 Yu Fan
Yu Fan Ya’nan Jin2,3
Ya’nan Jin2,3 Kaixuan Zhang
Kaixuan Zhang Meiliang Zhou
Meiliang Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 15 December 2021
Sec. Plant Systematics and Evolution
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.799904
This article is part of the Research Topic Evolution of Crop Genomes and Epigenomes View all 13 articles
Buckwheat (Fagopyrum genus, Polygonaceae), is an annual or perennial, herbaceous or semi-shrub dicotyledonous plant. There are mainly three cultivated buckwheat species, common buckwheat (Fagopyrum esculentum) is widely cultivated in Asia, Europe, and America, while Tartary buckwheat (F. tataricum) and F. cymosum (also known as F. dibotrys) are mainly cultivated in China. The genus Fagopyrum is taxonomically confusing due to the complex phenotypes of different Fagopyrum species. In this study, the chloroplast (cp) genomes of three Fagopyrum species, F. longistylum, F. leptopodum, F. urophyllum, were sequenced, and five published cp genomes of Fagopyrum were retrieved for comparative analyses. We determined the sequence differentiation, repeated sequences of the cp genomes, and the phylogeny of Fagopyrum species. The eight cp genomes ranged, gene number, gene order, and GC content were presented. Most of variations of Fagopyrum species cp genomes existed in the LSC and SSC regions. Among eight Fagopyrum chloroplast genomes, six variable regions (ndhF-rpl32, trnS-trnG, trnC, trnE-trnT, psbD, and trnV) were detected as promising DNA barcodes. In addition, a total of 66 different SSR (simple sequence repeats) types were found in the eight Fagopyrum species, ranging from 8 to 16 bp. Interestingly, many SSRs showed significant differences especially in some photosystem genes, which provided valuable information for understanding the differences in light adaptation among different Fagopyrum species. Genus Fagopyrum has shown a typical branch that is distinguished from the Rumex, Rheum, and Reynoutria, which supports the unique taxonomic status in Fagopyrum among the Polygonaceae. In addition, phylogenetic analysis based on the cp genomes strongly supported the division of eight Fagopyrum species into two independent evolutionary directions, suggesting that the separation of cymosum group and urophyllum group may be earlier than the flower type differentiation in Fagopyrum plants. The results of the chloroplast-based phylogenetic tree were further supported by the matK and Internal Transcribed Spacer (ITS) sequences of 17 Fagopyrum species, which may help to further anchor the taxonomic status of other members in the urophyllum group in Fagopyrum. This study provides valuable information and high-quality cp genomes for identifying species and evolutionary analysis for future Fagopyrum research.
As the organelle specialized for carrying out photosynthesis in plants, the chloroplast is descended from cyanobacteria, and occurs in eukaryotic autotrophs such as land plants and algae (Jin and Daniell, 2015; Gao et al., 2019). Chloroplasts are involved in photosynthesis and important biochemical processes including storage of starch, and the biosynthesis of sugars, several amino acids, lipids, vitamins, and pigments within plant cells, as well as sulfate reduction and nitrogen cycle supplying for the driving force of plants growth and development (Neuhaus and Emes, 2000; Jarvis and Soll, 2001; Leister, 2003; Bausher et al., 2006). As the center of photosynthesis, chloroplast has a complete genetic system, in which the genetic material is the cp genome (Zhao et al., 2019). Like nuclear DNA, chloroplasts have the same functions of replication, transcription, and inheritance, and cp genomes in plants are generally 10–20% of total genomes with an average length of about 120–170 kb (kilo-base pair) in tetrad ring structure (Shinozaki et al., 1986; Ruhlman and Jansen, 2014). The average cp genome size of land plants is 151 kb, with most species ranging from 130–170 kb in length, as well as the average GC content is 36.3%. The circle cp genome was separated by two inverted repeats (IRs, 20–28 kb) generating the large single copy (LSC, 16–27 kb) and the small single copy (SSC) (Jansen et al., 2007), which can provide abundant information for solving plant phylogenetic relationships and trends. Gene contents and sequences of cp genomes of angiosperm are generally conserved including 4 rRNAs, 30 tRNAs, and 80 unique proteins (Chumley et al., 2006). With the characteristics of parthenogenetic inheritance (maternal inheritance), relatively small genome and slow genome mutation rate (Palmer et al., 1988), analysis of the phylogenetic relationships of multiple chloroplast DNA can help to understand plant phylogeny, population genetic analysis, and taxonomic status at the molecular level (Alwadani et al., 2019). Although cp genomes of angiosperms are generally conserved in gene numbers and sequences (Jansen and Ruhlman, 2012), levels of structural variation in the genome different from various families and genera existed, such as gene duplication and large-scale rearrangement of genes, introns, and IR domains (Cosner et al., 2004; Lee et al., 2007; Cai et al., 2008; Guisinger et al., 2010; Martin et al., 2014).
The size of the cp genome was correlated with plant habits, environments, and other functional traits (Beaulieu et al., 2008; Li et al., 2018), making it a promising tool in studies of phylogeny, evolution, and population genetics of angiosperms (Tonti-Filippini et al., 2017). For example, the phylogenetic relationships among the main branches of flowering angiosperms were analyzed by using the coding genes from 64 cp genomes in Amborella Baill (Jansen et al., 2007); moreover, the relationship between genome evolution and phylogeny of Zingiberaceae was identified using the complete genome sequences of 14 chloroplasts of Curcuma Species (Liang et al., 2020).
Fagopyrum genus belongs to the Polygonaceae family, which are annual or perennial herb or semi-shrub plants (Zhang et al., 2021a). Wild buckwheats are mainly distributed in the regions of southwest China, which was recognized as the center of buckwheat origin and diversity (Ohnishi, 1995, 1998; Ohsako et al., 2002; Saski et al., 2005; Tang et al., 2010; Shao et al., 2011; Zhou et al., 2018). In 1742, Fagopyrum was established by Tourn, and named Fagopyrum Tourn ex Hall (Linnaeus, 1753). In 1992, the taxonomic status of buckwheat was confirmed, and the embryo position, morphology of cotyledon and perianth segments, characteristics of the pollen grain, and the basic number of chromosomes were taken as the basis for distinguishing Fagopyrum from Polygonum (Ye and Guo, 1992). With the continuous introduction of various buckwheat species, the classification based on morphological features gradually complicated, and plants from Fagopyrum were classified into 22–28 different species comprising two variants and two subspecies until 2021 (Zhang et al., 2021a). Due to the long-term change of buckwheat classification status, a consistent view of buckwheat was scarce, which limited the utilization of wild buckwheat varieties in plant improvement (Sharma and Jana, 2002; Neethirajan et al., 2011; Nagatomo et al., 2014). The controversies on buckwheat classification were including but were not limited to the following: (1) the genetic relationships among F. tataricum, F. esculentum, F. esculentum subsp. ancestrale, and F. cymosum. (2) The evolutionary paths between the cymosum group and urophyllum group are intersected or separated? (3) How to define the taxonomic status and phylogenetic relationship among Fagopyrum species in urophyllum group?
The rapid development of molecular biology and genomics provides favorable conditions for the study of cp genome of buckwheat plants, as well as the important genetic information for taxonomic status, phylogeny, and species identification. At present, five buckwheat cp genomes had been published, including F. tataricum, F. esculentum, F. esculentum subsp. ancestrale, F. cymosum, and F. luojishanense (Liu et al., 2008; Logacheva et al., 2008; Cho et al., 2015; Hou et al., 2015; Wang et al., 2017a; Zhang and Chen, 2018). However, the in-depth and conjoint study of Fagopyrum cp genome data sets was lacking, as well as the researches on buckwheat phylogeny and interspecific differences.
In this study, three cp genomes of Fagopyrum were sequenced, assembled, and annotated, then their cp genome data with five published ones were analyzed comprehensively, including characteristics of Fagopyrum cp genomes, codon usage, expansion of IR regions, SSRs analysis, and phylogenetic analysis of eight Fagopyrum species. Our objectives in this study were: (1) To present the complete sequence of cp genomes of three newly assembled buckwheat plants and to compare the global structure with five other previously published species (including one subspecies) within genus species comparisons; (2) SSR variations in the cp genome sequences of eight buckwheat plants were detected to develop a series of SSRs molecular markers that could be used to distinguish the relationship between different species; (3) The phylogenetic relationship and evolutionary path of buckwheat were reconstructed by combining genetic sequences based on eight cp genomes and six highly variable regions developed. (4) The taxonomic status of 17 buckwheat plants was discussed by using ITS and matK gene sequences.
In previous reports, we investigated in detail the survival status of Fagopyrum plants in southwest China (Cheng et al., 2020; Zhang et al., 2021a). The mature seeds of these plant materials are collected in the wild, then they are grown in the greenhouse of the institute of crop science, Chinese Academy of Agricultural Sciences (CAAS) in Beijing. The morphological details of eight Fagopyrum species were further observed. We mainly investigated the differences in plant type, leaf, inflorescence, seed and distribution (Cheng et al., 2020).
Further, the fresh leaves from three Fagopyrum species, F. longistylum, F. leptopodum, F. urophyllum were collected in Sichuan Province in 2020 (Supplementary Table 1). Voucher specimens of these samples were deposited in the Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing, China. Total genomic DNA was isolated from 2 g of silica-dried leaf sample using the modified CTAB method (Doyle, 1987). In addition, we downloaded the available complete cp genomes of five other Fagopyrum species and three Polygonaceae species from GenBank [F. tataricum, MT712164.1; F. cymosum (F. dibotrys), KY275181.1; F. esculentum, MT364821.1; F. esculentum subsp. ancestrale, EU254477.1; F. luojishanense, KY275182.1; Rumex hypogaeus, MT017652.1; Reynoutria japonica (also known as Polygonum cuspidatum) MW411186.1; Rheum officinale MN564925.1] for phylogeny study.
The total DNA was disrupted by ultrasonic wave, and DNA libraries were read of 350 bp with purified DNA constructed by Library Prep Kit from NEBNext®. Total DNA was sequenced in Hiseq 4000 PE150. After filtering the low-quality data, raw sequencing data were checked and spliced using SPAdes 3.6.1 (Bankevich et al., 2012). Contigs were used to screen the cp genome by Blast Software, using published F. esculentum cp genome (MT364821) as the reference genome (Altschul et al., 1997). Selected contigs of the cp genome were assembled using Sequencher 4.10 Software (GeneCodes Corp., Ann Arbor, MI, United States), and all reads were mapped to validate cp genome using Geneious 8.1 Software (Kearse et al., 2012). Polymerase Chain Reaction (PCR) was done with specific primers of gaps, which were born after assembling genomes. The PCR products were sequenced by ABI 3730, and were involved in manually correcting annotations. The circular structure map was constructed by Organellar Genome DRAW1 (Lohse et al., 2013).
Codon Usage analysis was done by codonW 1.4.4 (Peden, 2000), and the values of relative synonymous codon usage (RSCU) were used to evaluate codon preference.
