 Yuxuan He
Yuxuan He Likun Long
Likun Long Feiwu Li
Feiwu Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 14 December 2021
Sec. Technical Advances in Plant Science
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.794752
This article is part of the Research Topic Novel Technologies for Soybean Improvement View all 21 articles
Microribonucleic acids (miRNAs) play significant roles in the regulation of biological processes and in responses to biotic or abiotic environmental stresses. Therefore, it is necessary to quantitatively detect miRNAs to understand these complicated biological regulation mechanisms. This study established an ultrasensitive and highly specific method for the quantitative detection of miRNAs using simple operations on the ground of the ligation reaction of ribonucleotide-modified deoxyribonucleic acid (DNA) probes. This method avoids the complex design of conventional reverse transcription. In the developed assay, the target miRNA miR156b was able to directly hybridize the two ribonucleotide-modified DNA probes, and amplification with universal primers was achieved following the ligation reaction. As a result, the target miRNA could be sensitively measured even at a detection limit as low as 0.0001 amol, and differences of only a single base could be detected between miR156 family members. Moreover, the proposed quantitative method demonstrated satisfactory results for overexpression-based genetically modified (GM) soybean. Ligation-based quantitative polymerase chain reaction (PCR) therefore has potential in investigating the biological functions of miRNAs, as well as in supervising activities regarding GM products or organisms.
Micro ribonucleic acids (miRNAs) are a group of endogenous, non-coding, single-stranded small molecular RNA (18–25 nt in length) that participate in the post-transcriptional regulation of gene expression. miRNAs are emerging as novel biomarkers, with profound implications for research into the responses to abiotic (Aslam et al., 2020; Singroha et al., 2021) or biotic stress (Chopperla et al., 2020; Šečić et al., 2021), development and differentiation in cellular behavior (Krol et al., 2010; Miao et al., 2020), and multiple plant signaling pathways (Curaba et al., 2014). Since their first discovery, several miRNAs have been identified in a wide range of species. Following the identification of the functions of miRNAs in crops, a large number of transgenic crops related with miRNA have been developed and commercialized worldwide (Agrawal et al., 2015; Bally et al., 2020). Among the miRNAs in plant, microRNA156 (miR156) and its target mRNAs has been widely observed as a major regulators for crop development (Cao et al., 2015) and abiotic stresses (Visentin et al., 2020). The miR156 expresses at the highest accumulation at seedling stage (Miao et al., 2019). Moreover, the expression of the other miRNAs, such as miR159, miR33 might have a effect on miR156 level (Guo et al., 2017). In the whole growth stage of grapevine, miR156b/c/d exhibited typical temporal spatial-specific expression levels (Wang et al., 2016). The Arabidopsis AtmiR156b gene overexpressed in GM Brassica napus resulted in seed lutein and beta-carotene accumulation, these achievement suggest that miR156b have a potential applification in plant breeding for enhancing carotenoid production (Wei et al., 2010). To further explore the physiological functions of miRNA or detect the content in crop genomes, specific quantitative detection methods for miRNAs that have high sensitivity, efficiency, and reliability are urgently needed for miRNA mechanism applications (Papadopoulou et al., 2020). In early miRNA studies, northern blotting (De la Rosa and Reyes, 2019) and microarray (Li and Ruan, 2009) assays were conventionally used as “gold standard” methods to identify miRNA expression. However, these complex and relatively insensitive methods cannot overcome the complications associated with the unique characteristics of miRNAs such as their low abundances in tissue, short lengths, and the high sequence similarities among miRNA families. To resolve these problems, a series of cooperative amplification programs have been reported for miRNA detection. Locked nucleic acid (LNA)–modified probes with bicyclic high-affinity RNA analogs, which are rigid structures formed by linking the 2′ and 4′ carbons on the ribose, greatly improve the hybridization efficiency of northern blotting and are sensitive to miRNA determination (Shu et al., 2019). The Agilent Microarray SurePrint technology is advantageous owing to its low RNA input requirements (∼100 ng) and eliminates bias by omitting the fractionation or amplification steps (Dell’Aversana et al., 2017). However, many solid phase hybridization assays require sophisticated fabrication or expensive capture probe modifications (Zou et al., 2017). These issues increase their costs and restrict their extensive application. Modified miRNA detection methods based on nucleic acid amplification are undoubtedly the most attractive and prominent tools for miRNA analysis and include polymerase chain reaction (PCR), ligase chain reaction (LCR) (Zhang et al., 2013), rolling circle amplification (RCA) (Cheng et al., 2009; Liu et al., 2020), and isothermal exponential amplification reaction (IEXPAR) (Jia et al., 2010). For instance, with the design of a new hairpin/deoxyribonucleic acid (DNA) ring ternary probe in RCA, the target miRNA can bind the probes through toehold-mediated strand displacement to form ternary structures and generate many repeated metal ion-dependent deoxyribozyme (DNAzyme) sequences. Finally, fluorescently quenched hairpin signal probes can be cyclically cleaved by the DNAzyme sequences, which drastically enhances the fluorescence recovery of the target miRNA (Liu et al., 2019). Tian et al. (2019) rationally combined RCA with efficient loop-mediated isothermal amplification (LAMP), which directly templates the ligation of a padlock probe to trigger the RCA reaction. Thus, they significantly improved the amplification efficiency and sensitivity owing to the double stem-loop DNAs with functional sequences that were generated by RCA-produced DNA templates.
This study proposes the development of a facile and ultrasensitive method for miRNA detection by elegantly integrating sensitivity and specificity. Here, a miRNA quantification assay is achieved through real-time PCR integrated ligation reactions. As outlined in the schematic representation in Figure 1. After determining the target miRNA to be detected, the two DNA probes in this assay contain a specific sequence (blue), a stuffer sequence (yellow) in between and a universal sequence designed as amplification primer (green). Both of the specific sequences are complementary to the half-sequence of target miRNA at the 3′- and 5′-termini, respectively. Then, the 3′-termini of probe A is modified by two nucleotides and the 5′-termini of probe B is modified by a phosphate group. Therefore, the modified groups are immediately adjacent to each other following the hybridization of the probes with the half-sequence of target miRNA. As T4 RNA ligases specifically catalyze the formation of a 3′–5′ -phosphodiester bond between a 3′-hydroxyl group and a 5′-phosphoryl group in three nucleotidyl transfer steps (Zhuang et al., 2012), and T4 RNA ligase II exhibits significantly higher ligating activity and specificity for double stranded RNA than T4 RNA ligase I (Yin et al., 2003; Cheng et al., 2009). T4 RNA ligase II is employed to link the 3′-hydroxyl and 5′-phosphoryl groups in adjacent probes and thus form ligation production, which is used as an initial template for the next real-time PCR amplification with the universal sequences. The PCR products with fluorescence or SYBR Green I signal can then be detected directly. The detection limit of the proposed method was determined and its specificity was investigated with respect to its ability to distinguish miRNAs from their corresponding pre-miRNAs, and from homogeneous miRNAs in the same family. The robustness and feasibility of the method were investigated as a tool for genetically modified (GM) content analysis.
