 Haoyun Sun
Haoyun Sun Jingzhao Mei
Jingzhao Mei Weiwei Zhao
Weiwei Zhao Wenqian Hou
Wenqian Hou Yang Zhang
Yang Zhang Tao Xu
Tao Xu Shaoyuan Wu
Shaoyuan Wu Lei Zhang
Lei Zhang- 1Jiangsu Key Laboratory of Phylogenomics and Comparative Genomics, School of Life Sciences, Jiangsu Normal University, Xuzhou, China
- 2Department of Biochemistry and Molecular Biology, 2011 Collaborative Innovation Center of Tianjin for Medical Epigenetics, Tianjin Key Laboratory of Medical Epigenetics, Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
As a major plant-specific transcription factor family, SPL genes play a crucial role in plant growth, development, and stress tolerance. The SPL transcription factor family has been widely studied in various plant species; however, systematic studies on SPL genes in the genus Ipomoea are lacking. Here, we identified a total of 29, 27, 26, and 23 SPLs in Ipomoea batatas, Ipomoea trifida, Ipomoea triloba, and Ipomoea nil, respectively. Based on the phylogenetic analysis of SPL proteins from model plants, the Ipomoea SPLs were classified into eight clades, which included conserved gene structures, domain organizations and motif compositions. Moreover, segmental duplication, which is derived from the Ipomoea lineage-specific whole-genome triplication event, was speculated to have a predominant role in Ipomoea SPL expansion. Particularly, tandem duplication was primarily responsible for the expansion of SPL subclades IV-b and IV-c. Furthermore, 25 interspecific orthologous groups were identified in Ipomoea, rice, Arabidopsis, and tomato. These findings support the expansion of SPLs in Ipomoea genus, with most of the SPLs being evolutionarily conserved. Of the 105 Ipomoea SPLs, 69 were predicted to be the targets of miR156, with seven IbSPLs being further verified as targets using degradome-seq data. Using transcriptomic data from aboveground and underground sweet potato tissues, IbSPLs showed diverse expression patterns, including seven highly expressed IbSPLs in the underground tissues. Furthermore, the expression of 11 IbSPLs was validated using qRT-PCR, and two (IbSPL17/IbSPL28) showed significantly increased expression during root development. Additionally, the qRT-PCR analysis revealed that six IbSPLs were strongly induced in the roots under phytohormone treatments, particularly zeatin and abscisic acid. Finally, the transcriptomic data of storage roots from 88 sweet potato accessions were used for weighted gene co-expression network analysis, which revealed four IbSPLs (IbSPL16/IbSPL17/IbSPL21/IbSPL28) clusters with genes involved in “regulation of root morphogenesis,” “cell division,” “cytoskeleton organization,” and “plant-type cell wall organization or biogenesis,” indicating their potential role in storage root development. This study not only provides novel insights into the evolutionary and functional divergence of the SPLs in the genus Ipomoea but also lays a foundation for further elucidation of the potential functional roles of IbSPLs on storage root development.
Introduction
The SQUAMOSA promoter-binding protein-like (SPL) genes are of the plant-specific transcription factor families, which play fundamental roles in plant growth, development, and stress tolerance (Guo et al., 2008; Chen et al., 2010; Preston and Hileman, 2013; Chen et al., 2015; Wang and Wang, 2015). The SPL genes predominantly contain the SQUAMOSA promoter-binding (SBP) domain, which comprises three distinct motifs: two non-interleaved zinc-binding sites (Cys-Cys-Cys-His and Cys-Cys-His-Cys) and one nuclear localization signal (NLS) at the C-terminus (Guo et al., 2008). SPL genes (AmSBP1 and AmSBP2) were first discovered in snapdragon (Antirrhinum majus), following their role in flower development (Klein et al., 1996). Since then, numerous homologs have been identified and characterized in model species, such as Arabidopsis thaliana (Cardon et al., 1999), rice (Oryza sativa) (Xie et al., 2006), and tomato (Solanum lycopersicum) (Salinas et al., 2012). With the increasing number of sequenced genomes, SPL members have been increasingly annotated and reported in non-model plants, such as apple (Malus domestica) (Li et al., 2013), Jatropha curcas (Yu et al., 2020), pepper (Capsicum annuum) (Zhang et al., 2016), poplar (Populus trichocarpa) (Li and Lu, 2014), and soybean (Glycine max) (Tripathi et al., 2017).
SPL genes in model plants have been well studied, showing functional divergence. In A. thaliana, AtSPL3, AtSPL9, and AtSPL10 have been reported to regulate root development (Yu et al., 2015; Barrera-Rojas et al., 2020). Similar roles have also been reported in rice (Shao et al., 2019) and apple (Xu et al., 2017). Various studies have further found that SPL proteins participate in vegetative and reproductive phase transitions, for example, AtSPL3/4/5 promotes flowering by directly inducing AP1, FUL and LFY expression, which are flowering integrator genes (Yamaguchi et al., 2009). Additionally, SPL proteins are involved in fruit development and grain yield. For example, LeSPL-CNR regulates cell wall disassembly and carotenoid biosynthesis during fruit ripening in tomato (Orfila et al., 2002; Manning et al., 2006); OsSPL16 expression promotes cell division and grain filling, with positive results in grain width and yield in rice (Wang et al., 2015). SPL genes are also considered as miR156 targets, thus forming a functional miR156-SPL regulatory network (Wang and Wang, 2015). In the miR156-SPL network, AtSPL9 negatively affects anthocyanin accumulation (Gou et al., 2011), whereas OsSPL7 enhances disease resistance against bacterial blight (Liu et al., 2019). The functions of SPL genes have been comprehensively studied in Arabidopsis and other model plants; however, their functionality in Ipomoea are relatively scarce.
The genus Ipomoea, which includes 500–600 species, possesses the largest number of species in the family Convolvulaceae (Austin and Huáman, 1996). Ipomoea species are widely and globally distributed with great value in the fields of industry and agriculture (Austin and Huáman, 1996; Liu, 2017; Morita and Hoshino, 2018). For example, Japanese morning glory, Ipomoea nil (L.) Roth. (2n = 2x = 30), is cultivated as an ornamental plant due to its diverse flower color patterns (Morita and Hoshino, 2018). Sweet potato, Ipomoea batatas (L.) Lam. (2n = 6x = 90), is ranked as the seventh most important crop globally due to its strong adaptability, stable yields, and high nutritional value (Liu, 2017). The storage roots of sweet potato, which are mainly harvested, have significant nutrient content and yield (Zhang et al., 2020). The initiation and development of storage roots is known as a complex and genetically programmed process (Ravi et al., 2014). Although several studies have reported the formation and development of storage root at the morphological, physiological, and molecular level (Nakatani, 1991; Wang et al., 2005; Tanaka et al., 2008; Noh et al., 2010; Dong et al., 2019; Huan et al., 2020), the underlying mechanisms of storage root development have not yet been fully elucidated. Up to now, the genomes of four species (I. batatas, Ipomoea trifida, Ipomoea triloba, and I. nil) have been sequenced in Ipomoea (Hoshino et al., 2016; Yang et al., 2017; Wu et al., 2018). Among these species, I. trifida is the most closely related diploid to I. batatas, followed by I. triloba and I. nil (Wu et al., 2018). The reported haplotype-resolved genome assembly of I. batatas is of low-quality, making it difficult to accurately identify and characterize genes. Contrastingly, the genome assembly of the other three diploid relatives is of high-quality and can be used as robust references for I. batatas. Therefore, genome availability makes it possible to perform a genome-wide comparative analysis of SPL genes in Ipomoea.
In this study, genome-wide identification and characterization of SPL genes were performed in four publicly available Ipomoea species, including I. batatas, I. trifida, I. triloba, and I. nil. Following this, the phylogenetic relationships and evolutionary patterns of SPL genes were investigated in these four species. IbSPL gene expression patterns were determined using transcriptome and qRT-PCR in different organs or under various hormone treatments. Finally, weighted gene co-expression network analysis (WGCNA) was used to construct the co-expression network and infer the putative functions for the IbSPL genes in the storage root of sweet potato. This work not only provides insights into the evolutionary conservation and diversification of SPL genes in the genus Ipomoea but also lays the foundation for further research on IbSPL genes related to storage root development in sweet potato.
Materials and Methods
Identification of SQUAMOSA Promoter-Binding Protein-Like Genes in I. nil, I. triloba, I. trifida, and I. batatas
The genomes of four Ipomoea species (including I. nil, I. triloba, I. trifida, and I. batatas) were downloaded from the ‘‘Ipomoea nil Genome Project1 “ (Hoshino et al., 2016), ‘‘Sweetpotato Genomic Resource2 “ (Wu et al., 2018), and ‘‘Sweet potato genome browser3 “ (Yang et al., 2017), respectively. SPL genes in these four species were identified using the following three methods. First, the 16 A. thaliana SPL proteins4 were used as queries to find SPL homologs using BLASTP program with a threshold of e-value < 1e-3. Second, the Hidden Markov Model (HMM) of the SBP (PF03110) domain was downloaded from the Pfam database (El-Gebali et al., 2019) and used to identify putative SPL proteins using the hmmsearch (El-Gebali et al., 2019) program. Third, all the candidate SPL proteins obtained from the BLASTP and hmmsearch analysis were submitted to the SMART (Letunic and Bork, 2018) and ScanProsite databases (de Castro et al., 2006) to confirm an SBP domain presence. Proteins lacking the SBP domain were excluded, while the remaining were considered as the SPL proteins. Additionally, a manual examination was performed on the structures of identified IbSPLs to correct genome assembly errors.
The BUSCA online software (Savojardo et al., 2018) was used to predict the subcellular localization of Ipomoea SPL proteins. An in-house Perl script was used to analyze the physical and chemical properties of Ipomoea SPL proteins, such as protein length, molecular weight (MW, kD) and isoelectric point (pI).
Phylogenetic Analysis of SQUAMOSA Promoter-Binding Protein-Like Proteins
Phylogenetic analysis was performed on the SPL proteins from Chlamydomonas reinhardtii, A. thaliana, J. curcas, M. domestica, O. sativa, P. trichocarpa, S. lycopersicum, and the four Ipomoea species using the following steps: first, MAFFT software (v7.45) (Katoh and Standley, 2013) was used to obtain full-length SPL proteins’ multiple alignments; second, Gblocks program (v0.71b) (Castresana, 2000) was used to select conserved blocks from the multiple alignments; third, MEGA X software (Kumar et al., 2018) was used to construct a neighbor-joining phylogenetic tree with 1000 bootstrap replications, with the CRR1 protein from C. reinhardtii was set as an outgroup; finally, Evolview website was used to visualize the tree (Subramanian et al., 2019). Similarly for the Ipomoea SPL proteins, a neighbor-joining phylogenetic tree was constructed using the aforementioned.
Analysis of the Gene Structure, Protein Domain, and Motif
The exon/intron positions of all Ipomoea SPL genes were obtained from the downloaded GFF3 files of the genomic database. The domain organizations of Ipomoea SPL proteins were annotated based on the SMART database results (Letunic and Bork, 2018). The conserved sequences in each domain were shaded at four levels using GeneDoc. The motif compositions of Ipomoea SPL proteins were analyzed through MEME online database (Bailey et al., 2009), with the maximum number set to 10. Finally, the gene structure, domain organization, and motif composition were drawn using Tbtools (Chen et al., 2020).
Gene Duplication, Orthology, and Selection Analysis
MCScanX software (Wang et al., 2012) was used to identify collinear blocks within or between species to classify the SPL genes into five different types: singleton, dispersed, proximal, tandem, and segmental duplication. The synteny relationships of the collinearity blocks in each Ipomoea species were visualized using Circos (Krzywinski et al., 2009). OrthoMCL software (Li et al., 2003) was used to detect orthologous groups among the diverse SPL genes. For each orthologous gene pair, Ks (synonymous substitution rate), Ka (non-synonymous substitution rate), and Ka/Ks ratio (evolutionary constraint) were calculated using PAML (Yang, 2007).
Prediction of miR156-Targeted Genes
Publicly available datasets were used to identify miR156 sequences in Ipomoea. A total of 58 miRNA transcriptomes deposited in National Center for Biotechnology Information (NCBI) (Coordinators, 2018) (16 in PRJNA471495, 2 in PRJNA474012, 11 in PRJNA592001, 12 in PRJNA599544, 12 in PRJNA600587, and 5 in PRJNA638516) were collected (Supplementary Table 5; Kuo et al., 2019; Saminathan et al., 2019; Yang et al., 2020; Liu et al., 2021). Trimmomatic software (version 0.39) (Bolger et al., 2014) was used to filter the raw miRNA sequencing data, which removed low-quality reads and sequencing adaptors. Finally, using I. batatas as the reference genome, the miRDeep2 (version 1.1.4) pipeline (Kuang et al., 2019) was employed to identify miR156 sequences with default parameters.
