 Sang-Ho Kang
Sang-Ho Kang Woo-Haeng Lee2†
Woo-Haeng Lee2† Tae-Jin Oh
Tae-Jin Oh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 January 2022
Sec. Plant Biotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.773553
Senna occidentalis is an annual leguminous herb that is rich in anthraquinones, which have various pharmacological activities. However, little is known about the genetics of S. occidentalis, particularly its anthraquinone biosynthesis pathway. To broaden our understanding of the key genes and regulatory mechanisms involved in the anthraquinone biosynthesis pathway, we used short RNA sequencing (RNA-Seq) and long-read isoform sequencing (Iso-Seq) to perform a spatial and temporal transcriptomic analysis of S. occidentalis. This generated 121,592 RNA-Seq unigenes and 38,440 Iso-Seq unigenes. Comprehensive functional annotation and classification of these datasets using public databases identified unigene sequences related to major secondary metabolite biosynthesis pathways and critical transcription factor families (bHLH, WRKY, MYB, and bZIP). A tissue-specific differential expression analysis of S. occidentalis and measurement of the amount of anthraquinones revealed that anthraquinone accumulation was related to the gene expression levels in the different tissues. In addition, the amounts and types of anthraquinones produced differ between S. occidentalis and S. tora. In conclusion, these results provide a broader understanding of the anthraquinone metabolic pathway in S. occidentalis.
Senna occidentalis (L.) Link (subfamily, Caesalpinioideae; family, Leguminosae; also known as Cassia occidentalis L.) is a toxic leguminous annual plant widely distributed across tropical and subtropical regions in Europe, Australia, the southern and eastern United States, South Africa, South Asia, and India (Tokania et al., 2000). It is also found in a variety of environments, including roadsides, pastures, forests, coastal environs, and crop fields (Vashishtha et al., 2009; Pal et al., 2019). S. occidentalis is widely consumed in various countries and harvested from the wild for various pharmacological purposes, such as the treatment of hypertension, diabetes, dropsy, biliousness, fever, ringworm, eczema, rheumatism, eye pain, hematuria, typhoid fever, and tuberculosis (Burkill, 1995; Gidado et al., 2016). S. occidentalis contains polyphenols and anthraquinone derivatives that exhibit a variety of pharmacological properties, including antibacterial, antifungal, antioxidant, antiplasmodial, and antiparasitic activities (Yen and Chen, 1995; Bakhiet and Adam, 1996; Luximon-Ramma et al., 2002; Chukwujekwu et al., 2005; Duraipandiyan and Ignacimuthu, 2007; Morita et al., 2007).
In addition to phytochemicals such as kaempferol, sitosterol, tannin, and xanthine, S. occidentalis also produces anthraquinone derivatives, such as aloe-emodin, chrysophanol, obtusin, and physcion (Kudav and Kulkarni, 1974; Chukwujekwu et al., 2005; Verma et al., 2010; Li and Li, 2017). Anthraquinones have been used to treat various diseases and possess strong pharmacological activities, such as laxative, anti-cancer, anti-bacterial, anti-inflammatory, anti-injury, and antioxidant properties (Jung et al., 2016). About 700 anthraquinone derivatives have been reported, of which 200 are present in plants (Duval et al., 2016). Anthraquinones are generally classified as anthraquinone monomers, which can be subdivided into hydroxy anthraquinones, anthranones, anthranols, and bianthraquinones, depending on the structure of the parental nucleus (Zhao et al., 2016). The phenolic hydroxyl group of the anthracene nucleus has a great influence on its anti-cancer properties (Li and Jiang, 2018). In addition, anthraquinone monomers such as emodin, aloe-emodin, chrysophanol, and physcion have various physiological activities that might enable them to be used in treatments targeting cancer, viruses, and depression.
The secondary metabolites of an organism, such as anthraquinone derivatives, are produced through a variety of precursors and biosynthesis pathways. In particular, the most important gatekeepers in the biosynthesis of secondary metabolites are the enzymes that biosynthesize the backbones of the metabolites. The type III polyketide synthase (PKS) enzymes are found in bacteria, fungi, and plants (Austin and Noel, 2003; Austin et al., 2004; Funa et al., 2005; Seshime et al., 2005) and specialize in the production of important secondary metabolites (aromatic polyketides), which have potential nutraceutical, cosmeceutical, and pharmaceutical applications of increasing economic interest (Lauritano et al., 2017; Martínez et al., 2019). This enzyme has two independent active sites that catalyze a series of decarboxylation, condensation, and cyclization reactions (Tropf et al., 1995). The type III PKSs include chalcone synthase (CHS), stilbene synthase (STS), and acridone synthase (Dao et al., 2011), all of which have already been extensively studied in several plant species. One of the products of polyketide synthase, anthraquinone, is produced by bacteria, fungi, insects, and plants (Chiang et al., 2010; Shamim et al., 2014). Through several studies, two biosynthesis pathways for anthraquinone have now been proposed: the chorismite/O-succinyl benzoic acid pathway and the polyketide pathway (Burnett and Thomson, 1968; Bauch and Leistner, 1978). The former pathway was observed in the Rubiaceae plant family, while the latter is found in bacteria, fungi, and insects (Awakawa et al., 2009; Chiang et al., 2010).
Studies of anthraquinone biosynthesis in Senna tora have provided some information on secondary metabolites in the Senna genus. In a recent study, the anthraquinone scaffolds atrochrysone carboxylic acid and endocrocin anthrone were found in S. tora; these scaffolds are generated by the activity of chalcone synthase-like (CHS-L) enzymes (Kang et al., 2020b). Kang et al. identified 28 type III PKSs, among which 16 genes similar to CHS-L were systematically classified. Interestingly, the CHS-L gene group was only expanded in some species, in which it was identified as a separate gene subfamily, but was not expanded in another 11 species. The in vitro analysis of one CHS-L enzyme (CHS-L9) resulted in the generation of atrochrysone carboxylic acid and endocrocin anthrone, which are believed to be the backbone of anthraquinone, which confirmed that this enzyme is associated with the anthraquinone biosynthesis pathway in S. tora (Kang et al., 2020b).
This study aims to provide insights into the anthraquinone biosynthesis pathway through the transcriptomic analysis of S. occidentalis, which belongs to the same genus as S. tora. In our previous study, we presented the de novo transcriptome of S. tora, elucidating its anthraquinone biosynthesis pathway. Here, we reveal clear differences between S. occidentalis and S. tora through the transcriptomic and metabolomic analysis of S. occidentalis. Furthermore, we explore the variation in anthraquinone production in the different developmental stages and tissue types in these plants.
For the transcriptomic analysis, specimens of S. occidentalis (voucher number: IT 175099 from the National Agrobiodiversity Center, South Korea) grown in the field at the National Institute of Horticultural and Herbal Medicine (Eumseong, South Korea) were used. Leaf, root, and young and mature seed tissues were harvested from healthy plants and stored at −80°C until use for RNA extraction.
Total RNA was extracted from the leaf, root, and seed tissues using an RNeasy Plant Mini kit (Qiagen, Hilden, Germany). The RNA purity was measured using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States). Only samples with a minimum total RNA integrity value of 7 were used.
Poly(A)+ mRNA was twice purified from 1 μg total RNA using poly(T) oligo-attached magnetic beads and then fragmented. The randomly cleaved RNA fragments were reverse-transcribed into first-strand cDNA using reverse transcriptase, random hexamer primers, and dUTP. A single A base was added to these cDNA fragments, after which an adapter ligation was performed. The product was purified and enriched using polymerase chain reaction (PCR) to create the final-strand cDNA library. The quality of the amplified library was confirmed using capillary electrophoresis (bioanalyzer; Agilent Technologies). Quantitative PCR (qPCR) was conducted using SYBR Green PCR Master Mix (Thermo Fisher Scientific), after which equal amounts of the indexed tagged libraries were aggregated. The cBot-automated cluster creation system (Illumina, San Diego, CA, United States) was used to perform cluster generation in a flow cell. Sequencing was performed with a reading length of 2 × 100 base pairs (bp) by loading the flow cell into the HiSeq 2500 sequencing system (Illumina). Illumina short-read sequencing were performed in three biological replicates.
The library for Pacific Biosciences (PacBio; Menlo Park, CA, United States) Single Molecule Real Time (SMRT) sequencing was prepared from the aforementioned cDNAs. Cycle optimization was performed to determine the optimal number of cycles for the large PCRs. Three pools of cDNAs (1–2 kbp, 2–3 kbp, and 3–6 kbp) were prepared using the BluePipin size selection system. The SMRTbell library was configured using the SMRTbell Template Preparation Kit (PN 100-259-100). The DNA/Polymerase Binding Kit P6 (PacBio) was used for the DNA synthesis after the sequencing primers were annealed to the SMRTbell template. After the polymerase binding reaction, the library complex was combined with MagBeads and sequenced using the MagBead Kit. The MagBead-bound cDNA complex increases the number of reads per SMRT cell. This polymerization enzyme–SMRTbell–adapter complex was loaded into a zero-mode waveguide. The SMRTbell library was sequenced using eight SMRT cells (PacBio) with C4 chemistry (DNA Sequencing Reagent 4.0). Using the PacBio RS II sequencing platform, a 240-min movie was captured for each SMRT cell.
Raw transcriptomic data generated by the Illumina HiSeq were preprocessed to eliminate the nonsense sequences, including adapters, primers, and low-quality sequences (Phred quality score less than 20) using the NGS QC ToolKit (Patel and Jain, 2012). The raw data were further processed to remove ribosomal RNA using riboPicker v. 0.4.3 (Schmieder et al., 2012). The preprocessed reads were then assembled using Trinity (Haas et al., 2013), and the assembly statistics were calculated using an internal Perl script. The whole assembly was clustered to reduce the sequence redundancy (CD-HIT-EST v4.6.1) (Li and Godzik, 2006). Both the rank equality threshold and the alignment range (for short sequences) were set to 90% to create the clusters.
Genome and annotation data were downloaded for Glycine max (L.) Merr., Medicago truncatula Gaertn., Mimosa pudica L., and Chamaecrista fasciculata (Michx.) Greene. Gene models shorter than 10 amino acids or containing > 20% stop codons were removed, and OrthoMCL v. 2.0.31 was used to perform a Markov clustering algorithm to define the cluster structures (Li et al., 2003). An all-versus-all analysis was conducted using BLASTP v. 2.2.25 + (E-value threshold 10–5). The protein pairs obtained from the BLASTP analysis were analyzed using the OrthoMCL program.
Clean reads from each tissue were aligned with an abundant transcriptome assembly using Bowtie2 (Langmead and Salzberg, 2012). The aligned reads were quantified as fragments per kilobase of transcript per million reads (FPKM) for the non-redundant combined transcript sequences (90% sequence similarity determined using CD-HIT-EST). Read coefficients for the alignment were performed using RSEM (RNA-Seq by Extraction Maximization) v. 1.2.25 (Li and Dewey, 2011). The differential expression analysis was performed using the DESeq2 package (Anders and Huber, 2010). Differentially expressed genes (DEGs) were identified between the tissues using a combination of a more than twofold change in expression and a significance (P-value) threshold of 0.001, based on the three biological replicates.