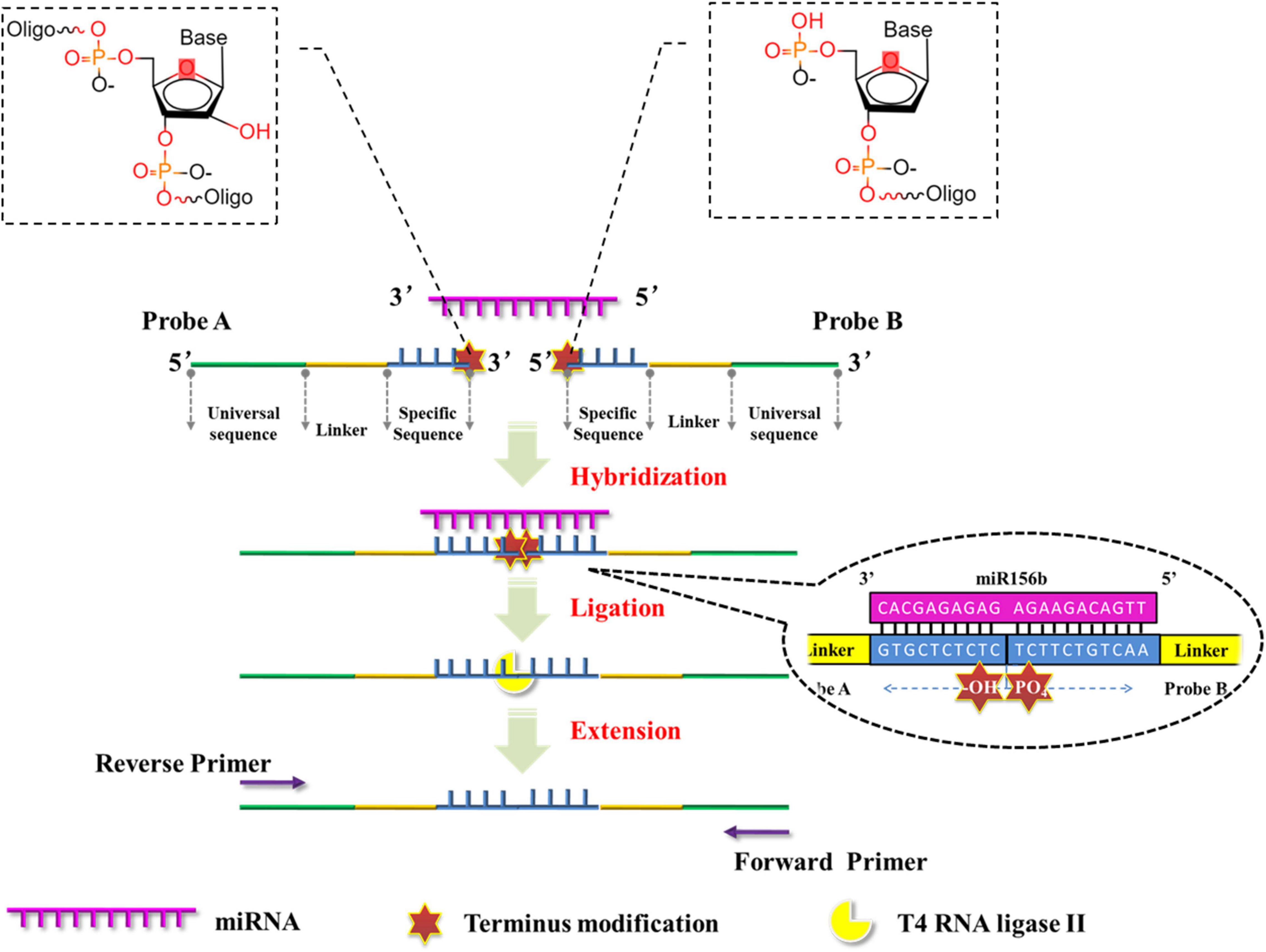
Figure 1. Schematic representation of miRNA quantitative polymerase chain reaction (PCR) based on the ligation reactions of ribonucleotide-modified deoxyribonucleic acid (DNA) probes.
All plant materials were kindly provided by Jilin Academy of Agricultural Sciences, Changchun, China, and germinated in soil (pH 7.19; soil: water 1: 4) under a photoperiod of 16 h of daylight at 23°C. A 181-bp stem-loop fragment of the GmmiR156b (GenBank No. NR048600.1) precursor was amplified using DNA samples obtained from the soybean cultivar Williams 82 and subcloned into the pTF101.1-35S vector, which carried the selection marker Bar gene and the cauliflower mosaic virus 35S promoter that could enhance the expression of miR156b. The recombinant construct pTF101.1-miR156b was then integrated into Agrobacterium tumefaciens strain EHA101 for cotyledon-node transformation (Flores et al., 2008). After the regenerated plants were transplanted into a greenhouse, glufosinate (150 mg/L) screening and LibertyLink strip (EnviroLogix Inc., Portland, ME, United States) detection were used to identify transgenic plants. Herbicide-resistant T4 generations containing a single copy number of the target sequence were employed as miR156b GM soybean event for further experimental analyses. In this GM soybean, the expression level of miR156b has been considered by the conventional stem-loop qPCR, which further confirmed the reliability of the plant materials. Total RNA and DNA samples were extracted from eight-week-old transgenic and wild-type (WT) soybean seedlings. All procedures were performed in accordance with the EasyPure Plant RNA kit (Transgen Biotech, Beijing, China) and Dneasy Plant Mini Kit (Qiagen, Hilden, Germany). The extracted concentration and quality were calculated using absorbance measurements with an ND-1000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, United States) at wavelengths of 260 and 280 nm. All the extracted samples were then stored at −80°C for further research.
All the miRNA sequences in the plant can be obtained from the online database PmiREN.1 The miR156 family comprises of typical miRNAs that are considered to have an explicit function applied in some transgenic soybeans; in this assay, the miR156b gene from the miR156 family was used as the model target. The structures of probes A and B are elucidated schematically in Figure 1. The specific sequences (blue) of probes A and B were designed to be complementary to half of the target miRNA sequences at the 3′- and 5′-terminals, respectively. A stuffer sequence (yellow) in between links up the specific sequence with the universal sequence (green). Probe A was modified with two nucleotides at its 3′-terminus, while probe B was modified with a phosphate group at its 5′-terminus. As the chemical features of the modified nucleotides, 3′-hydroxyl in probe A and 5′-phosphoryl groups in probe B (Figure 1) are immediately adjacent to each other following the DNA probes hybridize with the target miRNA, and the modified structure facilitates the formation of a 3′- to 5′-phosphodiester bond catalyzed by T4 RNA ligase II. Moreover, probe A-N has the same sequences as probe A except for the presence of 3′-hydroxyl groups in the two 3′-terminal ribonucleotides. Probe A-N was employed as an experimental control to verify the role of terminus modifications in the ligation reaction. In order to test the specificity of this proposed method in miRNA discrimination, for another miR156 member (miR156g), probes A-g and B-g were also designed in the same way following the replacement of specific sequences (Table 1).

Table 1. The sequences of the primers and probes in the ligation based quantitative polymerase chain reaction (PCR) amplification.
The forward and reverse primers for PCR amplification were designed according to the universal sequences in probes B and A, respectively. For the optimization of the ligation-based qPCR, primer pairs with different lengths were designed and orthogonally combined to screen the optimal primers for the amplification of the ligation product. The sequences of the primers and probes are shown in Table 1. All high-performance liquid chromatography–purified miRNAs, polyacrylamide gel electrophoresis–purified ribonucleotide-modified DNA probes, and primers were synthesized by Shanghai Sangon Biological Engineering Technology (Shanghai, China).
The mixture contained 2 nM each of template miRNA and probes A and B. The procedure was initially performed at 65°C for 8 min. In this step, both the DNA probes identify and hybridize with the target miRNA. Then, 2 U of T4 RNA ligase II and 10× ligation buffer (New England Biolabs, Shanghai, China) were added to the hybridization product, and a total final volume of 50 μl was incubated at 37°C for 1 h to perform the ligation reaction; the products were then immediately placed on ice until it cooled to room temperature (∼15 min).
The FastStart Universal SYBR® Green Master (Roche, Mannheim, Germany) was used for real-time quantitative PCR assays. The quantitative PCR assays for amplifying the target miRNA were performed with a 20-μl final volume containing 2× SYBR Green Mastermix (ROX), 0.2-μM forward and reverse primers, 5 μl of ligation production, and 4.2 μl of diethylpyrocarbonate-treated water (DEPC-H2O; Takara Biotechnology, Changchun, China). The reactions were incubated at 95°C for 5 min on a Bio-Rad CFX96 Real-time Thermal Cycler (Bio-Rad, Hercules, CA, United States), followed by 40 cycles of 95°C for 10 s, 60°C for 30 s, and then 72°C for 30 s. All reactions were performed in triplicate.