For the Ipomoea SPL genes, miR156 target sites were predicted using the psRNATarget server (Dai et al., 2018) with default settings. The predicted miR156-SPL interactions in I. batatas were validated using five degradomes (one in PRJNA592001 and four in PRJNA600587) (Yang et al., 2020; Liu et al., 2021) downloaded from public databases (Supplementary Table 5). After filtering out the low-quality reads and sequencing adaptors, the CleaveLand4 pipeline (Addo-Quaye et al., 2009) was used to identify miR156 cleavage sites. The identified targets with categories 0–3 and p-values < 0.05 were considered to be reliable miR156 target genes.
Promoter Analysis of Ipomoea SQUAMOSA Promoter-Binding Protein-Like Genes
The 2000 bp sequence upstream of the start codon for all Ipomoea SPL genes was retrieved using an in-house Perl script and submitted to the PlantCARE program (Lescot et al., 2002) to predict cis-acting elements as previously described (Chen et al., 2019). The distribution of cis-acting elements in each promoter was determined using TBtools (Chen et al., 2020).
Plant Materials and Hormone Treatments
Sweet potato (I. batatas cv. Xuyu34) plants used in this study were provided by the Xuzhou Academy of Agricultural Sciences, Xuzhou, Jiangsu, China. According to institutional, national, and international guidelines, these samples do not require specific permissions for research purposes. The plants were grown in greenhouses on the campus of Jiangsu Normal University, Xuzhou, China. For organ-specific expression analysis, the tissues of young leaves, mature leaves, flowers, and roots (10, 20, 40, 60, 80, 90, and 100 DAT roots with 0.3, 2, 7, 25, 37, 52, and 60 mm in diameter, respectively) were collected. For hormone treatments, stems with 4–5 leaves were cut and planted in 1/8 Hoagland solution to initiate adventitious root development for 10 days. Then stem cuttings with similar growth conditions were chosen and planted in 1/8 Hoagland solution separately containing 100 μM abscisic acid (ABA), indole-3-acetic acid (IAA), zeatin (ZT), and methyl-Jasmonate (MeJA). Stem cuttings without any hormone treatment were set as a control. Adventitious roots from the stem cuttings were collected at 0, 6, 12, 24, and 48 h post the treatments. Three biological replicates were collected for each sample. All samples were frozen in liquid nitrogen and finally stored at –80°C for subsequent use.
RNA Extraction and qRT-PCR Analysis
For analyzing expression patterns of IbSPL genes in different tissues or under phytohormone treatment, total RNA for each sample was extracted using the RNApure Plant Kit (CWBio, Beijing, China), following the manufacturers’ instructions. For investigating the miR156-SPL interactions, total RNA was extracted using TRIzol reagent (Invitrogen, CA, United States) according to the manufacturers’ instructions. The first cDNA strand was synthesized from 1.0 μg total pure RNA using the HiFiScript cDNA Synthesis Kit (CWBio, Beijing, China). The reverse transcription primer and qRT-PCR primer for miR156 were designed as previous study described (Zhou et al., 2020). Gene-specific primers for each IbSPL gene were designed using primer3 (Untergasser et al., 2012). qRT-PCR was performed via the Bio-rad CFX Connect™ Real-Time System (Bio-Rad, CA, United States) using 2 × Q3 SYBR qPCR Master Mix (Universal) premix (Tolo Biotechnology, Shanghai, China). IbARF gene was used as a reference gene for normalizing the expression levels (Park et al., 2012). The relative transcript abundance for each gene was calculated with mean ± SD of biological triplicate samples using the 2–ΔΔCT approach (Livak and Schmittgen, 2001). The primers used are listed in Supplementary Table 10.
Analysis of the Expression Patterns of IbSPLs Using Published Transcriptomic Data
To explore tissue- and developmental stage-specific expression patterns of IbSPL genes, publicly available transcriptome datasets from two previous studies (Ding et al., 2017; Wu et al., 2018; Supplementary Table 8) were used: one included eight different tissues from cultivar Xuzi3 and Yan252 under the BioProject accession number PRJCA000640 in National Genomics Data Center (NGDC) (National Genomics Data Center and Partners, 2020), and the other included eight different stages during root development from cultivar Beauregard under the BioProject accession number PRJNA491292 in NCBI (Coordinators, 2018). Transcriptome analysis was performed as described in our previous study (Zhang et al., 2020). The downloaded raw fastq files were filtered using Trimmomatic (version 0.39) (Bolger et al., 2014), and then were mapped to sweet potato genome Taizhong6 (Yang et al., 2017) using STAR (version 2.7.1a) software under the 2-pass mapping mode (Dobin et al., 2013). RSEM (Li and Dewey, 2011) was used to calculate Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values for each gene. Finally, a heatmap was plotted based on the normalized expression values of 29 IbSPL genes using the pheatmap package in R.
Construction of Co-expression Networks Involving IbSPL and Other I. batatas Genes in Sweet Potato Storage Root
Transcriptomic datasets of mature storage roots of 88 sweet potato accessions were obtained from a previous study (Supplementary Table 11; Ding et al., 2017) under the BioProject accession number PRJCA000642 in NGDC (National Genomics Data Center and Partners, 2020). Weighted co-expression network construction and module detection were performed using the R package WGCNA (version 1.4.9) (Langfelder and Horvath, 2008) with the following parameters: power = 9, minModuleSize = 30, cutHeight = 0.25, and network module export weight threshold = 0.05. The sub-network was subsequently visualized using Cytoscape (Smoot et al., 2011). eggNOG-mapper (version 2) (Huerta-Cepas et al., 2017) was used to assign the functional annotation to sweet potato genes, and Clusterprofiler (Yu et al., 2012) was used to perform GO enrichment analysis for genes co-expressed with IbSPLs (adjusted P-value < 0.05).
Statistical Analysis
The qRT-PCR results were analyzed using ANOVA (one-way analysis of variance) followed by LSD test. Statistically significant differences at p < 0.05 are indicated using different letters.
Results
Identification of SQUAMOSA Promoter-Binding Protein-Like Genes in Four Ipomoea Species
BLASTP and HMM were used to identify the SPL genes in Ipomoea species, while SMART and ScanProsite were used to validate the results. A total of 29, 27, 26, and 23 SPL genes were identified in I. batatas (Ib), I. trifida (Itf), I. triloba (Itb), and I. nil (In), respectively. The Ipomoea SPL genes were renamed according to their chromosomal location (Supplementary Table 1). The numbers of SPL genes and their total percentage in each species are displayed in Figure 1A. The results showed that the I. nil genome had the least number of SPL genes compared to the other three species.
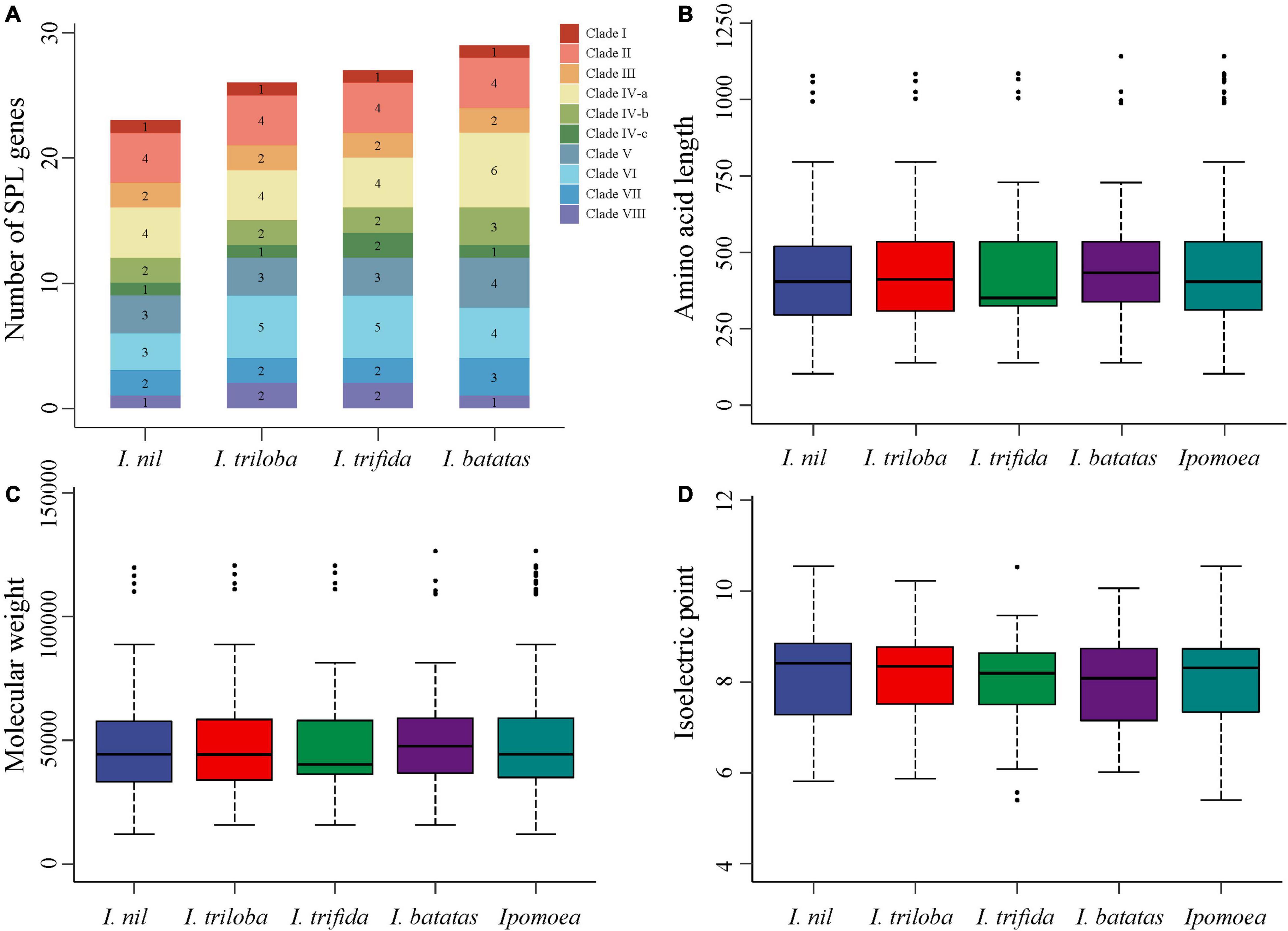
Figure 1. Comparison of SPL genes in four Ipomoea species. (A) Comparison of the number and ratio of SPL genes in four Ipomoea species, with different colors representing each clade. (B–D) Comparison of SPL protein lengths, MWs, and pIs in four Ipomoea species, respectively.
Subcellular localization analysis showed the nuclear localization of most Ipomoea SPL proteins (94, 89.52%) (Supplementary Table 1), suggesting their critical role in regulatory functions. Furthermore, the physical and chemical properties of SPL proteins were significantly differed within species but exhibited similar patterns among the four species (Figures 1B–D and Supplementary Table 1). Moreover, amino acid numbers in the Ipomoea SPL proteins ranged from 103 (InSPL9) to 1141 (IbSPL11), the MWs varied between 12.01 (InSPL9) and 126.51 (IbSPL11) kDa, and the pIs ranged from 5.40 (ItfSPL14) to 10.55 (InSPL15).
Comparative Phylogenetic Analysis of Ipomoea SQUAMOSA Promoter-Binding Protein-Like Genes
The evolutionary relationship between the Ipomoea SPL genes was explored via a rooted neighbor-joining phylogenetic tree, which was constructed using 105 SPL proteins from four Ipomoea species and 124 SPL proteins from seven other plant species (A. thaliana, J. curcas, M. domestica, O. sativa, P. trichocarpa, S. lycopersicum, and Chlamydomonas reinhardii) (Figure 2 and Supplementary Table 2). Based on the classification of SPLs from A. thaliana, S. lycopersicum, and O. sativa (Cardon et al., 1999; Xie et al., 2006; Salinas et al., 2012), Ipomoea SPL genes were classified into eight clades (I–VIII) (Figures 1, 2). All clades had at least one SPL gene in each Ipomoea species, indicating SPL conservation across Ipomoea genomes. However, the number of SPLs in certain clades was highly variable among Ipomoea species, suggesting a diversity of SPLs in the genus Ipomoea. Clade I was the smallest subfamily, containing only one member for each Ipomoea species while clade IV had the highest number of SPLs (>26%) in genus Ipomoea, with further divisions into three subclades: IV-a, IV-b, and IV-c. Members of the IV-b and IV-c subclades only comprised SPL genes from the Ipomoea species and no homologs of other species, indicating that the Ipomoea SPL genes in these two subclades were evolutionary conserved. Additionally, the phylogenetic analysis also indicated that most IbSPL genes were closer to ItfSPL genes than either ItbSPL or InSPL genes, supporting the fact that I. trifida is the most closely related diploid to hexaploid sweet potato (Wu et al., 2018). Moreover, the number of SPL genes in Ipomoea species (the average number of SPL genes in the four Ipomoea species was 26) greatly increased by approximately 2 times compared to that in S. lycopersicum (13), respectively. These results indicate the extensive expansion of Ipomoea SPL genes after the speciation of S. lycopersicum.