The divergence of 11 Polygonaceae genomes was counted by mVISTA in LAGAN mode (Frazer et al., 2004), and Rumex hypogaeus (MT017652), Polygonum cuspidatum (MW411186), and Rheum officinale (MN564925) were considered as the reference genomes. MAFFT was used to align all Fagopyrum species genome (Zhang et al., 2018), and the nucleotide diversity (Pi) of all complete cp genome was calculated using Launch DnaSP6 (Rozas et al., 2017), and the results were presented through a sliding window analysis with a window length of 600 bp and step size of 200 bp. Boundaries of inverted repeat (IR) regions, contraction, and expansion of eight cp genomes were determined using IRscope (Amiryousefi et al., 2018).
To identify the microsatellites, the Perl script MISA70 and the SSRs parameter were used to analyze the SSRs detection based on the following conditions (Beier et al., 2017); thresholds were set as eight repeat units for mononucleotide SSRs, four repeat units for dinucleotide SSRs, four repeat units for trinucleotide SSRs, and three repeat units for tetranucleotide, pentanucleotide, and hexanucleotide SSRs.
We used the 11 above-mentioned cp genomes to analyze the phylogenetic relationships among Fagopyrum species, including eight Fagopyrum species, and three Polygonaceae species (Rumex hypogaeus, Rheum officinale, and Reynoutria japonica) were used as outgroups. These cp sequences were aligned with the default parameters set using MAFFT program (Katoh and Standley, 2013) in GENEIOUS R8, and were manually adjusted in MEGA 6.0. The nucleotide sequence (matK and ITS) data were obtained from NCBI (Supplementary Table 9). The RAxML v7.2.8 program (Stamatakis, 2006) was used to perform the phylogenetic trees based on maximum likelihood analysis with 1000 bootstrap replicates. Bayesian inference was performed using the MrBayes v3.1.27 program (Ronquist and Huelsenbeck, 2003). Markov chain Monte Carlo simulations have two parallel runs with 2000,000 generations independently, and sampling trees every 100 generations. The initial 25% of trees were discarded as burn-in, and the remaining dates were used to construct a majority-rule consensus tree. Convergence diagnostics were monitored by examining the average standard deviation of split frequencies below 0.01.
The morphological characters of eight Fagopyrum species are further analyzed in this section. Buckwheat is a rare cereal crop that does not belong to Gramineae. Fagopyrum contains plants of both self-compatible (homostyly) and self-incompatible (heterostyly) species. Therefore, Fagopyrum species are good materials for studying the origin and spread of cultivated crops, as well as hot issues such as phylogenetic evolution of plants (Zhou et al., 2018). Morphological characteristics of eight typical different Fagopyrum species (including seven species and one subspecies) were systematically analyzed, and their differences were mainly concentrated in stems, leaves, flowers, and fruits (Figure 1 and Supplementary Table 1). In general, the morphology of Fagopyrum plants is relatively complex and their habits and features are various. In this study, three Fagopyrum species which cp genomes were not revealed were fully considered based on plant characteristics. F. leptopodum, which was commonly found in rocks and dry-hot valley areas, was considered to be a highly drought-resistant and barren resistant species. F. longistylum, a self-compatible but heteromorphic species, was a very rare phenomenon in plants. In addition, F. urophyllum, contained semi-woody branches and perennial rhizomes, which are considered as transitional species from herbaceous to woody plants (Ohnishi and Matsuoka, 1996; Zhang et al., 2021b).

Figure 1. The morphological characters of plants and flowers of eight Fagopyrum species. (A) F. tataricum; (B) F. cymosum; (C) F. esculentum; (D) F. esculentum subsp. ancestrale; (E) F. luojishanense; (F) F. urophyllum; (G) F. leptopodum; (H) F. longistylum.
The cp genomes of three wild Fagopyrum species were sequenced in this study, including two annual species (F. longistylum and F. leptopodum) and one perennial species (F. urophyllum). We obtained the complete cp genome sequences of 159,325 bp for F. longistylum, 159,350 bp for F. urophyllum, and 159,376 bp for F. leptopodum. Other published cp genomes of Fagopyrum were obtained from National Center for Biotechnology Information (NCBI), and all cp genomes ranged in size from 159,265 bp (F. luojishanense) to 159,599 bp (F. esculentum ssp. ancestrale) with 37.78–37.99% GC contents (Figure 2 and Table 1). Similar to other Polygonaceae, all cp genomes of cultivated and wild Fagopyrum species comprised a typical circular structure with four regions (Wu et al., 2020), and two inverted repeats (IR, IRa, and IRb) regions were separated by a LSC and a SSC (Figure 2). The LSC region in Fagopyrum accounted for 52.87–53.19% of the total cp genomes and ranged in size from 84,250 bp (F. urophyllum) to 84,885 bp (F. esculentum ssp. ancestrale); the SSC region in Fagopyrum accounted for 8.22–8.41% and ranged in size from 13,094 bp (F. luojishanense) to 13,406 bp (F. urophyllum); the Fagopyrum IR region accounted for 19.23–19.38% of the total size and ranged from 30,6845 bp (F. esculentum and F. esculentum ssp. ancestrale) to 30,870 bp (F. luojishanense). Moreover, the GC contents of all Fagopyrum cp genomes were similar, and the GC content of IR region was highest (41.26–41.48%), followed by the LSC region (36.01–36.32%) and the SSC region (31.97–32.99%).
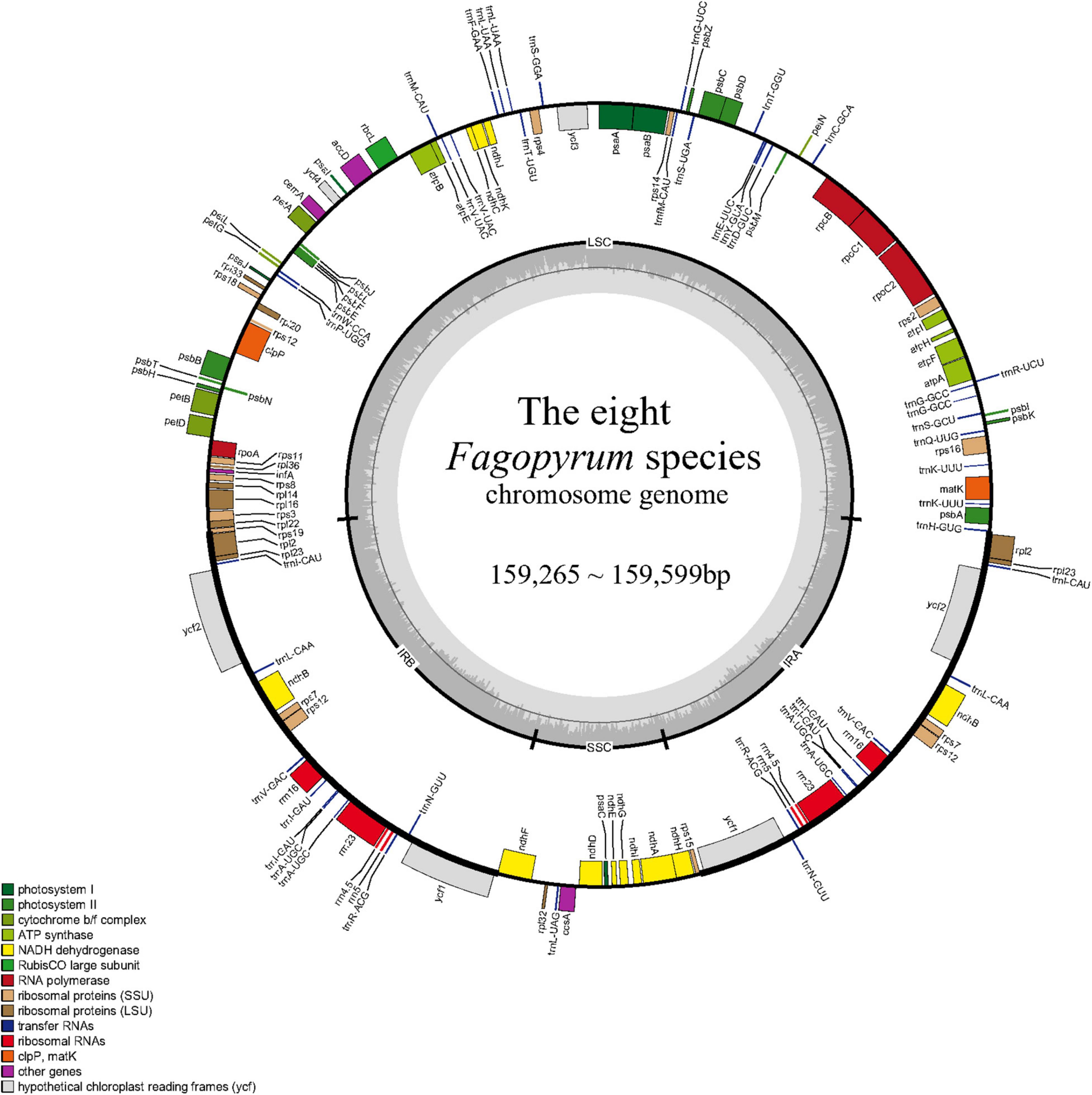
Figure 2. Gene map of the eight Fagopyrum species. The genes shown outside of the circle are transcribed clockwise, while those inside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. The dashed area in the inner circle indicates the GC content of the chloroplast genome.
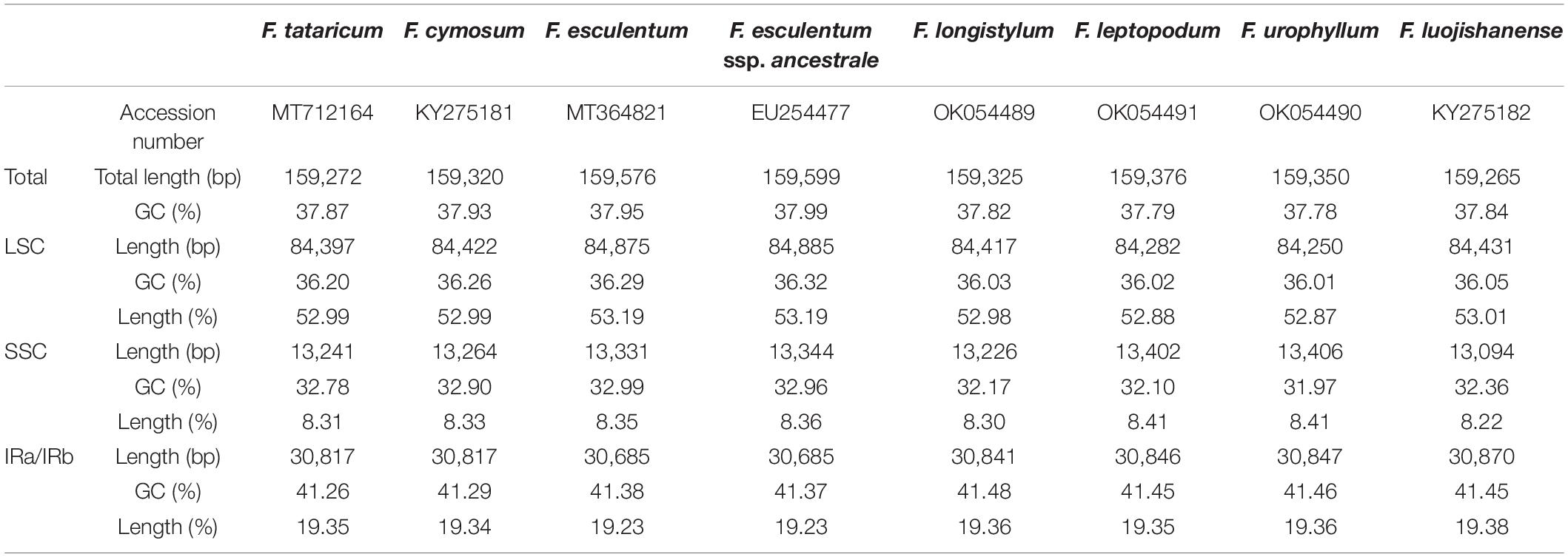
Table 1. Comparison of the complete chloroplast genomes for eight Fagopyrum species.
