
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 20 October 2021
Sec. Plant Metabolism and Chemodiversity
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.755494
This article is part of the Research TopicPlant Specialized Metabolism for Plant Protection: Genomics and BiotechnologyView all 13 articles
 Liu Yang1,2†Xiangxiao Meng2†
Liu Yang1,2†Xiangxiao Meng2† Shilin Chen2Jun Li2
Shilin Chen2Jun Li2 Wei Sun2Weiqiang Chen2Sifan Wang2Huihua Wan2Guangtao Qian3Xiaozhe Yi1Juncan Li1Yaqin Zheng1
Wei Sun2Weiqiang Chen2Sifan Wang2Huihua Wan2Guangtao Qian3Xiaozhe Yi1Juncan Li1Yaqin Zheng1 Ming Luo4Shanshan Chen2
Ming Luo4Shanshan Chen2 Xia Liu1*
Xia Liu1* Yaolei Mi2*
Yaolei Mi2*Histone deacetylases (HDACs) play crucial roles nearly in all aspects of plant biology, including stress responses, development and growth, and regulation of secondary metabolite biosynthesis. The molecular functions of HDACs have been explored in depth in Arabidopsis thaliana, while little research has been reported in the medicinal plant Cannabis sativa L. Here, we excavated 14 CsHDAC genes of C. sativa L that were divided into three relatively conserved subfamilies, including RPD3/HDA1 (10 genes), SIR2 (2 genes), and HD2 (2 genes). Genes associated with the biosynthesis of bioactive constituents were identified by combining the distribution of cannabinoids with the expression pattern of HDAC genes in various organs. Using qRT-PCR and transcription group analysis, we verified the expression of candidate genes in different tissues. We found that the histone inhibitor Trichostatin A (TSA) affected the expression of key genes in the cannabinoid metabolism pathway and the accumulation of synthetic precursors, which indirectly indicates that histone inhibitor may regulate the synthesis of active substances in C. sativa L.
Chromatin consists of nucleosomes, each of which is composed of an octamer of four key histones—H2A, H2B, H3, and H4, each of which is a dimer—around which is wrapped a strand of DNA (Yang and Nielsen, 2000; Lusser et al., 2001). Histone acetylases (HAT) are enzymes, which catalyzes the acetylation of histones, and histone deacetylase (HDAC) catalyzes the deacetylation of histones (Yang and Seto, 2007; Wang et al., 2009). When histones are acetylated, they release DNA due to the decrease in the affinity between histones and DNA, and subsequently promote the binding of transcription factors to DNA (Kurdistani and Grunstein, 2003). However, these effects are counteracted by histone deacetylases (Liu et al., 2012). HAT and HDAC coexist in various tissues and organs of animals and plants and interact to regulate the availability of DNA (Graessle et al., 2001; Haigis and Guarente, 2006). The classification and function of HDACs in humans and yeast have been studied in depth (Brownell et al., 1996; Song and Galbraith, 2006). In yeast, the HDAC gene family is divided into two subfamilies according to the characteristics of the conserved domain sequences RPD3/HDA1 (reduced potassium dependency 3) and SIR2 (silent information regulator 2; Frye, 2000; Guarente, 2000). Histone deacetylation can directly affect the level of transcription by altering the structure of chromatin (Brownell et al., 1996; Pandey et al., 2002). Both subfamilies have also been studied in plants (Zhang et al., 2020). For example, AtHDA1, AtSNLs, and AtHDA19 regulate flowering time by forming complexes (Lee et al., 2016); AtHDA19 and AtHSL1 together suppress the expression of seed maturation genes (Zhou et al., 2013). In rice, OsIDS1 physically interacts with the transcriptional corepressors OsTPR1 and OsHDA1 (Cheng et al., 2018). Additionally, another subfamily of HDAC, HD2 (Histone Deacetylase 2), was shown to be plant-specific (Gu et al., 2011; Nicolas-Francès et al., 2018). In Poplar lignin, the PtHDT1 mutant of Populus tomentosa, an HD2 subfamily member, has thinner stem nodes, more wood fiber cells, thicker wood fiber cell walls, and higher lignin than the wild type (Kornet and Scheres, 2009). PtHDT1 interacts with PtMYB to regulate the development of xylem and cambium (Zhang, 2018). In banana, MaERF1 recruits MaHDA1 to represses the expression of MaACO1, thereby negatively regulating ethylene biosynthesis (Han et al., 2016; Zheng et al., 2020). Therefore, HDAC families exert vital effects on the synthesis of secondary metabolites.
Cannabis sativa (Cannabinaceae) is an annual herb, mostly dioecious (Zuardi, 2006). This plant has been used in many areas including food, fiber, cosmetics, and medicine (Schachtsiek et al., 2018). Cannabinoids, the main components found in C. sativa. L, include CBD and THC (Marks et al., 2009; Vindenes, 2017). C. sativa is divided into hemp and marijuana, according to the content of tetrahydrocannabinol (THC; Morimoto et al., 1997; Vindenes, 2017). CBD has anti-vomiting, analgesic, anti-inflammatory, anti-spasmodic, anti-cancer, and other activities (De Meijer and Hammond, 2005). For instance, epidiolex (purified CBD) is an oral drug approved for treating epilepsy. However, THC is a neuroactive compound and is addictive (Pacher et al., 2006; Burstein, 2015). In China, the cultivation of hemp with a THC content higher than 0.3% is prohibited. Therefore, our lab is committed to cultivating high-quality varieties of C. sativa with high CBD and low THC content. This aim will be achieved by studying the molecular mechanism related to the production of CBD to increase natural yield and response to external environmental stress. The biosynthetic pathway of cannabinoids has been elucidated (Lydon et al., 1987; Booth et al., 2020; Figure 1). However, the molecular mechanism of regulation of cannabinoid synthesis has yet to be clearly resolved. Posttranscriptional modification plays a pivotal role in the biosynthetic pathway of active substances in medicinal plants (Appendino et al., 2008; VanBakel et al., 2011; Luo et al., 2019). We aimed to systemically summarize the members of the HDAC family at the genome-scale level and explore histone deacetylation the effects of posttranscriptional modification on cannabinoid synthesis. In this study, we identified 14 HDACs in C. sativa genome and studied their structural characteristics, subcellular localization, alternative splicing events, and differential expression in different tissues and organs. We treated C. sativa L seedlings with histone inhibitors and measured the expression of key genes and the accumulation of precursor substances (olivetolic acid-OA and geranyl diphosphate-GPP). Based on these studies, we explored the role of deacetylation in the cannabinoid synthesis pathway.
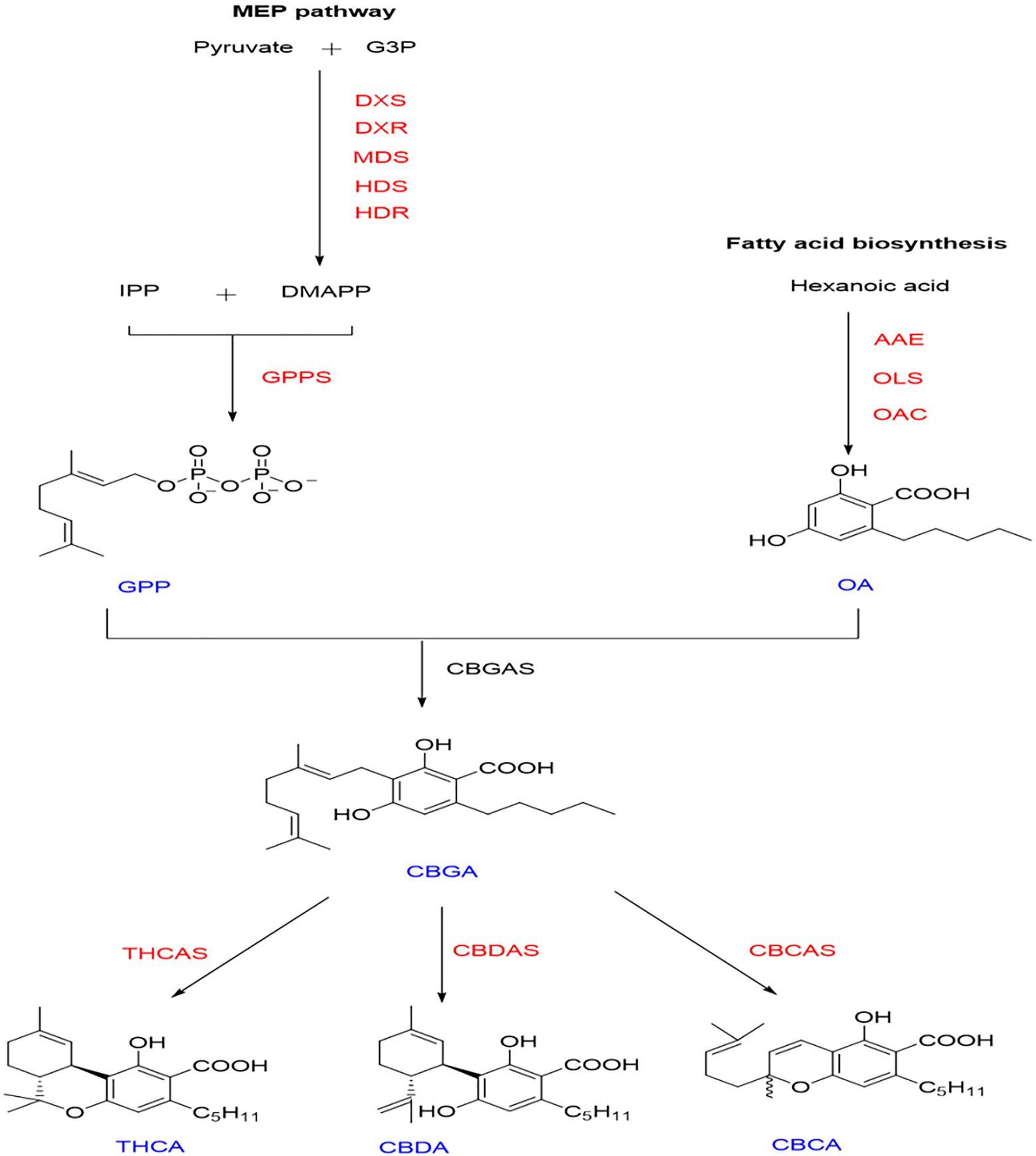
Figure 1. Synthetic pathway of cannabinoids. Intermediates and final products are shown in blue. Enzyme names are shown in red. Fatty acid pathway—AAE, Acyl activating enzyme; OAC, Olivetolic acid cyclase; and OLS, Olivetol synthase. Methylerythritol phosphate (MEP) pathway—DXS, 1-deoxy-d-xylulose 5 phosphate DXP synthase; DXR, DXP reductase; MDS, 2-C-methyl-pyrophosphate synthase; HDS, (E)-4-Hydroxy-3-methyl-but-2-enyIpyrophosphate (HMB-PP) synthase; HDR, HMB-PP reductase; and GPPS: Geranyl diphosphate synthase. Intermediates and final products—GPP, Geranyl diphosphate; OA, Olivetolic acid; CBGA, Cannabigerolic acid; THCA, Δ9-Tetrahydrocannabinolic acid A; CBDA, Cannabidiolic acid; and CBCA, Cannabichromenic acid.