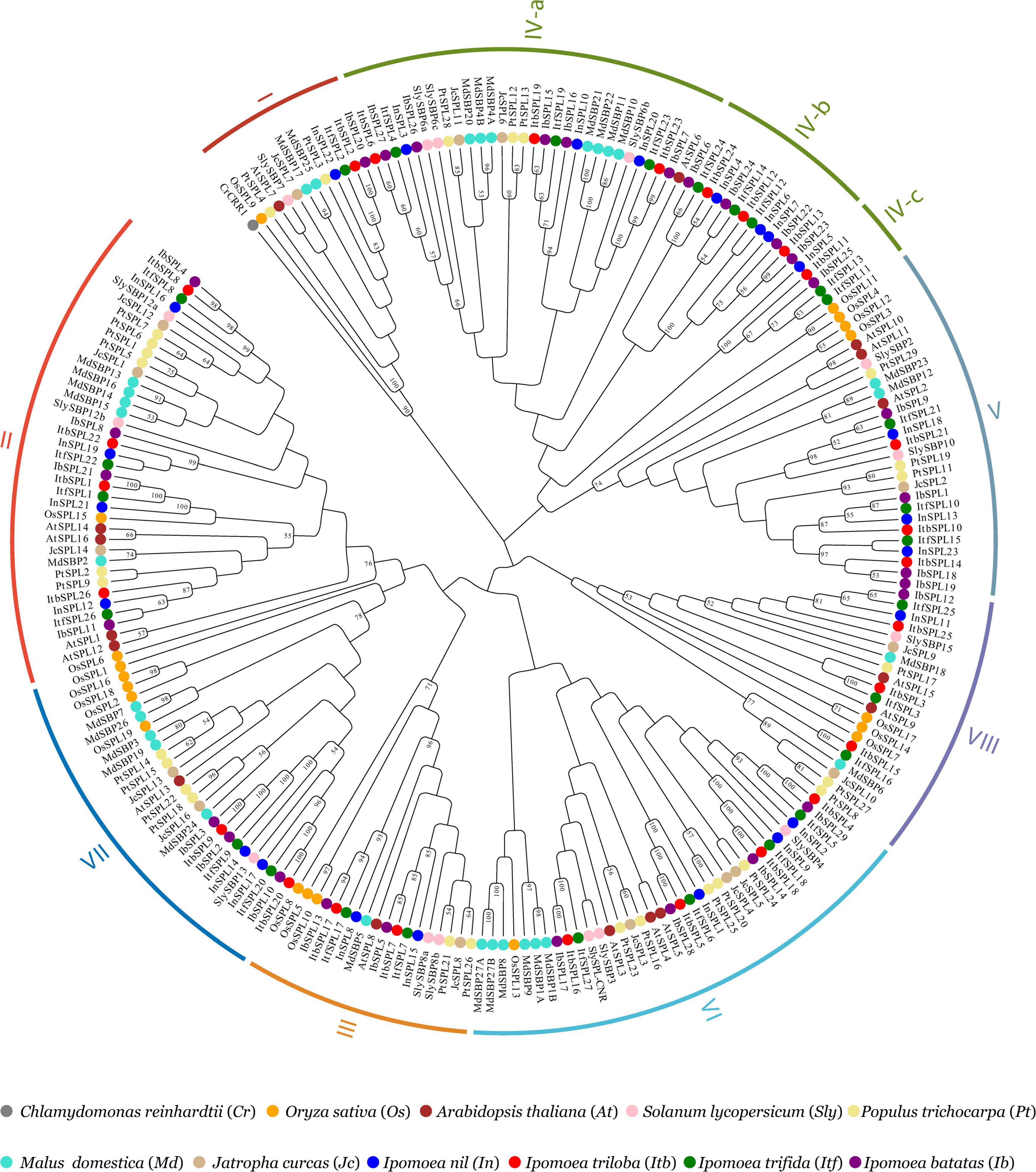
Figure 2. Phylogenetic analysis of SPL proteins from I. batatas, I. trifida, I. triloba, I. nil, C. reinhardtii, A. thaliana, J. curcas, M. domestica, P. trichocarpa, S. lycopersicum, and O. sativa. The rooted phylogenetic tree was constructed based on the conserved domain of 229 SPLs using the neighbor-joining method with 1000 bootstrap replications. The CrCRR1 protein from C. reinhardtii was used as an outgroup to root the tree. Numbers on the tree indicate bootstrap support (values <50% not shown). Each colored arcs indicates the different clades of the SPLs. SPL members from the same species are marked with the same colors: Blue, I. nil; red, I. triloba; green, I. trifida; purple, I. batatas; gray, C. reinhardtii; orange, O. sativa; brown, A. thaliana; pink, S. lycopersicum; tan, J. curcas; turquoise, M. domestica; khaki, P. trichocarpa.
Gene and Protein Structure of the Ipomoea SQUAMOSA Promoter-Binding Protein-Like Family
The structural diversity of the Ipomoea SPL genes was explored using intron/exon structure analysis (Supplementary Figure 1). Gene structure illustrations showed a high variation in the number of exons, ranging from 2 to 15 (Supplementary Figure 1a and Supplementary Table 1). For example, IbSPL20 contained the highest exons (15), whereas most Ipomoea SPL genes in Clade VI contained the least exons (2). Moreover, most Ipomoea SPL genes in the same clade exhibited similar gene structures, despite belonging to different species. SPL gene in clades I and II contained the highest exons, ranging from 10 to 15, while SPL genes in the remaining clades had 2–7 exons (except IbSPL19). These results suggested that the gain or loss of exon/intron had occurred during the Ipomoea SPL gene evolution, resulting in their functional divergence.
Ipomoea SPL protein features were investigated by analyzing the conserved domains using multiple sequence alignment. The results showed that the SPL members had the SBP domain, which comprised two non-interleaved zinc finger-like structures (Zn-1/2) and one NLS motif (Supplementary Figures 1c, 2). Based on the alignments of the Ipomoea SPLs, the Zn-2 motif showed higher conservation than the Zn-1 and NLS motifs, which was consistent with the results in Rosaceae (Jiang et al., 2021) and Oryza species (Zhong et al., 2019; Supplementary Figure 2). The Zn-2 motif in all Ipomoea SPLs was a Cys-Cys-His-Cys (C2HC) type (except IbSPL23) whereas the Zn-1 motif showed varied types: Cys-Cys-Cys-Cys (C4) type in clade I and Cys-Cys-Cys-His (C3H) type in the remaining clades. Moreover, other conserved domains were identified in specific clades. For instance, SPLs in clade I and II possessed a DEXDc domain (Supplementary Figure 3), which is involved in ATP-dependent DNA unwinding (Caruthers and McKay, 2002); SPLs in clade II contained Ankyrin repeats (Supplementary Figure 4), which are considered to be significant for mediating protein-protein interactions (Li et al., 2006).
To gain a better understanding of Ipomoea SPL protein characteristics, the MEME software was used to explore the motif compositions (Supplementary Figures 1d, 5). The results showed that Ipomoea SPL proteins within the same clade showed similar motif compositions, while those in different clades exhibited distinct variations in motif composition. In brief, all Ipomoea SPL proteins had two motifs (motif 2 and 3), which were a part of the SBP domain (Supplementary Figure 2); clade I and II had four motifs (motif 4, 5, 6, and 7), with motif 4 being the DEXDc domain (Supplementary Figure 3);clade II had motif 8, which consisted of Ankyrin repeats (Supplementary Figure 4); clade IV had motif 10, which had unknown functions.
Gene Duplication, Orthology Relationship, and Selective Pressure of Ipomoea SQUAMOSA Promoter-Binding Protein-Like Genes
To investigate the gene duplication modes of Ipomoea SPL genes, MCScanX (Wang et al., 2012) was used to perform gene collinearity analysis in each Ipomoea species. All Ipomoea SPL genes were estimated to be duplicated (absence of singleton mode), with segmental (51, 48.57%) mode as the dominant mode compared to the other duplication modes: dispersed (39, 37.14%), tandem (13, 12.38%), and proximal (2, 1.90%) (Figures 3A,B; Supplementary Table 1; Supplementary Figure 6). These results indicate that segmental duplication has played a predominant role in the evolution and expansion of Ipomoea SPL genes. Additionally, tandem duplication was found to be the predominant model in the IV-b and IV-c subclades, suggesting the expansion of SPL genes in these two clades via tandem duplication.
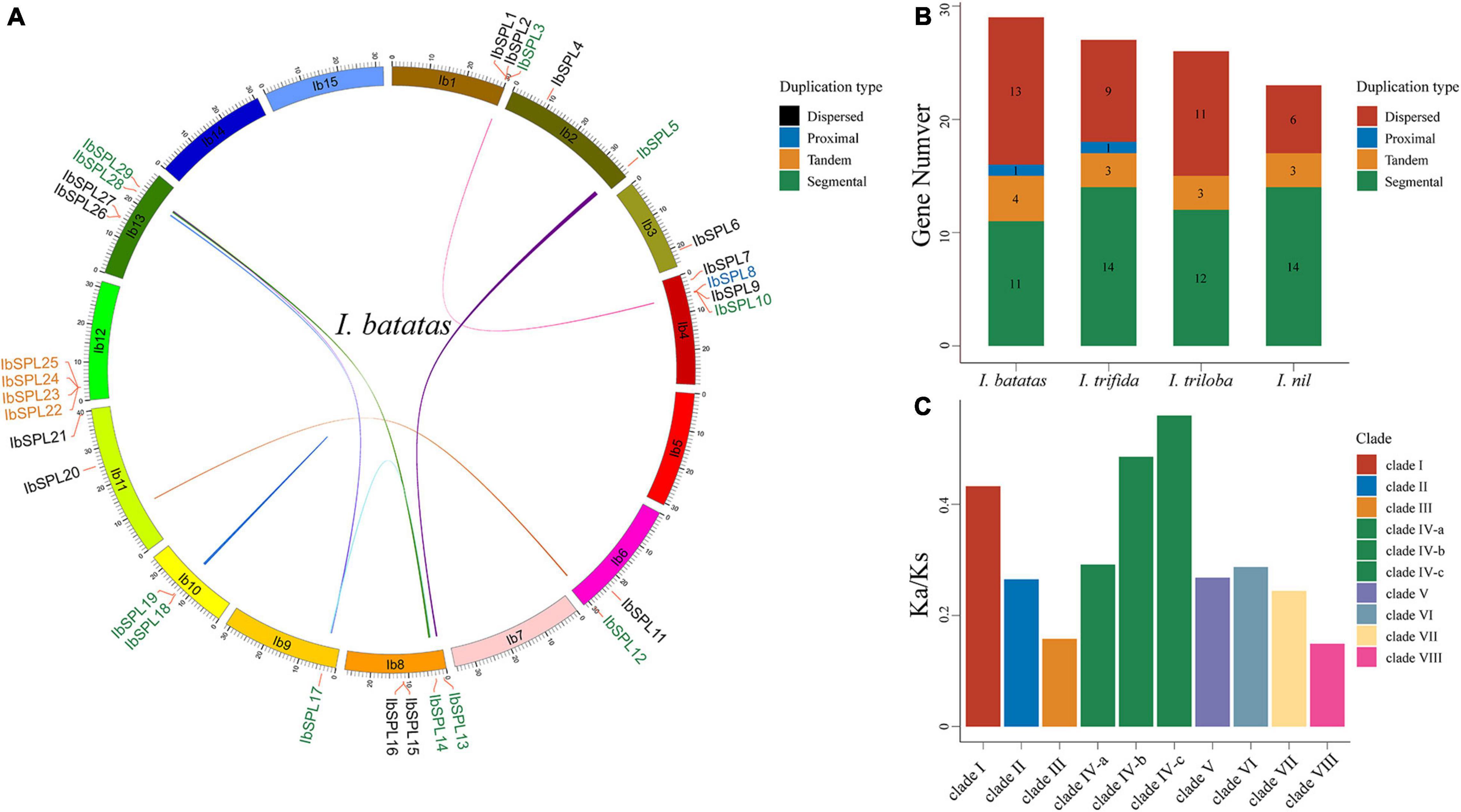
Figure 3. Syntenic relationships and selective pressure of Ipomoea SPLs. (A) The collinear relationship of SPL genes in I. batatas, wherein the colored lines indicate syntenic regions. IbSPL genes are classified into four duplicated types, each indicated by a different color. (B) The distribution of SPLs duplication modes in four Ipomoea species. (C) The mean Ka/Ks values in each clade.
The orthologous relationships among the SPL genes were determined using OrthoMCL (Li et al., 2003) across O. sativa, A. thaliana, S. lycopersicum, and the four Ipomoea species. A total of 25 (1, 2, 2, 8, 3, 5, 2, and 2) orthologous groups in the eight clades (clade I to VIII) were identified, respectively (Supplementary Table 3). Among these groups, nine groups had genes originating from O. sativa, suggesting that these SPL genes may have originated prior to the split of monocots and dicots; four groups had genes from A. thaliana but were absent in O. sativa, implying that they originated after the divergence of monocots and dicots; ten groups had genes that existed only in the genus Ipomoea, indicating their origination via a common ancestor of the Ipomoea lineage. Furthermore, the potential functions of certain Ipomoea SPL genes could be inferred from their orthologs in O. sativa, A. thaliana, and S. lycopersicum.