There was little difference in coding regions in eight Fagopyrum species. Overall, they encode a total of 108–113 chloroplast genes, including 76–79 protein-coding genes, 28–30 tRNAs, and 4 rRNAs (Figure 2 and Table 2). All the above-mentioned genes were furtherly categorized as three parts, of which 47 genes belong to photosynthesis related genes (including rubisco, photosystem I, assembly/stability of photosystem I, photosystem II, ATP synthase, cytochrome b/f complex, cytochrome c synthesis, and NADPH dehydrogenase), 60 genes belong to transcription and translation related genes (including transcription, ribosomal proteins, and translation initiation factor, ribosomal RNA, and transfer RNA), and the remaining genes belong to biomacromolecule metabolism related genes or other unknown functions (Table 2). Moreover, among these various 113 genes, 15 genes contained one intron comprising 9 protein-coding genes (atpF, petB, petD, ndhA, ndhB, rpoC1, rps12, rpl2, and rpl16) and 6 tRNA genes (trnA, trnG, trnI, trnK, trnL, and trnV), while 2 genes (ycf3, clpP) contained two introns. In addition, rps12 was identified as a noticeable trans-splicing gene of all Fagopyrum species, because the 5′ end of rps12 exon was located in the LSC region but the other end of that was located in the IR domain.

Table 2. Genes contained in the chloroplast genome of eight Fagopyrum species.
Codon is the connection between the nucleic acids and proteins, and codon usage reflects the preference for selective use of codons encoding specific amino acids with genetic information (Wanga et al., 2021). The codon usage frequency of 79 protein-coding genes for 8 Fagopyrum species were calculated, and 64 codons were involved in encoding proteins containing three termination codons, such as UAA, UAG, and UGA (Table 3). The relative synonymous codon usage (RSCU) analysis showed that 30 codons of 8 Fagopyrum species were > 1, and the UUA encoding leucine had the highest RSCU with 1.85–1.87 in 8 Fagopyrum species. While the lowest RSCU was 0.33–0.36 with the CGC encoding arginine.
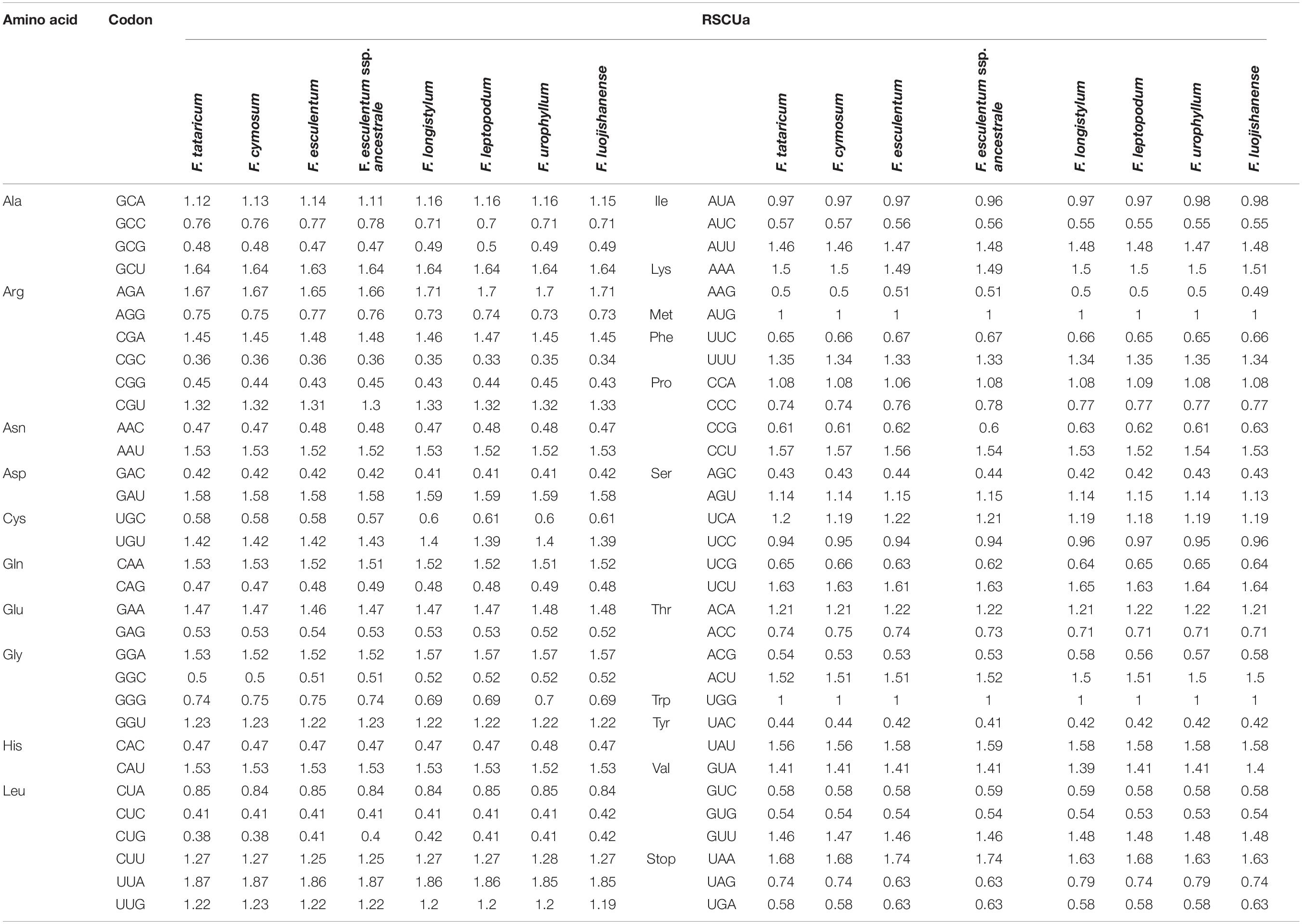
Table 3. Codon content of amino acids and stop codon of eight Fagopyrum species.
The genome of F. tataricum was served as the reference to conduct the mVISTA program for discovering Fagopyrum genome divergence, and three other genomes from Polygonaceae were regarded as the outgroups covering Rumex hypogaeus, Polygonum cuspidatum, and Rheum officinale. Results revealed that 11 cp genomes were relatively conserved (Figure 3). The three cultivated Fagopyrum species, four wild Fagopyrum species, and three outgroup members had higher similarity and low divergence, respectively. Furthermore, the divergence of LSC and SSC regions were higher than that of IR regions, and the non-coding regions exhibited greater variation than the coding regions.
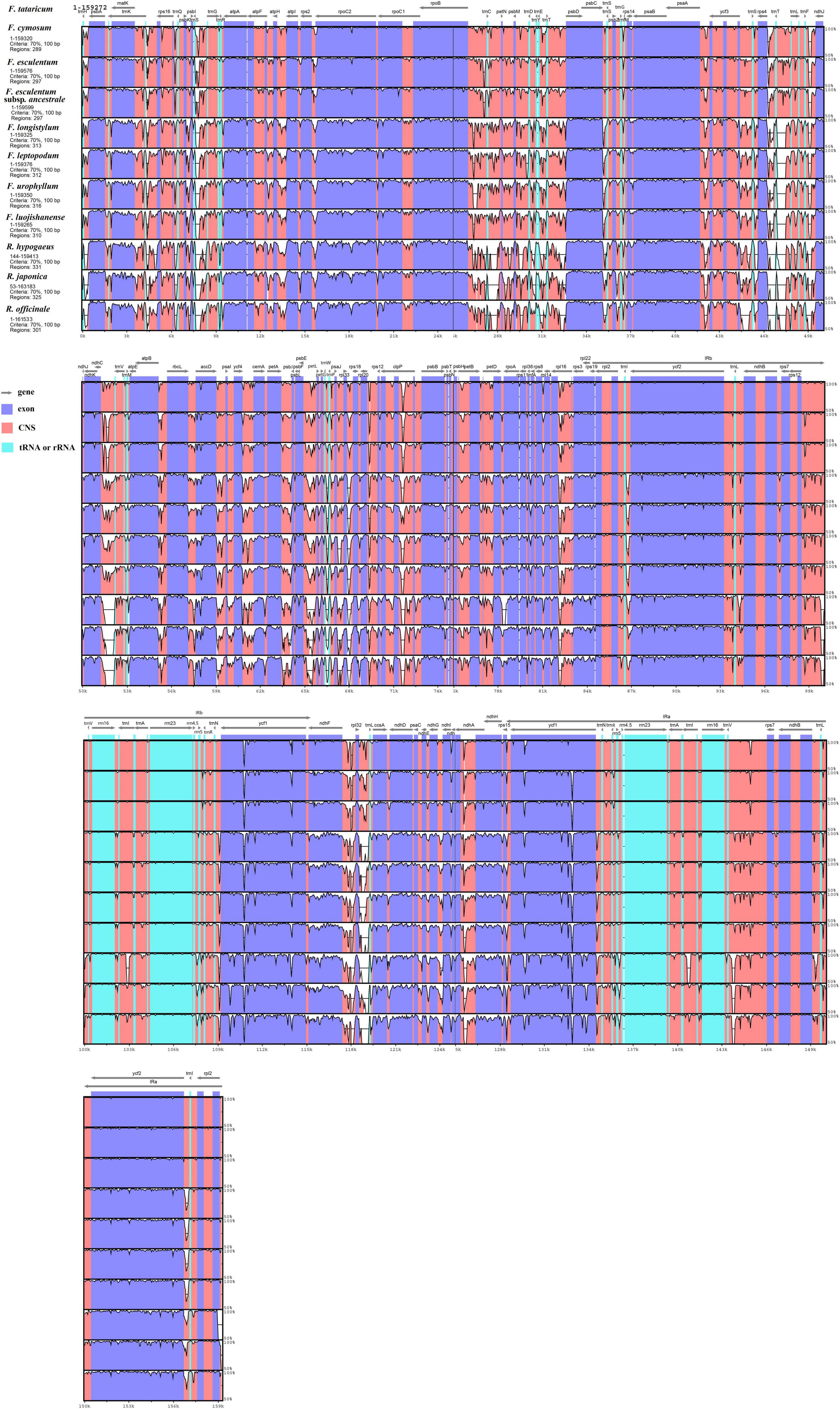
Figure 3. Sequence alignment of chloroplast genome among eight Fagopyrum species and three Polygonaceae species (Rumex hypogaeus, Reynoutria japonica, and Rheum officinale) with F. tataricum as a reference by using mVISTA. The Y-scale represents the percentage of identity ranging from 50 to 100%. Coding and non-coding regions are marked in purple and pink, respectively.