The precursor of miR156b, pre-miR156b, was prepared using an in vitro transcription reaction. First, the forward and reverse primers for pre-miR156b (pre-FP and pre-RP) were designed based on the sequences of pre-GmmiR156b,2 in which the 20 bases located at the 3′-terminus of pre-FP complemented each other with the 20 bases located at the 3′-terminus of pre-RP. A mixture containing 50 pmol of primer and 10 μl of Klenow buffer (Fermentas, Shanghai, China) was incubated at 75°C for 5 min and cooled on ice. After the 20 complementary bases hybridized completely with each other, deoxynucleotide triphosphate (dNTP; 250 μM final concentration; Takara Biotechnology, Changchun, China), Klenow buffer, and 5 U of Klenow DNA polymerase (exo-; Fermentas, Shanghai, China) were added and replenished with DEPC-H2O (Takara Biotechnology, Changchun, China) to a final volume of 20 μl. Following incubation at 37°C for 1 h, the 3′-terminus sequences of both pre-FP and pre-RP generated double-stranded DNA (dsDNA) by extension reactions. Then, the reaction mixture was heated at 75°C for 20 min to inactivate the Klenow DNA polymerase. The complete sequence consists of the T7 promoter, GGGA (guanine–guanine–guanine–adenine) spacer, and a pre-miR156b specific sequence from the 5′- to 3′-terminus sequence of this dsDNA. The in vitro transcription reaction for pre-miR156b was performed in a final volume of 50 μl containing 20 μl of dsDNA, 30 μl of in vitro transcription buffer, 100 U of ribonuclease inhibitor (Takara Biotechnology, Changchun, China), and 80 U of T7 RNA polymerase (Fermentas, Shanghai, China). The reaction mixture was incubated at 37°C for 4 h to amplify pre-miR156b, and then digested with 5 U of RNase-Free DNase I (Takara Biotechnology, Changchun, China). Finally, the above components were purified with an RNA cleanup and concentration kit (Shenggong, Shanghai, China) for 5 min. The concentration of the final pre-miR156b product was determined by analyzing its absorption at 260 nm with an ND-1000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE, United States).
For data analysis, a 10-fold dilution series of ligation products was used as a template for quantitative PCR assays. This was done to generate a plot of log copy numbers of miRNAs vs. the corresponding threshold of cycle (CT). The concentration of the target miRNA was detected through its linearity with CT, which spanned at least seven orders of magnitude. Slope and PCR amplification efficiency (E) were calculated according to the following equation: slope = −(1/log E). The relative detection rate of other miR156 family members was calculated as 1/2ΔCT, in which ΔCT = CT, other miRNA−CT, miR156b. The relative detection rate of miR156b was defined as 100% (Zhang et al., 2011).
The high specificity of T4 RNA ligase II is dependent on the 3′-hydroxyl and 5′-phosphoryl groups that are adjacent to either a duplex RNA or an RNA–DNA hybrid. Thus, the effect of the modification of two nucleotides on reaction efficiency was examined in the miR156b detection assay. After either probe A/probe B or probe A-N/probe B was hybridized with the target miRNA and ligated by the T4 RNA ligase II, the ligation product was used as an initial template for PCR amplification. As seen in the real-time fluorescence signal in Figure 2A, templates with concentrations as low as 2 nM could be accurately detected by the probe A/probe B set in the ligation step, with CT values ranging from 24.47 to 24.91 in triplicate. However, under the same experimental conditions, the ligation product using probe A-N/probe B had a CT value ranging from 27.48 to 28.25 after PCR amplification was completed. These results demonstrate that although T4 RNA ligase II could directly catalyze DNA nick ligation in a way, ribonucleotide modification at the 3′-terminus greatly improved catalytic efficiency, and was more conducive to ligation-based PCR amplification.
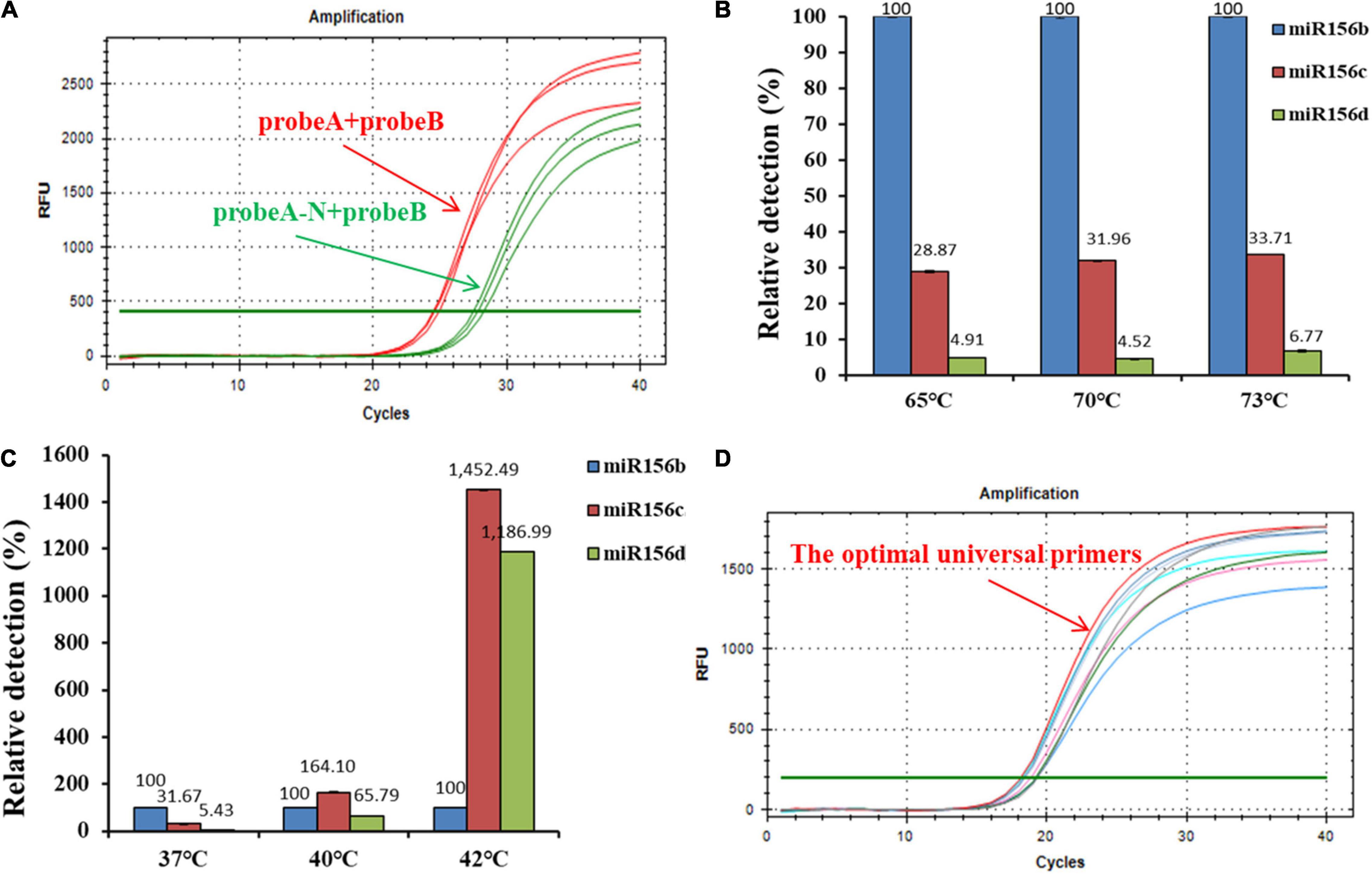
Figure 2. Optimization of ligation-based assays. (A) Analytical performance of deoxyribonucleic acid (DNA) probes modified with two ribonucleotides for reaction efficiency; probe A-N was a control probe without any ribonucleotide modification at its 3′-termini. (B) Effect of hybridization temperature on detection specificity. (C) Effect of ligation temperature on detection specificity. (D) Screening of optimal universal primers for amplification assays; red line: universal primer with the lowest GC content.
