 Ling Zhang1,2,3,4
Ling Zhang1,2,3,4 Shu Wang1,2
Shu Wang1,2 Chun Su1,2
Chun Su1,2 AJ Harris5
AJ Harris5 Liang Zhao1,2Na Su1,2Jun-Ru Wang1,2
Liang Zhao1,2Na Su1,2Jun-Ru Wang1,2 Lei Duan5*Zhao-Yang Chang1,2*
Lei Duan5*Zhao-Yang Chang1,2*- 1College of Life Science, Northwest A&F University, Yangling, China
- 2Herbarium of Northwest A&F University, Yangling, China
- 3College of Life Science, Tarim University, Alar, China
- 4Key Laboratory of Biological Resource Protection and Utilization of Tarim Basin Xinjiang Production & Construction Group, Alar, China
- 5Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
The genus Zygophyllum comprises over 150 species within the plant family Zygophyllaceae. These species predominantly grow in arid and semiarid areas, and about 20 occur in northwestern China. In this study, we sampled 24 individuals of Zygophyllum representing 15 species and sequenced their complete chloroplast (cp) genomes. For comparison, we also sequenced cp genomes of two species of Peganum from China representing the closely allied family, Nitrariaceae. The 24 cp genomes of Zygophyllum were smaller and ranged in size from 104,221 to 106,286 bp, each containing a large single-copy (LSC) region (79,245–80,439 bp), a small single-copy (SSC) region (16,285–17,146 bp), and a pair of inverted repeat (IR) regions (3,792–4,466 bp). These cp genomes contained 111–112 genes each, including 74–75 protein-coding genes (PCGs), four ribosomal RNA genes, and 33 transfer RNA genes, and all cp genomes showed similar gene order, content, and structure. The cp genomes of Zygophyllum appeared to lose some genes such as ndh genes and rRNA genes, of which four rRNA genes were in the SSC region, not in the IR regions. However, the SC and IR regions had greater similarity within Zygophyllum than between the genus and Peganum. We detected nine highly variable intergenic spacers: matK-trnQ, psaC-rps15, psbZ-trnG, rps7-trnL, rps15-trnN, trnE-trnT, trnL-rpl32, trnQ-psbK, and trnS-trnG. Additionally, we identified 156 simple sequence repeat (cpSSR) markers shared among the genomes of the 24 Zygophyllum samples and seven cpSSRs that were unique to the species of Zygophyllum. These markers may be useful in future studies on genetic diversity and relationships of Zygophyllum and closely related taxa. Using the sequenced cp genomes, we reconstructed a phylogeny that strongly supported the division of Chinese Zygophyllum into herbaceous and shrubby clades. We utilized our phylogenetic results along with prior morphological studies to address several remaining taxonomic questions within Zygophyllum. Specifically, we found that Zygophyllum kaschgaricum is included within Zygophyllum xanthoxylon supporting the present treatment of the former genus Sarcozygium as a subgenus within Zygophyllum. Our results provide a foundation for future research on the genetic resources of Zygophyllum.
Introduction
Zygophyllum was originally described by Linnaeus (1753) and is one of the 25 genera comprising the plant family Zygophyllaceae according to the Angiosperm Phylogeny Group (APG) IV (2016). This genus consists of approximately 150 species (Beier et al., 2003), which are widely distributed in northern and southern Africa, the Mediterranean region, and central Asia and Australia (White, 1983; Zyl and Marias, 1999; Retief, 2000). Among the Asian species, ca. 20 occur in northwestern China, where all species of Zygophyllum grow preferentially on sandy gravel terraces, hilly slopes, and gravel dunes and pediments, and where Zygophyllum xanthoxylon (Bunge) Maxim. is a dominant species in the desert and semidesert areas (Zeng et al., 2004). Species of Zygophyllum are highly adapted to dry conditions and provide critical ecosystem services within arid environments of Xinjiang, Inner Mongolia, and Gansu in China.
Within the family Zygophyllaceae, generic circumscriptions were first established by Sheahan and Chase (1996) by using the molecular phylogenetic analysis of the chloroplast (cp) marker, rbcL, and Sheahan and Chase (2000) later delimited the subfamily Zygophylloideae based on trnL-F and rbcL. A comprehensive phylogenetic analysis of trnL and morphological data for 43 species of Zygophylloideae showed that the genera, Augea (monotypic, southern Africa), Tetraena (monotypic, China), and Fagonia (ca. 30 species), are nested within Zygophyllum (ca. 150 species) (Beier et al., 2003). When Zygophyllum includes these genera, its monophyly is highly supported (Beier et al., 2003). In later studies, molecular phylogenies were used for divergence time dating and biogeographic analyses revealing that Asian Zygophyllum colonized the Asian interior from Africa in the early Oligocene, began diversification in the early Miocene, and underwent rapid radiation in the late Miocene (Wu et al., 2015, 2018).
Similar to the family as a complete, Zygophyllum of China has undergone considerable taxonomic change, especially from ca. 1980 to 2011. During this time, several new species were described and added to Zygophyllum, although some of these have not been widely accepted as truly distinctive (Liou, 1980, 1998; Zhao and Zhu, 2003; Liou and Zhou, 2008; Yang, 2011). Simultaneously, Sarcozygium Bunge, which contains one or two species and occurs in Xinjiang as well as Kazakhstan and Mongolia, was accepted as an independent genus (Liou, 1998; Yang, 2011), but in the Flora of China, it was merged to Zygophyllum (Liou and Zhou, 2008). Within Zygophyllum, species are typically delimited morphologically based especially on differences in the number of leaflets per leaf (1, 2–5 pairs), relative length of the stamens compared to the petals (equal, longer, or shorter), and features of the capsule (winged or not, ridged or not, and dehiscent or indehiscent) (Beier et al., 2003; Liou and Zhou, 2008). For example, Zygophyllum kaschgaricum Boriss. has a capsule that is linearly ovoid, spindle-like, or obovoid, and smaller than that of Z. xanthoxylon, whose capsule is nearly spherical. Similarly, Zygophyllum latifolium Schrenk has leaflets that are rotund to oblong, but the leaflets of Zygophyllum rosowii Bunge are ovate. However, these morphological characters alone are often insufficient to distinguish Chinese species due to more or less continuous variation. Thus, for Chinese species, a clear set of morphological characters to support the identification of plants in the field is needed. Moreover, species boundaries remain poorly resolved for the Chinese members of the genus, in part because they have been sparsely sampled in prior molecular phylogenetic studies and due to the lack of existing genetic materials.
Among Chinese Zygophyllum, complete cp genomes have been published for Z. xanthoxylon and Zygophyllum fabago L. (Xu et al., 2020), but additional cp genomes are needed to represent the genus in China and to support resolving species boundaries and phylogenetic relationships. The cp genomes have been shown to be a valuable tool for resolving species relationships and boundaries within a molecular phylogenetic framework (Mao et al., 2014; Shaw et al., 2014; Hollingsworth et al., 2016; Fu et al., 2017; Duan et al., 2020) and are also regarded as an important source for simple sequence repeat (cpSSR) markers that can be utilized in the studies of barcoding and population genetics (Powell et al., 1996; Wu et al., 2010; Perdereau et al., 2014; Park et al., 2016; Mohammad-Panah et al., 2017). The cp genome in most angiosperms is relatively small in size, maternally inherited, and highly conserved with low nucleotide substitution rates except within the extremely mutable intergenic spacer regions (Birky et al., 1983; Bendich, 2004; Moore et al., 2007; Liu et al., 2019). Thus, cp genomes are relatively easy to sequence due to their small size, and the phylogenetic analysis and interpretation are fairly straightforward due to the ease of alignment among conserved regions and lack of genetic anomalies associated with biparental inheritance, respectively. Moreover, obtaining the sequences of complete cp genomes is increasingly fast and cheap with the continual development of new sequencing technology. As a consequence, more and more complete cp genomes have been generated to resolve taxonomic and phylogenetic controversies.
Increasingly, the comparative analyses of the complete cp genome structures among closely related species have also yielded a wealth of information for taxonomically or phylogenetically recalcitrant lineages (Hu et al., 2017), such as Orychophragmus (Hu et al., 2015, 2016), Epimedium (Zhang et al., 2016), and Orchids (Niu et al., 2017). Broadly, the structures of cp genomes in angiosperms are highly conserved; they are circular double-helices consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of inverted repeat (IR) regions and are the sizes of cp genomes range from 100 to 220 kb in length (Ozeki et al., 1989; Marcelo et al., 2015). However, IRs expand and contract (Goulding et al., 1996; Downie and Jansen, 2015; He et al., 2017; Ye et al., 2018), yielding informative differences in plastome size and/or gene order. Additionally, cp gene gain and loss and pseudogenization are widespread within plastomes of angiosperms (Jansen et al., 2008; Delannoy et al., 2011; Quan and Zhou, 2011). Therefore, the structural analysis of plastomes is an additional, valuable tool to aid in species delimitation.
In this study, we sequenced 24 complete cp genomes of Chinese species of Zygophyllum representing 15 species to elucidate species boundaries and phylogenetic relationships. For additional comparison, we also included cp genomes of two species of Peganum, which were previously separated from Zygophyllaceae and are presently treated within Nitrariaceae (Angiosperm Phylogeny Group (APG) IV, 2016). The main objectives of this study were to (1) compare the sequences, structures, and gene organization of cp genomes within Chinese Zygophyllum with additional comparisons to representative Peganum; (2) detect highly variable regions and cpSSRs for the future development of genetic markers for Zygophyllum; and (3) integrate molecular phylogenetic evidence from the cp genomes with morphology for the delimitation of species of Zygophyllum in China.
Materials and Methods
Sample Preparation and DNA Sequencing
We collected fresh, healthy leaves from 24 individuals representing 15 species of Zygophyllum comprising six samples of Z. rosowii, two of Z. kaschgaricum, three each of Z. xanthoxylon and Z. fabago, and one each of the remaining 10 species. We collected all fresh samples in the field and stored them in silica sand. The density of our sampling within species reflected the levels of continuous variation that we observed in the field and ongoing taxonomic disputes. Specifically, for Z. rosowii and Z. fabago, we observed considerable intraspecific variation but relative uniformity across species, while Z. kaschgaricum and Z. xanthoxylon have poorly resolved boundaries. We selected and sequenced Peganum harmala L. and Peganum nigellastrum Bunge to represent the outgroup. Peganum belongs to Nitrariaceae now but was formerly treated within Zygophyllaceae (Liou, 1998). Following collections in the field, we stored the silica-dried leaves in a 4°C refrigerator until processing. In the field, we also collected voucher specimens, which were deposited in the herbarium of the Northwest A&F University (WUK) (Table 1).
