 Isabel García-García1*†
Isabel García-García1*† Belén Méndez-Cea1*†
Belén Méndez-Cea1*† David Martín-Gálvez2
David Martín-Gálvez2 José Ignacio Seco3
José Ignacio Seco3 Francisco Javier Gallego1
Francisco Javier Gallego1 Juan Carlos Linares3
Juan Carlos Linares3- 1Departamento de Genética, Fisiología y Microbiología, UD Genética, Facultad de CC Biológicas, Universidad Complutense de Madrid, Madrid, Spain
- 2Departamento de Biodiversidad, Ecología y Evolución, UD Zoología, Facultad de CC Biológicas, Universidad Complutense de Madrid, Madrid, Spain
- 3Departamento de Sistemas Físicos, Químicos y Naturales, Universidad Pablo de Olavide, Seville, Spain
Forest tree species are highly vulnerable to the effects of climate change. As sessile organisms with long generation times, their adaptation to a local changing environment may rely on epigenetic modifications when allele frequencies are not able to shift fast enough. However, the current lack of knowledge on this field is remarkable, due to many challenges that researchers face when studying this issue. Huge genome sizes, absence of reference genomes and annotation, and having to analyze huge amounts of data are among these difficulties, which limit the current ability to understand how climate change drives tree species epigenetic modifications. In spite of this challenging framework, some insights on the relationships among climate change-induced stress and epigenomics are coming. Advances in DNA sequencing technologies and an increasing number of studies dealing with this topic must boost our knowledge on tree adaptive capacity to changing environmental conditions. Here, we discuss challenges and perspectives in the epigenetics of climate change-induced forests decline, aiming to provide a general overview of the state of the art.
Climate Change-Induced Tree Dieback and Forests Decline
Rising temperatures are exacerbating drought stress for trees at global scales, causing events of decline in several species (usually known as forest dieback; Allen et al., 2010). Climatic projections indicate that mean global temperatures are expected to rise at least 1.5°C until 2100 in response to anthropogenic emissions (IPCC, 2018). Such projections of warming indicate the duration and severity of droughts are expected to amplify, probably leading to widespread forest dieback and tree mortality, affecting tree species currently not assumed to be prone to drought stress, for instance in tropical or boreal forests, or already affected by drought stress, such as those of the Mediterranean forests (Allen et al., 2010). Even though several mechanisms have been described as physiological drivers of dieback, including hydraulic failure and carbon starvation (McDowell et al., 2016; Choat et al., 2018), we lack forecasting tools with sufficient predictive power, since the likelihood of drought-induced tree dieback and mortality is not straightforward (Trugman et al., 2021). This mismatch enhances the need to understand the processes that modulate contrasting adaptive capacity among trees within a given declining forest in order to be able to provide, for instance, adaptive management strategies. Epigenetic processes are known to be able to modulate the patterns of gene expression in plants (Rapp and Wendel, 2005), some being involved in physiological and morphological responses to drought (e. g., Zheng et al., 2013; Liang et al., 2014; Chwialkowska et al., 2016). As drought-induced tree mortality has far-reaching consequences that affect a wide-range of fields, from environmental conservation to climate change mitigation efforts, and it is expected to be more frequent under a climate-change scenario, we need to improve our knowledge about the degree to which epigenetic mechanisms are linked to mortality predictions (e. g., Sow et al., 2018; Amaral et al., 2020; González-Benito et al., 2020).
Trees are capable of coping with short drought periods by avoiding water loss. Thereby, several morphological and physiological adaptations have been developed by forest tree species, for instance variations in tracheids size (Carvalho et al., 2015), reduced leaf area, embedded stomatas, and quick stomata close (Sánchez-Salguero et al., 2015; Kijowska-Oberc et al., 2020). However, long-lasting and severe drought periods are predicted to occur more often, so these strategies might not be enough for their survival (IPCC, 2018).
Tree growth can be used as a marker to study forest tree response to environment alterations, as it is affected by drought stress. Dendrochronology studies have evinced growth decline during drought stress periods, which eventually leads to forest dieback (Camarero et al., 2015; Cailleret et al., 2017). Thus far, drought-induced forests dieback has been observed in several conifer species found in the Mediterranean region, like Abies pinsapo Boiss. (Linares et al., 2010), Abies alba Mill. (Macías et al., 2006; Camarero et al., 2011, 2015), and Cedrus atlantica (Endl.) Manetti ex Carrière (Linares et al., 2011). The Mediterranean basin is considered a climate change hotspot, being one of the most vulnerable areas in the world. As this region was an important refuge in the last glaciation, a large number of endemic and relict species can be found there. Hence, variations in the environment could result in death of these drought-sensitive species which will trigger a huge loss of biodiversity.
In addition, large-distributed forest tree species, like Pinus sylvestris L., often live under different environment conditions (Bose et al., 2020), so individuals must show variations that allow them to cope with those circumstances (Camarero et al., 2015; DeSoto et al., 2020). Studying not only endemic species but also largely distributed ones and identifying these differences could be key to understand why some individuals survive while others die (Cailleret et al., 2017).
Surprisingly, there is a lack of data about epigenomic changes in natural populations in spite of their potential role in adaptation to a rapidly changing environment. The most common studies compare individuals under drought conditions with regularly watered individuals, grown in greenhouses (e.g., Gourcilleau et al., 2010; Xu et al., 2018; Li et al., 2020). While this approach allows to accurately control the factors affecting the plants and makes the experiments replicable, it may lack realism, especially when working with long-lived perennial plants due to restriction of their rooting volume, which hinders the extrapolation of the results to natural populations (Gibson et al., 2004). Therefore, studying epigenetic variation in natural populations would be of great interest to understand how trees respond to climate change in the nature.
On summary, as said before, growth of forest tree species is usually slow and the upcoming climate variations will likely be faster than the speed of allele frequencies shift of forest trees, which emphasizes a likely role of epigenetic modifications as a more rapid way of adaptation.
Epigenetic Mechanisms Under a Climate Changing Scenario
Forest trees are sessile organisms which must cope with environment variations since they are not able to migrate to ensure their survival. Hence, they possess other abilities to compensate for their lack of mobility, such as phenotypic plasticity (Petit and Hampe, 2006). Phenotypic plasticity is the capacity to develop phenotypic modifications in response to multiple environmental factors, increasing the probability of surviving (West-Eberhard, 2008). This mechanism is the result of molecular modifications that trigger changes in gene expression. One of the most important processes involved in phenotypic plasticity is epigenetic regulation, which produces changes in genes expression without modifying the DNA sequence (Iwasaki and Paszkowski, 2014). In fact, phenotypic plasticity is one of the main biological processes that allows trees to respond to rapid variations, like climate change (Alberto et al., 2013). Climate change exacerbates drought stress for trees at global scales, causing stand-level growth decline and episodes of mortality (Allen et al., 2010; Anderegg et al., 2013). As drought-induced forests decline and mortality are reported at an unprecedented rate (Jump and Peñuelas, 2005), a particularly urgent scientific challenge is to understand epigenetic mechanics linking an environmental cue, here increasing drought stress, received by adult trees and its resulting effects on the phenotype of coming generations (Alberto et al., 2013).
In particular, conifers, a group of forest tree species which include ecologically and economically important genera like Pinus, Picea, and Abies, seem to be particularly drought-sensitive (McDowell et al., 2016; DeSoto et al., 2020) and prone to a high rate of epigenetic regulation due to their large genome sizes with high amounts of non-coding DNA (Yakovlev et al., 2012). The study of growth patterns in declining conifer forests suggests a lower capacity of resilience in stressed/dying trees. In fact, trees that died after drought were less resilient after previous droughts, relative to surviving conspecific individuals. Specifically, drought-related mortality risk in conifers has been associated with a low capacity to recover after drought (DeSoto et al., 2020), whereas the epigenetic mechanisms involved on this recovery mechanisms remain largely unknown. Here, we hypothesize that epigenetic mechanisms are likely related to the fate of trees that survive after drought, while some conspecifics died.
Three relevant epigenetic mechanisms have been described: DNA methylation, histone modifications and non-coding RNAs (ncRNAs). Of these, DNA methylation is the most studied one in plants, both model and non-model (Zhang et al., 2018). Two main reasons are responsible of this fact. In one hand, DNA methylation has been reported to be implicated in significant biological processes, like silencing of Transposable Elements (TEs), gene transcription regulation, plant development, and response to biotic and abiotic stress (Zhang et al., 2018). On the other hand, several genomic tools have been developed to easily study at a global level the methylations present in the DNA. Therefore, in this review we will pay special attention to this epigenetic mark.
Regarding histone modifications, these epigenetic marks are involved in chromatin compaction, playing a role in determining its state, which can be decondensed (euchromatin) or condensed (heterochromatin). Euchromatin, also known as open chromatin, is characterized by the presence of a high number of genes, which are usually susceptible to transcription as they are accessible to transcription machinery, while heterochromatin, also known as closed chromatin, is characterized by repetitive and silenced sequences (Amaral et al., 2020). Acetylations, methylations, phosphorilations, and ubiquitylations are some of the panoply of modifications that histones can be subjected to Bannister and Kouzarides (2011), creating patterns that are associated to different transcriptional states. Unfortunately, these modifications are numerous and challenging to analyze in trees as standard chromatin immunoprecipitation (ChIP) procedures are hard to apply to woody cells and tissues (Li et al., 2014). Thus, very few studies regarding histone modifications in trees have been published to date.
