844) shows a closer relationship with A. hypogaea, while A. duranensis PI 468200, PI 468323, and PI263133 (Genbank no. MK144822) are grouped in another clade containing A. batizocoi, A. glabrata, A. hoehnei, Arachis kempff-mercadoi, and A. paraguariensis (Figure 4). This topology was generally consistent with that of the ML tree, in which the three botanical accessions of A. duranensis (ICG 8138, ICG 8123, and PI 262133) are distributed in different clades (Zhuang et al., 2019). Moreover, the 42 accessions of A. duranensis demonstrate clear variations in morphological features (Singh et al., 1996). In agreement with Bertioli’s work (Bertioli et al., 2019), some accession of A. duranensis may have served as the AA sub-genome maternal progenitor of A. hypogaea. However, the status of A. diogoi (former known as Arachis chacoensis) and A. cardenasii as another two potential progenitors is not supported by our study.
This does not contrary to the earlier view that A. monticola is the direct progenitor of cultivated peanuts, and that it plays a vital role in the transition of diploid wild species to tetraploid cultivated species (Simpson et al., 2001; Yin et al., 2020). Cultivated peanut (A. hypogaea) and wild A. monticola are allotetraploids (AABB), while other 30 described wild species are diploid (Stalker, 2017). The previous phylogeographical analyses often group these two species (A. hypogaea and A. monticola) together (Gimenes et al., 2002; Seijo et al., 2004). As former documented, A. monticola is a weedy subspecies of cultivated peanuts, and it is placed in one group with A. hypogaea in earlier phylogenetic studies (Koppolu et al., 2010; Stalker, 2017; Vishwakarma et al., 2017; Wang et al., 2019). Their close relationship can be further supported with the following evidence. Firstly, A. hypogaea is able to produce fertile hybrids when hybridized with A. monticola (Stalker and Moss, 1987). Secondly, this is in agreement with the results of previous studies focusing on somatic chromosomes, such as the virtually identical centromeric bands and in situ hybridization between A. hypogaea and A. monticola (Raina and Mukai, 1999). Thirdly, A. monticola may have been derived from a more ancient hybridization event according to the phylogenetic studies on the two FAD2A alleles, while the accessions of A. hypogaea may have evolved latter (Jung et al., 2003). During its evolution, A. monticola has accumulated more mutations in its plastome than most other cultivated peanuts do, which could be possibly traced back to different evolution rates or natural selection. Nevertheless, plastomics approach is very useful in inferring the maternal origin of cultivated peanuts and explaining the close phylogenetic relationship between A. monticola and A. hypogaea.
Conclusion
In summary, 33 Arachis plastomes were sequenced and analyzed in a comparative framework with the published plastomics data of cultivated and wild peanut species. These plastomes share similar structural organization with low nucleotide variations. The phylogenetic topology obtained in this study shows that plastomics could facilitate a better understanding of the phylogeny among deep lineages of Arachis. Based on our result, it is speculated that cultivated peanuts have experienced a multi-maternal hybridization event with a recent origin. Some wild species of the A. duranensis accessions might have contributed the maternal sub genomes to cultivated peanuts and A. monticola, which represents a transitional species between wild diploid species and tetraploid cultivated species. Owing to interspecific gene flow and recent speciation, the relationship among different Arachis species inferred based on phylogeny do not always go along with their genome types. As a result, more Arachis species with various genome types should be included in future study to fully elucidate the origin and evolutionary history of Arachis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