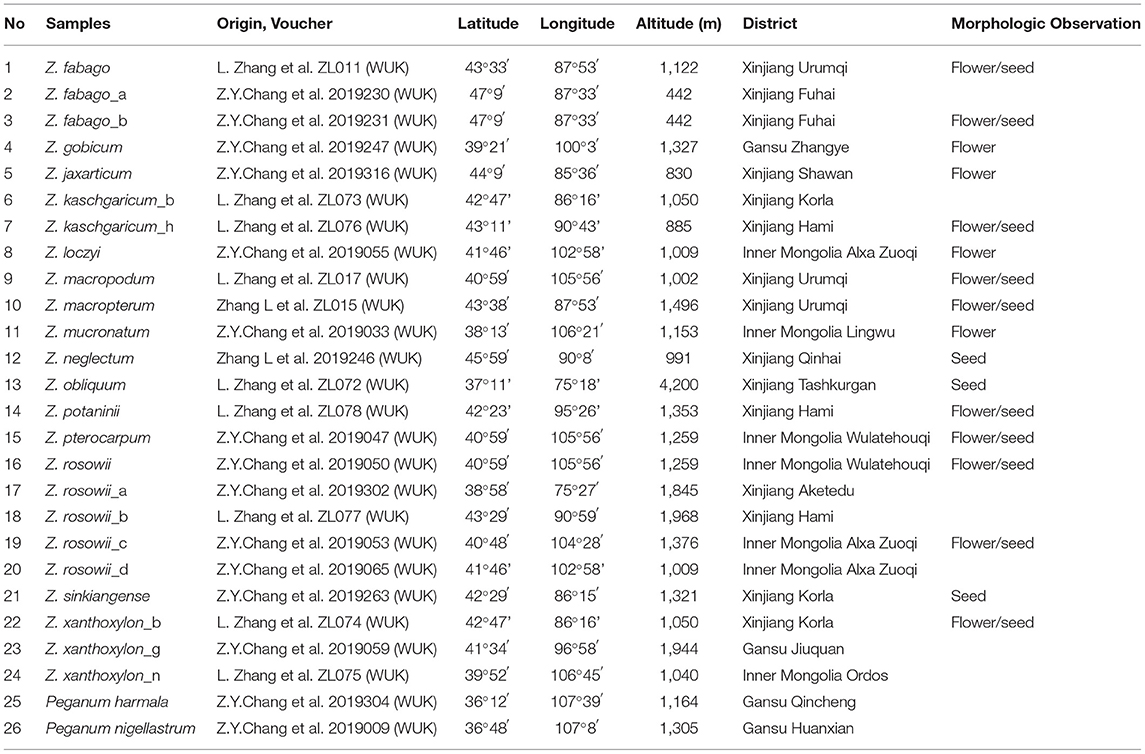
Table 1. Details of sampled accessions used in this study.
Using the silica-dried samples, we performed extractions of complete genomic DNA via a modified cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987) and assessed the quality and concentration of the extracted DNA with agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). The high-quality DNAs served as material to construct a library for sequencing on an Illumina NovaSeq 6000 system following the instructions of the manufacturer. The libraries consisted of fragments with an average of 270 bp and sequencing comprised 150 bp paired-end reads.
Genome Assembly, Annotation, and Sequence Analyses
Sequencing yielded a total of 205,535,340 bp of raw reads, which we cleaned and subsequently assembled in NOVOPlasty 2.7.2 (Dierckxsens et al., 2017) using the rbcL gene sequence of Z. rosowii (JF944812) as the seed and the complete cp genome sequence of Tetraena mongolica Maxim. (MH325021) as the reference based on the nested position of Tetraena (monotypic) within Zygophyllum (Beier et al., 2003) and the availability of the sequence. We annotated all protein-coding genes (PCGs), transfer RNAs (tRNAs), and ribosomal RNAs (rRNAs) of the cp genomes of Zygophyllum and Peganum using GeSeq (Tillich et al., 2017) on the Chlorobox web server (https://chlorobox.mpimp-golm.mpg.de/index.html) and PGA (Qu et al., 2019). We compared annotations from the two sources and made final adjustments manually in Geneious version 11.0.2 (Zufall, 2009). We detected the boundaries of the IR regions using Repeat Finder (Benson, 1999) implemented in Geneious 11.0.2, and we visualized the cp genomic maps with OGDRAW (Lohse et al., 2013) via Chlorobox under default settings with manual quality control. Using MAFFT version 7 (Katoh and Standley, 2013), we aligned the complete cp genomes of Zygophyllum and Peganum and then performed manual editing in Geneious version 11.0.2. Finally, we submitted all final cp genome sequences generated in this study to the NCBI database with the accession number (Table 1).
Comparison of cp Genomes and Identification of Polymorphic Regions
Based on our annotations, we compared the IR/SC boundary regions of the 15 species of Zygophyllum to each other, to other representatives of Zygophyllaceae [Teteaena mongolica Maxim. (subfamily Zygophylloideae), Guaiacum angustifolium Engelm. (subfamily Larreoideae), Tribulus terrestris L. (subfamily Balanitoideae)], and to P. harmala. Moreover, we more deeply compared the cp genomes among the 15 species of Zygophyllum using mVISTA in LAGAN mode (Frazer et al., 2004) with our newly annotated sequence of Z. fabago as the reference.
For determining the levels of polymorphism of sites within the cp genomes, we conducted a sliding window analysis of the 24 aligned plastomes of Zygophyllum in DnaSP version 5.10.01 (Librado and Rozas, 2009), and we determined nucleotide variability (Pi) using a step size of 200 bp and a window length of 600 bp. We performed the sliding window analysis for the complete cp genomes and for the LSC, SSC, and IR regions individually. We also detected and analyzed the distributions of cpSSRs using MISA (Beier et al., 2017) with the minimum number of repeats set to eight for mononucleotide repeats, six for dinucleotides, five for trinucleotides, four for tetranucleotides, three for pentanucletides, and three for hexanucletides. We defined SSRs as comprising one to six repetitive DNA bases, and we verified all repeats manually.
Phylogenetic Analyses
We reconstructed a phylogeny using all 24 complete cp genomes of Zygophyllum and two Peganum complete cp genomes sequenced in this study as well as four additional publicly available sequences representing the family that we downloaded from the NCBI (Supplementary Table 1). The additional representatives comprised Teteaena mongolica (MH325021), Larrea tridentata (DC.) Coville (KT272174.1), G. angustifolium (MK726011.1), and Teteaena terrestris (NC_046758). For phylogenetic analyses, we generated six datasets in Geneious version 11.0.2 (Zufall, 2009) consisting of the complete cp genomes and extracted sequences representing all coding sequences (CDSs); the LSC, the SSC, and the IR regions; and non-coding sequences (NCSs) and performed new alignments using MAFFT version 7 under default parameters with subsequent manual adjustments (Katoh and Standley, 2013).
Our phylogenetic analyses consisted of maximum likelihood (ML) and Bayesian inference (BI). ML analyses were implemented using RAxML-HPC BlackBox version 8.1.24 (Stamatakis, 2014) on the CIPRES Science Gateway (http://www.phylo.org/) (Miller et al., 2010) under the GTRGMMA+I model with 1,000 bootstrap replicates to determine branch support. For BI, we determined that GTR+I+G was the best fitting model of evolution for the complete cp genome, CDS, NCS, and LSC datasets based on jModelTest version 2.1.7 (Darriba et al., 2012). Trees based on the SSC and IRs were poorly based on the ML analyses (as described in the “Results” section), such that we did not perform BI. We performed BI using two independent runs in MrBayes version 3.2 (Ronquist et al., 2012) consisting of 10,000,000 generations each with sampling every 1,000 generations and the first 25% of trees discarded as burn-in. From the remaining trees, we determined posterior probability (PP) values after using Tracer version 1.7 (Rambaut et al., 2018) to verify the stationarity (ESS > 200) and convergence. We visualized the final ML and BI trees in Figtree version 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Microscopy Observation of Flower and Seed
For morphological and microscopic analysis, we sampled mature fruits and/or flowers in cases where they were available from the same specimens used for DNA extraction (Table 1). We fixed the flower samples in FAA solution (formalin/acetic acid/70% ethanol = 10:5:85) after collection and stored the dry fruits in paper bags at 4°C. We performed dissections of the flowers using tweezers and dissecting needles and observed them under a Nikon SMZ25 stereomicroscope. We also took photographs of flowers that were still attached to branches using a Nikon D5100 digital camera. For the fruits, we peeled away the pericarp and washed the seeds two times in an ultrasonic washer (2 min/time). Thereafter, we took photographs of the seeds when dry and wet. Using the available specimens, we were able to sample 10 flowers and 50 seeds for each unique species of Zygophyllum represented in this study.
Results
General Features of cp Genomes Sampled in This Study
Each of the complete cp genomes from 24 samples of Zygophyllum and two samples of Peganum yielded ca. 2G of the clean sequence data. Within Zygophyllum, the complete cp genomes ranged in size from 104,211 bp for Zygophyllum potaninii Maxim. to 106,286 bp for Zygophyllum sinkiangense Y. X. Liou, and the GC content had a narrow range from 33.7 to 34.1% (Table 2). All cp genomes in Zygophyllum included the LSC region of 79,245 to 80,439 bp, the SSC region of 16,285 to 17,146 bp, and the IR regions of 3,792 to 4,466 bp. The complete cp genome sequences of Peganum were 159,956 and 160,066 bp. All cp genomes sequenced in this study were deposited in GenBank (MW307829–MW771517) (Table 2).
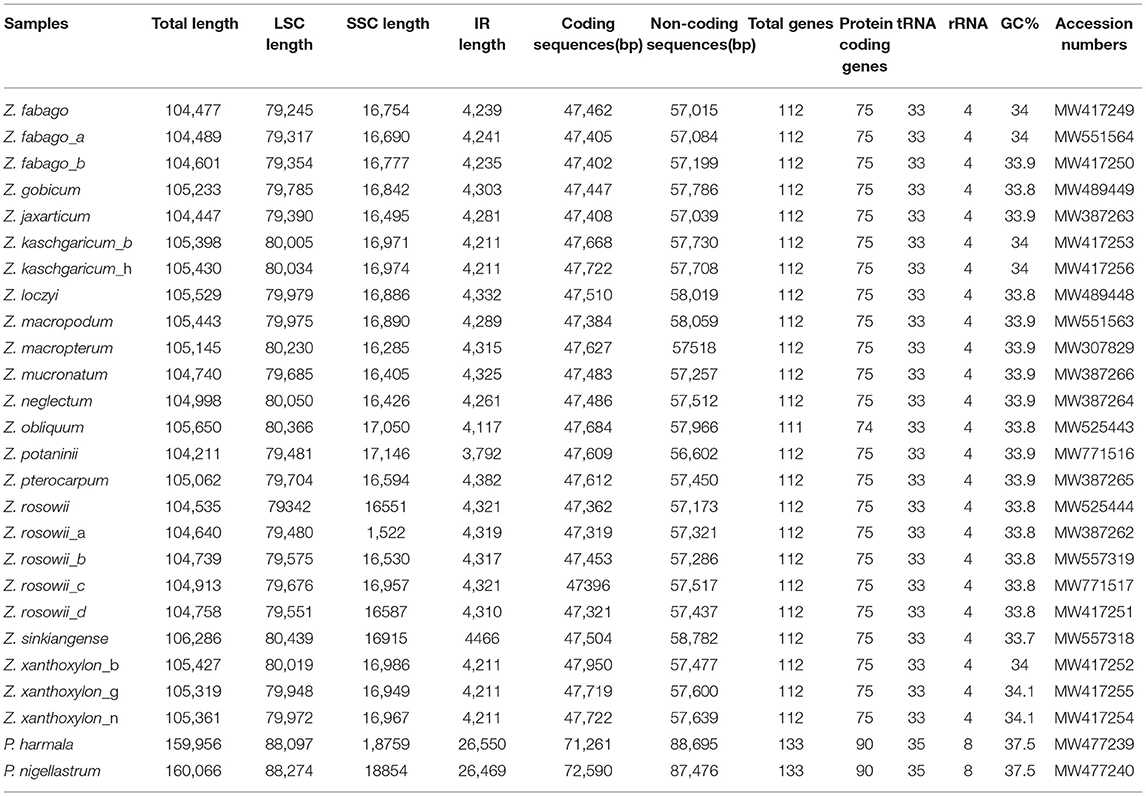
Table 2. Summary statistics for complete chloroplast genomes of the sampled accessions of Zygophyllum and Peganum.
We successfully, fully annotated all 26 cp genomes newly sequenced in this study using PGA. We found that the cp genomes of Zygophyllum contained a total of 111–112 genes, among which there were 33 tRNA genes, four rRNA genes, and 75 PCGs, except in Zygophyllum obliquum Popov., which lacked ndhH and, thus, had 74 PCGs (Table 2). In contrast, the cp genomes of the two species of Peganum contained 138 coding genes comprising 35 tRNA genes, eight rRNA genes, and 95 PCGs. The cp genomes of Zygophyllum possessed four categories of genes based on KOG (Tatusov et al., 2003): genes related to photosynthesis-, transcription- and translation-related genes, RNA genes, and other functionally unknown genes.
The Zygophyllum cp genome harbored 104 unique genes, consisting of 67 PCGs, 33 tRNA genes, and four rRNA genes (Table 3). The SSC region contained 10 PCGs (ccsA, ndhD, ndhH, ndhI, psaC, rpl32, rps7, rps12, rps15, and ycf68), six tRNAs (trnA-UGC, trnI-GAU, trnL-UAG, trnN-GUU, trnR-ACG, and trnV-GAC), and four rRNAs (rrn4.5, rrn5, rrn16, and rrn23), while the LSC region contained 58 PCGs and 21 tRNAs. The IR regions included four PCGs (rpl2, rpl23, rps19, and ycf15) and three tRNAs (trnH-GUG, trnI-CAU, and trnL-CAA). Within the SC regions, there were only three NADH dehydrogenase genes (ndhI, ndhD, and ndhH), and all were pseudogenized, while the IR regions did not contain any NADH dehydrogenase genes or any rRNAs. In the P. harmala cp genome, which is representative of both sequenced genomes of Peganum, we observed 133 genes consisting of 90 PCGs, 35 tRNA genes, and eight rRNA genes. The IR region in the P. harmala cp genome included 20 genes consisting of eight PCGs, six tRNAs, and four rRNA genes, and IR of T. terrestris and G. angustifolium contained 18 genes, while T. mongolica had seven genes. Thus, T. mongolica was more similar to Zygophyllum with respect to the gene content of the IR regions. Within the cp genomes of Zygophyllum, we identified 16 genes that harbored introns comprising eight PCGs and six tRNA genes (Table 3). Of the intron-containing genes, 14 possessed only 1 intron, while clpP and ycf3 possessed 2 introns in both Zygophyllum and Peganum.
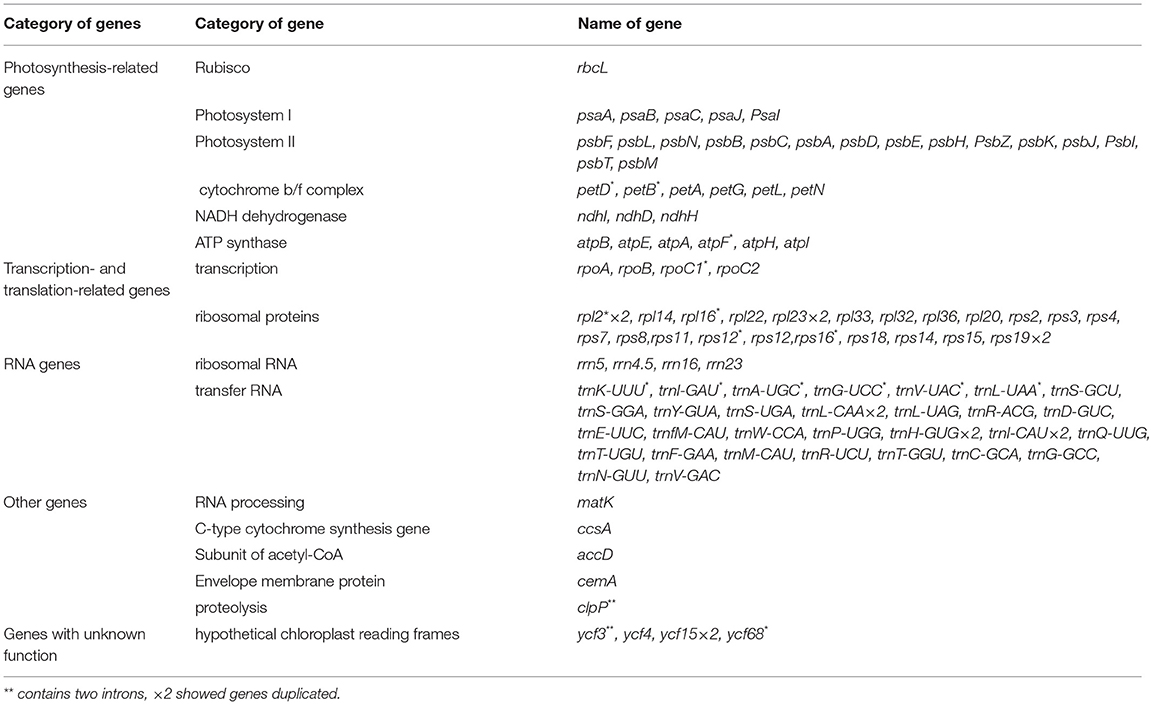
Table 3. Functional annotations of cp genes Zygophyllum based on KOG annotation. The ndhH gene is absent in Z. obliquum.
IR Boundary Analyses
We compared the IRs within Zygophyllum and to representative samples of three genera of Zygophyllaceae: Tribulus, Larrea, and Tetraena. We also summarized IRs of Zygophyllaceae to P. harmala of Nitrariaceae. The sizes of the IRs and the boundary locations were very similar within Zygophyllum and different among genera and families (Figure 1). Among Zygophyllum species, the boundary between the IR and LSC regions was placed in the intergenic region rpl22-trnH, with rpl22 being 86–137 bp and trnH being 77 bp away from the IRb/LSC boundary. The IRb/SSC boundary in Zygophyllum occurred between trnL-CAA and rpl32 with 14–588 bp from trnL-CAA to the IRb/SSC border and 39–256 bp from rpl32 to the IRb/SSC border (Figure 1). The IR/SC boundary positions in P. harmala, Teteaena terrestris, and G. angustifolium were more similar but differed greatly from Tetraena and Zygophyllum in Zygophylloideae.

Figure 1. Comparison of large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) borders among plastomes of Zygophyllaceae.
Comparative cp Genomic Analysis
We conducted comparisons of 15 cp genome sequences as representatives of Chinese Zygophyllum in mVISTA using our annotated genome of Z. fabago (Figure 2) as a reference to understand the distributions of conserved and divergent sequences among species (Figure 3). The comparative analysis revealed that NCSs were much more variable than CDSs. There was also considerable divergence among intergenic gene sequences (IGSs), such as psbZ-trnG, trnK-rps16, and trnS-trnG (Figure 3).
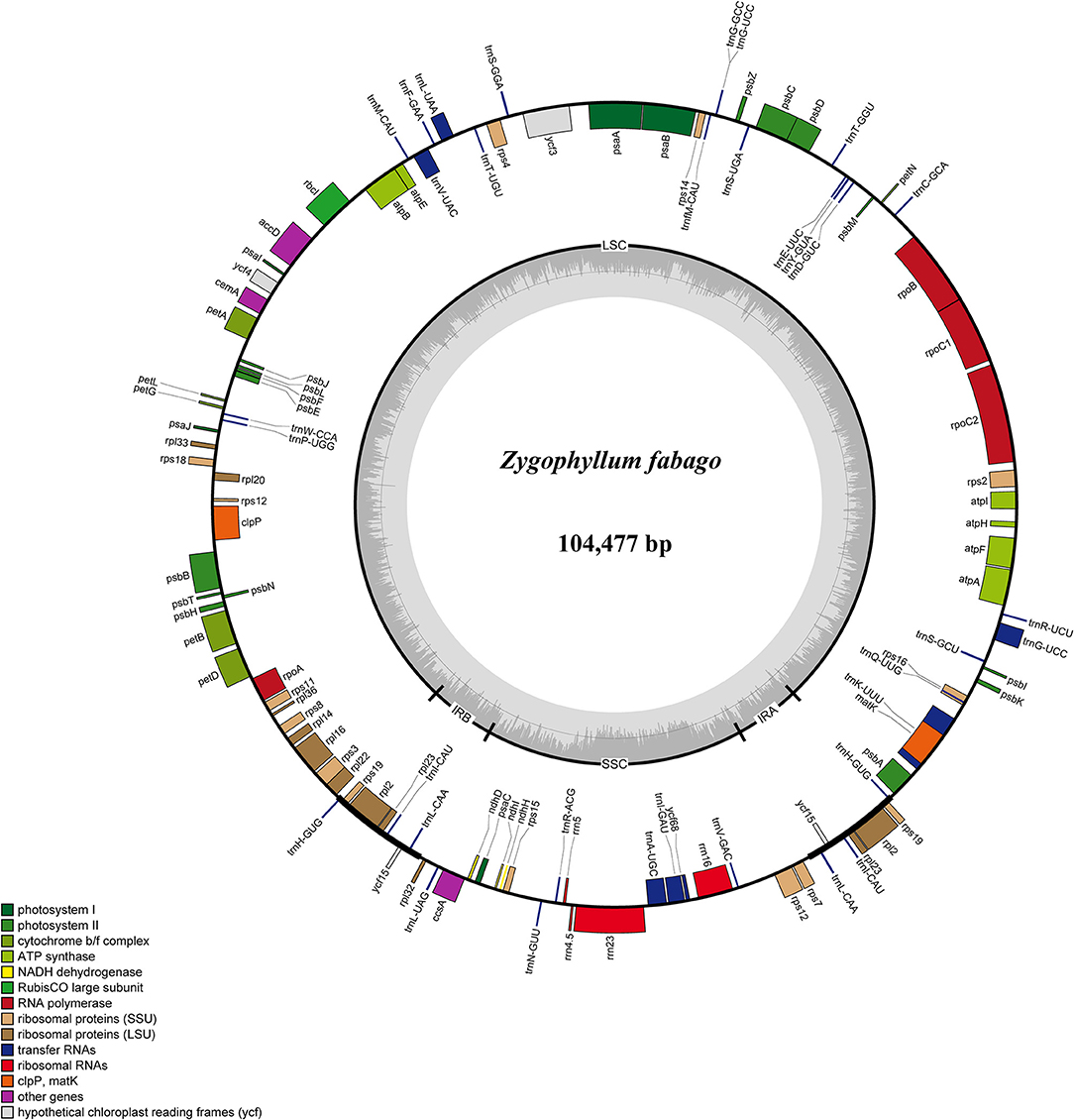
Figure 2. Gene map of the Zygophyllum fabago chloroplast (cp) genome. The genes inside the circle are transcribed clockwise, while those outside the circle are transcribed counterclockwise.
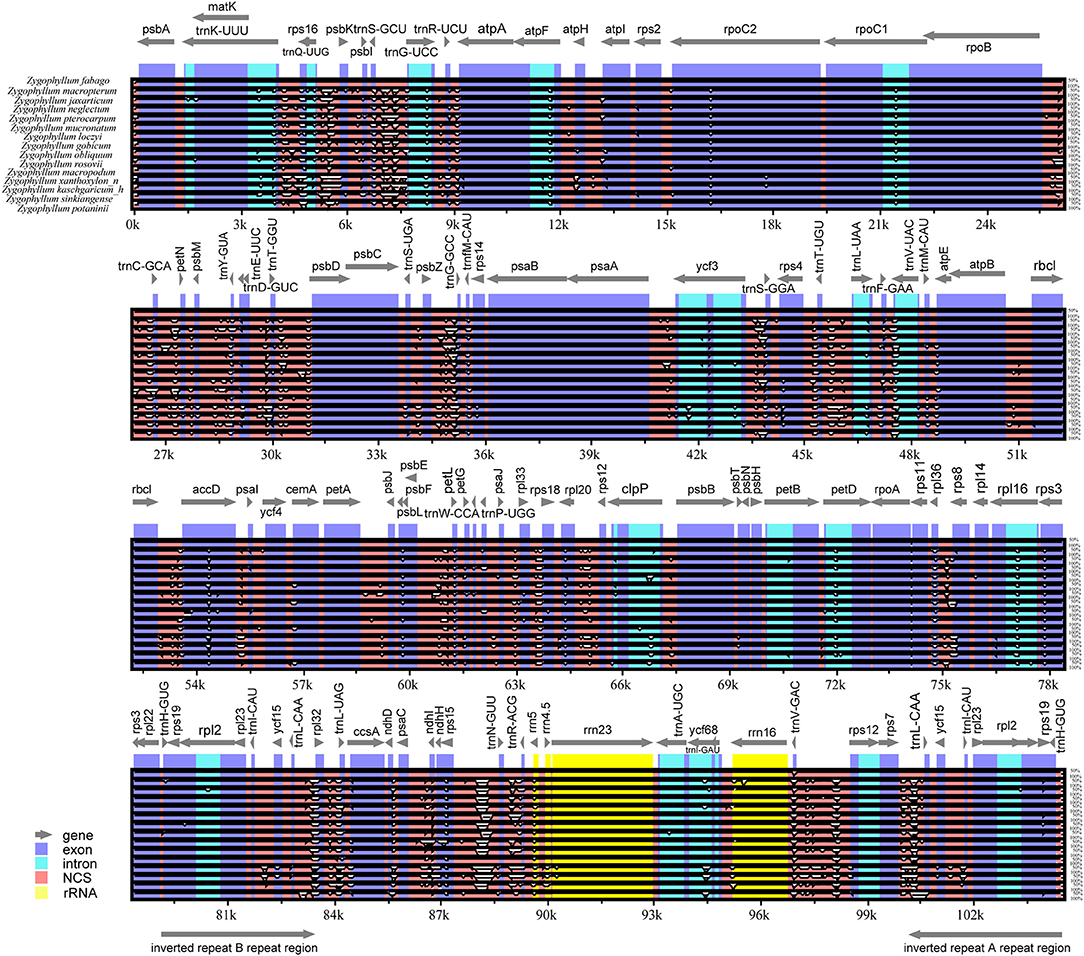
Figure 3. Comparison of plastomes of 15 species of Zygophyllum using Z. fabago as reference. Gray arrows and thick black lines above the alignment indicate gene orientation. Exons and introns are shown in blue (dark and light, respectively). Other intergenic spacers are shown in red. Ribosomal RNA genes are indicated in yellow. The y-axis indicates the percentage of identity, ranging from 50% to 100%.
Furthermore, we examined the numbers of variable sites and nucleotide variability (Pi) across the cp genomes and among regions using DnaSP version 5.10.01 (Table 4). The results showed that the IRs were less divergent than those of the LSC and SSC regions. The number of variable sites was 5,331 across the cp genomes, 3,840 in the LSC region, 1,307 in the SSC region, and 130 in the IR regions (Table 4). The IR regions showed the lowest nucleotide diversity (Pi = 0.00948), while the SSC region had the highest (Pi = 0.01692).

Table 4. Analyses of variable sites in cp genomes of Zygophyllum.
Highly Variable Regions and cpSSRs
To detect highly variable regions, or hypervariable regions, among the 24 complete cp genomes of Zygophyllum, we conducted a sliding window analysis using DnaSP version 5.10.01. The sliding window analysis revealed nine highly variable regions with Pi ranging from 0.00063 to 0.10663 across 24 complete cp genomes (Figure 4). The highly variable regions comprised the intergenic spacer regions: matK-trnQ, psaC-rps15, psbZ-trnG, rps7-trnL, rps15-trnN, trnE-trnT, trnL-rpl32, trnQ-psbK, and trnS-trnG. Among the nine highly variable regions, five regions (matK-trnQ, psbZ-trnG, trnE-trnT, trnQ-psbK, and trnS-trnG) were located in the LSC, and four regions (psaC-rps15, rps7-trnL, rps15-trnN, and trnL-rpl32) were in the SSC. None of the hypervariable regions were in the IRs, which had more conserved sequences.
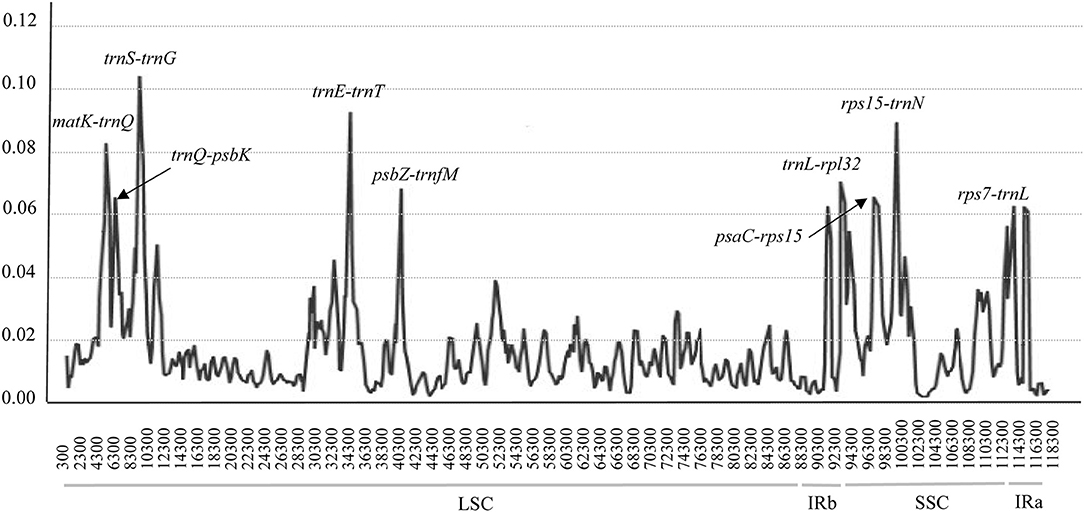
Figure 4. Hypervariable regions within cp genomes of species of Zygophyllum using a sliding window analysis. Window lengths: 600 bp; step size: 200 bp. Nine regions with the highest Pi values were marked out. LSC: large single-copy region, IR: inverted repeat region, SSC: small single-copy region. x-axis: position of the midpoint of a window; y-axis: nucleotide diversity of each window.
