 Qian-Qian Wang1
Qian-Qian Wang1 Meng-Jia Zhu2Xia Yu1Yuan-Yang Bi1
Meng-Jia Zhu2Xia Yu1Yuan-Yang Bi1 Zhuang Zhou1,3
Zhuang Zhou1,3 Ming-Kun Chen1Jiating Chen1Diyang Zhang1
Ming-Kun Chen1Jiating Chen1Diyang Zhang1 Ye Ai1
Ye Ai1 Zhong-Jian Liu1,3,4*Siren Lan1,2*
Zhong-Jian Liu1,3,4*Siren Lan1,2*- 1Key Laboratory of National Forestry and Grassland Administration for Orchid Conservation and Utilization at College of Landscape Architecture, Fujian Agriculture and Forestry University, Fuzhou, China
- 2College of Forestry, Fujian Agriculture and Forestry University, Fuzhou, China
- 3Zhejiang Institute of Subtropical Crops, Zhejiang Academy of Agricultural Sciences, Wenzhou, China
- 4Institute of Vegetable and Flowers, Shandong Academy of Agricultural Sciences, Jinan, China
Terpene synthases (TPSs) are essential for forming terpenes, which play numerous functional roles in attracting pollinators, defending plants, and moderating the interaction between plants. TPSs have been reported in some orchids, but genome-wide identification of terpenes in Cymbidium faberi is still lacking. In this study, 32 putative TPS genes were classified in C. faberi and divided into three subfamilies (TPS-a, TPS-b, and TPS-e/f). Motif and gene structure analysis revealed that most CfTPS genes had the conserved aspartate-rich DDxxD motif. TPS genes in the TPS-a and TPS-b subfamilies had variations in the RRX8W motif. Most cis-elements of CfTPS genes were found in the phytohormone responsiveness category, and MYC contained most of the numbers associated with MeJA responsiveness. The Ka/Ks ratios of 12/13 CfTPS gene pairs were less than one, indicated that most CfTPS genes have undergone negative selection. The tissue-specific expression patterns showed that 28 genes were expressed in at least one tissue in C. faberi, and TPS genes were most highly expressed in flowers, followed by leaves and pseudobulbs. In addition, four CfTPS genes were selected for the real-time reverse transcription quantitative PCR (RT-qPCR) experiment. The results revealed that CfTPS12, CfTPS18, CfTPS23, and CfTPS28 were mainly expressed in the full flowering stage. CfTPS18 could convert GPP to β-myrcene, geraniol, and α-pinene in vitro. These findings of CfTPS genes of C. faberi may provide valuable information for further studies on TPSs in orchids.
Introduction
Terpenes, which contain isoprene (C5), monoterpenes (C10), sesquiterpenes (C15), and diterpenes (C20), constitute the most prominent family of volatile components in plants (Nagegowda, 2010; Tholl, 2015). They play significant roles in attracting pollinators, defending plants against herbivores and pathogens, and moderating the interaction between plants (Wagner and Elmadfa, 2003; Dudareva et al., 2006). Terpenoids are dominant compounds in orchid floral scents (Ramya et al., 2018). For example, sesquiterpenes are the major floral scent compounds in Cymbidium goeringii (Ramya et al., 2019). The monoterpenes linalool, ocimene, and linalool oxide are the major scent components of Vanda Mini Palmer flowers (Mohd-Hairul et al., 2010). The floral scent in orchids is closely related to flower development stages and is vital for pollination ecology (Ramya et al., 2018).
Isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are precursors of terpene formation, and they are generated by cytosolic mevalonate acid (MVA) and the plastid methylerythritol phosphate (MEP) pathways (Dudareva et al., 2006). A plastid prenyltransferase synthesizes geranyl diphosphate (GPP), and the second type of plastid prenyltransferase produces geranylgeranyl diphosphate (GGPP) from the condensation of IPP and DMAPP. The condensation of DMAPP and IPP forms farnesyl diphosphate (FPP) in the cytosol (Figure 1). Plants have terpene synthases (TPSs) that catalyze monoterpene formation, sesquiterpene formation, and diterpene formation from GPP, FPP, and GGPP, respectively (Pichersky et al., 2006; Vranová et al., 2013).
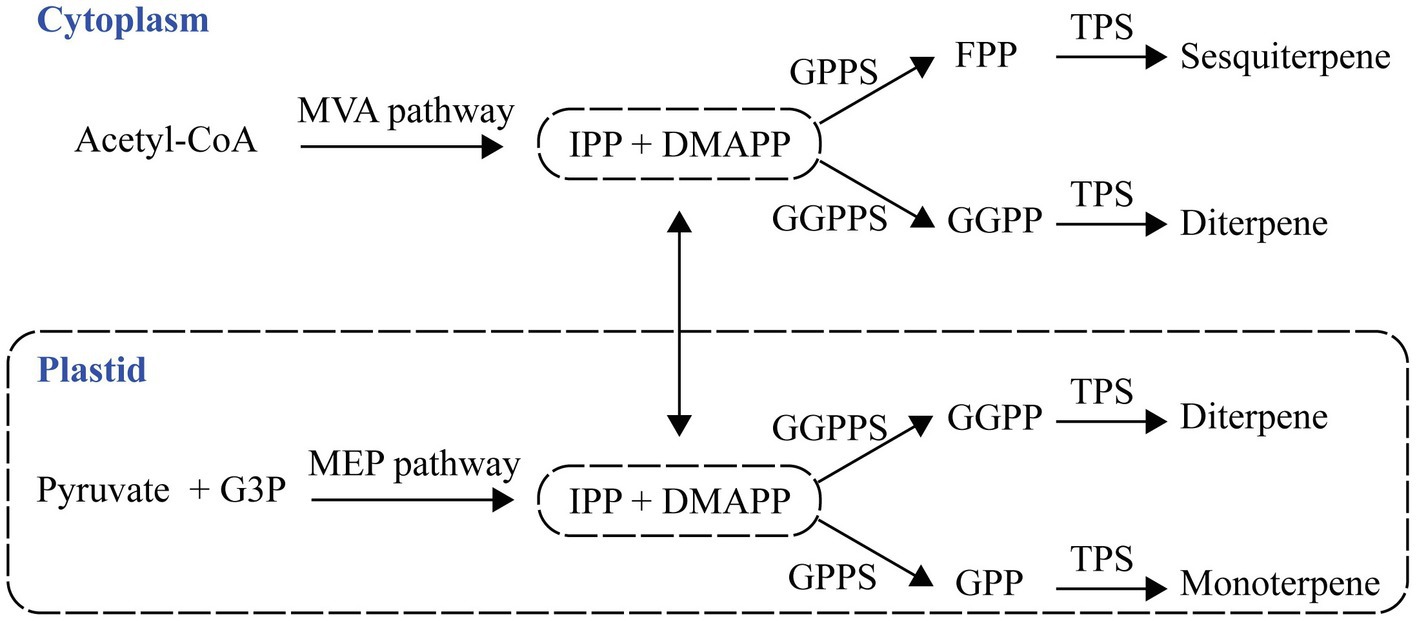
Figure 1. The pathway of plant terpenes biosynthesis. Terpenes are synthesized from two precursors generated by the cytosolic mevalonate acid (MVA) and plastid methylerythritol phosphate (MEP) pathways. G3P, glyceraldehyde 3-phosphate; IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GPP, geranyl diphosphate; GPPS, GPP synthase; FPP, farnesyl diphosphate; FPPS, FPP synthase; GGPP, geranylgeranyl diphosphate; GGPPS, GGPP synthase; and TPS, terpene synthase.
Most full-length TPSs contain two conserved domains defined in PFAM: PF01397 (N-terminal) containing a conserved RRX8W motif and PF03936 (C-terminal) containing a DDxxD motif and NSE/DTE motif (El-Gebali et al., 2019; Jiang et al., 2019). In addition, TPSs can be divided into seven major categories: TPS-a, TPS-b, TPS-c, TPS-d, TPS-e/f, TPS-g, and TPS-h (Chen et al., 2011). TPS-a, TPS-b, and TPS-g categories are angiosperm-specific. The TPS-a category can encode sesquiterpene synthases of these three categories and can be further classified into monocot-specific TPS-a-1 and dicot-specific TPS-a-2 groups. In recent reports, all characterized TPSs in TPS-b with RRX8W motifs are monoterpene synthases. The TPS-g category can encode monoterpene synthases without the RRX8W motif. In addition, TPS-d is a gymnosperm-specific category that encodes monoterpenes, sesquiterpenes, and diterpenes. TPS-e/f can encode kaurene or copalyl diphosphate synthases that function in gibberellic acid synthesis in vascular plants. TPS-c may represent the ancestral category, and TPS-h only appears in Selaginella moellendorffii in recent reports (Chen et al., 2011; Li et al., 2012).
Genome-wide TPS families have been identified in Arabidopsis thaliana, Vitis vinifera, Malus domestica, Camellia sinensis, Glycine max, Daucus carota, and Dendrobium. officinale (Aubourg et al., 2002; Martin et al., 2010; Nieuwenhuizen et al., 2013; Liu et al., 2014; Keilwagen et al., 2017; Yu et al., 2020; Zhou et al., 2020). Terpenoids are predominant components in orchid flower volatiles, but genome-wide TPS identification is limited in orchids (Ramya et al., 2018; Yu et al., 2020). Orchidaceae comprises five subfamilies, and in recent reports, a few TPS genes have been identified in Apostasia shenzhenica in Apostasioideae subfamily; Vanilla planifolia in Vanilloideae subfamily; and Phalaenopsis equestris and Dendrobium catenatum in Epidendroideae subfamily (Huang et al., 2021). However, genome-wide TPS genes identification in Orchidoideae was limited.
Cymbidium faberi is a plant of Orchidaceae with a long history of cultivation due to its characteristic flower fragrance and beautiful flower shape (Ramya et al., 2018). There are over 100 compounds in the C. faberi floral scent, and some terpenes have been identified in the blooming C. faberi flowers by headspace vapors (Omata et al., 1990). This study first analyzed the classification, phylogenetics, and expression patterns of TPS genes in C. faberi. Our results will provide valuable information for further studies on C. faberi and other orchids.
Materials and Methods
Plant Materials
The wild C. faberi collected in this study were cultivated in the greenhouse at Forest Orchid Garden in Fujian Agriculture and Forestry University (Fuzhou, Fujian Province, China) under natural light and temperatures. The temperature was about 20–25°C. Flowers, leaves, and pseudobulbs of C. faberi were sampled at the flowering stage. The buds about 1cm before anthesis, semi-open flowers about 3cm, and fully open flowers were also used in this study. All samples of C. faberi were frozen in liquid nitrogen for storage at −80°C until use.