ZW, XZ, and BH conceived the ideas. PD and LF contributed to the sampling. XT and LS performed the experiments. XT and YW analyzed the data. The manuscript was written and improved by XT, LS, JG, ZW, XZ, and BH. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by the China Postdoctoral Science Foundation (2020M672264), China Agricultural Research System (CARS-13), and Special Funds for Scientific and Technological Development from Henan Academy of Agricultural Sciences (2020CY07).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Yongsheng Chen from Peking University for his critical review of the manuscript and Ziqi Sun from Henan Academy of Agricultural Sciences for providing the cultivated peanut accessions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.804568/full#supplementary-material
Footnotes
References
Abdullah, Mehmood, F., Heidari, P., Rahim, A., Ahmed, I., and Poczai, P. (2021a). Pseudogenization of the chloroplast threonine (trnT-GGU) gene in the sunflower family (Asteraceae). Sci. Rep. 11:21122. doi: 10.1038/s41598-021-00510-4
Abdullah, Mehmood, F., Rahim, A., Heidari, P., Ahmed, I., and Poczai, P. (2021b). Comparative plastome analysis of Blumea, with implications for genome evolution and phylogeny of Asteroideae. Ecol. Evol. 11, 7810–7826. doi: 10.1002/ece3.7614
Abdullah, Mehmood, F., Shahzadi, I., Ali, Z., Islam, M., Naeem, M., et al. (2021c). Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion–deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. 59, 388–402. doi: 10.1111/jse.12585
Abdullah, Shahzadi, I., Mehmood, F., Ali, Z., Malik, M. S., Waseem, S., et al. (2019). Comparative analyses of chloroplast genomes among three Firmiana species: identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 19:100199. doi: 10.1016/j.plgene.2019.100199
Ahmed, I., Biggs, P. J., Matthews, P. J., Collins, L. J., Hendy, M. D., and Lockhart, P. J. (2012). Mutational Dynamics of Aroid Chloroplast Genomes. Genome Biol. Evol. 4, 1316–1323. doi: 10.1093/gbe/evs110
Alqahtani, A. A., and Jansen, R. K. (2021). The evolutionary fate of rpl32 and rps16 losses in the Euphorbia schimperi (Euphorbiaceae) plastome. Sci. Rep. 11:7466. doi: 10.1038/s41598-021-86820-z
Asaf, S., Waqas, M., Khan, A. L., Khan, M. A., Kang, S.-M., Imran, Q. M., et al. (2017). The Complete Chloroplast Genome of Wild Rice (Oryza minuta) and Its Comparison to Related Species. Front. Plant Sci. 8:304. doi: 10.3389/fpls.2017.00304
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Belamkar, V., Selvaraj, M. G., Ayers, J. L., Payton, P. R., Puppala, N., and Burow, M. D. (2011). A first insight into population structure and linkage disequilibrium in the US peanut minicore collection. Genetica 139:411. doi: 10.1007/s10709-011-9556-2
Bertioli, D. J., Abernathy, B., Seijo, G., Clevenger, J., and Cannon, S. B. (2020). Evaluating two different models of peanut’s origin. Nat. Genet. 52, 557–559. doi: 10.1038/s41588-020-0626-1
Bertioli, D. J., Cannon, S. B., Froenicke, L., Huang, G., Farmer, A. D., Cannon, E. K. S., et al. (2016). The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446. doi: 10.1038/ng.3517
Bertioli, D. J., Jenkins, J., Clevenger, J., Dudchenko, O., Gao, D., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51, 877–884. doi: 10.1038/s41588-019-0405-z