To further know the genetic diversity of various Fagopyrum species and exploit suitable polymorphic genes for identifying novel species, we calculate the nucleotide diversity (Pi) of eight Fagopyrum species. The Pi values were ranged from 0 to 0.10179 in the total cp genomes. The average Pi values of LSC and SSC regions were 0.0356 and 0.0445, respectively, but that of IR regions was 0.0084 (Supplementary Table 2). Most of the variations of Fagopyrum species cp genomes existed in the LSC and SSC regions. That is to say, two IR regions were more conserved than another two regions. A sliding window analysis showed that the Pi values of six regions were > 0.08, and these most divergent regions included ndhF-rpl32, trnS-trnG, trnC, trnE-trnT, psbD, and trnV (Figure 4 and Supplementary Table 2). Among them, three coding genes (ndhF, rpl31, and psbD) were highlighted, because coding genes were generally conserved. These polymorphic regions might be the critical loci for population genetic studies of Fagopyrum species.
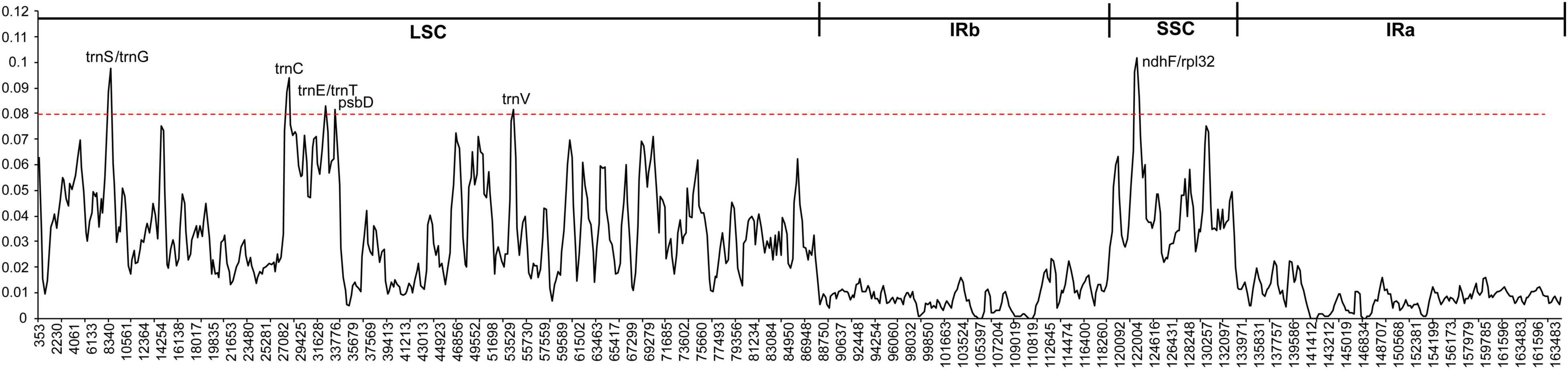
Figure 4. Comparison of nucleotide diversity (Pi) values among the eight Fagopyrum species. X-axis, position of the midpoint of each window; Y-axis, nucleotide diversity (Pi) of each window.
As we all know, contraction and expansion of the IR regions are strongly linked to the length of cp genomes (Liang et al., 2020), therefore the IR boundaries were detected to explain the differences in Fagopyrum cp genome size. In general, IRs of wild Fagopyrum species (F. longistylum, F. leptopodum, F. urophyllum, and F. luojishanense) were longer than cultivated Fagopyrum species (F. tataricum, F. cymosum, and F. esculentum). Among them, the size of the IR regions of the two F. esculentum was the shortest (30,685 bp) and that of F. luojishanense was the longest (30,870 bp) (Figure 5).
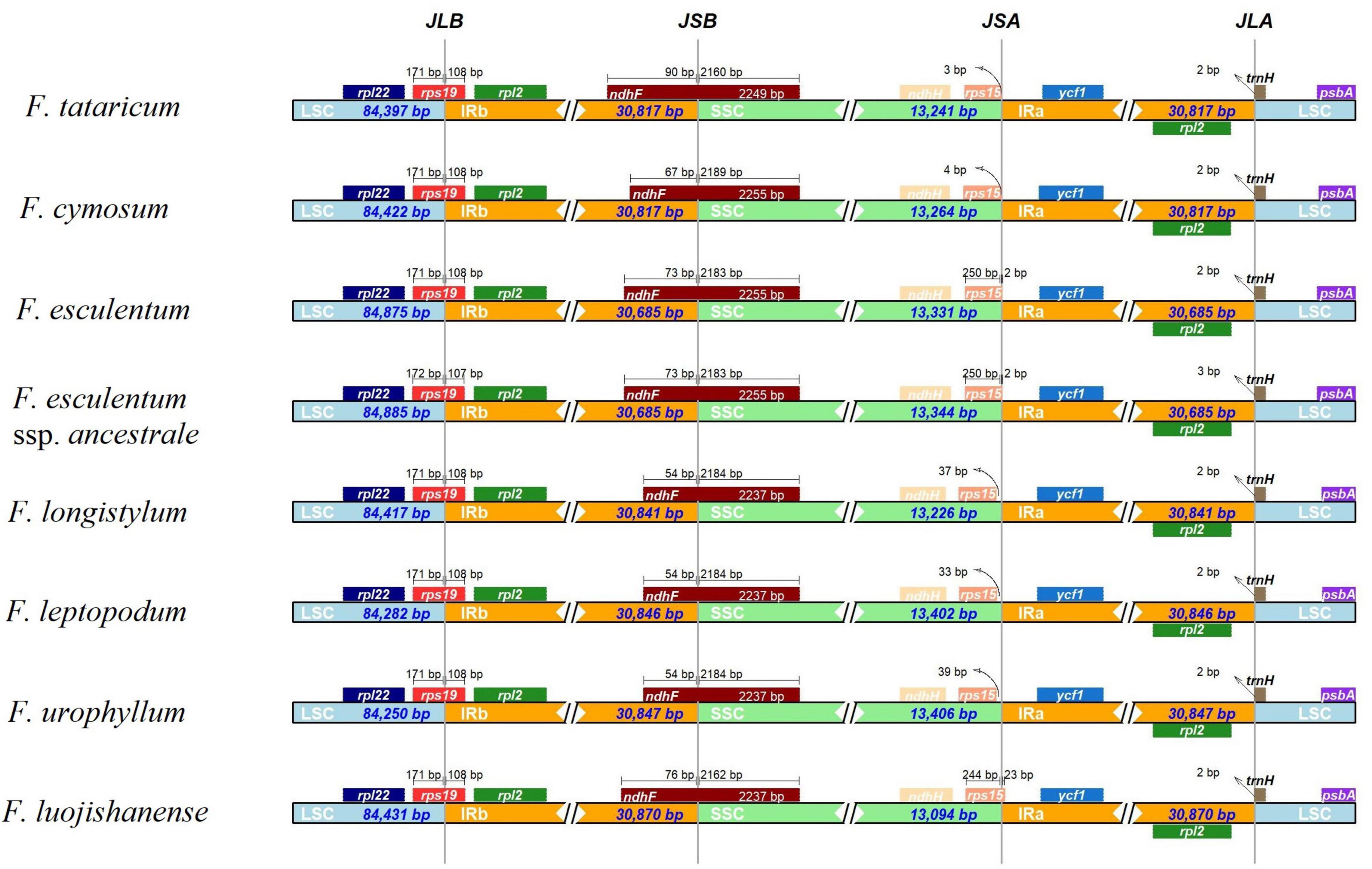
Figure 5. Comparison of the junctions between the LSC, SSC, and IR regions among eight Fagopyrum species chloroplast genomes. The figure is not scaled LSC, SSC, and IR.
Within the 8 Fagopyrum species, the rps19 genes were located in the boundaries of LSC/IRb regions (JLB) consistently, except for the location of rps19 from F. esculentum ssp. ancestrale in JLB was more forward than other members (1 bp). The SSC and IRb regions (JSB) were connected by ndhF genes, and the length of the ndhF in IRb from the JLB was 54–90 bp. In the JSA (SSC/IRa) regions, only JSA of three species were embedded in rps15 gene, including the two F. esculentum and F. luojishanense. Specifically, the rps15 gene was located on the right of the two F. esculentum with the distance of 2 bp, but that of F. luojishanense was 23 bp. The LSC/IRa (JLA) junctions in the cp genomes of 8 Fagopyrum species were identical. All in all, the IR boundaries of F. tataricum and F. cymosum were similar, as well as two F. esculentum species, and three wild species (F. longistylum, F. leptopodum, and F. urophyllum), respectively.
Simple sequence repeats, also known as microsatellites, consisted of short tandem repeats of 1–6 bp in length (Li B. et al., 2020). SSRs are widely distributed in the cp genome, and play a key role in the identification of plant genetic relationships and taxonomic status (Yang et al., 2019; Li Y. et al., 2020). In the cp genome sequence of the eight Fagopyrum species, SSRs were mainly located in the intergene region (∼57.72%), followed by the genic region (∼42.28%), while no SSR was observed in tRNAs and rRNAs (Figure 6A and Supplementary Table 3), which is consistent with the report of Wang et al. (2017b). Of note, the SSR numbers of F. leptopodum (133, ∼59.38%), F. longistylum (138, ∼60.26%), F. luojishanense (131, ∼58.48%), and F. urophyllum (143, ∼60.59%) in the intergene region were significantly higher than that of F. tataricum (110, ∼53.66%), F. cymosum (115, ∼56.65%), F. esculentum (119, ∼55.61%) and F. esculentum subsp. ancestrale (120, ∼56.34%). Most SSRs were located in LSC region (∼64.63%), followed by IR region (∼26.38%) and SSC region (∼8.99%) (Figure 6B and Supplementary Tables 4, 5). F. cymosum (129, ∼63.55%) had the least number of SSR in LSC region, followed by F. tataricum (130, ∼63.41%), F. esculentum (139, ∼64.95%) and F. esculentum subsp. ancestrale (138, ∼64.79%), in general, their number was significantly lower than F. leptopodum (146, ∼65.18%), F. luojishanense (145, ∼64.73%), F. longistylum (148, ∼64.35%), and F. urophyllum (156, ∼66.10%). Interestingly, as two typical cultivars, F. tataricum (58, ∼28.29%) and F. esculentum (56, ∼26.17%) showed significant expansion in SSR proportion in IR region. Further, a total of 24 gene located in different regions were found, which may be the result of co-evolution of cp genomes (Zhao et al., 2021). Among them, ndhB, ycf2, and ycf1 are in the IRb/IRa region, atpA, rbcL, rpl20, rpl22, rpoA, ycf4, cemA, petB, ycf3, petA, rpoB, atpF, rpoC1, rpl16, and rpoC2 are located in LSC region, and rps15, ndhF, ndhD are located in SSC region.
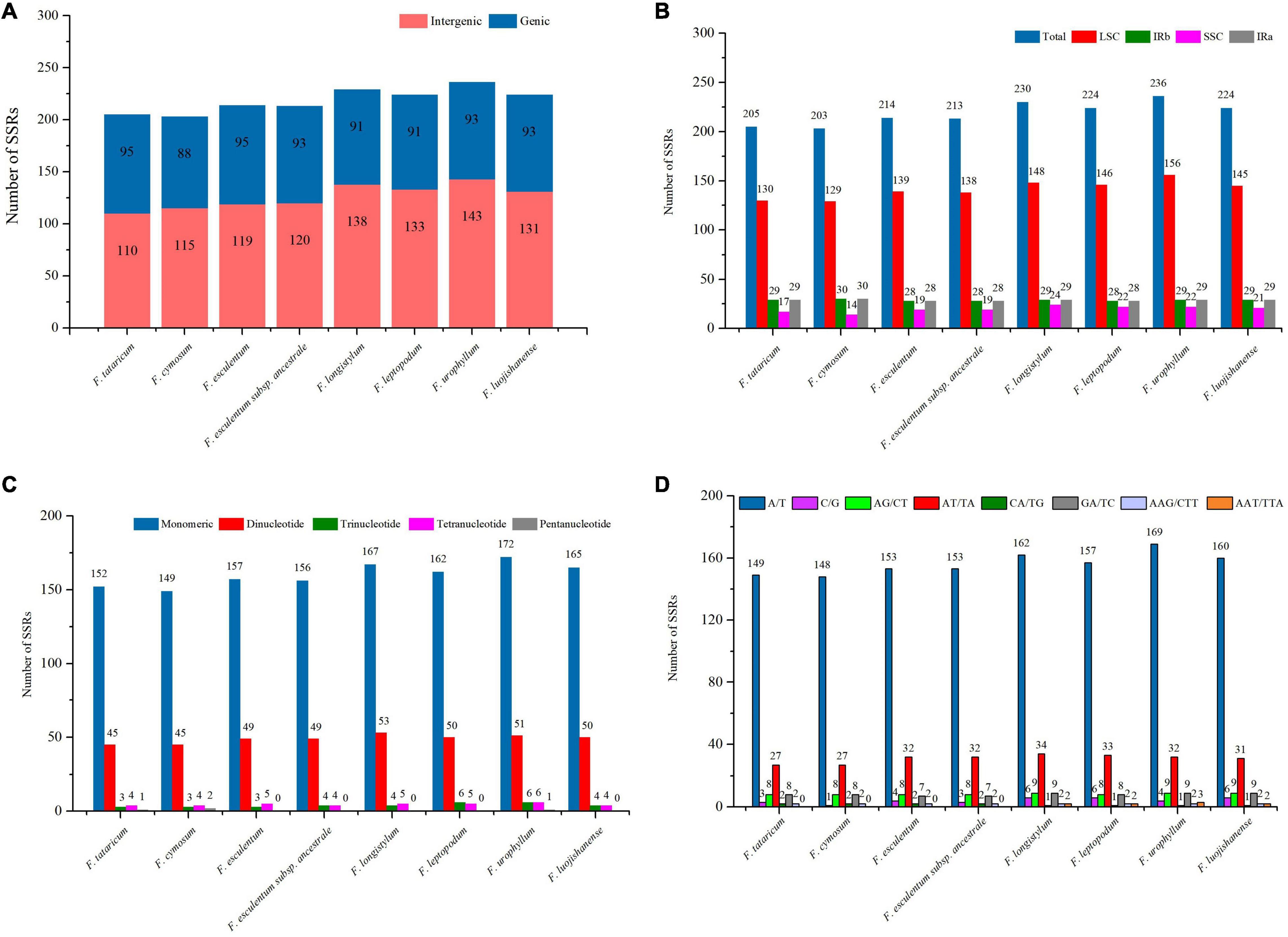
Figure 6. Analysis of SSRs in the eight Fagopyrum species cp genomes. (A) Distribution of SSRs in genic and intergenic. (B) Distribution of SSRs in LSC, IRa/IRb, and SSC. (C) The number of different SSR types detected in the eight Fagopyrum species cp genomes. (D) The number of different base SSR types in the eight Fagopyrum species cp genomes.
The distribution range of SSRs ranged from 8 to 16 bp in eight Fagopyrum species, with a total of 66 different types(Figure 6C and Supplementary Tables 4, 5). There were no hexanucleotide repeats have been found in these SSR sequences, and pentanucleotide repeats were only found in the cp genomes of F. urophyllum (ATTAT), F. tataricum (TTTTA), and F. cymosum (TCTAT/TTTTA). Among all Fagopyrum species, the number of mononucleotide repeats in the cymosum group was significantly lower than that in the urophyllum group. In general, this study supports that mononucleotide repeats may play a more important role in genetic variation in buckwheat than other SSR types (Huang et al., 2017; Liang et al., 2020). Although the chloroplast evolution of Fagopyrum species were relatively conserved, the cymosum group may be subjected to stronger selection and evolutionary pressure, resulting in the decline of SSR genetic diversity. Meanwhile, the number and types of SSR of the eight buckwheat plants in this study were further analyzed (Figure 6D and Supplementary Tables 5, 6). Further, the proportion of mononucleotide repeats for A/T and C/G types were 71.52 and 1.86%, respectively (Figure 6D and Supplementary Tables 5–7). This is similar to Zingiberales, Salicaceae, and Ranunculaceae, etc., indicating that mononucleotide repeats of A/T type may always be the most abundant base of simple repeat sequences (Huang et al., 2017; Liang et al., 2020; Park and Park, 2021). In addition, the number of mononucleotide repeats of A/T types or C/G types in the cymosum group was significantly lower than the urophyllum group, indicating that the number of SSR may still be similar in different subgroups of Fagopyrum species. The dinucleotides of eight Fagopyrum species were divided into four categories, which showed differences in some gene regions and repeated fragments among different groups. For example, repeat sequences of AG/CT and GA/TC types do not differ significantly between the cymosum group and urophyllum group. However, the proportion of CA/TG repeats in the cymosum group (∼0.96%) was much higher than that in the urophyllum group (∼0.44%). Similarly, AT/TA type accounted for the highest proportion of all dinucleotides (∼14.16%), which further confirmed the activity of A/T base in the cp genome. In this study, F. tataricum (27, ∼13.17%)/F. cymosum (27, ∼13.30%), F. esculentum (32, ∼14.95%)/F. esculentum subsp. ancestrale (32, ∼15.02%) had similar AT/TA types in number and proportion, which supported their genetic relationship to a certain extent. In addition, nucleotide repeats of AAT/TTA type did not exist in the four species of cymosum group (0), while F. longistylum (∼0.87%), F. leptopodum (∼0.89%), F. luojishanense (∼0.89%), and F. urophyllum (3, ∼1.27%) had a similar proportion. Therefore, there may exist two divergent evolutionary directions between the cymosum group and the urophyllum group. These results suggest that SSR can be used to identify genetic diversity, study evolution and develop molecular markers in buckwheat.
Chloroplast genome sequences of eight Fagopyrum species and three Polygonaceae plants, which were selected as the outgroup, were used to construct phylogenetic trees to elucidate their genetic relationships (Figure 7). The numbers on the branches show the bootstrap value of the maximum likelihood analysis. The results showed that all Fagopyrum species clustered together at a very high resolution, and the three Polygonaceae plants and the eight Fagopyrum species were divided into two main types, which confirmed the independent differentiation status of the Fagopyrum from other genera of Polygonaceae. Further, eight Fagopyrum species were classified into two typical subclades. Among them, F. tataricum and F. cymosum formed a subgroup different from F. esculentum, which further supports that they may have a relatively high degree of homology and a closer genetic relationship. And then, they gradually converged with F. esculentum and F. esculentum subsp. ancestrale to form a subbranch. In addition, F. longistylum first approximates to F. luojishanense, and then gradually forms with F. urophyllum and F. leptopodum. These results showed that there might be two different subgroups among the eight Fagopyrum species, and the cymosum group and the urophyllum group evolved independently. Further, we developed six molecular marker sequences based on Pi values (Supplementary Figures 1A–F and Supplementary Table 8). And, six cluster trees were constructed based on these sequences using the neighbor-joining method (NJ). Among them, trnS-trnG and trnV trees supported the topological structure of the cp genome, which can be further applied in the identification of genetic relationships in Fagopyrum species.
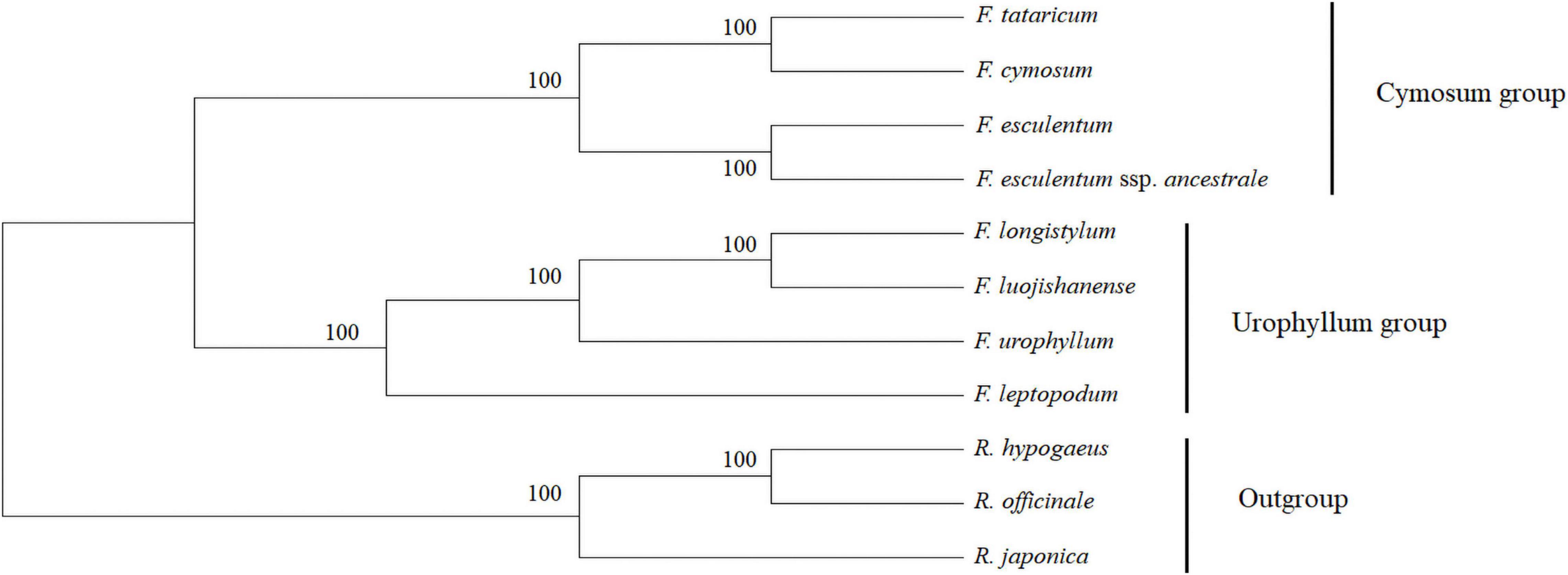
Figure 7. Phylogenetic tree obtained for eight Fagopyrum species inferred from ML analysis constructed by the complete chloroplast genomes. The number on the branches displayed the bootstrap support values.