Identification of CfTPS Genes in the C. faberi Protein Database
Two domains – PF01397 representing the TPS N-terminal domain and PF03936 representing the TPS C-terminal domain from PFAM1 – were used as queries to search the C. faberi protein database (El-Gebali et al., 2019). The C. faberi genome data will be published separately. An HMM search (built-in Tbtools) was used in this study with an e-value cut at 10−3. To avoid missing potential TPS genes, TPS sequences from A. thaliana in the TAIR database2 were also used to screen the C. faberi protein database using BLASTP (built-in Tbtools; Chen et al., 2018). The candidate TPS genes were checked manually by Pfam to verify putative full-length TPS genes, and TPS genes lacking either PF03936 or PF01397 were excluded. The grand average of hydrophobicity (GRAVY), molecular weight (Mw), isoelectric points (pI), aliphatic index (AI), and instability index (II) of the TPS proteins were predicted by the ExPASy database (Artimo et al., 2012).3 Subcellular localization was predicted by Plant-mPloc (Chou and Shen, 2010),4 AtSubP (Kaundal et al., 2010),5 and Ploc-mPlant (Cheng et al., 2017).6 Terzyme7 and BLATP8 were used to predict gene function (Priya et al., 2018).
Motifs and Gene Structure Analysis
Conserved motifs in the C. faberi TPS sequences were employed and analyzed by MEME software9 with default parameters (Bailey et al., 2009). We identified 20 motifs in this study. The exon-intron structure of the sequences was determined using GSDS software (Hu et al., 2015).10
Phylogenetic Analysis of TPS Genes
The transcriptomes of TPS sequences from P. equestris and A. shenzhenica were downloaded from their genome databases. TPS sequences from S. moellendorffii were downloaded from NCBI.11 TPS sequences from Picea abies were downloaded from UniProt,12 and TPS protein sequences from A. thaliana and Oryza sativa were downloaded from Phytozome.13 All these sequences were aligned with MAFFT (Rozewicki et al., 2019). The maximum likelihood (ML) method was used for the phylogenetic tree, which was constructed with RAxML on the CIPRES Science Gateway web server (RAxML-HPC2 on XSEDE; Miller et al., 2011). Bootstrap values were 1,000 replicates with the JTT model. The most appropriate protein evolution model for the alignment was predicted by ProTest (Darriba et al., 2011). The generated tree was redrawn and annotated by EVOLVIEW (He et al., 2016).14 The sequences of the CfTPS proteins used in this study are listed in Supplementary Table S7.
Promoter Element Analysis of TPS Genes
Tbtools software extracted the promoter sequences and 2,000bp regions upstream of 32 CfTPS genes (Chen et al., 2018). Afterward, the online software PlantCARE15 was used to identify the cis-active regulatory elements in the promoter regions of the CfTPS genes (Lescot et al., 2002).
Calculation of Ka and Ks Ratios
Gene pairs with similar genetic relationships were selected based on the phylogenetic tree. DNAMAN software was used to select the gene pairs with a consistency greater than 60%. Tbtools software was then used to calculate Ka (non-synonymous rate), Ks (synonymous substitution), and Ka/Ks (evolutionary constraint) values. Divergence time (T) was calculated by using the formula T=Ks/ (2×9.1×10−9)×10−6 million years ago (Mya; Zhang et al., 2018). In general, Ka/Ks<1.0 represents purifying or negative selection, Ka/Ks=1.0 represents neutral selection, and Ka/Ks>1.0 represents positive selection (Zhang et al., 2006).
Transcriptome Data and GO Classification Analysis
An RNA sequencing transcriptome database of leaves, pseudobulbs, petals, sepals, labellums, and gynostemium was established to study the expression patterns of CfTPS genes. Fragments per kilobase of transcript per million mapped reads (FPKM) values of CfTPS genes were used to evaluate translation abundance. DESeq was used to conduct gene differential expression analysis, and gene ontology (GO) classification analysis was performed based on the differentially expressed gene analysis. The heatmaps of CfTPS expression patterns were drawn by Tbtools software, and the color in the heatmap was expressed as the log2-transformed expression levels of each CfTPS gene (Chen et al., 2018).
Extraction of RNA and RT-qPCR Analysis
RNA was isolated from flowers of C. faberi at the flowering stage using the Biospin Plant Total RNA Extraction Kit (Bioer Technology, Hangzhou, China). First-strand DNA was synthesized with TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for quantitative PCR (qPCR; TransGen Biotech, Beijing, China). TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR was also used to remove genomic DNA. The real-time reverse transcription quantitative PCR (RT-qPCR) primers of CfTPS were designed by Primer Premier 5 software and can be found in Supplementary Table S12. Primer specificity was confirmed using Primer-BLAST on the NCBI website.16 PerfectStart™ Green qPCR SuperMix (TransGen Biotech, Beijing, China) was used for RT-qPCR analysis. The C. faberi reference gene GAPDH (GenBank Accession: JX560732; Tian et al., 2020) was used as the internal control and quantified by the 2-△△CT method (Livak and Schmittgen, 2001). There were three biological replicates in the RT-qPCR analysis.
CfTPS18 Enzyme Assays
The ORF of CfPS18 was synthesized and ligated into pET28a vector. Then, the recombinant plasmid was transformed into Escherichia coli BL21 (DE3) pLysS cells (Transgen, China). Primers are given in Supplementary Table S12. The positive clones were incubated with shaking at 200rpm, 37°C until OD600=0.6, and then at 37°C for 3h with shaking and 0.1mM IPTG. Recombinant CfTPS18 enzyme was induced at 16°C for 16h with 0.1mM IPTG. The precipitate was resuspended in extraction buffer [50mM NaH2PO4, 500mM NaCl, 10% (v/v) glycerol, and pH 7.0] and disrupted with a sonicator at 200W for 60s. The protein was purified with Ni-NTA agarose (Clontech). Purified CfTPS18 protein was examined by SDS-PAGE using Tris-HCl buffer (pH 7.5). The recombinant protein was performed in assay buffer [25mM HEPES, 10mM MgCl2, 100mM KCl, 5mM DTT, 10% (v/v) glycerol, and 30μM GPP], and 30μg of CfTPS18 protein at pH 7.2 and 30°C for 1h24.
GC-MS Analysis
The volatiles were exposed to SPME fiber with 50/30μm DVB/CAR/PDMS (divinylbenzene/carboxen/polydimethylsiloxane; Supelco Co., Bellefonte, PA, United States). The extract was analyzed using a gas chromatograph (Agilent 6,890N) and mass spectrometer (Agilent 5975B, Santa Clara, CA, United States) outfitted with a silica capillary column (DB-5MS; 60m×0.25mm×0.25μm). The temperature program was as follows: 55°C for 2min, 3°C min−1 up to 80°C, 5°C min−1 up to 180°C, 10°C min−1 up to 230°C, and finally, 20°C min−1 to 250°C. The ion source temperature was 230°C, and the electron energy was 70eV. The GC-MS interface zone temperature was 250°C, and the scan range was 50–500m/z. Reactions only added with GPP were used as the blank control. There were three biological replicates for the experiment. The retention time was compared with the NIST Mass Spectral Library.
Results
TPS Gene Identification and Protein Features in C. faberi
To retrieve the TPS genes in C. faberi, two domains, PF01397 and PF03936 in the PFAM, were used to search the C. faberi protein database (El-Gebali et al., 2019). BLASTP (built-in Tbtools) was also used to screen the C. faberi protein database (Chen et al., 2018). After removing artefacts, 32 TPS genes were obtained. The deduced proteins of these genes were in the range of 115 for CfTPS1 and CfTPS2 to 902 amino acids for CfTPS12 and had predicted molecular weights (Mw) in the range of 10.24 for CfTPS12 to 103.89 KDA for CfTPS31. The theoretical isoelectric point (pI) values were in the range of 4.86 for CfTPS1 and CfTPS2 to 9.28 for CfTPS26, and the deduced grand average of hydrophilic (GRAVY) values were in the range of −0.358 for CfTPS26 to 0.073 for CfTPS6, suggesting that most CfTPS proteins were hydrophilic except for CfTPS1, CfTPS2, and CfTPS6. Additionally, the aliphatic index (AI) of CfTPS-deduced proteins was in the range of 87.09 for CfTPS21 to 107.22 for CfTPS6, and the instability index (II) was in the range of 28.62 for CfTPS1 and CfTPS2 to 52.36 for CfTPS5. To retrieve information on the subcellular localization of CfTPS proteins, three predictors were used in this study: AtSubp, Plant-mPLoc, and pLoc-mPLant (Chou and Shen, 2010; Kaundal et al., 2010; Cheng et al., 2017). The results showed that 16 CfTPS proteins were marked in the chloroplast, and 16 CfTPS proteins were marked in chloroplast or cytoplasm, indicating that these three predictors produce different results and need to be further analyzed. We also annotated 32 CfTPS genes using BLASTP17 and Terzyme software (Supplementary Table S4; Priya et al., 2018). The results showed that 11 CfTPSs were annotated sesquiterpene synthases, 15 CfTPSs were annotated monoterpene synthases, and six CfTPSs were annotated diterpene synthases. The secondary structure predicted by the SOPMA program revealed that the average of α-helices, random coils, extended strands, and β-turns comprised 70.86, 21.37, 4.35, and 3.63% of the structure, respectively (Geourjon and Deléage, 1995). The results showed that α-helices were predominant in all CfTPS proteins (Table 1).
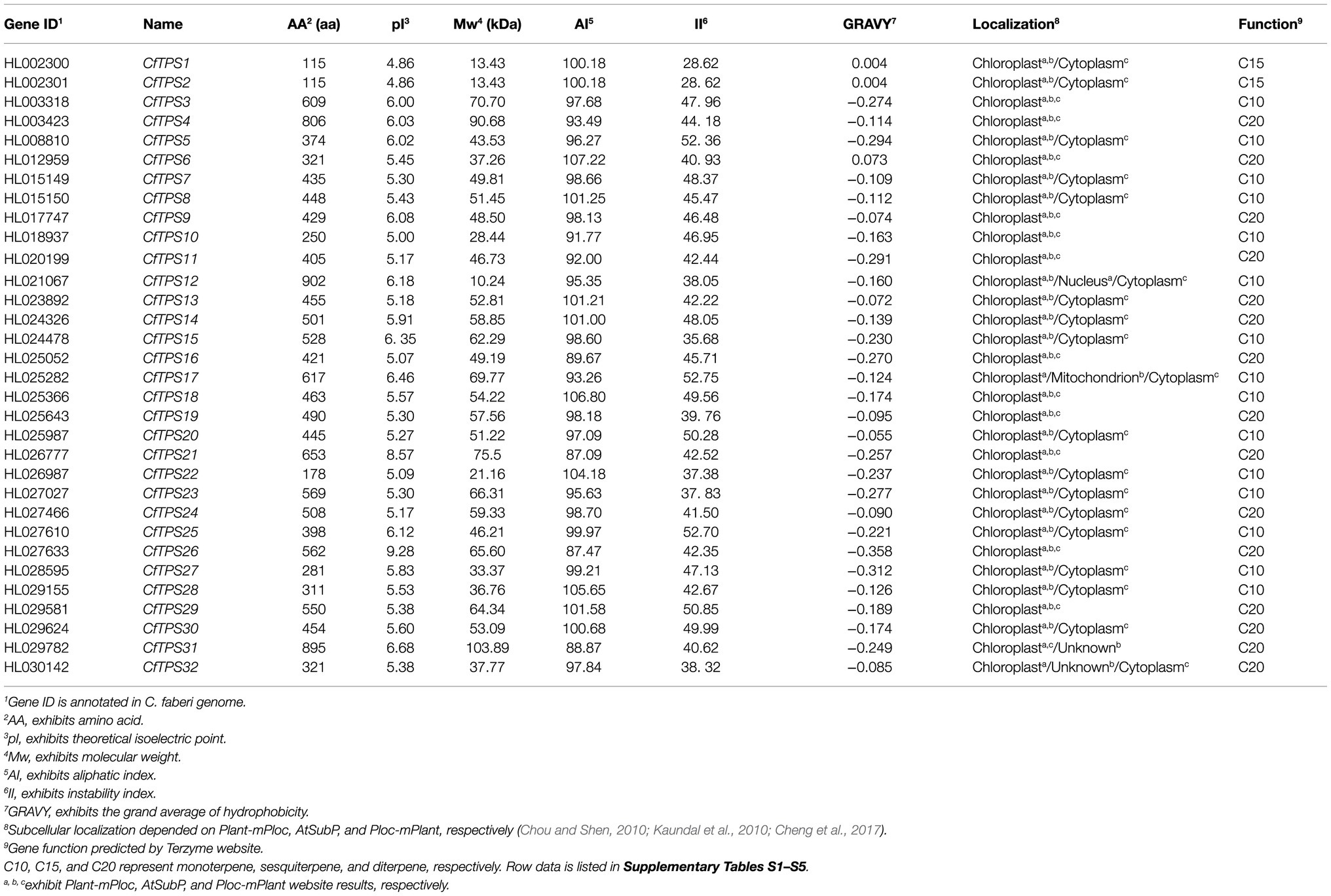
Table 1. A list of TPS genes in C. faberi, their characteristics, and functional annotation.
Motif and Gene Structure Analysis
To understand the intron-exon structure of CfTPS genes, we analyzed TPS gene structure with GSDS software (Hu et al., 2015). The exons in CfTPS genes ranged in numbers from 2 to 15, and the results showed that most of the genes in the same category contained a similar intron-exon structure.
To further analyze the motifs of the CfTPS genes, we identified 20 motifs using MEME software (Bailey et al., 2009). The numbers of C. faberi TPS motifs ranged in length from 4 to 16. CfTPS3 and CfTPS23 contained the most motifs, with 16, while CfTPS1, CfTPS2 had only four motifs. Motif 3 can be found in all CfTPS proteins except CfTPS9 and CfTPS16. Motif 4 was also the most common CfTPS protein (28/32). Twenty-five CfTPS genes contained the RRX8W motif (motif 1), and 30 CfTPS genes contained the DDxxD motif (motif 2; Figure 2C). Accordingly, different clusters have different forms of motifs. The same cluster’s CfTPS proteins generally contained similar motifs. These results of intron-exon structure and motif analysis verified the closeness of the phylogenetic tree in C. faberi (Figure 2).
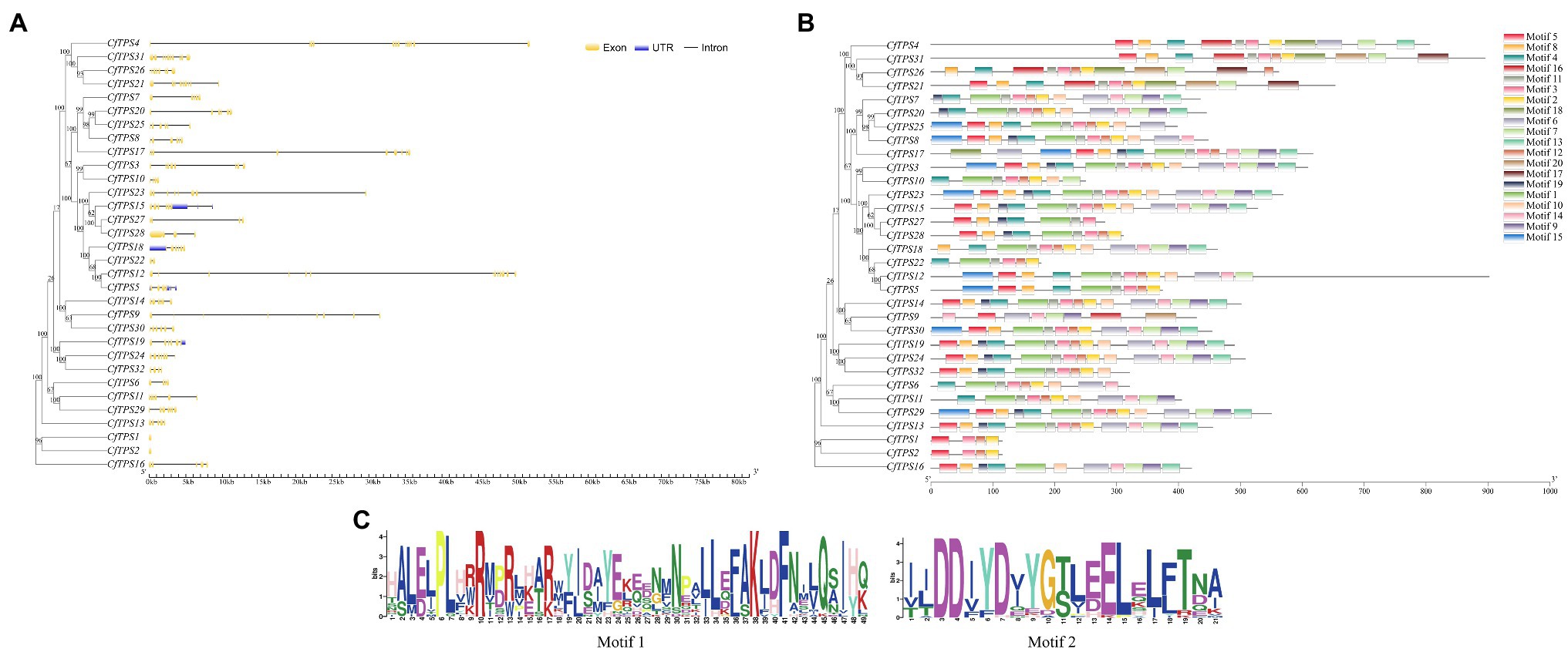
Figure 2. Phylogenetic tree and intron-exon structure of CfTPS genes. (A) Phylogenetic tree and intron-exon structure of CfTPS genes. The maximum likelihood (ML) method was used for the phylogenetic tree, which was constructed with RAxML on the CIPRES Science Gateway web server (RAxML-HPC2 on XSEDE; Miller et al., 2011). Bootstrap values were 1,000 replicates with the JTT model. The intron-exon structure was drawn by the GSDS website (Hu et al., 2015). Yellow boxes, blue boxes, and black lines exhibit exons, introns, and upstream or downstream-untranslated regions. (B) Phylogenetic tree and conserved motifs of CfTPS genes. Conserved motifs were determined by MEME software with default parameters (Kaundal et al., 2010). (C) Sequence logo of motif 1 (RRX8W) and motif 2 (DDxxD). Motif 1 shows the N-terminal motif RRX8W, and motif 2 shows the C-terminal motif DDxxD motif. Conserved motifs are available in Supplementary Table S6.
Phylogenetic Analysis of CfTPS Genes
A phylogenetic tree was constructed to analyze the evolutionary patterns of the CfTPS genes. Thirty-two CfTPS genes were used, and TPS protein sequences from six species (A. shenzhenica, P. equetris, O. sativa, A. thaliana, P. abies, and S. moellendorfii) were used. The maximum likelihood (ML) method was used for the phylogenetic tree, which was constructed with RAxML on the CIPRES Science Gateway web server (RAxML-HPC2 on XSEDE; Miller et al., 2011). Bootstrap values were 1,000 replicates with the JTT model. The phylogenetic tree indicated that TPS proteins belonged to seven categories. This classification result was consistent with a recent study: TPS-a, TPS-b, TPS-c, TPS-d, TPS-e/f, TPS-g, and TPS-h (Chen et al., 2011). Thirty-two CfTPS proteins belonged to three categories according to the phylogenetic tree (Figure 3): TPS-a, TPS-b, and TPS-e/f. Of these three categories, the TPS-a and TPS-b clades contained the most members and were the most expanded categories, with 13 and 15 genes, respectively, and were consistent with other plant species, such as A. thaliana, C. sinensis, V. planifolia, D. catenatum, P. equestris, and D. officinale (Aubourg et al., 2002; Yu et al., 2020; Zhou et al., 2020; Huang et al., 2021). The remaining four TPS genes belonged to the TPS-e/f subfamily.