Bertioli, D. J., Seijo, G., Freitas, F. O., Valls, J. F. M., Leal-Bertioli, S. C. M., and Moretzsohn, M. C. (2011). An overview of peanut and its wild relatives. Plant Genet. Resour. 9, 134–149. doi: 10.1017/s1479262110000444
Blazier, J. C., Jansen, R. K., Mower, J. P., Govindu, M., Zhang, J., Weng, M.-L., et al. (2016). Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 117, 1209–1220. doi: 10.1093/aob/mcw065
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, X., Li, H., Pandey, M. K., Yang, Q., Wang, X., Garg, V., et al. (2016). Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. U. S. A. 113:6785. doi: 10.1073/pnas.1600899113
Chen, X., Lu, Q., Liu, H., Zhang, J., Hong, Y., Lan, H., et al. (2019). Sequencing of Cultivated Peanut, Arachis hypogaea, Yields Insights into Genome Evolution and Oil Improvement. Mol. Plant 12, 920–934. doi: 10.1016/j.molp.2019.03.005
Choi, I.-S., Jansen, R., and Ruhlman, T. (2020). Caught in the Act: variation in plastid genome inverted repeat expansion within and between populations of Medicago minima. Ecol. Evol. 10, 12129–12137. doi: 10.1002/ece3.6839
da Cunha, F. B., Nobile, P. M., Hoshino, A. A., Moretzsohn, M. D. C., Lopes, C. R., and Gimenes, M. A. (2008). Genetic relationships among Arachis hypogaea L. (AABB) and diploid Arachis species with AA and BB genomes. Genet. Resour. Crop Evol. 55, 15–20. doi: 10.1016/j.gene.2021.145539
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Fávero, A. P., Simpson, C. E., Valls, J. F. M., and Vello, N. A. (2006). Study of the Evolution of Cultivated Peanut through Crossability Studies among Arachis ipaënsis, A. duranensis, and A. hypogaea. Crop Sci. 46, 1546–1552. doi: 10.2135/cropsci2005.09-0331
Feldman, M., Levy, A. A., Fahima, T., and Korol, A. (2012). Genomic asymmetry in allopolyploid plants: wheat as a model. J. Exp. Bot. 63, 5045–5059. doi: 10.1093/jxb/ers192
Ferguson, M. E., Bramel, P. J., and Chandra, S. (2004). Gene Diversity among Botanical Varieties in Peanut (Arachis hypogaea L.). Crop Sci. 44, 1847–1854. doi: 10.2135/cropsci2004.1847
Garcia, G. M., Stalker, H. T., and Kochert, G. (1995). Introgression analysis of an interspecific hybrid population in peanuts (Arachis hypogaea L.) using RFLP and RAPD markers. Genome 38, 166–176. doi: 10.1139/g95-021
Gibbons, R. W., Bunting, A. H., and Smartt, J. (1972). The classification of varieties of groundnut (Arachis hypogaea L.). Euphytica 21, 78–85. doi: 10.1007/bf00040550
Gill, N., Findley, S., Walling, J. G., Hans, C., Ma, J., Doyle, J., et al. (2009). Molecular and Chromosomal Evidence for Allopolyploidy in Soybean. Plant Physiol. 151, 1167–1174. doi: 10.1104/pp.109.137935
Gimenes, M. A., Lopes, C. R., Galgaro, M. L., Valls, J. F. M., and Kochert, G. (2002). RFLP analysis of genetic variation in species of section Arachis, genus Arachis (Leguminosae). Euphytica 123, 421–429. doi: 10.1007/s00122-005-0017-0
Guo, L., Guo, S., Xu, J., He, L., Carlson, J. E., and Hou, X. (2020). Phylogenetic analysis based on chloroplast genome uncover evolutionary relationship of all the nine species and six cultivars of tree peony. Ind. Crops Prod. 153:112567. doi: 10.1016/j.indcrop.2020.112567
Hassoubah, S., Farsi, R., Alrahimi, D., Nass, N., and Bahieldin, A. (2020). Comparison of Plastome SNPs/INDELs among different Wheat (Triticumsp.) Cultivars. Biosci. Biotechnol. Res. Asia 17, 27–44. doi: 10.13005/bbra/2807
He, G., Barkley, N. A., Zhao, Y., Yuan, M., and Prakash, C. S. (2014). Phylogenetic relationships of species of genus Arachis based on genic sequences. Genome 57, 327–334. doi: 10.1139/gen-2014-0037
He, G., and Prakash, C. (2001). Evaluation of genetic relationships among botanical varieties of cultivated peanut (Arachis hypogaea L.) using AFLP markers. Genet. Resour. Crop Evol. 48, 347–352.