In this study, we used the hemp variety Diku (DK), a female plant crossing Purple Kush with Dinamed Autoflowering CBD. This variety has a short growth cycle and is a variety with high CBD and low THC content. It is an excellent reference variety for cannabis breeding. The plants were cultivated in the experimental field of the Institute of Chinese Materia Medica of the Chinese Academy of Chinese Medical Sciences, China. Flowers, bracts, leaves, stems, seeds, and roots of hemp were collected to analyze the expression patterns of HDAC genes. Hemp seeds were cultured in MS medium for 3weeks. After the seedlings grew, they were transferred to an MS medium containing 0-μm (DMSO matrix), 0.25-μm, or 1.25-μm Trichostatin A (TSA) and cultured for 1week to analyze the effect of TSA on the HDAC gene of hemp. All sample materials collected in this study were frozen in liquid nitrogen immediately and then reserved at −80°C for further analysis.
The genomic data (GCA_900626175.1) of hemp variety female CRBRX, with a high-CBDA cultivar (15% CBDA and 0.3% THCA) used in this study, were downloaded from the NCBI1 database. Arabidopsis thaliana and soybean HDAC genetic data were obtained from the PlantTFDB database.2 We measured the transcriptome of five different tissues and organs of Diku. The transcriptome data of its flowers, bracts, stems, leaves, and seeds were available at NCBI (NCBI accession number: flowers: SAMN16122886~SAMN16122888; bracts: SAMN16122880~SAMN16122882; stems: SAMN16122883~SAMN16122885; leaves: SAMN16122889~SAMN1612281; and seeds: SAMN20474456~SAMN20474458).
We downloaded the full gene set and annotation files of C. sativa (GCA_900626175.1), Glycine max (GCF_000004515), and A. thaliana (GCA_000005425.2) from the NCBI Web site. We extracted the CDS sequence and protein sequence of hemp using the TBtools software (Chen et al., 2020). Using the protein sequence of AtHDAC and GmHDAC as query in BLASTp (score value of ≥100, e-value ≤ e−10), we ran the extracted hemp data against the AtHDAC sequence to identify potential HDACs in hemp. Then, we initially established identity-reliable domains by manually searching the NCBI reserved domain database.3 Finally, 14 HDAC family members were obtained. The basic characteristics, such as the protein molecular weight, isoelectric point, and amino acid number, were analyzed on the ExPASy Web site.4
CsHDAC, GmHDAC, and AtHDAC domain amino acid sequences were aligned using MEGA 7.0 software. The neighbor-joining (NJ) adjacency method was used to build a phylogenetic tree against the A. thaliana and G. max. The bootstrap test was repeated 1,000 times, and the BLASTp cutoff was set as value of E<1×10−10. The structure of the CsHDACs and their localization on the chromosomes were visualized using TBtools (Chen et al., 2020). The conserved motifs of CsHDAC proteins were carried out using MEME.5 The parameters are set as follows: The maximum number of themes is 20, and the best theme width is 10–200. Conserved structural domains were analyzed using NCBI-CDD.6 Subcellular localization was predicted using the wolf PSORT Web site.7 Sequences 2000bp upstream of the start codon of each CsHDAC gene were extracted, and the distribution of the cis-elements in the promoter regions was analyzed using PlantCARE software.8 We downloaded the chromosomal locations of 14 HDAC genes in the C. sativa genome from the NCBI.9 The HDAC gene locations were then represented using TBtools (Chen et al., 2020). Multiple collinear scanning toolkits (MCScanX) were used to detect gene duplication events (Wang et al., 2012).
Differential expression analysis of CsHDACs and all isoforms of CsHDACs were performed, and heat maps were created using TBtools software. In short, SpliceMap was used to detect the junctions of Illumina short reads obtained from samples, and IDP was then used to predict and detect isoforms by integrating both Illumina short reads and SMRT long reads. The results of IDP show the sites where isoforms are located on the genome. Integrative Genomics Viewer was used to visualize the isoform structure that may exist for each HDAC gene family member.10 Specific steps were referred to in our previous research (Gao et al., 2019; Xu et al., 2020). We took the original annotated genes as reference isoforms (ref isoforms). The other isoforms were divided into intron retained (IR), an exon skipping (ES), and alternative 3′ splice site (AA). Isoforms that did not belong to the aforementioned three types were defined as “others.”
According to the manufacturer’s instructions, we isolated total RNA from samples (Tiangen Biotech, Beijing, China). Using a Fast Quant RT Kit, the first cDNA strand was synthesized (Tiangen Biotech). The specific primers for qRT-PCR were designed using NCBI-Primer blast (Table 1). CFX96™ real-time system (Bio-Rad, Hercules, CA, United States) was used for qRT-PCR. Program: 95°C for 2min, followed by 40cycles of 95°C for 15s, 56°C for 35s, and 72°C for 15s. A melting curve was generated and analyzed. The reference gene in this study was EF1-α (Nicot et al., 2005). Each sample was in triplicate experiments.

Table 1. qRT-PCR primers for genes.
After adding liquid nitrogen in a mortar, leaves of hemp seedlings were crushed into a uniform powder. An ultrasonic ice water bath was used to dissolve approximately 0.1-g sample powder in 10.0-ml 95% aqueous methanol for 30min. Approximately 12,000rpm was applied for 10min at 4°C to centrifuge the solution; then, the supernatant was filtered by a 0.22-μm hydrophilic organic nylon microporous membrane.
The Agilent UPLC 1290II–G6400 triple quadrupole mass spectrometer (QQQ; Agilent Technologies, Santa Clara, CA, United States) was used to determine the relative quality of synthetic precursors. We used the peak area to express the relative mass under each condition.
A bidirectional solvent delivery system, an autosampler, and a column compartment were used in the UPLC. A column (2.1 * 100mm, 1.8μm) of C18 was used for chromatographic separations at a temperature of 40°C. Mobile phase A contained water containing 0.1% formic acid; phase B is 100% methanol. Elution was proceeded linearly at a flow rate of 0.3ml/min. In gradients, these settings were used as: 5% B→70%B (0–2min); 70% B→100% B (2–10min); 100% B (10–13min); 100% B→5% B(13–14min); and 5% B (14–15min). Autosampler was set to 4°C and 3-μl sample volume was injected. The experiment was performed in triplicate. The MRM diagrams of OA, GPP, and their standard products are shown in Supplementary Figure S1.
All the data were analyzed by the Prism 8 Statistics programs, and the means were compared by the least significant difference test at the 0.05 and 0.01 level of significance.
In our study, 14 HDAC genes were obtained, and RPD3/HDA1 subfamily contained 10 members, named CsHDA1–CsHDA10; two members of HD2 subfamily named CsHDT1–CsHDT2; and two members of SIR2 subfamily named CsSRT1–CsSRT2. Their sequences ranged in length from 432 to 1953 bp. The molecular weight of their translated protein ranged from 21702.17 to 56624.95Da, and they had between 203 and 504 amino acids. The other characteristics, such as theoretical IP and subcellular localization, were shown in Table 2.