To understand the divergence of Ipomoea SPL genes, the Ka, Ks, and Ka/Ks ratios for all orthologous groups were calculated using PAML software (Yang, 2007; Figure 3C; Supplementary Table 4). As a result, the mean Ka/Ks values of all clades were lower than 1.0, suggesting the evolution of Ipomoea SPL genes under the pressure of purifying selection. Genes in clade VIII showed the lowest mean Ka/Ks values (0.15) compared to those in the other clades, indicating their evolution under strong positive selection. Contrastingly, genes in subclades IV-b and IV-c exhibited the highest Ka/Ks values, implying that these two subclades have generally diverged much more rapidly than the other clades.
miR156 Target Site of Ipomoea SQUAMOSA Promoter-Binding Protein-Like Genes
A total of nine IbmiR156 members were identified in I. batatas (Figure 4A) using the publicly available miRNA transcriptomes (Supplementary Table 5). To explore the roles of miR156-mediated post-transcriptional regulation of SPLs in the genus Ipomoea, the transcripts of all the 105 Ipomoea SPL genes were searched for the target site of miR156 using psRNATarget (Dai et al., 2018). As a result, a total of 69 SPL genes were found to be potential miR156 targets, including 10 InSPLs, 19 ItbSPLs, 18 ItfSPLs, and 22 IbSPLs (Figure 4B and Supplementary Table 1). Among the miR156 target SPL genes, most of which (84%) the sites recognized by miR156 were located downstream of the SBP domain in the CDS region, then followed by the 3′-UTR (Figure 4B).
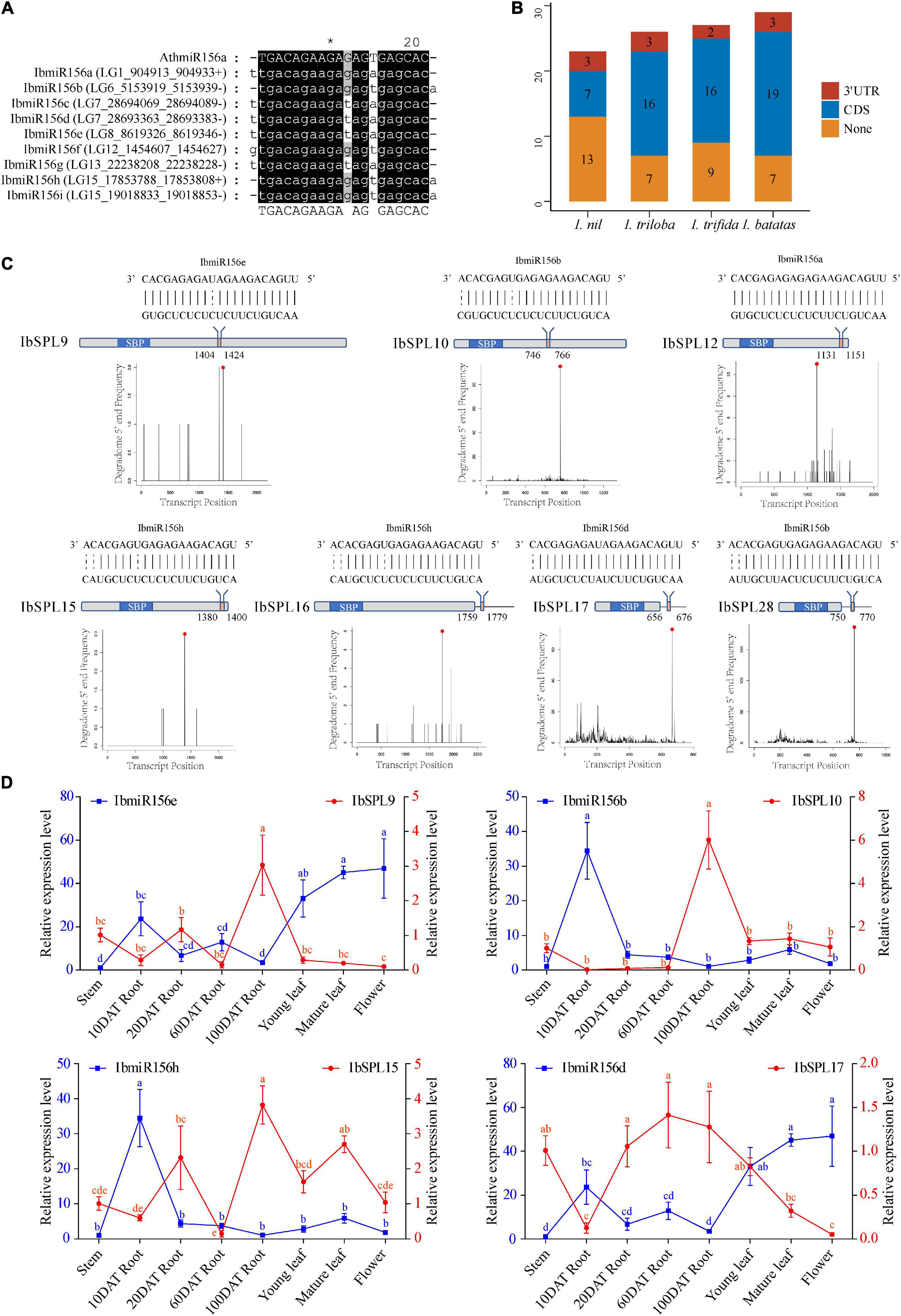
Figure 4. miR156 target site of the Ipomoea SPL genes. (A) Multiple alignments of identified IbmiR156 sequences. (B) The summary of miR156-targeted SPL genes in the Ipomoea genus. (C) Schematic diagram of IbSPL genes targeted by IbmiR156. The top diagram represents the complementary sequences between IbmiR156 and their targets. The gray box indicates the CDS region, the blue box represents the SBP domain, and the red box indicates the IbmiR156 target site. The diagram below is the target plots (t-plots) of IbmiR156 targets confirmed by degradome sequencing. (D) Expression correlation between IbmiR156 and IbSPLs. The lines represent the abundance of IbmiR156 and IbSPLs in different tissues. The Y-axis on the left and right indicates the relative expression levels of the IbmiR156 and IbSPLs, respectively. Relative expression was calculated using the 2–ΔΔCT method. The error bars indicate the standard deviations of the three biological replicates. Different letters indicate statistically significant differences at p < 0.05.
The predicted miR156-SPL interactions in I. batatas were validated using publicly available degradomes (Supplementary Table 5). The results showed that seven miR156-SPL interactions predicted by psRNATarget were confirmed by degradome sequencing (Figure 4C and Supplementary Table 1). Notably, the miR156 target sites of IbSPL9, IbSPL10, IbSPL12, and IbSPL15 were located in the CDS region, while the target sites of IbSPL16, IbSPL17, and IbSPL28 were located in the 3′-UTR region. The four IbmiR156-IbSPL pairs (IbmiR156b-IbSPL10, IbmiR156d-IbSPL17, IbmiR156e-IbSPL9, and IbmiR156h-IbSPL15) validated through degradomes data, were further selected for expression analysis by qRT-PCR. To understand the regulatory mechanisms of selected miR156 genes, correlation in the expression pattern of miR156 and their target SPLs was determined in different tissues (Figure 4D). The expression pattern of IbmiR156e was higher in flower, followed by mature leaf, young leaf, 10DAT root, 60DAT root, 20DAT root, and stem; conversely, the opposite trend was observed for its target IbSPL9. The expression of IbmiR156d and IbSPL17 also showed negative correlation in different tissues. While the expression of IbmiR156b-IbSPL10 and IbmiR156h-IbSPL15 were partially negatively correlated in some tissues.
Cis-Acting Elements in the Promoters of Ipomoea SQUAMOSA Promoter-Binding Protein-Like Genes
To understand the regulatory mechanisms and potential functions of Ipomoea SPL genes, cis-acting elements were analyzed in the 2000 bp upstream sequence from the start codon for all SPL genes by using the PlantCARE database (Lescot et al., 2002). A total of 4088 putative cis-acting elements were identified and divided into four categories: light responsiveness, plant growth, phytohormone, and abiotic/biotic stress response (Supplementary Tables 6, 7). As shown in Supplementary Figure 7a, SPL genes in the same clades exhibited similar cis-acting element compositions in the promoter, indicating their conserved biological functions. Among these categories, the abiotic/biotic stress response category covered the largest portion (43.66%), followed by the light response (25.93%), phytohormone response (20.77%), and plant growth (9.64%) categories (Supplementary Figure 7b). In the abiotic/biotic stress response category, MYB/MYC (responds to abiotic stress signals), STRE (metal-responsive element), WUN-motif (wound-responsive element), and LTR (low-temperature-responsive element) elements were found. In the light responsiveness category, Box 4, G-box, GT1-motif, TCT-motif, GATA-motif, and MRE elements were found, with the Box 4 motif as the most common (24%) element. As for the phytohormone response category, the ABRE element (responds to ABA), CGTCA-motif (responds to MeJA), ERE (responds to ethylene), TCA-element (responds to salicylic acid), and as-1 (responds to auxin) were commonly found, appearing in more than 60 Ipomeoa SPL genes. In the plant growth category, ARE elements essential for the anaerobic induction, CAT-box related to meristem expression and O2-site involved in zein metabolism regulation were the three major elements. Therefore, analysis of the cis-acting elements suggested that Ipomoea SPL genes participate in various biological processes.
Expression Profiles of IbSPL Genes in Different Tissues
Among the four Ipomoea species, I. batatas is the most important crop cultivated globally. To explore the putative roles of IbSPL genes, the tissue-specific expression patterns of IbSPLs were analyzed in eight tissues (four aboveground and four underground tissues) of two sweet potato cultivars (Xuzi3 and Yan252) using publicly available transcriptomic data (Supplementary Table 8; Ding et al., 2017). FPKM values were calculated to evaluate gene expression levels (Supplementary Table 9). As shown in Figure 5A, the expression patterns of IbSPLs were classified into three groups. The first group included seven IbSPL genes (IbSPL6/IbSPL15/IbSPL22/IbSPL23/IbSPL24/IbSPL25/IbSPL26), with lowest expression levels [log2(FPKM) < 2] in all tissues. The second group included five IbSPL genes (IbSPL4/IbSPL8/IbSPL11/IbSPL17/IbSPL20), with relatively high expression levels in all tissues. The third group included the remaining 17 IbSPL genes, with high expression in some aboveground tissues, especially in shoots or young leaves. Additionally, the gene expression profiles in underground tissues (fibrous and tuberous root) were investigated, with seven IbSPL genes highly expressed in underground tissues (mean FPKM > 10), such as IbSPL1, IbSPL4, IbSPL11, IbSPL17, IbSPL20, IbSPL21, and IbSPL28, implying their potential functionality in root development. Furthermore, these results showed that IbSPL genes within the same clades exhibit distinct expression patterns, such as IbSPL14, IbSPL17, IbSPL28, and IbSPL29 in clade VI.