Non-coding RNAs are also rather relevant as an epigenomic mechanism, as they are mobile within and between cells and are not lost during cell divisions, unlike the majority DNA methylations and histone modifications (Yakovlev et al., 2012). They are classified by their length: if they have more than 200 nucleotides, they are called as long ncRNAs (lncRNAs) whereas if they have less than 200 nucleotides, they are called short ncRNAs. These short ncRNAs include micro RNAs (miRNAs), short interfering RNAs (siRNAs), and Piwi-interacting RNAs (piRNAs), and they are involved in genomic imprinting, X-chromosome inactivation, repression of gene transcription, and transposon repression (Wei et al., 2016). Therefore, given the elevated content of TEs in conifer genomes, ncRNAs stand as a major epigenetic mark that should be studied in depth.
Lastly, DNA methylation positions itself as a highly relevant epigenetic mark in conifers due to their large content in TEs and its role in silencing them. In addition, there are numerous studies that demonstrate its participation in the control of gene expression in a large number of traits, in plants in general and in conifers in particular, which will be addressed later. DNA methylation is a covalent change which consists in the addition of a methyl group to a cytosine residue, in most cases to its fifth carbon position, and can be, in some cases, inherited by the offspring. In plants, DNA methylation happens not only in CpG sites but also in CHG and CHH contexts, where H can be A, T, or C (Law and Jacobsen, 2010). CpG sites are the most frequent mark in/near protein-coding gene sequences (Ashapkin et al., 2020) especially of those with constitutive expression. Although this methylation is known to affect gene expression, its role is still unclear, as it can lead to both silencing and increased expression (Zemach and Ziberman, 2010). Regarding CHG and CHH sites, different studies have highlighted that they are mainly present in repetitive sequences and TEs, both abundant in tree genomes, and their methylation usually reduces or even blocks DNA transcription, allowing the maintenance of the genome integrity (Zemach et al., 2010a; Bewick and Schmitz, 2017).
It has been proved that DNA methylation is responsible of phenotypic variation in plants (Martin et al., 2009; Mougninot et al., 2021; Zhao et al., 2021). Considering that different types of stress, such as abiotic stress, can trigger methylation, eventually leading to variation of gene expression, this field of research has become a useful tool for understanding how tree species deal with the environment variations. Several studies have highlighted that epigenetics seems to act like a memory process to respond to different environmental stresses, which is called “stress memory” (Kinoshita and Seki, 2014). Thus, a new question arises: could it be possible to inherit this stress-induced DNA methylations? Most of the time, epigenetic marks are erased to start over in the progeny, however it is possible for this reset to be incomplete, allowing some marks to pass on to the offspring. Moreover, it is known that some germline cells come from somatic cells in plants, so epigenetic marks that appear in those cells could be passed on to the offspring. Hence, epigenetic marks can be transmitted to the offspring by meiosis in some cases. Studies carried out with Arabidopsis thaliana L. (Cervera et al., 2002; Boyko et al., 2010; Lang-Mladek et al., 2010) or Linaria vulgaris Mill. (Cubas et al., 1999) evidenced epigenetic modifications related to environment variations can be inherited. However, the mechanisms involved in this process are not well understood to date.
All in all, epigenetic modifications could be a rapid way to achieve short-term adaptations, since epigenetic marks bring new phenotypes which might affect fitness and, thus, the underlying evolutionary processes. Particularly, it could be key for forest trees survival, as they are sessile long-lived organism with long generation times, so the possible beneficial effects of natural selection on them are limited and slow and will soon be surpassed by the effects of climate change.
Techniques for Epigenomic Analysis
Over the years, different techniques have been developed to study DNA methylation. Their first requirement is high-quality DNA, which highlights the importance of selecting an appropriated method for sample conservations during and after each collection, as well as an extraction method as efficient as possible for each type of samples. Once the DNA is obtained, there is a wide variety of methods than can be used to analyze its methylation patters (Table 1). The most remarkable ones are shown below.
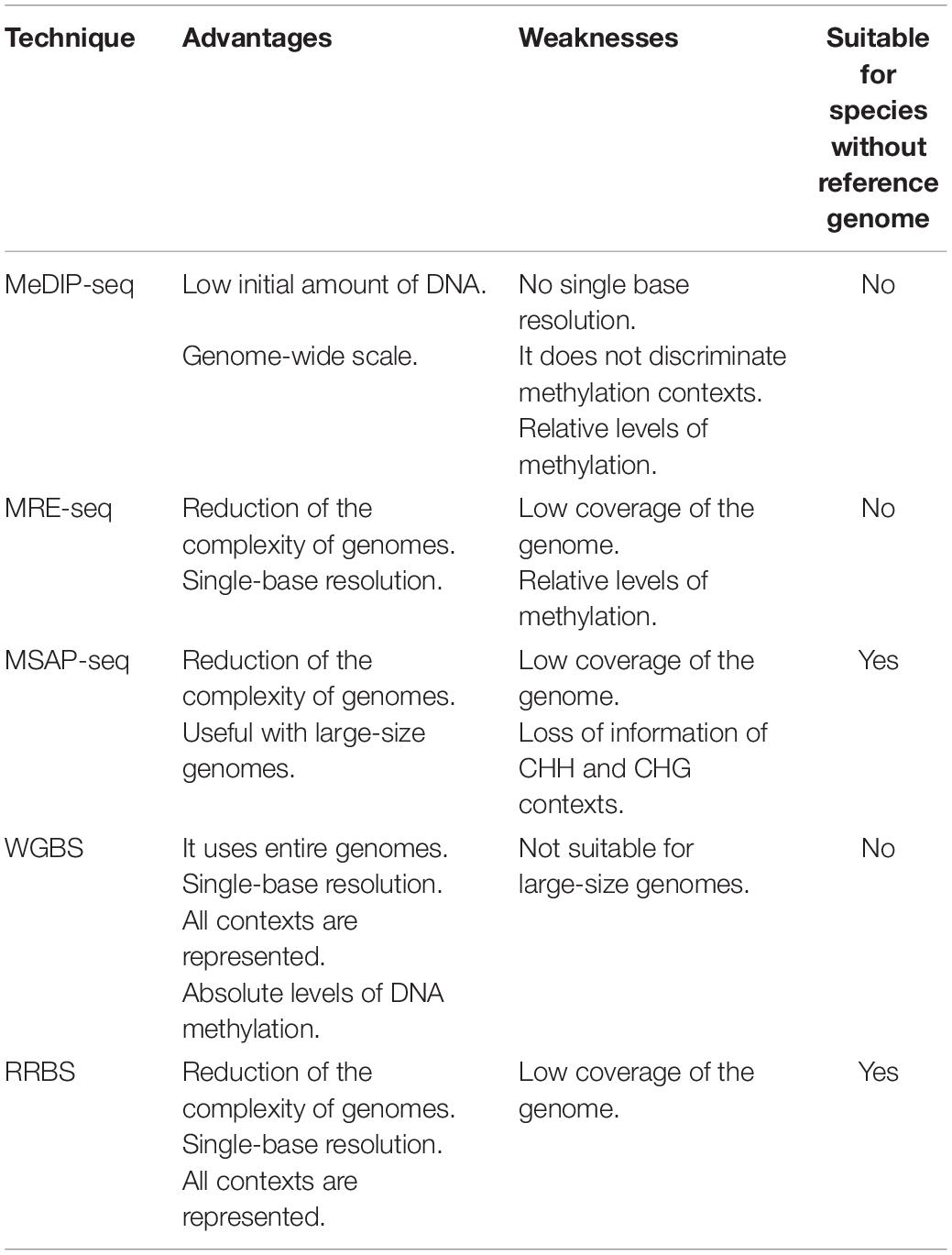
Table 1. Summary of the advantages and weaknesses of the most relevant DNA methylation techniques and their suitability for species without reference genomes.
At the beginning, methods based on antibodies affinity to methylcytosine residues were used. One of them is the well-known methyl-DNA immunoprecipitation (MeDIP) technique, which captures the methylated DNA fragments with an antibody against methylated cytosines (Thu et al., 2009). Then a new approach of this technique, called MeDIP-seq, was developed (Zhao et al., 2014). The novelty of this method consisted in a final sequencing step based in next-generation sequencing (NGS). Both techniques require a previous DNA sonication step to obtain random DNA fragments. MeDIP-seq is frequently used in combination with MRE-seq (methylation sensitive restriction enzyme sequencing) (Maunakea et al., 2010). MRE-seq consists of a DNA fragmentation using several restriction enzymes which are methylation sensitive followed by a final sequencing step. Although MeDIP-seq and MRE-seq can be used independently, their combination can increase the accuracy of DNA-methylation studies (Li et al., 2015).