All assembled unigenes were annotated through their comparison with the National Center for Biotechnology Information (NCBI) non-redundant (Nr) protein database, the Swiss-Prot protein database, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways database using the BLAST program (Altschul et al., 1990) with an E-value cutoff of 10–5. Whenever the results from the different databases conflicted, the results from the Swiss-Prot database were selected first, followed by the Nr database, and the KEGG database. A functional classification was performed using Gene Ontology (GO) terms (Ashburner et al., 2000) with the Blast2GO program (Conesa et al., 2005), with an E-value threshold of 10–5. AgriGO (Du et al., 2010) was used to identify any enrichment of the GO categories.
To investigate the putative transcription factor (TF) families in S. occidentalis, the unigenes were mapped for all TF protein sequences available in the Plant Transcription Factor Database (PlantTFDB v. 4.02) using BLASTX, with an E-value threshold of 10–5.
Following the manufacturer’s instructions, an RNeasy Plant Mini Kit (Qiagen) was used to extract the total RNA. The quality of the extracted RNA was checked on an ethidium bromide–stained agarose gel, and the concentration was calculated according to the optical density (OD) of the specimen measured at 260 and 280 nm (DropSense96c Spectrophotometer; PerkinElmer, Waltham, MA, United States). Using the SuperScript III first strand RT-PCR kit (Thermo Fisher Scientific) with oligo(dT)20 primers, 1 μg of RNA was used for the cDNA synthesis. After obtaining cDNA from S. occidentalis, qRT-PCR was performed using gene-specific primers (Supplementary Table 1). The RT-PCR analysis was optimized and performed using a Roche LightCycler 480 II instrument (Roche, Basel, Switzerland) and SYBR Green Real-Time PCR Master Mix (Bio-Rad Laboratories, Hercules, CA, United States) under an initial denaturation of 95°C for 30 s, followed by 40 cycles of denaturation for 15 s and annealing and elongation at 55°C for 10 s. The relative expression of certain genes was quantified using the 2–ΔΔCt calculation (Livak and Schmittgen, 2001), where ΔΔCt is the difference in the threshold number of cycles. The internal reference gene for the data normalization was ELONGATION FACTOR2. The reliability of the amplification parameters was analyzed from a 1:15 dilution of the cDNA sample. The average threshold cycle value for the gene of interest was calculated from three experimental replications.
Young- and mature-stage seed samples were extracted with methanol using sonication for 30 min at 60°C. After extraction, the samples were centrifuged at 12,000 rpm for 3 min at 25°C, and the supernatant was filtered with 0.2-μm Acrodisc MS Syringe Filters with a WWPTFE membrane (Pall Corporation, Port Washington, NY, United States). A quantitative analysis of the anthraquinones was performed using a Triple TOF 5600 + spectrometer with a DuoSpray ion source (AB Sciex, Framingham, MA, United States) coupled with a Nexera X2 UHPLC (Shimadzu, Kyoto, Japan) equipped with a binary solvent manager, a sample manager, a column heater, and a photodiode array detector. UHPLC was performed on an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 × 100 mm, Waters Corporation, Milford, MA, United States). The mobile phases consisted of 5 mM ammonium acetate in water (eluent A) and 100% acetonitrile (eluent B). The gradient profile was as follows: 0–1 min, 20% eluent B; 1–3.5 min, 20–30% eluent B; 3.5–8 min, 30–50% eluent B; 8–12 min, 50–100% eluent B; and 11–17 min, 100% eluent B. The flow rate was 0.5 mL/min. A total of 5 μL of each sample was injected. To detect the peaks from the test samples, the following MS parameters in ESI-negative mode were used: nebulizing gas, 50 psi; heating gas, 50 psi; curtain gas, 25 psi; desolation temperature, 500°C; and ion spray voltage floating, 4.5 kV.
The RNA-Seq and Iso-Seq sequences generated from the Illumina and PacBio RS II sequencing of the four tissue samples of S. occidentalis were deposited in the NCBI Sequence Read Archive database under the project number PRJNA631110.
De novo transcriptome analysis is a great method for generating genetic information for an organism without performing additional genomic sequencing. It can lead to the discovery of new genes and molecular markers and can elucidate tissue-specific expression patterns. We sequenced cDNA libraries from the leaf, root, and seeds of S. occidentalis using the Illumina HiSeq 2500 system and the PacBio RS II platform. The Illumina sequencing platform produced 26,917,977,630 raw reads and averaged 2,243,164,802 reads per tissue (Supplementary Table 2). In total, more than 2 billion reads were of high quality (Q30-values over 81%). The Trinity assembler for the four different libraries generated 121,592 unigenes over 300 bp in length (Table 1). The length of the transcripts varied from 300 to 18,086 bp, with an average length of 827 bp, an N50 length of 918 bp, and a GC content of 39.0%.
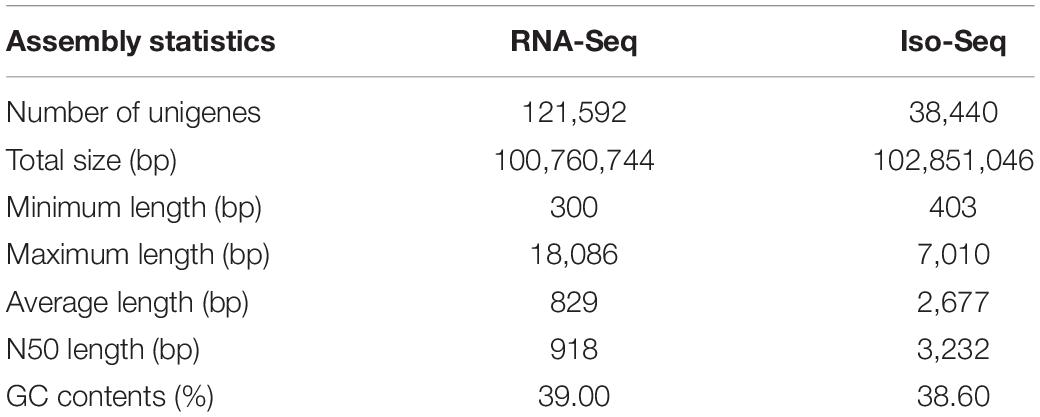
Table 1. Assembly statistics of the S. occidentalis transcriptome by RNA-seq and Iso-Seq.
The PacBio RS II sequencing platform produced 713,759 raw reads and 78,043 high-quality isoforms from three different libraries, which included 44,867, 34,858, and 42,274 high-quality isoforms for each of the sequencing sizes (< 2 kbp, 2–3 kbp, and > 3 kbp, respectively) (Supplementary Table 3). A total of 38,440 non-redundant unigenes were identified following the removal of redundant isoforms by the CD-HIT-EST program. The total size of the assembly was 102 Mbp, with a minimum unigene length of 403 bp and a maximum length of 7,010 bp. In total, our analysis generated two unigene sets: 121,592 unigenes from RNA-Seq and 38,440 unigenes from Iso-Seq (Table 1). Overall, the Iso-Seq produced longer reads than the RNA-Seq. The minimum length, average length, and N50 length were all confirmed to be longer for the Iso-Seq reads (Supplementary Figure 1); however, the GC contents for the assemblies produced by both sequencing methods were similar (Table 1).
To characterize transcripts and understand the complexity and diversity of an organism, we need to annotate the function of the genes. For the functional annotation, the assembled 121,592 unigenes obtained using RNA-Seq from four samples (leaf, root, and young- and mature-stage seed tissues) were screened using an FPKM criterion of ≥ 1. To obtain the best annotation, the resulting 61,147 assembled RNA-Seq unigenes and 38,440 Iso-Seq unigenes from S. occidentalis were aligned with five public protein databases. We used the BLASTX program to search the unigene sequences against NCBI Nr, Clusters of Orthologous Groups of proteins (COGs), KEGG, GO, and Swiss-Prot databases with an E-value threshold of 1e–5, enabling the unigenes to be annotated with the function of matching known proteins. The Venn diagram in Supplementary Figure 2 displays the best BLASTX hits unique to the NCBI Nr, COG, KEGG, GO, and Swiss-Prot databases. A total of 43,809 of the RNA-Seq sequences were matched to the NCBI Nr database (66.28%), 53,965 to Swiss-Prot (81.64%), 50,952 to GO (77.08%), 24,171 to KEGG (36.57%), and 3,324 to COG (5.03%) (Supplementary Figure 2A), with the Swiss-Prot database providing the most matches. In contrast, 36,027 of the Iso-Seq sequences were matched to the NCBI Nr database (99.74%), 29,759 to Swiss-Prot (82.39%), 26,134 to GO (72.35%), 12,712 to KEGG (35.19%), and 6,473 to COG (17.92%) (Supplementary Figure 2B), with the most matches in the NCBI Nr database. New genes, long non-coding RNAs, or non-significant genes that could represent less-conserved untranslated regions were not evaluated in this annotation and require further analysis.
To further functionally characterize the S. occidentalis transcripts, we classified the functions of the RNA-Seq and Iso-Seq unigenes using a GO analysis. The distribution of the RNA-Seq and Iso-Seq unigene sets in different GO categories is shown in Supplementary Figure 3. The three main categories of GO annotation for RNA-Seq included 26,579 unigenes associated with GO terms for biological process (42.77%), 20,088 for molecular function (32.32%), and 15,482 for cellular component (24.91%). The most widely distributed GO terms in the main categories were 4,470 unigenes annotated with organic substance metabolic process functions in biological processes (17% of unigenes), with 3,739 and 3,736 unigenes associated with the GO terms of organic cyclic compound binding and heterocyclic compound binding, respectively, in molecular function (each 19% of unigenes), and 3,418 unigenes annotated as cell part and cells in cellular component (22% of unigenes). Conversely, for the Iso-Seq data, 58,931 unigenes were associated with GO terms for biological process (47.03%), 29,411 for molecular function (23.47%), and 36,973 for cellular component (29.50%). The most abundant GO categories in each main category were organic substance metabolic process in biological process (16% of unigenes), nucleotide binding and nucleoside phosphate binding in molecular function (16% of unigenes), and cell parts and cells in cellular component (24% of unigenes). This showed that the GO term patterns for RNA-Seq and Iso-Seq were similar (Supplementary Figure 3).
Members of TF families, such as ARF, bHLH, and C2H2, bind to specific regulatory elements in the promoters of their target genes to regulate their expression. TFs thus play a key regulatory role in the expression of genes involved in the biosynthesis and signaling of plant secondary metabolites and the response to environmental stress. The number of genes encoding different TFs varies between species as they are evolved to perform species-specific or developmentally specific functions (Yang et al., 2012). In this study, 6,422 unigenes (3,179 RNA-Seq and 3,243 Iso-Seq unigenes) were allocated to 58 TF groups. Among them, bHLH genes (544, 17.11%) were found to be the most abundant in the RNA-Seq dataset, followed by WRKYs (205, 6.45%), MYBs (185, 5.82%), and bZIPs (179, 5.63%). Similarly, bHLH genes (639, 19.70%) were also the most abundant TF group in the Iso-Seq dataset, although the next most abundant were C3Hs (305, 9.40%), bZIPs (201, 6.20%), and WRKYs (193, 5.95%) (Supplementary Figure 4).