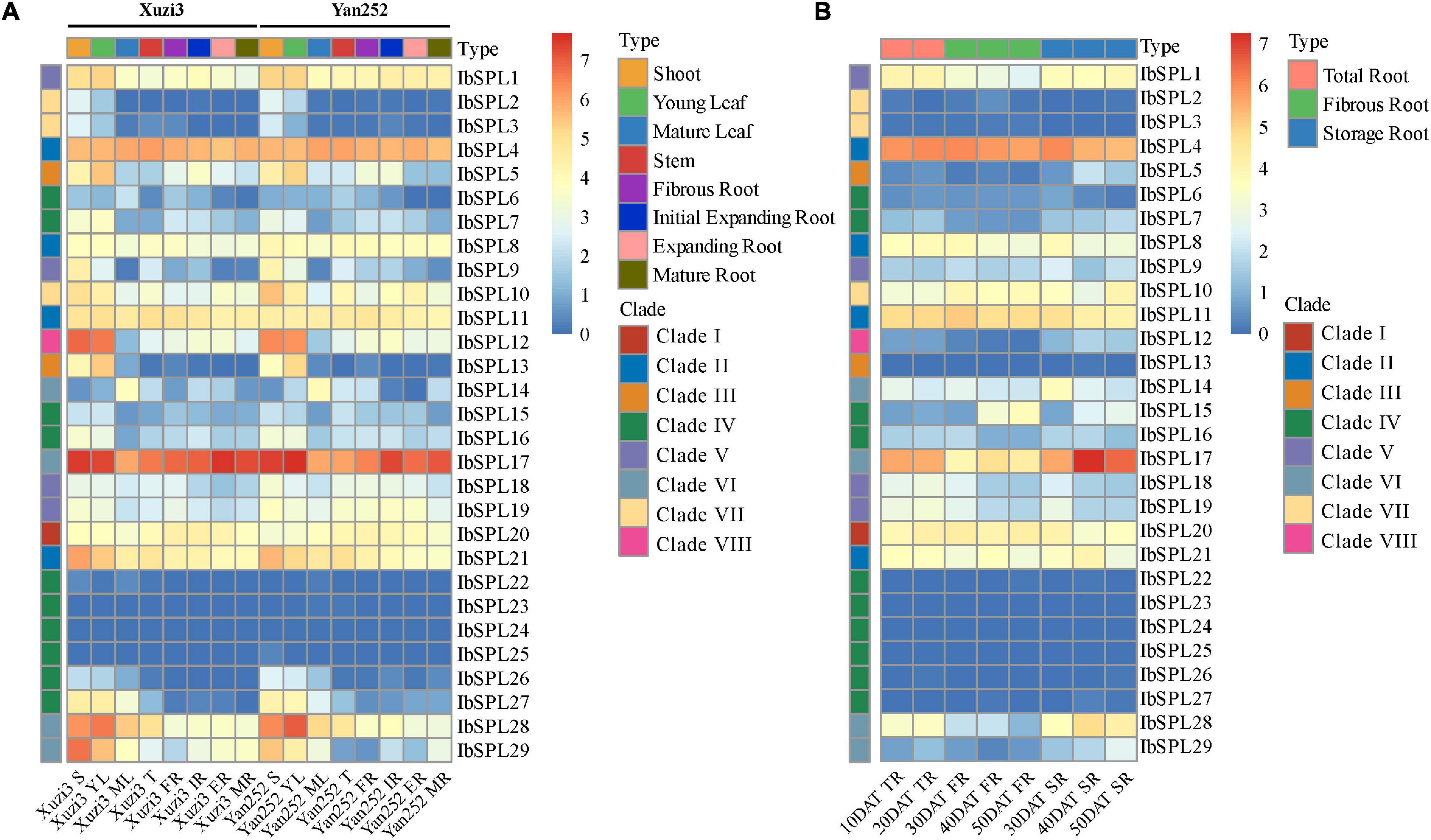
Figure 5. Expression profiles of IbSPL genes in different tissues and developmental stages using transcriptome analysis. (A) Heatmap representing the expression patterns of IbSPL genes in eight different tissues of two sweet potato cultivars (Xuzi3 and Yan252). Xuzi3: sweet potato cultivar Xuzi3, Yan252: sweet potato cultivar Yan252, S: shoot, YL: young leaf, ML: mature leaf, T: stem, FR: fibrous root, IR: initial tuberous root, ER: expanding storage root, MR: mature storage root. (B) Heatmap representing the expression patterns of IbSPL genes in eight different stages of root development. 10DAT_TR: undifferentiated root (10 days after transplanting), 20DAT_TR: undifferentiated root (20 days after transplanting), 30DAT_FR: fibrous root (30 days after transplanting), 40DAT_FR: fibrous root (days after transplanting), 50DAT_FR: fibrous root (50 days after transplanting), 30DAT_SR: storage root (30 days after transplanting), 40DAT_SR: storage root (40 days after transplanting), 50DAT_SR: storage root (50 days after transplanting). Heatmaps were generated based on the log2(FPKM + 1) values for each IbSPL gene.
To further investigate the expression profiles of IbSPL genes in underground tissues, publicly available transcriptomic data of eight different stages during root development from the cultivar ‘Beauregard’ were used (Figure 5B and Supplementary Tables 8, 9). The results showed that the overall expression patterns of IbSPL genes in the roots of the cultivar ‘Beauregard’ were similar to those of cultivar ‘Xuzi3’ and ‘Yan252.’ Specifically, the expression levels of IbSPL17, IbSPL28, and IbSPL29 were the highest in storage roots compared with undifferentiated and fibrous roots. IbSPL1 showed the highest expression in undifferentiated roots whereas IbSPL10 showed the highest expression in fibrous roots. However, the expression levels of IbSPL4, IbSPL8, IbSPL11, IbSPL20, and IbSPL21 showed no distinct variation in all tissues. These results, therefore, imply that these genes may play important roles in root development.
To confirm the expression patterns of IbSPLs derived from the transcriptomic data, a total of 11 IbSPL genes (IbSPL1, IbSPL4, IbSPL5, IbSPL12, IbSPL16, IbSPL17, IbSPL20, IbSPL21, IbSPL27, IbSPL28, and IbSPL29) highly expressed in aboveground or underground tissues were selected for qRT-PCR analysis in 11 tissues of cultivar ‘Xuyu34’ (Figure 6). The results showed consistent expression patterns of IbSPL genes between the transcriptomic data and qRT-PCR results. Moreover, the expression levels of IbSPLs differed in various tissues. For example, IbSPL27 and IbSPL29 were highly expressed in young leaves, while IbSPL12, IbSPL16, and IbSPL21 were highly expressed in flower. Moreover, the gradual increase in IbSPL17 and IbSPL28 expressions indicated their differential roles in storage root development in sweet potato.
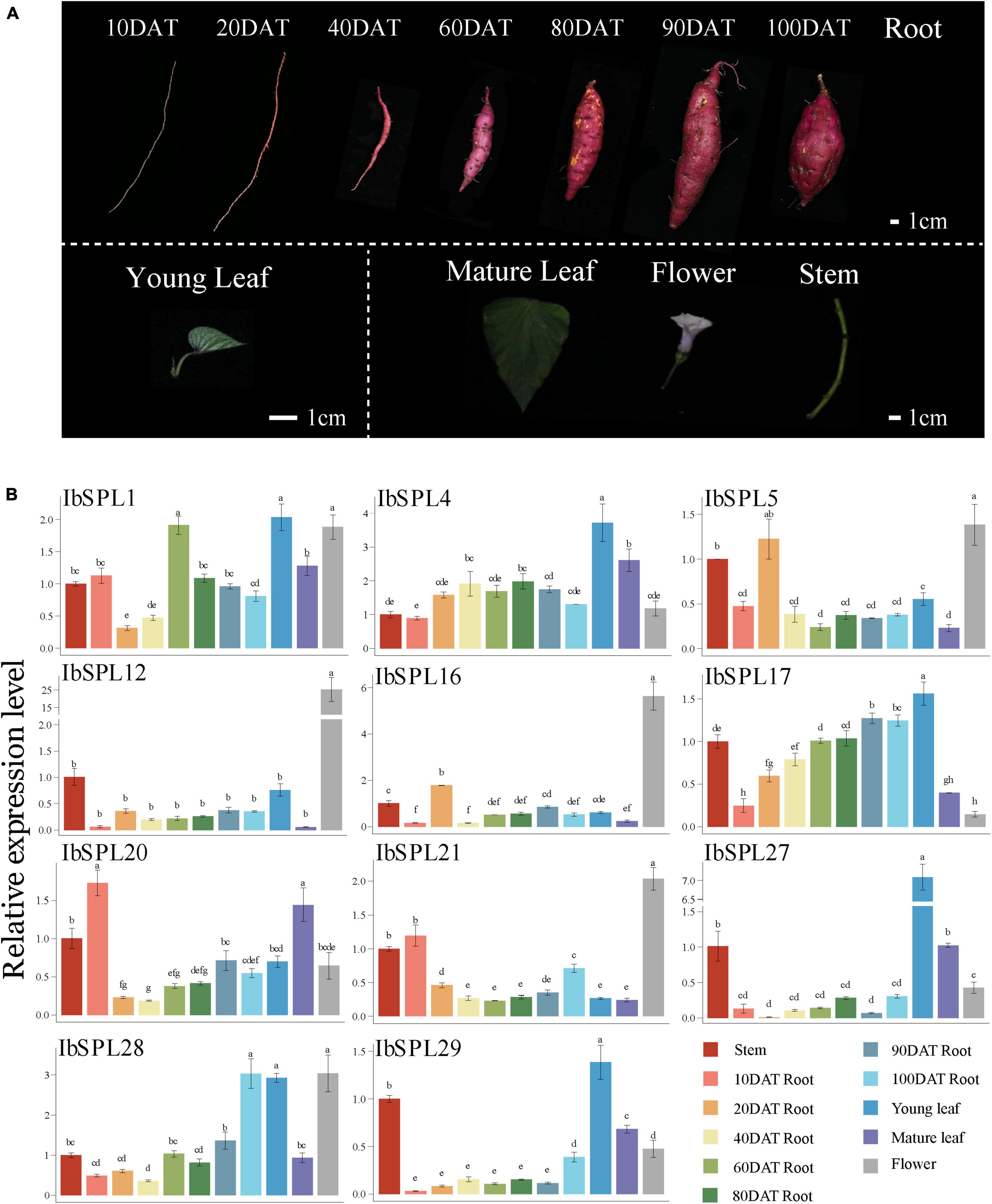
Figure 6. Expression profiles of 11 IbSPL genes in different tissues and at different developmental stages using qRT-PCR. (A) Different tissues from cultivar Xuyu34 are shown, including stem, leaf, flower, and root. Black bar is scale of 1 cm. (B) Expression patterns of 11 IbSPL genes determined using qRT-PCR. The x-axis indicates different tissues. The y-axis represents the relative expression calculated using the 2–ΔΔCT method. The error bars indicate the standard deviations of the three biological replicates. Different letters indicate statistically significant differences at p < 0.05.
IbSPL Genes in Response to Exogenous Phytohormones
The promoter analysis revealed that IbSPL genes could be regulated by various phytohormones, which are known regulators of plant growth and development. To reveal the potential roles of IbSPLs in hormone signaling pathways, six IbSPL genes (IbSPL1, IbSPL4, IbSPL16, IbSPL17, IbSPL21, and IbSPL28) highly expressed in roots were selected to perform qRT-PCR analysis under exogenous phytohormone treatments, which included IAA, MeJA, ZT, and ABA. The expression analysis indicated that the six IbSPL genes exhibited highly divergent response patterns under phytohormone treatment in the adventitious root (Figure 7). Under IAA treatment, IbSPL16 and IbSPL21 were rapidly upregulated after 6 h of treatment, while the other four genes were upregulated after 12 or 24 h of treatment. Under MeJA treatment, all IbSPL genes were significantly upregulated after 48 h of treatment, with IbSPL4 and IbSPL21 upregulated around 45.3 and 178.1 folds, respectively. Under ZT treatment, all IbSPL genes were highly upregulated after 6 h of treatment, with a subsequent decline followed by approximately 10-fold increased expression than CK. Notably, IbSPL21 showed a particularly positive response to ZT treatment. Under ABA treatment, all examined IbSPL genes, particularly IbSPL1, IbSPL4, IbSPL16, IbSPL17, and IbSPL21, were rapidly upregulated after 6 h of treatment, whereas IbSPL28 showed significantly upregulation after 24 h of treatment.