Subsequently, an improvement based on NGS allowed to modify the methyl-sensitive amplified polymorphism (MSAP) technique, giving rise to MSAP-seq. Originally, the MSAP technique appeared as a modification of the amplified fragment length polymorphism method (AFLP). This technique was described by Reyna-López et al. (1997) in fungi and then Xiong et al. (1999) adapted it to work with plants. MSAP starts with a genome fragmentation using restriction enzymes with differential sensitivity to cytosine methylation. MspI and HpaII are one of the enzyme isoschizomer pairs used in MSAP. Both recognize the same target 5’-CCpGG-3’, but HpaII will not cleave if the inner cytosine residue is methylated. Hence, this pair is used to identify differences in methylation marks between DNA samples in CpG contexts mainly. Following the cleavage step, a PCR amplification is carried out with the aim to obtain an overrepresentation of previously obtained fragments. Then, amplicons are separated and visualized via gel electrophoresis. MSAP-seq replaces this last sept by a sequencing one. This method was first described by Chwialkowska et al. (2016) in barley and was later improved to work with large genome species (Chwialkowska et al., 2017). It allows studies in non-model species with lack of a reference genome. However, restriction enzymes seem to cut less efficiently in CHH and CHG contexts than in CpG islands, which may lead to mistakes in methylation calling, which in turn might imply a loss in methylated states. This could be a remarkable weakness of this technique considering that the larger genome size is, the greater amount of cytosine methylations there is in the other contexts.
Finally, new approaches based on sodium bisulfite treatments have been developed to study DNA methylation, since bisulfite sequencing was developed in 1992 (Frommer et al., 1992). Bisulfite methods are useful to study genome-wide methylation and provide high single-base resolution. These techniques convert unmethylated cytosines to uracils, as methylated cytosines are protected against the deamination effect produced by the bisulfite treatment. After that, uracils are turned into thymines in a PCR amplification. Then, these changes can be easily identified by sequencing. To date, there are two bisulfite techniques: whole genome bisulfite sequencing (WGBS) and reduced representation bisulfite sequencing (RRBS), which is subdivided in three protocols based on RAD-seq (Baird et al., 2008) and genotyping by sequencing (GBS; Elshire et al., 2011) techniques: epiRADseq (Schield et al., 2016), bsRADseq (Trucchi et al., 2016), and epiGBS (van Gurp et al., 2016).
WGBS works with the entire genome, while the RRBS method targets specific regions. In RRBS methylation sensitive restriction enzymes are used to fragment the whole genome, and then DNA fragments are filtered by size. The characteristics of WGBS make it suitable for studies with model organisms with short length genome size and for species with a reference genome (Paun et al., 2019). On the other hand, the genome fragmentation carried out with RRBS methods constitutes a useful tool to reduce the complexity of genomes (Davey et al., 2011), which allows to work with large-size genomes, with or without a reference genome.
Although this review is focused on DNA methylation, here is a brief description of other techniques used to study the remaining epigenetic marks: histone modifications and ncRNAs.
One of the most common techniques to study in vivo histone modifications is chromatin immunoprecipitation (ChIP). First, samples are treated with formaldehyde to cross-link the chromatin interactions (protein–protein and protein–DNA). Then, small fragments are obtained by sonication or nuclease digestion. Next, specific antibodies are used to retain the DNA-histone modification complex of interest. Finally, different methods can be carried out to determine these regions such as PCR, real-time PCR, Southern blot, and Western blot. The development of NGS techniques has led to the addition of a final sequencing step to the ChIP protocol, creating the technique known as ChIP-seq. However, the presence of polysaccharide and phenolic compounds in plants may difficult these experiments, that could turn them into a challenge in some cases (Muhammad et al., 2020).
The most common technique used to study ncRNA sequences is RNA-seq (Wang et al., 2009). Then, sequencing data can be combined with RT-qPCRs to determine expression levels, or with fluorescence in situ hybridization (FISH) experiments to localize the ncRNAs in a cellular context, depending on the aim of the study. An in silico approach consists in looking for previously described or computationally predicted ncRNAs in the genomic region under study. Several databases of ncRNAs sequences are available online, such as CANTATAdb 2.01 (Szcześniak et al., 2015) which has over 200,000 lncRNAs from 39 plant species from which 2 are trees. As usual, forest trees are poorly represented in this kind of databases, mainly due to the lack of reference genomes.
In Search of a Reference
Forest trees span a huge range of genome sizes, including species with less than 1 Gb, such as the Populus genus, to the conifer giga-genomes, whose size varies from ∼6.5 to ∼37 Gb, being ∼15 Gb the most common 1C-value (Ahuja and Neale, 2005). Compared to the ∼0.6 Gb average of most angiosperms (Dodsworth et al., 2015), conifer genomes are remarkably large and, therefore, constitute a challenging work scenario for genomic and epigenomic studies. In addition, a great number of experimental techniques and bioinformatic pipelines are designed work with the 3.3 Gb-long human genome, making it difficult to adapt them to genomes which are on average five times bigger. For instance, techniques aimed at sequencing complete genomes, such as WGBS, might not be suitable for giga-genomes as optimal depth and coverage values will be hard to achieve.
To complicate matters further, approximately 75% of conifer genomes consists of highly repetitive DNA, mainly represented by non-coding DNA, such as mobile elements, along with rDNA repeat units (Ahuja and Neale, 2005). In addition, gene duplications have created large gene families, which are not necessarily found in certain regions but dispersed across the genomes (Kinlaw and Neale, 1997).
On the other hand, luckily, polyploidy is not a common phenomenon in conifers, and it has only been reported in the Cupressaceae family (Ahuja, 2005). All in all, however, the global genetic and, thus, epigenetic landscapes of these tree species do not provide a research-friendly environment.
While many plant genomes are being sequenced and assembled every year due to the improvement of next generation sequencing techniques and their decrease in cost (Mackay et al., 2012), few conifer reference genomes are available. The first gymnosperm giga-genome ever published was that of Picea abies (L.) H. Karst. (Nystedt et al., 2013). Since then, six more conifer genomes have been sequenced: Pinus taeda L. (Neale et al., 2014), Picea glauca (Moench) Voss (Warren et al., 2015), Pinus lambertiana Douglas (Stevens et al., 2016), Pseudotsuga menziesii (Mirb.) Franco (Neale et al., 2017), Larix sibirica Ledeb. (Kuzmin et al., 2019), and Abies alba Mill. (Mosca et al., 2019). The N50 statistic is used as an indicator for genome assembly quality. It is defined as the length of the contig that reaches 50% of the total genome length, being the contigs sorted by length in descending order. Therefore, the higher this value is, the better the assembly is, as the median contig size will be longer. The total length of the previously mentioned genomes ranges from ∼12 to ∼32 Gb, with N50 scaffold sizes between ∼5 to ∼2,500 Kb, which shows that further refinement is still needed. Nevertheless, over 45,000 genes have been successfully annotated in each of the cited examples.
The recent upsurge in popularity and number of users of third generation sequencing technologies, also known as long-read sequencing, could provide a substantial boost to the improvement of the quality of the existing reference genomes. Long reads, alone or in combination with short reads, have a high potential for assembling and re-assembling genomes (Amarasinghe et al., 2020). The reads obtained from third generation sequencing methods, such as PacBio single-molecule real-time (SMRT) and Oxford Nanopore, constitute a less fragmented representation of large genomes, facilitating their assembly in longer scaffolds. For instance, the quality of the reference genome of barley (Hordeum vulgare L.), which is not a forest tree but whose genome shares some characteristics with the conifer ones, such as being highly repetitive and ∼5 Gb-long, was superior for long-read assemblies than for the one based on short reads, obtaining higher contig N50 values (Mascher et al., 2021).
On the other hand, sequencing of transcriptomes is more extensive in conifers, due to their notably reduced size compared to whole genomes, which reduces costs and increases the probability of capturing the majority of the sequences. The One Thousand Plant Transcriptomes Initiative database2 collects over 1,300 transcriptomes of green plants, of which 76 belong to 72 different conifer species, coming from diverse tissue samples, including leaves, branches, and young shoots. Hardwood Genomics Project3 and TreeGenes4 are two other examples of databases specialized in trees that include transcriptomes as well as genomes, annotations and genetic markers.
However, the information coming merely from transcriptome sequencing is clearly insufficient to address the study of epigenetic mechanisms in conifers. Some of the most common epigenomic methodologies, such as identification of differentially methylated regions (DMRs) and differentially methylated promoters (DMPs), need the information of the sequences surrounding the coding regions. Then, the transcripts identified in transcriptomic experiments are useless without a reference to provide their genomic context.
Regarding complete methylomes, it has been described that CpG and CHG methylation is positively correlated with genome size (Ausin et al., 2016; Takuno et al., 2016), and high methylation levels are also known to be characteristic of genomes with a high content of transposable elements, as they constitute the main target of DNA methylation (Baduel and Colot, 2021). The lack of methylation maps of conifers is therefore not surprising, given the vast amount of data that needs to be analyzed to create them. Withal, the Norway spruce (P. abies) DNA methylation map was successfully obtained (Ausin et al., 2016). As previously mentioned, the genome of this species is fully sequenced, which highlights the importance of having reference genomes in order to be able to conduct this type of high throughput epigenetic studies.
Bioinformatic Tools
Bioinformatic pipelines for the analysis of DNA methylation data obtained after bisulfite treatments consist of four main steps: quality check and read trimming, read mapping, methylation calling, and identification of differentially methylated regions (Wreczycka et al., 2017).
Quality check and trimming of raw reads can be performed with well-known software, including FastQC (Andrews, 2010), and Trimmomatic (Bolger et al., 2014) and Cutadapt (Martin, 2011), respectively. Generally, these steps include removing adapter sequences, filtering out low-quality reads and trimming reads of low-quality bases. Specific bisulfite-sequencing tools are also available, such as BSeQC (Lin et al., 2013), which evaluates quality and automatically trims technical biases that may interfere in accurate methylation calling. For example, it can detect 5’ bisulfite conversion failure and remove the reads affected by this bias, which causes artificially high methylation rates at that end of the reads.
Bisulfite conversion rates should also be checked, especially if non-CG methylation levels are the focus of the study. Non-CG context, as previously mentioned, play an important role in silencing repetitive sequences and TEs, which are highly abundant in conifer genomes, so their study could be of great interest. In particular, CHH contexts can be quite problematic due to their low methylation levels (Takuno et al., 2016), which require a high bisulfite conversion rate to ensure that they are not underestimated. Thus, a spike-in control, such as unmethylated lambda phage DNA, could be useful to check that the bisulfite conversion is working at optimal levels.
Read mapping is a particularly challenging step. After bisulfite treatment, unmethylated cytosines are converted to uracils and then to thymines in the following PCR amplification, while methylated cytosines are not affected. This creates a need for special reference genomes, modified to take into consideration these nucleotide changes. In addition to the increased mapping searching sequence, asymmetric cytosine to thymine alignments and multiple CpG heterogeneous methylation must be taken into consideration by the mapping software (Xi and Li, 2009). Nonetheless, several bisulfite mapping tools have been developed, such as BSMAP (Xi and Li, 2009) and Bismark (Krueger and Andrews, 2011). Recently, Grehl et al. (2020) compared the efficiency and impact of the mapping results on the identification of DMRs of eight mappers and concluded that these two are the best options, as they showed the highest precision with the shortest run time and lowest memory requirements, respectively.
Both tools also provide methylation level files showing methylation calling status for each cytosine position. The coverage files produced by the Bismark methylation extractor function can be directly used as inputs for DMR-finding programs, such as the popular edgeR package (Robinson et al., 2010), or can easily be edited to fit other tools’ input requirements, such as DSS (Park and Wu, 2016) and swDMR (Wang et al., 2015).
In addition to these tools that allow users to perform each step of the analysis separately, others combine all the procedures in one pipeline. Bicycle (Graña et al., 2017) and SMAP (Gao et al., 2015) are some of the recommended all-in-one software for WGBS and RRBS data analysis, respectively (Rauluseviciute et al., 2019).
Despite these apparently wide variety of tool choices, there are several issues to take into consideration when it comes to analyzing high-throughput bisulfite sequencing data. First, the need of a good quality reference genome to perform the read mapping step, which is not a frequent situation, as previously explained. Besides, some tools, like swDMR, require an annotation file as input, whose quality relies on the genome quality itself. For instance, if a DMR falls in a region that is not described, likely due to genome fragmentation caused by large number of scaffolds or small scaffold sizes, this DMR will provide little information regarding its biological meaning. In addition, the amount of Cs that would need to be analyzed will in all probability be quite high in comparison to other non-conifer species. Therefore, the computational requirements in epigenomic experiments on forest tree species are likely to be very demanding, particularly in terms of RAM memory usage, storage space and run time.
Researchers that choose non-bisulfite-based techniques are likely to face the above-mentioned problems derived from genome size and lack of references, as they also include a next-generation sequencing step. Nonetheless, as in the case of bilsulfite-based methods, there are many bioinformatic tools specifically designed to work with this kind of data. The MeDUSA pipeline (Wilson et al., 2012) and Batman software (Down et al., 2008) are suitable for MeDIP-seq data analysis, while the MSEQER pipeline (Chwialkowska et al., 2017) is designed to work with MSAP-seq data.
Previous Studies
As it is said above, studies of epigenetic marks in plants are mainly focused on DNA methylation (Kurdyukov and Bullock, 2016). Nevertheless, the role of histone acetylations in root growth and development (Ma et al., 2016) and in photosynthesis (Li et al., 2017) has been described in different species of poplar, as well as that of miRNAs in growth and phosphorus nutrition (Schönberger et al., 2016). It is worth noting that this kind of studies have been carried out on poplars because they are considered a forest tree model organism. As mentioned above, poplar has a small genome (∼0.5 Gb) with a low proportion of repeated sequences. This genome has been fully sequenced and annotated, so its genomic resources are of high quality. Except in this particular case, histone modifications and ncRNAs remain mostly undescribed in conifer species and represent a major challenge to advance in the knowledge of epigenomics in trees.
The first plant methylome published was that of A. thaliana L. (Cokus et al., 2008), and it was developed using a large-scale bisulfite sequencing experiment, while the first conifer one, of P. abies, did not come until 2016 (Ausin et al., 2016).
Over the years, some studies have focused on changes in DNA methylation during normal plant development (e.g., Bitonti et al., 2002; Zemach et al., 2010b; Perrin et al., 2020) and heteroblastic development (Hasbún et al., 2016). Others are about phenotypic plasticity and stress memory (e.g., Herrera and Bazaga, 2013; Le Gac et al., 2018). What it is common to the vast majority of them is the use of model species, like A. thaliana (e.g., Korotko et al., 2021; Markus et al., 2021), and important commercial species such as crops (e.g., Feng et al., 2010; Wang et al., 2010; Karan et al., 2012; Bhanu et al., 2020; Merce et al., 2020) or fruit trees (e.g., Avramidou et al., 2015; Badad et al., 2021; Rothkegel et al., 2021).
The knowledge about forest tree epigenetics is scarce. Nevertheless, some studies related with epigenetic memory during embryogenesis have been developed on forest trees of species such as P. abies (Kvaalen and Johnsen, 2008; Yakovlev et al., 2014, 2016), P. glauca (Webber et al., 2005), or P. sylvestris (Dormling and Johnsen, 1992), and more studies are coming (e.g., Lafon-Placette et al., 2018; Vanden Broeck et al., 2018).
Furthermore, studies have generally focused on the involvement of DNA methylation in abiotic stresses response and adaptation in forest trees and other plants (Table 2). Modifications in response to water have been found in the methylome of Populus species (Gourcilleau et al., 2010; Raj et al., 2011), as well as variations in the methylation pattern of Pinus radiata D. Don. in response to heat stress (Lamelas et al., 2020). Again, it is remarkable the huge amount of poplar (Populus spp.) studies which have been carried out over the years, thanks to the availability of a quality reference genome and its wide phenotypic variation.
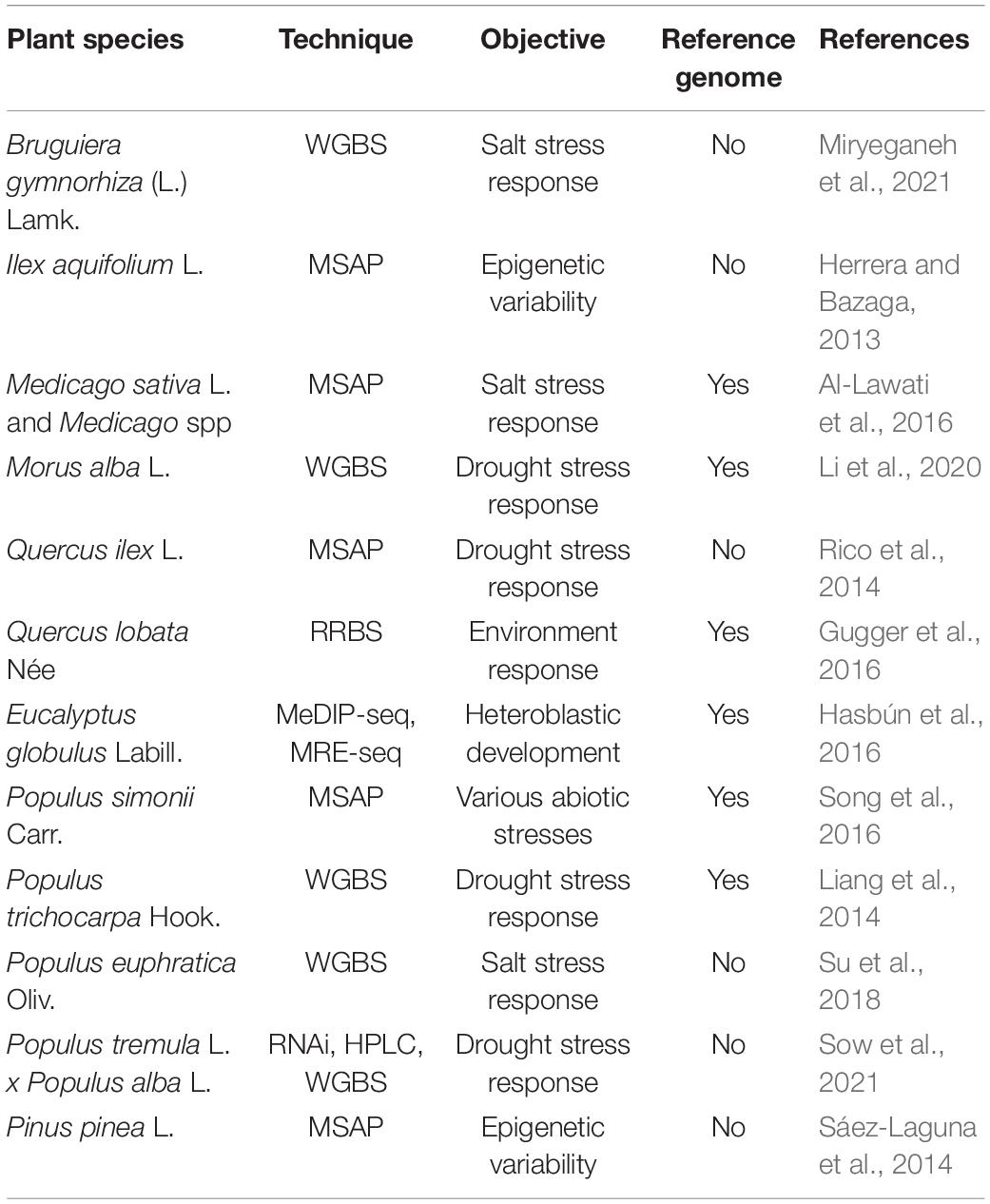
Table 2. Brief description of some DNA methylation studies carried out with the main objective of studying plant species response to several abiotic stresses.
Studies in natural populations are of particular importance to understand how epigenetic modifications can contribute to adaptation to the new climatic conditions caused by climate change. One of these studies was carried out with P. sylvestris (Alakärppä et al., 2018), which is a widely distributed conifer, likely to show variations in its epigenetic marks as it can be found under quite different environmental scenarios, and a link between local adaptation and environment condition, and modifications in DNA methylation has been suggested. Another work with samples of natural populations of Populus simonii Carr. (Ci et al., 2016) was conducted to analyze population epigenetic structure, which showed a substructure population associated to different methylation patterns.
In the literature, several reviews about epigenetics in plants and forest trees can be found (Paun et al., 2019; Amaral et al., 2020; Ashapkin et al., 2020; Zhao et al., 2021). Most of them are focused on the epigenetic response to specific abiotic stresses, such as drought, salt, or heavy metals, and they are useful as an introduction to this challenging field. On top of that, this review should prove valuable to researchers who wish to design an experiment to study methylation, as it covers the whole process: from choosing an appropriate technique to performing the bioinformatic analysis of the data.
To sum up, epigenomics open a research field to identify some of the processes related with phenotypic plasticity and its study will bring us a better understanding of adaptive forest tree response to a changing environment. Fortunately, substantial efforts are being made to improve DNA methylation techniques to make them suitable and cost-effective for non-model species, specifically with trees or species with large genomes (e.g., Chwialkowska et al., 2017; Pereira et al., 2020; Werner et al., 2020).
Conclusion
It is expected that the growth rates of forest tree species become on average slow, while the upcoming climate change trends become faster than the rate of allele frequencies shift of forest trees. This emphasizes the putative role of epigenetic modifications as a fast adaptive mechanism to cope with current warmer and drier climate scenario.
Phenotypic plasticity is the principal strategy to compensate for trees’ lack of ability to move when it comes to dealing with rapid environmental variations, such as ongoing climate change. It is based on modifications of genetic regulation which depend on epigenetic mechanisms such as histone modifications, DNA methylation and non-coding RNAs. Despite of the importance of these processes, epigenomic studies in forest tree species are challenging and scarce, mainly due to their complex genomes and lack of references.
Throughout this review, we have gathered enough information to give a general overview about the key points for methylation experiments design when dealing with forest trees. We have also highlighted opportunities for future advances in this framework and the relevance of dedicating efforts to improve the techniques for epigenomic analysis and the bioinformatic tools to make them suitable for large-size genomes.
Author Contributions
IG-G and BM-C wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
IG-G is recipient of a predoctoral fellowship (FPU18/01153), BM-C is recipient of a UCM predoctoral fellowship (CT42/18-CT43/18) and this work was supported by MICINN RTI2018-096884-B-C33.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ http://cantata.amu.edu.pl/
- ^ http://www.onekp.com
- ^ https://hardwoodgenomics.org/
- ^ https://treegenesdb.org/
References
Ahuja, M. R. (2005). Polyploidy in gymnosperms: Revisited. Silvae Genet. 54, 59–69. doi: 10.1515/sg-2005-0010
Ahuja, M. R., and Neale, D. B. (2005). Evolution of Genome Size in Conifers. Silvae Genet. 54, 126–137. doi: 10.1515/sg-2005-0020
Alakärppä, E., Salo, H. M., Valledor, L., Cañal, M. J., Häggman, H., and Vuosku, J. (2018). Natural variation of DNA methylation and gene expression may determine local adaptations of Scots pine populations. J. Exp. Bot. 69, 5293–5305. doi: 10.1093/jxb/ery292
Alberto, F. J., Aitken, S. N., Alia, R., Gonzalez-Martinez, S. C., Hanninen, H., Kremer, A., et al. (2013). Potential for evolutionary responses to climate change evidence from tree populations. Glob. Change Biol. 19, 1645–1661. doi: 10.1111/gcb.12181
Al-Lawati, A., Al-Bahry, S., Victor, R., Al-Lawati, A. H., and Yaish, M. W. (2016). Salt stress alters DNA methylation levels in alfalfa (Medicago spp). Genet. Mol. Res. 15, 1–16. doi: 10.4238/gmr.15018299
Allen, C. D., Macalady, A. K., Chenchouni, H., Bachelet, D., McDowell, N., Vennetier, M., et al. (2010). A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For. Ecol. Manage. 259, 660–684. doi: 10.1016/j.foreco.2009.09.001
Amaral, J., Ribeyre, Z., Vigneaud, J., Sow, M. D., Fichot, R., Messier, C., et al. (2020). Advances and Promises of Epigenetics for Forest Trees. Forests 11:976. doi: 10.3390/f11090976
Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., and Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21:30. doi: 10.1186/s13059-020-1935-5
Anderegg, W. R. L., Kane, J. M., and Anderegg, L. D. L. (2013). Consequences of widespread tree mortality triggered by drought and temperature stress. Nat. Clim. Change 3, 30–36. doi: 10.1038/nclimate1635
Andrews, S. (2010). FastQC. A quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Ashapkin, V. V., Kutueva, L. I., Aleksandrushkina, N. I., and Vanyushin, B. F. (2020). Epigenetic mechanisms of plant adaptation to biotic and abiotic stresses. Internat. J. Mole. Sci. 21:7457. doi: 10.3390/ijms21207457
Ausin, I., Feng, S., Yu, C., Liu, W., Kuo, Y., Jacobsen, E. L., et al. (2016). DNA methylome of the 20-gigabase Norway spruce genome. Proc. Natl. Acad. Sci. USA 113, E8106–E8113. doi: 10.1073/pnas.1618019113
Avramidou, E. V., Ganopoulos, I. V., Doulis, A. G., Tsaftaris, A. S., and Aravanopoulos, F. A. (2015). Beyond population genetics: natural epigenetic variation in wild cherry (Prunus avium). Tree Genet. Genomes 11:95. doi: 10.1007/s11295-015-0921-7
Badad, O., Lakhssassi, N., Zaid, N., El Baze, A., Zaid, Y., Meksem, J., et al. (2021). Genome Wide MeDIP-Seq Profiling of Wild and Cultivated Olives Trees Suggests DNA Methylation Fingerprint on the Sensory Quality of Olive Oil. Plants 10:1405. doi: 10.3390/plants10071405
Baduel, P., and Colot, V. (2021). The epiallelic potential of transposable elements and its evolutionary significance in plants. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 376:20200123. doi: 10.1098/rstb.2020.0123
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS One 3:e3376. doi: 10.1371/journal.pone.0003376
Bannister, A., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Bewick, A. J., and Schmitz, R. J. (2017). Gene body DNA methylation in plants. Curr. Opin. Chem. Biol. 36, 103–110. doi: 10.1016/j.pbi.2016.12.007
Bhanu, B. D., Ulaganathan, K., and Shanker, A. K. (2020). Water Stress Responsive Differential Methylation of Organellar Genomes of Zea mays Z59. Am. J. Plant Sci. 11:1077. doi: 10.4236/ajps.2020.117077
Bitonti, M. B., Cozza, R., Chiappetta, A., Giannino, D., Castiglione, M. R., Dewitte, W., et al. (2002). Distinct nuclear organization, DNA methylation pattern and cytokinin distribution mark juvenile, juvenile-like and adult vegetative apical meristems in peach (Prunus persica (L.) Batsch). J. Exp. Bot. 53, 1047–1054. doi: 10.1093/jexbot/53.371.1047
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinform. 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bose, A. K., Gessler, A., Bolte, A., Bottero, A., Buras, A., Cailleret, M., et al. (2020). Growth and resilience responses of Scots pine to extreme droughts across Europe depend on predrought growth conditions. Glob. Change Biol. 26, 4521–4537. doi: 10.1111/gcb.15153
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., et al. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS One 5:9514. doi: 10.1371/journal.pone.0009514
Cailleret, M., Jansen, S., Robert, E. M., Desoto, L., Aakala, T., Antos, J. A., et al. (2017). A synthesis of radial growth patterns preceding tree mortality. Glob. Change Biol. 23, 1675–1690. doi: 10.1111/gcb.13535
Camarero, J. J., Bigler, C., Linares, J. C., and Gil-Pelegrín, E. (2011). Synergistic effects of past historical logging and drought on the decline of Pyrenean silver fir forests. For. Ecol. Manag. 262, 759–769. doi: 10.1016/j.foreco.2011.05.009
Camarero, J. J., Gazol, A., Sangüesa-Barreda, G., Oliva, J., Vicente-Serrano, S. M., and Gibson, D. (2015). To die or not to die: early warnings of tree dieback in response to a severe drought. J. Ecol. 103, 44–57. doi: 10.1111/1365-2745.12295
Carvalho, A., Nabais, C., Vieira, J., Rossi, S., and Campelo, F. (2015). Plastic Response of Tracheids in Pinus pinaster in a Water-Limited Environment: Adjusting Lumen Size instead of Wall Thickness. PLoS One 10:e0136305. doi: 10.1371/journal.pone.0136305
Cervera, M. T., Ruiz-García, L., and Martínez-Zapater, J. M. (2002). Analysis of DNA methylation in Arabidopsis thaliana based on methylation-sensitive AFLP markers. Mol. Genet. Genomics 268, 543–552. doi: 10.1007/s00438-002-0772-4
Choat, B., Brodribb, T. J., Brodersen, C. R., Duursma, R. A., Lopez, R., and Medlyn, B. E. (2018). Triggers of tree mortality under drought. Nature 558, 531–539. doi: 10.1038/s41586-018-0240-x
Chwialkowska, K., Korotko, U., Kosinska, J., Szarejko, I., and Kwasniewski, M. (2017). Methylation Sensitive Amplification Polymorphism Sequencing (MSAP-Seq) - A Method for High-Throughput Analysis of Differentially Methylated CCGG Sites in Plants with Large Genomes. Front. Plant Sci. 8:2056. doi: 10.3389/fpls.2017.02056
Chwialkowska, K., Nowakowska, U., Mroziewicz, A., Szarejko, I., and Kwasniewski, M. (2016). Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (Hordeum vulgare L.). J. Exp. Bot. 67, 1109–1121. doi: 10.1093/jxb/erv552
Ci, D., Song, Y., Du, Q., Tian, M., Han, S., and Zhang, D. (2016). Variation in genomic methylation in natural populations of Populus simonii is associated with leaf shape and photosynthetic traits. J. Exp. Bot. 67, 723–737. doi: 10.1093/jxb/erv485
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510. doi: 10.1038/nrg3012
DeSoto, L., Cailleret, M., Sterck, F., Jansen, S., Kramer, K., Robert, E. M. R., et al. (2020). Low growth resilience to drought is related to future mortality risk in trees. Nat. Comm. 11:545. doi: 10.1038/s41467-020-14300-5
Dodsworth, S., Leitch, A. R., and Leitch, I. J. (2015). Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 35, 73–78. doi: 10.1016/j.gde.2015.10.006
Dormling, I., and Johnsen, Ø (1992). Effects of the parental environment on full- sib families of Pinus sylvestris. Can. J. For. Res. 22, 88–100. doi: 10.1139/x92-013
Down, T. A., Rakyan, V. K., Turner, D. J., Flicek, P., Li, H., Kulesha, E., et al. (2008). A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat. Biotechnol. 26, 779–785. doi: 10.1038/nbt1414
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS One 6:e19379. doi: 10.1371/journal.pone.0019379
Feng, S., Cokus, S. J., Zhang, X., Chen, P. Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and divergence of methylation patterning in plants and animals. PNAS 107, 8689–8694. doi: 10.1073/pnas.1002720107
Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., et al. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. PNAS 89, 1827–1831. doi: 10.1073/pnas.89.5.1827
Gao, S., Zou, D., Mao, L., Zhou, Q., Jia, W., Huang, Y., et al. (2015). SMAP: a streamlined methylation analysis pipeline for bisulfite sequencing. Gigascience 4:29. doi: 10.1186/s13742-015-0070-9
Gibson, D. J., Connolly, J., Hartnett, D. C., and Weidenhamer, D. (2004). Designs for greenhouse studies of interactions between plants. J. Ecol. 87, 1–16. doi: 10.1046/j.1365-2745.1999.00321.x
González-Benito, M. E., Ibáñez, M. Á., Pirredda, M., Mira, S., and Martín, C. (2020). Application of the MSAP Technique to Evaluate Epigenetic Changes in Plant Conservation. Int. J. Mol. Sci. 21:7459. doi: 10.3390/ijms21207459
Gourcilleau, D., Bogeat-Triboulot, M. B., Le Thiec, D., Lafon-Placette, C., Delaunay, A., and El-Soud, et al. (2010). DNA methylation and histone acetylation: genotypic variations in hybrid poplars, impact of water deficit and relationships with productivity. Ann. For. Sci. 67:208. doi: 10.1051/forest/2009101
Graña, O., López-Fernández, H., Fdez-Riverola, F., Pisano, D., and Glez-Peña, D. (2017). bicycle: a bioinformatics pipeline to analyze bisulfite sequencing data. Bioinform 34, 1414–1415. doi: 10.1093/bioinformatics/btx778
Grehl, C., Wagner, M., Lemnian, I., Glaser, B., and Grosse, I. (2020). Performance of Mapping Approaches for Whole-Genome Bisulfite Sequencing Data in Crop Plants. Front. Plant Sci. 11:176. doi: 10.3389/fpls.2020.00176
Gugger, P. F., Fitz-Gibbon, S., PellEgrini, M., and Sork, V. L. (2016). Species-wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol. Ecol. 25, 1665–1680. doi: 10.1111/mec.13563
Hasbún, R., Iturra, C., Bravo, S., Rebolledo-Jaramillo, B., and Valledor, L. (2016). Differential Methylation of Genomic Regions Associated with Heteroblasty Detected by M&M Agorithm in the Nonmodel Species Eucalyptus globulus Labill. Int. J. Genom. 2016:4395153. doi: 10.1155/2016/4395153
Herrera, C. M., and Bazaga, P. (2013). Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linn. Soc. 171, 441–452. doi: 10.1111/boj.12007
IPCC (2018). Global warming of 1.5°C. An IPCC special report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty. Geneva: World Meteorological Organization.
Iwasaki, M., and Paszkowski, J. (2014). Epigenetic memory in plants. EMBO J. 33, 1–12. doi: 10.15252/embj.201488883
Jump, A. S., and Peñuelas, J. (2005). Running to stand still: adaptation and the response of plants to rapid climate change. Ecol. Lett. 8, 1010–1020. doi: 10.1111/j.1461-0248.2005.00796.x
Karan, R., DeLeon, T., Biradar, H., and Subudhi, P. K. (2012). Salt Stress Induced Variation in DNA Methylation Pattern and Its Influence on Gene Expression in Contrasting Rice Genotypes. PLoS One 7:e40203. doi: 10.1371/journal.pone.0040203
Kijowska-Oberc, J., Staszak, A. M., Kamiński, J., and Ratajczak, E. (2020). Adaptation of Forest Trees to Rapidly Changing Climate. Forests 11:123. doi: 10.3390/f11020123
Kinlaw, C. S., and Neale, D. B. (1997). Complex gene families in pine genomes. Trends Plant Sci. 2, 356–359. doi: 10.1016/S1360-1385(97)84624-9
Kinoshita, T., and Seki, M. (2014). Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 55, 1859–1863. doi: 10.1093/pcp/pcu125
Korotko, U., Chwialkowska, K., Sanki-Sawczenko, I., and Kwasmiewski, M. (2021). DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana. Int. J. Mol. Sci. 22:1555. doi: 10.3390/ijms22041555
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-seq applications. Bioinform 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Kurdyukov, S., and Bullock, M. (2016). DNA methylation analysis: choosing the right method. Biology 5:3. doi: 10.3390/biology5010003
Kuzmin, D. A., Feranchuk, S. I., Sharov, V. V., Cybin, A. N., Makolov, S. V., Putintseva, Y. A., et al. (2019). Stepwise large genome assembly approach: a case of Siberian larch (Larix sibirica Ledeb). BMC Bioinf. 20:37. doi: 10.1186/s12859-018-2570-y
Kvaalen, H., and Johnsen, Ø (2008). Timing of bud set in Picea abies is regulated by a memory of temperature during zygotic and somatic embryogenesis. New Phytol. 177, 49–59. doi: 10.1111/j.1469-8137.2007.02222.x
Lafon-Placette, C., Le Gac, A. L., Chauveau, D., Segura, V., Delaunay, A., Lesage-Descauses, M. C., et al. (2018). Changes in the epigenome and transcriptome of the poplar shoot apical meristem in response to water availability affect preferentially hormone pathways. J. Exp. Bot. 69, 537–551. doi: 10.1093/jxb/erx409
Lamelas, L., Valledor, L., Escandón, M., Pinto, G., Cañal, M. J., and Meijon, M. (2020). Integrative analysis of the nuclear proteome in Pinus radiata reveals thermopriming coupled to epigenetic regulation. J. Exp. Bot. 71, 2040–2057. doi: 10.1093/jxb/erz524
Lang-Mladek, C., Popova, O., Kiok, K., Berlinger, M., Rakic, B., Aufsatz, W., et al. (2010). Transgenerational inheritance and resetting of stress-induced loss of epigenetic gene silencing in Arabidopsis. Mol. Plant 3, 594–602. doi: 10.1093/mp/ssq014
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Le Gac, A. L., Lafon-Placette, C., Chauveau, D., Segura, V., Delaunay, A., Fichot, R., et al. (2018). Winter-dormant shoot apical meristem in poplar trees shows environmental epigenetic memory. J. Exp. Bot. 69, 4821–4837. doi: 10.1093/jxb/ery271
Li, D., Zhang, B., Xing, X., and Wang, T. (2015). Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods 72, 29–40. doi: 10.1016/j.ymeth.2014.10.032
Li, R., Hu, F., Li, B., Zhang, Y., Chen, M., Fan, T., et al. (2020). Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 10:8013. doi: 10.1038/s41598-020-64975-5
Li, W., Lin, Y. C., Li, Q., Shi, R., Lin, C.-Y., Chen, H., et al. (2014). A robust chromatin immunoprecipitation protocol for studying transcription factor–DNA interactions and histone modifications in wood-forming tissue. Nat. Protoc. 9, 2180–2193. doi: 10.1038/nprot.2014.146
Li, Y., Dong, X. M., Jin, F., Shen, Z., Chao, Q., and Wang, B. C. (2017). Histone Acetylation Modifications Affect Tissue-Dependent Expression of Poplar Homologs of C4 Photosynthetic Enzyme Genes. Front. Plant Sci. 8:950. doi: 10.3389/fpls.2017.00950
Liang, D., Zhang, Z., Wu, H., Huang, C., Shuai, P., Ye, C. Y., et al. (2014). Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 15:S9. doi: 10.1186/1471-2156-15-S1-S9
Lin, X., Sun, D., Rodriguez, B., Zhao, Q., Sun, H., Zhang, Y., et al. (2013). BSeQC: quality control of bisulfite sequencing experiments. Bioinform. 29, 3227–3229. doi: 10.1093/bioinformatics/btt548
Linares, J. C., Camarero, J. J., and Carreira, J. A. (2010). Competition modulates the adaptation capacity of forests to climatic stress: insights from recent growth decline and death in relict stands of the Mediterranean fir Abies pinsapo. J. Ecol. 98, 592–603. doi: 10.1111/j.1365-2745.2010.01645.x
Linares, J. C., Taïqui, L., and Camarero, J. J. (2011). Increasing Drought Sensitivity and Decline of Atlas Cedar (Cedrus atlantica) in the Moroccan Middle Atlas Forests. Forests 2, 777–796. doi: 10.3390/f2030777
Ma, X., Zhang, C., Zhang, B., Yang, C., and Li, S. (2016). Identification of genes regulated by histone acetylation during root development in Populus trichocarpa. BMC Genom. 17:96. doi: 10.1186/s12864-016-2407-x
Macías, M., Andreu, L., Bosch, O., Camarero, J. J., and Gutiérrez, E. (2006). Increasing aridity is enhancing silver fir Abies alba (Mill.) water stress in its south-western distribution limit. Clim. Change 79, 289–313. doi: 10.1007/s10584-006-9071-0
Mackay, J., Dean, J. F., Plomion, C., Peterson, D. G., Cánovas, F. M., Pavy, N., et al. (2012). Towards decoding the conifer giga-genome. Plant Mol. Biol. 80, 555–569. doi: 10.1007/s11103-012-9961-7
Markus, C., Pecinka, A., and Merotto, A. Jr. (2021). Insights into the Role of Transcriptional Gene Silencing in Response to Herbicide-Treatments in Arabidopsis thaliana. Int. J. Mol. Sci. 22:3314. doi: 10.3390/ijms22073314
Martin, A., Troadec, C., Boualem, A., Rajab, M., Fernandez, R., Morin, H., et al. (2009). A transposon-induced epigenetic change leads to sex determination in melon. Nature 461, 1135–1138. doi: 10.1038/nature08498
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mascher, M., Wicker, T., Jenkins, J., Plott, C., Lux, T., Koh, C. S., et al. (2021). Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33, 1888–1906. doi: 10.1093/plcell/koab077
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’Souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
McDowell, N. G., Williams, A. P., Xu, C., Pockman, W. T., Dickman, L. T., Sevanto, S., et al. (2016). Multi-scale predictions of massive conifer mortality due to chronic temperature rise. Nat. Clim. Change 6, 295–300. doi: 10.1038/nclimate2873
Merce, C., Bayer, P. E., Fernandez, C. T., Batley, J., and Edwards, D. (2020). Induced Methylation in Plants as a Crop Improvement Tool: Progress and Perspectives. Agronomy 10:1484. doi: 10.3390/agronomy10101484
Miryeganeh, M., Marlétaz, F., Gavriouchkina, D., and Saze, H. (2021). De novo genome assembly and in natura epigenomics reveal salinity-induced DNA methylation in the mangrove tree Bruguiera gymnorhiza. New Phytol. 2021:17738. doi: 10.1111/nph.17738
Mosca, E., Cruz, F., Gómez-Garrido, J., Bianco, L., Rellstab, C., Brodbeck, S., et al. (2019). A Reference Genome Sequence for the European Silver Fir (Abies alba Mill.): A Community-Generated Genomic Resource. G3 9, 2039–2049. doi: 10.1534/g3.119.400083
Mougninot, P., Aparicio, N. L., Gourcilleau, D., Latutrie, M., Marin, S., Hemptinne, J. L., et al. (2021). Phenotypic Response to Light Versus Shade Associated with DNA Methylation Changes in Snapdragon Plants (Antirrhinum majus). Genes 12:227. doi: 10.3390/genes12020227
Muhammad, I. I., Kong, S. L., Akmar Abdullah, S. N., and Munusamy, U. (2020). RNA-seq and ChIP-seq as Complementary Approaches for Comprehension of Plant Transcriptional Regulatory Mechanism. Int. J. Mol. Sci. 21:167. doi: 10.3390/ijms21010167
Neale, D. B., McGuire, P. E., Wheeler, N. C., Stevens, K. A., Crepeau, M. W., Cardeno, C., et al. (2017). The Douglas-Fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 7, 3157–3167. doi: 10.1534/g3.117.300078
Neale, D. B., Wegrzyn, J. L., Stevens, K. A., Zimin, A. V., Puiu, D., Crepeau, M. W., et al. (2014). Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15:R59. doi: 10.1186/gb-2014-15-3-r59
Nystedt, B., Street, N. R., Wetterbom, A., Zuccolo, A., Lin, Y.-C., Scofield, D. G., et al. (2013). The Norway spruce genome sequence and conifer genome evolution. Nature 497, 579–584. doi: 10.1038/nature12211
Park, Y., and Wu, H. (2016). Differential methylation analysis for BS-seq data under general experimental design. Bioinform 32, 1446–1453. doi: 10.1093/bioinformatics/btw026
Paun, O., Verhoeven, K. J. F., and Richards, L. (2019). Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. New Phytol. 221, 738–742. doi: 10.1111/nph.15388
Pereira, W. J., Pappas, M. D. C. R., Grattapaglia, D., and Pappas, G. J. Jr. (2020). A cost-effective approach to DNA methylation detection by Methyl Sensitive DArT sequencing. PLoS One 15:e0233800. doi: 10.1371/journal.pone.00233800
Perrin, A., Daccord, N., Roquis, D., Celton, J. M., Vergne, E., and Bucher, E. (2020). Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees. Epigenomes 4:4. doi: 10.3390/epigenomes4010004
Petit, R. J., and Hampe, A. (2006). Some Evolutionary Consequences of Being a Tree. Annu. Rev. Ecol. Evol. Syst. 37, 187–214. doi: 10.1146/annurev.ecolsys.37.091305.110215
Raj, S., Bräutigam, K., Hamanishi, E. T., Wilkins, O., Thomas, B. R., Schroeder, W., et al. (2011). Clone history shapes Populus drought responses. PNAS 108, 12521–12526. doi: 10.1073/pnas.1103341108
Rapp, R. A., and Wendel, J. F. (2005). Epigenetics and plant evolution. New Phytol. 168, 81–91. doi: 10.1111/j.1469-8137.2005.01491.x
Rauluseviciute, I., Drabløs, F., and Rye, M. B. (2019). DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis. Clin. Epigenet. 11:193. doi: 10.1186/s13148-019-0795-x
Reyna-López, G. A., Simpson, J., and Ruiz-Herrera, J. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 253, 703–710. doi: 10.1007/s004380050374
Rico, L., Ogaya, R., Barbeta, A., and Peñuelas, J. (2014). Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biol. 16, 419–427. doi: 10.1111/plb.12049
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinform. 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rothkegel, K., Espinoza, A., Sanhueza, D., Lillo-Carmona, V., Riveros, A., Campos-Vargas, R., et al. (2021). Identification of DNA Methylation and Transcriptomic Profiles Associated With Fruit Mealiness in Prunus persica (L.) Batsch. Front. Plant sci. 12:684130. doi: 10.3389/fpls.2021.684130
Sáez-Laguna, E., Guevara, M. A., Díaz, L. M., Sánchez-Gómez, D., Collada, C., Sánchez-Gómez, D., et al. (2014). Epigenetic Variability in the Genetically Uniform Forest Tree Species Pinus pinea L. PLoS One 9:e103145. doi: 10.1371/journal.pone.0103145
Sánchez-Salguero, R., Ortíz, C., Covelo, F., Ochoa, V., García-Ruíz, R., Seco, J. I., et al. (2015). Regulation of Water Use in the Southernmost European Fir (Abies pinsapo Boiss.): Drought Avoidance Matters. Forests 6, 2241–2260. doi: 10.3390/f6062241
Schield, D. R., Wlash, M. R., Card, D. C., Andrew, A. l., Adams, R. H., and Castoe, T. A. (2016). EpiRADseq: scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 7, 60–69. doi: 10.1111/2041-210X.12435
Schönberger, B., Chen, X., Mager, S., and Ludewig, U. (2016). Site-Dependent Differences in DNA Methylation and Their Impact on Plant Establishment and Phosphorus Nutrition in Populus trichocarpa. PLoS One 11:e0168623. doi: 10.1371/journal.pone.0168623
Song, Y., Ci, D., Tian, M., and Zhang, D. (2016). Stable methylation of a non-coding RNA gene regulates gene expression in response to abiotic stress in Populus simonii. J. Exp. Bot. 67, 1477–1492. doi: 10.1093/jxb/erv543
Sow, M. D., Allona, I., Ambroise, C., Conde, D., Fichot, R., Gribkova, S., et al. (2018). “Epigenetics in Forest Trees: State of the Art and Potential Implications for Breeding and Management in a Context of Climate Change,” in Advances in Botanical Research, eds M. Mirouze, E. Bucher, and P. Gallusci (Cambridge: Academic Press), 387–453.
Sow, M. D., Le Gac, A. L., Fichot, R., Lanciano, S., Delaunay, A., Le Jan, I., et al. (2021). RNAi suppression of DNA methylation affects the drought stress response and genome integrity in transgenic poplar. New Phytol. 2021:17555. doi: 10.1111/nph.17555
Stevens, K. A., Wegrzyn, J. L., Zimin, A., Puiu, D., Crepeau, M., Cardeno, C., et al. (2016). Sequence of the Sugar Pine Megagenome. Genetics 204, 1613–1626. doi: 10.1534/genetics.116.193227
Su, Y., Bai, X., Yang, W., Wang, W., Chen, Z., Ma, J., et al. (2018). Single-base-resolution methylomes of Populus euphratica reveal the association between DNA methylation and salt stress. Tree Genet. Genomes 14:86. doi: 10.1007/s11295-018-1298-1
Szcześniak, M. W., Rosikiewicz, W., and Makalowska, I. (2015). CANTATAdb: A collection of plant long non-coding RNAs. Plant Cell Physiol. 57:e8. doi: 10.1093/pcp/pcv201
Takuno, S., Ran, J. H., and Gaut, B. (2016). Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2:15222. doi: 10.1038/nplants.2015.222
Thu, K. L., Vucic, E. A., Kennett, J. Y., Heryet, C., Brown, C. J., Lam, W. L., et al. (2009). Methylated DNA immunoprecipitation. J. Vis. Exp. 23:935. doi: 10.3791/935
Trucchi, E., Mazzarella, A. B., Gilfillan, G. D., Lorenzo, M. T., Schönswetter, P., and Paun, O. (2016). BsRADseq: screening DNA methylation in natural populations of non-model species. Mol. Ecol. 25, 1697–1713. doi: 10.1111/mec.13550
Trugman, A., Anderegg, L. D. L., Anderegg, W. R. L., Das, A. J., and Stephenson, N. L. (2021). Why is tree drought mortality so hard to predict? Trends Ecol. Evol. 36, 520–532. doi: 10.1016/j.tree.2021.02.001
van Gurp, T., Wagemaker, N. C. A. M., Wouters, B., Vergeer, P., Ouborg, J. N. J., and Verhoeven, K. J. F. (2016). epiGBS: reference-free reduced representation bisulfite sequencing. Nat. Methods 13, 322–324. doi: 10.1038/nmeth.3763
Vanden Broeck, A., Cox, K., Brys, R., Castiglione, S., Cicatelli, A., Guarino, F., et al. (2018). Variability in DNA methylation and generational plasticity in the lombardy poplar, a single genotype worldwide distributed since the eighteenth century. Front. Plant Sci. 9:1635. doi: 10.3389/fpls.2018.01635
Wang, W. S., Pan, Y. J., Zhao, X. Q., Dwivedi, D., Zhu, L. H., Ali, J., et al. (2010). Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 62, 1951–1960. doi: 10.1093/jxb/erq391
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wang, Z., Xianfeng, L., Jiang, Y., Shao, Q., Liu, Q., Chen, B. Y., et al. (2015). swDMR: A Sliding Window Approach to Identify Differentially Methylated Regions Based on Whole Genome Bisulfite Sequencing. PLoS One 10:e0132866. doi: 10.1371/journal.pone.0132866
Warren, R. L., Keeling, C. I., Yuen, M. M., Raymond, A., Taylor, G. A., Vandervalk, B. P., et al. (2015). Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 83, 189–212. doi: 10.1111/tpj.12886
Webber, J., Ott, P., Owens, J., and Binder, W. (2005). Elevated temperature during reproductive development affects cone traits and progeny performance in Picea glauca x engelmannii complex. Tree Physiol. 25, 1219–1227. doi: 10.1093/treephys/25.10.1219
Wei, J.-W., Huang, K., Yang, C., and Kang, C.-S. (2016). Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 37, 3–9. doi: 10.3892/or.2016.5236
Werner, O., Prudencio, A. S., de la Cruz-Martínez, E. I., Nieto-Liglde, M., Martínez-Gómez, P., and Ros, R. M. (2020). A cost reduced variant of epi-genotyping by sequencing for studying DNA methylation in non-model organisms. Front. Plant Sci. 11:694. doi: 10.3389/fpls.2020.00694
West-Eberhard, M. J. (2008). “Phenotypic Plasticity,” in Encyclopedia of Ecology, eds S. Erik Jørgensen and B. D. Fath (Cambridge: Academic Press), 2701–2707. doi: 10.1016/B978-008045405-4.00837-5
Wilson, G. A., Dhami, P., Feber, A., Cortazar, D., Suzuki, Y., Schulz, R., et al. (2012). Resources for methylome analysis suitable for gene knockout studies of potential epigenome modifiers. GigaScience 1:3. doi: 10.1186/2047-217X-1-3
Wreczycka, K., Gosdschan, A., Yusuf, D., Grüning, B., Assenov, Y., and Akalin, A. (2017). Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 216, 105–115. doi: 10.1016/j.jbiotec.2017.08.007
Xi, Y., and Li, W. (2009). BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinform. 10:232. doi: 10.1186/1471-2105-10-232
Xiong, Z., Xu, C. G., Saghai Maroof, M. A., and Zhang, Q. (1999). Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol. Gen. Genet. 261, 439–446. doi: 10.1007/s004380050986
Xu, J., Zhou, S., Gong, X., Song, Y., van Nocker, S., Ma, F., et al. (2018). Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 16, 672–687. doi: 10.1111/pbi.12820
Yakovlev, I., Fossdal, C. G., Skrøppa, T., Olsen, J. E., Jahren, A. H., and Johnsen, O. (2012). An adaptive epigenetic memory in conifers with important implications for seed production. Seed Sci. Res. 22, 63–76. doi: 10.1017/s0960258511000535
Yakovlev, I. A., Carneros, E., Lee, Y., Olsen, J. E., and Fossdal, C. G. (2016). Transcriptional profiling of epigenetic regulators in somatic embryos during temperature induced formation of an epigenetic memory in Norway spruce. Planta 243, 1237–1249. doi: 10.1007/s00425-016-2484-8
Yakovlev, I. A., Lee, Y., Rotter, B., Olsen, J. E., Skrøppa, T., Johnsen, O., et al. (2014). Temperature-dependent differential transcriptomes during formation of an epigenetic memory in Norway spruce embryogenesis. Tree Genet. Genomes 10, 355–366. doi: 10.1007/s11295-013-0691-z
Zemach, A., McDaniel, I. E., Silva, P., and Ziberman, D. (2010a). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zemach, A., Kim, M. Y., Silva, P., Rodrigues, J. A., Dotson, B., Brooks, M. D., et al. (2010b). Local DNA hypomethylation activates genes in rice endosperm. PNAS 107, 18729–18734. doi: 10.1073/pnas.1009695107
Zemach, A., and Ziberman, D. (2010). Evolution of eukaryotic DNA methylation and the pursuit of saver sex. Curr. Biol. 20, R780–R785.
Zhang, H., Lang, Z., and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhao, J. G., Lu, Z. G., Wang, L., and Jin, B. (2021). Plant responses to heat stress: physiology, transcription, noncoding RNAs and epigenetics. Int. J. Mol. Sci. 22:117. doi: 10.3390/ijms22010117
Zhao, M. T., Whyte, J. J., Hopkins, G. M., Kirk, M. D., and Prather, R. S. (2014). Methylated DNA immunoprecipitation and high-throughput sequencing (MeDIP-seq) using low amounts of genomic DNA. Cell Reprogram 16, 175–184. doi: 10.1089/cell.2014.0002
Keywords: epigenetics, climate change, forest tree species, abiotic stress, methylation
Citation: García-García I, Méndez-Cea B, Martín-Gálvez D, Seco JI, Gallego FJ and Linares JC (2022) Challenges and Perspectives in the Epigenetics of Climate Change-Induced Forests Decline. Front. Plant Sci. 12:797958. doi: 10.3389/fpls.2021.797958
Received: 19 October 2021; Accepted: 13 December 2021;
Published: 04 January 2022.
Edited by:
Naganand Rayapuram, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Rodrigo J. Hasbun, University of Concepcion, ChilePao-Yang Chen, Institute of Plant and Microbial Biology, Academia Sinica, Taiwan
Copyright © 2022 García-García, Méndez-Cea, Martín-Gálvez, Seco, Gallego and Linares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel García-García, aXNhYmVsMDZAdWNtLmVz; Belén Méndez-Cea, YmVsZW5tZW5AdWNtLmVz
†These authors have contributed equally to this work and share first authorship