The nine hypervariable regions ranged from 917 bp (trnL-rpl32) to 2004 bp (matK-trnQ) in length (Table 5). There were 965 variable sites in the nine highly variable regions, including 545 parsimony informative sites. The Pi values of nine hypervariable variable sites ranged from 0.03495 to 0.08242 and were highest in trnL-rpl32 and trnS-trnG (Table 5).

Table 5. Nine hypervariable regions of cp genomes of Zygophyllum.
We detected and analyzed the occurrence, type, and distributions of SSRs of the 24 Zygophyllum complete cp genomes. In total, we found 156 cpSSRs, ranging from 86 cpSSRs in Z. xanthoxylon_b, Z. xanthoxylon_g, and Z. fabago to 124 cpSSRs in Zygophyllum macropterum (Figure 5, Supplementary Table 2). Among these cpSSR repeats, mononucleotide repeats (58–75%) and dinucleotide repeats (10–20%) were the most common, followed by tetranucleotide repeats (8–13%). Based on this, the mononucleotide repeats may be the most suitable targets for future studies of genetic diversity or species delimitation in the Zygophyllum. The proportions of trinucleotide, pentanucleotide, and hexanucleotide repeats were relatively low for each sample (Supplementary Table 2). Among mononucleotide repeats, polyA (20–38% of all SSRs) and polyT (35–48% of all SSRs) repeats were the most common, while polyC and polyG repeats were less frequent (Supplementary Table 2). For 156 cpSSRs, seven were shared across all 24 accessions of Zygophyllum sampled in this study (Supplementary Table 2).
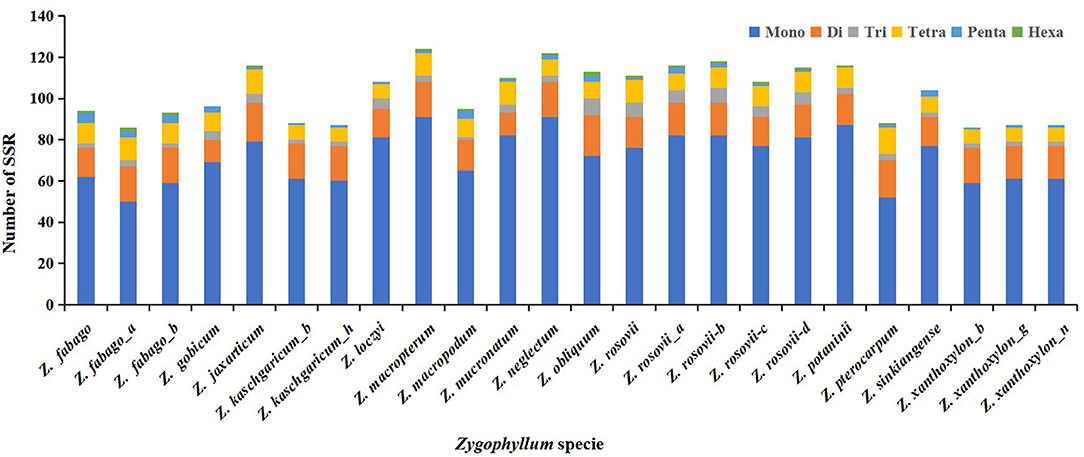
Figure 5. Number of simple sequence repeats (SSRs) from 24 Zygophyllum samples.
We found that SSRs were non-randomly distributed in the cp genomes of Zygophyllum. Of all cpSSRs, 74–84% were located within the LSC region, while only 8–18% and 5–14% were located within the SSC and IR regions, respectively (Figure 6, Supplementary Table 3). Similarly, most SSRs also occurred within the NCS regions (92–97%) compared to the CDS regions (3–8%) (Supplementary Table 3).
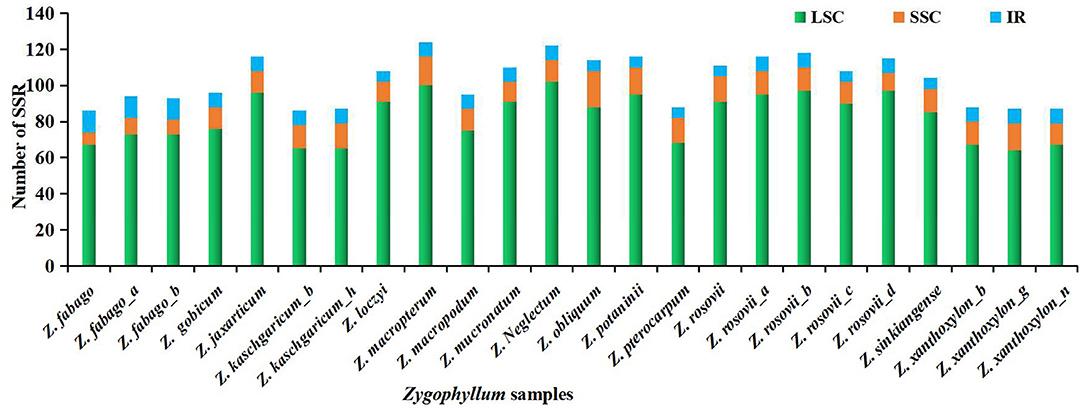
Figure 6. Number and distribution of SSRs in 24 complete cp genomes of Zygophyllum samples.
Phylogenetic and Morphological Analyses
To investigate evolutionary relationships between species of Zygophyllum in China and preliminarily assess species boundaries, we performed the phylogenetic analyses using ML and BI on the complete cp genomes as well as independently for all CDSs, all NCS regions, the LSC, the SSC, and IR regions. All trees resulting from the analyses of the complete cp genomes (Figure 7), CDSs (Supplementary Figure 1), NCSs, (Supplementary Figure 2), and LSCs (Supplementary Figure 4) showed similar topologies, except that the ML and BI trees of the NCSs differed slightly, with the ML tree being more similar to those obtained from the other datasets (Supplementary Figures 2, 3). Trees based on the SSC and IRs were poorly supported and were not further considered in this study (Supplementary Figures 5, 6).
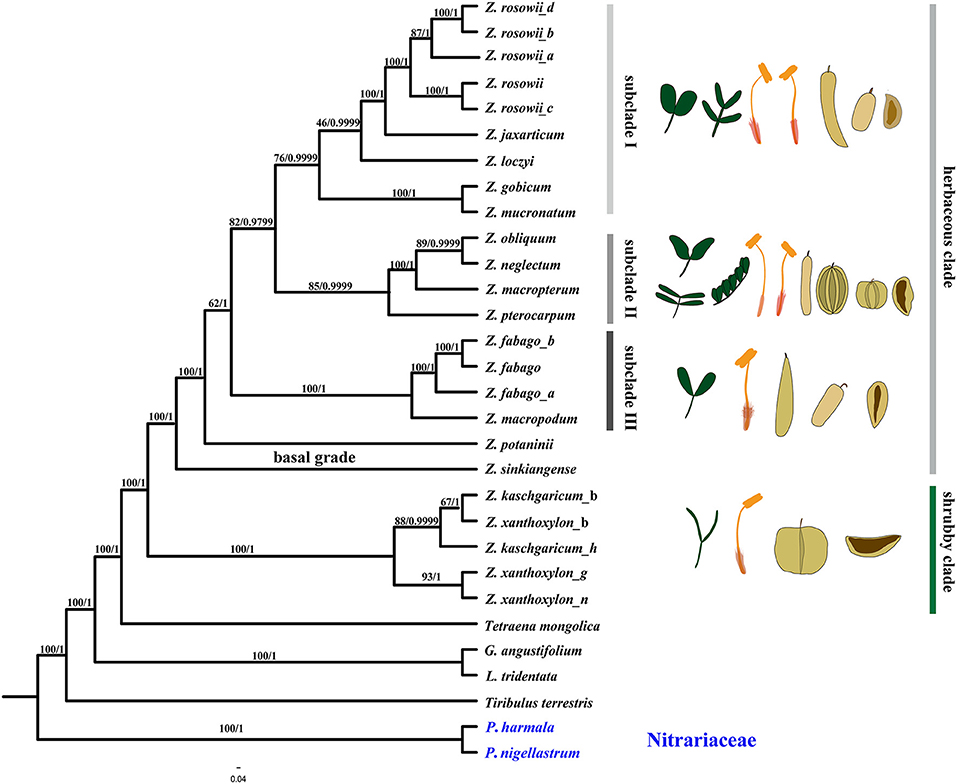
Figure 7. Phylogenetic relationships of Zygophyllum and its allies obtained from an ML and BI analysis of the complete cp genome. Numbers above the branches are the bootstrap values and Bayesian posterior probabilities, respectively.
The reconstructed phylogenies (Figure 7, Supplementary Figures 1–4) from the complete cp genome, the LSC, CDS, and NCS datasets showed that species of Zygophyllum clustered into two main clades (Figure 7), while the two Peganum species as the outgroup had a far phylogenetic relationship to Zygophyllum. One clade comprised Z. kaschgaricum and Z. xanthoxylon with high support [maximum likelihood bootstrap (MLBS) = 100%, PP = 1.0] and represents shrubby species that were formerly classified into Sarcozygium (Sheahan and Chase, 2000; Liou and Zhou, 2008). This clade was sister to the rest of the Chinese Zygophyllum, which is composed exclusively of herbaceous species. Within the shrubby clade, the accessions of Z. kaschgaricum and Z. xanthoxylon did not form mutually monophyletic groups (Figure 7).
The herbaceous clade contained a basal grade and three distinct subclades. Subclade I comprised nine accessions representing Zygophyllum gobicum, Zygophyllum jaxarticum, Zygophyllum loczyi, Zygophyllum mucronatum, and Z. rosowii. All these species in the subclade I have wingless capsular fruits and stamens longer than the petals except Z. loczyi, which has stamens that are shorter than the petals. Subclade II consisted of Z. macropterum, Zygophyllum neglectum, Zygophyllum pterocarpum, and Z. obliquum, all of which have orange petals, stamens equal to or shorter than petals, and dark yellow seeds. Subclade III contained Z. fabago and Zygophyllum macropodum, which are morphologically united by their wingless capsules, petals that are orange-red at the base and white apically, and the stamens that are longer than the petals (Figure 8). All species of the herbaceous clade have seeds that are transparent when dry except for Z. potaninii and Z. sinkiangense.

Figure 8. The morphological variation of species in Zygophyllum. Morphological characteristics of species of Zygophyllum representing the clade of woody shrubs (Z. xanthoxylon_b: 1, 6, 12, 17) and herbaceous species of the basal grade (Z. potaninii: 10,16; Z. sinkiangense: 11) and subclades I (Z. rosowii: 4, 8, 15, 20), II (Z. pterocarpum: 3, 9, 13, 18; Z. neglectum:5), and III (Z. fabago: 2, 7, 14, 19).
Species of the basal grade differed from the other herbaceous species by having seeds that were opaque whether wet or dry (as opposed to transparent when dry). Moreover, between these two species, Z. potaninii has seeds that are dark brown with papillae, while Z. sinkiangense has brownish-yellow seeds with rough hairs (Figure 8). This is in contrast to the seeds of the other sampled herbaceous species, which are pale or dark yellow and have dots on the surface. The leaves Z. potaninii and Z. sinkiangense are also much bigger and fleshier than those of other species of the herbaceous clade.
All phylogenies reconstructed in this study revealed that the herbaceous species of Chinese Zygophyllum formed a clade, but subclades within the herbaceous group differed slightly among trees. For example, Z. pterocarpum, which has winged capsules, clustered with species without capsular wings (Supplementary Figures 1–4) in the phylogenetic trees from CDS, NCS, and LSC datasets but species both wide-winged and wingless capsules in the phylogeny based on complete cp genomes. Additionally, the positions of Z. potaninii and Z. sinkiangense at the base of the tree were poorly resolved. A clear intrageneric treatment of the herbaceous species will require more molecular data as well as more morphological data from fruits, seeds, flowers, and vegetative organs.
Discussion
Comparison of the cp Genomes of Zygophyllum
All sampled cp genomes of Zygophyllum and Peganum displayed a quadripartite structure that is typical for most vascular plants (Wicke et al., 2011; Tonti-Filippini et al., 2017), and the GC content was consistent with cp genomes reported for many other angiosperm taxa (Gruenstaeudl et al., 2017; Yuan et al., 2017; Mower and Vickrey, 2018). However, the complete cp genomes of Zygophyllum were unusually small in size compared to other closely related genera, especially Nitraria (159,414 bp; Abla et al., 2019), Tetraena terrestris (158,148 bp), L. tridentata (136,149 bp), G. angustifolium (130,809 bp), and Peganum, but they were somewhat similar in size to Tetraena mongolica (106,259 bp). Generally, the sizes of cp genomes in vascular plants range from 100 to 220 kb sequence in higher plants (Marcelo et al., 2015), so that Zygophyllum and Tetraena mongolica exhibit genomes on the small end of the range, although much smaller cp genomes have been found in nonphotosynthetic, parasitic plants, including the smallest within Rhizanthella gardneri (Orchidaceae) with only 59,190 bp (Delannoy et al., 2011). As with the complete cp genome of Zygophyllum, its IR regions (3,792–4,466 bp; Table 1) were much smaller than those of many other land plants (10–15 kb) and seed plants (20–30 kb) (Mower and Vickrey, 2018).
Beyond the genome size, the number of genes within the Zygophyllum cp genomes (111–112) was also generally less than observed in other land plants (Zhang et al., 2016; He et al., 2017; Hu et al., 2017; Yuan et al., 2017), aside from other closely related species sampled in this study (i.e., four species of Zygophyllaceae and two species of Peganum). Notably, Zygophyllum appears to have lost several genes within the NADH dehydrogenase complex (i.e., ndh genes) and undergone pseudogenization within the SC regions. This is relatively rare in plants that are not parasitic (Yu et al., 2017) but has been detected in Najas (Peredo et al., 2013), some species of orchids (Kim et al., 2015), Pinaceae (Braukmann et al., 2009) and gnetophytes (Wu et al., 2009). In orchids, ndh genes appear to have been lost several times independently, and it was speculated that this may be due to shifts between autotrophism and mycoparasitism in the family, although not all modern species exhibiting losses are mycoparasites (Kim et al., 2015). However, in Pinaceae and gnetophytes, which have no known parasitic species, the explanation is more likely that ndh genes became encoded in the nucleus and were subsequently lost in the cpDNA (Braukmann et al., 2009), and this could be a possibility for Zygophyllum, which is also non-parasitic. The loss of several genes including ndh in Zygophyllum may partially explain its unusually small genome size.
In addition to the loss of ndh genes, the IR regions of Zygophyllum also lacked genes that were typically duplicated. Instead, these genes appeared in the SC regions. For example, four rRNA genes, which were commonly duplicated in the plant group, occurred only in the SSC of Zygophyllum as was also the case for the parasitic orchid, R. gardneri (Delannoy et al., 2011), which has the smallest cp genome known among land plants. Notably, we observed that the plastomes of Peganum, which were much larger than those of Zygophyllum, contained the typically duplicated rRNA genes. Moreover, in Peganum, six tRNA genes were duplicated, but only three were duplicated in Zygophyllum. Thus, the smaller number of genes within the IRs of Zygophyllum were also likely to play a role in the genus having a relatively small plastome size.
Based on the IR boundary analysis, we found that the IRs were the most highly conserved regions within the cp genomes of Zygophyllum and that the IR/SC boundaries were highly similar within the genus, as is common in other plant groups (Park et al., 2017; Xu et al., 2017). However, between Zygophyllum and other sampled genera, the boundaries between the IRs and the SSC/LSC were considerably different. These differences likely arose due to shifts in the boundaries following the losses of genes within the IRs of Zygophyllum, such as losses of ndhF gene and rRNA genes. Nevertheless, the IR and the SSC/LSC boundaries of species of Zygophyllum were more similar to other Zygophyllaceae than to P. harmala, thus supporting the current taxonomic treatment of the latter within the related family Nitrariaceae.
We detected highly variable regions and cpSSRs within the cp genomes of 15 species of Zygophyllum through the sliding window analysis and by using MISA, respectively. The results of the sliding windows analysis showed that the average mutation rate of the intergenic regions within the SC regions was much higher than in the IR regions, as has been observed in other taxa (Perry and Wolfe, 2002). Higher mutation rates typically yield higher variability within intergenic spacers due to reduced selective pressures (Elspeth et al., 2005; Wang et al., 2005). Coincidentally, we detected nine highly variable regions with high Pi values in the intergenic spacer regions. Using MISA, we also found 156 cpSSRs, among which mononucleotide repeats were most abundant, and most cpSSRs were located in NCS regions. Of the mononucleotide repeats, polyA and polyT were most common within Zygophyllum. The prevalence of polyA and polyT cpSSRs has also been observed in other plant lineages such as Lagerstroemia (Zhang et al., 2020), Primula (Ren et al., 2018), Fritillaria (Bi et al., 2018), and Allium (Xie et al., 2019). The 9 highly variable regions and 156 cpSSRs will represent robust genetic resources for future identification and population studies of species of Zygophyllum in China.
Phylogenetic and Taxonomic Implication
Our phylogenetic and comparative cp genomic results strongly supported the treatment of Zygophyllaceae as separate from Nitrariaceae, represented here by Peganum, (Figure 7) as in APG IV (Angiosperm Phylogeny Group (APG) IV, 2016). We also found a higher resolution for relationships between species of Zygophyllum in China compared to prior studies based on one or a few gene regions (Liou, 1998; Beier et al., 2003; Bellstedt et al., 2008). Notably, all well-supported ML and BI trees representing the 24 plastome sequences of Zygophyllum and 2 sequences of Peganum supported mutually monophyletic herbaceous and shrubby clades. The shrubby clade of Z. kaschgaricum and Z. xanthoxylon was sister to the herbaceous clade. The shrubby clade comprises the former species of Sarcozygium, which is now sometimes regarded as subgenus within Zygophyllum, such as the Flora of USSR (Bobrov, 1949) and which has been resolved within other, prior molecular phylogenetic studies (Beier et al., 2003; Bellstedt et al., 2008, Wu et al., 2015, 2018).
While species of the herbaceous and shrubby clades differ in several morphological characters other than their habits (i.e., herbaceous species have 10 stamens and five carpels, while shrubs have 10 stamens and three carpels), they share features, some of which distinguish them within Zygophyllaceae and support the including of the shrubby clade within the genus. For example, fruits of all species are capsules and most (except the basal grade within the herbaceous clade, Z. potaninii and Z. sinkiangense) have seeds that are transparent when wet or dry (Figure 8). Moreover, the two clades have cp genomic features that are much more similar to one another than to other genera in Zygophyllaceae (Figure 3). Prior studies have also shown that Z. xanthoxylon of the shrubby clade and Z. fabago of the herbaceous clade were both clustered with south African and Australian Zygophyllum (Bellstedt et al., 2008) based on molecular phylogeny, thus, supporting treatment of the shrubby species within the genus.
Within the shrubby clade, the accessions of Z. kaschgaricum and Z. xanthoxylon comprised a mixed group suggesting that they should be regarded as one species, Z. xanthoxylon as in the studies by Zhao and Zhu (2003) and Yang (2011). This is also consistent with our examination of morphology, in which we found more-or-less continuous variation in fruit size and shape. Neither molecular phylogeny nor morphological results supported the treatment of Z. kaschgaricum as a separate species as described in the study by Liou and Zhou (2008).
The herbaceous clade comprised a basal grade and three subclades. While these relationships were largely highly supported, we found that the relationships of Z. pterocarpum, Z. potaninii, and Z. sinkiangense were poorly resolved, and we observed few obvious morphological synapomorphies to support the subclades. Within the herbaceous clade, whether fruits were broad or narrow and winged or wingless, species varied broadly in the number of leaflets (4–10). Thus, resolving relationships within the herbaceous clade and establishing meaningful taxonomic boundaries within it will likely require additional morphological and molecular data.