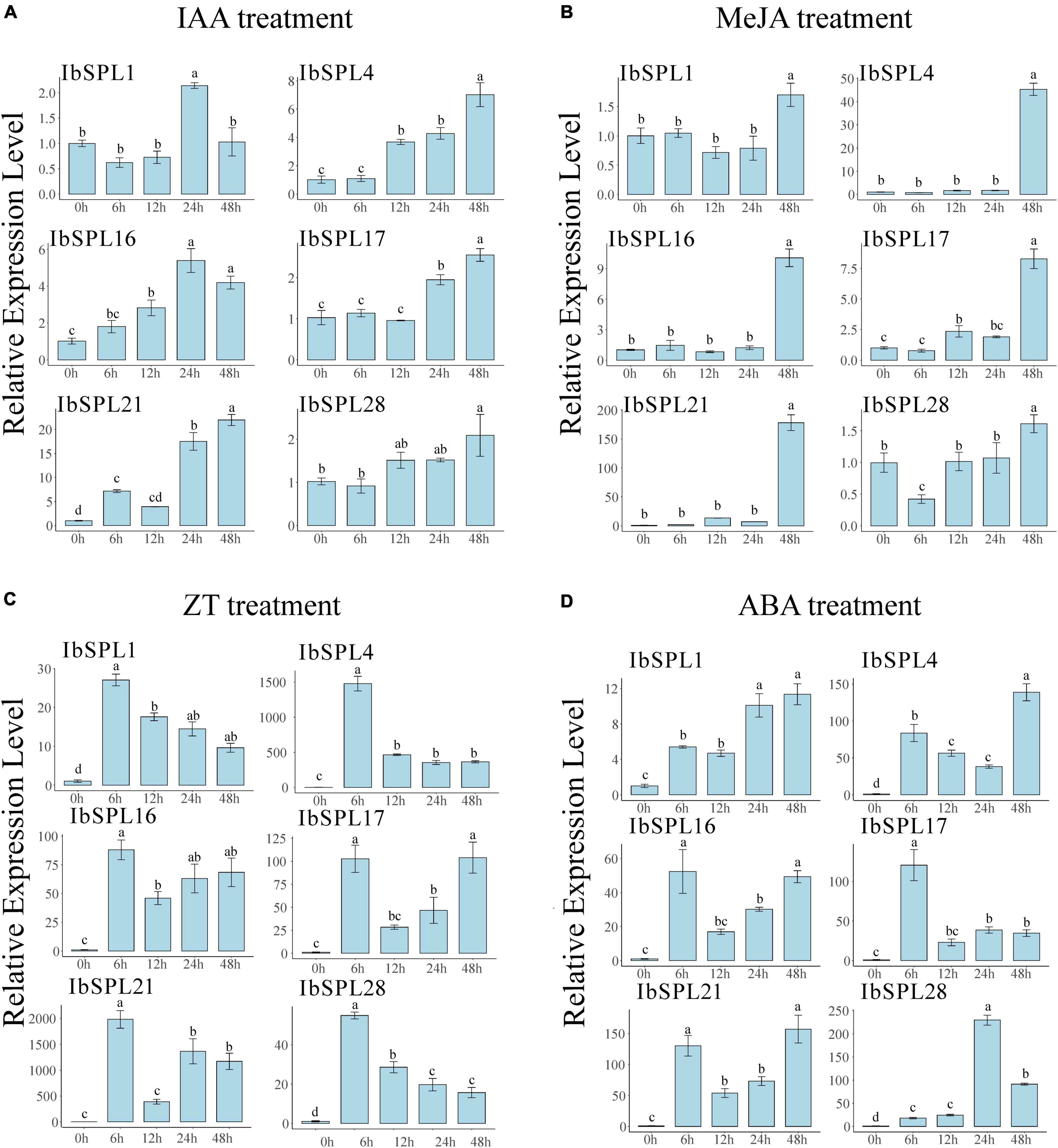
Figure 7. Expression patterns of six IbSPL genes under phytohormone treatment. (A) Expression patterns of the six IbSPL genes under IAA treatment. (B) Expression patterns of the six IbSPL genes under MeJA treatment. (C) Expression patterns of the six IbSPL genes under ZT treatment. (D) Expression patterns of the six IbSPL genes under ABA treatment. Samples were collected at 0, 6, 12, 24, and 48 h after treatment. The x-axis indicates different samples, while the y-axis represents the relative expression calculated using the 2–ΔΔCT method. The error bars indicate the standard deviations of the three biological replicates. Different letters indicate statistically significant differences at p < 0.05.
Regulatory Sub-Networks Involving IbSPLs and Other I. batatas Genes in the Storage Root of Sweet Potato
To identify the regulatory sub-networks involving IbSPLs in the storage root of sweet potato, WGCNA was performed based on transcriptomic data of mature storage roots from 88 sweet potato accessions (Supplementary Figure 8 and Supplementary Table 11). A total of 19 modules were obtained from this analysis, with three modules (turquoise, blue and yellow) containing totally eight IbSPL genes (Figure 8A). In the yellow module, IbSPL20 showed co-expression with only one gene. In the blue module, IbSPL9 and IbSPL10 were co-expressed with 17 and 107 genes, respectively. In the turquoise module, IbSPL1, IbSPL16, IbSPL17, IbSPL21, and IbSPL28 had 12, 147, 370, 732, and 198 co-expressed genes, respectively (Supplementary Table 12). Interestingly, IbSPL16, IbSPL17, IbSPL21, and IbSPL28 in the turquoise module share 101 co-expressed genes (Figure 8B), indicating functionality in similar biological processes.
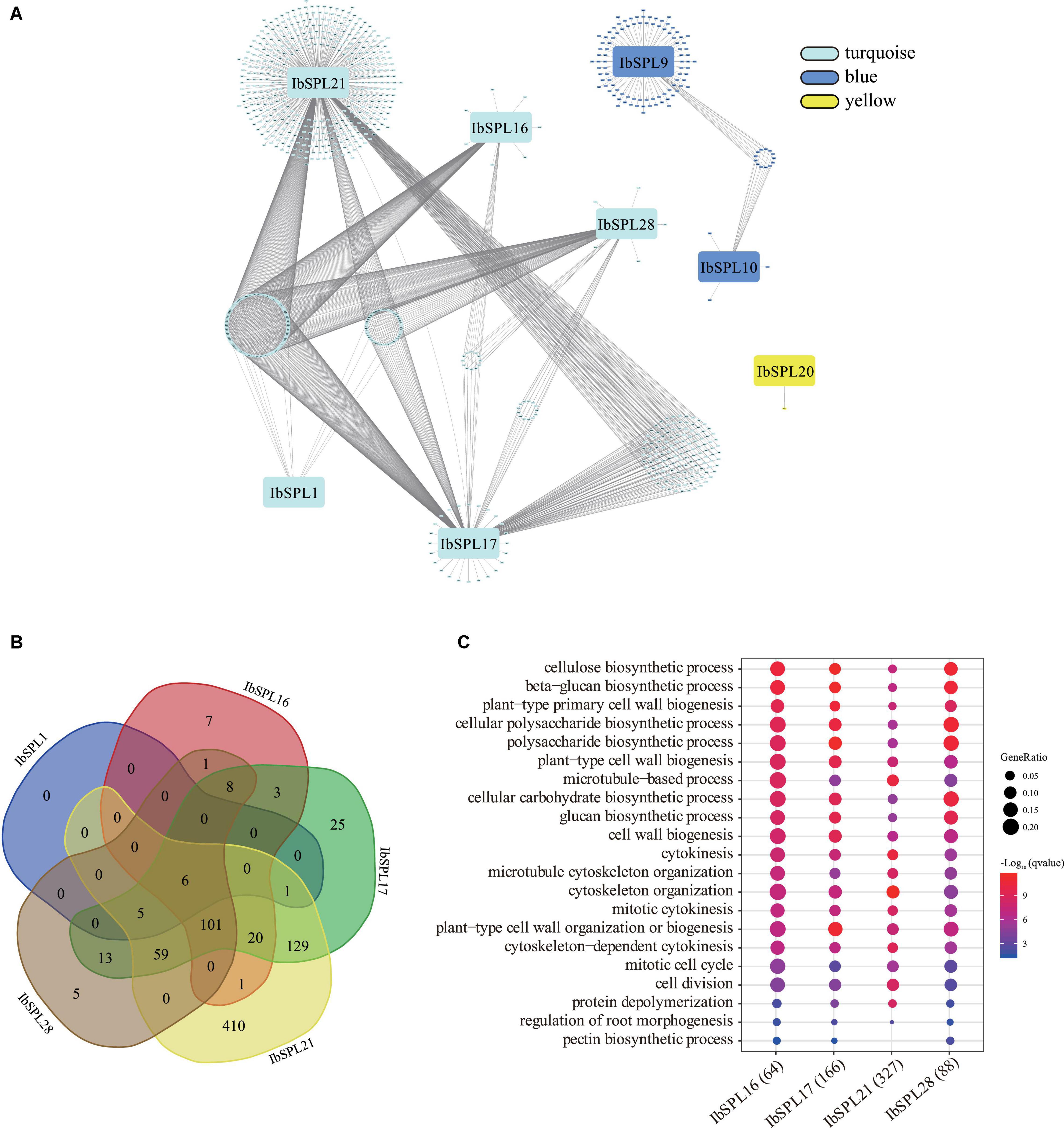
Figure 8. Regulatory sub-networks involving IbSPLs and other I. batatas genes in the storage root of sweet potato. (A) Co-expression sub-network visualization for IbSPLs in the storage root of sweet potato. Only the edges with a weight above a threshold of 0.05 are displayed. (B) Venn diagram of the number of co-expressed genes for IbSPL1, IbSPL16, IbSPL17, IbSPL21, and IbSPL28 in the turquoise module. (C) GO enrichment analysis of co-expressed genes for IbSPL16, IbSPL17, IbSPL21, and IbSPL28.
To further explore the putative functions of the SPLs in the storage root, GO enrichment analysis was performed on the co-expressed genes. For IbSPL1, IbSPL20, and IbSPL9, GO enrichment results could not be obtained due to the small number of co-expressed genes. For IbSPL10 in the blue module, co-expressed genes related to “response to chitin” and “response to organonitrogen compound” were enriched (Supplementary Table 13). For IbSPL16, IbSPL17, IbSPL21, and IbSPL28 in the turquoise module, the similar GO terms were enriched, such as “regulation of root morphogenesis,” “cell division,” “cytoskeleton organization,” “plant-type cell wall organization or biogenesis,” and “cellulose biosynthetic process” (Figure 8C and Supplementary Table 13). It is known that the cytoskeleton, cell division, and cell wall organization/biogenesis are important biological processes involved in storage root development and formation (Dong et al., 2019). Therefore, these results indicated that IbSPL16/IbSPL17/IbSPL21/IbSPL28 may play a key role in storage root development in sweet potato.
Discussion
SPL genes are important plant-specific transcription factors with a highly conserved SBP domain. Since its discovery in A. majus, SPL gene members have been increasingly identified in plants (Klein et al., 1996; Cardon et al., 1999; Xie et al., 2006; Salinas et al., 2012; Li et al., 2013; Zhang et al., 2016; Tripathi et al., 2017). However, comprehensive molecular, evolutionary and functional analysis of the SPL genes in the genus Ipomoea are lacking. The genus Ipomoea has significant nutritional and economic value for humans, including the seventh most important crop I. batatas and ornamental plant I. nil. Up to now, four Ipomoea species have been sequenced: I. nil, I. triloba, I. trifida, and I. batatas. Utilizing these genomes, this study systematically analyzed the Ipomoea SPL genes, including molecular characteristics, evolutionary process, post-transcriptional regulation, and physiological function.
Comparative Analysis of SQUAMOSA Promoter-Binding Protein-Like Genes in the Genus Ipomoea
Recently, some important transcription factor gene families have been investigated in Ipomoea species, such as bZIPs (Yang et al., 2019), WRKYs (Li Y. et al., 2019), GRPs (Lu et al., 2019), MADS (Zhu et al., 2020), and DEAD-box (Wan et al., 2020). However, these studies have focused only on the gene families in a single Ipomoea species, and comparative analysis of gene families in the genus Ipomoea are scarce. This study dissects the evolutionary dynamics of SPL genes in the genus Ipomoea, identifying SPL genes in four Ipomoea species: 29 IbSPLs, 27 ItfSPLs, 26 ItbSPLs, and 23 IniSPLs (Figure 1). Notably, the number of SPL genes in sweet potato is approximately equal to that of the diploid wild relatives (I. nil, I. triloba, and I. trifida), owing to the haplotype-resolved genome assembly of hexaploid sweet potato (Yang et al., 2017). Following the classifications of A. thaliana, S. lycopersicum, and O. sativa (Cardon et al., 1999; Xie et al., 2006; Salinas et al., 2012), the Ipomoea SPL genes were also divided into eight clades (Figure 2), with clade IV comprising the highest members (32) and clade I comprising the lowest (four). Gene and protein structure analysis revealed that most Ipomoea SPLs from the same phylogenetic clade share similar intron/exon structures, domain organizations, and motif compositions (Supplementary Figure 1), indicating that SPLs within the same clade may have similar functions in Ipomoea species. Interestingly, apart from the conserved domain and motifs present in the SPL proteins, other domains or motifs were found in clades I, II, and IV, such as DEXDc domain (motif 4) and Ankyrin repeats (motif 8), which were also observed in papaya (Xu et al., 2020) and barley (Tripathi et al., 2018). These results suggest that the SPLs in these clades may have undergone evolutionary functional differentiation and/or neofunctionalization.
Ipomoea species were found to possess more SPL genes than dicotyledonous model plants, such as A. thaliana and S. lycopersicum (Cardon et al., 1999; Salinas et al., 2012), implying the genus-specific expansion of the SPL gene family in Ipomoea species. The expansion of gene families is a result of evolutionary duplication events (Moore and Purugganan, 2005). This study showed that segmental duplication plays a major role in the evolution and expansion of Ipomoea SPL genes (Figure 3B), which is consistent with findings in cotton (Cai et al., 2018), Rosacea species (Abdullah et al., 2018) and Euphorbiaceae species (Li J. et al., 2019). Previous studies have reported that a whole-genome triplication event occurred 46.1 million years ago (Mya) in the progenitor of the genus Ipomoea (Yang et al., 2017), and thereby the derivation of the segmental duplication of SPLs from this event was speculated. Furthermore, tandem duplication was found to be the most frequent event in the IV-b and IV-c subclades (Supplementary Table 1) and orthologs were found to be absent in dicots (Supplementary Table 3), implying that the SPL members in these two subclades were tandem duplicated from a recent event. Therefore, the SPL genes were speculated to have undergone replication expansion in the progenitor of the genus Ipomoea and that various SPL members were retained due to their important role in growth and development during the Ipomoea species differentiation. Additionally, the Ka/Ks ratios were less than 1 for all Ipomoea SPL ortholog gene pairs, indicating that the Ipomoea SPL genes were under strong purifying selection (Supplementary Table 4).