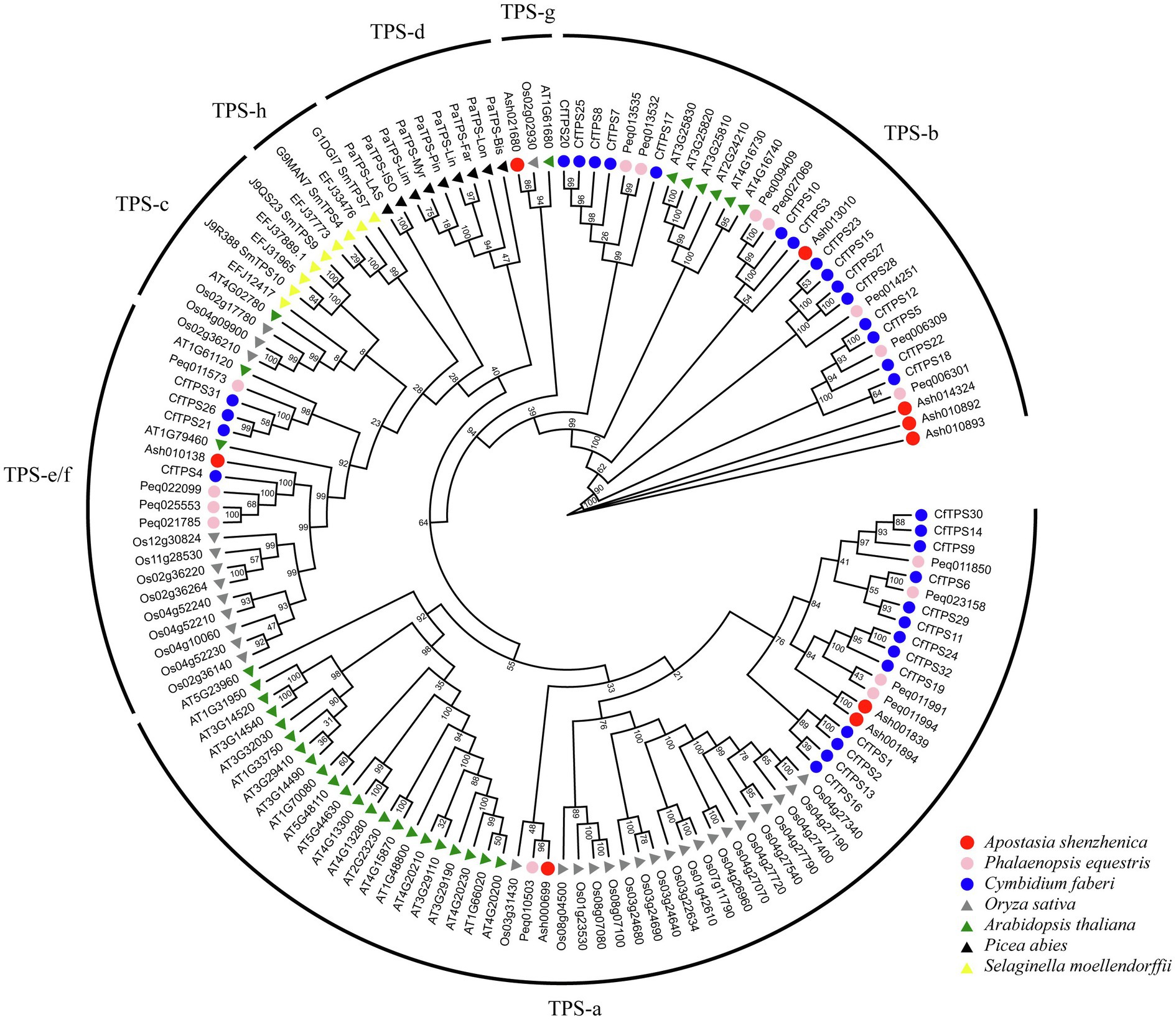
Figure 3. Phylogenetic tree of CfTPS genes based on the TPS protein sequences of seven plant species. The ML method was used for the phylogenetic tree, which was constructed with RAxML on the CIPRES Science Gateway web server (RAxML-HPC2 on XSEDE; Miller et al., 2011). The bootstrap values were 1,000 replicates with the JTT model. The generated tree was redrawn and annotated by the EVOLVIEW website (He et al., 2016). The TPS family was classified into seven categories: TPS-a, TPS-b, TPS-c, TPS-d, TPS-e/f, TPS-g, and TPS-h (Chen et al., 2011). TPS protein sequences in Cymbidium faberi are available in Supplementary Table S7.
We aligned the multiple sequences using MAFFT to analyze the TPS RRX8W motif in the N-terminus, DDxxD, and NSE/DTE motifs in the C-terminus (Rozewicki et al., 2019). The alignment showed that almost all the CfTPS proteins in the TPS-a and TPS-b subfamilies had the highly conserved aspartate-rich motif DDxxD, except CfTPS9 and CfTPS16 in the TPS-a subfamily and CfTPS17, CfTPS27, and CfTPS28 in the TPS-b subfamily (Jiang et al., 2019). TPS genes in TPS-a and TPS-b had variations in the RRX8W and RxR motif. However, in the TPS-a and TPS-b categories, the RRX8W motif was not found in nine CfTPS genes. The RRX8W motif was absent in the TPS-e/f subfamily, and the NSE/DTE motif was found in 16 CfTPS genes (Figure 4). Among them, DDxxD and NSE/DTE are important in the metal-dependent ionization of the prenyl diphosphate substrate, and the RRX8W motif is essential in the cyclization of monoterpene synthase (Bohlmann et al., 1998; Chen et al., 2011; Jiang et al., 2019). In addition, the members in the TPS-a subfamily detected in both dicot and monocot plants encode only sesquiterpenes (Jiang et al., 2019). The secondary “R” in this family is not conserved (Martin et al., 2010). TPS-b encodes monoterpenes containing the conserved RRX8W motif (Chen et al., 2011; Jiang et al., 2019).

Figure 4. RRX8W, DDxD, and NSE/DTE motifs in the CfTPS protein amino acid sequences. Multiple sequence alignments were constructed by MAFFT, and Jalview software was used to visualize the sequences (Troshin et al., 2011; Rozewicki et al., 2019).
Promoter Analysis of CfTPS Genes
To retrieve the potential function of CfTPS genes, we obtained a 2,000-bp region upstream of the 32 CfTPS genes and analyzed them using the online software Plantcare (Lescot et al., 2002). In total, we found 784 cis-acting regulatory elements in the promoter areas of CfTPS genes. We classified them into three categories according to the function of these elements: plant growth and development, stress responsiveness, and phytohormone responsiveness. The plant growth and dependent category (166/784) contained nine cis-acting regulatory elements and consists of AAGAA motifs, As-1 elements, A1-rich elements, etc. Most of them had As-1 elements (66/166), which are related to shoot expression. The stress responsiveness category (216/874) contained ARE, DRE, LTR, ST-RE, ABRE, etc. STRE (48/216) is an essential element in the promoter that is related to stress. Interestingly, most of the cis-elements (401/784) were in the phytohormone responsiveness category, which contained CGTCA motifs, EREs, MYC motifs, TGAGG motifs, etc. Among them, most cis-elements were MYC (137/401), which is associated with MeJA responsiveness (Dombrecht et al., 2007). The results indicated that the CfTPS gene expression patterns might be regulated by MeJA treatment (Figure 5).
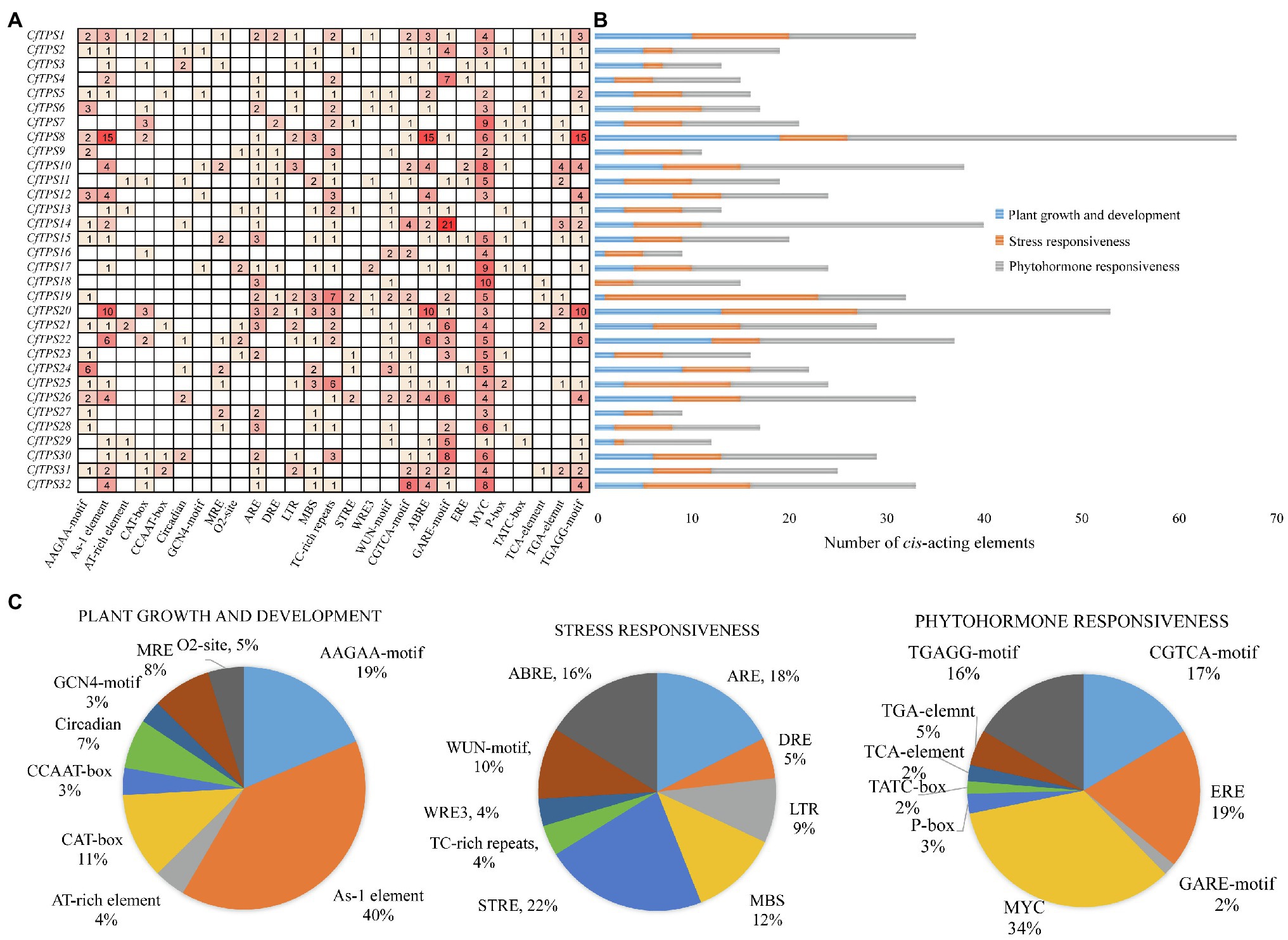
Figure 5. Promoter analysis of CfTPS genes. (A) The numbers in the box exhibit the number of cis-acting elements in CfTPS. (B) Blue, orange, and gray colors exhibit cis-acting elements in plant growth and development, stress responsiveness, and phytohormone responsiveness. (C) The proportion of different cis-acting elements in plant growth and development, stress responsiveness, and phytohormone responsiveness.
Ka/Ks Analysis in C. faberi
The Ka/Ks ratio can show positive selection (Ka/Ks>1), negative or purifying selection (Ka/Ks<1), and neutral selection (Ka/Ks=1) during the evolution (Zhang et al., 2006). In this study, 13 gene pairs with similar genetic relationships were selected for Ka/Ks calculation. The results showed that the Ka/Ks ratios of 12/13 CfTPS genes were less than one, indicated that most CfTPS genes underwent negative selection (Table 2). The divergence time of 13 CfTPS gene pairs was in the range of 0.75 for CfTPS1 and CfTPS2 to 60.46 for CfTPS3 and CfTPS10.
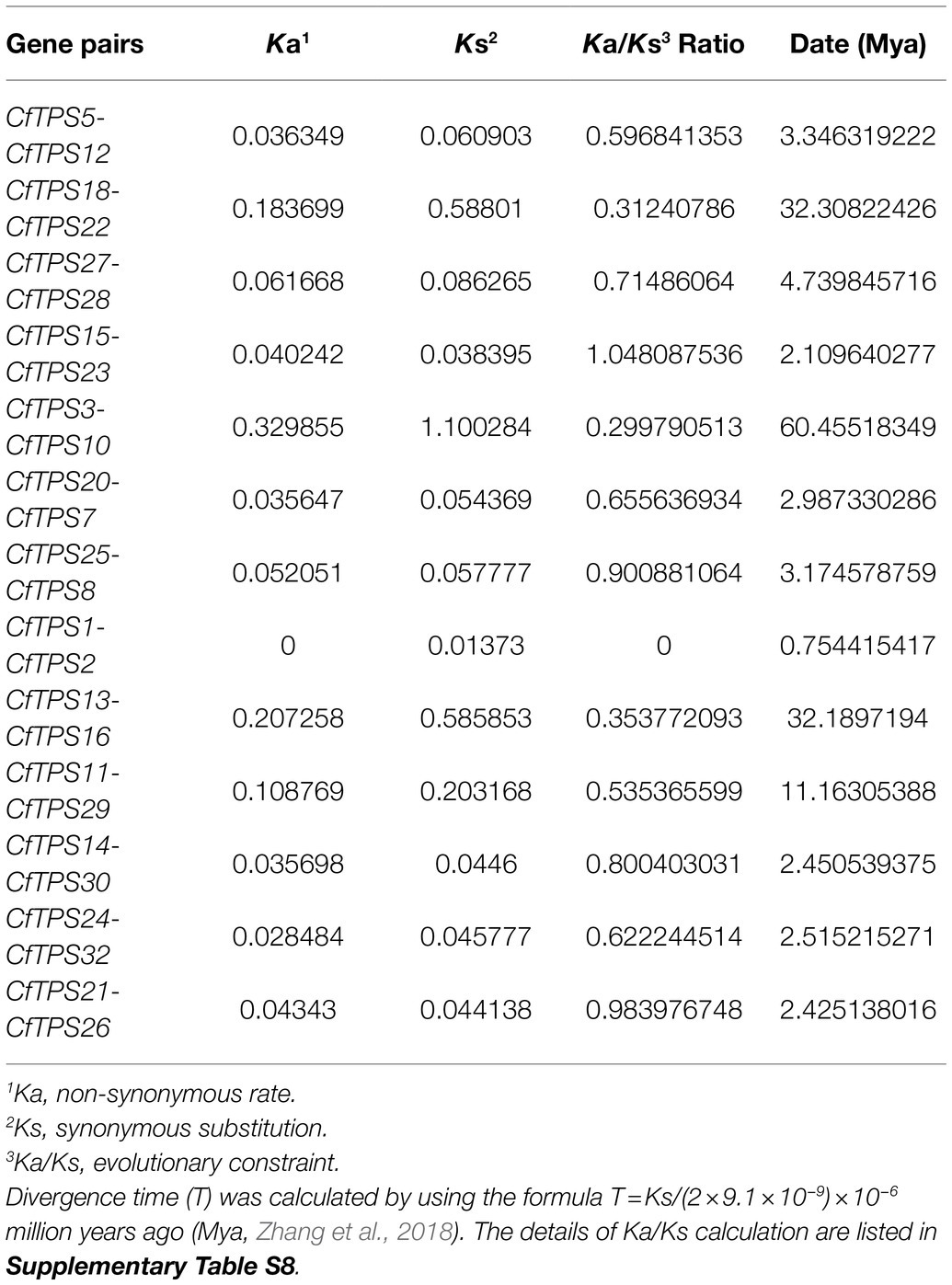
Table 2. Ka/Ks analysis of TPS genes in C. faberi.
Expression Patterns in Different C. faberi Organs and GO Classification of TPS Genes
An RNA sequencing transcriptome database of leaves, pseudobulbs, petals, sepals, labellums, and gynostemium was established to study the expression patterns of CfTPS genes. Eighteen genes were expressed in both leaves and flowers, and 20 genes were expressed in the labellums. Twenty-five genes were found in sepals and 24 in gynostemium. Four genes were not found to be expressed in any of the tissues. CfTPS12, CfTPS15, and CfTPS23 exhibited a high level of expression in leaves and pseudobulbs. Notably, CfTPS3, CfTPS12, CfTPS15, CfTPS17, CfTPS18, CfTPS28, and CfTPS31 displayed high transcript abundance in floral organs, suggesting that these TPS genes might be related to flower scent in C. faberi (Figure 6).
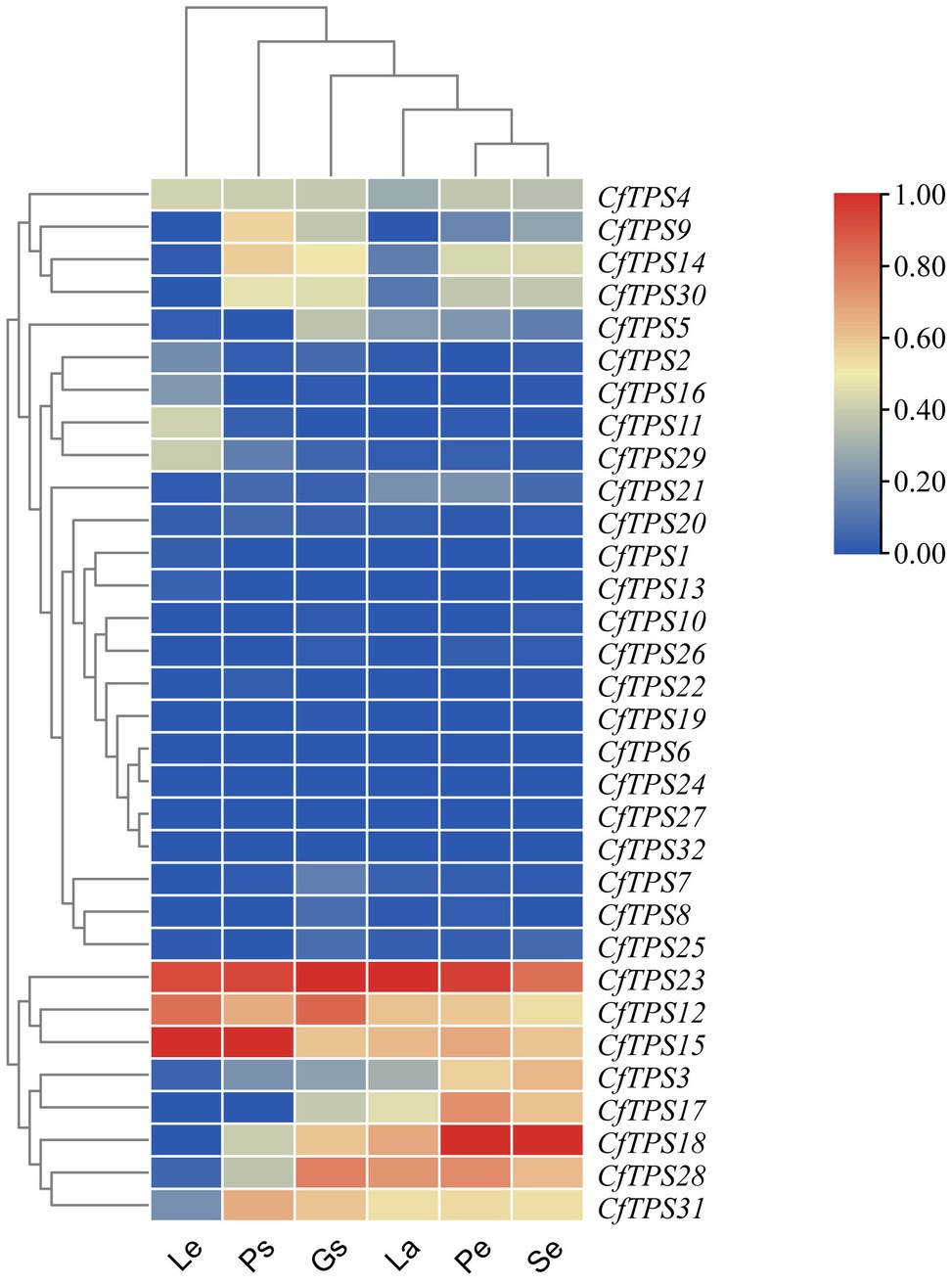
Figure 6. Expression patterns of CfTPS genes in different organs. The heatmap was produced in Tbtools (Chen et al., 2018). The tissues were leaves (L), pseudobulbs (Ps), petals (Pe), sepals (Se), labellum (La), and gynostemium (Gs) in wild C. faberi. The fragments per kilobase of transcript per million fragments (FPKM) values are listed in Supplementary Table S9.
Gene ontology classification analysis was performed based on the differentially expressed gene analysis. According to the classification results, molecular function contained most genes, and genes were mostly enriched in lyase activity, magnesium binding, and terpene synthase activity (Figure 7).
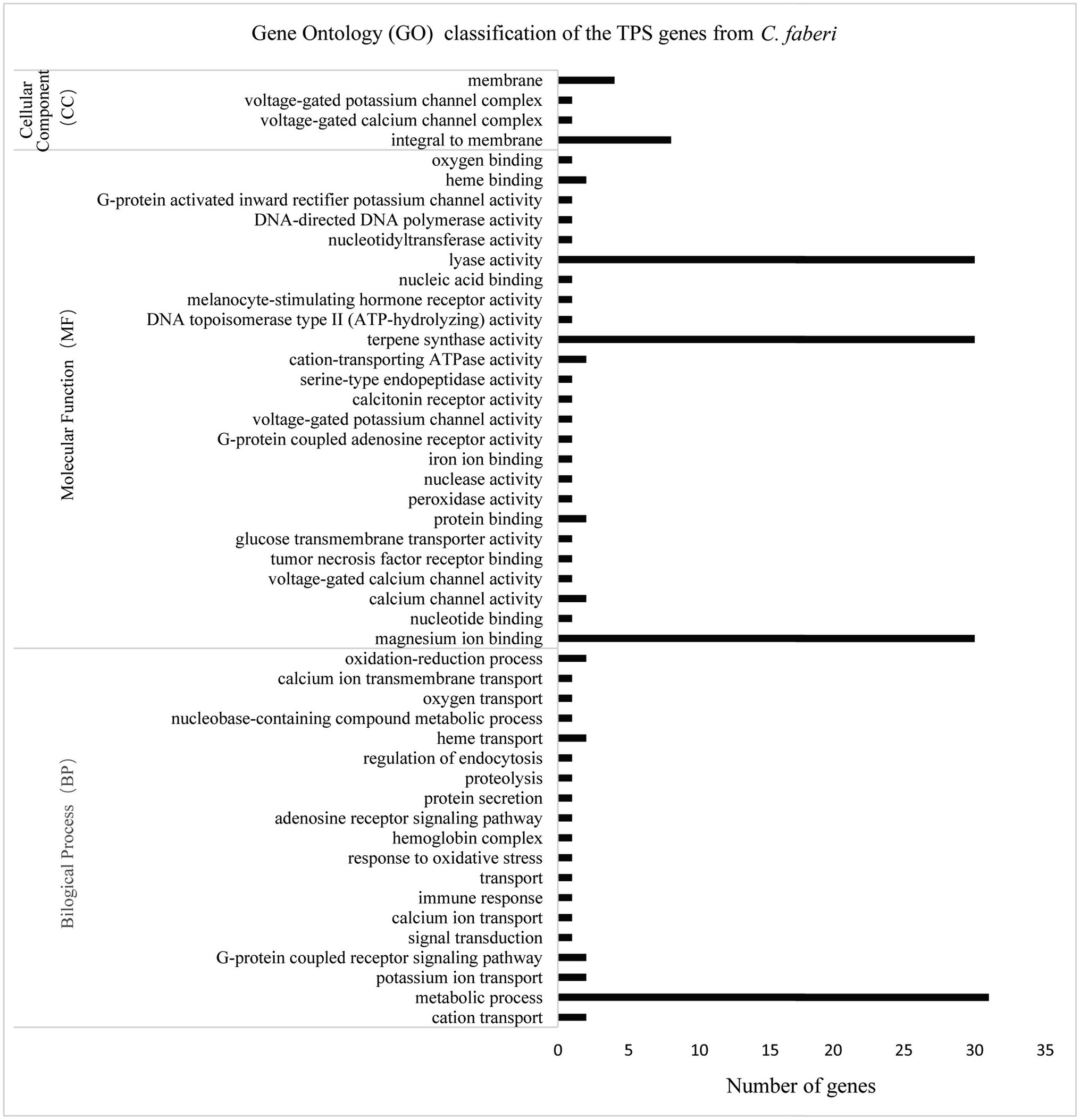
Figure 7. Gene ontology (GO) classification of CfTPS genes. GO annotation details are listed in Supplementary Table S10.
Expression Patterns of CfTPS in Flowers at Three Floral Developmental Stages
Real-time reverse transcription quantitative PCR was performed to investigate the expression patterns of CfTPS genes in flowers at three floral developmental stages. Four putative functional genes, CfTPS12, CfTPS18, CfTPS23, and CfTPS28, which are highly expressed in floral organs, were used. They were expressed differently among the three floral stages. Interestingly, these genes were mainly expressed in the full flowering stage (Figure 8). Notably, CfTPS18 had the highest transcript levels during the floral developmental stages, consistent with the expression patterns displayed in Figure 5. Gene annotation indicated that CfTPS12, CfTPS18, CfTPS23, and CfTPS28 likely encoded monoterpene synthases and clustered in TPS-b subfamily (Table 1).
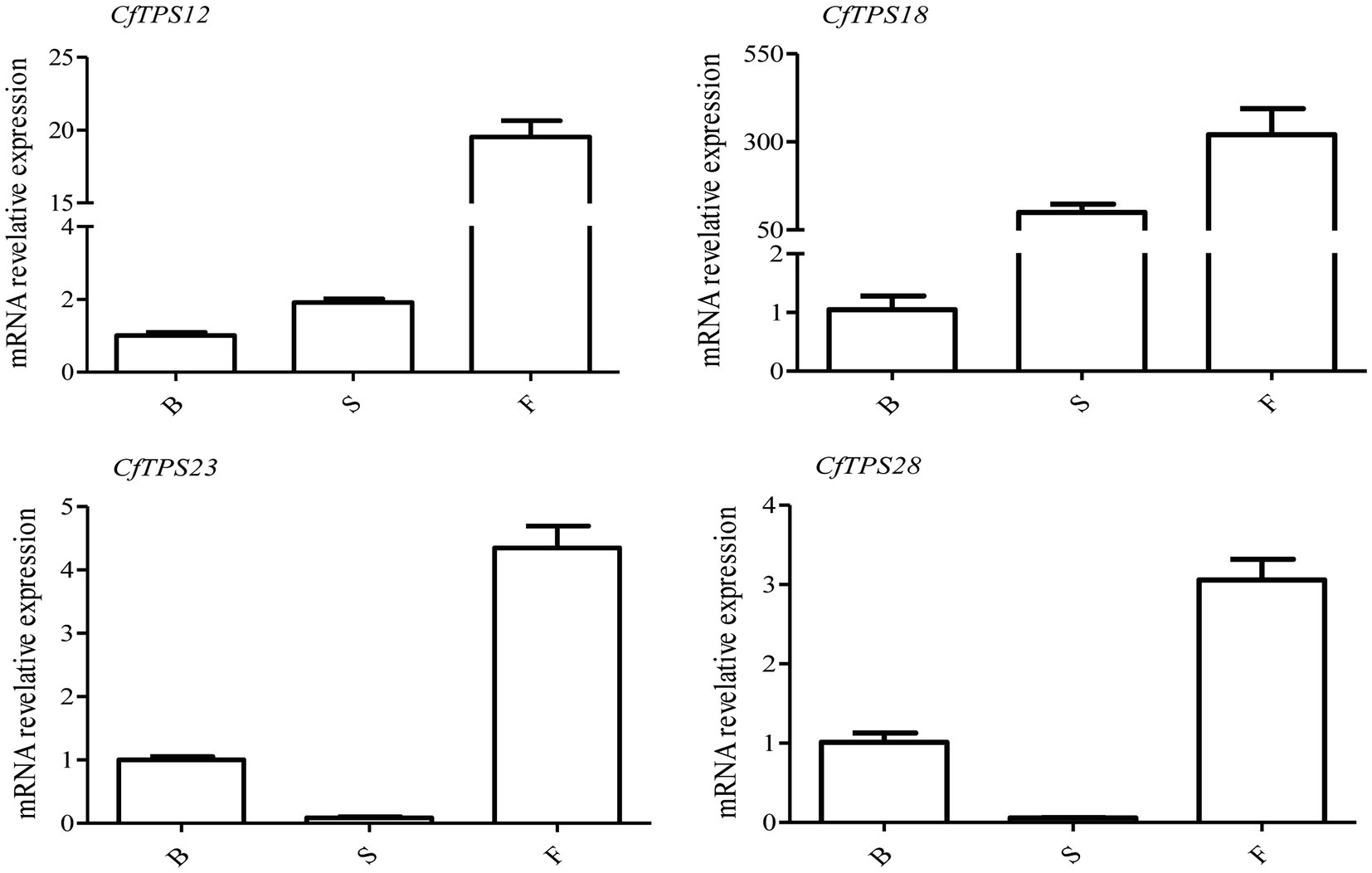
Figure 8. Real-time reverse transcription quantitative PCR (RT-qPCR) validation of transcriptomic data of the CfTPS genes at three flowering stages. B, budding flowers; S, semi-open flowers; and F, fully open flowers. The error bars indicate three RT-qPCR biological replicates. The values were standardized by the C. faberi reference gene GAPDH (GenBank Accession: JX560732; Tian et al., 2020). The expression values of CfTPS genes are listed in Supplementary Table S11, and the RT-qPCR primers of CfTPS are listed in Supplementary Table S12.
Functional Characterization of CfTPS18 in E. coli
To investigate the enzyme activity, the full-length ORF sequence of CfTPS18 was cloned to vector pET28a and ectopically expressed in E. coli. Recombinant CfTPS18 enzyme was induced with 0.1mM isopropyl-β-d-galactopyranoside and purified with Ni-NTA agarose (Clontech). SDS-PAGE analysis of CfTPS18 recombinant protein can be found in Supplementary Figure S1. After using GPP as substrate in the reactions, the products were analyzed by GC-MS. The result indicated that CfTPS18 could convert GPP into β-myrcene, geraniol, and α-pinene which were validated by the NIST Mass Spectral Library (Figure 9). The blank control with only GPP added to the reactions could not produce monoterpenes. Thus, CfTPS18 was considered a monoterpene synthase.
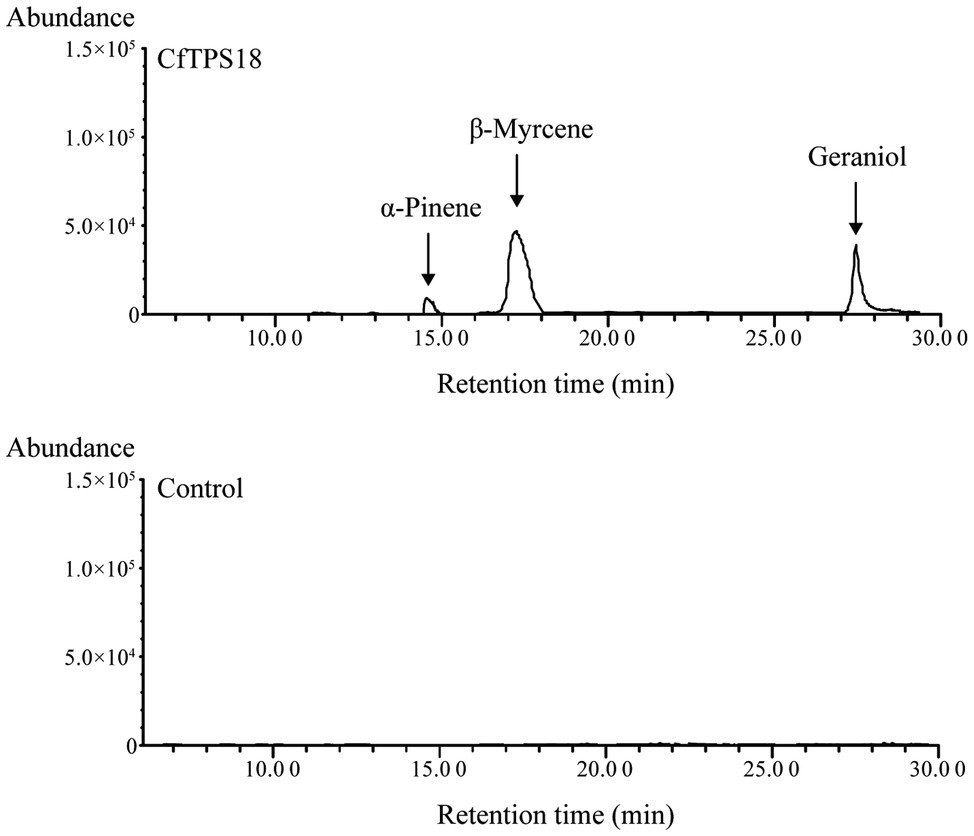
Figure 9. Enzymatic assays after incubating recombinant CfTPS18. Reactions only performed with GPP were used as the blank control. The retention time was compared with the NIST Mass Spectral Library.
Discussion
Orchids are among the largest angiosperm families in angiosperms and demonstrate a diversity of epiphytic and terrestrial growth forms (Hsiao et al., 2011). Cymbidium faberi is one of the longest cultivated orchids planted in China and has high ornamental value due to its characteristic flower scent and beautiful flower shape (Omata et al., 1990; Hsiao et al., 2011; Sun et al., 2016). Terpenoids play an essential role in floral scent and can attract pollinating insects and defend against environmental stresses (Wagner and Elmadfa, 2003; Dudareva et al., 2006; Sun et al., 2016). Plants have enzymes called TPSs, which encode the synthesis of monoterpene (C10), sesquiterpene (C15), or diterpenes (C20) from DMAPP, GPP, and GGPP, respectively (Pichersky et al., 2006). Terpenoids are dominant in the flower scent of orchids. In this study, we systematically retrieved and classified TPS genes in C. faberi.
We identified 32 CfTPS genes in the C. faberi genome according to the TPS N-terminal and C-terminal domains (Table 1). We classified them into three categories: TPS-a, TPS-b, and TPS-e/f. TPS-b was the most expanded category, which was consistent with patterns in D. officinale, V. planifolia, and D. catenatum, and C. faberi have more genes in TPS-b than P. equestris (Yu et al., 2020; Huang et al., 2021). However, it was not consistent with A. thaliana, O. sativa, and S. bicolor, which have a dominant subfamily TPS-a (Aubourg et al., 2002; Chen et al., 2011). These results suggest that orchids have more TPS genes in TPS-b than other angiosperm dicot species and are related to emit floral scent to attract pollinators (Huang et al., 2021). According to the phylogenetic tree, most of the TPS-b genes are present in dicots. TPS-a genes can be further split into monocot-specific TPS-a-1 and dicot-specific TPS-a-2 clades. The TPS gene family is a medium-sized family, and the numbers of TPS genes range from approximately 20–100 (Chen et al., 2011). For instance, 14 SmTPS, 23 GmTPS, 32 AtTPS, 34 DoTPS, 23 CsTPS, and 69 VvTPS were found in S. moellendorfii, G. max, A. thaliana, D. officinale, C. sinensis, and V. vinifera, respectively (Li et al., 2012; Yu et al., 2020; Zhou et al., 2020). In addition, TPS occupied 0.26 genes/M in A. thaliana (125M), 0.14 genes/M in V. vinifera (487M), 0.13 genes/M in S. moellendorfii (106M), 0.02 genes/M in D. officinale (1.35G), 0.02 genes/M in G. max (1.011G), and 0.01 genes/M in C. faberi (3.77G; Jaillon et al., 2007; Schmutz et al., 2010; Poczai et al., 2014; Zhang et al., 2016). These results indicate that the TPS family has undergone expansion throughout the evolutionary history of land plants, and different species may show a difference in the expansion mechanism (Chen et al., 2011; Jiang et al., 2019).
We also annotated 32 CfTPS genes, and the results showed that all TPSs in the TPS-a clade encode sesquiterpenes, and all TPSs in the TPS-b clade encode monoterpenes (Table 1). All the CfTPS in the TPS-e/f clade were annotated as diterpene synthases. This is consistent with a recent study in which most TPS genes in the TPS-a subfamily were determined to be sesquiterpene synthases. TPS-e/f encoded monoterpene, sesquiterpene, and diterpene in a recent study (Chen et al., 2011). Sesquiterpenes, diterpenes, and monoterpenes are important to emit floral scents to attract pollinators and defend against environmental stress (Huang et al., 2021).
Each full-length TPS had two conserved domains, including the N-terminal domain containing the RRX8W motif and the C-terminal domain containing two highly conserved aspartate-rich motifs: DDxxD and NSE/DTE (Starks et al., 1997; Jiang et al., 2019). DDxxD and NSE/DTE are significant in the metal-dependent ionization of the prenyl diphosphate substrate, and the RRX8W motif is essential in the cyclization of monoterpene synthase (Bohlmann et al., 1998; Chen et al., 2011; Jiang et al., 2019). The secondary “R” in the TPS-a subfamily is not conserved (Martin et al., 2010). TPS-b contains the conserved RRX8W motif, which is related to monoterpene formation. TPS-c does not include the DDxxD motif (Chen et al., 2011; Jiang et al., 2019). Motif RxR was also conserved in TPS-a and TPS-b subfamilies. In this study, multiple sequence alignments showed that 28/32 CfTPS had highly conserved DDxxD motifs, 16/32 CfTPS had RRX8W motifs, 16/32 CfTPS had NSE/DTE motifs, and 9/32 CfTPS had RxR motifs. The RRX8W motif was not found in 11 CfTPSs in the TPS-a and TPS-b clades, and it was absent in the TPS-e/f subfamily. The results indicated that motif loss might have appeared during family evolution in C. faberi and that different subfamilies have different motif features.
Different elements were observed in promoter areas of CfTPS genes. Most of the cis-elements were in the phytohormone responsiveness group, and the number of MYCs associated with MeJA responsiveness contained most of this group. The results indicated that the expression patterns of CfTPS may be regulated by MeJA treatment and may respond to multiple environmental stresses (Chaiprasongsuk et al., 2018). In recent studies, MeJA was shown to regulate TPS gene expression in D. officinale and C. sinensis (Yu et al., 2020; Zhao et al., 2020; Zhou et al., 2020). However, this needs to be further investigated in C. faberi. The Ka/Ks ratio analysis indicated that the TPS gene family in C. faberi mainly underwent negative selection, making it more stable during the evolution. GO annotation analysis of CfTPS genes indicated that molecular function contained most genes, and genes were mostly enriched in lyase activity, magnesium binding, and terpene synthase activity. Expression pattern analysis indicated that CfTPS genes were mainly expressed in the floral organs of C. faberi, indicating that they were related to floral scent. We selected four CfTPS genes with the highest transcript levels in floral organs for qRT-PCR analysis at three flowering stages. The results showed that they all belonged to the TPS-b clade and were mainly expressed in the full flowering stage. According to our annotation, four genes were all annotated as monoterpene synthases, which may play essential roles in floral scent and attracting pollinators in C. faberi. Enzymatic assays suggested that CfTPS18 could convert GPP to β-myrcene, geraniol, and α-pinene. In recent reports, DoTPS10 in D. officinale can convert GPP to linalool in vitro (Yu et al., 2020). DoGES1 in D. officinale can catalyze geraniol in vitro and in vivo (Zhao et al., 2020). Linalool and geraniol belong to monoterpenes which play essential roles in floral scents. TPS that catalyze terpenes can also be found in V. vinifera, M. domestica, and Litsea cubeba (Martin et al., 2010; Nieuwenhuizen et al., 2013; Chen et al., 2020). To better understand terpenes production and function in C. faberi, more studies of expression profiles should be developed.
Conclusion
In this study, 32 CfTPS genes were identified from the genomes of C. faberi. We analyzed their conserved motifs, exon-intron structure, phylogeny, Ka/Ks ratios, and cis-acting regulatory elements. We also analyzed the expression patterns of CfTPS genes in leaves, pseudobulbs, petals, sepals, labellums, and gynostemium in wild C. faberi. Four putatively functional genes highly expressed in floral organs were used to analyze the expression patterns at three flowering stages. Enzymatic assays indicated that CfTPS18 could convert GPP to β-myrcene, geraniol, and α-pinene. The results will provide valuable information for further studies on floral scents in C. faberi and other orchids.
Data Availability Statement
The original contributions presented in the study are included in the article/ Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
SL, Z-JL, and DZ contributed to conceptualization and validation. Q-QW, M-JZ, and ZZ prepared the original draft. Q-QW, Y-YB, and XY analyzed the data. M-KC, YA, and JC contributed to the visualization. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by The National Key Research and Development Program of China (2019YFD1001000), The National Natural Science Foundation of China (no. 31870199), The Key Laboratory of National Forestry and Grassland Administration for Orchid Conservation and Utilization Construction Funds (Grant 115/118990050, 115/KJG18016A), Natural Science Foundation of Zhejiang Province (Grant nos. LY20C160005, LY19C150003), Key Research and Development Program of Zhejiang Province (Grant no. 2021C02043), and Wenzhou Agricultural New Variety Breeding Cooperative Group Project (Grant no. 2019ZX004-3).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.751853/full#supplementary-material
Abbreviations
AI, Aliphatic index; bp, Base pair; DMAPP, Dimethylallyl diphosphate; FPP, Farnesyl diphosphate; FPKM, Fragments per kilobase of transcript per million fragments mapped; GGPP, Geranylgeranyl diphosphate; GPP, Geranyl diphosphate; GRAVY, Grand average of hydrophobicity; II, Instability index; IPP, Isopentenyl diphosphate; MeJA, Methyl jasmonate; MEP, Methylerythritol phosphate; Mw, Molecular weight; MVA, Mevalonate acid; NCBI, National Center for Biotechnology Information; NJ, Neighbor-joining; pI, Isoelectric point; RT-qPCR, Real-time reverse transcription quantitative PCR; TPS, Terpene synthase.