To study the relationships between S. occidentalis and other plant species, we categorized the transcripts into gene families and compared them with the gene families from four Fabaceae species: Chamaecrista fasciculata, Mimosa pudica, Glycine max, and Medicago truncatula. We found that 6,948 gene families were shared by all five species, whereas 1,900 gene families (containing 3,231 genes) were specific to S. occidentalis (Figure 1A). These S. occidentalis–specific genes included those encoding serine threonine-protein kinase, UDP-glucose 4-epimerase, transposons, and GATA TF family member proteins. A GO analysis of these novel gene families revealed that most of them are involved in metabolic processes and cell proliferation (Figure 1B). In addition, we showed that the S. occidentalis sequences showed a 38% similarity to those of Glycine max, 14% to Phaseolus vulgaris, and 9% to Medicago truncatula, suggesting that S. occidentalis was most closely related to Glycine max in the Fabaceae family (Supplementary Figure 5).
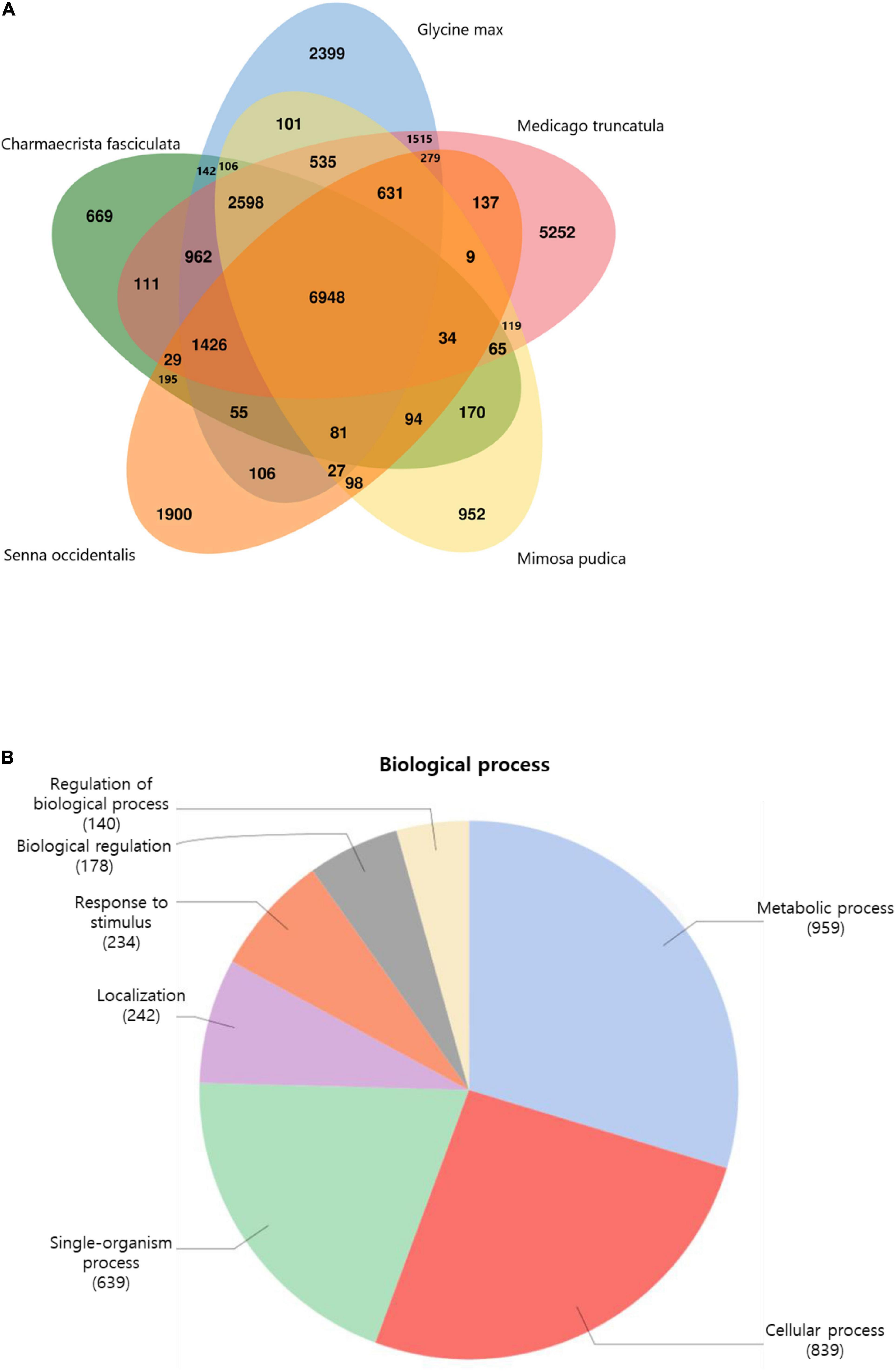
Figure 1. Histogram of gene family distribution between legume plants. (A) The gene family distribution between Senna occidentalis (Iso-Seq dataset) and other plants. (B) Gene Ontology analysis of the novel gene families of S. occidentalis associated with the biological process categories.
The expression of genes varies depending on the species, developmental stage, or the environment to which the organism is exposed, and many genes are simultaneously expressed depending on the specific circumstances. Analyzing the expression levels of the genes encoding TFs is essential for understanding the transcriptional regulation of an organism. Accordingly, we studied tissue-specific TF gene expression in the leaves, roots, and developing seeds of S. occidentalis (Supplementary Table 4). Different TF expression patterns were observed in each S. occidentalis tissue. First, 68, 173, 99, and 50 TF-encoding genes were found to be expressed in the leaves, roots, and young- and mature-stage seeds, with 41, 102, 79, and 31 specific to each tissue, respectively. The TFs specifically expressed in the young seeds included members of the ARF, B3, CPP, LBD, Nin, and YABBY families, while in the mature-stage seeds, GRF and HD-ZIP family members were specifically expressed. Auxin response factors are TFs that play an important role in the expression of genes regulating the pathways for auxin, a plant hormone (Guilfoyle et al., 1998). In addition, the B3 TF family regulates various functions of plant growth and development (Peng and Weselake, 2013), while YABBY TFs are involved in a variety of activities, such as the response to plant hormone signals, stress tolerance, and the formation and development of reproductive organs (Zhang et al., 2020). On the contrary, GRF genes, which were specifically expressed in the mature seeds, are plant-specific TFs originally shown to function in stem and leaf development (Omidbakhshfard et al., 2015); however, recent research has emphasized their importance in other central developmental processes, including seed formation. The expression of GRF genes was also observed in rice (Oryza sativa L.) and maize (Zea mays L.), suggesting that it is involved in seed development (Zhang et al., 2008; Liu et al., 2012).
To compare the differential expression of the S. occidentalis genes in mature-stage seed development compared with young-stage development, we used the DESeq method. The transcripts with a log2 fold change (FC) > 1 and a p-value < 1e–3 were considered to be DEGs. A pair-wise comparison of the transcripts from the young and mature stages of seed development resulted in 8,874 identified DEGs in the RNA-Seq dataset. As the seeds matured, 2,443 genes were upregulated and 6,431 genes were downregulated. These DEGs belong to diverse functional groups, including CHSs, dehydrogenases, carboxypeptidases, hormone-associated genes, and development-associated genes. A heatmap was constructed to cluster the top 25 up-regulated and 25 down-regulated genes based on the similarity and diversity of their expression profiles using normalized FPKM-values within a range of 6 to 16 (Figure 2). In the young stages of seed development, the expression of genes related to cellular components such as oleosin and tubulin alpha-4 are remarkably upregulated, and the expression of sugar-related genes such as galactomannan galactosyltransferase and beta-glucosidase were expressed at higher levels than in the mature stages. On the other hand, the genes involved in the development of seeds, such as those encoding embryonic proteins and late embryogenesis abundant proteins, were expressed at a higher level in the mature stage of seed development. In addition, it was confirmed that CHS was expressed at a high level in the mature-stage seed, likely to create substances for the protection of the seeds.
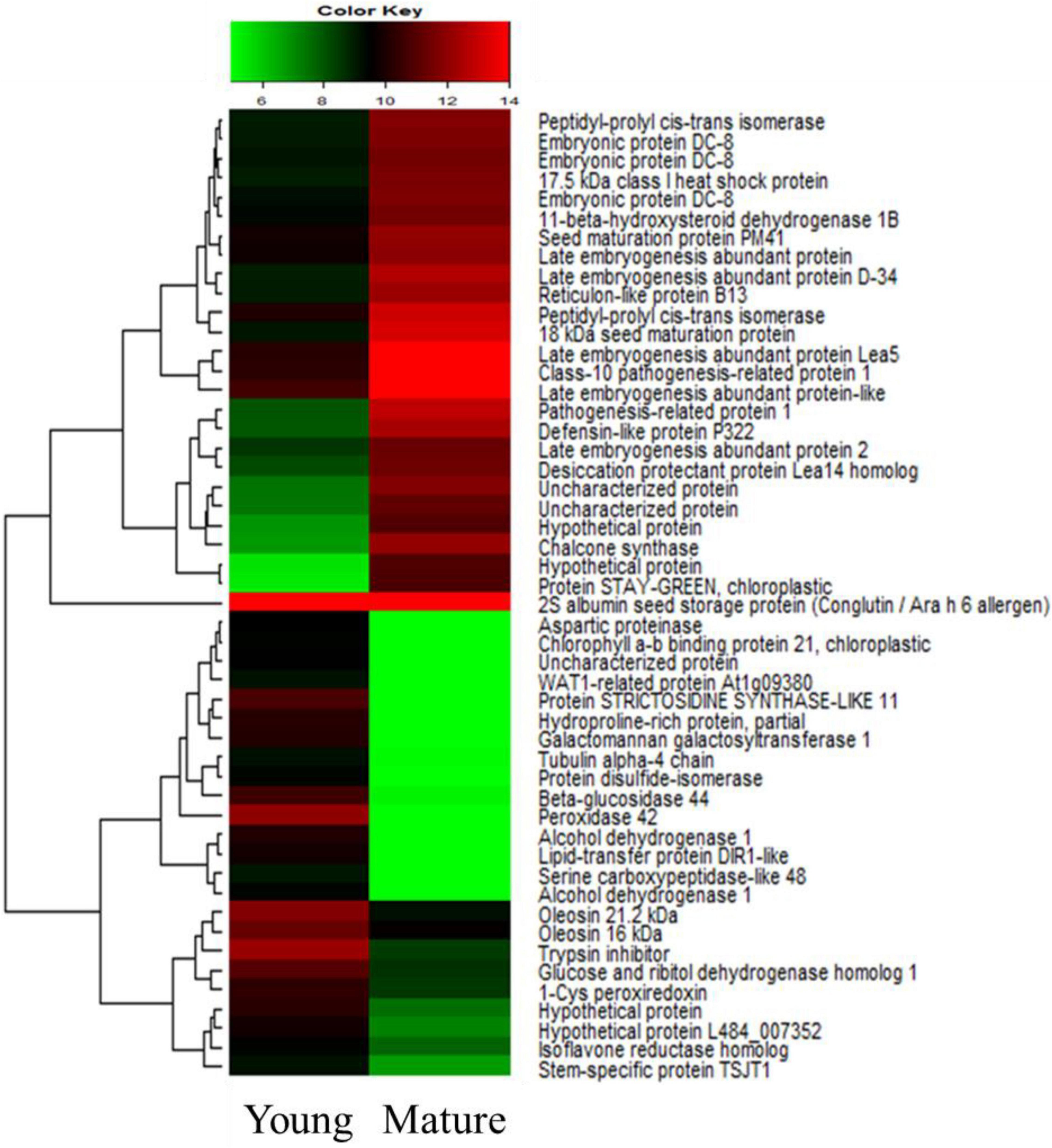
Figure 2. Heatmap of the top 25 up-regulated and 25 down-regulated genes according to the seed development stage of Senna occidentalis. The color bar indicates the value of the expression level in a sample.
Similarly, the tissue-specific gene expression was determined in the leaves, roots, and young- and mature-seeds tissues. Transcripts exhibiting tissue-specific expression were identified and the top 10 transcripts were selected (Supplementary Figure 6). A qRT-PCR analysis was performed to accurately identify the differential expression of selected transcripts belonging to the carbohydrate metabolic pathway, the secondary metabolite pathway, and the associated TFs (Figure 3). The tissue-specific gene expression data were congruous in various tissues. Glyceraldehyde 3-phosphate dehydrogenase has a catalytic role in glycolysis and plays a crucial role in plant metabolism and development (Muñoz-Bertomeu et al., 2010). The glucan 1,3-beta-glucosidase gene exhibited the highest expression level in the leaf, followed by the root, mature seed, and young seed. The gene encoding the late embryogenesis abundant protein showed the highest expression level in the mature seed, followed by the root, young seed, and leaves. The genes encoding the defensin-like protein and pathogenesis-related-like protein were most highly expressed in the mature seed, followed by the root, young seed, and leaves. The gene encoding strictosidine synthase-like protein 11 was most highly expressed in the young seed, followed by the root. Strictosidine synthase is an enzyme that biosynthesizes strictosidine through the Pictet–Spengler reaction using tryptamine and secologanin as substrates and is important in alkaloid biosynthesis (Maresh et al., 2008).
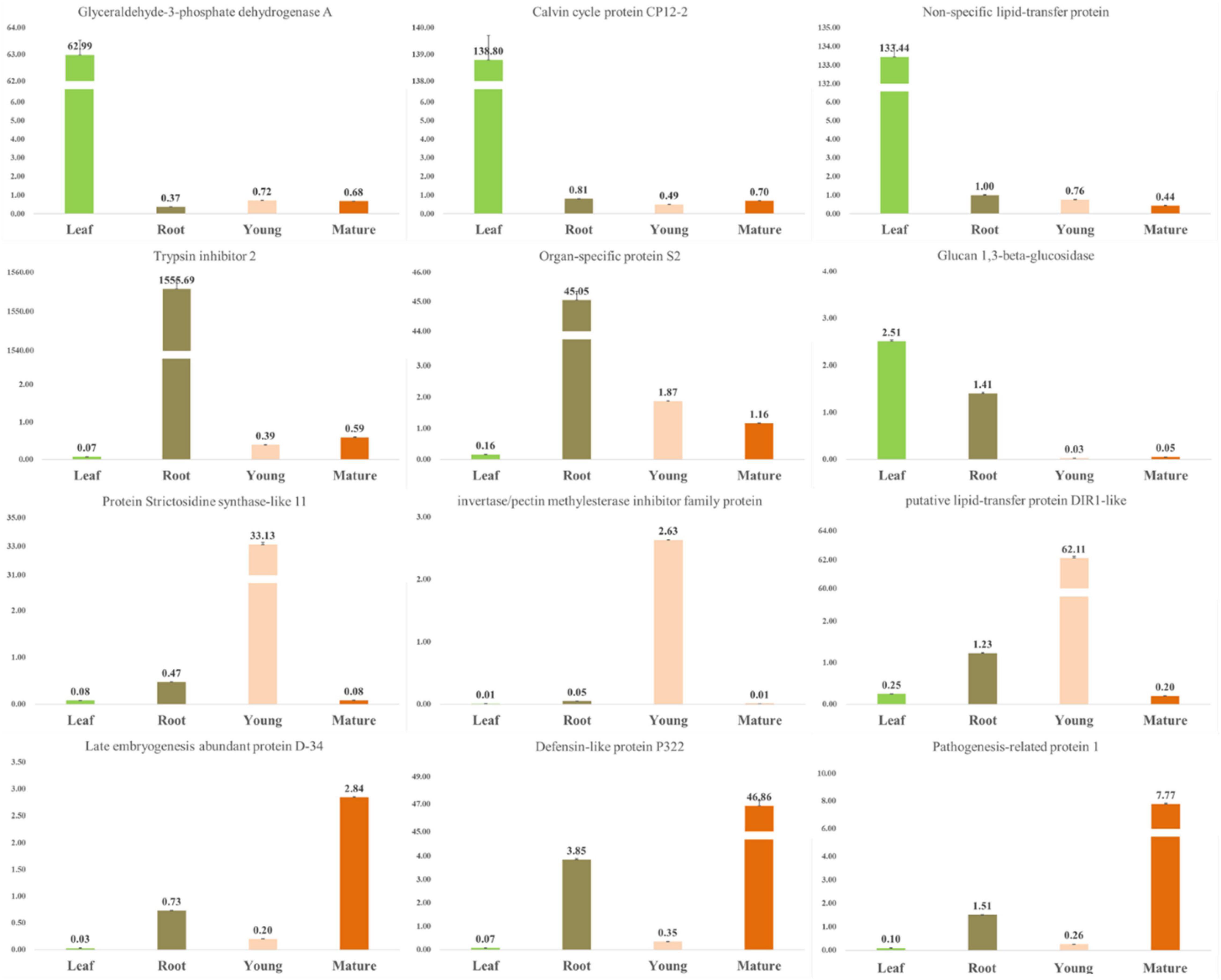
Figure 3. qRT-PCR validation of tissue-specific unigene expression levels. All RT-PCR experiments were performed at least three times in each independent biological experiment (three replicates). Error bars represent the SEM.
To determine the biological function of the DEGs in seed development, a GO classification analysis was performed using the Blast2GO program. A total of 62,149 unigenes were categorized into 25 functional groups, including three major ontologies, with annotations added based on the GO database. In terms of the biological process terms, most of the DEGs were classified into the organic substance metabolic process function; for molecular functions, organic cyclic compound binding was the most common term, while cell parts and cells dominated the cellular components category. The orthologous S. occidentalis genes were subjected to a GO enrichment analysis using the AgriGO program to determine whether the transcripts were enriched in functions essential for seed development. The carbohydrate metabolic process GO term was highly enriched in the DEGs downregulated in the seeds (Supplementary Figure 7). In addition, the lipid metabolic process GO term (biological process) was enriched in the young seed (Supplementary Figure 8).
To identify the specific metabolic pathway responsible for the changes in gene expression during the process of seed development in S. occidentalis, a MapMan (Thimm et al., 2004) analysis was performed using the expression data of genes showing at least a twofold difference in expression between the young and mature seeds. We designed a diagram of the biological process required for the analysis, and based on this, the log2 normalized expression value was displayed in the diagram (Figure 4). The results of the MapMan v. 3.5.1R2 mapping of the annotated genes revealed an enrichment in functions associated with the cell wall, lipids, and secondary metabolism, such as phenylpropanoids, terpenes, and flavonoids. In terms of the cell wall, genes related to pectin-esterase and UDP-sugar metabolism were all shown to be downstream, and in terms of cell wall breakdown, genes related to the pectin-lyases and polygalacturonase were downstream except for two genes. In the case of lipids, the results were varied. All five genes annotated as GDSL lipase were downstream, whereas some beta-oxidation and phospholipase genes were upstream. In addition, the fatty acid synthase genes largely functioned downstream, but six genes were shown to be upstream. Genes involved in the secondary metabolism were largely downstream, while some genes associated with phenylpropanoids, phenolics, and terpenes functioned upstream.
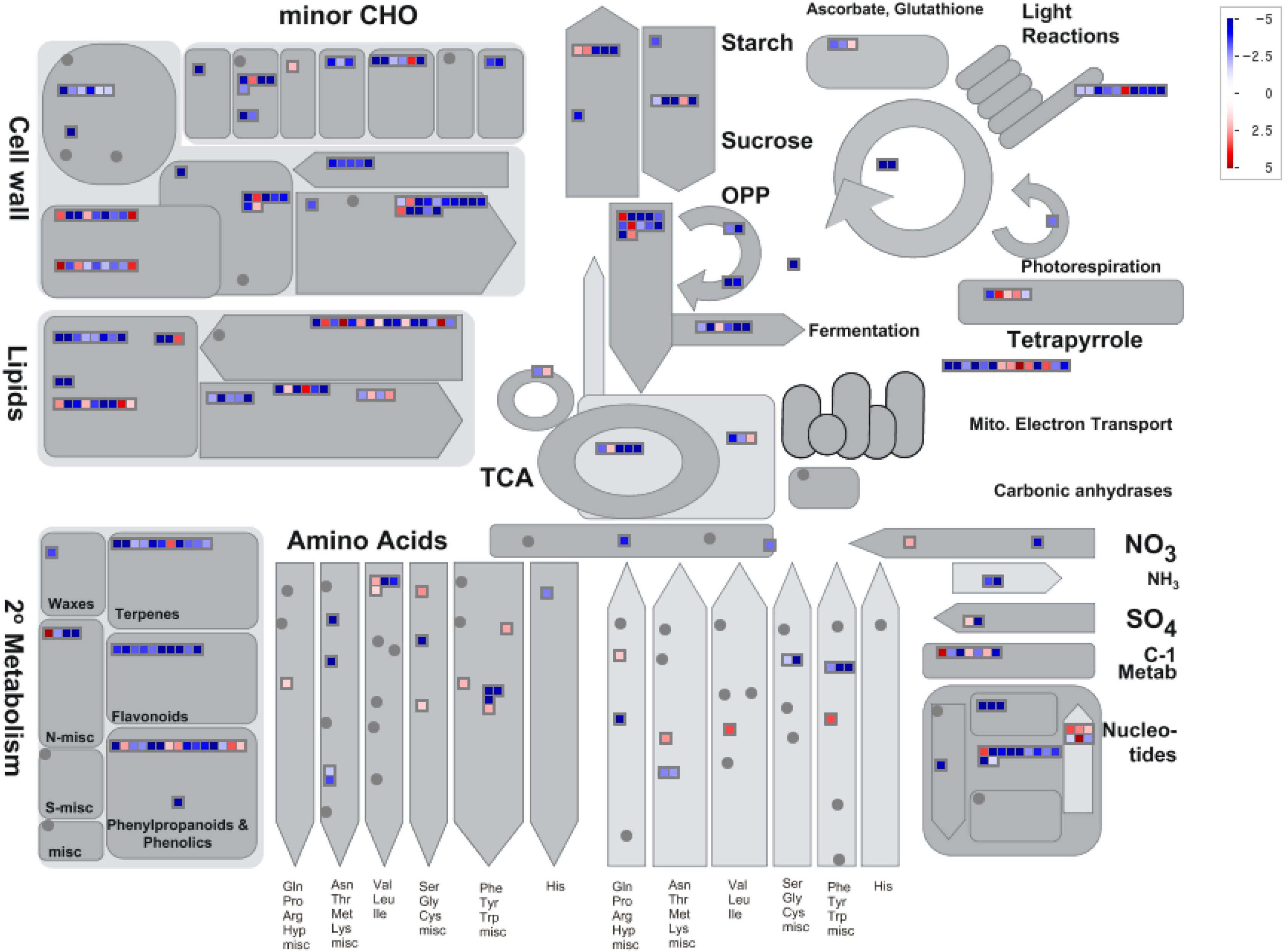
Figure 4. MapMan metabolism overview maps showing differences in transcript levels between young- and mature-stage seeds. MapMan software was used to provide a snapshot of the modulated genes in the main metabolic pathways. Log2 fold-change values are represented. Upregulated and downregulated transcripts are shown in red and blue, respectively.
Senna occidentalis is used in traditional medicine in many countries around the world. Each of its medicinal uses (e.g., as a laxative, analgesic, expectorant, diuretic, etc.) derives from its various secondary metabolites, the best known of which are anthraquinones, alkaloids, flavonoids, and terpenoids. In addition, toxicological studies have shown that S. occidentalis has myotoxic, hepatotoxic, and neurotoxic effects that can harm laboratory animals (Gotardo et al., 2017). The major toxic component of this plant’s seeds is dianthrone, a compound derived from anthraquinone (Haraguchi et al., 1998, 2003). Anthraquinone derivatives are biosynthesized through the isochorismate pathway, which is shared with the biosynthesis pathways of the phenylpropanoids and the MEP/DOXP, MEV, as well as the shikimate biosynthesis pathways, shared with the carotenoid and flavonoid biosynthesis pathways. In addition, the polyketide pathway, involving PKSs such as CHS, is an important part of anthraquinone biosynthesis (Kang et al., 2020b).
To analyze anthraquinone biosynthesis, we determined the levels of seven derivatives of the anthraquinone biosynthesis pathway in the young- and mature-stage seeds and compared them with those identified in S. tora in previous studies (Table 2; Kang et al., 2020a). Three of the derivatives (obtusin, chryso-obtusin, and obtusifolin) isolated from S. tora could not be isolated from S. occidentalis or were present only in trace amounts. Citreorosein was isolated from S. occidentalis at a concentration of 6.91 ± 2.65 μg/g in the mature-stage seeds. Physcion was isolated from both S. tora and S. occidentalis, but was more abundant in the latter. The differences in the concentrations of each of the seven derivatives between the two species is caused by the differences in their compositions of the genes involved in the anthraquinone biosynthesis pathway. The total amount tended to decrease over time, as more was isolated from the young seeds than the mature seeds; however, aurantio-obtusin maintained a constant concentration in S. tora regardless of the stage of seed development, while citreorosein accumulated in higher quantities in the mature-stage S. occidentalis seeds.
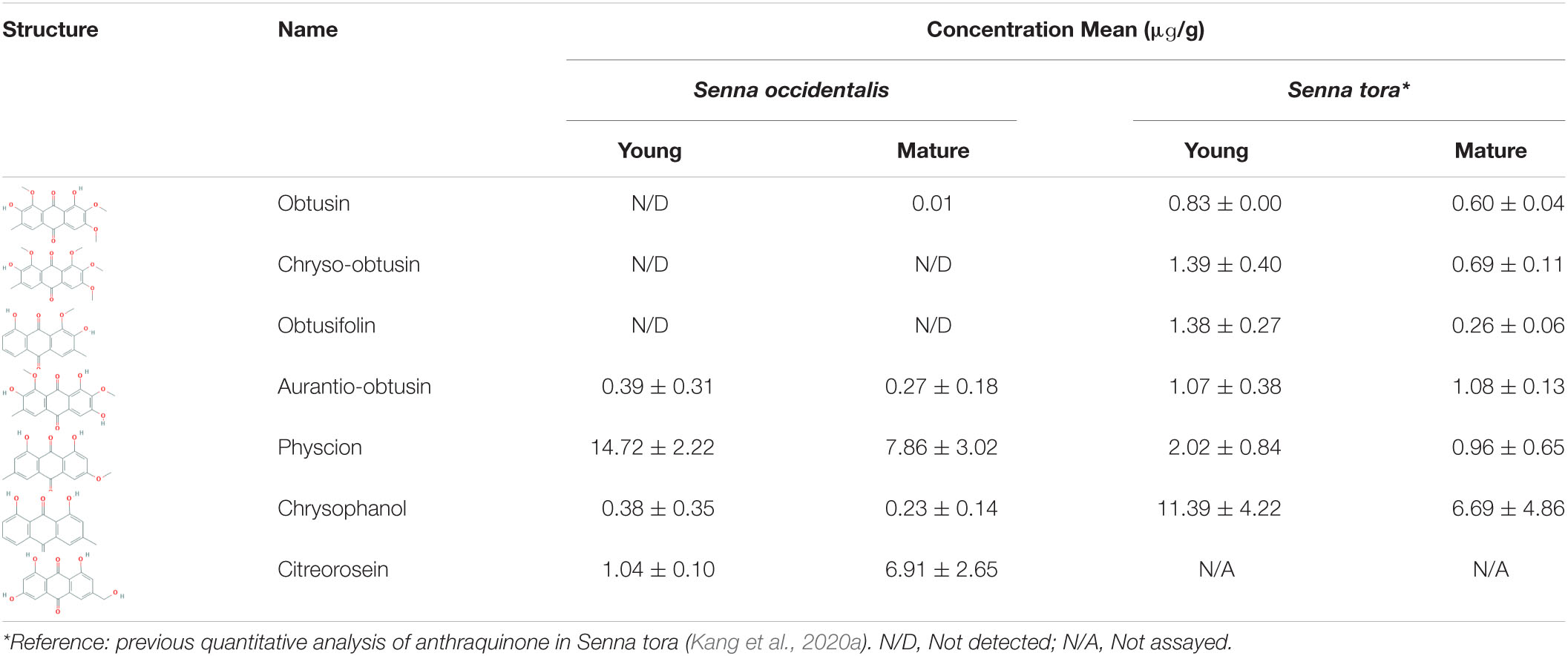
Table 2. Summary of the anthraquinone derivative concentration between S. occidentalis and S. tora.
To observe the differential gene expression levels in the leaves, roots, and seeds, the expression levels were normalized to the FPKM-values, and the transcripts were hierarchically clustered based on the log2 (FPKM + 1) value (Figure 5). In our study, 582 RNA-Seq unigenes were involved in the S. occidentalis secondary metabolite pathways, which were classified into five groups (the MEP/DOXP, MEV, shikimate, carotenoid, and flavonoid/polyketide pathways) based on the anthraquinone biosynthesis pathway of S. tora (Figure 5 and Supplementary Table 5; Kang et al., 2020a). For the MEP/DOXP pathway, which led to the production of the precursor dimethylallyl diphosphate, 28 and 32 unigenes from the RNA-Seq and Iso-Seq datasets, respectively, were identified. In addition, there were 27 and 30 unigenes for RNA-Seq and Iso-Seq, respectively, in the MEV pathway, which produces the same precursor as the MEP/DOXP pathway. For genes belonging to both pathways, more than one unigene could be identified in each tissue and seed development stage, and all the unigenes (except those with consistently high expression levels) showed low expression levels in the mature-stage seeds. Equally, 27 and 30 unigenes belonging to the shikimate pathway were identified in the RNA-Seq and Iso-Seq datasets, respectively. Multiple Iso-Seq unigenes were identified for some genes, whereas in the RNA-Seq dataset, PHYLLO (2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase) was not detected. Again, the expression level of each gene showed a tendency to be downregulated in the mature-stage seeds, except those genes with high expression levels in all tissues and throughout seed development. The precursors of the S. occidentalis secondary metabolites are believed to be generated in the young stages of seed development, and these precursors downregulate gene expression as the seed moves into the late developmental stages.
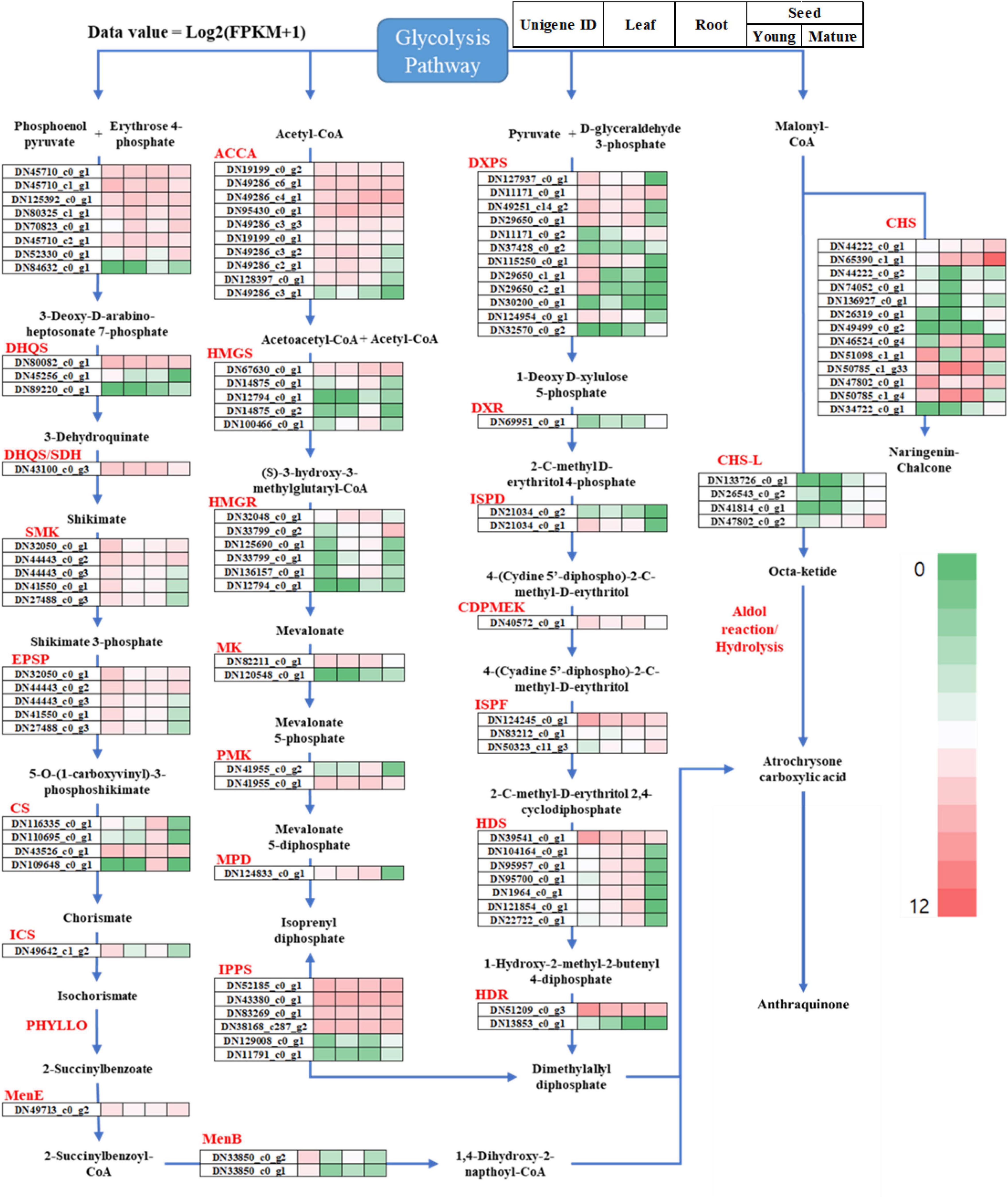
Figure 5. Overview of the putative anthraquinone biosynthesis pathway genes in Senna occidentalis. The gene expression levels were normalized to the fragments per kilobase of transcript per million reads (FPKM) values to compare the changes in expression between different tissues of S. occidentalis. The total gene expression levels were clustered based on log2 (FPKM + 1).
Based on previous studies, many researchers predicted that anthraquinone might be biosynthesized in plants through the polyketide biosynthesis pathway, the most important enzyme of which is CHS, a type III PKS (Pandith et al., 2016). It was also suggested that PKS could form an anthraquinone precursor, octaketide, using acetyl-CoA and malonyl-CoA, which would then be cyclized by PKC-encoding polyketide cyclase to form three-ring structures with A, B, and C rings (Rama Reddy et al., 2015). In fact, CHS-L was recently found to catalyze the production of atrochrysone carboxylic acid, a precursor of anthraquinones found in S. tora (Kang et al., 2020b). Of the 16 CHS-L proteins identified in that species, Sto07g228250 (CHS-L9) was found to biosynthesize atrochrysone carboxylic acid and endocrocin anthrone using malonyl-CoA as substrates. Based on this, we identified 28 RNA-Seq unigenes encoding type III PKSs such as CHS and STS (Supplementary Table 5). These unigenes averaged 609 bp in length, with the shortest being 301 bp and the longest being 1,715 bp. To identify the CHS-L unigene involved in the biosynthesis of anthraquinone, we created a phylogenetic tree to compare these sequences against the CHS-L gene of S. tora (Sto07g228250) and the CHS2 gene of Medicago sativa (L.). To enhance the accuracy of the analysis, only the 17 genes that encode proteins longer than 100 amino acids were used; the remaining 11 genes were excluded from the analysis (Figure 5). DN133726_c0_g1, DN26543_c0_g1, DN41814_c0_g1, and DN47802_c0_g2 were found to share sequence similarity with CHS-L (Sto07g228250), with the remaining 13 genes being classified as CHS. Among the CHS-L orthologs, DN47802_c0_g2 was more highly expressed in the mature stage than in the young stage of seed development, with the other genes showing a similar pattern or an even higher expression difference. In addition, DN47802_c0_g2 was not expressed in the leaves or roots, or was only present in very small amounts. In the case of the CHS genes, DN65390_C1_g1 showed very high expression levels in the mature-stage seed, and DN50785_c1_g33 and DN50785_c1_g4 showed the highest expression in the root.
The anthraquinone biosynthesis pathway exists as two major pathways, the chorismite/O-succinyl benzoic acid pathway and the polyketide pathway, as described above. However, the rest of this pathway has not been elucidated in plants such as Senna and rhubarb (Rheum sp. L.) after the backbone of anthraquinone has been formed. The full anthraquinone pathway is not yet understood, except in bacteria and fungi, which produce anthranoid or xanthone compounds (Zhou et al., 2019). Many of the genes in bacteria and fungi may have similarities with those in plants, however. In addition, as the flavonoid biosynthesis pathway in plants shares the polyketide pathway, several similarities may exist between the anthraquinone and flavonoid biosynthesis pathways (Liu et al., 2020). Based on the principal component of S. occidentalis and previous studies of the anthraquinone biosynthesis pathway, we propose a putative anthraquinone biosynthesis pathway including the anthraquinone derivatives, as shown in Figure 6. The polyketide backbone formation, oxidation, methylation, and glycosylation that occur in the biosynthesis of anthraquinone derivatives exist in all the above processes, suggesting that the biosynthesis of compounds can share the same functional enzymes.
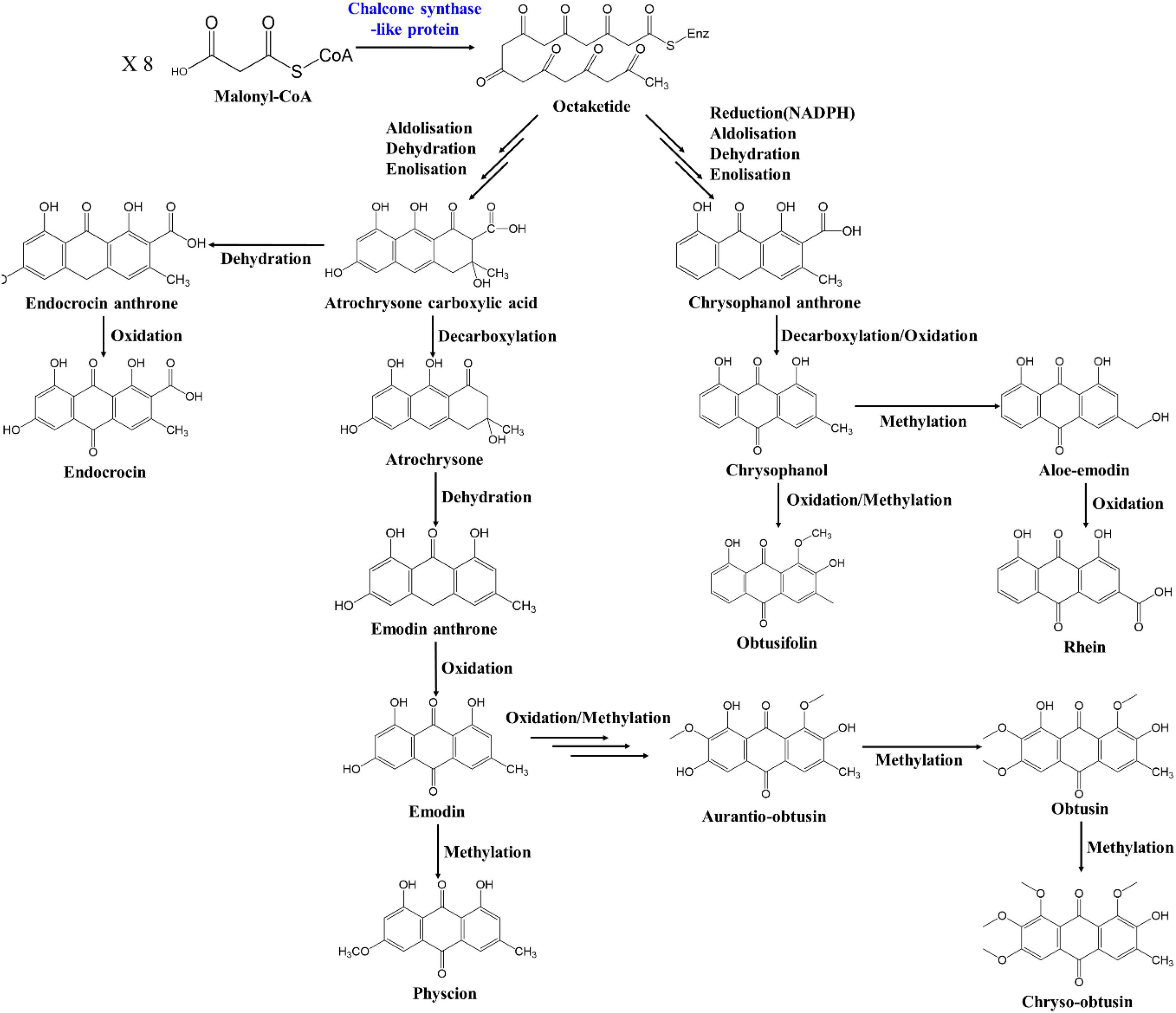
Figure 6. Putative anthraquinone biosynthesis pathways in Senna occidentalis.
Plants produce secondary metabolites by first producing the backbone molecular structure and then converting this backbone into a functional substance using specific structure-modifying enzymes. The modifying enzymes involved in this pathway include cytochrome P450 (CYP) and glycosyltransferase (GT). Among these, the CYP superfamily is a group of heme proteins that play important roles in promoting plant growth, development, and protection from stress through several biosynthesis and detoxification pathways (Ohkawa et al., 1998; Li et al., 2012). Here, we identified 180 CYPs (Supplementary Table 5) in the RNA-Seq of S. occidentalis and 50 CYPs in the Iso-Seq dataset. The overall average length of the putative CYPs was 1,213 bp, the longest was 3,076 bp, and the shortest was 301 bp. Our analysis of these unigenes based on their annotation using NCBI Nr revealed that the most commonly identified CYP groups were CYP71, CYP94, CYP82, and CYP89, with 39, 18, 14, and 10 unigene representatives, respectively. In addition, six cytochrome P450 reductases, an important component of the catalytic activity of CYP, were also identified. Among these unigenes, some of the CYP unigenes were specifically expressed at distinct stages of seed development. DN50163_c9_g1, DN50163_c8_g3, and DN95508_c0_g1 appeared to be expressed in both young and mature seeds, but showed higher expression levels in the mature-stage seeds. Conversely, DN125836_c0_g1, DN17292_c0_g1, DN50624_c1_g1, DN95253_c0_g1, and DN41954_c0_g2 showed higher expression levels in the young-stage seeds than the mature-stage seeds.
Glycosylation is widespread in nature and is involved in almost all important processes. In general, it refers to saccharide polymerization or the binding of sugar molecules to other biomolecules, including proteins, lipids, nucleic acids, and small natural molecules. These glycosylated compounds possess a wide range of functions, including energy storage, the maintenance of cellular structural integrity, information storage and transfer, molecular recognition, the regulation of cell–cell interactions, viral immune responses, and chemical defense (Liang et al., 2015). Glycosylation occurs at the end of secondary metabolite biosynthesis and improves the solubility and stability of the compounds. These glycosylated compounds are structurally the most diverse biomolecules, and their biosynthesis requires complex biological processes regulated by many enzymatic systems. This glycosylation is catalyzed by GTs, which mediate the formation of stereo-specific glycosidic bonds and regions between the sugar moieties and various important biomolecules. In nature, UDP-glycosyltransferases (UGTs) normally facilitate glycosylation, generating a product with a glucose moiety at the hydroxyl group (Ross et al., 2001). Here, we identified 157 putative GT genes in the S. occidentalis transcripts. In addition, nine, three, four, and two unigenes were specifically expressed in the leaves, roots, young-stage seeds, and mature-stage seeds, respectively. In particular, DN80158_c0_g1 showed the highest expression level among the tissue-specific GTs, while DN48605_c0_g1 was the most highly expressed unigene in the mature-stage seeds. Specifically, this unigene showed a rapid change in expression level from 8.32 in the young-stage seeds to 208.66 in the mature-stage seeds. After the anthraquinone derivative is formed, glycosylation is required for them to accumulate them in the seed, which suggests that this gene may be vital for such a role.
The biosynthesis of the anthraquinone backbone is similar in S. occidentalis and S. tora, although more unigenes related to this function were identified in S. tora than in S. occidentalis (Supplementary Table 6). In addition, the PHYLLO unigene, 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase in the shikimate pathway, could not be identified in RNA-Seq dataset in either species, but two and four PHYLLO unigenes were found in the Iso-Seq datasets for S. occidentalis and S. tora, respectively. Similar patterns were observed in the expression levels in the RNA-Seq data from the two species; however, isopentenyl diphosphate delta-isomerase, which is responsible for the conversion of isoprenyl diphosphate and dimethylallyl diphosphate in the MEV and MEP/DOXP pathways, was more highly expressed in S. occidentalis than S. tora. In addition, the expression of chorismite synthase in the shikimate pathway was equally high in the young-stage seeds of S. occidentalis and S. tora, but S. occidentalis showed higher levels in the mature-stage seeds.
In the RNA-Seq data for S. occidentalis, 28 type III PKSs were identified, whereas only 27 were found in S. tora. On the other hand, the Iso-Seq data showed 23 type III PKSs in S. tora and 22 in S. occidentalis. Some of the type III PKSs in S. tora were classified as CHS-Ls, similar to CHSs, which form the backbone of anthraquinone compounds called atrochrysone carboxylic acid. Following our classification of the type III PKSs in S. occidentalis based on the S. tora genome (Kang et al., 2020b), only 4 of the 28 genes were classified as CHS-Ls in S. occidentalis, while in S. tora 16 genes were considered CHS-Ls. The CHSs in S. tora were more highly expressed in the young stages of seed development, whereas in S. occidentalis they showed high expression in the young and mature stages. This suggests that the types and amounts of anthraquinone derivatives that accumulate in S. occidentalis will be different from those in S. tora. In the RNA-Seq dataset, the expression patterns of S. occidentalis and S. tora suggest that the polyketide pathway was preferred over the chorismite/O-succinylbenzoyl acid pathway due to the absence of PHYLLO and the inconsistency of the expression patterns and product patterns of the pathway-associated unigenes. In addition, CYP and UGT were differently expressed in S. occidentalis and S. tora. A total of 180 and 194 genes were identified in the RNA-Seq datasets of S. occidentalis and S. tora, respectively, whereas 65 and 71 genes were found in the Iso-Seq dataset. There were no significant differences in the CYP expression patterns between species, but a significant difference was observed in the UGT genes. The modifying enzyme genes involved in anthraquinone biosynthesis have not yet been identified yet.
This study suggested that CHS-L proteins are responsible for producing anthraquinones in S. occidentalis, as a similar function was determined for orthologous genes in S. tora in our previous study (Kang et al., 2020b). Variation in the anthraquinone production between different developmental stages, tissues, and species, including S. occidentalis and S. tora, should be further explored. In addition, the expression of this metabolite’s structure-modifying enzymes, CYP and UGT, was demonstrated in S. occidentalis and S. tora, providing an insight into secondary metabolite formation and the resistance to stress in plants. Hence, our transcript analysis for S. occidentalis and S. tora could lead to the identification of various secondary metabolite biosynthesis pathways and their regulatory mechanisms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
S-HK conceived of and supervised the project. S-HK and J-SS contributed to the sample preparation, sequencing, and metabolite analysis. S-HK and W-HL performed the transcriptome data analysis. NT, SC, and J-PH participated in the methodology. S-HK and W-HL wrote the manuscript. S-HK and T-JO revised the manuscript. All authors read and approved the manuscript.
This work was carried out with the support of the National Institute of Agricultural Sciences [project no. PJ015713] and the BioGreen21 Agri-Tech Innovation Program [project no. PJ015710], Rural Development Administration, South Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.773553/full#supplementary-material
Supplementary Figure 1 | The length distribution of transcripts in S. occidentalis.
Supplementary Figure 2 | The distribution of annotated unigenes using various public protein databases. Venn diagrams display the proportion of unigenes annotated using the NCBI Nr, KEGG, Swiss-Prot, and GO databases.
Supplementary Figure 3 | Histogram of Gene Ontology classifications. The results are summarized in the three main categories: biological process, molecular function, and cellular component.
Supplementary Figure 4 | Distribution of the transcription factor (TF) families in S. occidentalis. Distribution of transcripts (3,179 for RNA-Seq and 3,243 for Iso-Seq) that encode for TFs.
Supplementary Figure 5 | Species distribution of the top BLAST hits. The most frequently hit species using the RNA-Seq and Iso-Seq datasets were calculated based on sequence alignments with the lowest E-value obtained from BLAST.
Supplementary Figure 6 | Heatmaps representing the top 10 genes with tissue-specific expression in the S. occidentalis leaf, root, and young- and mature-stage seeds. Red and green represent high and low abundance, respectively.
Supplementary Figure 7 | AgriGO analysis of the downregulated genes during seed development. The downregulated genes associated with molecular terms are represented by increasingly red colors. A Gene Ontology (GO) term enrichment analysis was performed using the single enrichment analysis (SEA) tool in AgriGO. Box colors indicate the levels of statistical significance: yellow = 0.05; orange = e–05; and red = e–09.
Supplementary Figure 8 | AgriGO analysis of tissue-specific genes during seed development. Tissue-specific genes (A, leaf; B, root; and C, young_seed) associated with molecular terms are represented by increasingly red colors. A Gene Ontology (GO) term enrichment analysis was performed using the single enrichment analysis (SEA) tool in AgriGO. Box colors indicates the levels of statistical significance: yellow = 0.05; orange = e–05; and red = e–09.
Supplementary Table 1 | Gene-specific primers used for tissue-specific qRT-PCR.
Supplementary Table 2 | General properties of the reads produced using the Illumina HiSeq 2500 sequencing platform.
Supplementary Table 3 | General properties of the reads produced using the PacBio sequencing platform.
Supplementary Table 4 | Distribution of the tissue-enriched and tissue-specific transcription factors in each tissue.
Supplementary Table 5 | Genes associated with the secondary metabolite pathway in S. occidentalis.
Supplementary Table 6 | Comparison of the number of genes involved in the anthraquinone biosynthesis pathway in S. tora and S. occidentalis.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Austin, M. B., Izumikawa, M., Bowman, M. E., Udwary, D. W., Ferrer, J. L., Moore, B. S., et al. (2004). Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J. Biol. Chem. 279, 45162–45174. doi: 10.1074/jbc.M406567200
Austin, M. B., and Noel, J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110. doi: 10.1039/b100917f
Awakawa, T., Yokota, K., Funa, N., Doi, F., Mori, N., Watanabe, H., et al. (2009). Physically discrete beta-lactamase-type thioesterase catalyzes product release in atrochrysone synthesis by iterative type I polyketide synthase. Chem. Biol. 16, 613–623. doi: 10.1016/j.chembiol.2009.04.004
Bakhiet, A. O., and Adam, S. E. I. (1996). Toxicity to bovans chicks of Cassia italica seeds. Phytother. Res. 10, 156–160. doi: 10.1002/(SICI)1099-1573(199603)10:2<156::AID-PTR795<3.0.CO;2-M
Bauch, H. J., and Leistner, E. (1978). Aromatic metabolites in cell suspension cultures of Galium mollugo L. Planta Med. 33, 105–123. doi: 10.1055/s-0028-1097365
Burkill, H. M. (1995). The useful plants of West tropical African, Vol. 3. Richmond: Royal Botanic Gardens Kew.
Burnett, A. R., and Thomson, R. H. (1968). Naturally occurring quinones. Part XV Biogenesis of the anthraquinones in Rubia tinctorum L (madder). J. Chem. Soc. 1968, 2437–2441.
Chiang, Y. M., Szewczyk, E., Davidson, A. D., Entwistle, R., Keller, N. P., Wang, C. C., et al. (2010). Characterization of the Aspergillus nidulans monodictyphenone gene cluster. Appl. Environ. Microbiol. 76, 2067–2074. doi: 10.1128/AEM.02187-09
Chukwujekwu, J. C., Staden, J. V., Smith, P., and Meyer, J. J. M. (2005). Antibacterial, anti-inflammatory and antimalarial activities of some Nigerian medicinal plants. S. Afr. J. Bot. 71, 316–325. doi: 10.1016/S0254-6299(15)30105-8
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dao, T., Linthorst, H., and Verpoorte, R. (2011). Chalcone synthase and its function in plant resistance. Phytochem. Rev. 10, 397–412. doi: 10.1007/s11101-011-9211-7
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70.
Duraipandiyan, V., and Ignacimuthu, S. (2007). Antibacterial and antifungal activity of Cassia fistula L.: an ethnomedicinal plant. J. Ethnopharmacol. 112, 590–594. doi: 10.1016/j.jep.2007.04.008
Duval, J., Pecher, V., Poujol, M., and Lesellier, E. (2016). Research advances for the extraction, analysis and uses of anthraquinones: a review. Ind Crops Prod. 94, 812–833. doi: 10.1016/j.indcrop.2016.09.056
Funa, N., Awakawa, T., and Horinouchi, S. (2005). Pentaketide resorcyclic acid synthesis by type III polyketide synthases in Aspergillus oryzae. Biochem. Biophys. Res. Commun. 331, 253–260.