Many SPL genes are miR156 targets, thus forming a functional regulatory network of miR156-SPL, which plays an important role in plant growth and development (Gou et al., 2011; Wang and Wang, 2015; Liu et al., 2019). More than half of the SPL gene family members have been reported to be targeted by miR156 in various plant species, such as rice (Xie et al., 2006), tomato (Salinas et al., 2012), and apple (Li et al., 2013). In this study, two-thirds of SPL genes in each Ipomoea species were predicted to be miR156 targets (Figure 4B). Phylogenetic analysis showed that all SPL genes in clade III lacked miR156 binding sites, which is consistent with the reported results of A. thaliana, rice, and tomato (Preston and Hileman, 2013). Additionally, two binding site types of miR156 were identified in SPL genes: one located in CDS and the other located in the 3′-UTR (Figure 4B), which is consistent with the observation in other plants, such as rice (Xie et al., 2006), tomato (Salinas et al., 2012), apple (Li et al., 2013), and papaya (Xu et al., 2020). The degradome data of I. batatas further confirmed seven IbSPL genes as the targets of miR156 (Figure 4C and Supplementary Table 1). Among these IbSPL genes, the miR156 binding site for IbSPL17 and IbSPL28 is located in the 3′-UTR, which is consistent with the miR156 binding site of their orthologous genes (AtSPL3 and LeSPL-CNR) in A. thaliana and tomato (Gandikota et al., 2007; Chen et al., 2015). This suggests the high conservation of the miR156-SPL regulatory module in plants. Additionally, our results showed a negative correlation in the expression pattern of miR156 and their target SPL genes, suggesting that SPLs might be regulated by miR156 at the post-transcriptional level. However, most of the miR156-SPL interactions in this study were predicted using in silico analysis, requiring further experimental verification for the miR156-SPL interactions in the genus Ipomoea.
IbSPL Genes Are Putatively Involved in Storage Root Development
Sweet potato, the seventh most important crop globally, has strong adaptability, stable yield, and high nutritional value (Liu, 2017). The storage root of sweet potato is economically useful for its nutrient content and yield, and thus, dissecting the mechanisms underlying storage root formation and development is significant to improve sweet potato nutrient content and yield. Considering the key regulatory roles of SPL genes in root architecture (Yu et al., 2015; Barrera-Rojas et al., 2020) and biomass enhancement (Wang et al., 2015), the expression patterns of IbSPL genes in different tissues or at different developmental stages were evaluated using the public transcriptome data. Most of the IbSPLs were found to be highly expressed in aboveground tissues, especially in shoots or young leaves; however, only some IbSPLs were found to be highly expressed in underground tissues (Figure 7). qRT-PCR analysis of the expression levels of two IbSPL genes (IbSPL17/IbSPL28) revealed a significant increase with storage root development (Figure 6). This study provides evidence that SPL genes have important functions during storage root development in sweet potato.
The formation and development of storage roots is a complex physiological process that includes the cessation of root elongation, genesis and development of the primary and secondary vascular cambium, increase in radial growth and accumulation of starch and storage proteins (Ravi et al., 2009). These processes are closely related to the endogenous phytohormones, such as IAA, cytokinins (CTKs), JA, and ABA (Nakatani, 1991; Tanaka et al., 2008; Ravi et al., 2009; Dong et al., 2019). For instance, IAA is involved in early stages of storage root formation and primary storage root thickening (Noh et al., 2010); ABA plays a significant role in storage root bulking by activating cell division (Huan et al., 2020) and CTKs play a key role in storage root initiation and expansion as a pre-requirement for cambial cell proliferation (Dong et al., 2019). Moreover, storage root yields are positively correlated with ABA and CTK contents (Wang et al., 2005). In the present study, different kinds of hormone-responsive elements were found by analyzing the IbSPL promoters (Supplementary Figure 7), implying that IbSPL genes may participate in hormone signaling pathways. qRT-PCR analysis further confirmed that the expression of the tested IbSPLs (IbSPL1, IbSPL4, IbSPL16, IbSPL17, IbSPL21, and IbSPL28) was strongly induced under exogenous phytohormone treatments, particularly ZT and ABA, suggesting their crucial roles in root development.
Storage root formation and development is maintained by coordinated cellular behaviors, such as cell division, expansion, and differentiation. Previous studies have revealed that cell wall biosynthesis and cytoskeleton organization are critical in these cellular behaviors (Bashline et al., 2014; Brasil et al., 2017). The regulatory sub-networks in this study were analyzed using WGCNA, which indicated that eight IbSPL genes were co-expressed with at least one other I. batatas genes (Figure 8A). GO enrichment analysis of co-expressed genes speculated the role of IbSPL genes in stress responses, root morphogenesis, and cell division (Supplementary Table 13). Moreover, the genes co-expressed with IbSPL16/IbSPL17/IbSPL21/IbSPL28 in the turquoise module were all significantly enriched for “regulation of root morphogenesis,” “cell division,” “cytoskeleton organization,” “plant-type cell wall organization or biogenesis,” and “cellulose biosynthetic process.” These enriched processes are essential for cell morphogenesis and cell cycles, implying their key roles in storage root development. In the future, functional characterization is needed to elucidate the specific roles of IbSPLs in storage root development.
Conclusion
In summary, a genome-wide analysis of the SPL gene family in four Ipomoea species, including I. batatas, I. trifida, I. triloba, and I. nil was performed. A total of 105 Ipomoea SPL genes were identified and divided into eight clades. Genes in one clade were found to harbor similar gene structures, domain organizations, motif compositions, and cis-acting elements, suggesting potential functional similarity. Moreover, segmental duplication was predominantly responsible for the expansion of the Ipomoea SPL gene family. On combining the results from the expression patterns and regulatory sub-networks, IbSPL16/IbSPL17/IbSPL21/IbSPL28 were found to play an important role in storage root development. Therefore, this study not only provides novel insights into the evolutionary and functional divergence of the SPL genes in the genus Ipomoea but also lays a foundation for further elucidation of the potential functional roles of IbSPL genes during storage root development.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
LZ, SW, and TX conceived and designed the research. HS, JM, WZ, LZ, WH, and YZ performed the research and analyzed the data. LZ and HS wrote the manuscript. All authors have read and approved the manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (No. BK20190995), the Shanghai Municipal Afforestation and City Appearance and Environmental Sanitation Administration (No. G212402), the Natural Science Foundation of Xuzhou City (No. KC19070), Postgraduate Research and Practice Innovation Program of Jiangsu Province (No. KYCX20_2319), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Funders had no role in the design of the study and collection, analysis, and interpretation of data, and in writing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Meng Kou (Xuzhou Academy of Agricultural Sciences) for providing the sweet potato (I. batatas cv. Xuyu34) plants.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.801061/full#supplementary-material
Footnotes
- ^ http://viewer.shigen.info/asagao
- ^ http://sweetpotato.uga.edu
- ^ http://public-genomes-ngs.molgen.mpg.de/SweetPotato
- ^ https://www.arabidopsis.org
References
Abdullah, M., Cao, Y., Cheng, X., Shakoor, A., Su, X., Gao, J., et al. (2018). Genome-Wide Analysis Characterization and Evolution of SBP Genes in Fragaria vesca, Pyrus bretschneideri, Prunus persica and Prunus mume. Front. Genet. 9:64. doi: 10.3389/fgene.2018.00064
Addo-Quaye, C., Miller, W., and Axtell, M. J. (2009). CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131. doi: 10.1093/bioinformatics/btn604
Austin, D. F., and Huáman, Z. (1996). A synopsis of Ipomoea (Convolvulaceae) in the Americas. Taxon 45, 3–38.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Barrera-Rojas, C. H., Rocha, G. H. B., Polverari, L., Pinheiro Brito, D. A., Batista, D. S., Notini, M. M., et al. (2020). miR156-targeted SPL10 controls Arabidopsis root meristem activity and root-derived de novo shoot regeneration via cytokinin responses. J. Exp. Bot. 71, 934–950. doi: 10.1093/jxb/erz475
Bashline, L., Lei, L., Li, S., and Gu, Y. (2014). Cell wall, cytoskeleton, and cell expansion in higher plants. Mol. Plant 7, 586–600. doi: 10.1093/mp/ssu018
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brasil, J. N., Costa, C. N. M., Cabral, L. M., Ferreira, P. C. G., and Hemerly, A. S. (2017). The plant cell cycle: Pre-Replication complex formation and controls. Genet. Mol. Biol. 40(1 Suppl. 1), 276–291. doi: 10.1590/1678-4685-GMB-2016-0118
Cai, C., Guo, W., and Zhang, B. (2018). Genome-wide identification and characterization of SPL transcription factor family and their evolution and expression profiling analysis in cotton. Sci. Rep. 8:762. doi: 10.1038/s41598-017-18673-4
Cardon, G., Hohmann, S., Klein, J., Nettesheim, K., Saedler, H., and Huijser, P. (1999). Molecular characterisation of the Arabidopsis SBP-box genes. Gene 237, 91–104. doi: 10.1016/s0378-1119(99)00308-x
Caruthers, J. M., and McKay, D. B. (2002). Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12, 123–133. doi: 10.1016/s0959-440x(02)00298-1
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mole. Biol. Evol. 17, 540–552.
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, W., Kong, J., Lai, T., Manning, K., Wu, C., Wang, Y., et al. (2015). Tuning LeSPL-CNR expression by SlymiR157 affects tomato fruit ripening. Sci. Rep. 5:7852. doi: 10.1038/srep07852
Chen, X., Zhang, Z., Liu, D., Zhang, K., Li, A., and Mao, L. (2010). SQUAMOSA promoter-binding protein-like transcription factors: star players for plant growth and development. J. Integr. Plant Biol. 52, 946–951. doi: 10.1111/j.1744-7909.2010.00987.x
Chen, Y., Zhu, P., Wu, S., Lu, Y., Sun, J., Cao, Q., et al. (2019). Identification and expression analysis of GRAS transcription factors in the wild relative of sweet potato Ipomoea trifida. BMC Genom. 20:911. doi: 10.1186/s12864-019-6316-7
Coordinators, N. R. (2018). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46, D8–D13. doi: 10.1093/nar/gkx1095
Dai, X., Zhuang, Z., and Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46, W49–W54. doi: 10.1093/nar/gky316
de Castro, E., Sigrist, C. J., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365. doi: 10.1093/nar/gkl124
Ding, N., Wang, A., Zhang, X., Wu, Y., Wang, R., Cui, H., et al. (2017). Identification and analysis of glutathione S-transferase gene family in sweet potato reveal divergent GST-mediated networks in aboveground and underground tissues in response to abiotic stresses. BMC Plant Biol. 17:225. doi: 10.1186/s12870-017-1179-z
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dong, T., Zhu, M., Yu, J., Han, R., Tang, C., Xu, T., et al. (2019). RNA-Seq and iTRAQ reveal multiple pathways involved in storage root formation and development in sweet potato (Ipomoea batatas L.). BMC Plant Biol. 19:136. doi: 10.1186/s12870-019-1731-0
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Gandikota, M., Birkenbihl, R. P., Hohmann, S., Cardon, G. H., Saedler, H., and Huijser, P. (2007). The miRNA156/157 recognition element in the 3’ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 49, 683–693. doi: 10.1111/j.1365-313X.2006.02983.x
Gou, J. Y., Felippes, F. F., Liu, C. J., Weigel, D., and Wang, J. W. (2011). Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23, 1512–1522. doi: 10.1105/tpc.111.084525
Guo, A. Y., Zhu, Q. H., Gu, X., Ge, S., Yang, J., and Luo, J. (2008). Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family. Gene 418, 1–8. doi: 10.1016/j.gene.2008.03.016
Hoshino, A., Jayakumar, V., Nitasaka, E., Toyoda, A., Noguchi, H., Itoh, T., et al. (2016). Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun. 7:13295. doi: 10.1038/ncomms13295
Huan, L., Jin-Qiang, W., and Qing, L. (2020). Photosynthesis product allocation and yield in sweet potato with spraying exogenous hormones under drought stress. J. Plant Physiol. 253, 153265. doi: 10.1016/j.jplph.2020.153265
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Jiang, X., Chen, P., Zhang, X., Liu, Q., and Li, H. (2021). Comparative analysis of the SPL gene family in five Rosaceae species: Fragaria vesca, Malus domestica, Prunus persica, Rubus occidentalis, and Pyrus pyrifolia. Open Life Sci. 16, 160–171. doi: 10.1515/biol-2021-0020
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Klein, J., Saedler, H., and Huijser, P. (1996). A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol. Gen. Genet. 250, 7–16. doi: 10.1007/BF02191820
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuang, Z., Wang, Y., Li, L., and Yang, X. (2019). miRDeep-P2: accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics 35, 2521–2522. doi: 10.1093/bioinformatics/bty972
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kuo, Y.-W., Lin, J.-S., Li, Y.-C., Jhu, M.-Y., King, Y.-C., and Jeng, S.-T. (2019). MicroR408 regulates defense response upon wounding in sweet potato. J. Exp. Bot. 70, 469–483.