The ability to distinguish similar miRNAs is essential for miRNA detection. The central goal of the developed ligation-based qPCR assay was to establish the appropriate reaction conditions to improve the specificity and reduce the unexpected ligation and hybridization with non-target miRNAs. To ensure high ligation reaction efficiency, the hybridization and ligation temperatures were optimized. In this ligation reaction, miR156b, miR156c, and miR156d were selected as experimental templates based on the similarity of the sequences. The results of the real-time fluorescence signal (Figures 2B,C) show that the hybridization temperature (from 65 to 73°C) had no consequence on the specificity to identify target miR156b, whereas the specificity of the ligation reaction decreased with increasing ligation temperature (from 37 to 42°C). When the ligation temperature was >40°C, the fluorescence signals of interference generated by miR156c were significantly enhanced compared to those of miR156d and miR156b. When the ligation temperature was set at 40 or 42°C, with equivalent molar amounts of the miRNA template, the cycle value of miR156b amplification was later than that at 37°C (Supplementary Figure 1). Data indicate that the initial amount of miR156 used for amplification was reduced in the real-time PCR assay. Based on the above mentioned comprehensive experimental results, 37°C was considered as the optimal ligation temperature to ensure detection specificity.
The amplification efficiency of quantitative real-time PCR depends on the oligonucleotide primer sequences. In order to produce well-defined real-time fluorescence signals, several combinations of forward and reverse primers with different lengths were designed for PCR amplification. As depicted in Figure 2D and Supplementary Table 1, real-time fluorescence curves were generated by the three primer pair combinations. The CT values of target miR156b ranged from 18.15 to 19.33. Although there was no significant difference between the experimental data of each group, the primer pair with the smallest CT values was still employed as the optimal universal primer pair (forward primer: 5′-CGACGGTAGCAAGCATAGAGTGTGTGA-3′; reverse primer: 5′-GGGATACTGGAACCTGATGATGACTAACTAACTACC-3′) for subsequent experiments. As the GC content was closely related to the annealing temperature, it is speculated that the lower GC content can promote the binding of primer and target template to a certain extent.
Using the systematically optimized reaction parameters, a standard curve for evaluating the dynamic range and sensitivity of the ligation-based qPCR method was established via the 10-fold series dilution of ligation products as quantitative templates, spanning over at least seven orders of magnitude (miR156b inputs in the range of 1 fmol–0.0001 amol). This calibration method is superior to the delta threshold of cycle (CT) method in terms of its detection limits (Taverniers et al., 2004). The quantitative PCR assay targeting miR156b further demonstrated an excellent linearity between the CT value and the log of the target concentration in the range of 1 fmol–0.0001 amol. The correlation equation was CT = −3.157 log CmiRNA + 25.766. The proposed ligation-based quantitative system showed a corresponding PCR efficiency of 107.4%, and the squared regression coefficient (R2) of the standard curve was 0.983 for the three replicates (Figure 3). These results show that there was a good linear relationship between the target substance and PCR CT value, over a wide range of concentrations. Notably, miR156b could be detected at concentrations as low as 0.0001 amol, demonstrating that the developed quantitative method has good sensitivity.
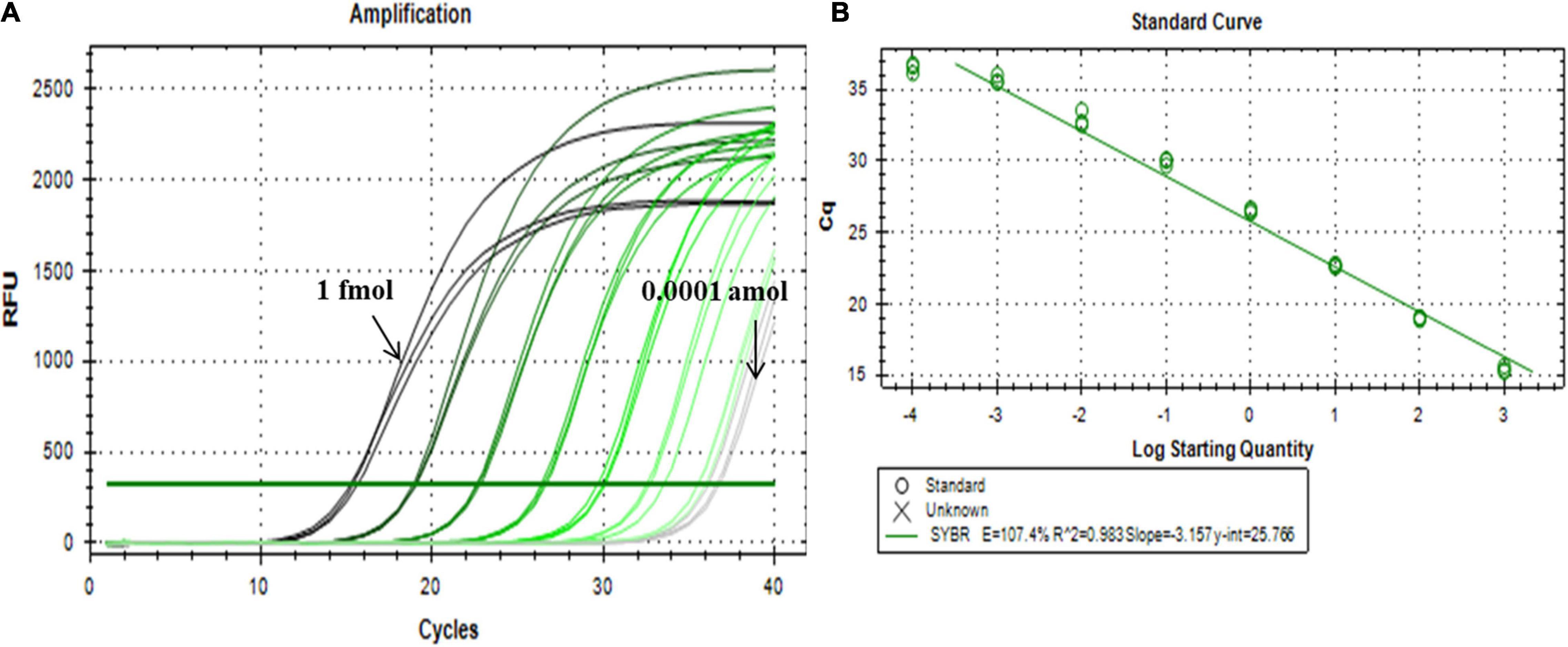
Figure 3. Dynamic range and sensitivity of the synthetic miR156b assay over eight orders of magnitude. (A) From left to right, the concentrations of synthetic miR156b is 1 fmol, 100 amol, 10 amol, 1 amol, 0.1 amol, 0.01 amol, 0.001 amol, and 0.0001 amol. (B) Dynamic relationship between CT value and log of miR156b concentration; E: polymerase chain reaction (PCR) efficiency, R2: squared regression coefficient of the standard curve.
The miR156 family contains seven highly homologous miRNAs with only one to a few mismatched nucleotides. To evaluate the specificity of the proposed qPCR method for miRNA detection, probes A and B were selected to verify whether the said method could identify other miR156 family members with different sequences. As shown in Figure 4, miR156b was identified, and each miR156 sequence obtained a different relative detection rate. Compared to miR156b, both miR156c and miR156f differed by only a single adenine nucleotide; the single-base nucleotide in miR156c was located at the 3′-terminus of probe B. Non-specific detection (31.48%) was higher than that of miR156f (11.89%), whose mature sequence was consistent with miR156b except for the star sequence (Figures 4C,D). This difference may further affect the secondary structure. The analytical data stored in the online database PmiREN (see text footnote 1) also confirms this aspect of miRNA. These results demonstrate that the secondary structure can affect specificity by influencing the binding between the probe and target miRNA to some extent. Both miR156a and miR156d had a mismatched nucleotide at the same position on the 3′-terminus of probe A, but another differential nucleotide in miR156a was located at the 5′-terminus of the mature miRNA sequence, while that of miR156d was at the 3′-terminus of the mature miRNA sequence. The higher non-specific detection determined for miR156a (9.61%) indicates that the method’s specificity was affected not only by the number of differential nucleotides, but also by the location. In addition, two negative controls were established to verify the effect of the ligation between DNA probes on the fluorescence signals, one was a quantitative reaction without any ligation product (NTC), the other was a negative control used for the miRNA-free ligation product as the amplification template (blank). Neither of the negative controls generated notable fluorescence signals. Therefore, it can be inferred that the two DNA probes ligate each other in the absence of the target miRNA to produce a minimal effect on the quantitative results. Collectively, the proposed ligation-based qPCR method can characterize with a high specificity, discriminate differences as small as a single nucleotide, and detect secondary structural differences among miRNA targets.
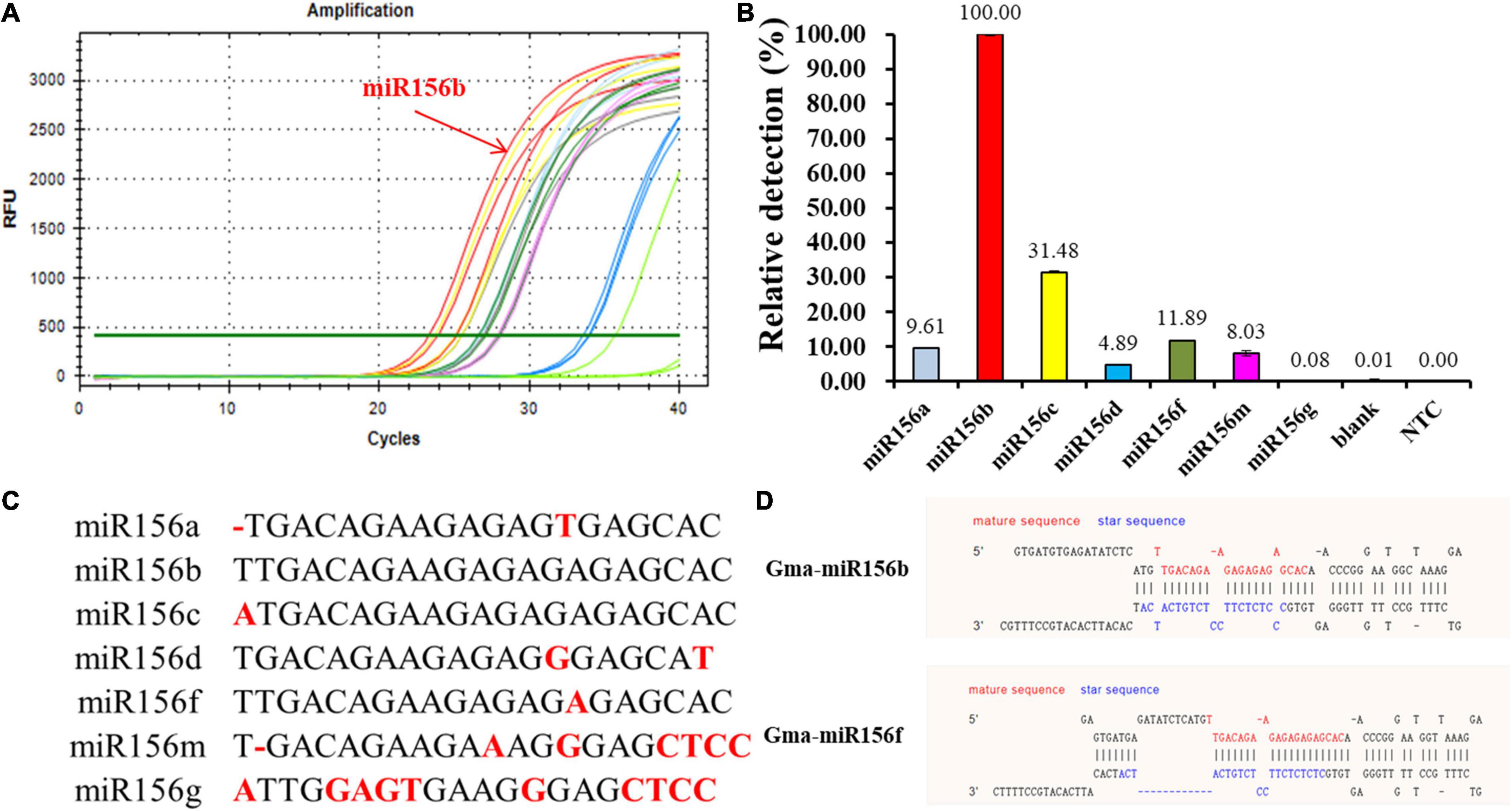
Figure 4. Evaluation of specificity among miR156b family. (A) Amplification plot of synthetic miR156 family members; red line: miR156b; the other color lines: the highly homologous miR156 members. (B) Relative detection rate of miR156 family members. Blank was a negative control used miRNA-free ligation product as the amplification template; NTC was a quantitative reaction without any ligation product. (C) The alignment of all miR156 sequences. Nucleotides that differ from those in miR156b are highlighted. (D) The structural difference between GmmiR156b and GmmiR156f.
A different miRNA (miR156g) was randomly selected to verify the universal practicability of the developed ligation-based quantitative PCR methods. To this end, specific probes A-g and B-g were designed with modified ribonucleotides containing the target-specific sequence that is complementary to the half sequence of miR156g. The stuffer sequence and universal sequences for PCR amplification were the same as those for probes A and B. Distinct results can be observed in Figures 5A–C, where equivalent amounts of miR156b and miR156g with different probes hybridized with respective or adverse probes produced well-defined signals. When using miR156b-specific probes (probe A and probe B) to perform the ligation reaction, and calculate the relative detection level with the formula:
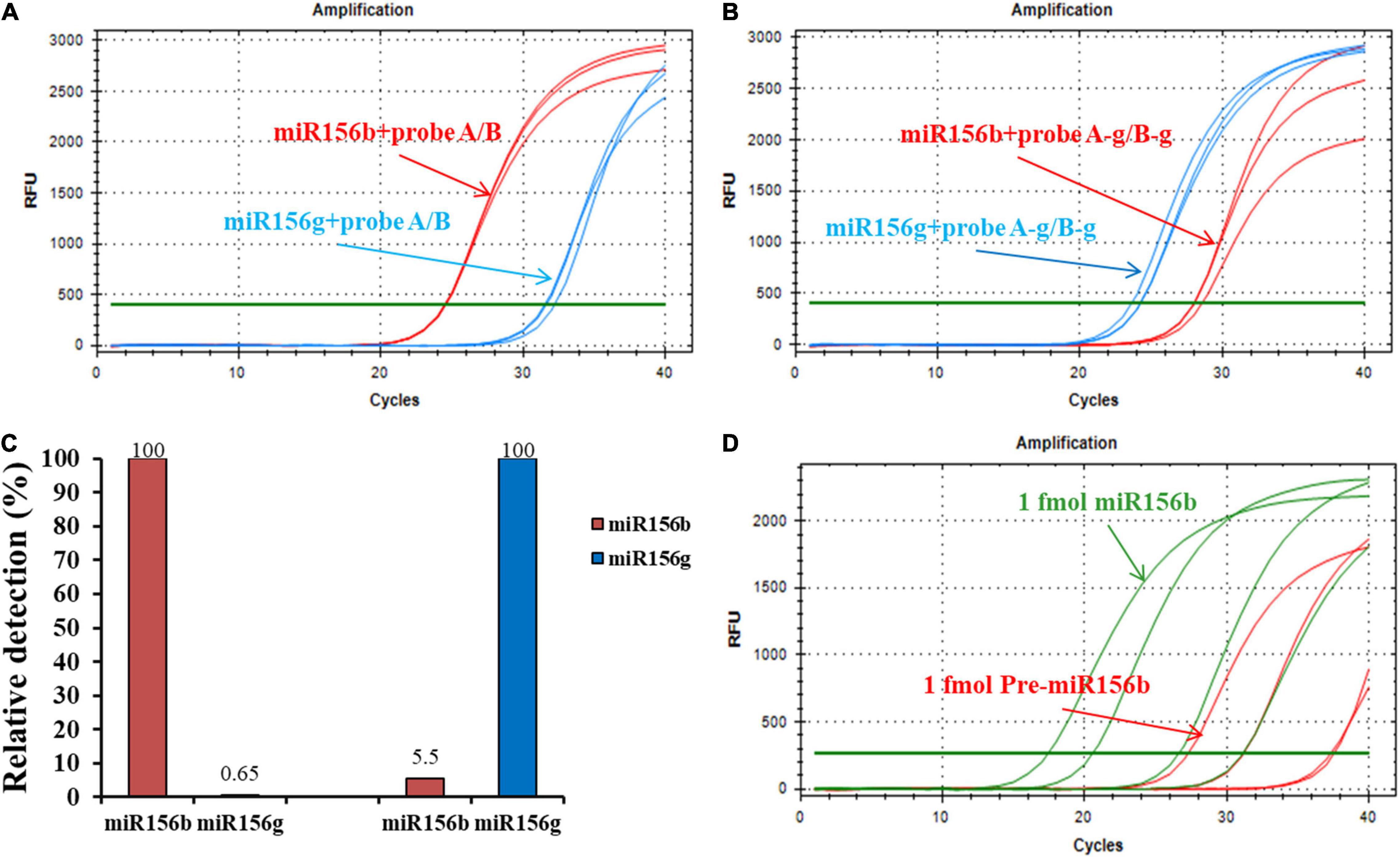
Figure 5. Generality and practicability of the ligation-based assays for detection of different miRNAs. (A) Amplification plot of miR156b and miR156g with probe A/B. (B) Amplification plot of miR156b and miR156g with probe A-g/B-g. (C) Relative detections for miR156b and miR156g with different target miRNAs. (D) Specificity of developed method in distinguishing miRNAs from their corresponding pre-miRNAs. From left to right in red line, the concentrations of synthetic miR156b is 1 fmol, 100 amol, 10 amol, 1 amol; from left to right in green line, the concentrations of synthetic pre-miR156b is 1 fmol, 100 amol, 10 amol, 1 amol.
The non-specific detection rate using miR156g reached 0.64%. For the target miRNA, the non-specific detection using miR156b was 5.5% while using miR156g-specific probes. These results demonstrate that the method has good generality and practicability for the detection of different miRNAs. The high specificity of ligation-based method depends on the complementary degree of the probes and the target sequence, but not the target miRNA sequence itself.
Pre-miRNA is the precursor of mature miRNA; it has stem-loop hairpin secondary structures that can be further cleaved by the dicer-like 1 (DCL1) and HYPONASTIC LEAVES1 (HYL1) into an miRNA: miRNA* (* represents secondary structure) duplex in the nucleus (Sun, 2012). To further investigate the pre-miRNA that were present in the sample on their specificity for miRNA detection, the pre-miRNA of miR156b (pre-miR156b) was prepared using the in vitro transcription reaction. The template concentration for the ligation reaction for pre-miR156b was determined to be in the range of 1 fmol–1 amol. The amplification plots of miR156b and pre-miR156b with the ligation-based qPCR assay showed that pre-miRNA produced negligible signals at concentrations below 10 amol (Figure 5D). In the template concentration range from 1 fmol to 10 amol, each CT value of pre-miR156b was nearly ten cycles later than the corresponding miR156b, equating to 0.1% of the non-specific detection. These experimental data indicate that the developed ligation-based quantitative method has high specificity in distinguishing miRNAs from their corresponding pre-miRNAs.
As shown in Figure 6A, when soybean DNA was used as a template, no signal was produced in the reaction. This confirmed that the RNA sample reaction was not affected, even if some DNA was present. An equivalent amount of RNA extracted from GM and WT seedlings generated well-defined fluorescence signals in the ligation-based qPCR (Figure 6B), but it was evident that miR156b was expressed in GM soybean much more than in WT soybean. The synthesis miRNA156b range from 1 fmol to 0.0001 amol was selected for standard curve construction. As shown in Figures 6C,D, there was good linearity between CT and the log of total RNA from 10 to 0.4 μg in the quantified assays. The data show that PCR efficiency was 101.7% and the R2 of the standard curve was 0.979. Based on this standard curve, miR156b levels were determined in both the GM and WT samples, revealing that the fold change of miR156b expression level in the GM plants was 5.08 times higher using the established ligation-based quantitative method. These results are quite similar to the that of the developer’s research data (4.82-fold change). Therefore, the established ligation-based method has the potential to detect and quantify GM samples using a miRNA mechanism.
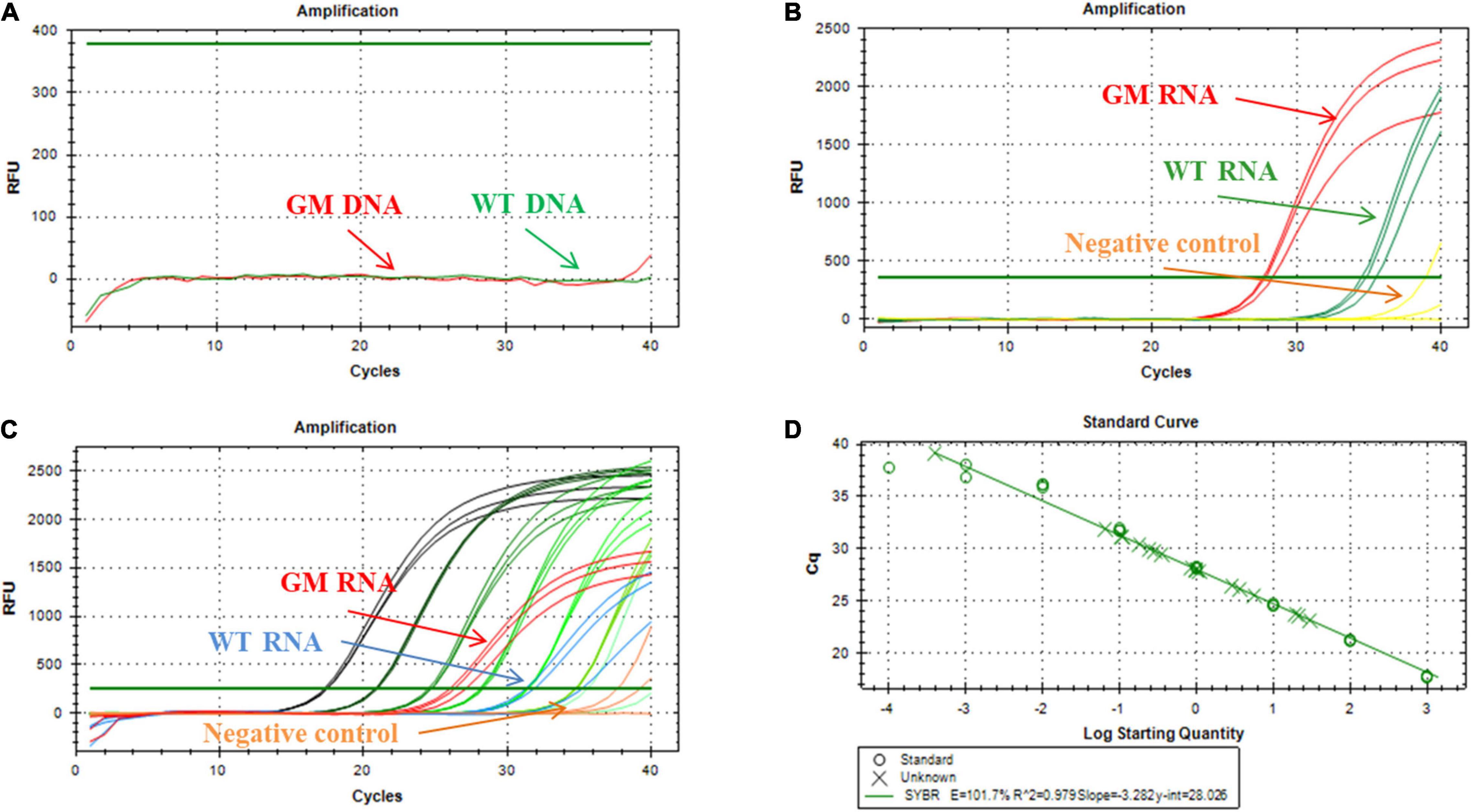
Figure 6. Application of ligation-based assay for miRNA detection in genetically modified (GM) soybean. (A) Amplification plot of miR156b using deoxyribonucleic acid (DNA) as template, the total DNA was extracted from WT and GM soybean, respectively. (B) Amplification plot of miR156b using RNA as template, the total RNA was extracted from WT and GM soybean, respectively; red line: GM plants, green line: WT plants, yellow line: negative control (NTC). (C) Dynamic range and quantification of miR156b assay; red line: total RNA samples of GM plants, blue line: total RNA samples of WT plants, yellow line: negative control. (D) Correlation of total RNA input to CT values for miR156b detection. The total RNA input ranged from 10 to 0.4 μg per ligation reaction.
In summary, this study experimentally demonstrated an ultrasensitive fluorescence method for miRNA quantification by elegantly integrating sensitivity and specificity. Under the hybridization procedure followed by DNA nick ligation catalyzed by T4 RNA ligase II, the initial template for miRNA amplification was obtained and quantified by quantitative real-time PCR. After optimizing the influential conditions, the proposed method could accurately discriminate among various miRNAs even when the difference was as small as a single nucleotide mismatch or the secondary structure difference leading by the nucleotides location site. High specificity was also achieved in the detection of mature miRNAs against their precursor sequences. In addition, the practicability of the proposed ligation-based quantitative PCR was further confirmed in that the total DNA and RNA samples from GM and WT soybeans generated well-defined fluorescence signals in a satisfactory dynamic range. The fold change of miR156b expression level compared well with data obtained by the conventional stem-loop qPCR, which further confirmed the reliability of the method. In contrast to the advantages and disadvantages of previously described methods for miRNA detection with data side-by-side (Table 2), the DNA probes and universal primers used in this method can be simply designed, without the need for complex structures such as the stem-loop reaction (Mei et al., 2012) or RCA assay (Liu et al., 2019), operators only need to substitute the specific sequences complementary to half of the target miRNA. Moreover, this method uses SYBR Green I and a small amount of ribonucleotide-modified DNA probes, meaning that it is also more cost efficient than that of the TaqMan probe. This characteristic greatly improves the method’s efficiency and convenience in the quantification and detection of various miRNAs. Combining the detection limit and specificity results investigated with its ability to distinguish miRNA from the other similar sequences, the simple operation and feasibility of this ligation-based PCR method implies that it can become a potential tool in GM content analysis.
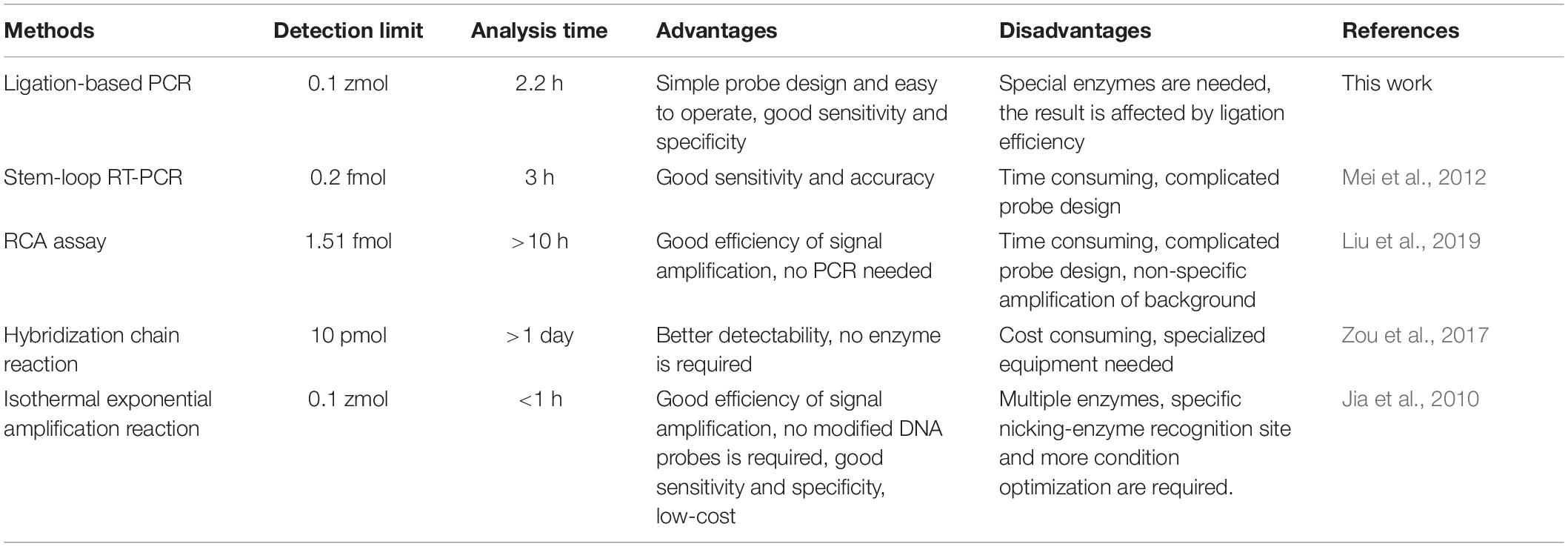
Table 2. The comparison among ligation-based polymerase chain reaction (PCR) and the other similar methods for miRNA detection.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YH contributed to the conception and design. LL and WY performed the main laboratory and wrote the manuscript. LD and WX carried out the data analysis and interpretation. CL provided the direction. FL drafted the manuscript and critically revised it for important intellectual content. All authors read and approved the manuscript.
Postdoc Foundation of Jilin Agricultural Technological Innovation Project, Jilin Academy of Agricultural Sciences, China (CXGC2020RCB002); High-level Talents Research Start-up Foundation of Jilin Agricultural Technological Innovation Project, Jilin Academy of Agricultural Sciences, China (CXGC2021RCG002); and the IAQST Foundation from Jilin Academy of Agricultural Sciences, Changchun, China (ZBS20Y02) supported this research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.794752/full#supplementary-material
Agrawal, A., Rajamani, V., Reddy, V. S., Mukherjee, S. K., and Bhatnagar, R. K. (2015). Transgenic plants over-expressing insect-specific microRNA acquire insecticidal activity against Helicoverpa armigera: an alternative to Bt-toxin technology. Transgenic Res. 24, 791–801. doi: 10.1007/s11248-015-9880-x
Aslam, M., Sugita, K., Qin, Y., and Rahman, A. (2020). Aux/IAA14 regulates microRNA-mediated cold stress response in Arabidopsis roots. Intern. J. Mol. Sci. 21:8441. doi: 10.3390/ijms21228441
Bally, J., Fishilevich, E., Doran, R. L., Lee, K., de Campos, S. B., German, M. A., et al. (2020). Plin-amiR, a pre-microRNA-based technology for controlling herbivorous insect pests. Plant Biotechnol. J. 18, 1925–1932. doi: 10.1111/pbi.13352
Cao, D., Li, Y., Wang, J., Nan, H., Wang, Y., Lu, S., et al. (2015). GmmiR156b overexpression delays flowering time in soybean. Plant Mol. Biol. 89, 353–363. doi: 10.1007/s11103-015-0371-5
Cheng, Y., Zhang, X., Li, Z., Jiao, X., Wang, Y., and Zhang, Y. (2009). Highly sensitive determination of microRNA using target-primed and branched rolling-circle amplification. Angew. Chem. Intern. Edn. 48, 3268–3272. doi: 10.1002/anie.200805665
Chopperla, R., Mangrauthia, S. K., Rao, T. B., Balakrishnan, M., Balachandran, S. M., Prakasam, V., et al. (2020). A comprehensive analysis of MicroRNAs expressed in susceptible and resistant rice cultivars during Rhizoctonia solani AG1-IA infection causing sheath blight disease. Intern. J. Mol. Sci. 21:7974. doi: 10.3390/ijms21217974
Curaba, J., Singh, M. B., and Bhalla, P. L. (2014). miRNAs in the crosstalk between phytohormone signalling pathways. J. Exper. Bot. 65, 1425–1438. doi: 10.1093/jxb/eru002
De la Rosa, C., and Reyes, J. L. (2019). “Northern blot analysis of microRNAs and other small RNAs in plants,” in Plant MicroRNAs. Methods in Molecular Biology, Vol. 1932, ed. S. de Folter (New York, NY: Humana Press), 121–129. doi: 10.1007/978-1-4939-9042-9_9
Dell’Aversana, C., Giorgio, C., and Altucci, L. (2017). MicroRNA expression profiling using agilent one-color microarray. Methods Mol. Biol. 1509, 169–183. doi: 10.1007/978-1-4939-6524-3_16
Flores, T., Karpova, O., Su, X., Zeng, P., and Bilyeu, K. (2008). Silencing of GmFAD3 gene by siRNA leads to low α-linolenic acids (18:3) of fad3-mutant phenotype in soybean [Glycine max (Merr.)]. Transgenic Res. 17, 839–850. doi: 10.1007/s11248-008-9167-6
Guo, C., Xu, Y., Shi, M., Lai, Y., Wu, X., Wang, H., et al. (2017). Repression of miR156 by miR159 regulates the timing of the juvenile-to-adult transition in Arabidopsis. Plant Cell 29, 1293–1304. doi: 10.1105/tpc.16.00975
Jia, H., Li, Z., Liu, C., and Cheng, Y. (2010). Ultrasensitive detection of microRNAs by exponential isothermal amplification. Angew. Chem. Intern. Edn. 49, 5498–5501. doi: 10.1002/anie.201001375
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Li, W., and Ruan, K. (2009). MicroRNA detection by microarray. Anal. Bioanal. Chem. 394, 1117–1124. doi: 10.1007/s00216-008-2570-2
Liu, C., Han, J., Zhou, L., Zhang, J., and Du, J. (2020). DNAzyme-based target-triggered rolling-circle amplification for high sensitivity detection of microRNAs. Sensors 20:2017. doi: 10.3390/s20072017
Liu, X., Zou, M., Li, D., Yuan, R., and Xiang, Y. (2019). Hairpin/DNA ring ternary probes for highly sensitive detection and selective discrimination of microRNA among family members. Anal. Chim. Acta 1076, 138–143. doi: 10.1016/j.aca.2019.05.027
Mei, Q., Li, X., Meng, Y., Wu, Z., Guo, M., Zhao, Y., et al. (2012). A facile and specific assay for quantifying MicroRNA by an optimized RT-qPCR approach. PLoS One 7:e46890. doi: 10.1371/journal.pone.0046890
Miao, C., Wang, D., He, R., Liu, S., and Zhu, J.-K. (2020). Mutations in MIR396e and MIR396f increase grain size and modulate shoot architecture in rice. Plant Biotechnol. J. 18, 491–501. doi: 10.1111/pbi.13214
Miao, C., Wang, Z., Zhang, L., Yao, J., Hua, K., Liu, X., et al. (2019). The grain yield modulator miR156 regulates seed dormancy through the gibberellin pathway in rice. Nat. Commun. 10:3822. doi: 10.1038/s41467-019-11830-5
Papadopoulou, N., Devos, Y., Alvarez-Alfageme, F., Lanzoni, A., and Waigmann, E. (2020). Risk assessment considerations for genetically modified RNAi Plants: EFSA’s activities and perspective. Front. Plant Sci. 11:445. doi: 10.3389/fpls.2020.00445
Šečić, E., Kogel, K.-H., and Ladera-Carmona, M. J. (2021). Biotic stress-associated microRNA families in plants. J. Plant Physiol. 263:153451. doi: 10.1016/j.jplph.2021.153451
Shu, P., Wu, C., Liu, W., Ruan, X., Liu, C., Hou, L., et al. (2019). The spatiotemporal expression pattern of microRNAs in the developing mouse nervous system. J. Biol. Chem. 294, 3444–3453. doi: 10.1074/jbc.RA118.004390
Singroha, G., Sharma, P., and Sunkur, R. (2021). Current status of microRNA-mediated regulation of drought stress responses in cereals. Physiol. Plant. 172, 1808–1821. doi: 10.1111/ppl.13451
Sun, G. (2012). MicroRNAs and their diverse functions in plants. Plant Mol. Biol. Rep. 80, 17–36. doi: 10.1007/s11103-011-9817-6
Taverniers, I., Bockstaele, E. V., and Loose, M. D. (2004). Cloned plasmid DNA fragments as calibrators for controlling GMOs: different real-time duplex quantitative PCR methods. Anal. Bioanal. Chem. 378, 1198–1207. doi: 10.1007/s00216-003-2372-5
Tian, W., Li, P., He, W., Liu, C., and Li, Z. (2019). Rolling circle extension-actuated loop-mediated isothermal amplification (RCA-LAMP) for ultrasensitive detection of microRNAs. Biosens. Bioelectron. 128, 17–22. doi: 10.1016/j.bios.2018.12.041
Visentin, I., Pagliarani, C., Deva, E., Caracci, A., Turečková, V., Novák, O., et al. (2020). A novel strigolactone-miR156 module controls stomatal behaviour during drought recovery. Plant Cell Environ. 43, 1613–1624. doi: 10.1111/pce.13758
Wang, B., Wang, J., Wang, C., Shen, W., Jia, H., Zhu, X., et al. (2016). Study on expression modes and cleavage role of miR156b/c/d and its target gene Vv-SPL9 during the whole growth stage of grapevine. J. Heredity 107, 626–634. doi: 10.1093/jhered/esw030
Wei, S., Yu, B., Gruber, M. Y., Khachatourians, G. G., Hegedus, D. D., and Hannoufa, A. (2010). Enhanced seed carotenoid levels and branching in transgenic Brassica napus expressing the Arabidopsis miR156b gene. J. Agric. Food Chem. 58, 9572–9578. doi: 10.1021/jf102635f
Yin, S., Ho, C. K., and Shuman, S. (2003). Structure-function analysis of T4 RNA ligase 2. J. Biol. Chem. 278, 17601–17608. doi: 10.1074/jbc.M300817200
Zhang, J., Li, Z., Wang, H., Wang, Y., Jia, H., and Yan, J. (2011). Ultrasensitive quantification of mature microRNAs by real-time PCR based on ligation of a ribonucleotide-modified DNA probe. Chem. Commun. 47, 9465–9467. doi: 10.1039/c1cc13466c
Zhang, P., Zhang, J., Wang, C., Liu, C., Wang, H., and Li, Z. (2013). Highly sensitive and specific multiplexed microRNA quantification using size-coded ligation chain reaction. Anal. Chem. 86, 1076–1082. doi: 10.1021/ac4026384
Zhuang, F., Fuchs, R. T., Sun, Z., Zheng, Y., and Robb, G. B. (2012). Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 40:e54. doi: 10.1093/nar/gkr1263
Keywords: MiRNAs, ribonucleotide-modified DNA probe, ligation reaction, quantitative method, MiR156b
Citation: He Y, Long L, Yan W, Dong L, Xia W, Li C and Li F (2021) Establishment and Application of Ligation Reaction-Based Method for Quantifying MicroR-156b. Front. Plant Sci. 12:794752. doi: 10.3389/fpls.2021.794752
Received: 14 October 2021; Accepted: 25 November 2021;
Published: 14 December 2021.
Edited by:
Xianzhong Feng, Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Fabrizio Cillo, Institute for Sustainable Plant Protection, National Research Council (CNR), ItalyCopyright © 2021 He, Long, Yan, Dong, Xia, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feiwu Li, bGlmZWl3dTMzOTRAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.