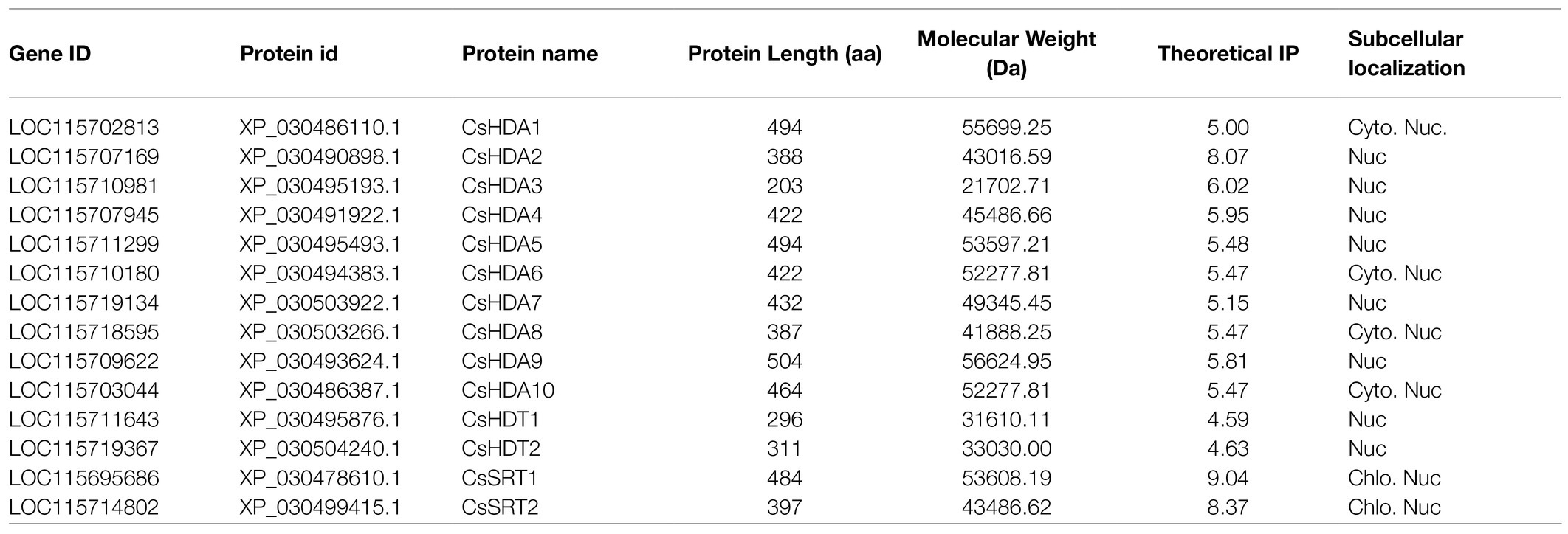
Table 2. Description of the HDACs in C. sativa.
To demonstrate the evolutionary relationships of CsHDAC in plants, we constructed a phylogenetic tree using amino acid sequences from model plant Arabidopsis, model food crop soybean, and hemp. The CsHDAC family was categorized into three subfamilies based on the difference in sequence features and the number of HDAC domains, SIR2, HD2, and RPD3/HDA1, with 2, 2, and 10 members, respectively (Zhang et al., 2020). Clades I–IV represented the RPD3/HDA1 subfamily, which is a type of Zn+-dependent histone deacetylase. Clade V represented the HD2 subfamily, which are plant-specific HDACs. Clade VI represented the SIR2 subfamily, a type of nicotinamide adenine dinucleotide (NAD+)-dependent HDACs. The 10 members of RPD3/HDA1 subfamilies were further divided into four subgroups based on the similarities of the HDAC domain amino acid sequences, with Subgroup I including CsHDA1 and CsHDA7, Subgroup II including CsHDA6 and CsHDA10, Subgroup III including CsHDA2, and Subgroup IV including CsHDA3-5 and CsHDA8-9 (Figure 2).
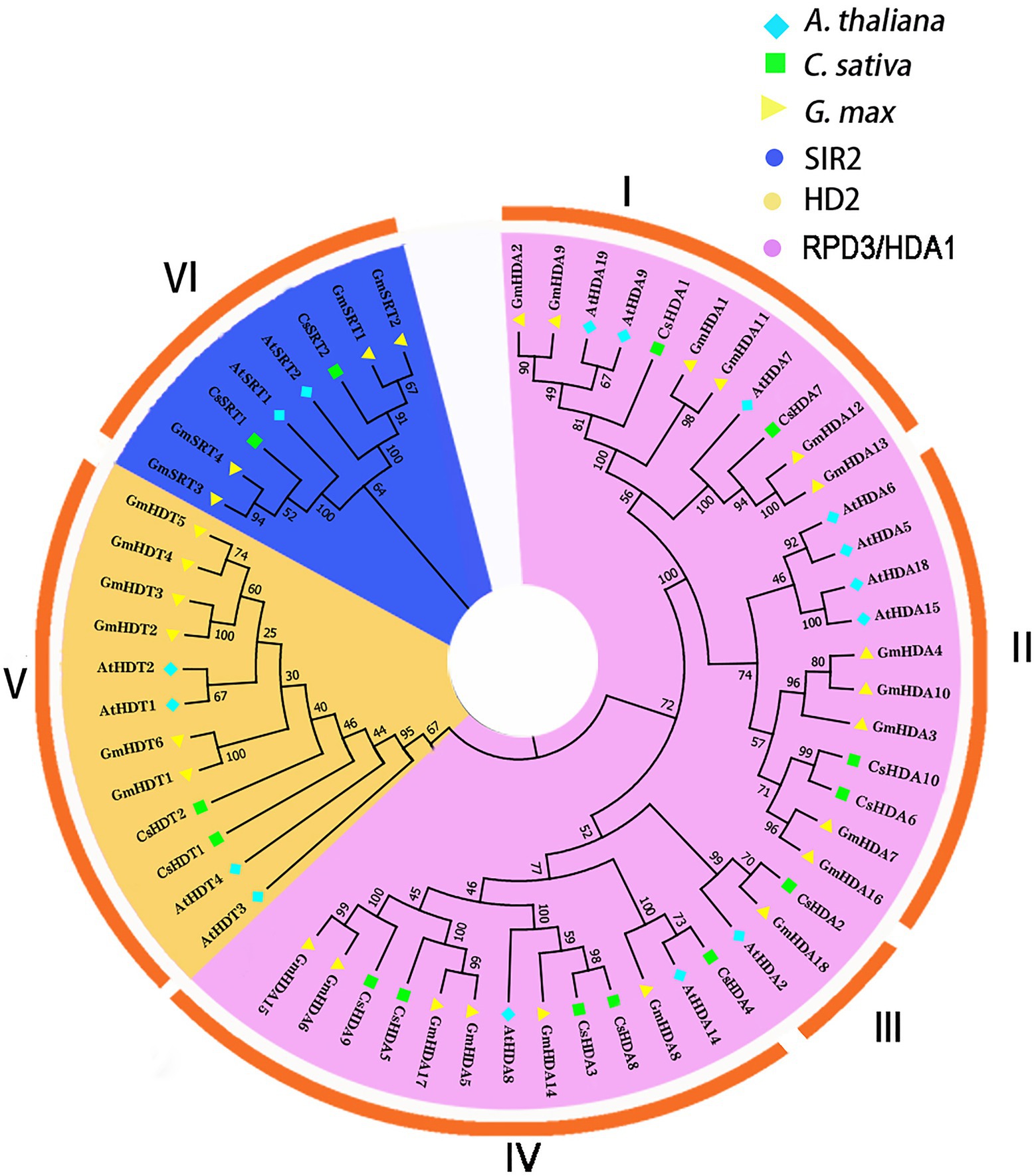
Figure 2. An unrooted phylogenetic tree was constructed with the HDAC domain amino acid sequences of Arabidopsis thaliana, Cannabis sativa, and Glycine max. A neighbor-joining tree was constructed using MEGA7 with 1,000 bootstrap replicates. The tree was divided into six groups that contained the RPD3/HDA1, SIR2, and HD2 subfamilies (I–IV: RPD3/HDA1; V: HD2; and VI: SIR2). The blue rhombus represents A. thaliana; the green square indicates C. sativa; and the yellow triangle represents G. max. The purple background shows the SIR2 subfamily; the yellow background refers to the HD2 subfamily; and the blue background represents the RPD3/HDA1 subfamily.
The conserved motifs of proteins may be related to their transcriptional activity, protein interactions, and nuclear localization (Ikeda and Ohme-Takagi, 2009; Causier et al., 2012). Thus, we studied the conserved motifs of all CsHDAC proteins combined with the evolutionary relationship of hemp (Figure 3A), and 20 conserved motifs were predicted (Figure 3B). The RPD3/HDA1 subfamily included numerous conserved motifs, while the SIR2 and HD2 subfamilies contained relatively fewer conserved motifs. According to the analysis of the conserved motifs, motifs 1 and 3 partly represented the distribution of the HDAC-conserved domain and were shared by all 10 members of the RPD3/HDA1 subfamily. Motifs 10, 13, and 14 were highly conserved and were included only in the Class II subgroup. The conserved motifs 16 and 17 were identified as a characteristic NPL domain, a structure that is unique to the HD2 subfamily. Motif 18 corresponded to the SIR2 conserved domain, which only existed in the SIR2 subfamily (Figure 3E). Multiple sequence comparison of important conserved motifs is shown in Supplementary Figure S2. Other conserved motifs were also found in CsHDACs, while the action mechanism of these motifs has not been analyzed. Overall, the conserved motif composition and gene structure within each family of CsHDAC members had a high similarity, and the results of the phylogenetic analysis advocated the validity of the population classification.