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9:559. doi: 10.1186/1471-2105-9-559
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., and Bork, P. (2018). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496. doi: 10.1093/nar/gkx922
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12:323. doi: 10.1186/1471-2105-12-323
Li, C., and Lu, S. (2014). Molecular characterization of the SPL gene family in Populus trichocarpa. BMC Plant Biol. 14:131. doi: 10.1186/1471-2229-14-131
Li, J., Gao, X., Sang, S., and Liu, C. (2019). Genome-wide identification, phylogeny, and expression analysis of the SBP-box gene family in Euphorbiaceae. BMC Genomics 20, (Suppl. 9):912. doi: 10.1186/s12864-019-6319-4
Li, J., Hou, H., Li, X., Xiang, J., Yin, X., Gao, H., et al. (2013). Genome-wide identification and analysis of the SBP-box family genes in apple (Malus x domestica Borkh.). Plant Physiol. Biochem. 70, 100–114. doi: 10.1016/j.plaphy.2013.05.021
Li, J., Mahajan, A., and Tsai, M. D. (2006). Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178. doi: 10.1021/bi062188q
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, Y., Zhang, L., Zhu, P., Cao, Q., Sun, J., Li, Z., et al. (2019). Genome-wide identification, characterisation and functional evaluation of WRKY genes in the sweet potato wild ancestor Ipomoea trifida (H.B.K.) G. Don. under abiotic stresses. BMC Genet. 20:90. doi: 10.1186/s12863-019-0789-x
Liu, M., Shi, Z., Zhang, X., Wang, M., Zhang, L., Zheng, K., et al. (2019). Inducible overexpression of Ideal Plant Architecture1 improves both yield and disease resistance in rice. Nat. Plants 5, 389–400. doi: 10.1038/s41477-019-0383-2
Liu, Q. (2017). Improvement for agronomically important traits by gene engineering in sweetpotato. Breed Sci. 67, 15–26. doi: 10.1270/jsbbs.16126
Liu, Y., Su, W., Wang, L., Lei, J., Chai, S., Zhang, W., et al. (2021). Integrated transcriptome, small RNA and degradome sequencing approaches proffer insights into chlorogenic acid biosynthesis in leafy sweet potato. PLoS One 16:e0245266. doi: 10.1371/journal.pone.0245266
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, Y., Sun, J., Yang, Z., Zhao, C., Zhu, M., Ma, D., et al. (2019). Genome-wide identification and expression analysis of glycine-rich RNA-binding protein family in sweet potato wild relative Ipomoea trifida. Gene 686, 177–186. doi: 10.1016/j.gene.2018.11.044
Manning, K., Tor, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Moore, R. C., and Purugganan, M. D. (2005). The evolutionary dynamics of plant duplicate genes. Curr. Opin. Plant Biol. 8, 122–128. doi: 10.1016/j.pbi.2004.12.001
Morita, Y., and Hoshino, A. (2018). Recent advances in flower color variation and patterning of Japanese morning glory and petunia. Breed Sci. 68, 128–138. doi: 10.1270/jsbbs.17107
Nakatani, M. (1991). Changes in endogenous level of zeatin riboside, abscisic acid and indole acetic acid during formation and thickening of tuberous root in sweet potato. Jpn. J. Crop 1991:60.
National Genomics Data Center, M., and Partners (2020). Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 48, D24–D33. doi: 10.1093/nar/gkz913
Noh, S. A., Lee, H. S., Huh, E. J., Huh, G. H., Paek, K. H., Shin, J. S., et al. (2010). SRD1 is involved in the auxin-mediated initial thickening growth of storage root by enhancing proliferation of metaxylem and cambium cells in sweetpotato (Ipomoea batatas). J. Exp. Bot. 61, 1337–1349. doi: 10.1093/jxb/erp399
Orfila, C., Huisman, M. M., Willats, W. G., van Alebeek, G. J., Schols, H. A., Seymour, G. B., et al. (2002). Altered cell wall disassembly during ripening of Cnr tomato fruit: implications for cell adhesion and fruit softening. Planta 215, 440–447. doi: 10.1007/s00425-002-0753-1
Park, S. C., Kim, Y. H., Ji, C. Y., Park, S., Jeong, J. C., Lee, H. S., et al. (2012). Stable internal reference genes for the normalization of real-time PCR in different sweetpotato cultivars subjected to abiotic stress conditions. PLoS One 7:e51502. doi: 10.1371/journal.pone.0051502
Preston, J. C., and Hileman, L. C. (2013). Functional Evolution in the Plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) Gene Family. Front. Plant Sci. 4:80. doi: 10.3389/fpls.2013.00080
Ravi, V., Chakrabarti, S. K., Makeshkumar, T., and Saravanan, R. (2014). Molecular Regulation of Storage Root Formation and Development in Sweet Potato. Horticult. Rev. 42, 157–208.
Ravi, V., Naskar, S. K., Makeshkumar, T., Babu, B., and Krishnan, B. P. (2009). Molecular physiology of storage root formation and development in sweet potato (Ipomoea batatas (L.) Lam.). J. Root Crops 35, 1–27.
Salinas, M., Xing, S., Hohmann, S., Berndtgen, R., and Huijser, P. (2012). Genomic organization, phylogenetic comparison and differential expression of the SBP-box family of transcription factors in tomato. Planta 235, 1171–1184. doi: 10.1007/s00425-011-1565-y
Saminathan, T., Alvarado, A., Lopez, C., Shinde, S., Gajanayake, B., Abburi, V. L., et al. (2019). Elevated carbon dioxide and drought modulate physiology and storage-root development in sweet potato by regulating microRNAs. Funct. Integr. Genomics 19, 171–190. doi: 10.1007/s10142-018-0635-7
Savojardo, C., Martelli, P. L., Fariselli, P., Profiti, G., and Casadio, R. (2018). BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 46, W459–W466. doi: 10.1093/nar/gky320
Shao, Y., Zhou, H. Z., Wu, Y., Zhang, H., Lin, J., Jiang, X., et al. (2019). OsSPL3, an SBP-Domain Protein, Regulates Crown Root Development in Rice. Plant Cell 31, 1257–1275. doi: 10.1105/tpc.19.00038
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P.-L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432.
Subramanian, B., Gao, S., Lercher, M. J., Hu, S., and Chen, W. H. (2019). Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 47, W270–W275. doi: 10.1093/nar/gkz357
Tanaka, M., Kato, N., Nakayama, H., Nakatani, M., and Takahata, Y. (2008). Expression of class I knotted1-like homeobox genes in the storage roots of sweetpotato (Ipomoea batatas). J. Plant Physiol. 165, 1726–1735. doi: 10.1016/j.jplph.2007.11.009
Tripathi, R. K., Bregitzer, P., and Singh, J. (2018). Genome-wide analysis of the SPL/miR156 module and its interaction with the AP2/miR172 unit in barley. Sci. Rep. 8:7085. doi: 10.1038/s41598-018-25349-0
Tripathi, R. K., Goel, R., Kumari, S., and Dahuja, A. (2017). Genomic organization, phylogenetic comparison, and expression profiles of the SPL family genes and their regulation in soybean. Dev. Genes Evol. 227, 101–119. doi: 10.1007/s00427-017-0574-7
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115.
Wan, R., Liu, J., Yang, Z., Zhu, P., Cao, Q., and Xu, T. (2020). Genome-wide identification, characterisation and expression profile analysis of DEAD-box family genes in sweet potato wild ancestor Ipomoea trifida under abiotic stresses. Genes Genomics 42, 325–335. doi: 10.1007/s13258-019-00910-x
Wang, H., and Wang, H. (2015). The miR156/SPL Module, a Regulatory Hub and Versatile Toolbox, Gears up Crops for Enhanced Agronomic Traits. Mol. Plant 8, 677–688. doi: 10.1016/j.molp.2015.01.008
Wang, Q. M., Zhang, L. M., and Wang, Z. L. (2005). Formation and Thickening of Tuberous Roots in Relation to the Endogenous Hormone Concentrations in Sweetpotato. Scientia Agricult. Sinica 38, 2414–2420.
Wang, S., Li, S., Liu, Q., Wu, K., Zhang, J., Wang, S., et al. (2015). The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 47, 949–954. doi: 10.1038/ng.3352
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wu, S., Lau, K. H., Cao, Q., Hamilton, J. P., Sun, H., Zhou, C., et al. (2018). Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 9:4580. doi: 10.1038/s41467-018-06983-8
Xie, K., Wu, C., and Xiong, L. (2006). Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 142, 280–293. doi: 10.1104/pp.106.084475
Xu, X., Li, X., Hu, X., Wu, T., Wang, Y., Xu, X., et al. (2017). High miR156 Expression Is Required for Auxin-Induced Adventitious Root Formation via MxSPL26 Independent of PINs and ARFs in Malus xiaojinensis. Front Plant Sci. 8:1059. doi: 10.3389/fpls.2017.01059
Xu, Y., Xu, H., Wall, M. M., and Yang, J. (2020). Roles of transcription factor SQUAMOSA promoter binding protein-like gene family in papaya (Carica papaya) development and ripening. Genomics 112, 2734–2747. doi: 10.1016/j.ygeno.2020.03.009
Yamaguchi, A., Wu, M. F., Yang, L., Wu, G., Poethig, R. S., and Wagner, D. (2009). The microRNA-regulated SBP-Box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 17, 268–278. doi: 10.1016/j.devcel.2009.06.007
Yang, J., Moeinzadeh, M. H., Kuhl, H., Helmuth, J., Xiao, P., Haas, S., et al. (2017). Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants 3, 696–703. doi: 10.1038/s41477-017-0002-z
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Sun, J., Chen, Y., Zhu, P., Zhang, L., Wu, S., et al. (2019). Genome-wide identification, structural and gene expression analysis of the bZIP transcription factor family in sweet potato wild relative Ipomoea trifida. BMC Genet. 20:41. doi: 10.1186/s12863-019-0743-y
Yang, Z., Zhu, P., Kang, H., Liu, L., Cao, Q., Sun, J., et al. (2020). High-throughput deep sequencing reveals the important role that microRNAs play in the salt response in sweet potato (Ipomoea batatas L.). BMC Genomics 21:164. doi: 10.1186/s12864-020-6567-3
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Yu, N., Niu, Q. W., Ng, K. H., and Chua, N. H. (2015). The role of miR156/SPLs modules in Arabidopsis lateral root development. Plant J. 83, 673–685. doi: 10.1111/tpj.12919
Yu, N., Yang, J. C., Yin, G. T., Li, R. S., and Zou, W. T. (2020). Genome-wide characterization of the SPL gene family involved in the age development of Jatropha curcas. BMC Genomics 21:368. doi: 10.1186/s12864-020-06776-8
Zhang, H. X., Jin, J. H., He, Y. M., Lu, B. Y., Li, D. W., Chai, W. G., et al. (2016). Genome-Wide Identification and Analysis of the SBP-Box Family Genes under Phytophthora capsici Stress in Pepper (Capsicum annuum L.). Front. Plant Sci. 7:504. doi: 10.3389/fpls.2016.00504
Zhang, L., Yu, Y., Shi, T., Kou, M., Sun, J., Xu, T., et al. (2020). Genome-wide analysis of expression quantitative trait loci (eQTLs) reveals the regulatory architecture of gene expression variation in the storage roots of sweet potato. Horticult. Res. 7:90. doi: 10.1038/s41438-020-0314-4
Zhong, H., Kong, W., Gong, Z., Fang, X., Deng, X., Liu, C., et al. (2019). Evolutionary Analyses Reveal Diverged Patterns of SQUAMOSA Promoter Binding Protein-Like (SPL) Gene Family in Oryza Genus. Front. Plant Sci. 10:565. doi: 10.3389/fpls.2019.00565
Zhou, R., Yu, X., Ottosen, C. O., Zhang, T., Wu, Z., and Zhao, T. (2020). Unique miRNAs and their targets in tomato leaf responding to combined drought and heat stress. BMC Plant Biol. 20:107. doi: 10.1186/s12870-020-2313-x
Keywords: Ipomoea, SPL transcription factor, evolutionary patterns, root development, expression profiles
Citation: Sun H, Mei J, Zhao W, Hou W, Zhang Y, Xu T, Wu S and Zhang L (2022) Phylogenetic Analysis of the SQUAMOSA Promoter-Binding Protein-Like Genes in Four Ipomoea Species and Expression Profiling of the IbSPLs During Storage Root Development in Sweet Potato (Ipomoea batatas). Front. Plant Sci. 12:801061. doi: 10.3389/fpls.2021.801061
Received: 24 October 2021; Accepted: 17 December 2021;
Published: 21 January 2022.
Edited by:
Hai Du, Southwest University, ChinaReviewed by:
Moyang Liu, Shanghai Jiao Tong University, ChinaSatyabrata Nanda, Centurion University of Technology and Management, India
Copyright © 2022 Sun, Mei, Zhao, Hou, Zhang, Xu, Wu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, bGVpemhhbmdAanNudS5lZHUuY24=; Shaoyuan Wu, c2hhb3l1YW53dUBvdXRsb29rLmNvbQ==; Tao Xu, eHV0YW9feXJAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship