 Nan Chai1†
Nan Chai1† Jie Xu
Jie Xu Yulin Cheng
Yulin Cheng Mingyang Li
Mingyang Li Daofeng Liu
Daofeng Liu- 1Chongqing Engineering Research Center for Floriculture, Key Laboratory of Horticulture Science for Southern Mountainous Regions of Ministry of Education, College of Horticulture and Landscape Architecture, Southwest University, Chongqing, China
- 2Key Laboratory of Plant Hormones and Development Regulation of Chongqing, School of Life Sciences, Chongqing University, Chongqing, China
Botrytis elliptica, the causal agent of gray mold disease, poses a major threat to commercial Lilium production, limiting its ornamental value and yield. The molecular and metabolic regulation mechanisms of Lilium's defense response to B. elliptica infection have not been completely elucidated. Here, we performed transcriptomic and metabolomic analyses of B. elliptica resistant Lilium oriental hybrid “Sorbonne” to understand the molecular basis of gray mold disease resistance in gray mold disease. A total of 115 differentially accumulated metabolites (DAMs) were detected by comparing the different temporal stages of pathogen infection. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed the differentially expressed genes (DEGs) and DAMs were enriched in the phenylpropanoid and flavonoid pathways at all stages of infection, demonstrating the prominence of these pathways in the defense response of “Sorbonne” to B. elliptica. Network analysis revealed high interconnectivity of the induced defense response. Furthermore, time-course analysis of the transcriptome and a weighted gene coexpression network analysis (WGCNA) led to the identification of a number of hub genes at different stages, revealing that jasmonic acid (JA), salicylic acid (SA), brassinolide (BR), and calcium ions (Ca2+) play a crucial role in the response of “Sorbonne” to fungal infection. Our work provides a comprehensive perspective on the defense response of Lilium to B. elliptica infection, along with a potential transcriptional regulatory network underlying the defense response, thereby offering gene candidates for resistance breeding and metabolic engineering of Lilium.
Introduction
Lilium is one of the most economically important genera of ornamental monocots, whose species are used worldwide as cut flowers, garden plants, and potted plants. However, both the ornamental value and yield of commercial Lilium are often restricted by gray mold (Cui et al., 2018b). Lilium is highly susceptible to gray mold disease, and its effect is compounded by high humidity and low temperature (Hsieh et al., 2001). Gray mold disease, also known as leaf blight disease, is caused by the necrotrophic pathogens Botrytis cinerea and Botrytis elliptica. Among these, B. cinerea has been widely studied as a model necrotrophic fungus, as it has a broad host range and can infect more than 200 plant species (Hsiang and Chastagner, 1991; Gonzalez et al., 2017), whereas B. elliptica has a narrow host range and especially infects Lilium (Huang et al., 2001; Van Baarlen et al., 2004). At the early stages of gray mold infection, hygrophanous lesions appear on Lilium leaves in the form of oval or circular spots that change in color from yellowish brown to reddish brown over time. Then, the disease spreads rapidly throughout the whole plant, and its control becomes difficult (Ma et al., 2018). Development of disease resistant Lilium cultivars is currently the most economical and effective way to prevent gray mold disease incidence and spread. Thus, understanding the defense mechanism of Lilium against B. elliptica will help accelerate the process of breeding gray mold resistance traits in Lilium (Peng et al., 2017; Liu et al., 2020).
To defend against pathogen attack, plants have evolved two layers of immunity: pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and effector-triggered immunity (ETI) (Liu et al., 2020). pathogen-associated molecular patterns trigger PTI and confer basic resistance to the attacked plant, while resistance (R) genes, whose encoded products can specifically recognize the cognate effector or pathogen avirulence proteins, function to regulate ETI (Zhao et al., 2021). Both PTI and ETI induce the production of defense related secondary metabolites, pathogen-related transcription factors (TFs), and pathogenesis-related (PR) proteins and activate hormone signal transduction as well as calcium (Ca2+) signaling (Liu et al., 2020). Phenolic metabolites, being important components of the plant immune system, play crucial roles in how plants respond to various pathogenic infections. Phenylpropanoids are precursors of a wide range of phenolic compounds, such as flavonoids, isoflavonoids, and cumarins (Shetty et al., 2011). Increased accumulation of phenylalanine in plants via its exogenous treatment significantly reduces their susceptibility to pathogens (Martinez et al., 2017; Doppler et al., 2019; Kumar et al., 2020). Flavonoids another important type of phenolic metabolite, have been reported to engage in antibacterial activity and can inactivate cell envelope transport proteins and disrupt microbial membranes and the respiratory chain (Long et al., 2019). Some flavonoid metabolites have been to promote phytohormone signaling and strengthen host resistance to necrotrophic B. cinerea in Arabidopsis thaliana (Hong et al., 2015). Numerous defense-related genes, such as those encoding TFs and PR proteins, control these various immune responses (Kumar et al., 2016; Sun et al., 2020).
The interaction between lilies and gray mold has been investigated at the molecular level in a few studies. Cui et al. (2018b) identified 23 LrWRKY genes from the resistant species Lilium regale, and showed that the overexpression of LrWRKY4 and LrWRKY12 enhanced B. cinerea resistance in transgenic Arabidopsis. Several resistance genes and pathogen-related microRNAs (miRNAs) were identified in B. elliptica-infected L. regale through RNA-seq and miRNA-seq (Gao et al., 2017; Cui et al., 2018a). More recently, Fu et al. (2020) performed comparative RNA-seq analysis of the expression profiles of the monolignol pathway genes from L. regale after its inoculation with B. cinerea, and were the first to report CCoAOMT as a potential molecular target in Lilium. Nevertheless, the molecular and metabolic regulatory mechanisms underlying the defense response of Lilium to B. elliptica remain largely unknown. Next-generation sequencing (NGS) has accelerated the pace of genetic studies by facilitating de novo genome assemblies from sequence reads obtained using the Illumina technology (Unamba et al., 2015; Mazumdar and Chattopadhyay, 2016; Almeida et al., 2018). Considering the large size and highly heterozygous nature of the Lilium genome, NGS is the most suitable approach for conducting molecular research on Lilium in the absence of a reference genome sequence. Additionally, untargeted metabolomic analysis is a newly developed method used for qualitative and quantitative analyses of various metabolites in plants (Abu-Nada et al., 2007; Parker et al., 2009; Lloyd et al., 2011). Combing transcriptome and metabolome investigations thus offers a feasible way to reliably reveal the various signals conveyed by Lilium after its infection with B. elliptica.
Here, we performed transcriptomic and metabolomic analyses of the Lilium oriental hybrid cultivar “Sorbonne”, which is known for its high volume of sales and strong resistance to gray mold (Zhang et al., 2015; Gao et al., 2018). The objectives of this study were to identify the major metabolic pathways that operate in Lilium after inoculation with B. elliptica, and to define a plausible transcriptional regulatory mechanism responsible for that response. The results of this study provide key insights into the transcriptional and metabolic mechanisms underlying the defense response of Lilium to B. elliptica infection.
Materials and Methods
Plant Cultivation and Pathogen Inoculation
The high resistant Lilium oriental hybrid cultivar “Sorbonne” (Gao et al., 2018), was used in this study. Bulbs were stored in a refrigerator (4°C) for 28 days, and then placed at the bottom of 15-cm deep pots filled with turf: vermiculite: perlite substrate (1:1:1, v/v/v). The pots containing the bulbs were placed in a greenhouse for 45 days at 25°C under 12-h light/12-h dark photoperiod, with photosynthetic photon flux density of 240 μmol·m.2·s-1 (Jang et al., 2018).
Botrytis elliptica strain 36423, isolated from symptomatic Lilium plants, was purchased from the Agricultural Cultural Collection of China (http://www.accc.org.cn/). The mycelium was cultured on potato dextrose agar (PDA, pH 5.8; Coolaber, China) medium in Petri dishes (9 cm diameter) at 25°C in the dark for 1 week.
Fully expanded, but not senescent, leaves were collected from Lilium plants at the flower bud stage, and inoculated with B. elliptica, according to the “detached leaves inoculation methods” of Gao et al. (2018). Before inoculation, all utensils and water were autoclaved at 121°C for 20 min, and the detached leaves were wiped clean using cotton soaked with sterile water. To inoculate the detached leaves, B. elliptica mycelium discs (5-mm diameter) were collected from the PDA plates using a sterilizing puncher, and then used to inoculate the abaxial surface of the detached lily leaves in vitro. Each leaf was inoculated with six mycelium discs (inoculated treatment), while those uninoculated with B. elliptica served as the control treatment. The inoculated and uninoculated leaves were placed in Petri dishes (15 cm diameter) lined with moist filter paper. The filter paper and cotton surrounding the leaf petiole were soaked with sterile water to maintain humidity within a range of 90–100%.
The detached leaves were sampled and photographed at 6, 8, 12, 24, 36, 48, and 72 h post inoculation (hpi). The area of each lesion was measured with the ImageJ software (https://imagej.nih.gov/ij/). Superoxide dismutase (SOD) activity was assayed by monitoring the photoreduction of nitro blue tetrazolium (NBT), as describe previously (Li, 2000). Based on the results of the SOD activity assay and the phenotype of disease lesions, leaves in the control, and inoculated treated treatments were sampled at 6, 24, and 48 hpi (hereafter referred to as control_6 h, control_24 h, and control_48 h, and inoculated_6 h, inoculated_24 h, and inoculated_48 h, respectively) for transcriptome and metabolome sequencing. Three biological replicates were performed for each treatment, with each replicate containing three technical repeats.
Metabolomics
The leaf samples were freeze-dried under vacuum using the Scientz-100F lyophilizer (Scientz, China), and then crushed at 30 Hz for 1.5 min using a grinder (MM 400; Retsch, Germany) to obtain a fine powder. Then, 100 mg of each powdered sample was extracted in 0.6 ml of 70% methanol. The samples were stored at 4°C overnight, during which time they were vortexed six times to hasten the extraction. Then, each sample was centrifuged at 10 000 × g for 10 min. The supernatant was filtered through a microporous membrane (pore size: 0.22 μm), and stored in sample bottles for ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) analysis.
To analyze the extracts, UPLC (Shim-pack UFLC SHIMADZU CBM30A; https://www.shimadzu.com.cn/) and MS/MS (Applied Biosystems 4500 QTRAP; http://www.appliedbiosystems.com.cn/) were performed using a C18 chromatographic column (Waters ACQUITY UPLC HSS T3 C18; 1.8 μm, 2.1 × 100 mm) with solvent A (0.04% acetic acid in ultrapure water) and solvent B (0.04% acetic acid in acetonitrile) as the mobile phase. A 4-μl aliquot of each sample was injected into the column, and eluted using the following gradient program: 0 min with 95% A and 5% B; 0–10 min with 95–5% A and 5–95% B; 10–11 min with 5% A and 95% B; 11–11.1 min with 5–95% A and 95–5% B; and 11.1–14 min, 95% A and 5% B. The flow rate and column temperature were maintained at 0.35 mL/min and 40°C, respectively. The mass spectrometer parameters were set as follows: temperature of electrospray ionization: 550°C; voltage: 5.5 kV; curtain gas: 30 psi; collision-activated dissociation: high. Each ion pair was scanned and detected based on optimized declustering potential and collision energy in the triple quadrupole system.
Qualitative and quantitative analyses of the metabolites were performed using the multiple reaction monitoring (MRM), KEGG compound database, and MetWare database. Metabolites were identified based on their molecular weight, Mass Spectrometry (MS2) fragments, MS2 fragments isotope distribution, and retention time (RT). Through the MetWare self-developed intelligent secondary spectrum matching method, the secondary spectrum and RT of the metabolites in the project samples are intelligently matched one by one with the MetWare database. The MS tolerance and MS2 tolerance are set to 2 and 5 ppm, respectively. The peak area integral of all the mass spectrum peaks was derived after obtaining the metabolic substance spectrum analysis data of different samples, followed by an integral correction performed for the mass spectrum peak of the same metabolite occurring in different samples (Fraga et al., 2010). Quality control (QC) samples, i.e., samples prepared from a mixture of sample extracts, were used to analyze the reproducibility of the instrument under the same treatment method.
The “MetaboAnalystR 1.0.1” package in the R computing platform v 3.5.0 (R Core Team, 2018, Austria) was used to statistically analyze the metabolomics data and to generate plots. Orthogonal partial least squares discriminant analysis (OPLS-DA) was conducted using MetaboAnalystR to identify differentially accumulated metabolites (DAMs), with Variable Importance in Projection (VIP) score ≥ 1 and absolute Log2fold change (FC) ≥ 1. Pathways with significantly regulated metabolites mapped to it were then subjected to metabolite set enrichment analysis; their respective statistical significance was determined using the hypergeometric test and its p-value.
Transcriptomics
A total of 18 cDNA libraries were sequenced using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB; USA). Clean reads obtained from each cDNA library were assembled de novo into unigenes using the Trinity (2.6.6.) platform (Haas et al., 2013). Functional annotations of the unigenes were determined using NCBI non-redundant (NR) protein, UniProt, KEGG, Gene Ontology (GO), and Clusters of Orthologous Groups of proteins (COG) databases (Kanehisa et al., 2008). Transcription factors and gene coding sequence (CDS) were used ITAK web online (http://itak.feilab.net/cgi-bin/itak/index.cgi) and TransDecoder (5.3.0, https://github.com/TransDecoder) software, respectively. Differentially expressed genes (DEGs) were analyzed using the DESeq2 (v1.22.2) software package in R, and subjected to the Benjamini–Hochberg method for multiple hypothesis testing (i.e., |log2FC| ≥ 1, FDR [false discovery rate] <0.05) (Love et al., 2014, Varet et al., 2016). Heat maps were constructed using the R package “pheatmap” (v1.0.12) and TBtools software v0.66836 (Chen et al., 2020). Venn diagrams were generated using Venn v1.6. Weighted gene coexpression network analysis (WGCNA) was performed using the R package “WGCNA” (Zhang and Horvath, 2005), and visualized using Cytoscape v1.7.251 (https://cytoscape.org/index.html).
Quantitative Real-Time PCR (qRT-PCR)
To validate the RNA-seq data, the expression of 10 defense-related DEGs, including four DEGs involved in plant–pathogen interactions (Cui et al., 2018a) and six DEGs involved in the phenylpropanoid and flavonoid biosynthesis pathways, was analyzed by qRT-PCR. The qRT-PCR was performed using the SsoFast EvaGreen Supermix (Bio-Red, USA) on the CFX96 Real-Time PCR Detection System (Bio-Rad, USA) under the following conditions: 95°C for 30 s, followed by 39 cycles of 95°C for 5 s and 57°C for 5 s, melt curve 65–95°C, increment 0.5°C for 5s. The specificity of the primers was verified based on the unimodality of the melt curve. Actin (ACT) and elongation factor 1 (EF1) served as reference genes (Cui et al., 2018a). Three biological replicates were performed for each treatment, with each replicate containing three technical repeats. Data were analyzed using the 2−ΔΔCt method (Livak and Schmittgen, 2001). Primers used for qRT-PCR are listed in Supplementary Table 1.
Results
B. elliptica-Induced Lesions and Altered SOD Activity in Lilium Hybrid “Sorbonne”
Water-soaked lesions of the same diameter (0.5 cm) as the plug used for inoculation were observed at 12 hpi (Figure 1A). Over time, the lesions first became rotten and brown (36 hpi) and then turned necrotic (48–72 hpi). The area of lesions expanded with time after inoculation (Figure 1B). Changes in SOD activity were also observed in inoculated leaves (Figure 1C). Significant differences were detected in SOD activity between the control_6 h and inoculated_6 h samples. Superoxide dismutase activity declined before 8 hpi but then increased over time. The sampling time points for metabolomics and transcriptomics were determined based on both the disease symptoms and SOD activity. Given that transcriptional changes usually precede the associated physiological and phenotypic changes, here we considered 6 hpi as the early stage of infection. As the regular ellipse-shaped lesions formed, the 24- and 48-hpi time points were considered as the middle and late stages stage of infection, respectively, for sampling.
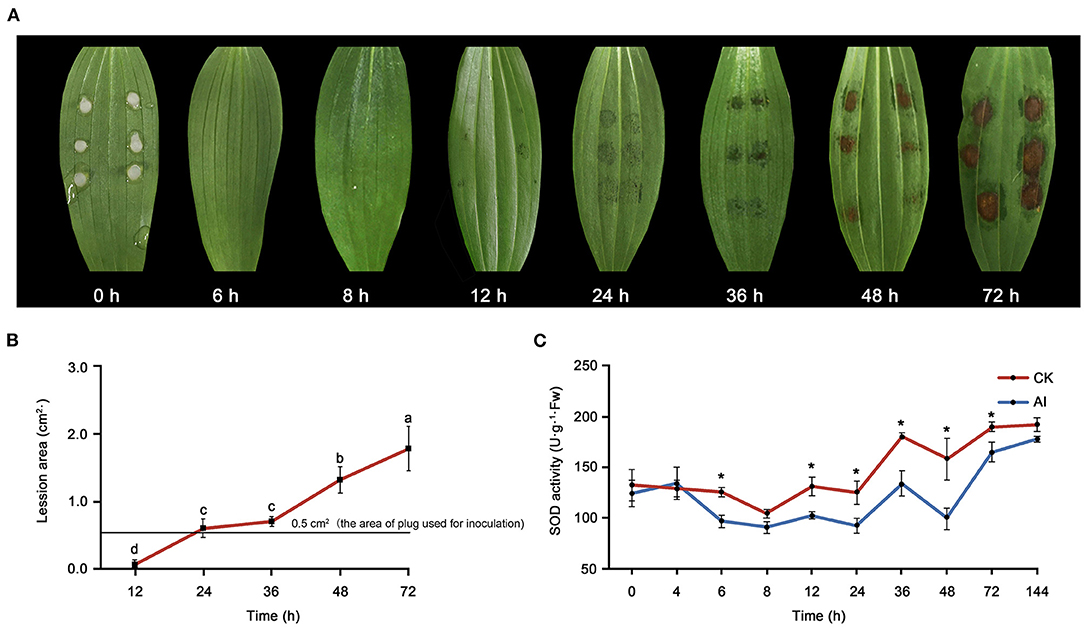
Figure 1. Evaluation of the response of Lilium oriental high resistant hybrid “Sorbonne” to Botrytis elliptica inoculation. (A) Progression over time (0–72 hpi) of symptoms on B. elliptica inoculated Sorbonne leaves. Each leaf is representative of nine repetitions. (B) Change in lesion area over time. Data represent mean ± standard error (SE) of triplicate assays. (C) Changes in super oxide dismutase (SOD) activity over time. Data represent mean ± SE of triplicate assays. The line charts were generated based on IBM SPSS Statistics 20. The “*” represents the significant differences in the SOD activity.
Global Metabolomic Changes
To determine the metabolic changes induced by B. elliptica infection, we performed non-targeted metabolome analysis using control_6 h, control_24 h, control_48 h, inoculated_6 h, inoculated_24 h, and inoculated_48 h samples. A total of 524 metabolites common to all samples were identified based on their chromatographic and mass spectrometric parameters (Supplementary Table 2). Orthogonal partial least squares discriminant analysis and principal components analysis (PCA) uncovered differences in the metabolites of all samples (Supplementary Figures 1, 2). High correlation was observed among the QC samples (Supplementary Figure 3). Pearson correlation coefficients were consistently high (r > 0.819) across all three biological replicates (Supplementary Figure 4). Heat map, based on the hierarchical clustering analysis of metabolite levels, revealed significant differences in metabolites levels between inoculated and control treatments, and these differences became more pronounced over time (Supplementary Figure 5).
The DAMs were similarly identified by comparing the different temporal stages of pathogen infection, with cutoff values of log2 FC ≥ 1 and VIP score ≥ 1. Venn diagram shows the overlap of DAMs among the three stages of infection (Supplementary Figure 6; Supplementary Table 3). A total of 115 DAMs were detected, of which 8, 12, and 62 DAMs were expressed only at the 6-, 24- hpi, and 48-hpi time points. Only eight DAMs, including benzyl salicylate, eriodictyol, tryptamine, esculetin, butin, caffeic acid, dihydrokaempferol, and N'-feruloyl putrescine, were common to all three infection stages (Supplementary Figure 7). Among these eight DAMs, five were involved in phenolic acid metabolism and are known as important secondary metabolites for pathogen resistance (Supplementary Figure 7). Moreover, the accumulation of 15 metabolites was up-regulated and that of 6 metabolites was down-regulated in inoculated_6 h compared with control_6 h (Supplementary Figure 8). Similarly, 39 and 4 metabolites were up-regulated and down-regulated, respectively, in inoculated_24 h compared with control_24 h, and 85 and 5 metabolites were up-regulated and down-regulated, respectively, in inoculated_48 h compared with control_48 h (Supplementary Figure 8). Furthermore, according to KEGG enrichment analysis, the DAMs were enriched in phenylpropanoid biosynthesis, flavonoid biosynthesis, metabolic pathway, and biosynthesis of secondary metabolites at all three infection stages. Phenylpropanoid biosynthesis was significantly enriched at 6 hpi (Supplementary Figure 9A); tryptophan and indole alkaloid biosynthesis were significantly enriched at 24 hpi (Supplementary Figure 9B); and isoflavonoid biosynthesis, fructose and mannose metabolism, and galactose metabolism were significantly enriched at 48 hpi (Supplementary Figure 9C).
Global Transcriptomic Changes
Samples were used for non-targeted metabolome analysis were subjected to RNA-seq to profile the genome-wide changes in gene expression upon the inoculation of leaves with B. elliptica. After removing low-quality reads, 435.16 Gb clean reads were obtained, with an average GC content of 49.87% (Supplementary Table 4). A total of 430,835 transcripts were obtained, averaging 701 bp in length, with N50 and N90 values of 1,213 and 266 bp, respectively (Supplementary Table 5). Using KEGG, NR, Swiss-Prot, GO, and Trembl databases, a total of 283,213 unigenes, with an average length of 534 bp, were functionally annotated (Supplementary Tables 5, 6). In the NCBI NR database, Asparagus officinalis (8.94%), Elaeis guineensis (8.72%), Phoenix dactylifera (7.39%), Vitis vinifera (3.63%), and Cajanus cajan (3.45%) gave the top BLASTx hits (Supplementary Figure 10). Hierarchal clustering analysis was performed to examine the significant changes in unigene expression. The results showed a stage-specific transcriptome profile after inoculation with B. elliptica (Supplementary Figure 11). To validate the RNA-seq data, the expression profiles of 10 defense-related DEGs were evaluated in inoculated and control leaves at 6, 12, 24, 36, and 48 hpi by qRT-PCR (Supplementary Figure 12). All RNA-seq data can be downloaded from NCBI (BioProjects: PRJNA742853).