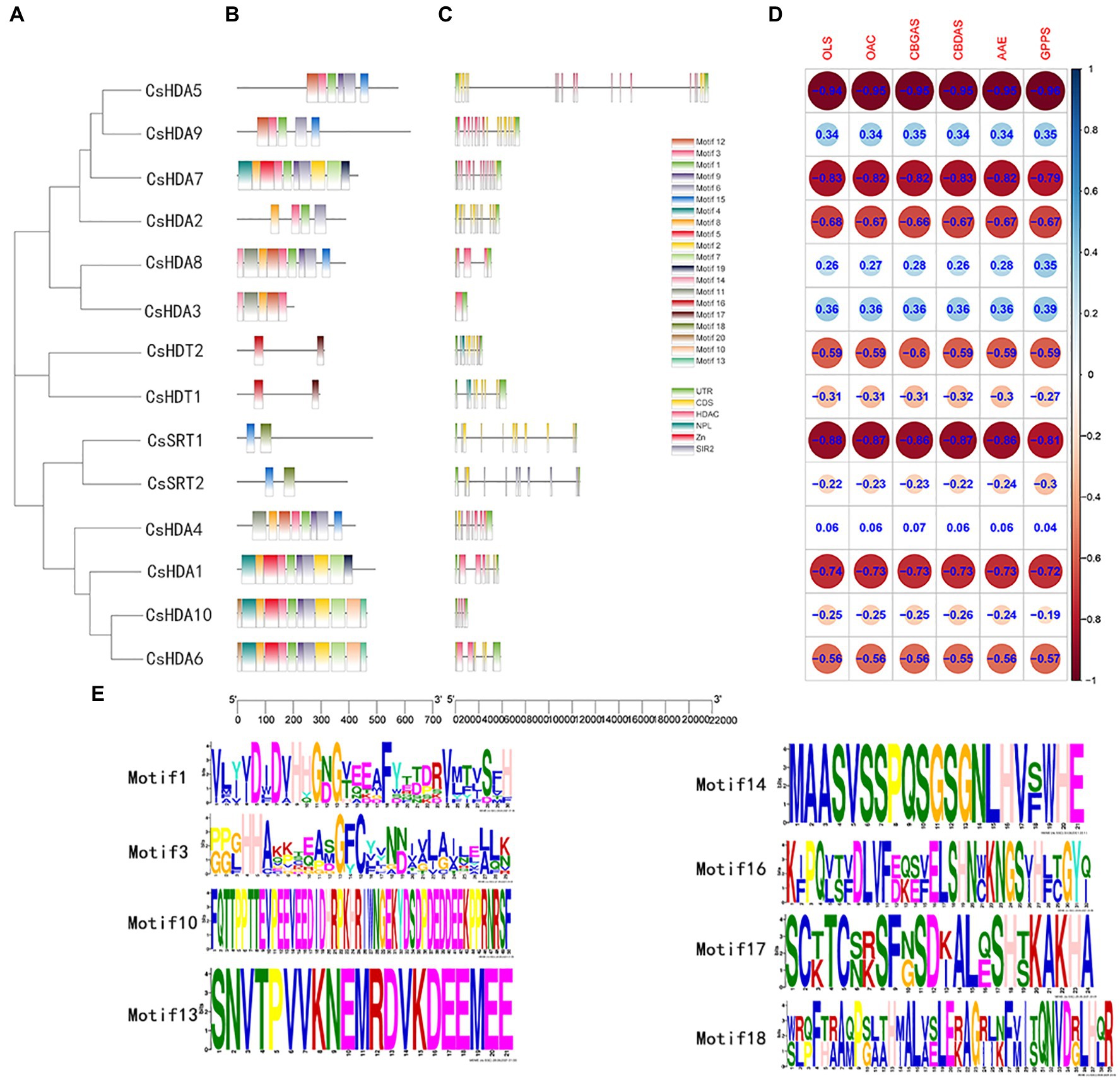
Figure 3. The gene structures and the distribution of conserved motifs of CsHDACs. (A) Construction of phylogenetic tree based on the full-length sequence of CsHDAC protein. (B) The distribution of the motifs of each CsHDAC protein. (C) The gene structure and distribution of the conserved domains of CsHDACs. Green boxes, unrelated area; yellow boxes, exons; black lines, introns; pink boxes, HDAC domains; blue boxes; blue boxes, NPL domains; red box, zinc finger domain; and gray boxes, SIR2 domains. (D) Co-expression analysis of CsHDACs and key genes in the cannabinoid synthesis pathway. (E) The sequence information for representative motifs.
We investigated the exons and introns from their number and distribution to study the structural composition of the CsHDAC genes (Figure 3C). Two members of the SIR2 subfamily had eight exons, and two members of the HD2 subfamily contained 10 exons. The coding sequences of the entire RPD3/HDA1 subfamily were interrupted by introns, with the number of exons in the range of 1–17. The CsHDAC genes had three main domains, which were specifically distributed into three subfamilies (Figure 3B). The SIR2, RPD3/HDA1, and HD2 subfamilies consisted of the SIR2, HDAC, and NPL domains, respectively. Members of the HD2 subfamily of C. sativa had conserved MEFWG pentapeptide at the N-terminus and possibly had a zinc finger at the C-terminus. Generally, the number of exons within each subgroup was similar. The domain of each subfamily was conserved, although the structures of members within a particular subfamily were different. In addition, we analyzed the co-expression analysis of CsHDACs and key genes in the cannabinoid synthesis pathway (Figure 3D; Supplementary Table S1). We found that CsHDA1, CsHDA5, CsHDA7, and CsSRT1 have obvious correlations with key genes.
Chromosome mapping of the CsHDAC genes was performed using the latest C. sativa genome database. A total of 14 CsHDAC genes were unevenly distributed on six chromosomes (Figure 4A). Among them, chromosomes 1, 2, 3, 4, 6, and 8 harbored 5, 1, 4, 1, 3, and 1 genes, respectively. While chromosomes 5, 7, 9, and 10 did not contain any HDAC gene. We further studied the homologous genes of CsHDAC within and between species. No homologous genes are found on the chromosomes of C. sativa. The results of collinearity analysis between C. sativa and A. thaliana showed three orthologous genes (Figure 4B).
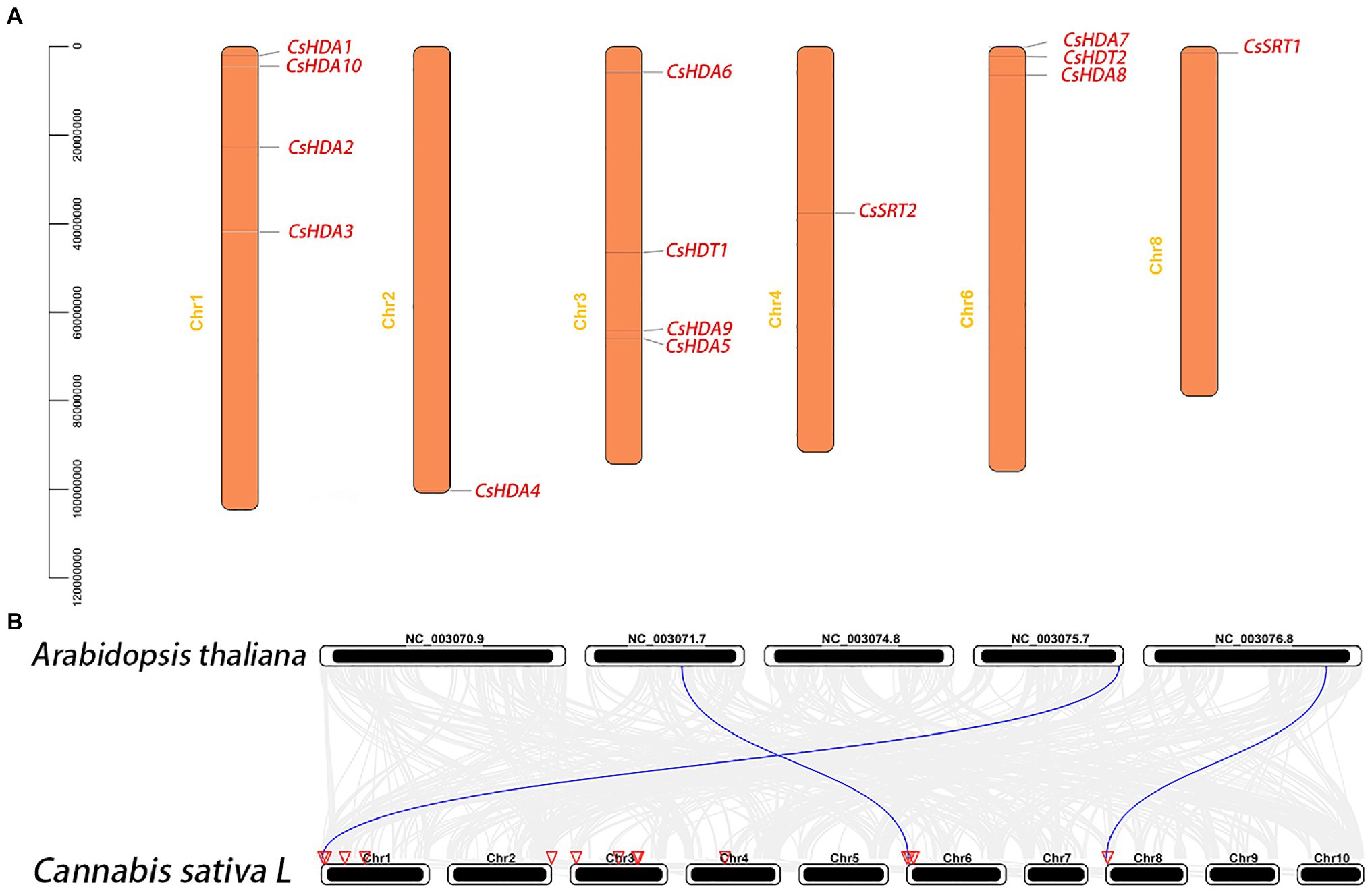
Figure 4. (A) Chromosomal locations of the CsHDAC genes. The y axis shows the length of the chromosome (unit: bp). (B) Synteny analysis of HDAC genes between Cannabis sativa and A. thaliana. The red triangle side indicates the position of the HDAC gene on the chromosome. The blue lines represent the syntenic HDAC gene pairs.