The most widely used chloroplast gene matK and nuclear marker ITS were selected to further speculate the genetic relationship of eighteen Fagopyrum species (including one variety: F. gracilipes var. odontopterum) (Supplementary Figures 2A,B and Supplementary Table 9). In general, the two ML trees based on ITS and matK supported the above-mentioned cp genome tree results: F. tataricum and F. cymosum in the two phylogenetic trees are first clustered into one branch, then clustered with F. esculentum, and then gradually clustered into other wild species. Therefore, phylogenetic trees based on different markers in this study all supported the conclusion that F. tataricum and F. cymosum in the cymosum group has a more close relationship than F. esculentum, which consisted with the previous study (Zhang et al., 2021a). Similarly, F. luojishanense and F. longistylum of the urophyllum group may be closely related, and then cluster with F. leptopodum and F. urophyllum. These results further supported the chloroplast phylogenetic tree results. Therefore, the relationship of Fagopyrum plants was further inferred, F. luojishanense, F. longistylum, F. gracilipes, F. gracilipes var. odontopterum and other wild species may have a close relationship. According to the clustering results, F. gracilipes var. odontopterum as the division of F. gracilipes is considered reasonable. The F. lineare and F. leptopodum may be closely related to each other. They are both short plants, thin stem nodes, and highly adaptable in these Fagopyrum plants. Moreover, the two evolutionary trees supported F. caudatum and F. pugense were closely related. In general, these sequences of molecular markers with stable phylogenetic relationships of Fagopyrum plants will be considered as “references” to further infer taxonomic status among other species. However, it should be pointed out that the phylogenetic trees based on matK and ITS sequences could not completely define the relationships of some Fagopyrum species. For example, the genetic relationship between F. macrocarpum and F. qiangcai is still unstable. Therefore, it is necessary to further analyze the taxonomic status of Fagopyrum plants through extensive molecular marker sequences or complete genome sequencing.
In this study, we compared the complete cp genomes of eight Fagopyrum species, which showed a typical circular tetrad structure. It consisted of a LSC region (84,494.9 bp in average), a SSC region (13,288.5 bp in average), and two reverse repeats (IR) regions (30,801 bp in average). The structures, genome lengths and proportion of these cp genomes were highly conserved. Among the eight cp genomes, the gene spacer is the largest variable region, which is consistent with most angiosperms (Wicke et al., 2011). The total GC ranges from 37.78 to 37.99%, which are higher than that of Euonymus, and Curcuma (Liang et al., 2020; Li et al., 2021). The GC ratios of the cp genome of angiosperms are usually between 34 and 40%, which plays an important role in the transmission of gene information (Zhu et al., 2017). The cp genome differences of different species are obvious through changes in base composition. These GC contents of the Fagopyrum species are the highest in IRa/IRb region, and the uneven distribution of GC ratio and gene conversion between IR sequences, which may be the reason why the IR region is more conserved than the LSC and SSC region (Khakhlova and Bock, 2006; Fan et al., 2018).
The contraction or expansion of the IR boundary is one of the main driving forces of cp genome length and structure difference, and the change of IR/SC connection location is a typical evolutionary phenomenon in plants (He et al., 2017). Interestingly, we found significant expansion of the LSC region in F. esculentum and F. esculentum ssp. ancestrale, which may be direct evidence of both cp genome length expansion and IRb region contraction. In addition, a significant contraction was observed in the SSC region of F. luojishanense (∼13,094 bp), which had the largest IRa/IRb region (∼30,870 bp), resulting in the C terminal of rps15 crossing into the IRb region (∼23 bp). Furthermore, we found that the loss of functional genes in cymosum members were significantly higher than that in urophyllum group. And, this phenomenon was more obvious in many transfer RNAs. Therefore, we hypothesized that this deletion may result from the apparent activity of the highly structured chloroplast genome in cymosum group. For example, trnfM-CAU lost in F. esculentum and F. esculentum ssp. ancestrale. The chloroplast genome structures of urophyllum members were more conserved, and there were little difference in the numbers and positions of encoded genes. In addition, trnfM-CAU/trnM-CAU, trnG-UCC/trnG-GCC in cymosum group were significant differences in gene location in cp genomes. tRNAs are one of the most important and versatile molecules responsible for the maintenance and maintenance of protein translation mechanisms (Mohanta et al., 2019). Differences in the number and distribution of tRNAs in the cp genome may result in significantly influences of post-translational modification processes on genes in the photosynthetic system, especially rpoA, rpoB, and rpoC genes (Little and Hallick, 1988; Zhang, 2020). In addition, deletion of rpl23 gene in cp genomes of two cultivated species (F. tataricum and F. esculentum) were observed. This phenomenon illustrated a typical case of protein (gene) substitution in the evolution of chloroplast ribosomes in Fagopyrum plants, and nuclear genome could progressively exert stronger over the chloroplast translational system (Bubunenko et al., 1994). It is worth noting that F. esculentum, as a Fagopyrum plant which is mostly distributed in the middle and high latitude areas of the northern hemisphere with long sunshine, is observed the most loss of functional genes, such as trnT-UGU, rpl23, trnI-CAU, etc.
DNA barcoding is widely used in species identification, germplasm management, genetic diversity analysis, phylogeny, and evolution (Gregory, 2005; Liu et al., 2019). In previous studies, the phylogeny of structural Fagopyrum plants was mainly based on SSR markers (Ma et al., 2009; Yang et al., 2020), single-copy nuclear gene (Ohnishi and Matsuoka, 1996; Ohsako and Ohnishi, 1998). The taxonomic analysis and genetic identification of Fagopyrum species are hampered by the lack of genomic information. Cp genome sequences are relatively conserved, which is less affected by non-parallel evolutionary in functional genes of nuclear genes in phylogenetic tree construction. Therefore, the cp genome sequences are often used in angiosperms phylogenetic prediction in recent years (Zhang et al., 2017; Zhao et al., 2020). To determine divergence packaging, the mVISTA program was used to compare the cp genome sequences of eight Fagopyrum species. The results showed that the cp genomes of eight Fagopyrum species were rich in the variable sites, and some regions with high variable frequency could be directly used as potential molecular markers for species identification (Song et al., 2017; Xu et al., 2017). In general, the proportion of variable loci in the non-coding region was higher than that in the coding region. Meanwhile, sequence differentiation in the IR region was slower and more conserved than that in LSC and SSC region. These results are consistent with most cp genome studies in plants, and we speculate that this may be due to higher gene conversion between the two IR regions (Khakhlova and Bock, 2006; Jansen and Ruhlman, 2012; Huang et al., 2014). In addition, the nucleotide diversity (Pi) of eight Fagopyrum species were assessed by sliding window analysis. These results of Pi values were generally consistent with mVISTA analysis, and the nucleotide diversity in the non-coding region was higher than that in the coding region. Six variable regions (ndhF-rpl32, trnS-trnG, trnC, trnE-trnT, psbD, and trnV) were identified as highly variable sites at the species level of Fagopyrum. These variable regions were further used to identify the genetic relationship of eight Fagopyrum species. And, the results showed that trnS-trnG, and trnV trees showed highly consistent results with cp genomes, so that they were further recommended as potential molecular markers in genetic development analysis and assisted breeding in Fagopyrum plants.
Simple repeat sequences play important role in the combination and arrangement of cp genome structures, which are highly variable in different species of the same genus, Thus, SSRs have been widely used in population genetics and species biodiversity studies (Thiel et al., 2003; Zhou et al., 2019). In this study, it was found that the SSR polymorphism levels of the four major components of these cp genomes were inconsistent. SSRs were mainly found in the LSC region of the eight Fagopyrum species, which was closely related to the interval length. The distribution density of SSRs in the eight Fagopyrum species were uneven, and there may be more SSRs in some sections and gene locus. For example, matK, rpoC2, clpP, ycf1, ycf2, ycf3, and other gene regions showed higher SSR density, which was consistent with Zingiberales and other plants (Liang et al., 2020). Although the cp genome evolution of Fagopyrum plants is generally co-evolutionary, some functional gene regions may respond to important biological effects and thus be subjected to more significant evolutionary pressures (Williams et al., 2019). At present, only a few “star genes,” such as matK, rbcL, ycf1, and ycf2, have been found as common positive selection sites (Liang et al., 2020; Li et al., 2021), other studies on the response evolution and biological role of chloroplast functional genes are still scarce. Nevertheless, it is desirable to select some segments or polymorphism of repeating sequence fragments from the cp genome as new tools for studying systematic differentiation.
A total of 110 (∼F. tataricum) ∼143 SSR markers (∼F. urophyllum) were found in the cp genomes of eight Fagopyrum plants, including mononucleotides, dinucleotides, tetranucleotides, trinucleotides, pentucleotide. Notably, there were no hexonotides found in all Fagopyrum species, which is inconsistent with Euonymus, Zanthoxylum, Curcuma, Wurfbainia Villosa, Amomum, Kaempferia, etc. (Liang et al., 2020; Li et al., 2021; Zhao et al., 2021). A/T and AT/TA repeats are the main SSR types, which may be because A/T bases are more easily changed than G/C bases (Li et al., 2021). However, these AT-rich regions did not contribute significantly to the expansion of cp genome size (Figure 6D). Compared with the gene regions, most of the SSRs were distributed in the intergene region (IGS region), which was more obvious in the members of the urophyllum group. It should be noted that there were significant differences in SSR markers in some gene regions between the urophyllum group and the cymosum group. For example, CA (4) existed only in cymosum group members, while AAT (4), AG (5), GA (5), TCAA (3), and TTA (4) were all found in urophyllum group members. These markers can be further applied to the identification of the two subgroups. In addition, many unique SSR markers were found in some Fagopyrum species, which can be used in the identification of different species. For example, AAAT (3) only existed in tartary buckwheat, AATT (4), A (16), TCTAT (3) only exist in F. cymosum, AATG (4) only existed in F. longistylum. Interestingly, there are still some unique SSR markers in F. esculentum and F. esculentum ssp. ancestrale, which will be effectively used in the identification of cultivated and wild ancestor species. For example, TTGA (3) was found in F. esculentum, while GTA (5), and C (12) were unique to F. esculentum ssp. ancestrale.
Interestingly, we observed significant differences in repeat sequences among some photosystem genes between members of the cymosum group and urophyllum group (Supplementary Table 7). For example, ycf1 and two ribosome large subunit genes (rpl32, rps15) at the IR boundary showed significant SSR expansion in the cymosum group. This may contribute to the light adaptation of cymosum group members, which is conducive to planting (Fan et al., 2018; Liang et al., 2020). Photosystem subunit genes (psaJ, psbK, psbZ) showed significant SSR expansion in F. esculentum and F. esculentum subsp. ancestrale. They are more adapted to the long-sunshine of the northern hemisphere (Ikeuchi et al., 1991; Sugimoto and Takahashi, 2003). In addition, the urophyllum group members have a narrower distribution range, mainly growing in mountainous areas of southwest China. However, they are more adaptable to complex geographical environments, such as mountain areas and sandy areas, which are too harsh for the cultivated species (Zhou et al., 2018). In general, the process of artificial domestication or natural selection pressure leads to a significant decline in genetic diversity in the genome (Louwaars, 2018; Zhu et al., 2019). However, this was not significantly reflected in cp genomes of F. cymosum, F. esculentum, and F. tataricum. Therefore, we speculate that these domestication intervals may exist mainly in the nuclear genome. In conclusion, SSR markers of eight Fagopyrum species were systematically reported for the first time, which can provide a reference for the subsequent study of molecular evolution and phylogeny of Fagopyrum genus and Polygonaceae family.