Henriquez, C. L., Abdullah, Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 251:72. doi: 10.1007/s00425-020-03365-7
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U. S. A. 104:19369. doi: 10.1073/pnas.0709121104
Jarvis, A., Ferguson, M. E., Williams, D. E., Guarino, L., Jones, P. G., Stalker, H. T., et al. (2003). Biogeography of Wild Arachis. Crop Sci. 43, 1100–1108. doi: 10.2135/cropsci2003.1100
Jeon, J.-H., and Kim, S.-C. (2019). Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae). Genes 10:23. doi: 10.3390/genes10010023
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., Depamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Jung, S., Tate, P. L., Horn, R., Kochert, G., Moore, K., and Abbott, A. G. (2003). The Phylogenetic Relationship of Possible Progenitors of the Cultivated Peanut. J. Hered. 94, 334–340. doi: 10.1093/jhered/esg061
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kochert, G., Stalker, H. T., Gimenes, M., Galgaro, L., Lopes, C. R., and Moore, K. (1996). RFLP and Cytogenetic Evidence on the Origin and Evolution of Allotetraploid Domesticated Peanut, Arachis hypogaea (Leguminosae). Am. J. Bot. 83, 1282–1291.
Konate, M., Sanou, J., Miningou, A., Okello, D., Desmae, H., Janila, P., et al. (2020). Past, Present and Future Perspectives on Groundnut Breeding in Burkina Faso. Agronomy 10:704. doi: 10.3390/agronomy10050704
Koppolu, R., Upadhyaya, H. D., Dwivedi, S. L., Hoisington, D. A., and Varshney, R. K. (2010). Genetic relationships among seven sections of genus Arachis studied by using SSR markers. BMC Plant Biol. 10:15. doi: 10.1186/1471-2229-10-15
Krapovickas, A., Gregory, W. C., Williams, D. E., and Simpson, C. E. (2007). Taxonomy of the genus Aeachis (Leguminosae). Bonplandia 16, 7–205.
Kuang, D.-Y., Wu, H., Wang, Y.-L., Gao, L.-M., Zhang, S.-Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/g11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Leal-Bertioli, S. C. M., Santos, S. P., Dantas, K. M., Inglis, P. W., Nielen, S., Araujo, A. C. G., et al. (2014). Arachis batizocoi: a study of its relationship to cultivated peanut (A. hypogaea) and its potential for introgression of wild genes into the peanut crop using induced allotetraploids. Ann. Bot. 115, 237–249. doi: 10.1093/aob/mcu237
Lee, C., Choi, I.-S., Cardoso, D., De Lima, H. C., De Queiroz, L. P., Wojciechowski, M. F., et al. (2021). The chicken or the egg? Plastome evolution and an independent loss of the inverted repeat in papilionoid legumes. Plant J. 107, 861–875. doi: 10.1111/tpj.15351
Li, P., Zhang, S., Li, F., Zhang, S., Zhang, H., Wang, X., et al. (2017). A Phylogenetic Analysis of Chloroplast Genomes Elucidates the Relationships of the Six Economically Important Brassica Species Comprising the Triangle of U. Front. Plant Sci. 8:111. doi: 10.3389/fpls.2017.00111
Li, X., Yang, J.-B., Wang, H., Song, Y., Corlett, R. T., Yao, X., et al. (2021). Plastid NDH Pseudogenization and Gene Loss in a Recently Derived Lineage from the Largest Hemiparasitic Plant Genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 62, 971–984. doi: 10.1093/pcp/pcab074
Liu, S., Wang, Z., Wang, H., Su, Y., and Wang, T. (2020). Patterns and Rates of Plastid rps12 Gene Evolution Inferred in a Phylogenetic Context using Plastomic Data of Ferns. Sci. Rep. 10:9394. doi: 10.1038/s41598-020-66219-y
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Mehmood, F., Abdullah, Shahzadi, I., Ahmed, I., Waheed, M. T., and Mirza, B. (2020). Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 112, 1522–1530. doi: 10.1016/j.ygeno.2019.08.024
Middleton, C. P., Senerchia, N., Stein, N., Akhunov, E. D., Keller, B., Wicker, T., et al. (2014). Sequencing of Chloroplast Genomes from Wheat, Barley, Rye and Their Relatives Provides a Detailed Insight into the Evolution of the Triticeae Tribe. PLoS One 9:e85761. doi: 10.1371/journal.pone.0085761
Moner, A. M., Furtado, A., and Henry, R. J. (2020). Two divergent chloroplast genome sequence clades captured in the domesticated rice gene pool may have significance for rice production. BMC Plant Biol. 20:472. doi: 10.1186/s12870-020-02689-6
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U. S. A. 104:19363. doi: 10.1073/pnas.0708072104
Moretzsohn, M. C., Gouvea, E. G., Inglis, P. W., Leal-Bertioli, S. C. M., Valls, J. F. M., and Bertioli, D. J. (2013). A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann. Bot. 111, 113–126. doi: 10.1093/aob/mcs237
Moretzsohn, M. D. C., Hopkins, M. S., Mitchell, S. E., Kresovich, S., Valls, J. F. M., and Ferreira, M. E. (2004). Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol. 4:11. doi: 10.1186/1471-2229-4-11
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2014). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nock, C. J., Hardner, C. M., Montenegro, J. D., Ahmad Termizi, A. A., Hayashi, S., Playford, J., et al. (2019). Wild Origins of Macadamia Domestication Identified Through Intraspecific Chloroplast Genome Sequencing. Front. Plant Sci. 10:334. doi: 10.3389/fpls.2019.00334
Pandey, M. K., Pandey, A. K., Kumar, R., Nwosu, C. V., Guo, B., Wright, G. C., et al. (2020). Translational genomics for achieving higher genetic gains in groundnut. Theor. Appl. Genet. 133, 1679–1702. doi: 10.1007/s00122-020-03592-2
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427. doi: 10.1038/nature11798
Prabhudas, S. K., Prayaga, S., Madasamy, P., and Natarajan, P. (2016). Shallow Whole Genome Sequencing for the Assembly of Complete Chloroplast Genome Sequence of Arachis hypogaea L. Front. Plant Sci. 7:1106. doi: 10.3389/fpls.2016.01106
Qu, X.-J., Moore, M. J., Li, D.-Z., and Yi, T.-S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15:50. doi: 10.1186/s13007-019-0435-7
Raina, S. N., and Mukai, Y. (1999). Genomic in situ hybridization inArachis (Fabaceae) identifies the diploid wild progenitors of cultivated (A. hypogaea) and related wild (A. monticola) peanut species. Plant Syst. Evol. 214, 251–262.
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Schwarz, E. N., Ruhlman, T. A., Sabir, J. S. M., Hajrah, N. H., Alharbi, N. S., Al-Malki, A. L., et al. (2015). Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 53, 458–468. doi: 10.1111/jse.12179
Seijo, J. G., Lavia, G. I., Fernández, A., Krapovickas, A., Ducasse, D., and Moscone, E. A. (2004). Physical mapping of the 5S and 18S–25S rRNA genes by FISH as evidence that Arachis duranensis and A. ipaensis are the wild diploid progenitors of A. hypogaea (Leguminosae). Am. J. Bot. 91, 1294–1303. doi: 10.3732/ajb.91.9.1294
Sigmon, B. A., Adams, R. P., and Mower, J. P. (2017). Complete chloroplast genome sequencing of vetiver grass (Chrysopogon zizanioides) identifies markers that distinguish the non-fertile ‘Sunshine’ cultivar from other accessions. Ind. Crops Prod. 108, 629–635. doi: 10.1016/j.indcrop.2017.07.029
Simpson, C. E., Krapovickas, A., and Valls, J. F. M. (2001). History of Arachis Including Evidence of A. hypogaea L. Progenitors. Peanut Sci. 28, 78–80. doi: 10.3146/i0095-3679-28-2-7
Singh, A. K., Gurtu, S., and Jambunathan, R. (1994). Phylogenetic relationships in the genus Arachis based on seed protein profiles. Euphytica 74, 219–225.
Singh, A. K., and Moss, J. P. (1982). Utilization of wild relatives in genetic improvement of Arachis hypogaea L. Theor. Appl. Genet. 61, 305–314.
Singh, A. K., Subrahmanyam, P., and Gurtu, S. (1996). Variation in a wild groundnut species, Arachis duranensis Krapov. & W.C. Gregory. Genet. Resour. Crop Evol. 43, 135–142.
Song, Y., Yu, W.-B., Tan, Y., Liu, B., Yao, X., Jin, J., et al. (2017). Evolutionary Comparisons of the Chloroplast Genome in Lauraceae and Insights into Loss Events in the Magnoliids. Genome Biol. Evol. 9, 2354–2364. doi: 10.1093/gbe/evx180
Stalker, H. T., Dhesi, J. S., Parry, D. C., and Hahn, J. H. (1991). Cytological and Interfertility Relationships of Arachis Section Arachis. Am. J. Bot. 78, 238–246.
Stalker, H. T., and Moss, J. P. (1987). Speciation, Cytogenetics, and Utilization of Arachis Species. Adv. Agron. 41, 1–40.
Subrahmanyam, P., Anaidu, R., Reddy, L. J., Kumar, P. L., and Ferguson, M. E. (2001). Resistance to groundnut rosette disease in wild Arachis species. Ann. Appl. Biol. 139, 45–50. doi: 10.1111/j.1744-7348.2001.tb00129.x
Sugiura, C., and Sugita, M. (2004). Plastid transformation reveals that moss tRNAArg-CCG is not essential for plastid function. Plant J. 40, 314–321.
Tallury, S. P., Hollowell, J. E., Isleib, T. G., and Stalker, H. T. (2014). Greenhouse Evaluation of Section Arachis Wild Species for Sclerotinia Blight and Cylindrocladium Black Rot Resistance. Peanut Sci. 41, 17–24. doi: 10.3146/ps13-02.1
Tan, D., Zhang, Y., and Wang, A. (2010). A review of geocarpy and amphicarpy in angiosperms, with special reference to their ecological adaptive significance. Chin. J. Plant Ecol. 34, 72–88.
Tian, X., Ye, J., and Song, Y. (2019). Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae. PeerJ 7:e7662. doi: 10.7717/peerj.7662
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tonti-Filippini, J., Nevill, P. G., Dixon, K., and Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. 90, 808–818. doi: 10.1111/tpj.13491
Turmel, M., Otis, C., and Lemieux, C. (2002). The chloroplast and mitochondrial genome sequences of the charophyte ⁢em>Chaetosphaeridium globosum⁢/em>: insights into the timing of the events that restructured organelle DNAs within the green algal lineage that led to land plants. Proc. Natl. Acad. Sci. U. S. A. 99:11275.
Tyagi, S., Jung, J.-A., Kim, J. S., and Won, S. Y. (2020). A comparative analysis of the complete chloroplast genomes of three Chrysanthemum boreale strains. PeerJ 8:e9448. doi: 10.7717/peerj.9448
Upadhyaya, H. D., Dwivedi, S. L., Nadaf, H. L., and Singh, S. (2011). Phenotypic diversity and identification of wild Arachis accessions with useful agronomic and nutritional traits. Euphytica 182:103.
Varshney, R. K., Mahendar, T., Aruna, R., Nigam, S. N., Neelima, K., Vadez, V., et al. (2009). High level of natural variation in a groundnut (Arachis hypogaea L.) germplasm collection assayed by selected informative SSR markers. Plant Breed. 128, 486–494. doi: 10.1111/j.1439-0523.2009.01638.x
Vishwakarma, M. K., Kale, S. M., Sriswathi, M., Naresh, T., Shasidhar, Y., Garg, V., et al. (2017). Genome-Wide Discovery and Deployment of Insertions and Deletions Markers Provided Greater Insights on Species, Genomes, and Sections Relationships in the Genus Arachis. Front. Plant Sci. 8:2064. doi: 10.3389/fpls.2017.02064
Wang, C.-L., Ding, M.-Q., Zou, C.-Y., Zhu, X.-M., Tang, Y., Zhou, M.-L., et al. (2017). Comparative Analysis of Four Buckwheat Species Based on Morphology and Complete Chloroplast Genome Sequences. Sci. Rep. 7:6514. doi: 10.1038/s41598-017-06638-6
Wang, J., Li, C., Shi, D., Liu, Y., Tang, R., He, L., et al. (2021). Verifying high variation regions based on sect. Arachis chloroplast genome and revealing the interspecies genetic relationship. Chin. J. Oil Crop Sci. 43:495.
Wang, J., Li, C., Yan, C., Zhao, X., and Shan, S. (2018). A comparative analysis of the complete chloroplast genome sequences of four peanut botanical varieties. PeerJ 6:e5349. doi: 10.7717/peerj.5349
Wang, J., Li, Y., Li, C., Yan, C., Zhao, X., Yuan, C., et al. (2019). Twelve complete chloroplast genomes of wild peanuts: great genetic resources and a better understanding of Arachis phylogeny. BMC Plant Biol. 19:504. doi: 10.1186/s12870-019-2121-3
Wheeler, T. J., and Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi: 10.1093/bioinformatics/btt403
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xu, J.-H., Liu, Q., Hu, W., Wang, T., Xue, Q., and Messing, J. (2015). Dynamics of chloroplast genomes in green plants. Genomics 106, 221–231. doi: 10.1016/j.ygeno.2015.07.004
Xue, S., Shi, T., Luo, W., Ni, X., Iqbal, S., Ni, Z., et al. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 6:89. doi: 10.1038/s41438-019-0171-1
Yin, D., Ji, C., Song, Q., Zhang, W., Zhang, X., Zhao, K., et al. (2020). Comparison of Arachis monticola with Diploid and Cultivated Tetraploid Genomes Reveals Asymmetric Subgenome Evolution and Improvement of Peanut. Adv. Sci. 7:1901672. doi: 10.1002/advs.201901672
Yin, D., Wang, Y., Zhang, X., Ma, X., He, X., and Zhang, J. (2017). Development of chloroplast genome resources for peanut (Arachis hypogaea L.) and other species of Arachis. Sci. Rep. 7:11649.
Yu, J., Xu, F., Wei, Z., Zhang, X., Chen, T., and Pu, L. (2020). Epigenomic landscape and epigenetic regulation in maize. Theor. Appl. Genet. 133, 1467–1489. doi: 10.1007/s00122-020-03549-5
Zhang, D., Luo, K., Wu, F., Wang, Y., and Zhang, J. (2017). Advances in cleistogamy of angiosperms. Pratacultural Sci. 34, 1215–1227. doi: 10.1111/tpj.12693
Zheng, S., Poczai, P., Hyvönen, J., Tang, J., and Amiryousefi, A. (2020). Chloroplot: an Online Program for the Versatile Plotting of Organelle Genomes. Front. Genet. 11:576124. doi: 10.3389/fgene.2020.576124
Zheng, X., Wang, J., Feng, L., Liu, S., Pang, H., Qi, L., et al. (2017). Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 7:1555. doi: 10.1038/s41598-017-01518-5
Zheng, Z., Sun, Z., Fang, Y., Qi, F., Liu, H., Miao, L., et al. (2018). Genetic Diversity, Population Structure, and Botanical Variety of 320 Global Peanut Accessions Revealed Through Tunable Genotyping-by-Sequencing. Sci. Rep. 8:14500. doi: 10.1038/s41598-018-32800-9
Zhuang, W., Chen, H., Yang, M., Wang, J., Pandey, M. K., Zhang, C., et al. (2019). The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 51, 865–876. doi: 10.1038/s41588-019-0402-2
Keywords: Arachis, whole plastid genome, genetic structure, phylogenomics, maternal hybridization event
Citation: Tian X, Shi L, Guo J, Fu L, Du P, Huang B, Wu Y, Zhang X and Wang Z (2021) Chloroplast Phylogenomic Analyses Reveal a Maternal Hybridization Event Leading to the Formation of Cultivated Peanuts. Front. Plant Sci. 12:804568. doi: 10.3389/fpls.2021.804568
Received: 29 October 2021; Accepted: 30 November 2021;
Published: 17 December 2021.
Edited by:
Hai Du, Southwest University, ChinaReviewed by:
Abdullah, Quaid-i-Azam University, PakistanXu Zhang, Wuhan Botanical Garden, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Tian, Shi, Guo, Fu, Du, Huang, Wu, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinyou Zhang, aGFhc3pAMTI2LmNvbQ==; Zhenlong Wang, d3psQHp6dS5lZHUuY24=