Cis-acting element analysis was performed on the CsHDAC gene family members, and the results were visualized (Figure 5). The HDAC gene family contained 13 cis-type elements, and each member of the family contained multiple response elements. Among them, HDAC9 contained many light-responsive elements, suggesting that HDAC9 may be highly sensitive to light stimuli. HDA10 contained the largest number of cis-elements, with six cis-elements, whereas HDA8 had the least number of cis-elements, with only two cis-elements.
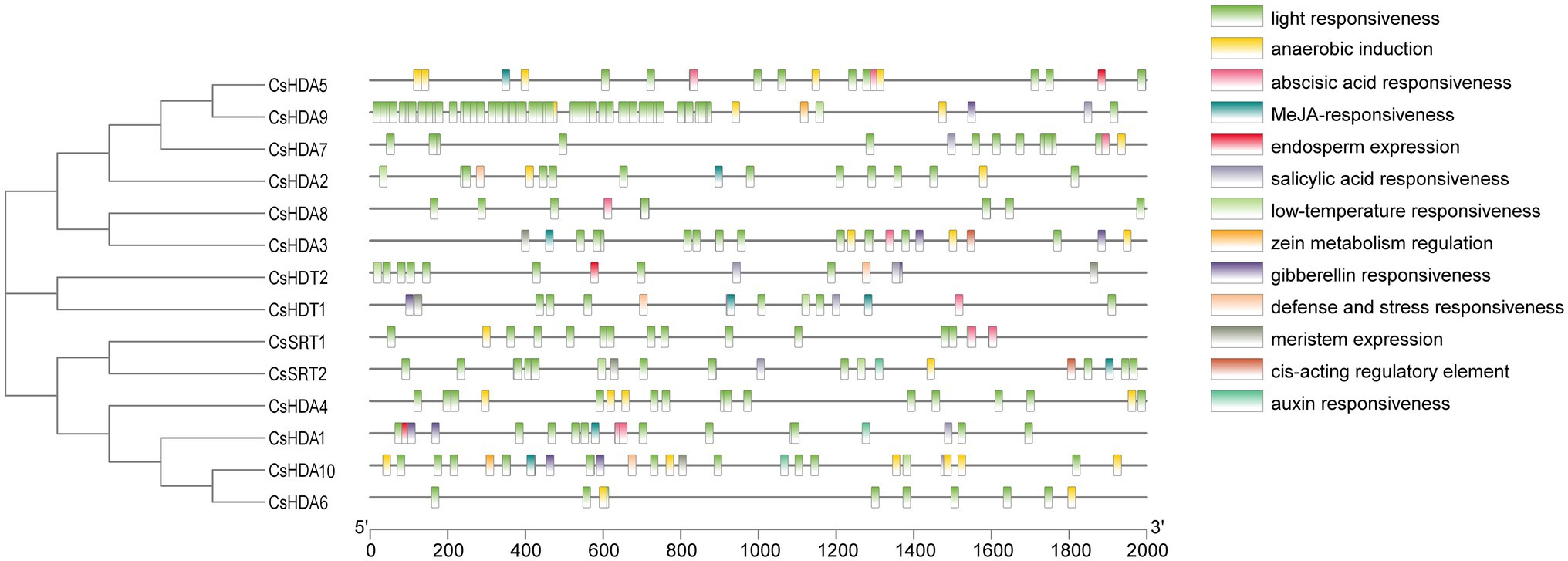
Figure 5. CIS-acting elements of the CsHDAC genes.
Alternative splicing, which causes polymorphisms in the function and structure of proteins, is extensive in eukaryotic organisms (Chaudhary et al., 2019). Therefore, we performed hypomorphic detection and prediction on the basis of data obtained via second-generation sequencing and third-generation sequencing and identified 21 splicing events in eight HDAC genes (Figure 6A). Compared with the eight selected reference isoforms, splicing events included seven intron retentions (IR), three alternative 3′ splice site (A3SS; AA), one exon skipping (ES), and two other isoforms (“others”). One gene may generate different alternative splicing events. For instance, NC_044372.1: 65918457–65940183.9 (CsHDA5) contained two cutting types: IR and “others.” The expression of different transcripts of the same gene differed. In other words, some transcripts dominated the expression of the gene in all tissues. For instance, rna-XM_030622750.1 (CsSRT1) had higher expression in all tissues than the other transcripts. Additionally, different transcripts may be enriched in various tissues. For instance, the rna-XM_030643555.1 (CsSRT2) had the highest expression in roots, while another splice variant, rna-XM_030643556.1 (CsSRT2), had the highest expression in leaves.
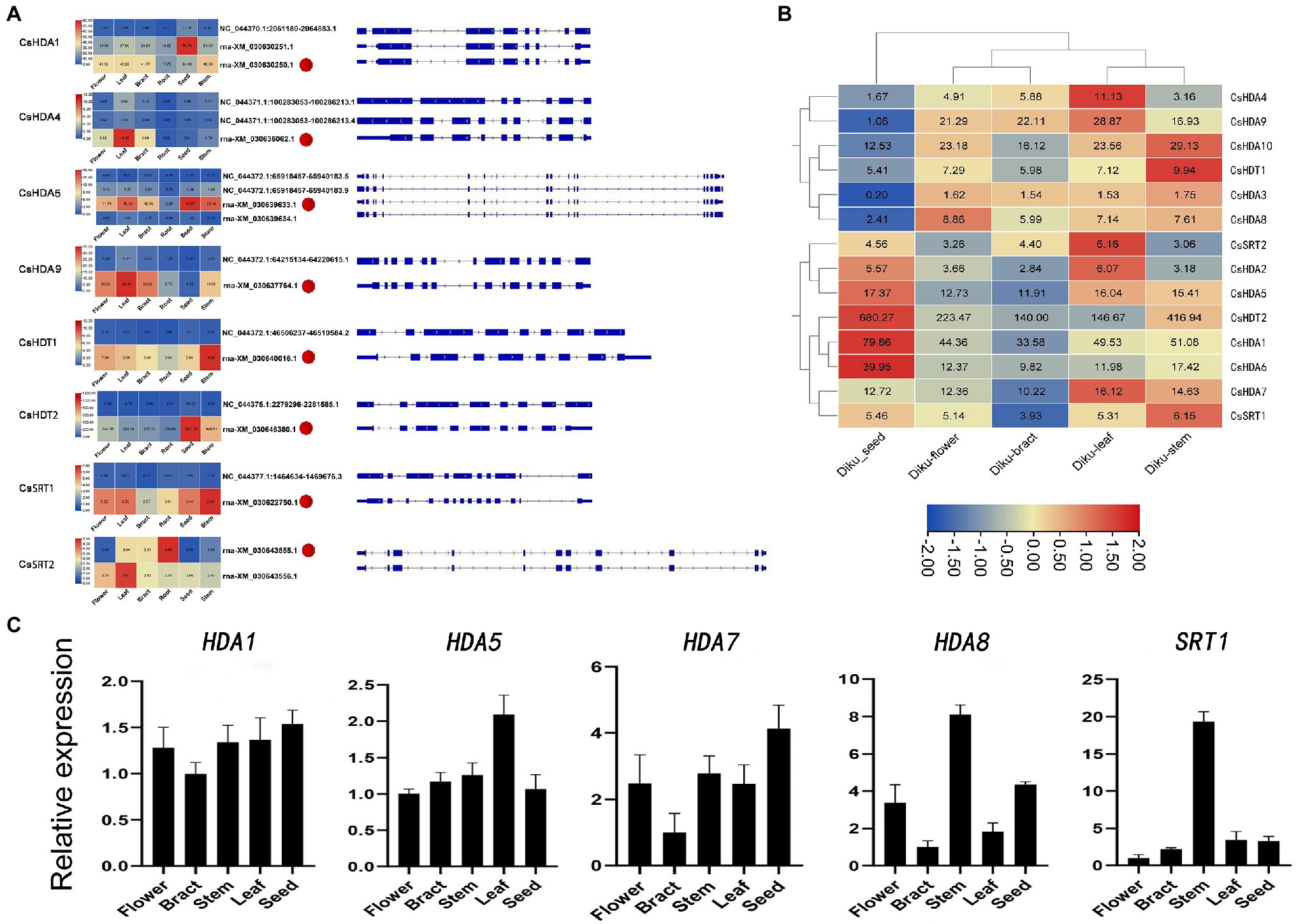
Figure 6. (A) Alternative splicing isoforms and the expression pattern of eight CsHDAC genes. Red dots refer to the selected reference isoforms. (B) The gene expression profile of CsHDACs in the seed, flower, bract, leaf, and stem. The row scale on the values has been performed, and the FPKM value of each gene was presented in the figure. (C) Expression patterns of the selected CsHDACs genes in the flower, bract, stem, leaf, and seed. The characters on the x axis indicate different organs and tissues. The y axis shows the relative level of gene expression. The EF1-α gene was used as an internal reference.
To explore the expression patterns of the CsHDAC gene family in different organs, we drew heat maps based on the FPKM calculated from RNA-seq data of seeds, flowers, stems, leaves, and bracts of DK (Figure 6B). CsHDA1, 5, 7, and 13 were commonly expressed in all tissues. CsHDA1 and CsHDT2 were highly expressed in all tissues, especially in seeds. The expression of CsHDA6 was also relatively higher in seeds. CsHDA10 was highly expressed in the stem, while CsHDA4 was enriched in leaves. Genes in flowers and bracts had a similar expression pattern, and the majority was expressed at lower levels than in other tissues. Expression patterns of CsHDAC genes in different tissues can be used as the foundation for identifying functional genes in C. sativa. According to previously published research, MaDHAC1, AtHDA6, and AtHD2 were associated with plant secondary metabolism (Han et al., 2016). Therefore, we selected candidate homologous CsHDAC members by constructing NJ trees and performing motif analysis between these genes and CsHDACs (Supplementary Figure S3) and verified them via qRT-PCR analysis. CsHDA1 and CsHDA5 were evenly enriched in all tissues. CsHDA7, CsHDA8, and CsSRT1 were less strongly expressed in the bracts (Figure 6C).
TSA can broadly inhibit the HDAC subfamily (Li et al., 2014; Su et al., 2015). To study whether HDAC is related to the synthesis of cannabinoids, we compared the changes in gene expression in hemp seedlings before and after TSA treatment. HDAC gene expression decreased significantly with increasing concentration of TSA. TSA could effectively inhibit the expression of all the CsHDAC genes in the exception of CsHDA10 (Figure 7A). In this study, two kinds of synthetic precursors were unambiguously identified as OA and GPP, respectively, by comparing the retention times, adductions, and productions with those of authentic standards. We also tested the expression of representative genes in the cannabinoid metabolic pathway and the changes in the synthetic precursors OA and GPP. The expression of genes in the fatty acid synthesis pathway, OAC, OLS, and AAE, was generally downregulated (Figure 7C). However, the expression of PP1 (HMB-PP reductase1), PP2 (HMB-PP reductase2), and geranyl diphosphate synthase (GPPS) in the MEP pathway increased (Figure 7B). Correspondingly, the content of synthetic precursor OA was reduced, while GPP accumulated (Figure 7D). The above results indicated that TSA inhibited the expression of CsHDAC genes and altered the contents of representative genes and synthetic precursors in the cannabinoid synthesis pathway.
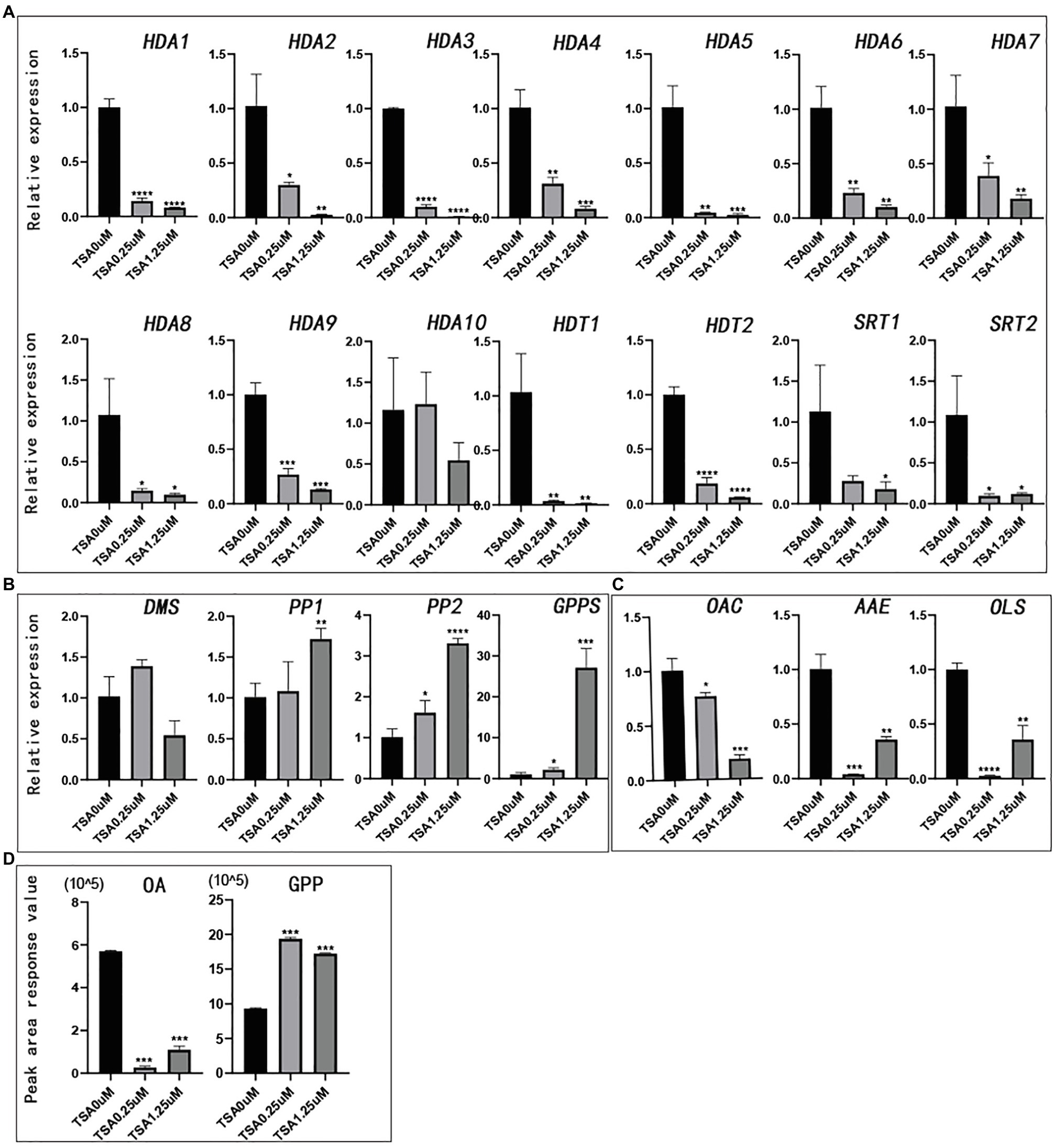
Figure 7. The relative expression of genes and the peak area of synthetic precursors OA and GPP after TSA treatment. The control was performed with 0μm TSA (DMSO matrix) treatment. n=3. (A) The relative expression of partial histone deacetylase genes; (B) The relative expression of key genes in the MEP pathway; (C) The relative expression of key genes in the fatty acid pathway; and (D) Peak area of synthetic precursors OA and GPP. *value of p <.05, **value of p <0.01, ***value of p <0.001, and ****value of p <0.0001.
Histone deacetylases are widely present in yeast, animals, and plants. The HDAC gene family has been isolated and characterized in several plants, involving 18 genes in Arabidopsis (Hollender and Liu, 2008), 18 genes in rice (Fu et al., 2007), 28 genes in soybean (Yang et al., 2018), 11 genes in litchi (Peng et al., 2017), and 15 genes in tomato (Zhao et al., 2015). In this study, we identified 14 CsHDAC members and surveyed their gene structures, expression profiles, phylogenetic relationships, and conserved motifs. CsHDACs can be classified into three subfamilies, RPD3/HDA1, HD2, and SIR2. The RPD3/HDA1 was the most highly represented CsHDAC subfamily in Arabidopsis (77%) and soybean (64%), suggesting that the RPD3/HDA1 subfamily may have undergone significant expansion during the process of evolution. The HD2 subfamily will be more likely to have a high affinity with the DNA-binding domain or mediate protein-protein interactions if it contains a C-terminal zinc finger (Wu et al., 2000; Dangl et al., 2001). For example, the interaction between AtHDT2 and AtDNMT2 (DNA methyl transfer) affected the expression of cold stress genes (Song, 2010), and PtHDT1 interacted with PtMYB to regulate lignin biosynthesis (Zhang, 2018). In our study, CsHDT2 had a C2H2 zinc finger in the C-terminus, suggesting that it may participate in protein-protein interactions. Motif 18 was unique to SIR2. We compared the SIR2 subfamily of Arabidopsis, soybean, and hemp, and found that they all contained motif 18, indicating that motif 18 was an important structural feature, that can be used to identify the SIR2 subfamily, and motif 18 could be the SIR2 subfamily structural basis with similar functions (König et al., 2014).
The expression of genes in tissues can help us infer the function of genes. In our study, we predicted and verified the expression of members of the CsHDAC family in different tissues. It was found that the expression of CsHDA7 in flowers and bracts was relatively low when compared its expression in other organs, which was consistent with the contention that HDACs usually bind inhibitory regulators to exert a negative regulatory effect. CsHDA1 was evenly distributed in all tissues, and in previous studies, its homologous gene AtHDA6 significantly contributed to a variety of aspects of plant development and growth, such as abiotic stress responses, jasmine and ethylene signal transmission, and leaf and flower development (Probst et al., 2004; Yu et al., 2011). Both genes were predicted to be localized in the nucleus, suggesting that they were involved in transcriptional regulation. Therefore, CsHDA1 may be a vital regulator throughout the life course of hemp.
Histone deacetylase is involved in the development of plants, the response to environmental changes, and the synthesis of secondary metabolites (Waterborg, 2011). HDAC usually forms a complex with transcription repressors to regulate target gene expression. In previous research, AtHDA15 interacted with a negative regulator, AtHY5, to repress photomorphogenesis (Jing et al., 2020). OsHDA1 physically interacted with OsIDS1 and the transcriptional corepressor OsTPR1 (topless-related 1) contributing to the repression of OsLEA1 and OsSOS1 expression (Cheng et al., 2018). MaHDAC1 and the transcription repressor MaERF11 inhibited the expression of genes related to maturation (Han et al., 2016). Combined with previous studies on the regulation of secondary metabolites by HDACs, tissue-specific expression, and co-expression analysis, we initially screened five candidate genes that may be related to cannabinoid regulation. Furthermore, we compared the domain amino acid sequences of CsHDACs and MaHDA1, constructed the NJ tree, and performed BLAST sequence alignment. The results showed that CsHDA7 and MaHDA1 had 86.98% amino acid similarity (Supplementary Figure S5), suggesting that CsHDA7 and MaHDA1 have a similar effect.
TSA has been shown to inhibit the activity of histone deacetylase. Similarly, the expression of CsHDACs was significantly depressed by TSA during this study. The synthesis of cannabinoids originated from the MEP pathway and fatty acid pathway. Both pathways differed after TSA treatment regardless of gene expression or metabolite accumulation, indicating that TSA can affect the biosynthesis of cannabinoids in several ways. However, it is still unclear whether TSA plays a role via inhibiting the expression of related genes or by mediating the structure of chromatin by repressing the activity of histone deacetylase in the cannabinoid synthesis pathway (Supplementary Figure S4).
We identified 14 CsHDAC family members and analyzed their structural characteristics, evolutionary relationships, and spatial expression patterns. By combining RNA-seq with qRT-PCR analysis, we predicted that five HDACs were associated with cannabinoid synthesis. Furthermore, the reduction of HDAC through TSA treatment directly contributed to the expression of related genes in cannabinoid synthesis and the changes in synthetic precursors of cannabinoids. However, both genes and the synthetic precursors involved in the fatty acid and MEP pathways behaved opposite with the TSA treatment, implying the complexity of HDACs in cannabinoid synthesis. Therefore, the function of candidate genes in cannabinoid synthesis should be thoroughly studied in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material .
ML, WS, and YM contributed to conception of the study. LY wrote the manuscript. LY, XM, XY, SC, and JCL conducted the bioinformatics analysis. LY, YZ, and SW contributed to data visualization. LY and YM performed the qRT-PCR experiment. JL, LY, and GQ performed the metabolic experiment. WC completed the data extraction. ML and WS revised the manuscript. SC, WS, and XL contributed to supervision. All authors have read and agreed to the published version of the manuscript.
This research received funding from the Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences (CI2021A04806 and CI2021A04008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.755494/full#supplementary-material
1. ^https://www.ncbi.nlm.nih.gov
2. ^http://planttfdb.cbi.pku.edu.cn/
3. ^https://www.ncbi.nlm.nih.gov/structure/cdd/wrpsb.cgi
4. ^http://web.expasy.org/protparam/
5. ^http://meme-suite.org/tools/meme
6. ^https://www.ncbi.nlm.nih.gov
7. ^http://www.genscript.com/wolf PSORT. HTML
8. ^http://www.plantcare.co.uk/
Appendino, G., Gibbons, S., Giana, A., Pagani, A., Grassi, G., Stavri, M., et al. (2008). Antibacterial cannabinoids from Cannabis sativa: A structure-activity study. J. Nat. Prod. 71, 1427–1430. doi: 10.1021/np8002673
Booth, J. K., Yuen, M., Jancsik, S., Madilao, L. L., Page, J. E., and Bohlmann, J. (2020). Terpene synthases and terpene variation in Cannabis sativa. Plant Physiol. 184, 130–147. doi: 10.1104/pp.20.00593
Brownell, J. E., Zhou, J., Ranalli, T., Kobayashi, R., Edmondson, D. G., Roth, S. Y., et al. (1996). Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84, 843–851. doi: 10.1016/S0092-8674(00)81063-6
Burstein, S. (2015). Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 23, 1377–1385. doi: 10.1016/j.bmc.2015.01.059
Causier, B., Ashworth, M., Guo, W., and Davies, B. (2012). The TOPLESS interactome: A framework for gene repression in Arabidopsis. Plant Physiol. 158, 423–438. doi: 10.1104/pp.111.186999
Chaudhary, S., Jabre, I., Reddy, A., Staiger, D., and Syed, N. (2019). Perspective on alternative splicing and proteome complexity in plants. Trends Plant Sci. 24, 496–506. doi: 10.1016/j.tplants.2019.02.006
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, X., Zhang, S., Tao, W., Zhang, X., Liu, J., Sun, J., et al. (2018). Indeterminate spikelet1 recruits histone deacetylase and a transcriptional repression complex to regulate rice salt tolerance1. Plant Physiol. 178, 824–837. doi: 10.1104/pp.18.00324
Dangl, M., Brosch, G., Haas, H., Loidl, P., and Lusser, A. (2001). Comparative analysis of HD2 type histone. Deacetylases in higher plants. Planta 275, 280–285. doi: 10.1007/s004250000506
De Meijer, E. P. M., and Hammond, K. M. (2005). The inheritance of chemical phenotype in Cannabis sativa L. Genitics. 145, 189–198. doi: 10.1007/s10681-005-1164-8
Frye, R. A. (2000). Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798. doi: 10.1006/bbrc.2000.3000
Fu, W., Wu, K., and Duan, J. (2007). Sequence and expression analysis of histone deacetylases in rice. Biochem. Biophys. Res. Commun. 356, 843–850. doi: 10.1016/j.bbrc.2007.03.010
Gao, H., Li, F., and Xu, Z. (2019). Genome-wide analysis of methyl jasmonate-regulated isoform expression in the medicinal plant Andrographis paniculata. Ind. Crops. Prod. 135, 39–48. doi: 10.1016/j.indcrop.2019.04.023
Graessle, S., Loidl, P., and Brosch, G. (2001). Histone acetylation: plants and fungi as model systems for the investigation of histone deacetylases. Cell. Mol. Life Sci. 58, 704–720. doi: 10.1007/PL00000894
Gu, X., Jiang, D., Yang, W., Jacob, Y., Michaels, S. D., and He, Y. (2011). Arabidopsis homologs of retinoblastoma-associated protein 46/48 associate with a histone deacetylase to act redundantly in chromatin silencing. PLoS Genet. 7:e1002366. doi: 10.1371/journal.pgen.1002366
Guarente, L. (2000). Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14, 1021–1026.
Haigis, M. C., and Guarente, L. P. (2006). Mammalian sirtuins - emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20, 2913–2921. doi: 10.1101/gad.1467506
Han, Y. C., Kuang, J. F., Chen, J. Y., Liu, X. C., Xiao, Y. Y., Fu, C. C., et al. (2016). Banana transcription factor MaERF11 recruits histone deacetylase MaHDA1 and represses the expression of MaACO1 and expansins during fruit ripening. Plant Physiol. 171, 1070–1084. doi: 10.1104/pp.16.00301
Hollender, C., and Liu, Z. (2008). Histone deacetylase genes in Arabidopsis development. J. Integr. Plant Biol. 50, 875–885. doi: 10.1111/j.1744-7909.2008.00704.x
Ikeda, M., and Ohme-Takagi, M. (2009). A novel group of transcriptional repressors in Arabidopsis. Plant Cell Physiol. 50, 970–975. doi: 10.1093/pcp/pcp048
Jing, Y., Guo, Q., and Lin, R. (2020). The SNL-HDA19 histone deacetylase complex antagonizes HY5 activity to repress photomorphogenesis in Arabidopsis. New Phytol. 229, 3221–3236. doi: 10.1111/nph.17114
König, A. C., Hartl, M., and Pham, P. A. (2014). The Arabidopsis class II sirtuin is a lysine deacetylase and interacts with mitochondrial energy metabolism. Plant Physiol. 164, 1401–1414. doi: 10.1104/pp.113.232496
Kornet, N., and Scheres, B. (2009). Members of the GCN5 histone acetyltransferase complex regulate PLETHORA-mediated root stem cell niche maintenance and transit amplifying cell proliferation in Arabidopsis. Plant Cell 21, 1070–1079. doi: 10.1105/tpc.108.065300
Kurdistani, S. K., and Grunstein, M. (2003). Histone acetylation and deacetylation in yeast. Nay. Rev. Mol. Cell Biol. 4, 276–284. doi: 10.1038/nrm1075
Lee, K., Park, O. S., Jung, S. J., and Seo, P. J. (2016). Histone deacetylation-mediated cellular dedifferentiation in Arabidopsis. J. Plant Physiol. 191, 95–100. doi: 10.1016/j.jplph.2015.12.006
Li, H., Soriano, M., Cordeverer, J., Muiño, J. M., Riksen, T., Fukuoka, H., et al. (2014). Thehistone decetylase inhibitor trichostatin A promotes Totipotency in the malle gaming. Plan Cell. 26, 195–2009. doi: 10.1105/tpc.113.116491
Liu, X., Luo, M., and Wu, K. (2012). Epigenetic interplay of histone modifications and DNA methylation mediated by HDA6. Plant Signal. Behav. 7, 633–635. doi: 10.4161/psb.19994
Luo, X., Reiter, M. A., d’Espaux, L., Wong, J., Denby, C. M., Lechner, A., et al. (2019). Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 567, 123–126. doi: 10.1038/s41586-019-0978-9
Lusser, A., Kölle, D., Loidl, P., and Lusser, A. (2001). Trends in plant science histone acetylation lessons from the plant kingdom. Trends Plant Sci. 6, 59–65. doi: 10.1016/S1360-1385(00)01839-2
Lydon, J., Teramura, A. H., and Coffman, C. B. (1987). UV-B radiation effects on photosynthesis, growth and cannabinoid production of two Cannabis sativa CHEMOTYPES. Photochem. Photobiol. 46, 201–206. doi: 10.1111/j.1751-1097.1987.tb04757.x
Marks, M. D., Tian, L., Wenger, J. P., Omburo, S. N., Soto-Fuentes, W., He, J., et al. (2009). Identification of candidate genes affecting Δ9- tetrahydrocannabinol biosynthesis in Cannabis sativa. J. Exp. Bot. 60, 3715–3726. doi: 10.1093/jxb/erp210
Morimoto, S., Komatsu, K., Taura, F., and Shoyama, Y. (1997). Enzymological evidence for cannabichromenic acid biosynthesis. J. Nat. Prod. 60, 854–867. doi: 10.1021/np970210y
Nicolas-Francès, V., Grandperret, V., Liegard, B., Jeandroz, S., Vasselon, D., Aimé, S., et al. (2018). Evolutionary diversification of type-2 HDAC structure, function and regulation in Nicotiana tabacum. Plant Sci. 269, 66–74. doi: 10.1016/j.plantsci.2018.01.007
Nicot, N., Hausman, J. F., Hoffmann, L., and Evers, D. (2005). Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56, 2907–2914. doi: 10.1093/jxb/eri285
Pacher, P., Bátkai, S., and Kunos, G. (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 58, 389–462. doi: 10.1124/pr.58.3.2
Pandey, R., Mu, A., Ller, È., Napoli, C. A., Selinger, D. A., Pikaard, C. S., et al. (2002). Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 30, 5036–5055. doi: 10.1093/nar/gkf660
Peng, M., Ying, P., Liu, X., Li, C., Xia, R., Li, J., et al. (2017). Genome-wide identification of histone modifiers and their expression patterns during fruit abscission in litchi. Front. Plant Sci. 8:639. doi: 10.3389/fpls.2017.00639
Probst, A. V., Fagard, M., Proux, F., Mourrain, P., Boutet, S., Earley, K., et al. (2004). Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing anddetermines nuclear organization of rDNA repeats. Plant Cell 16, 1021–1034. doi: 10.1105/tpc.018754
Schachtsiek, J., Warzecha, H., Kayser, O., and Stehle, F. (2018). Current perspectives on biotechnological cannabinoid production in plants. Planta Med. 84, 214–220. doi: 10.1055/s-0043-125087
Song, Y. (2010). Interaction and function study of Arabidopsis DNA methyltransferase DNMT2 and histone deacetylase family HD2. Biochem. Biophys. Res. Commun. 396, 187–192. doi: 10.7666/d.Y1704966
Song, C. P., and Galbraith, D. W. (2006). AtSAP18, an orthologue of human SAP18, is involved in the regulation of salt stress and mediates transcriptional repression in Arabidopsis. Plant Mol. Biol. 60, 241–257. doi: 10.1007/s11103-005-3880-9
Su, L. C., Ding, B., Liu, S., Limei, L., Yuting, Z., and Ling, L. (2015). Isolation and characterization of an osmotic stress and ABA induced histone deacetylasm. Arachis Hygogaea. Font. PlantSci. 6, 512–522. doi: 10.3389/fpls.2015.00512
VanBakel, H., Stout, J. M., Cote, A. G., Tallon, C. M., Sharpe, A. G., Hughes, T. R., et al. (2011). The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12:R102. doi: 10.1186/gb-2011-12-10-r102
Vindenes, V. (2017). Increasing plant concentrations of THC and implications on health related disorders. Handbook of Cannabis and Related Pathologies. 3, 24–32.
Wang, Y. P., Tang, H. B., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, Z. B., Zang, C., Cui, K., Schones, D. E., Barski, A., Peng, W., et al. (2009). Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031. doi: 10.1016/j.cell.2009.06.049
Waterborg, J. H. (2011). “Plant histone acetylation: In the beginning …” in Biochimica et Biophysica Acta Gene Regulatory Mechanisms. ed. G. Grafi, vol. 1809 (Elsevier), 353–359.
Wu, K., Tian, L., Mallik, K., Brown, D., and Miki, B. (2000). Functional analysis of HD2 histone deacetylase homologues in Arabidopsis thaliana. Plant J. 22, 19–27. doi: 10.1046/j.1365-313x.2000.00711.x
Xu, Z. C., Ranran, G., Pu, X. D., Xu, R., Wang, J., Zheng, S., et al. (2020). Comparative genome analysis of Scutellaria baicalensis and Scutellaria barbata reveals the evolution of active flavonoid biosynthesis. Geno. Proteo. Bioinfo. 18, 230–240. doi: 10.1016/j.gpb.2020.06.002
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yang, X. J., and Seto, E. (2007). HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318. doi: 10.1038/sj.onc.1210599
Yang, C., Shen, W., Chen, H., Chu, L., and Xu, Y. (2018). Characterization and subcellular localization of histone deacetylases and their roles in response to abiotic stresses in soybean. BMC Plant Biol. 18, 1–13. doi: 10.1186/s12870-018-1454-7
Yu, C. W., Liu, X., Luo, M., Chen, C., Lin, X., Tian, G., et al. (2011). Histonedeacetylase6 interacts with flowering locus D and regulatesflowering in Arabidopsis. Plant Physiol. 156, 173–184. doi: 10.1104/pp.111.174417
Zhang, Y. (2018). HDT1 histone deacetylase and MYB211 ellipse regulate the growth and development of plant cambium and xylem. doi: 10.26949/d.cnki.gblyu.2018.000130
Zhang, Z., Wang, B., Wang, S., Lin, T., and Yang, L. (2020). Genome-wide target mapping shows histone deacetylase complex1 regulates cell proliferation in cucumber fruit. Plant Physiol. 182, 167–184. doi: 10.1104/pp.19.00532
Zhao, L., Lu, J., Zhang, J., Wu, P. Y., Yang, S., and Wu, K. (2015). Identification and characterization of histone deacetylases in tomato (Solanum lycopersicum). Front. plant science 5:760. doi: 10.3389/fpls.2014.00760
Zheng, Y., Ge, J., Bao, C., Chang, W., and Liu, J. (2020). Histone deacetylase HDA9 and WRKY53 transcription factor are mutual antagonists in regulation of plant stress response. Mol. Plant 13, 598–611. doi: 10.1016/j.molp.2019.12.011
Zhou, Y., Tan, B., Luo, M., Li, Y., Liu, C., and Chen, C. (2013). Histone deacetylase19 interacts with HSL1 and participates in the repression of seed maturation genes in Arabidopsis seedlings. Plant Cell 25, 134–148. doi: 10.1105/tpc.112.096313
Keywords: histone deacetylase, Cannabis sativa, gene family, bioinformatics, genome-wide
Citation: Yang L, Meng X, Chen S, Li J, Sun W, Chen W, Wang S, Wan H, Qian G, Yi X, Li J, Zheng Y, Luo M, Chen S, Liu X and Mi Y (2021) Identification of the Histone Deacetylases Gene Family in Hemp Reveals Genes Regulating Cannabinoids Synthesis. Front. Plant Sci. 12:755494. doi: 10.3389/fpls.2021.755494
Edited by:
Lei Zhang, Second Military Medical University, ChinaReviewed by:
Dongming Ma, Guangzhou University of Chinese Medicine, ChinaCopyright © 2021 Yang, Meng, Chen, Li, Sun, Chen, Wang, Wan, Qian, Yi, Li, Zheng, Luo, Liu and Mi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Liu, bHJ4MTEyNUAxMjYuY29t; Yaolei Mi, eGlhb21pMjAwNjNAc2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.