For a long time, the taxonomic status of Fagopyrum genus has changed frequently, and no consensus has been reached among different species (Linnaeus, 1753; Miller, 1754; Meisner, 1826; Gross, 1913; Stewad, 1930; Zhang et al., 2021b). In this study, the phylogenetic trees based on cp genomes of eight Fagopyrum species and Rumex, Rheum, and Reynoutria supported the independent evolution of Fagopyrum plants. Therefore, it is reliable that Fagopyrum has a separate taxonomic status in the Polygonaceae.
Furthermore, the cymosum members (F. tataricum, F. cymosum, F. esculentum, F. esculentum subsp. ancestrale) had significant independent cluster branches into the urophyllum group. Therefore, we infer that the evolutionary processes of the two groups of Fagopyrum species may be independent rather than overlapping. Similarly, the separation of the cymosum group and the urophyllum group may be earlier than the flower type differentiation of Fagopyrum plants, and then two pollination modes of self-pollination (self-compatibility) and cross-pollination (self-incompatibility) are produced. In addition, this study concluded that the genetic relationship in the cymosum group is clear, the F. cymosum and F. tataricum are more closely related than F. esculentum, although their pollination patterns are not consistent. However, the taxonomic status of the members of the urophyllum group are more complicated, as the urophyllum group consists of 18 species. Although there were significant differences in differentiation rates between nuclear and cp genomes, ITS clearly supported the clustering results of the urophyllum group in the evolutionary tree of cp genomes. Four urophyllum group members can further anchor the taxonomic status of other wild species members, which is further supported by the previous study (Cheng et al., 2020; Zhang et al., 2021a). It should be noted that the taxonomic status of some members of the urophyllum Group cannot be significantly anchored by a single molecular marker, which may require further molecular evidence.
The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) repository, accession number were: F. longistylum (OK054489), F. urophyllum (OK054490), F. leptopodum (OK054491).
KZ, MZ, JC, and YF conceived and designed the work. YT and MD collected the samples. YF, YJ, and KZ performed the experiments and analyzed the data. YF and YJ wrote the manuscript. MD, MZ, and JC revised the manuscript. All the authors have read and agreed to the published version of the manuscript.
This work was financially supported by the National Key R&D Program of China (2019YFD1000700 and 2019YFD1000703) and National Science Foundation of China (31560578).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.799904/full#supplementary-material
Supplementary Figure S1 | Phylogenetic tree based on the ndhF-rpl32 (A), psbD (B), trnC (C), trnE-trnT (D), trnS-trnG (E), and trnV (F) sequences of eight Fagopyrum species constructed from NJ analysis.
Supplementary Figure S2 | Phylogenetic tree based on the ITS (A) and matK (B) sequences of eighteen Fagopyrum species constructed from ML analysis.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Alwadani, K. G., Janes, J. K., and Andrew, R. L. (2019). Chloroplast genome analysis of box-ironbark Eucalyptus. Mol. Phylogenet. Evol. 136, 76–86. doi: 10.1016/j.ympev.2019.04.001
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bausher, M. G., Singh, N. D., Lee, S. B., Jansen, R. K., and Daniell, H. (2006). The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 6:21. doi: 10.1186/1471-2229-6-21
Beaulieu, J. M., Leitch, I. J., Patel, S., Pendharkar, A., and Knight, C. A. (2008). Genome size is a strong predictor of cell size and stomatal density in angiosperms. New Phytol. 179, 975–986. doi: 10.1111/j.1469-8137.2008.02528.x
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bubunenko, M. G., Schmidt, J., and Subramanian, A. R. (1994). Protein substitution in chloroplast ribosome evolution. A eukaryotic cytosolic protein has replaced its organelle homologue (L23) in spinach. J. Mol. Biol. 240, 28–41. doi: 10.1006/jmbi.1994.1415
Cai, Z., Guisinger, M., Kim, H. G., Ruck, E., Blazier, J. C., McMurtry, V., et al. (2008). Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 67, 696–704. doi: 10.1007/s00239-008-9180-7
Cheng, C., Fan, Y., Tang, Y., Zhang, K., Joshi, D. C., Jha, R., et al. (2020). Fagopyrum esculentum ssp. ancestrale-a hybrid species between diploid F. cymosum and F. esculentum. Front. Plant Sci. 11:1073. doi: 10.3389/fpls.2020.01073
Cho, K. S., Yun, B. K., Yoon, Y. H., Hong, S. Y., Mekapogu, M., Kim, K. H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS One 10:e0125332. doi: 10.1371/journal.pone.0125332
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Cosner, M. E., Raubeson, L. A., and Jansen, R. K. (2004). Chloroplast DNA rearrangements in Campanulaceae: phylogenetic utility of highly rearranged genomes. BMC Evol. Biol. 4:27. doi: 10.1186/1471-2148-4-27
Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., and Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9:689. doi: 10.3389/fpls.2018.00689
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, L. Z., Liu, Y. L., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Bio. 2:278. doi: 10.1038/s42003-019-0531-2
Gregory, T. R. (2005). DNA barcoding does not compete with taxonomy. Nature 434:1067. doi: 10.1038/4341067b
Gross, M. H. (1913). Remarques sur les polygonees del’Asie orientale. Bull. Torrey Bot. Club 23, 7–32.
Guisinger, M. M., Chumley, T. W., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2010). Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J. Mol. Evol. 70, 149–166. doi: 10.1007/s00239-009-9317-3
He, L., Qian, J., Li, X., Sun, Z., Xu, X., and Chen, S. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22:249. doi: 10.3390/molecules22020249
Hou, L. L., Zhou, M. L., Zhang, Q., Qi, L. P., Yang, B. X., Tang, Y., et al. (2015). Fagopyrum luojishanense, a new species of polygonaceae from sichuan, China. Novon J. Bot. Nomenclat. 24, 22–26. doi: 10.3417/2013047
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huang, Y., Wang, J., Yang, Y., Fan, C., and Chen, J. (2017). Phylogenomic analysis and dynamic evolution of chloroplast genomes in salicaceae. Front. Plant Sci. 8:1050. doi: 10.3389/fpls.2017.01050
Ikeuchi, M., Eggers, B., Shen, G. Z., Webber, A., Yu, J. J., Hirano, A., et al. (1991). Cloning of the psbK gene from Synechocystis sp. PCC 6803 and characterization of photosystem II in mutants lacking PSII-K. J. Biol. Chem. 266, 11111–11115.
Jansen, R. K., and Ruhlman, T. A. (2012). Plastid Genomes of Seed Plants, Genomics of Chloroplasts, and Mitochondria. Dordrecht: Springer. 103–126. doi: 10.1007/978-94-007-2920-9_5
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jarvis, P., and Soll, J. (2001). Toc, Tic, and chloroplast protein import. Biochimica et biophysica acta. 1541, 64–79. doi: 10.1016/s0167-4889(01)00147-1
Jin, S., and Daniell, H. (2015). The engineered chloroplast genome just got smarter. Trends Plant Sci. 20, 622–640. doi: 10.1016/j.tplants.2015.07.004
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. Cell Mol. Biol. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Lee, H. L., Jansen, R. K., Chumley, T. W., and Kim, K. J. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 24, 1161–1180. doi: 10.1093/molbev/msm036
Leister, D. (2003). Chloroplast research in the genomic age. Trends Genet. 19, 47–56. doi: 10.1016/s0168-9525(02)00003-3
Li, B., Lin, F., Huang, P., Guo, W., and Zheng, Y. (2020). Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 53:21. doi: 10.1186/s40659-020-00289-0
Li, X., Li, Y., Zang, M., Li, M., and Fang, Y. (2018). Complete chloroplast genome sequence and phylogenetic analysis of quercus acutissima. Int. J. Mol. Sci. 19:2443. doi: 10.3390/ijms19082443
Li, Y., Chen, X., Wu, K., Pan, J., Long, H., and Yan, Y. (2020). Characterization of simple sequence repeats (SSRs) in ciliated protists inferred by comparative genomics. Microorganisms 8:662. doi: 10.3390/microorganisms8050662
Li, Y., Dong, Y., Liu, Y., Yu, X., Yang, M., and Huang, Y. (2021). Comparative analyses of euonymus chloroplast genomes: genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within celastrineae. Front. Plant Sci. 11:593984. doi: 10.3389/fpls.2020.593984
Liang, H., Zhang, Y., Deng, J., Gao, G., Ding, C., Zhang, L., et al. (2020). The complete chloroplast genome sequences of 14 curcuma species: insights into genome evolution and phylogenetic relationships within zingiberales. Front. Genet. 11:802. doi: 10.3389/fgene.2020.00802
Little, M. C., and Hallick, R. B. (1988). Chloroplast rpoA, rpoB, and rpoC genes specify at least three components of a chloroplast DNA-dependent RNA polymerase active in tRNA and mRNA transcription. J. Biol. Chem. 263, 14302–14307.
Liu, M., Li, X. W., Liao, B. S., Luo, L., and Ren, Y. Y. (2019). Species identification of poisonous medicinal plant using DNA barcoding. Chin. J. Nat. Med. 17, 585–590. doi: 10.1016/S1875-5364(19)30060-3
Liu, J. L., Tang, Y., Xia, M. Z., Shao, J. R., Cai, G. Z., Luo, Q., et al. (2008). Fagopyrum crispatifolium a new species of Polygonaceae from Sichuan, China. J. Syst. Evol. 46, 929–932.