Conclusion
In this study, we reported the complete cp genome sequences for 24 individuals of Zygophyllum (Zygophyllaceae), representing 15 species distributed in China, and for 2 closely related species, P. harmala and P. nigellastrum (Nitrariaceae). Through the comparisons of these newly sequenced cp genomes to additional sequences from species of Zygophyllaceae in public databases, we found that the genomes of Zygophyllum were much smaller, highly conserved in gene content and order, but differed markedly from other genera within the family and from Peganum. Based on the comparisons with all other sampled genera, the cp genomes of Zygophyllum appeared to have experienced considerable gene loss, ndh genes, and rRNA genes. Within Zygophyllum, IRs were the most highly conserved regions and contained none of the nine highly variable regions and numerous SSRs that we identified. We expect that these variable regions and repeat markers, which occur within the LSC and SSC, will be useful in future phylogenetic, phylogeographic, population genetic, and genetic relationship studies on Zygophyllum. Our phylogenomic results based on the complete cp genomes, CDSs, LSC, and NCS supported the monophyly of Zygophyllum in China and its division into an herbaceous and shrubby clade. The shrubby clade is highly supported and contains two species, Z. xanthoxylon and Z. kaschgaricum, formerly regarded in the genus, Sarcozygium. Our phylogenetic results and comparative analyses of cp genomic structures supported the treatment of Sarcozygium in Zygophyllum. The sampled accessions of the two members of the shrubby clade did not form mutually monophyletic groups. Therefore, based on this and morphological evidence, we recommend that Z. kaschgaricum be merged into Z. xanthoxylon. Overall, these results demonstrated the power of cp plastomes to improve the phylogenetic resolution of species and to resolve taxonomic questions.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: accession numbers in Table 2.
Data Accessibility
All the sequences were deposited as GenBank: Accessions MW307829, MW387262–MW387266, MW417249–MK417256, MW477239–MW477240, MW489448–MW489449, MW525443–MW525444, MW551563–MW551564, MW557318–MW557319, MW771516–MW771517.
Author Contributions
LD, Z-YC, and LZ designed the study and collected all samples. LZ, SW, and CS led data analysis and the making of figures and tables with assistance from J-RW, NS, and LZ. The manuscript was drafted by LZ, AH, LD, and Z-YC edited the manuscript for structure, language, and scientific content. All authors approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 31660048 and 32070229).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Ai-Jun Qiu, Pei-Pei Jiao, Liang Xiao, Zhen-Lei Wang, and Dong Wang for the collection of samples in Xinjiang, Inner Mongolia, and Gansu. We also thank Pei-Liang Liu and Hui Wang for their helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.723622/full#supplementary-material
Supplementary Figure 1. Phylogenetic tree constructed using Maximum Likelihood (ML) and Bayesian Inference (BI) methods, based on the CDS sequences from different species.
Supplementary Figure 2. Phylogenetic tree constructed using Maximum Likelihood (ML) based on the NCS sequences from different species.
Supplementary Figure 3. Phylogenetic tree constructed using Bayesian Inference (BI) methods, based on the NCS sequences from different species.
Supplementary Figure 4. Phylogenetic tree constructed using Maximum Likelihood (ML) and Bayesian Inference (BI) methods, based on the LSC sequences from different species.
Supplementary Figure 5. Phylogenetic tree constructed using Maximum Likelihood (ML) based on the SSC sequences from different species.
Supplementary Figure 6. Phylogenetic tree constructed using Maximum Likelihood (ML) IR sequences from different species.
Supplementary Table 1. Information of reported chloroplast genomes in outgroup species.
Supplementary Table 2. The number, type, and distribution of SSRs of 24 complete cp genomes in Zygophyllum.
Supplementary Table 3. The size and location of SSRs from 24 complete cp genomes in Zygophyllum.
References
Abla, M., Zha, X., Wang, Y., Wang, X. Y., and Feng, J. (2019). Characterization of the complete chloroplast genome of Nitraria tangutorum, a desert shrub. J. Genet. 98:s12041-019-1135-9 doi: 10.1007/s12041-019-1135-9
Angiosperm Phylogeny Group (APG) IV (2016). An update of the angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Beier, B. A., Chase, M. W., and Thulin, M. (2003). Phylogenetic relationships and taxomony of subfamily Zygophylloideae (Zygophyllaceae) based on molecular and morphological data. Plant Syst. Evol. 240, 11–39. doi: 10.1007/s00606-003-0007-0
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bellstedt, D. U., van Zyl, L., Marais, E. M., Bytebier, B., de Villiers, C. A., and Makwarela, A. M. (2008). Phylogenetic relationships, character evolution and biogeography of southern African members of Zygophyllum (Zygophyllaceae) based on three plastid regions. Mol. Phylogenet. Evol. 47, 932–949. doi: 10.1016/j.ympev.2008.02.019
Bendich, A. J. (2004), Circular chloroplast chromosomes: the grand illusion. Plant Cell. 16, 1661–1666. doi: 10.1105/tpc.160771
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, Y., Zhang, M. F., Jing, X., Ran, D., and Zhang, X. H. (2018). Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on fritillaria. Sci, Rep. 8:1184. doi: 10.1038/s41598-018-19591-9
Birky, C. W., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527. doi: 10.1093/genetics/103.3.513
Bobrov, E.G. (1949). “Zygophyllaceae,” in Flora of the USSR, eds B. K. Shishkin, E. G. Bobrov (Leningrad: Academy of Sciences of the USSR), 146–198.
Braukmann, T. W. A., Kuzmina, M., and Stefanovic, S. (2009). Loss of all plastid ndh genes in Gnetales and conifers: extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 55, 323–337. doi: 10.1007/s00294-009-0249-7
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat.Methods. 9:772. doi: 10.1038/nmeth.2109
Delannoy, E., Fujii, S., Francs-Small, C. C. D., Brundrett, M., and Small, I. (2011). Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mole. Biol. Evol. 28, 2077–2086. doi: 10.1093/molbev/msr028
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Downie, S. R., and Jansen, R. K. (2015). A comparative analysis of whole plastid genomes from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 40, 336–351. doi: 10.1600/036364415X686620
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 9, 11–15. doi: 10.1016/0031-9422(80)85004-7
Duan, L., Harris, A. J., Su, C., Zhang, Z. R., Arslan, E., Ertugrul, K. et al. (2020). Chloroplast phylogenomics reveals the intercontinental biogeographic history of the Liquorice genus (Leguminosae: Glycyrrhiza). Front. Plant Sci. 11:793. doi: 10.3389/fpls.2020.00793
Elspeth, M. H., Gwilym, P. L., and Julie, A. H. (2005). A phylogenetic reappraisal of the peltophorum group (Caesalpinieae: leguminosae) based on the chloroplast trnL-F, rbcL and rps16 sequence data. Amer. J. Bot. 92, 1359–1371. doi: 10.3732/ajb.92.8.1359
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fu, C. N., Li, H. T., Richard, M., Zhang, T., Ma, P. F., Jing, Y. et al. (2017). Comparative analyses of plastid genomes from fourteen cornales species: inferences for phylogenetic relationships and genome evolution. BMC Genom. 18:956. doi: 10.1186/s12864-017-4319-9
Goulding, S. E., Olmstead, R. G., Morden, C. W., and Wolfe, K. H. (1996). Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet. 252, 195–206. doi: 10.1007/BF02173220
Gruenstaeudl, M., Nauheimer, L., and Borsch, T. (2017). Plastid genome structure and phylogenomics of Nymphaeales: conserved gene order and new insights into relationships. Plant Syst. Evol. 303, 1251–1270. doi: 10.1007/s00606-017-1436-5
He, L., Qian, J., Li, X., Sun, Z., Xu, X., and Chen, S. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules. 22:249. doi: 10.3390/molecules22020249
Hollingsworth, P. M., Li, D., Der Bank, M. V., and Twyford, A. D. (2016). Telling plant species apart with DNA: from barcodes to genomes. Philos. Trans. R. Soc. Lond,. B,. Biol. Sci. 371:20150338. doi: 10.1098/rstb.2015.0338
Hu, H., Al-Shehbaz, I., Sun, Y., Hao, G., Wang, Q., and Liu, J. (2015). Species delimitation in Orychophragmus (Brassicaceae) based on chloroplast and nuclear DNA barcodes. Taxon 64, 714–726. doi: 10.12705/644.4
Hu, H., Hu, Q., Al-Shehbaz, I., Luo, X., Zeng, T., Guo, X. et al. (2016). Species Delimitation and Interspecific Relationships of the Genus Orychophragmus (Brassicaceae) Inferred from Whole Chloroplast Genomes. Front. Plant Sci. 7:1826. doi: 10.3389/fpls.2016.01826
Hu, Y., Woeste, K. E., and Zhao, P. (2017). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., and Boore, J. L. (2008). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci., India, Sect. B. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., Williams, N. H., Whitten, W. M. et al. (2015). Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE. 10:e0142215. doi: 10.1371/journal.pone.0142215
Librado, P., and Rozas, J. (2009). Dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Linnaeus, C. (1753). Species plantarum, exhibentes plantas rite cognitas, ad genera relates, cum differentiis specieficis, nominibus tricialibus, synonymis selectis, locis natalibus, secundum systema sexuale digetas. Holmiae: Impensis Laurentii Salvii 1753:669. doi: 10.5962/bhl.title.669
Liou, Y. X. (1980). The new species of Zygophyllum in China. Acta phytotaxono. sin. 18, 454–456 (in Chinese with English abstract).
Liou, Y. X. (1998). “Zygophyllaceae,” in Flora Republicae Popular is Sinicae, eds L.R. Xu, C. C. Huang, (Beijing: Science Press), 116–145.
Liou, Y. X., Zhou, L. F. (2008). “Zygophyllaceae,” in Flora of China, eds Z.Y. Wu, P. Reven, D. Y. Hong, Beijing: Beijing Science PressandMissouri Botanical Garden Press.
Liu, B. B., Hong, D. Y., Zhou, S. L., Xu, C., Dong, W., Johnson, G. et al. (2019). Phylogenomic analyses of the Photinia complex support the recognition of anew genus Phippsiomeles and the resurrection of a redefined Stranvaesia in Maleae (Rosaceae). J. Syst. Evol. 57, 678–694. doi: 10.1111/jse.12542
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). Organellar Genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–581. /nar/ gkt289 doi: 10.1093/nar/gkt289
Mao, Y. R., Zhang, Y. H., Nakamura, N., Guan, B. C., and Qiu, Y. X. (2014). Developing DNA barcodes for species identification in Podophylloideae (Berberidaceae). J. Syst. Evol. 52, 487–499. doi: 10.1111/jse.12076
Marcelo, R., Leila, D., Fraga, H. P., and Guerra, M. P. (2015). Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 6:00586 doi: 10.3389/fpls.2015.00586
Miller, M. A., Pfeiffer, W. T., and Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. GCE 14, 1–8. doi: 10.1109/GCE.2010.5676129
Mohammad-Panah, N., Shabanian, N., Khadivi, A., Rahmani, M. S., and Emami, A. (2017). Genetic structure of galloak (Quercus infectoria) characterized by nuclear and chloroplast SSR markers. Tree Genet Genomes. 13, 70–82. doi: 10.1007/s11295-017-1146-8
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Mower, J. P., and Vickrey, T. L. (2018). Structural diversity among plastid genomes of land plants. Adv. Bot. Res. 85, 263–292. doi: 10.1016/bs.abr.2017.11.013
Niu, Z., Xue, Q., Zhu, S., Sun, J., Liu, W., and Ding, X. (2017). The complete plastome sequences of four orchid species: insights into the evolution of the Orchidaceae and the utility of plastomic mutational hotspots. Front. Plant Sci. 8:715. doi: 10.3389/fpls.2017.00715
Ozeki, H., Umesono, K., Inokuchi, H., Kohchi, T., and Ohyam, A. K. (1989). The chloroplast genome of plants: a unique origin. Genome 31, 169–174. doi: 10.1139/g89-029
Park, I., Kim, W. J., Yang, S., Yeo, S. M., Li, H. L., and Moon, B. C. (2017). The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE. 12:e0184257. doi: 10.1371/journal.pone.0184257
Park, S., Sang, I. P., Gil, J., Um, Y., Jung, C. S., Lee, J. H. et al. (2016). Development of Simple sequence repeat (SSR)markers Based on Chloroplast DNA to distinguish 3 Angelica species. Horticult. Abstr. 10:226.
Perdereau, A. C., Kelleher, C. T., Douglas, G. C., and Hodkinson, T. R. (2014). High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biol. 14:202. doi: 10.1186/s12870-014-0202-x
Peredo, E. L., King, U. M., and Les, D. H. (2013). The plastid genome of Najas flexilis: adaptation to submersed environments is accompanied by the complete loss of the NDH complex in an aquatic angiosperm. PLoS ONE. 8:e68591. doi: 10.1371/journal.pone.0068591
Perry, A. S., and Wolfe, K. H. (2002). Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mole. Evol. 55, 501–508. doi: 10.1007/s00239-002-2333-y
Powell, W., Machray, G. C., and Provan, J. (1996). Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222. doi: 10.1016/S1360-1385(96)86898-0
Qu, X. J., Moore, M. J., Li, D. Z., and Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Method. 15, 1–12. doi: 10.1186/s13007-019-0435-7
Quan, X., and Zhou, S. L. (2011). Molecular identification of species in prunus sect. persica (Rosaceae), with emphasis on evaluation of candidate barcodes for plants. J Syst. Evol. 49, 138–145. doi: 10.1111/j.1759-6831.2010.00112.x
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ren, T., Yang, Y., Tao, Z., and Liu, Z. L. (2018). Comparative plastid genomes of Primula species: Sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 19:1050. doi: 10.3390/ijms19041050
Retief, E. (2000). “Zygophyllaceae,” in Seed Plants of Southern Africa: Families and Genera, ed O. A. Leistner, Strelitzia, 563–565.
Ronquist, F., Teslenko, M., Van der Mark, P., Ayres, D. L., Darling, A., and Hohna, S. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M. S., and Morris, A. B. (2014). Chloroplast DNA sequenceutility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Sheahan, M. C., and Chase, M. W. (1996). A phylogenetic analysis of Zygophyllaceae R.Br. based on morphological, anatomical and rbcL DNA sequence data. Bot. J. Linn. Soc. 122, 279–300. doi: 10.1111/j.1095-8339.1996.tb02077.x
Sheahan, M. C., and Chase, M. W. (2000). Phylogenetic relationships within Zygophyllaceae based on DNA sequences of three plastid regions, with special emphasis on Zygophylloideae. Syst. Bot. 25, 371–384. doi: 10.2307/2666648
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V. et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinform. 4:41. doi: 10.1186/1471-2105-4-41
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R. et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6-W11. doi: 10.1093/nar/gkx391
Tonti-Filippini, J., Nevill, P. G., Dixon, K., Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. 90, 808–818. doi: 10.1111/tpj.13491
Wang, A. L., Yang, M., and Liu, J. Q. (2005). Molecular phylogeny, recent radiation and evolution of gross morphology of the rhubarb genus rheum (Polygonaceae) inferred from chloroplast DNA trnL-F sequences. Ann. Bot. 96, 489–498. doi: 10.1093/aob/mci201
White, F. (1983). The Vegetation of Africa. a descriptive memoir to accompany the UNESCO AETFAT/UNSO vegetation map of Africa. Nat. Resour. Res. (Paris, Fr.) vol. 20. UNESCO, Paris.
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, C. S., Lai, Y. T., Lin, C. P., Wang, Y. N., and Chaw, S. M. (2009). Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Mol Phylog Evol. 52, 115–124. doi: 10.1016/j.ympev.2008.12.026
Wu, S. D., Li, L., Lin, L., Yu, S. X., and Zhang, L. J. (2015). Evolution of Asian interior arid-zone biota: evidence from the diversification of Asian Zygophyllum (Zygophyllaceae). PLoS ONE 10:e0138697. doi: 10.1371/journal.pone.0138697
Wu, S. D., Zhang, L. J., Lin, L., Yu, S. X., Chen, Z. D., and Wang, W. (2018). Insights into the historical assembly of global dryland floras: the diversification of Zygophyllaceae. BMC Evol. Biol. 18, 1277. doi: 10.1186/s12862-018-1277-z
Wu, Y. X., Wang, J., and Liu, J. Q. (2010). Development and characterization of microsatellite markers in Populus euphratica (Populaceae). Mol. Ecol. Res. 8, 1142–1144. doi: 10.1111/j.1755-0998.2008.02202.x
Xie, D. F., Yu, H. X., Price, M., Xie, C., Deng, Y. Q., Chen, J. P. et al. (2019). Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 10:460. doi: 10.3389/fpls.2019.00460
Xu, C., Dong, W., Li, W., Lu, Y., Xie, X., Jin, X. et al. (2017). Comparative analysis of six lagerstroemia complete chloroplast genomes. Front. Plant Sci. 8:15. doi: 10.3389/fpls.2017.00015
Xu, H., Fu, W., Xie, W., Wang, Y., and Zhang, Y. (2020). The complete chloroplast genomes of two species of Zygophyllum (Zygophyllaceae). Mitochondrial DNA Part B. 5, 3494–3495. doi: 10.1080/23802359.2020.1825132
Yang, C. Y. (2011). “Zygophyllaceae,” in Flora of Xinjiangensis, ed G. M. Shen (Beijing: Urumqi/Xinjiang science and Technology Press), 308–329.
Ye, W. Q., Yap, Z. Y., Li, P., Comes, H. P., and Qiu, Y. X. (2018). Plastome organization, genome-based phylogeny and evolution of plastid genes in podophylloideae (berberidaceae). Mol. Phylogenet. Evol. 7:127. doi: 10.1016/j.ympev.2018.07.001
Yu, S., Yu, W. B., Tan, Y. H., Liu, B., Yao, X., Jin, J. J. et al. (2017). Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids. Genome Biol. Evol. 9, 2354–2364. doi: 10.1093/gbe/evx180
Yuan, C., Zhong, W., Mou, F., Gong, Y., Pu, D., Ji, P. et al. (2017). The complete chloroplast genome sequence and phylogenetic analysis of Chuanminshen (Chuanminshen violaceum Sheh et Shan). Physiol. Mol. Biol. Pla. 23, 35–41. doi: 10.1007/s12298-016-0395-6
Zeng, Y. J., Wang, Y. R., Zhuang, G. H., and Yang, Z. S. (2004). Seed germination responses of Reaumuria soongorica and Zygophyllum xanthoxylum to drought stress and sowing depth. Acta Ecol Sinica. 24, 1629-1634 (in Chinese with English abstract).
Zhang, F. J., Zhang, F., Wang, T., Shu, X., Wang, N., Zhuang, W. et al. (2020). Complete Chloroplast Genomes and Comparative Analyses of L.chinensis, L.anhuiensis, and L.aurea (Amaryllidaceae). Int. J. Mol. Sci. 21:16. doi: 10.3390/ijms21165729
Zhang, Y., Du, L., Ao, L., Chen, J., Li, W., Hu, W. et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhao, Y. Z., and Zhu, Z. Y. (2003). The endemic genera of desert region in the center of Asia. Acta Botanica Yunnanica. 25, 113 (in Chinese with English abstract).
Zufall, R. A. (2009). Beyond simple homology searches: multiple sequence alignments and phylogenetic trees. Curr Protoc. Essent Lab Tech. 1, 11–3. doi: 10.1002/9780470089941.et1103s01
Keywords: chloroplast genome, comparative analysis, phylogeny, Zygophyllum, Zygophyllaceae
Citation: Zhang L, Wang S, Su C, Harris AJ, Zhao L, Su N, Wang J-R, Duan L and Chang Z-Y (2021) Comparative Chloroplast Genomics and Phylogenetic Analysis of Zygophyllum (Zygophyllaceae) of China. Front. Plant Sci. 12:723622. doi: 10.3389/fpls.2021.723622
Received: 11 June 2021; Accepted: 24 August 2021;
Published: 24 September 2021.
Edited by:
Damien Daniel Hinsinger, INRA Center Versailles-Grignon, FranceReviewed by:
Gang Yao, South China Agricultural University, ChinaFabiola Areces-Berazain, University of Puerto Rico, Puerto Rico
Copyright © 2021 Zhang, Wang, Su, Harris, Zhao, Su, Wang, Duan and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhao-Yang Chang, Y3p5YmJnQG53c3VhZi5lZHUuY24=; Lei Duan, ZHVhbmxlaUBzY2JnLmFjLmNu