Time-Course RNA-Seq Analysis
The DEGs were identified by comparing the RNA-seq data of inoculated leaf samples (inoculated_6 h, inoculated_24 h, and inoculated_48 h) with those of control leaf samples (control_6 h, control_24 h, and control_48 h) using cutoff values of log2(FC) ≥ 1 and Padj ≤ 0.05. All DEGs identified at the three temporal stages were analyzed using the K-means clustering algorithm. The DEGs could be grouped into six clusters and classified into four types (Figure 2A): up-regulated (clusters I, II); down-regulated (clusters IV, V); first up-regulated, then down-regulated (clusters VI); and first down-regulated, then up-regulated (cluster III) (Figure 2A). Gene ontology enrichment analysis showed that one cytomembrane-related term, two chloroplast-related terms, two cytoderm-related terms, three signal transduction receptor-related terms, and six photosynthesis-related terms were enriched among the down-regulated genes (Figure 2B). This suggests that certain biological processes, such as photosynthesis, chlorophyll synthesis, cell wall biogenesis, anchored component of membrane, and signal transduction, might be inhibited by the destruction of leaf tissue by the necrotrophic pathogen. The up-regulated gene clusters were found to be related to chitinase, oxidoreductase activity, cell recognition, secondary metabolites, and the phenylpropanoid biosynthetic process, pointing to their potential positive role in the production of resistant metabolites and proteins in B. elliptica-inoculated leaves. The results of KEGG enrichment analysis were consistent with those of GO enrichment analysis: Clusters I and II were mainly enriched in the phenylpropanoid biosynthesis, flavonoid biosynthesis, plant hormone signal transduction, and plant–pathogen interaction pathways; clusters IV and V were mainly enriched in the pathways of photosynthesis and starch and sucrose metabolism (Figure 2C). Taken together, the up-regulated clusters resolved by the K-means clustering algorithm potentially play a pivotal part in the resistance to B. elliptica, while the down-regulated clusters related to photosynthesis, cell wall biosynthesis, and other metabolic pathways seem to be negatively affected by B. elliptica.
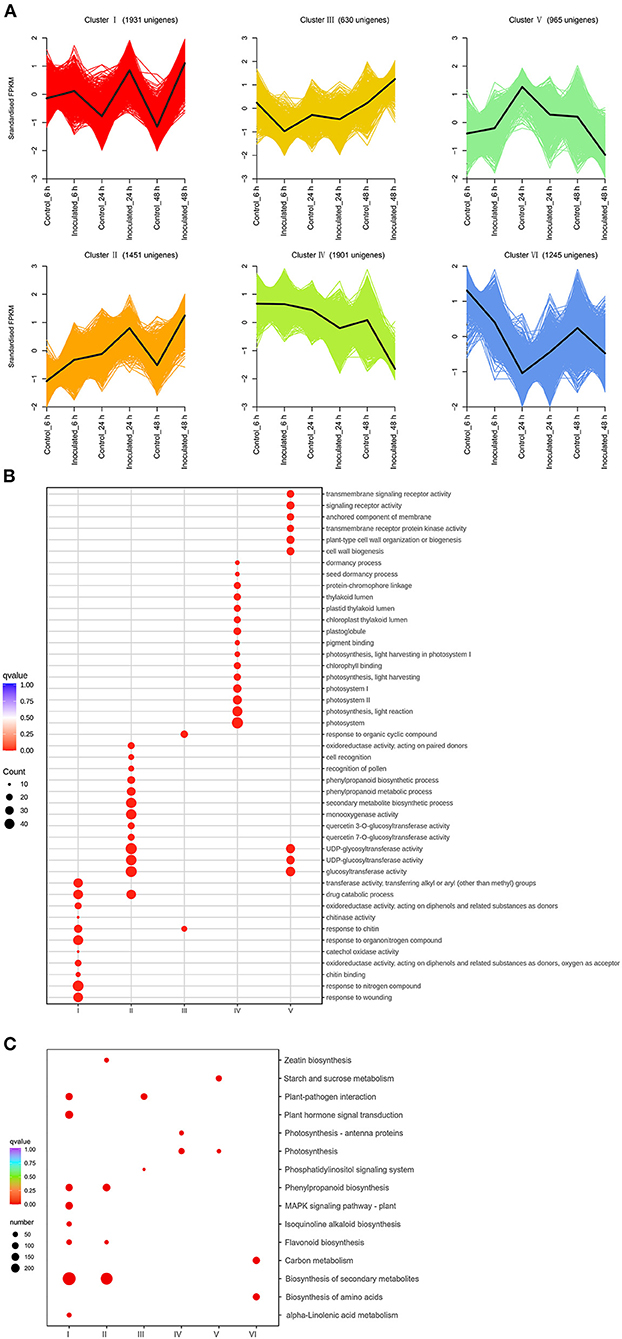
Figure 2. Transcript abundance of all unigenes identified in “Sorbonne”. (A) K-means clustering analysis of differentially expressed genes (DEGs), according to their expression profiles. (B) Comparison of the gene ontology (GO) enrichment of all unigene clusters. The sizes of dots are proportional to the number of genes per GO term. Only the GO terms with more than 10 DEGs were identified; accordingly, the GO enrichment of cluster VI was eliminated. (C) Enrichment of KEGG annotations of DEGs in Lilium “Sorbonne” with B. elliptica infection. Only the representative and significant pathways among the five clusters are shown in the figure. The size of the circle indicates the quantity, and its color corresponds to the q-value.
A Venn diagram was used to demonstrate the DEGs identified at all three stages. Overall, 5,295 DEGs were detected, of which 125, 434, and 3,089 were uniquely expressed at 6, 24, and 48 hpi, respectively, and 72 DEGs were common to all three stages (Supplementary Figure 13; Supplementary Table 7). Kyoto Encyclopedia of Genes and Genomes enrichment analysis was performed to further characterize these DEGs. At 6 hpi, 401 DEGs (86 up-regulated, 315 down-regulated) were significantly enriched in “biosynthesis of secondary metabolites”, “biosynthesis of amino acid”, and “carbon metabolism” pathways (Supplementary Figures 14, 15A). At 24 hpi, 1,887 DEGs (309 up-regulated, 1,578 down-regulated) were significantly enriched in six pathways: “biosynthesis of secondary metabolites”, “plant hormone signal transduction”, “phenylpropanoid biosynthesis”, “flavonoid biosynthesis”, “isoquinoline alkaloid biosynthesis”, and “MAPK signal pathway” (Supplementary Figures 14, 15B). At 48 hpi, 4,726 DEGs (2,772 up-regulated, 1,954 down-regulated) were significantly enriched in five pathways: “biosynthesis of secondary metabolites”, “plant hormone signal transduction”, “plant–pathogen interaction”, “flavonoid biosynthesis”, and “carbon metabolism” (Supplementary Figures 14, 15C). Taken together, these results indicate that “biosynthesis of secondary metabolites”, “flavonoid biosynthesis”, “phenylpropanoid biosynthesis”, “plant hormone transduction signal”, “plant-pathogen interaction”, and “MAPK signal pathway” were the chief pathways underpinning the responses of “Sorbonne” to B. elliptica infection at the transcript level.
Role of Phenylpropanoid and Flavonoid Biosynthesis Pathways in the Lilium Defense Response
The phenylpropanoid and flavonoid biosynthesis pathways could be integrated into a transcriptional cascade and metabolic network, which together play a critical role in how the Lilium hybrid “Sorbonne” responds to B. elliptica infection. All the DEGs and DAMs participating in the phenylpropanoid and flavonoid biosynthesis pathways that exhibited up- or down-regulated trends in the different infection stages were selected and mapped to this network (Figure 3; Supplementary Figure 16).
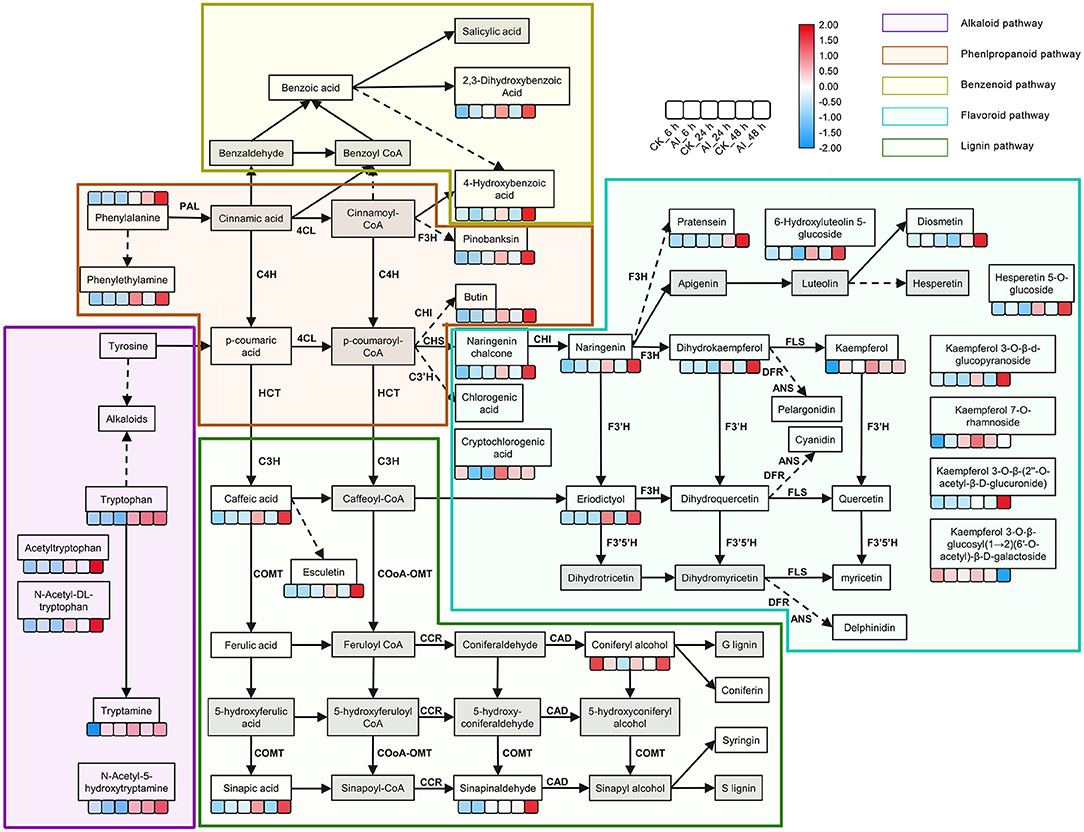
Figure 3. Summary of metabolite changes in Lilium “Sorbonne” caused by B. elliptica infection. The metabolites not detected are indicated by a gray-filled grid. Dotted lines indicated some metabolites, and enzymes are not shown. Metabolites with statistically significant (P ≤ 0.05) changes are shown in the heat map, in which blue and red colors indicate low and high accumulation, respectively. Data on the number of metabolites in the heat map were log-transformed. The six boxes represent control (uninoculated) and inoculated samples at different stages of infection (i.e., different time points post-inoculation): control_6 h, inoculated_6 h, control_24 h, inoculated_24 h, control_48 h, and inoculated_48 h (left to right). Different background colors represent different metabolic pathways: purple, alkaloid pathway; brown, phenylpropanoid pathway; yellow green, benzenoid pathway; blue, flavonoid pathway; green, lignin pathway. Gene names, annotations, and RNA-seq data are summarized in Supplementary Figure 15. PAL, phenylalanine ammonia-lyase; C4H, cinnamic acid 4-hydroxylase; 4CL, p-coumaroylCoA ligase; HCT, hydroxycinnamoyl transferase; CSE, caffeoylshikimate esterase; C3′H, coumaroylquinate 3′-monooxygenase; COMT, caffeic acid O-methyltransferase; CCoAOMT, caffeoyl coenzyme A 3-O-methyltransferase; CCR, cinnamoyl-CoA:NADPH oxidoreductase; CAD, cinnamyl alcohol dehydrogenase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, naringenin 3-dioxygenase; F3′H, flavonoid 3′-monooxygenase; FLS, flavonol synthase; DFR, dihydroflavonol 4-reductase; ANS, anthocyanidin synthase.