Logacheva, M. D., Samigullin, T. H., Dhingra, A., and Penin, A. A. (2008). Comparative chloroplast genomics and phylogenetics of Fagopyrum esculentum ssp. ancestrale - A wild ancestor of cultivated buckwheat. BMC Plant Biol. 8:59. doi: 10.1186/1471-2229-8-59
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). Organellar Genome DRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Louwaars, N. P. (2018). Plant breeding and diversity: A troubled relationship? Euphytica: Netherlands J. Plant Breed. 214:114. doi: 10.1007/s10681-018-2192-5
Ma, K. H., Kim, N. S., Lee, G. A., Lee, S. Y., Lee, J. K., Yi, J. Y., et al. (2009). Development of SSR markers for studies of diversity in the genus Fagopyrum. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 119, 1247–1254. doi: 10.1007/s00122-009-1129-8
Martin, G. E., Rousseau-Gueutin, M., Cordonnier, S., Lima, O., Michon-Coudouel, S., Naquin, D., et al. (2014). The first complete chloroplast genome of the Genistoid legume Lupinus luteus: evidence for a novel major lineage-specific rearrangement and new insights regarding plastome evolution in the legume family. Ann. Bot. 113, 1197–1210. doi: 10.1093/aob/mcu050
Mohanta, T. K., Khan, A. L., Hashem, A., Allah, E., Yadav, D., and Al-Harrasi, A. (2019). Genomic and evolutionary aspects of chloroplast tRNA in monocot plants. BMC Plant Biol. 19:39. doi: 10.1186/s12870-018-1625-6
Nagatomo, Y., Usui, S., Ito, T., Kato, A., Shimosaka, M., and Taguchi, G. (2014). Purification, molecular cloning and functional characterization of flavonoid C-glucosyltransferases from Fagopyrum esculentum M. (buckwheat) cotyledon. Plant J. Cell Mol. Biol. 80, 437–448. doi: 10.1111/tpj.12645
Neethirajan, S., Hirose, T., Wakayama, J., Tsukamoto, K., Kanahara, H., and Sugiyama, S. (2011). Karyotype analysis of buckwheat using atomic force microscopy. Microsc Microanal. 17, 572–577. doi: 10.1017/S1431927611000481
Neuhaus, H. E., and Emes, M. J. (2000). Nonphotosynthetic metabolism in plastids. Ann. Rev. Plant Physiol. Plant Mol. Biol. 51, 111–140. doi: 10.1146/annurev.arplant.51.1.111
Ohnishi, O. (1995). Discovery of new Fagopyrum species and its implication for the studies of evolution of Fagopyrum and of the origin of cultivated buckwheat. Proc. Intl. Symp. Buckwheat 1995, 175–190.
Ohnishi, O. (1998). Search for the wild ancestor of buckwheat I Description of new Fagopyrum (Polygonaceae) species and their distribution in China. Fagopyrum 15, 18–28.
Ohnishi, O., and Matsuoka, Y. (1996). Search for the wild ancestor of buckwheat ii. taxonomy of Fagopyrum (polygonaceae) species based on morphology, isozymes and cpdna variability. Genes Genetic Syst. 71, 383–390.
Ohsako, T., and Ohnishi, O. (1998). New Fagopyrum species revealed by morphological and molecular analyses. Genes Genetic Syst. 73, 85–94.
Ohsako, T., Yamane, K., and Ohnishi, O. (2002). Two new Fagopyrum (po1ygonaceae) species F. gracilipedoides and F. jinshaense from Yunnan. Genes Genetic Syst. 77, 399–408.
Palmer, J. D., Jansen, R. K., Michaels, H. J., Chase, M. W., and Manhart, J. R. (1988). Chloroplast DNA variation and plant phylogeny. Ann. Missouri. Bot. Garden 75, 1180–1206.
Park, K. T., and Park, S. (2021). Phylogenomic Analyses of Hepatica Species and Comparative Analyses Within Tribe Anemoneae (Ranunculaceae). Front. Plant Sci. 12:638580. doi: 10.3389/fpls.2021.638580
Peden, J. F. (2000). Analysis of codon usage. Univ. Nottingham. 90, 73–74. doi: 10.1006/expr.1997.4185
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruhlman, T. A., and Jansen, R. K. (2014). The plastid genomes of flowering plants. Methods Mol. Biol. 1132, 3–38. doi: 10.1007/978-1-62703-995-6_1
Saski, C., Lee, S. B., Daniell, H., Wood, T. C., Tomkins, J., Kim, H. G., et al. (2005). Complete chloroplast genome sequence of Glycine max and comparative analysis with other legume genomes. Plant Mol. Biol. 59, 309–322. doi: 10.1007/s11103-005-8882-0
Shao, J. R., Zhou, M. L., Zhu, X. M., Wang, D. Z., and Bai, D. Q. (2011). Fagopyrum wenchuanense and Fagopyrum qiangcai, two new species of polygonaceae from sichuan, china. Novon 21, 256–261.
Sharma, R., and Jana, S. (2002). Species relationships in Fagopyrum revealed by PCR-based DNA fingerprinting. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik. 105, 306–312. doi: 10.1007/s00122-002-0938-9
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049.
Song, Y., Wang, S., Ding, Y., Xu, J., Li, M. F., Zhu, S., et al. (2017). Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 7:3427. doi: 10.1038/s41598-017-02083-7
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stewad, A. N. (1930). The Polygoneae of eastern Asia. Contrib. Gray Herbarium Harvard Universit. 88, 1–129.
Sugimoto, I., and Takahashi, Y. (2003). Evidence that the PsbK polypeptide is associated with the photosystem II core antenna complex CP43. J. Biol. Chem. 278, 45004–45010. doi: 10.1074/jbc.M307537200
Tang, Y., Zhou, M. L., Bai, D. Q., Shao, J. R., Zhu, X. M., Wang, D. Z., et al. (2010). Fagopyrum pugense (Polygonaceae), a new species from Sichuan, China. Novon J. Bot. Nomenclature 20, 239–242.
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Tonti-Filippini, J., Nevill, P. G., Dixon, K., and Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. Cell Mol. Biol. 90, 808–818. doi: 10.1111/tpj.13491
Wang, C. L., Ding, M. Q., Zou, C. Y., Zhu, X. M., Tang, Y., Zhou, M. L., et al. (2017b). Comparative analysis of four buckwheat species based on morphology and complete chloroplast genome sequences. Sci. Rep. 7:6514. doi: 10.1038/s41598-017-06638-6
Wang, C. L., Li, Z. Q., Ding, M. Q., Tang, Y., Zhu, X., and Liu, J. (2017a). Fagopyrum longzhoushanense, a new species of Polygonaceae from Sichuan. China Phytotaxa. 291, 73–80.
Wanga, V. O., Dong, X., Oulo, M. A., Mkala, E. M., Yang, J. X., Onjalalaina, G. E., et al. (2021). Complete chloroplast genomes of Acanthochlamys bracteata (China) and Xerophyta (Africa) (Velloziaceae): comparative genomics and phylogenomic placement. Front. Plant Sci. 12:691833. doi: 10.3389/fpls.2021.691833
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Williams, A. M., Friso, G., van Wijk, K. J., and Sloan, D. B. (2019). Extreme variation in rates of evolution in the plastid Clp protease complex. Plant J. 98, 243–259. doi: 10.1111/tpj.14208
Wu, C. X., Zhai, C. C., and Fan, S. J. (2020). Characterization of the complete chloroplast genome of Rumex nepalensis (Polygonaceae). Mitochondrial DNA B Resour. 5, 2458–2459. doi: 10.1080/23802359.2020.1778568
Xu, C., Dong, W., Li, W., Lu, Y., Xie, X., Jin, X., et al. (2017). Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 8:15. doi: 10.3389/fpls.2017.00015
Yang, B., Li, L., Liu, J., and Zhang, L. (2020). Plastome and phylogenetic relationship of the woody buckwheat Fagopyrum tibeticum in the Qinghai-Tibet Plateau. Plant Divers. 43, 198–205. doi: 10.1016/j.pld.2020.10.001
Yang, Y., Zhang, Y., Chen, Y., Gul, J., Zhang, J., Liu, Q., et al. (2019). Complete chloroplast genome sequence of the mangrove species Kandelia obovata and comparative analyses with related species. PeerJ 7:e7713. doi: 10.7717/peerj.7713
Ye, N. G., and Guo, G. Q. (1992). Classification, origin and evolution of genus Fagopyrum in China. Taiyuan: Agricult. Publ. House 1992, 19–28.
Zhang, D., Gao, F., Li, W. X., Jakovlic, I., Zou, H., Zhang, J., et al. (2018). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, K., Fan, Y., Weng, W. F., Tang, Y., and Zhou, M. L. (2021a). Fagopyrum longistylum (Polygonaceae), a new species from Sichuan. China. Phytotaxa 482, 173–182.
Zhang, K., He, M., Fan, Y., Zhao, H., Gao, B., Yang, K., et al. (2021b). Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 22:23. doi: 10.1186/s13059-020-02217-7
Zhang, S., Jin, J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhang, T. (2020). The butterfly effect: natural variation of a chloroplast tRNA-modifying enzyme leads to pleiotropic developmental defects in rice. Plant Cell 32, 2073–2074. doi: 10.1105/tpc.20.00342
Zhang, Y., and Chen, C. (2018). The complete chloroplast genome sequence of the medicinal plant Fagopyrum dibotrys (Polygonaceae). Mitochondrial DNA Part B Res. 3, 1087–1089. doi: 10.1080/23802359.2018.1483761
Zhao, K., Li, L., Lu, Y., Yang, J., Zhang, Z., Zhao, F., et al. (2020). Characterization and comparative analysis of two rheum complete chloroplast genomes. Biomed. Res. Int. 2020, 1–11. doi: 10.1155/2020/6490164
Zhao, K., Li, L., Quan, H., Yang, J., Zhang, Z., Liao, Z., et al. (2021). Comparative Analyses of Chloroplast Genomes From 14 Zanthoxylum Species: Identification of Variable DNA Markers and Phylogenetic Relationships Within the Genus. Front. Plant Sci. 11:605793. doi: 10.3389/fpls.2020.605793
Zhao, Z., Gao, A., and Huang, J. (2019). Sequencing and analysis of chloroplast genome of Clausena lansium (lour.) Skeels. Anhui. Agric. Sci. 47, 115–118. doi: 10.3969/j.issn.0517-6611.2019.11.032
Zhou, M. L., Tang, Y., Deng, X. Y., Ruan, C., Tang, Y. X., and Wu, Y. M. (2018). Classification and Nomenclature of Buckwheat Plants, Buckwheat Germplasm in the World. Cambridge: Academic Press. 9–20.
Zhou, T., Ruhsam, M., Wang, J., Zhu, H., Li, W., Zhang, X., et al. (2019). The complete chloroplast genome of Euphrasia regelii, Pseudogenization of ndh genes and the phylogenetic relationships within Orobanchaceae. Front. Genet. 10:444. doi: 10.3389/fgene.2019.00444
Zhu, G., Li, W., Wang, G., Li, L., Si, Q., Cai, C., et al. (2019). Genetic basis of fiber improvement and decreased stress tolerance in cultivated versus semi-domesticated upland cotton. Front. Plant Sci. 10:1572. doi: 10.3389/fpls.2019.01572
Keywords: Fagopyrum, Polygonaceae, chloroplast genome, comparative analysis, phylogenetic relationship
Citation: Fan Y, Jin Y, Ding M, Tang Y, Cheng J, Zhang K and Zhou M (2021) The Complete Chloroplast Genome Sequences of Eight Fagopyrum Species: Insights Into Genome Evolution and Phylogenetic Relationships. Front. Plant Sci. 12:799904. doi: 10.3389/fpls.2021.799904
Received: 22 October 2021; Accepted: 18 November 2021;
Published: 15 December 2021.
Edited by:
Wei Hu, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, ChinaReviewed by:
Yun-peng Du, Beijing Academy of Agriculture and Forestry Sciences, ChinaCopyright © 2021 Fan, Jin, Ding, Tang, Cheng, Zhang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Cheng, Y2hlbmdqaWFucGluZzYzQHFxLmNvbQ==; Kaixuan Zhang, zhangkaixuan@caas.cn; Meiliang Zhou, zhoumeiliang@caas.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.