Gidado, N. M., Tanko, Y., Sada, N. H., and Mohammed, A. (2016). Effects of Senna occidentalis leaf supplement on blood glucose level, liver enzymes and total protein in alloxan-induced diabetic wistar rats. Bayero J. Pure Appl. Sic. 9, 68–75. doi: 10.4314/bajopas.v9i1.11
Gotardo, A. T., Haraguchi, M., Raspantini, P. C. F., Dagli, M. L. Z., and Górniak, S. L. (2017). Toxicity of Senna occidentalis seeds in laying hens and its effects on egg production. Avian Pathol. 46, 332–337. doi: 10.1080/03079457.2016.1278199
Guilfoyle, T. J., Ulmasov, T., and Hagen, G. (1998). The ARF family of transcription factors and their role in plant hormone-responsive transcription. Cell Mol. Life Sci. 54, 619–627. doi: 10.1007/s000180050190
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haraguchi, M., Calore, E. E., Dagli, M. L., Cavaliere, M. J., Calore, N. M., Weg, R., et al. (1998). Muscle atrophy induced in broiler chicks by parts of Senna occidentalis seeds. Vet. Res. Commun. 22, 265–271. doi: 10.1023/a:1006051618056
Haraguchi, M., Dagli, M. L. Z., Raspantini, P. C. F., and Górniak, S. L. (2003). The effects of low doses of Senna occidentalis seeds on broiler chickens. Vet. Res. Commun. 27, 321–328. doi: 10.1023/a:1024088209712
Jung, H. A., Ali, M. Y., Jung, H. J., Jeong, H. O., Chung, H. Y., and Choi, J. S. (2016). Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinessterases. J. Ethnopharacol. 191, 152–160. doi: 10.1016/j.jep.2016.06.037
Kang, S. H., Pandey, R. P., Lee, C. M., Sim, J. S., Jeong, J. T., Choi, B. S., et al. (2020b). Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 11:5875. doi: 10.1038/s41467-020-19681-1
Kang, S. H., Lee, W. H., Lee, C. M., Sim, J. S., Won, S. Y., Han, S. R., et al. (2020a). De novo transcriptome sequence of Senna tora provides insight into anthraquinone biosynthesis. PLoS One 15:e0225564. doi: 10.1371/journal.pone.0225564
Kudav, N. A., and Kulkarni, A. B. (1974). Chemical investigations on Cassia occidentalis Linn: II. Isolation of islandicin, helminthosporin, Xanthorin & NMR spectral studies of cassiollin & its derivatives. Indian J. Chem. 2013, 1042–1044.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lauritano, C., De Luca, D., Ferrarini, A., Avanzato, C., Minio, A., Esposito, F., et al. (2017). De novo transcriptome of the cosmopolitan dinoflagellate Amphidinium carterae to identify enzymes with biotechnological potential. Sci. Rep. 7:11701. doi: 10.1038/s41598-017-12092-1
Li, B., and Dewey, C. N. (2011). RSEM accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12:323. doi: 10.1186/1471-2105-12-323
Li, D. M., Wang, Y., and Han, K. L. (2012). Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord Chem. Rev. 256, 1137–1150. doi: 10.1016/j.ccr.2012.01.016
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, S. F., and Li, S. L. (2017). Cycloartane triterpenoid and its glucoside isolated from Cassia occidentalis. Chin. J. Nat. Med. 15, 950–954. doi: 10.1016/S1875-5364(18)30012-8
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, Y., and Jiang, J. G. (2018). Health functions and structure-activity relationships of natural anthraquinones from plants. Food Funct. 9, 6063–6080. doi: 10.1039/c8fo01569d
Liang, D. M., Liu, J. H., Wu, H., Wang, B. B., Zhu, H. J., and Qiao, J. J. (2015). Glycosyltransferases: mechanisms and applications in natural product development. Chem. Soc. Rev. 44, 8350–8374. doi: 10.1039/c5cs00600g
Liu, J., Hua, W., Yang, H. L., Zhan, G. M., Li, R. J., Deng, L. B., et al. (2012). The BnGRF2 gene (GRF2-like gene from Brassica napus) enhances seed oil production through regulating cell number and plant photosynthesis. J. Exp. Bot. 63, 3727–3740. doi: 10.1093/jxb/ers066
Liu, J., Leng, L., Liu, Y., Gao, H., Yang, W., Chen, S., et al. (2020). Identification and quantification of target metabolites combined with transcriptome of two rheum species focused on anthraquinone and flavonoids biosynthesis. Sci. Rep. 10:20241. doi: 10.1038/s41598-020-77356-9
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luximon-Ramma, A., Bahorun, T., Soobrattee, M. A., and Aruoma, O. I. (2002). Antioxidant activities of phenolic, proanthocyanidin, and flavonoid components in extracts of Cassia fistula. J. Agric. Food Chem. 50, 5042–5047. doi: 10.1021/jf0201172
Maresh, J. J., Giddings, L. A., Friedrich, A., Loris, E. A., Panjikar, S., Trout, B. L., et al. (2008). Strictosidine synthase: mechanism of a Pictet-Spengler catalyzing enzyme. J. Am. Chem. Soc. 130, 710–723. doi: 10.1021/ja077190z
Martínez, K. A., Lauritano, C., Druka, D., Romano, G., Grohmann, T., Jaspars, M., et al. (2019). Amphidinol 22, A new cytotoxic and antifungal amphidinol from the dinoflagellate Amphidinium carterae. Mar. Drugs. 17:385. doi: 10.3390/md17070385
Morita, H., Oshimi, S., Hirasawa, Y., Koyama, K., Honda, T., Ekasari, W., et al. (2007). Cassiarins A and B, novel antiplasmodial alkaloids from Cassia siamea. Org. Lett. 9, 3691–3693. doi: 10.1021/ol701623n
Muñoz-Bertomeu, J., Cascales-Miñana, B., Alaiz, M., Segura, J., and Ros, R. A. (2010). critical role of plastidial glycolytic glyceraldehyde-3-phosphate dehydrogenase in the control of plant metabolism and development. Plant Signal. Behav. 5, 67–69. doi: 10.4161/psb.5.1.10200
Ohkawa, H., Imaishi, H., Shiota, N., Yamada, T., Inui, H., and Ohkawa, Y. (1998). Molecular mechanisms of herbicide resistance with special emphasis on cytochrome P450 monooxygenases. Plant Biotechnol. 15, 173–176. doi: 10.5511/plantbiotechnology.15.173
Omidbakhshfard, M. A., Proost, S., Fujikura, U., and Mueller-Roeber, B. (2015). Growth-regulating factors (GRFs): A small transcription factor family with important functions in plant biology. Mol. Plant 8, 998–1010. doi: 10.1016/j.molp.2015.01.013
Pal, S., Kumar, P., Ramakrishna, E., Kumar, S., Porwal, K., Kumar, B., et al. (2019). Extract and fraction of Cassia occidentalis L. (a synonym of Senna occidentalis) have osteogenic effect and prevent glucocorticoid-induced osteopenia. J. Ethnopharmacol. 235, 8–18. doi: 10.1016/j.jep.2019.01.029
Pandith, S. A., Dhar, N., Rana, S., Bhat, W. W., Kushwaha, M., Gupta, A. P., et al. (2016). Functional promiscuity of two divergent paralogs of type III plant polyketide synthases. Plant Physiol. 171, 2599–2619. doi: 10.1104/pp.16.00003
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Peng, F. Y., and Weselake, R. J. (2013). Genome-wide identification and analysis of the B3 superfamily of transcription factors in Brassicaceae and major crop plants. Theor. Appl. Genet. 126, 1305–1319. doi: 10.1007/s00122-013-2054-4
Rama Reddy, N. R., Mehta, R. H., Soni, P. H., Makasana, J., Gajbhiye, N. A., Ponnuchamy, M., et al. (2015). Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS One 10:e0129422. doi: 10.1371/journal.pone.0129422
Ross, J., Li, Y., Lim, E., and Bowles, D. J. (2001). Higher plant glycosyltransferase. Genome Biol. 2:3004. doi: 10.1186/gb-2001-2-2-reviews3004
Schmieder, R., Lim, Y. W., and Edwards, R. (2012). Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28, 433–435. doi: 10.1093/bioinformatics/btr669
Seshime, Y., Juvvadi, P. R., Fujii, I., and Kitamoto, K. (2005). Discovery of a novel superfamily of type III polyketide synthases in Aspergillus oryzae. Biochem. Biophys. Res. Commun. 331, 253–260.
Shamim, G., Ranjan, S. K., Pandey, D. M., and Ramani, R. (2014). Biochemistry and biosynthesis of insect pigments. Eur. J. Entomol. 111, 149–164. doi: 10.14411/eje.2014.021
Thimm, O., Bläsing, O., Gibon, Y., Nagel, A., Meyer, S., Krüger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313x.2004.02016.x
Tropf, S., Kärcher, B., Schröder, G., and Schröder, J. (1995). Reaction mechanisms of homodimeric plant polyketide synthase (Stilbenes and chalcone synthase). A single active site for the condensing reaction is sufficient for synthesis of stilbenes, chalcones, and 6’-deoxychalcone. J. Biol. Chem. 270, 7922–7928. doi: 10.1074/jbc.270.14.7922
Vashishtha, V. M., John, T. J., and Kumar, A. (2009). Clinical & pathological features of acute toxicity due to Cassia occidentalis in vertebrates. Indian J. Med. Res. 130, 23–30.
Verma, L., Khatri, A., Kaushik, B., Patil, U. K., and Pawar, R. S. (2010). Antidiabetic activity of Cassia occidentalis (Linn) in normal and alloxan-induced diabetic rats. Indian J. Pharmacol. 42, 224–228. doi: 10.4103/0253-7613.68422
Yang, C. Q., Fang, X., Wu, X. M., Mao, Y. B., Wang, L. J., and Chen, X. Y. (2012). Transcriptional regulation of plant secondary metabolism. J. Integr. Plant Biol. 54, 703–712. doi: 10.1111/j.1744-7909.2012.01161
Yen, G. C., and Chen, H. Y. (1995). Antixodiant activity of various tea extracts in relation to their antimutagenicity. J. Agric. Food Chem. 43, 27–32. doi: 10.1021/jf00049a007
Zhang, D. F., Li, B., Jia, G. Q., Zhang, T. F., Dai, J. R., Li, J. S., et al. (2008). Isolation and characterization of genes encoding GRF transcription factors and GIF transcriptional coactivators in maize (Zea mays L.). Plant Sci. 175, 809–817. doi: 10.1016/j.plantsci.2008.08.002
Zhang, T., Li, C., Li, D., Liu, Y., and Yang, X. (2020). Roles of YABBY transcription factors in the modulation of morphogenesis, development, and phytohormone and stress responses in plants. J. Plant Res. 133, 751–763. doi: 10.1007/s10265-020-01227-7
Zhao, P. P., Tong, J. M., Tian, Y. F., and Zhang, S. F. (2016). Research progress in pharmacological effects of anthraquinones. J. Chengde Med. Coll. 33, 152–155.
Keywords: Senna occidentalis, anthraquinone, secondary metabolite, transcriptome analysis, transcription factor
Citation: Kang S-H, Lee W-H, Sim J-S, Thaku N, Chang S, Hong J-P and Oh T-J (2022) De novo Transcriptome Assembly of Senna occidentalis Sheds Light on the Anthraquinone Biosynthesis Pathway. Front. Plant Sci. 12:773553. doi: 10.3389/fpls.2021.773553
Received: 10 September 2021; Accepted: 04 November 2021;
Published: 03 January 2022.
Edited by:
Clive Lo, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Dongming Ma, Guangzhou University of Chinese Medicine, ChinaCopyright © 2022 Kang, Lee, Sim, Thaku, Chang, Hong and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang-Ho Kang, aG9zYW5nOTNAa29yZWEua3I=; Tae-Jin Oh, dGpvaDM3ODJAc3VubW9vbi5hYy5rcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.