Footnotes
2. ^https://www.arabidopsis.org/
4. ^http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/
5. ^http://bioinfo3.noble.org/AtSubP/
6. ^https://www.swmath.org/software/25191
7. ^http://www.nipgr.ac.in/terzyme.html
8. ^http://blast.ncbi.nlm.nih.gov
11. ^https://www.ncbi.nlm.nih.gov/genbank/
13. ^https://phytozome.jgi.doe.gov/
15. ^http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
References
Artimo, P., Jonnalagedda, M., Arnold, K., Baratin, D., Csardi, G., De Castro, E., et al. (2012). ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, 597–603. doi: 10.1093/nar/gks400
Aubourg, S., Lecharny, A., and Bohlmann, J. (2002). Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol. Gen. Genomics 267, 730–745. doi: 10.1007/s00438-002-0709-y
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37, 1–7. doi: 10.1093/nar/gkp335
Bohlmann, J., Meyer-Gauen, G., and Croteau, R. (1998). Plant terpenoid synthases: molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. U. S. A. 95, 4126–4133. doi: 10.1073/pnas.95.8.4126
Chaiprasongsuk, M., Zhang, C., Qian, P., Chen, X., Li, G., Trigiano, R. N., et al. (2018). Biochemical characterization in Norway spruce (Picea abies) of SABATH methyltransferases that methylate phytohormones. Phytochemistry 149, 146–154. doi: 10.1016/j.phytochem.2018.02.010
Chen, C., Chen, H., He, Y., and Xia, R. (2018). TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv [Preprint]. doi: 10.1101/289660
Chen, Y. C., Li, Z., Zhao, Y. X., Gao, M., Wang, J. Y., Liu, K. W., et al. (2020). The Litsea genome and the evolution of the laurel family. Nat. Commun. 11, 1–14. doi: 10.1038/s41467-020-15493-5
Chen, F., Tholl, D., Bohlmann, J., and Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Cheng, X., Xiao, X., and Chou, K. C. (2017). PLoc-mPlant: predict subcellular localization of multi-location plant proteins by incorporating the optimal GO information into general PseAAC. Mol. BioSyst. 13, 1722–1727. doi: 10.1039/C7MB00267J
Chou, K. C., and Shen, H. B. (2010). Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 5:e11335. doi: 10.1371/journal.pone.0011335
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Dombrecht, B., Gang, P. X., Sprague, S. J., Kirkegaard, J. A., Ross, J. J., Reid, J. B., et al. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19, 2225–2245. doi: 10.1105/tpc.106.048017
Dudareva, N., Negre, F., Nagegowda, D. A., and Orlova, I. (2006). Plant volatiles: recent advances and future perspectives. Crit. Rev. Plant Sci. 25, 417–440. doi: 10.1080/07352680600899973
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Geourjon, C., and Deléage, G. (1995). Sopma: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11, 681–684. doi: 10.1093/bioinformatics/11.6.681
He, Z., Zhang, H., Gao, S., Lercher, M. J., Chen, W. H., and Hu, S. (2016). Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 44, W236–W241. doi: 10.1093/nar/gkw370
Hsiao, Y. Y., Pan, Z. J., Hsu, C. C., Yang, Y. P., Hsu, Y. C., Chuang, Y. C., et al. (2011). Research on orchid biology and biotechnology. Plant Cell Physiol. 52, 1467–1486. doi: 10.1093/pcp/pcr100
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Huang, L. M., Huang, H., Chuang, Y. C., Chen, W. H., Wang, C. N., and Chen, H. H. (2021). Evolution of terpene synthases in Orchidaceae. Int. J. Mol. Sci. 22:6947. doi: 10.3390/ijms22136947
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Jiang, S. Y., Jin, J., Sarojam, R., and Ramachandran, S. (2019). A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 11, 2078–2098. doi: 10.1093/gbe/evz142
Kaundal, R., Saini, R., and Zhao, P. X. (2010). Combining machine learning and homology-based approaches to accurately predict subcellular localization in Arabidopsis. Plant Physiol. 154, 36–54. doi: 10.1104/pp.110.156851
Keilwagen, J., Lehnert, H., Berner, T., Budahn, H., Nothnagel, T., Ulrich, D., et al. (2017). The terpene synthase gene family of carrot (Daucus carota L.): identification of QTLs and candidate genes associated with terpenoid volatile compounds. Front. Plant Sci. 8:1930. doi: 10.3389/fpls.2017.01930
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van De Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, G., Köllner, T. G., Yin, Y., Jiang, Y., Xu, Y., Gershenzon, J., et al. (2012). Correction for Li et al., nonseed plant Selaginella moellendorfii has both seed plant and microbial types of terpene synthases. Proc. Natl. Acad. Sci. U. S. A. 109, 20774–20774. doi: 10.1073/pnas.1204300109
Liu, J., Huang, F., Wang, X., Zhang, M., Zheng, R., Wang, J., et al. (2014). Genome-wide analysis of terpene synthases in soybean: functional characterization of GmTPS3. Gene 544, 83–92. doi: 10.1016/j.gene.2014.04.046
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Martin, D. M., Aubourg, S., Schouwey, M. B., Daviet, L., Schalk, M., Toub, O., et al. (2010). Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 10:226. doi: 10.1186/1471-2229-10-226
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2011). “The CIPRES science gateway: A community resource for phylogenetic analyses.” in Proceedings of the TeraGrid 2011 Conference: Extreme Digital Discovery, TG’11.
Mohd-Hairul, A. R., Namasivayam, P., Lian, G. E. C., and Abdullah, J. O. (2010). Terpenoid, benzenoid, and phenylpropanoid compounds in the floral scent of Vanda Mimi palmer. J. Plant Biol. 53, 358–366. doi: 10.1007/s12374-010-9123-x
Nagegowda, D. A. (2010). Plant volatile terpenoid metabolism: biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett. 584, 2965–2973. doi: 10.1016/j.febslet.2010.05.045
Nieuwenhuizen, N. J., Green, S. A., Chen, X., Bailleul, E. J. D., Matich, A. J., Wang, M. Y., et al. (2013). Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol. 161, 787–804. doi: 10.1104/pp.112.208249
Omata, A., Nakamura, S., Yomogida, K., Moriai, K., Ichikawa, Y., and Watanabe, I. (1990). Volatile components of to-Yo-ran flowers (Cymbidium faberi and Cymbidium virescens). Agric. Biol. Chem. 54, 1029–1033. doi: 10.1080/00021369.1990.10870040
Pichersky, E., Noel, J. P., and Dudareva, N. (2006). Biosynthesis of plant volatiles: nature’s diversity and ingenuity. Science 311, 808–811. doi: 10.1126/science.1118510
Poczai, P., Cernák, I., Varga, I., and Hyvönen, J. (2014). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Genet. Resour. Crop. Evol. 61, 796–815. doi: 10.1134/S1022795411020074
Priya, P., Yadav, A., Chand, J., and Yadav, G. (2018). Terzyme: a tool for identification and analysis of the plant terpenome. Plant Methods 14, 1–18. doi: 10.1186/s13007-017-0269-0
Ramya, M., An, H. R., Baek, Y. S., Reddy, K. E., and Park, P. H. (2018). Orchid floral volatiles: biosynthesis genes and transcriptional regulations. Sci. Hortic. 235, 62–69. doi: 10.1016/j.scienta.2017.12.049
Ramya, M., Park, P. H., Chuang, Y. C., Kwon, O. K., An, H. R., Park, P. M., et al. (2019). RNA sequencing analysis of cymbidium goeringii identifies floral scent biosynthesis related genes. BMC Plant Biol. 19:337. doi: 10.1186/s12870-019-1940-6
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M., and Katoh, K. (2019). MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10. doi: 10.1093/nar/gkz342
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Starks, C. M., Back, K., Chappell, J., and Noel, J. P. (1997). Structural basis for cyclic terpene biosynthesis by tobacco 5-epi- aristolochene synthase. Science 277, 1815–1820. doi: 10.1126/science.277.5333.1815
Sun, Y., Wang, G., Li, Y., Jiang, L., Yang, Y., and Guan, S. (2016). De novo transcriptome sequencing and comparative analysis to discover genes related to floral development in Cymbidium faberi Rolfe. Springerplus 5:1458. doi: 10.1186/s40064-016-3089-1
Tholl, D. (2015). Biosynthesis and biological functions of terpenoids in plants. Adv. Biochem. Eng. Biotechnol. 148, 63–106. doi: 10.1007/10_2014_295
Tian, C., Liu, S., Jiang, L., Tian, S., and Wang, G. (2020). The expression characteristics of methyl jasmonate biosynthesis–related genes in Cymbidium faberi and influence of heterologous expression of CfJMT in Petunia hybrida. Plant Physiol. Biochem. 151, 400–410. doi: 10.1016/j.plaphy.2020.03.051
Troshin, P. V., Procter, J. B., and Barton, G. J. (2011). Java bioinformatics analysis web services for multiple sequence alignment-JABAWS:MSA. Bioinformatics 27, 2001–2002. doi: 10.1093/bioinformatics/btr304
Vranová, E., Coman, D., and Gruissem, W. (2013). Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700. doi: 10.1146/annurev-arplant-050312-120116
Wagner, K. H., and Elmadfa, I. (2003). Biological relevance of terpenoids: overview focusing on mono-, di- and tetraterpenes. Ann. Nutr. Metab. 47, 95–106. doi: 10.1159/000070030
Yu, Z., Zhao, C., Zhang, G., Teixeira da Silva, J. A., and Duan, J. (2020). Genome-wide identification and expression profile of TPS gene family in Dendrobium officinale and the role of DoTPS10 in linalool biosynthesis. Int. J. Mol. Sci. 21, 1–22. doi: 10.3390/ijms21155419
Zhang, J., Li, Y., Liu, B., Wang, L., Zhang, L., Hu, J., et al. (2018). Characterization of the Populus Rab family genes and the function of PtRabE1b in salt tolerance. BMC Plant Biol. 18:124. doi: 10.1186/s12870-018-1342-1
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K. S., and Yu, J. (2006). KaKs_calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zhang, G. Q., Xu, Q., Bian, C., Tsai, W. C., Yeh, C. M., Liu, K. W., et al. (2016). The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6, 1–10. doi: 10.1038/srep19029
Zhao, C., Yu, Z., Teixeira da Silva, J. A., He, C., Wang, H., Si, C., et al. (2020). Functional characterization of a Dendrobium officinale geraniol synthase DoGES1 involved in floral scent formation. Int. J. Mol. Sci. 21:7005. doi: 10.3390/ijms21197005
Keywords: terpenes, terpene synthase, orchids, floral scent, expression analysis, Cymbidium
Citation: Wang Q-Q, Zhu M-J, Yu X, Bi Y-Y, Zhou Z, Chen M-K, Chen J, Zhang D, Ai Y, Liu Z-J and Lan S (2021) Genome-Wide Identification and Expression Analysis of Terpene Synthase Genes in Cymbidium faberi. Front. Plant Sci. 12:751853. doi: 10.3389/fpls.2021.751853
Edited by:
Xinchao Wang, Tea Research Institute, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Xinlu Chen, The University of Tennessee, Knoxville, United StatesMingle Wang, Huazhong Agricultural University, China
Copyright © 2021 Wang, Zhu, Yu, Bi, Zhou, Chen, Chen, Zhang, Ai, Liu and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong-Jian Liu, empsaXVAZmFmdS5lZHUuY24=; Siren Lan, bGt6eEBmYWZ1LmVkdS5jbg==