Phenylalanine is the first metabolite in the phenylpropanoid pathway. Although our non-targeted metabolome analysis indicated a significant difference in its accumulation between the inoculated and control treatments at 48 hpi, we identified significant expression of phenylalanine ammonia-lyase (PAL), a structural gene that catalyzes phenylalanine, clustering at 24 hpi. The expression of PAL (DN129152_c1_g2) in the inoculated treatment was 3.5- and 6.5-fold higher at 24 and 48 hpi, respectively, compared with the control (Supplementary Figure 16; Supplementary Table 2), implying that PAL participates in the defense response. Moreover, the levels of phenylalanine derivatives, such as phenethylamine and 4-hydroxybenzoic acid, were significantly higher in the inoculated leaves at 24 and 48 hpi compared with the control, indicating that the phenylpropanoid pathway was activated by the infection. Besides, the same expression pattern was exhibited by the structural gene (DN134629_c3_g2) that catalyzes 4-hydroxybenzoic acid. The accumulated level of caffeic acid, a precursor of the lignin pathway, was 1.6-, 3.8-, and 11-fold higher in inoculated leaves at 6, 24, and 48 hpi, respectively, compared with the control (Supplementary Table 2). The level of esculetin, a downstream metabolite of caffeic acid, increased rapidly in leaves after inoculation (Supplementary Table 2). Among the downstream metabolites of caffeic acid, ferulic acid and coniferyl alcohol, which act as precursors of guaiacyl, were down-regulated at 6 hpi but up-regulated at 24 hpi and 48 hpi. Sinapinaldehyde and sinapic acid, precursors of syringyl lignin, were up-regulated in all three stages. The accumulation of syringaresinol-hex and syringaresinol, downstream metabolites of S-lignin, increased significantly in leaves post inoculation (Supplementary Table 2).
The accumulation of naringenin chalcone, naringenin, and dihydrokaempferol, which represent metabolites in the flavonoid biosynthesis pathway, increased significantly at different stages of infection. Additionally, the expression of chalcone synthase (CHS), a structural gene that promotes the synthesis of the three abovementioned metabolites, was induced at all three stages (Supplementary Figure 16; Supplementary Table 2). Among all the DAMs detected in the flavonoid biosynthesis pathway, the greatest difference in accumulation occurred in eriodictyol, which was undetectable in all the control treatments and was 5- and 37-fold higher at 24 and 48 hpi, respectively, than at 6 hpi in the inoculated treatments (Supplementary Table 2). The accumulation level of kaempferol in the inoculated treatment was 190- and 14-fold higher than that in the control treatment at 6 and 24 hpi, respectively, but its level was similar between the inoculated and control treatments at 48 hpi (Supplementary Table 2). The expression of UDP-glycosyltransferase (DN130728_c1_g2) in the inoculated treatment increased significantly at 6 hpi, which could potentially explain the accumulation of kaempferol derivatives at 24 and 48 hpi. The accumulation of four downstream metabolites of kaempferol, namely kaempferol 3-O-β-d-glucopyranoside, kaempferol 3-O-β-glucosyl(1 → 2)(6′-O-acetyl)-β-D-galactoside, kaempferol 7-O-rhamnoside, and kaempferol 3-O-β-(2″-O-acetyl-β-D-glucuronide), was significantly increased in the inoculated treatment at 48 hpi compared with the corresponding control (Figure 3). In addition, the accumulation of the derivatives of luteolin (6-hydroxyluteolin 5-glucoside) and hesperetin (hesperetin 5-O-glucoside) in the inoculated treatment increased significantly at 24 and 48 hpi. The level of diosmetin, a downstream product of luteolin, was five-fold higher in the inoculated treatment than in the control treatment.
Potential Transcriptional Regulatory Mechanisms
To investigate the gene regulatory network of Lilium Sorbonne after its inoculation with B. elliptica, we performed WGCNA of all DEGs identified from the RNA-seq analysis of the 18 cDNA libraries. After preprocessing the RNA-seq data, 14 gene co-expression modules, each comprising 80–2,659 genes, were discovered (Figure 4A). Module-trait and sample relationship analyses showed the eigengenes of these modules were correlated with the different infection stages. Unlike most modules in which the trend of genes was that of almost no difference between the inoculated and control treatments, a few modules did differ significantly during the defense response (Figure 4B). Next, we identified key genes (i.e., hub genes) that played crucial roles during the infection, based on the WGCNA for these notable modules. The levels of gene expression in the black, dark-green, and green modules showed peak up-regulation at 6, 24, and 48 hpi, respectively, which correspond to the early, middle, and late stages of infection, respectively (Figure 5). Gene expression in the dark-gray module was higher in the inoculated treatment than in the control treatment at all-time points, indicating that genes in this module play a prominent role in the defense response against B. elliptica (Figure 6).
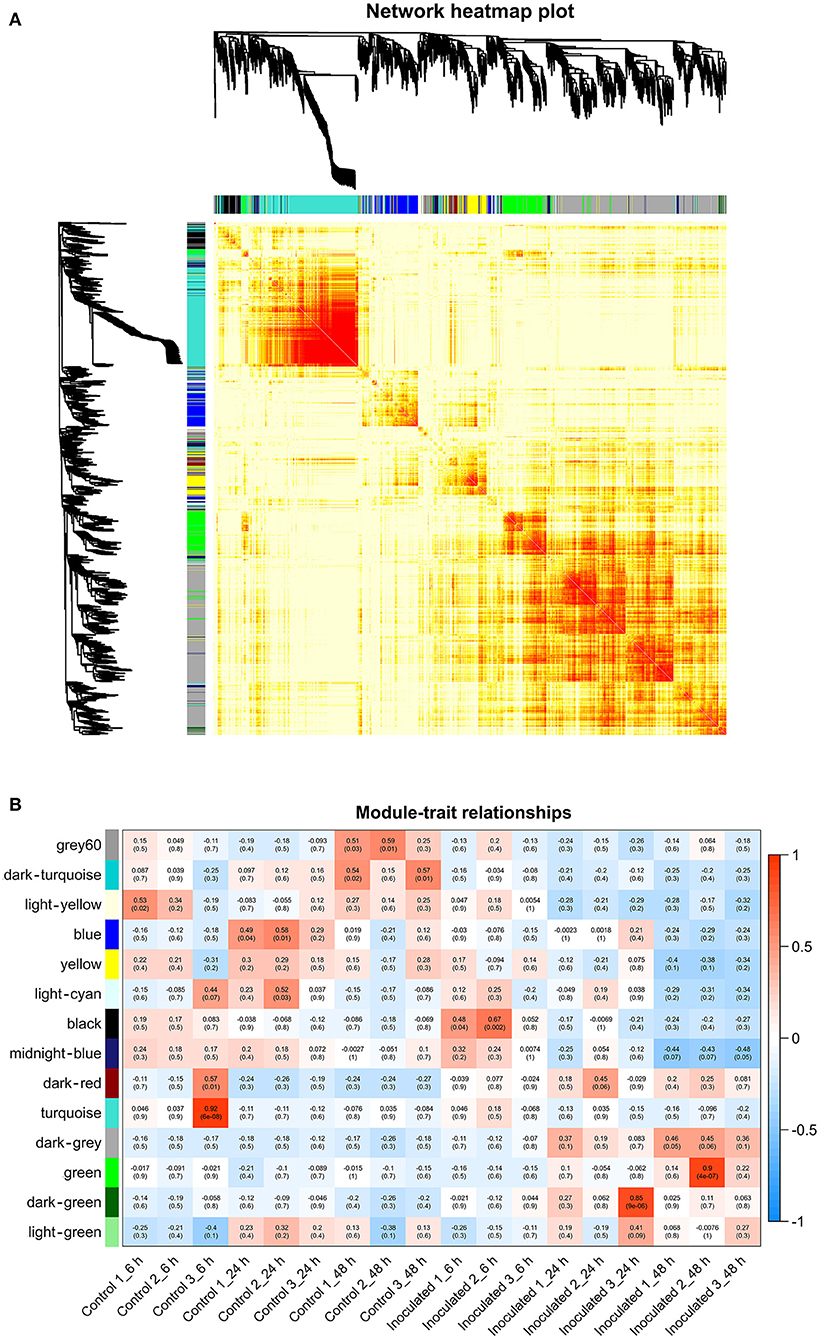
Figure 4. Results of weighted gene coexpression network analysis (WGCNA) of “Sorbonne” transcripts. (A) Dendrogram of genes, based on co-expression network analysis. The gene dendrogram was obtained by hierarchical clustering analysis, with the module color indicated by the color of the row underneath. A total of 14 distinct modules were identified. (B) Association between modules and plant defense traits. The color of each module is the same as that in (A). Each row in the table corresponds to a module, and each column corresponds to a sample. Values in each cell indicate the number of corresponding correlations and their P-values.
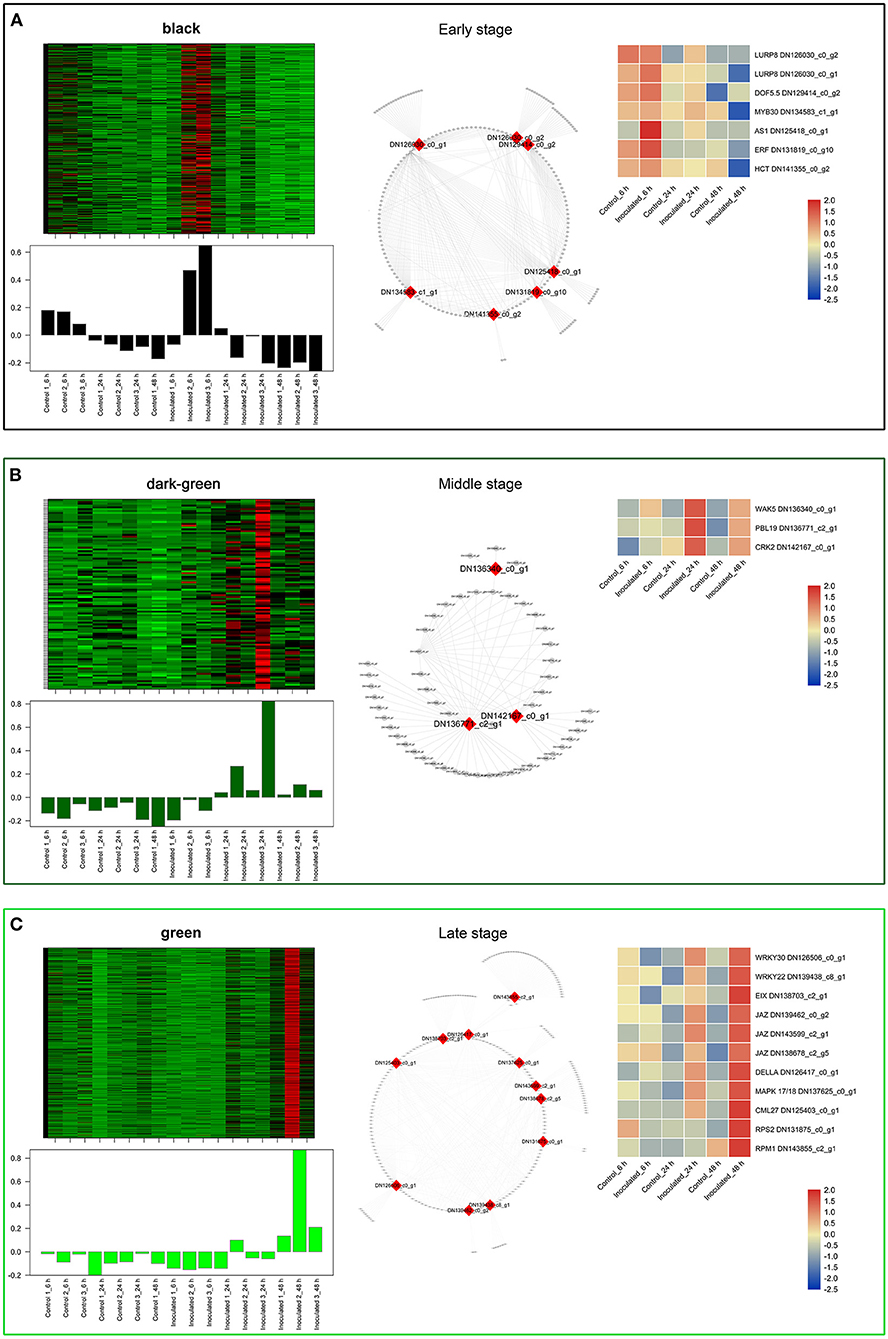
Figure 5. “Sorbonne” DEGs identified at different stages of B. elliptica infection. (A–C) Co-expression network of the black module (A), dark-green module (B), and green module (C). Heat maps and bar graphs show the co-expressed genes in each module (left). Red rectangles on the heat map denote high expression levels; green rectangles denote low expression levels. The network of top hub genes is indicated by red squares in the network (middle). Heat maps (right) show the expression patterns of hub genes.
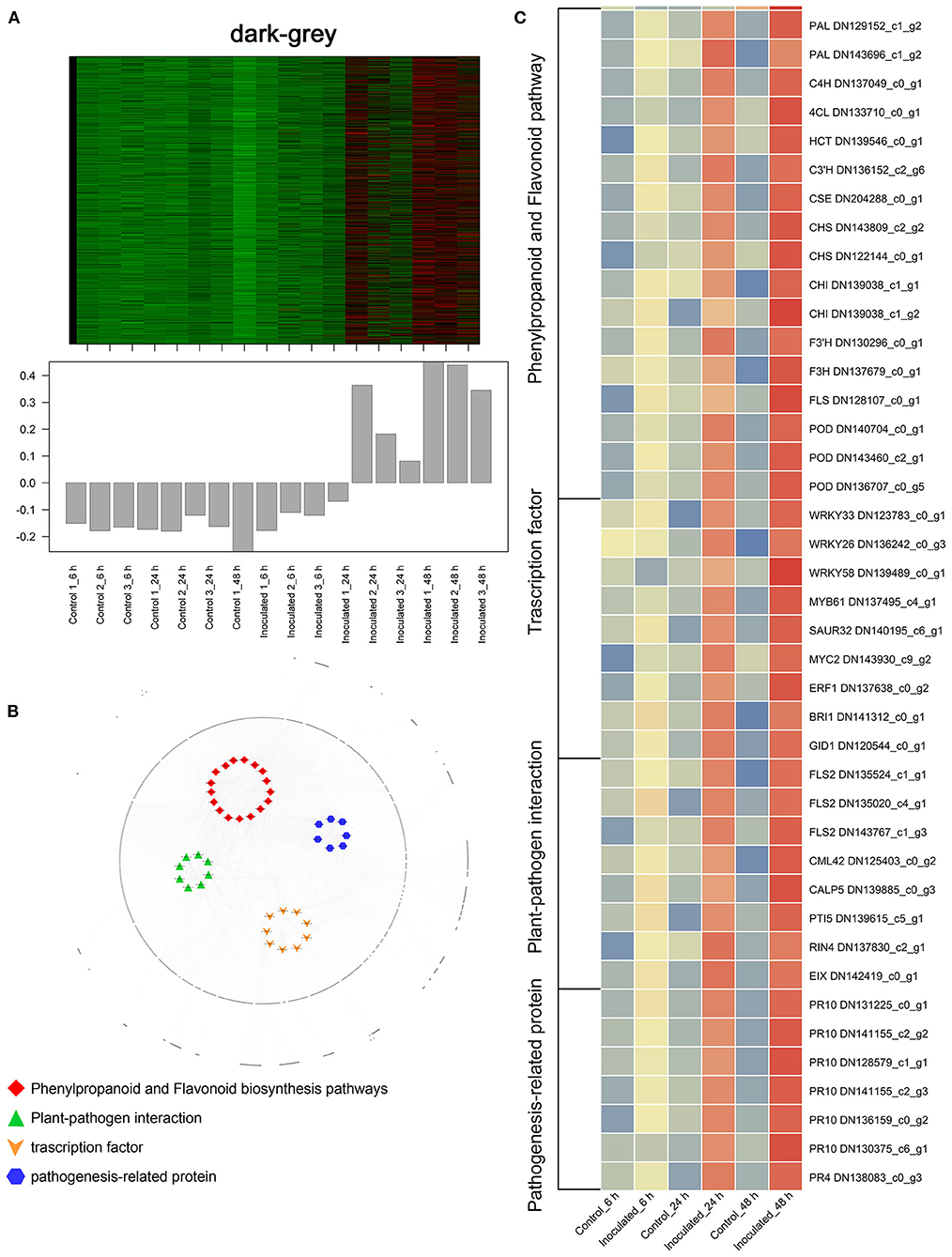
Figure 6. “Sorbonne” DEGs common to all three stages of B. elliptica infection. (A) Heat maps and bar graphs show the co-expressed genes in each module. Red rectangles denote high expression levels; green rectangles denote low expression levels. (B) Network of hub genes is indicated by larger font size in the network. Small dots around the hub genes represent other co-expressed genes, and their relationships are connected by lines. Red squares represent the phenylpropanoid and flavonoid pathways; green triangles represent plant–pathogen interactions; orange arrows represent transcription factors (TFs); blue hexagons represent pathogenesis-related (PR) proteins. (C) Heat maps showing the expression patterns of hub genes.
In the black module (Figure 5A), seven hub genes related to plant defense were identified, such as two LURP homologs (DN126030_c0_g1, DN126030_c0_g2), one MYB30 homolog (DN134583_c1_g1), and one AS1 homolog (DN125418_c0_g1). Among these, DOF5.5 (DN129414_c0_g2) showed a regulatory relationship with the three defense response-related hub genes. In addition, one WRKY70 gene (DN139965_C0_G2) showed significant difference in expression between inoculated and control treatments at 6 hpi. Additionally, an ERF homolog (DN131819_c0_g10) and a HCT homolog (DN141355_c0_g2) were up-regulated at the early stage of infection. In the dark-green module (Figure 5B), we identified three hub genes related to plant–pathogen interactions: a PBL19 homolog (DN136771_c2_g1), a CRK2 homolog (DN142167_c0_g1), and a WAK5 homolog (DN136340_c0_g1). Notably, PBL19 showed a strong regulatory relationship with other two hub genes, CRK2 and WAK5. A CKX9 homolog (DN129547_c0_g1), a I2′H homolog (DN140336_c1_g1), and a CAD homolog (DN127300_c1_g1) were significantly up-regulated in the dark-green module. At the late stage of the infection (Figure 5C), 11 hub genes were identified, including a WRKY22 homolog (DN139438_c8_g1), a WRKY30 homolog (DN126506_c0_g1), three JAZ homologs (DN139462_c0_g2, DN143599_c2_g1, DN138678_c2_g5,), a DELLA homolog (DN126417_c0_g11), a MAPK17/18 homolog (DN137625_c0_g), and a CML27 homolog (DN125403_c0_g1). Furthermore, both an RPS2 homolog (DN131875_c0_g1) and an RPM1 homolog (DN143855_c2_g1) displayed strong regulatory relationships with other genes in the green module. Genes in the dark-gray module (2,659, i.e., more than half of all identified DEGs) were up-regulated at all three infection stages (Figure 6A). Analysis of DEGs revealed 41 hub genes, which included a MYB61 homolog (DN137495_c4_g1), a WRKY33 homolog (DN123783_c0_g1), MYC2 homolog (DN143930_c9_g2), and a BRI1 homolog (DN141312_c0_g1), among others (Figures 6B,C). A number of hub genes were structural genes involved in the phenylpropanoid and flavonoid biosynthesis pathways (e.g., two PAL homologs [DN129152_c1_g2, DN143696_c1_g2], an HCT homolog [DN139546_c0_g1], two CHS homologs [DN143809_c2_g2, DN122144_c0_g1], an F3′H homolog [DN130296_c0_g1], and a FLS homolog [DN128107_c0_g1], and plant–pathogen interactions (e.g., three FLS2 homologs [DN135524_c1_g1, DN135020_c4_g1, DN143767_c1_g3] and an RIN4 homolog [DN137830_c2_g1], in addition to several PR genes. Lastly, a gene called Pti5 (DN139615_c5_g1), which encoded an AP2-EREBP family TF, also exerted a great influence on plant–pathogen interactions.
Discussion
The Central Role of Phenylpropanoid and Flavonoid Biosynthesis Pathways in the Defense Response of Lilium
Phenolic compounds, such as metabolites of the phenylpropanoid and flavonoid pathways, act as fungitoxic and antimicrobial defense compounds in host–pathogen interactions (Martinez et al., 2017; Xu et al., 2018, 2019). Caffeic acid is a key metabolite in the phenylpropanoid pathway. In our study, significantly more caffeic acid accumulated over time in the inoculated treatment than in the control treatment. Consistent with this result, the structural genes involved in the synthesis of caffeic acid (HCT and CSE) were significantly up-regulated in the inoculated treatment (Figure 3; Supplementary Figures 12, 16). In apple, caffeic acid has been shown to effectively promote lignin accumulation and inhibit gray mold infection (Zhang et al., 2020). Although the accumulation of lignin was not detected in our study, we detected significant accumulation of the precursors precursor of G-lignin (coniferyl alcohol) and S-lignin (sinapic acid and sinapinaldehyde) (Figure 3). The structural genes for the synthesis of lignin, COMT, CCR, and CAD, were also significantly up-regulated in the inoculated treatment (Supplementary Figure 16). Additionally, two transcripts, DN141508_c1_g3 and DN141508_c1_g4, encoding the CCoAOMT enzyme, were significantly up-regulated at all stages of infection (Supplementary Table 7). CCoAOMT is reportedly a lignin synthase that can enhance the resistance of Lilium to B. cinerea infection (Fu et al., 2020). Our results showed that caffeic acid may enhance the disease resistance of Lilium by modulating the lignin biosynthetic pathway.
In our network, the flavonoid pathway begins with naringenin chalcone to produce naringenin, which is then converted into apigenin, dihydrokaempferol, and eriodictyol (Figure 3). Subsequently, apigenin and dihydrokaempferol are converted into luteolin and kaempferol, respectively (Figure 3). The entire pathway was specifically activated in Lilium after infection with B. elliptica, as evident from the up-regulation of several structural genes, including CHS, CHI, F3′H, and FLS (Supplementary Figures 12, 16). Eriodictyol was recently identified as a novel antibacterial compound; however, its role in plant–pathogen interactions has not yet been determined (Ho et al., 2018). In the current study, eriodictyol accumulation showed significant differences between the inoculated and control treatments at all stages of infection. Interestingly, evidence shows that eriodictyol synthase F3′H (DN130296_c0_g1) enhances host plant resistance against invading pathogens (Mizuno et al., 2014; Hutabarat et al., 2016). Further analysis of eriodictyol may thus open new opportunities for the cultivation of resistant Lilium. Despite no significant difference in the accumulation of kaempferol at the late stage of infection, the expression of its biosynthetic gene FLS (DN128107_c0_g1) and conjugated compounds, which can be stored in vacuoles for long periods, increased with time post-inoculation. FLS and F3′H are responsible for the synthesis of kaempferol and quercetin, and were recently shown to be critical for disease resistance in plants (Zhang et al., 2016). Together, these findings suggest the possibility that kaempferol and eriodictyol are the main metabolites of the flavonoid pathway that shape the response of Lilium to B. elliptica infection, and their levels are affected by a series of enzymes such as CHS, F3H, and FLS.
Analysis of the time-course RNA-seq data and WGCNA results also support the central role of phenylpropanoid and flavonoid biosynthesis pathways in the defense response of Lilium. At the early stage of infection, the MYB30 gene was significantly up-regulated in inoculated leaves (Figure 5A). The MYB TFs have been shown to confer resistance to rice plants by regulating the phenylpropanoid pathway (He et al., 2020). Consistently, the accumulation of phenylpropanoid pathway-related metabolites and the expression of MYB30 significantly increased at the early stage of pathogen infection. At the middle stage, CAD and I2′H, which represent structural genes in the lignin and flavonoid pathways, respectively, were significantly up-regulated in the dark-green module (Figure 5B). At all three stages, MYB61 was highly up-regulated, which affected the expression of structural genes involved in the phenylpropanoid and flavonoid pathways, such as PAL, CSE, CHS, CHI, and POD (Figure 6). This finding is consistent with the significant accumulation of caffeic acid, dihydrokaempferol, and kaempferol in the inoculated treatments at the three stage of infection.
Role of Hormone Signaling Pathways in B. elliptica–“Sorbonne” Interaction
Analysis of the transcriptome time series and WGCNA results let us further elucidate important signaling pathways underlying the defense responses at transcriptional level. In B. elliptica-inoculated leaves with no lesions, the defense regulator gene DOF5.5 was significantly up-regulated, indicating its involvement in the plant response to infection, similar to that of MYB30 and AS1 (Figure 5A). The DOF TFs function as upstream regulators of the salicylic acid (SA) signaling pathway, and enhance the resistance to pathogens in plants (Kang et al., 2003, 2016; Yu et al., 2019). In Arabidopsis, changes in MYB30 expression levels modulated the SA content and SA-associated gene expression levels (Raffaele et al., 2006). Interestingly, the WGCNA results showed \ a regulatory relationship between DOF5.5 and AS1. The AS1 gene acts a positive regulator of SA-independent extracellular defenses but a negative regulator of the defense response by selectively binding to the promoters of genes controlled by the immune activator, jasmonic acid (JA) (Nurmberg et al., 2007). Additionally, WRKY70 differed significantly between the inoculated and control treatments at the early stage of infection. The AtWRKY70 is an important node of convergence for the SA- and JA-mediated defense signaling pathways (Li et al., 2017).
With the expansion of necrotic lesions on B. elliptica-inoculated “Sorbonne” leaves (middle stage of infection), genes including PBL19, CRK2, and WAK5, which are all members of the Interleukin-1 Receptor-Associated Kinase (IRAK) gene family, were instrumental in the defense against B. elliptica (Figure 5B). The IRAK family proteins are conserved upstream signaling molecules that can regulate various stress adaptation programs in plants (Srideepthi et al., 2020). Our results demonstrated a strong correlation between the expression of PBL19 and CKX9. OsPBL1 exhibits 67% amino acid sequence identity to a positive regulator of ETI, and the expression of OsPBL1 in transgenic Arabidopsis increased after an exogenous treatment of cytokinin and SA (Lee and Kim, 2015). CKX5, the only known gene involved in cytokinin catabolism, was recently proven to respond to B. cinerea infection in various ways that are differently modulated by JA and ethylene biosynthesis pathways in Arabidopsis (Li et al., 2021).
At the late stage of B. elliptica infection (green module), necrotic lesions on leaves expanded, and JAZ, WRKY30, and WRKY22 genes played central roles in plant defense (Figure 5C). The JAZ genes encode transcriptional repressors of JA-responsive genes and major components of the JA receptor complex (Thatcher et al., 2016). WRKY30 and WRKY22 are known to enhance disease resistance in plants via the SA and JA defense systems (Peng et al., 2012; Han et al., 2013; Jiang et al., 2015; Kloth et al., 2016).
Furthermore, several differentially expressed TF-encoding genes were identified in the dark-gray module, indicating that these TFs may be significantly involved in the defense response at all three stages (Figure 6). The results revealed a major TF involved in SA and JA signaling, WRKY33 (Figure 5C). WRKY33 showed a strong regulatory relationship with MYC2 and NPR1, and was highly expressed in the inoculated leaves. AtWRKY33 plays a major role in the crosstalk between JA and SA signaling pathways and metabolic responses in response to B. cinerea infection (Birkenbihl et al., 2012; Liu et al., 2017). MYC2 positively regulates JA-mediated flavonoid biosynthesis, while NPR1 modulates SA and JA antagonism in plants (Dombrecht et al., 2007; Knoth et al., 2009). In addition, the FLS2, BAK1, BRI1, and MYB61 genes were significantly up-regulated across all infection stages in our results. FLS2 is a phylogenetically related cell surface pattern recognition receptor and a coreceptor for BAK1. BAK1 is a coreceptor for the brassinolide (BR) receptor BRI1 (Tian et al., 2014), which is responsible for initiating the events of BR signal transduction. While the BR signaling pathway contributes to the growth–defense tradeoff by suppressing the expression of defensin and glucosinolate biosynthesis genes (Liu et al., 2018; Liao et al., 2020). MYB61 was also positively regulated by BRI1 in our results, but whether MYB61 is involved in BR signaling remains unknown.
Taken together, our analysis indicates that SA and JA signaling pathways play pivotal roles in B. elliptica– Sorbonne” interaction. This supports the proposal that a large number of transcripts related to B. elliptica resistance in L. regale were involved in the JA and phenylpropanoid pathways (Cui et al., 2018a). Moreover, as reported in the interactions between other hosts and Botrytis spp. (e.g., Arabidopsis–B. cinerea and Arabidopsis–Alternaria brassicicola), the SA and JA signaling pathways play a crucial role in the response of “Sorbonne” to fungal infection (Zheng et al., 2006; Ederli et al., 2015, 2021; Liao et al., 2020). Besides, the BR-related genes were significantly up-regulated during across all infection stages in our results. The crosstalk between BR and JA signaling affects the growth-defense tradeoff in Arabidopsis–B. cinerea interaction (Liu et al., 2018; Liao et al., 2020), while exogenous BR before the inoculation of B. cinerea enhances the defense response in rose petals (Liu et al., 2018). The role of BR pathway in Lilium defense response to the gray mold remains to be further elucidated.
Other Signal Transduction Pathways That Contribute to the Lilium Defense Response Against B. elliptica
In addition to phenolics and hormone signaling pathways, our results showed that other signal transduction pathways play a prominent role in the defense response. At the early stage of stage, LURP was significantly up-regulated in the plant response to infection, and regulated the transcript levels of POD and HSP70 (Figure 5A). POD and HSP70 are important for plant resistance (Baig, 2018). AtLURP1 shows an unusually pronounced transcriptional up-regulation in response to infection (Knoth and Eulgem, 2008). Together, these data suggest that LURP plays a pivotal role in the response of “Sorbonne” to B. elliptica infection at the early stage.
At the middle stage, the PBL19, CRK2, and WAK5 genes were instrumental in the defense response of “Sorbonne” against B. elliptica (Figure 5B). The pbl13-2 knockout mutant shows higher level of reactive oxygen species (ROS) and greater flagellin-induced activation of mitogen-activated protein kinases (MAPKs) than the wild type (Lin et al., 2015). Moreover, overexpression of CRKs enhances PTI in transgenic Arabidopsis (Yeh et al., 2015). Wall-associated kinases (WAKs) localize to the cell wall and participate in pathogen recognition and signal transduction in plants (Kurt et al., 2020). Collectively, the transduction of various signals contributed to the defense response of Lilium against B. elliptica at the middle stage of infection.
At the late stage of infection, CML27, DELLA, and JAZ played central roles in plant defense. CML27 encodes a Ca2+-binding protein involved in the Ca2+ signaling pathway. Ca2+ regulates diverse cellular processes and functions as a secondary messenger, enabling plants to sense and quickly respond to extracellular stimuli (Edel et al., 2017). AtRPS2 and AtRPM1 activate Ca2+-dependent protein kinases (CPKs) for mediating bifurcate immune responses (Gao et al., 2013). In our results, RPS2 and RPM1 were significantly up-regulated among the hub genes, indicating the important role of the Ca2+ signaling pathway in the defense response of “Sorbonne”.
In all three stages (Figure 6), MYB61 was highly expressed, implying that it is central to the defense response. B. elliptica infects Lilium via the stomata, and MYB61 regulates stomatal aperture (Hsieh et al., 2001; Gao et al., 2018; Romero-Romero et al., 2018), which may explain its role in the defense response. In the current study, the expression levels of PR10 and PR4 were strongly correlated with that of Pti5 (Figure 6). Overexpression of the tomato TF genes, Pti4/5/6, in Arabidopsis showed that Pti4/5/6 activate the expression of a wide array of PR genes in vivo, resulting in enhanced defense against certain fungal pathogens (Gu et al., 2002; Gonzalez-Lamothe et al., 2008). The expression levels of RIN4, RPM1, RPS2, NPR1, and TGA were significantly up-regulated at all infection stages. In Arabidopsis, the phosphorylation and cleavage of RIN4 activates RPM1 and RPS2, which encode R proteins involved in ETI (Li et al., 2019; Ray et al., 2019; Yang et al., 2021; Zhao et al., 2021). NPR1-mediated DNA binding of TGA2 is critical for the activation of defense related genes. Both NPR1 and TGA1 act as master redox-sensitive transcriptional regulators of PR genes in plants (Fu and Dong, 2013). Altogether, our results suggest the transduction of diverse signals jointly shape the defense response of Lilium hybrid “Sorbonne” to B. elliptica.
Conclusion
In this study, we used a non-targeted metabolomic analysis complemented by NGS to understand the defense response of Lilium hybrid “Sorbonne” to B. elliptica infection. Multivariate data analysis demonstrated that the phenylpropanoid and flavonoid pathways play a central role in the plant defense response. Network analysis revealed high interconnectivity among factors involved in the induced defense response. Furthermore, we performed WGCNA to investigate the DEGs, and identified a number of hub genes at different stages of infection, indicating that JA, SA, BR, and Ca2+ also play important roles in the defense response. Thus, our study provides a comprehensive understanding of the defense response of “Sorbonne” to B. elliptica infection. Further investigation is needed to elucidate the regulatory mechanisms underlying the defense response of Lilium.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: National Center for Biotechnology Information (NCBI) BioProject database under accession number PRJNA742853 (https://www.ncbi.nlm.nih.gov/sra/PRJNA742853).
Author Contributions
DL and NC: conceptualization. ML and NC: methodology. NC, JX, and RZ: formal analysis and investigation. JX, NC, and ZS: writing—original draft preparation. DL, YC, and SS: writing—review and editing. YC: funding acquisition. ML: resources. DL and SS: supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFD1000407).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank Prof. Yin Fang (Nanyang Technological University) for helpful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.730620/full#supplementary-material
References
Abu-Nada, Y., Kushalappa, A. C., Marshall, W. D., Al-Mughrabi, K., and Murphy, A. (2007). Temporal dynamics of pathogenesis-related metabolites and their plausible pathways of induction in potato leaves following inoculation with Phytophthora infestans. Eur. J. Plant Pathol. 118, 375–391. doi: 10.1007/s10658-007-9150-8
Almeida, A. M. R., Pineyro-Nelson, A., Yockteng, R. B., and Specht, C. D. (2018). Comparative analysis of whole flower transcriptomes in the Zingiberales. Peerj 6, e5490. doi: 10.7717/peerj.5490
Baig, A. (2018). Role of Arabidopsis LOR1 (LURP-one related one) in basal defense against Hyaloperonospora arabidopsidis. Physiol. Mol. Plant Pathol. 103, 71–77. doi: 10.1016/j.pmpp.2018.05.003
Birkenbihl, R. P., Diezel, C., and Somssich, I. E. (2012). Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol. 159, 266–285. doi: 10.1104/pp.111.192641
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cui, Q., Liu, Q., Gao, X., Yan, X., Jia, G.-X., and Willenborg, C. (2018a). Transcriptome-based identification of genes related to resistance against Botrytis elliptica in Lilium regale. Canad. J. Plant Sci. 98, 1058–1071. doi: 10.1139/cjps-2017-0254
Cui, Q., Yan, X., Gao, X., Zhang, D. M., He, H. B., and Jia, G. X. (2018b). Analysis of WRKY transcription factors and characterization of two Botrytis cinerea-responsive LrWRKY genes from Lilium regale. Plant Physiol. Biochem. 127, 525–536. doi: 10.1016/j.plaphy.2018.04.027
Dombrecht, B., Xue, G. P., Sprague, S. J., Kirkegaard, J. A., Ross, J. J., Reid, J. B., et al. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19, 2225–2245. doi: 10.1105/tpc.106.048017
Doppler, M., Kluger, B., Bueschl, C., Steiner, B., Buerstmayr, H., Lemmens, M., et al. (2019). Stable isotope-assisted plant metabolomics: investigation of phenylalanine-related metabolic response in wheat upon treatment with the fusarium virulence factor deoxynivalenol. Front. Plant Sci. 10, 18. doi: 10.3389/fpls.2019.01137
Edel, K. H., Marchadier, E., Brownlee, C., Kudla, J., and Hetherington, A. M. (2017). The evolution of calcium-based signalling in plants. Curr. Biol. 27, R667–R679. doi: 10.1016/j.cub.2017.05.020
Ederli, L., Dawe, A., Pasqualini, S., Quaglia, M., Xiong, L. M., and Gehring, C. (2015). Arabidopsis flower specific defense gene expression patterns affect resistance to pathogens. Front. Plant Sci. 6, 79. doi: 10.3389/fpls.2015.00079
Ederli, L., Salerno, G., and Quaglia, M. (2021). In the tripartite combination Botrytis cinerea-Arabidopsis-Eurydema oleracea, the fungal pathogen alters the plant-insect interaction via jasmonic acid signalling activation and inducible plant-emitted volatiles. J. Plant Res. 134, 523–533. doi: 10.1007/s10265-021-01273-9
Fraga, C. G., Clowers, B. H., Moore, R. J., and Zink, E. M. (2010). Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 82, 4165–4173. doi: 10.1021/ac1003568
Fu, Y., Zhu, Y., Yang, W., Xu, W., Li, Q., Chen, M., et al. (2020). Isolation and functional identification of a Botrytis cinerea-responsive caffeoyl-CoA O-methyltransferase gene from Lilium regale Wilson. Plant Physiol. Biochem. 157, 379–389. doi: 10.1016/j.plaphy.2020.10.030
Fu, Z. Q., and Dong, X. (2013). Systemic acquired resistance: turning local infection into global defense. Annu. Rev. Plant Biol. 64, 839–863. doi: 10.1146/annurev-arplant-042811-105606
Gao, X., Cui, Q., Cao, Q. Z., Liu, Q., He, H. B., Zhang, D. M., et al. (2017). Transcriptome-wide analysis of Botrytis elliptica responsive micrornas and their targets in Lilium regale Wilson by high-throughput sequencing and degradome analysis. Front. Plant Sci. 8, 753. doi: 10.3389/fpls.2017.00753
Gao, X., Cui, Q., Cao, Q. Z., Zhao, Y. Q., Liu, Q., He, H. B., et al. (2018). Evaluation of resistance to Botrytis elliptica in Lilium hybrid cultivars. Plant Physiol. Biochem. 123, 392–399. doi: 10.1016/j.plaphy.2017.12.025
Gao, X. Q., Chen, X., Lin, W. W., Chen, S. X., Lu, D. P., Niu, Y. J., et al. (2013). Bifurcation of Arabidopsis NLR immune signaling via Ca2+-dependent protein kinases. PLoS Pathog. 9, 14. doi: 10.1371/journal.ppat.1003127
Gonzalez, M., Brito, N., and Gonzalez, C. (2017). The Botrytis cinerea elicitor protein BcIEB1 interacts with the tobacco PR5-family protein osmotin and protects the fungus against its antifungal activity. New Phytol. 215, 397–410. doi: 10.1111/nph.14588
Gonzalez-Lamothe, R., Boyle, P., Dulude, A., Roy, V., Lezin-Doumbou, C., Kaur, G. S., et al. (2008). The transcriptional activator Pti4 is required for the recruitment of a repressosome nucleated by repressor SEBF at the potato PR-10a gene. Plant Cell 20, 3136–3147. doi: 10.1105/tpc.108.061721
Gu, Y. Q., Wildermuth, M. C., Chakravarthy, S., Loh, Y. T., Yang, C. M., He, X. H., et al. (2002). Tomato transcription factors Pti4, Pti5, and Pti6 activate defense responses when expressed in Arabidopsis. Plant Cell 14, 817–831. doi: 10.1105/tpc.000794
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Han, M., Ryu, H.-S., Kim, C.-Y., Park, D.-S., Ahn, Y.-K., and Jeon, J.-S. (2013). OsWRKY30 is a transcription activator that enhances rice resistance to the Xanthomonas oryzae pathovar oryzae. J. Plant Biol. 56, 258–265. doi: 10.1007/s12374-013-0160-0
He, J., Liu, Y., Yuan, D., Duan, M., Liu, Y., Shen, Z., et al. (2020). An R2R3 MYB transcription factor confers brown planthopper resistance by regulating the phenylalanine ammonia-lyase pathway in rice. Proc. Natl. Acad. Sci. U.S.A. 117, 271–277. doi: 10.1073/pnas.1902771116
Ho, K. V., Lei, Z. T., Sumner, L. W., Coggeshall, M. V., Hsieh, H. Y., Stewart, G. C., et al. (2018). Identifying antibacterial compounds in black walnuts (Juglans nigra) using a metabolomics approach. Metabolites 8, 18. doi: 10.3390/metabo8040058
Hong, G., Wang, J., Hochstetter, D., Gao, Y., Xu, P., and Wang, Y. (2015). Epigallocatechin-3-gallate functions as a physiological regulator by modulating the jasmonic acid pathway. Physiol. Plant. 153, 432–439. doi: 10.1111/ppl.12256
Hsiang, T., and Chastagner, G. A. (1991). Growth and virulence of fungicide-resistant isolates of 3 species of Botrytis. Canad. J. Plant Pathol. 13, 226–231. doi: 10.1080/07060669109500934
Hsieh, T. F., Huang, J. W., and Hsiang, T. (2001). Light and scanning electron microscopy studies on the infection of oriental lily leaves by Botrytis elliptica. Eur. J. Plant Pathol. 107, 571–581. doi: 10.1023/A:1017947328718
Huang, J. B., Hsieh, T. F., Chastagner, G. A., and Hsiang, T. (2001). Clonal and sexual propagation in Botrytis elliptica. Mycol. Res. 105, 833–842. doi: 10.1017/S0953756201004051
Hutabarat, O. S., Flachowsky, H., Regos, I., Miosic, S., Kaufmann, C., Faramarzi, S., et al. (2016). Transgenic apple plants overexpressing the chalcone 3-hydroxylase gene of Cosmos sulphureus show increased levels of 3-hydroxyphloridzin and reduced susceptibility to apple scab and fire blight. Planta 243, 1213–1224. doi: 10.1007/s00425-016-2475-9
Jang, J. Y., Subburaj, S., Lee, G. J., and Kim, H. S. (2018). In vitro screening for Botrytis leaf blight resistance in Lilium species. Sci. Hortic. 239, 133–140. doi: 10.1016/j.scienta.2018.05.009
Jiang, W., Wu, J., Zhang, Y., Yin, L., and Lu, J. (2015). Isolation of a WRKY30 gene from Muscadinia rotundifolia (Michx) and validation of its function under biotic and abiotic stresses. Protoplasma 252, 1361–1374. doi: 10.1007/s00709-015-0769-6
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kang, H. G., Foley, R. C., Onate-Sanchez, L., Lin, C. G. T., and Singh, K. B. (2003). Target genes for OBP3, a Dof transcription factor, include novel basic helix-loop-helix domain proteins inducible by salicylic acid. Plant J. 35, 362–372. doi: 10.1046/j.1365-313X.2003.01812.x
Kang, W. H., Kim, S., Lee, H. A., Choi, D., and Yeom, S. I. (2016). Genome-wide analysis of Dof transcription factors reveals functional characteristics during development and response to biotic stresses in pepper. Sci. Rep. 6, 12. doi: 10.1038/srep33332
Kloth, K. J., Wiegers, G. L., Busscher-Lange, J., Van Haarst, J. C., Kruijer, W., Bouwmeester, H. J., et al. (2016). AtWRKY22 promotes susceptibility to aphids and modulates salicylic acid and jasmonic acid signalling. J. Exp. Bot. 67, 3383–3396. doi: 10.1093/jxb/erw159
Knoth, C., and Eulgem, T. (2008). The oomycete response gene LURP1 is required for defense against Hyaloperonospora parasitica in Arabidopsis thaliana. Plant J. 55, 53–64. doi: 10.1111/j.1365-313X.2008.03486.x
Knoth, C., Salus, M. S., Girke, T., and Eulgem, T. (2009). The synthetic elicitor 3,5-dichloroanthranilic acid induces NPR1-dependent and NPR1-independent mechanisms of disease resistance in Arabidopsis. Plant Physiol. 150, 333–347. doi: 10.1104/pp.108.133678
Kumar, V., Hatan, E., Bar, E., Davidovich-Rikanati, R., Doron-Faigenboim, A., Spitzer-Rimon, B., et al. (2020). Phenylalanine increases chrysanthemum flower immunity against Botrytis cinerea attack. Plant J. 104, 226–240. doi: 10.1111/tpj.14919
Kumar, Y., Zhang, L. M., Panigrahi, P., Dholakia, B. B., Dewangan, V., Chavan, S. G., et al. (2016). Fusarium oxysporum mediates systems metabolic reprogramming of chickpea roots as revealed by a combination of proteomics and metabolomics. Plant Biotechnol. J. 14, 1589–1603. doi: 10.1111/pbi.12522
Kurt, F., Kurt, B., and Filiz, E. (2020). Wall associated kinases (WAKs) gene family in tomato (Solanum lycopersicum): insights into plant immunity. Gene Reports 21, 100828. doi: 10.1016/j.genrep.2020.100828
Lee, K. J., and Kim, K. (2015). The rice serine/threonine protein kinase OsPBL1 (ORYZA SATIVA ARABIDOPSIS PBS1-LIKE 1) is potentially involved in resistance to rice stripe disease. Plant Growth Regul. 77, 67–75. doi: 10.1007/s10725-015-0036-z
Li, B. B., Wang, R. L., Wang, S. Y., Zhang, J., and Chang, L. (2021). Diversified regulation of cytokinin levels and signaling during Botrytis cinerea infection in Arabidopsis. Front. Plant Sci. 12, 14. doi: 10.3389/fpls.2021.584042
Li, H. S. (2000). Principles and Techniques of Plant Physiological Biochemical Experiment. Beijing: Higher Education Press.
Li, J., Zhong, R., and Palva, E. T. (2017). WRKY70 and its homolog WRKY54 negatively modulate the cell wall-associated defenses to necrotrophic pathogens in Arabidopsis. PLoS ONE 12, 22. doi: 10.1371/journal.pone.0183731
Li, Z. W., Huang, J. Z., Wang, Z. Y., Meng, F., Zhang, S. Y., Wu, X. Q., et al. (2019). Overexpression of Arabidopsis nucleotide-binding and leucine-rich repeat genes RPS2 and RPM1(D505V) confers broad-spectrum disease resistance in rice. Front. Plant Sci. 10, 16. doi: 10.3389/fpls.2019.00417
Liao, K., Peng, Y.-J., Yuan, L.-B., Dai, Y.-S., Chen, Q.-F., Yu, L.-J., et al. (2020). Brassinosteroids antagonize jasmonate-activated plant defense responses through BRI1-EMS-SUPPRESSOR1 (BES1). Plant Physiol. 182, 1066–1082. doi: 10.1104/pp.19.01220
Lin, Z. J. D., Liebrand, T. W. H., Yadeta, K. A., and Coaker, G. (2015). PBL13 is a serine/threonine protein kinase that negatively regulates Arabidopsis immune responses. Plant Physiol. 169, 2950–2962. doi: 10.1104/pp.15.01391
Liu, S. A., Ziegler, J., Zeier, J., Birkenbihl, R. P., and Somssich, I. E. (2017). Botrytis cinerea B05.10 promotes disease development in Arabidopsis by suppressing WRKY33-mediated host immunity. Plant Cell Environ. 40, 2189–2206. doi: 10.1111/pce.13022
Liu, X., Cao, X., Shi, S., Zhao, N., Li, D., Fang, P., et al. (2018). Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 19, 62. doi: 10.1186/s12863-018-0668-x
Liu, Y., Xin, J., Liu, L., Song, A., Guan, Z., Fang, W., et al. (2020). A temporal gene expression map of Chrysanthemum leaves infected with Alternaria alternata reveals different stages of defense mechanisms. Hortic Res 7, 23. doi: 10.1038/s41438-020-0245-0
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lloyd, A. J., William Allwood, J., Winder, C. L., Dunn, W. B., Heald, J. K., Cristescu, S. M., et al. (2011). Metabolomic approaches reveal that cell wall modifications play a major role in ethylene-mediated resistance against Botrytis cinerea. Plant J. 67, 852–868. doi: 10.1111/j.1365-313X.2011.04639.x
Long, L., Liu, J., Gao, Y., Xu, F.-C., Zhao, J.-R., Li, B., et al. (2019). Flavonoid accumulation in spontaneous cotton mutant results in red coloration and enhanced disease resistance. Plant Physiol. Biochem. 143, 40–49. doi: 10.1016/j.plaphy.2019.08.021
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 38. doi: 10.1186/s13059-014-0550-8
Ma, S., Hu, Y., Liu, S., Sun, J., Irfan, M., Chen, L. J., et al. (2018). Isolation, identification and the biological characterization of Botrytis cinerea. Int. J. Agric. Biol. 20, 1033–1040. doi: 10.17957/IJAB/15.0600
Martinez, G., Regente, M., Jacobi, S., Del Rio, M., Pinedo, M., and De La Canal, L. (2017). Chlorogenic acid is a fungicide active against phytopathogenic fungi. Pestic. Biochem. Physiol. 140, 30–35. doi: 10.1016/j.pestbp.2017.05.012
Mazumdar, A. B., and Chattopadhyay, S. (2016). Sequencing, de novo assembly, functional annotation and analysis of Phyllanthus amarus leaf transcriptome using the illumina platform. Front Plant Sci 6, 1199. doi: 10.3389/fpls.2015.01199
Mizuno, H., Yazawa, T., Kasuga, S., Sawada, Y., Ogata, J., Ando, T., et al. (2014). Expression level of a flavonoid 3'-hydroxylase gene determines pathogen-induced color variation in sorghum. BMC Res. Notes 7, 761. doi: 10.1186/1756-0500-7-761
Nurmberg, P. L., Knox, K. A., Yun, B. W., Morris, P. C., Shafiei, R., Hudson, A., et al. (2007). The developmental selector AS1 is an evolutionarily conserved regulator of the plant immune response. Proc. Natl. Acad. Sci. U.S.A. 104, 18795–18800. doi: 10.1073/pnas.0705586104
Parker, D., Beckmann, M., Zubair, H., Enot, D. P., Caracuel-Rios, Z., Overy, D. P., et al. (2009). Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant J. 59, 723–737. doi: 10.1111/j.1365-313X.2009.03912.x
Peng, A. H., Chen, S. C., Lei, T. G., Xu, L. Z., He, Y. R., Wu, L., et al. (2017). Engineering canker-resistant plants through CRISPR/Cas9-targeted editing of the susceptibility gene CsLOB1 promoter in citrus. Plant Biotechnol. J. 15, 1509–1519. doi: 10.1111/pbi.12733
Peng, X., Hu, Y., Tang, X., Zhou, P., Deng, X., Wang, H., et al. (2012). Constitutive expression of rice WRKY30 gene increases the endogenous jasmonic acid accumulation, PR gene expression and resistance to fungal pathogens in rice. Planta 236, 1485–1498. doi: 10.1007/s00425-012-1698-7
R Core Team. (2018). MetaboAnalyst 5.0 - user-friendly, streamlined metabolomics data analysis. Available online at: https://www.metaboanalyst.ca/docs/RTutorial.xhtml
Raffaele, S., Rivas, S., and Roby, D. (2006). An essential role for salicylic acid in AtMYB30-mediated control of the hypersensitive cell death program in Arabidopsis. FEBS Lett. 580, 3498–3504. doi: 10.1016/j.febslet.2006.05.027
Ray, S. K., Macoy, D. M., Kim, W. Y., Lee, S. Y., and Kim, M. G. (2019). Role of RIN4 in regulating PAMP-triggered immunity and effector-triggered immunity: current status and future perspectives. Mol. Cells 42, 503–511. doi: 10.14348/molcells.2019.2433
Romero-Romero, J. L., Inostroza-Blancheteau, C., Orellana, D., Aquea, F., Reyes-Diaz, M., Gil, P. M., et al. (2018). Stomata regulation by tissue-specific expression of the Citrus sinensis MYB61 transcription factor improves water-use efficiency in Arabidopsis. Plant Physiol. Biochem. 130, 54–60. doi: 10.1016/j.plaphy.2018.06.034
Shetty, R., Frette, X., Jensen, B., Shetty, N. P., Jensen, J. D., Jorgensen, H. J. L., et al. (2011). Silicon-induced changes in antifungal phenolic acids, flavonoids, and key phenylpropanoid pathway genes during the interaction between miniature roses and the biotrophic pathogen Podosphaera pannosa. Plant Physiol. 157, 2194–2205. doi: 10.1104/pp.111.185215
Srideepthi, R., Krishna, M. S. R., Suneetha, P., Krishna, R. S., and Karthikeyan, S. (2020). Genome-wide identification, characterization and expression analysis of non-RD receptor like kinase gene family under Colletotrichum truncatumstress conditions in hot pepper. Genetica 148, 283–296. doi: 10.1007/s10709-020-00104-4
Sun, L., Di, D. W., Li, G. J., Li, Y. L., Kronzucker, H. J., and Shi, W. M. (2020). Transcriptome analysis of rice (Oryza sativa L.) in response to ammonium resupply reveals the involvement of phytohormone signaling and the transcription factor OsJAZ9 in reprogramming of nitrogen uptake and metabolism. J. Plant Physiol. 246, 11. doi: 10.1016/j.jplph.2020.153137
Thatcher, L. F., Cevik, V., Grant, M., Zhai, B., Jones, J. D., Manners, J. M., et al. (2016). Characterization of a JAZ7 activation-tagged Arabidopsis mutant with increased susceptibility to the fungal pathogen Fusarium oxysporum. J. Exp. Bot. 67, 2367–2386. doi: 10.1093/jxb/erw040
Tian, R., Yang, Y., and Wang, X. (2014). Research progress on BAK1 of a receptor kinase. Acta Botan. Bor. Occident. Sin. 34, 636–644. doi: 10.7606/j.issn.1000-4025.2014.03.0636
Unamba, C. I. N., Nag, A., and Sharma, R. K. (2015). Next generation sequencing technologies: the doorway to the unexplored genomics of non-model plants. Front. Plant Sci. 6, 1074. doi: 10.3389/fpls.2015.01074
Van Baarlen, P., Staats, M., and Van Kan, J. A. L. (2004). Induction of programmed cell death in lily by the fungal pathogen Botrytis elliptica. Mol. Plant Pathol. 5, 559–574. doi: 10.1111/j.1364-3703.2004.00253.x
Varet, H., Brillet-Gueguen, L., Coppee, J.-Y., and Dillies, M.-A. (2016). SARTools: a DESeq2-and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS ONE 11, e157022. doi: 10.1371/journal.pone.0157022
Xu, D., Deng, Y., Han, T., Jiang, L., Xi, P., Wang, Q., et al. (2018). In vitro and in vivo effectiveness of phenolic compounds for the control of postharvest gray mold of table grapes. Postharvest Biol. Technol. 139, 106–114. doi: 10.1016/j.postharvbio.2017.08.019
Xu, D., Deng, Y., Xi, P., Yu, G., Wang, Q., Zeng, Q., et al. (2019). Fulvic acid-induced disease resistance to Botrytis cinerea in table grapes may be mediated by regulating phenylpropanoid metabolism. Food Chem. 286, 226–233. doi: 10.1016/j.foodchem.2019.02.015
Yang, Q., Guo, J., Zeng, H., Xu, L., Xue, J., Xiao, S., et al. (2021). The receptor-like cytoplasmic kinase CDG1 negatively regulates Arabidopsis pattern-triggered immunity and is involved in AvrRpm1-induced RIN4 phosphorylation. Plant Cell. 33. doi: 10.1093/plcell/koab104
Yeh, Y. H., Chang, Y. H., Huang, P. Y., Huang, J. B., and Zimmerli, L. (2015). Enhanced Arabidopsis pattern-triggered immunity by overexpression of cysteine-rich receptor-like kinases. Front. Plant Sci. 6, 12. doi: 10.3389/fpls.2015.00322
Yu, Y. H., Bian, L., Wan, Y. T., Jiao, Z. L., Yu, K. K., Zhang, G. H., et al. (2019). Grape (Vitis vinifera) VvDOF3 functions as a transcription activator and enhances powdery mildew resistance. Plant Physiol. Biochem. 143, 183–189. doi: 10.1016/j.plaphy.2019.09.010
Zhang, B., and Horvath, S. (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17. doi: 10.2202/1544-6115.1128
Zhang, C. Y., Liu, H. C., Jia, C. G., Liu, Y. J., Wang, F. L., and Wang, J. Y. (2016). Cloning, characterization and functional analysis of a flavonol synthase from Vaccinium corymbosum. Trees Struct. Funct. 30, 1595–1605. doi: 10.1007/s00468-016-1393-6
Zhang, M. F., Jiang, L. M., Zhang, D. M., and Jia, G. X. (2015). De novo transcriptome characterization of Lilium ‘Sorbonne’ and key enzymes related to the flavonoid biosynthesis. Mol. Genet. Genomics 290, 399–412. doi: 10.1007/s00438-014-0919-0
Zhang, M. Y., Wang, D. J., Gao, X. X., Yue, Z. Y., and Zhou, H. L. (2020). Exogenous caffeic acid and epicatechin enhance resistance against Botrytis cinerea through activation of the phenylpropanoid pathway in apples. Sci. Hortic. 268, 6. doi: 10.1016/j.scienta.2020.109348
Zhao, G. D., Guo, D. Z., Wang, L. J., Li, H., Wang, C., and Guo, X. Q. (2021). Functions of RPM1-interacting protein 4 in plant immunity. Planta 253, 11. doi: 10.1007/s00425-020-03527-7
Keywords: Lilium, Botrytis elliptica, gray mold, phenylpropanoid pathway, flavonoid pathway, transcriptome, metabolic profiling
Citation: Chai N, Xu J, Zuo R, Sun Z, Cheng Y, Sui S, Li M and Liu D (2021) Metabolic and Transcriptomic Profiling of Lilium Leaves Infected With Botrytis elliptica Reveals Different Stages of Plant Defense Mechanisms. Front. Plant Sci. 12:730620. doi: 10.3389/fpls.2021.730620
Received: 25 June 2021; Accepted: 27 August 2021;
Published: 22 September 2021.
Edited by:
Victoria Pastor, University of Jaume I, SpainReviewed by:
Jiancai Li, Shanghai Institutes for Biological Sciences (CAS), ChinaMara Quaglia, University of Perugia, Italy
Copyright © 2021 Chai, Xu, Zuo, Sun, Cheng, Sui, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingyang Li, bGlteUBzd3UuZWR1LmNu; Daofeng Liu, bGl1MTk4MzAyMjJAMTYzLmNvbQ==
†These authors have contributed equally to this work