 Hui Li
Hui Li Guanghui Chen
Guanghui Chen Hongying Pang1
Hongying Pang1 Qiao Wang
Qiao Wang Xinren Dai
Xinren Dai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 02 July 2021
Sec. Plant Biotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.698602
This article is part of the Research Topic Wood Development and Physiology in a Changing Climate View all 17 articles
Enormous distinctions of the stem structure and cell types between gymnosperms and angiosperms tree species are expected to cause quite different wood physical and mechanical attributes, however, the molecular mechanisms underlying the differing wood morphology are still unclear. In this study, we compared the transcriptomes obtained by RNA-Seq between Populus alba × P. glandulosa clone 84K, and Larix kaempferi (Lamb.) Carr trees. Available genome resource served as reference for P. alba × P. glandulosa and the Iso-Seq results of a three-tissues mixture (xylem, phloem, and leaf) were used as the reference for L. kaempferi to compare the xylem-specifically expressed genes and their alternative splicing model. Through screening, we obtained 13,907 xylem-specifically expressed genes (5,954 up-regulated, 7,953 down-regulated) in the xylem of P. alba × P. glandulosa, and 2,596 xylem-specifically expressed genes (1,648 up-regulated, 948 down-regulated) in the xylem of L. kaempferi. From the GO and KEGG analyses, some genes associated with two wood formation-related pathways, namely those for phenylpropanoid biosynthesis, and starch and sucrose metabolism, were successfully screened. Then the distributions and gene expression models between P. alba × P. glandulosa and L. kaempferi in those pathways were compared, which suggested differential wood formation processes between the angiosperm and gymnosperm trees. Furthermore, a Weight Gene Co-expression Network Analysis (WGCNA) for total xylem-specifically expressed genes in two species was conducted, from which wood formation-related modules were selected to build a co-expression network for the two tree species. The genes within this co-expression network showed different co-expression relationships between the angiosperm and gymnosperm woody species. Comparing the alternative splicing events for wood formation-related genes suggests a different post-transcriptional regulation process exists between the angiosperm and gymnosperm trees. Our research thus provides the foundation for the in-depth investigation of different wood formation mechanisms of angiosperm and gymnosperm species.
Seed-bearing plants are the primary plant species on our planet and are composed of two main phyla, gymnosperms and angiosperms, which appeared ca. 300 million years ago (Pavy et al., 2012). Gymnosperms include seed-bearing plants that are woody, herbaceous, or climbing (vines), consisting of four extant sub-phyla: Cycadophyta (cycads), Ginkgophyta (Ginkgo biloba L.), Coniferophyta (conifers), and Gnetophyta (gnetophytes). Among them, the conifers are the most numerous gymnosperms found on earth, comprising 50 genera and 550 species, and they are widely distributed throughout the Northern Hemisphere (Carvalho et al., 2013). For the angiosperms, taxonomists have identified about 352,000 species, making it the most diversiform group on earth (www.theplantlist.org). All of these angiosperm species originated from one single ancestor ca. 167–199 million years ago (Bell et al., 2010), and diverged into eight extant clades, namely the Amborellales, Nymphaeales, Austrobaileyales, Monocots, Magnoliids, Ceratophyllales, Chloranthales and Eudicots. Among them, the Eudicots is the primary clade, containing about 262,000 species (Zeng et al., 2014).
Wood represents a renewable natural resource for feedstocks used in several bio-economy products, such as pulp, paper, and biomaterials, and potentially biofuels as well. It is also a major carbon sink in natural ecosystems. The stem of woody plants is mainly composed of secondary xylem (Li et al., 2010). The most striking divergent characteristics between gymnosperm and angiosperm wood are in their anatomical structure and chemical composition (Jokipii-Lukkari et al., 2018). In gymnosperm trees, tracheids provide both water transport and mechanical support; in angiosperms, vessel elements are responsible for carrying water, with fibers providing mechanical support for the stem. To some extent, however, the tracheids in gymnosperms are similar to the vessel in angiosperms (Courtois-Moreau et al., 2009; Dieset, 2011). Vessel elements appear only in angiosperm species and are shaped by the need for rapid and efficient water transport capacity (Sperry et al., 2006). Wood formation in tree species requires the complex coordination of two highly ordered processes, cell differentiation and secondary cell wall (SCW) thickening, which are initiated from the vascular cambium and result in thick-walled xylem cells (Zhao et al., 2014). Secondary walls of xylem cells are composed of high-content cellulose and lignin (Li et al., 2010). Importantly, the amount and chemical structure of lignin in SCWs differs between gymnosperms and angiosperms, and these differences are closely related to plant evolution (Vanholme et al., 2010; Nawawi et al., 2016). The lignin in gymnosperms is exclusively polymerized from guaiacyl (G) units, while both G units and syringyl (S) units are the major components of lignin in angiosperms (Sarkanen, 1971; Higuchi et al., 1977; Boerjan et al., 2003; Weng et al., 2008).
In the last few decades, significant progress has been made in uncovering the molecular players involved in SCW biosynthesis in tree species, including hormonal signals, receptor kinases, and the transcriptional network which controls SCW formation. Both NAC and MYB master switches and their downstream transcription factors (TFs) have been shown to play critical roles during SCW formation (Yamamoto et al., 1997; Aspeborg et al., 2005; Li et al., 2012; Zhong and Ye, 2014; Ye and Zhong, 2015; Zhang et al., 2018; Du et al., 2019; Wang et al., 2019). Genes encoding the enzymes for the biosynthesis of SCW cellulose, hemicelluloses, and lignin have been identified and most of them are now functionally characterized in Populus and Eucalyptus (Suzuki et al., 2006; Shi et al., 2009; Lu et al., 2013; Yuan et al., 2014; Ye and Zhong, 2015; Kim et al., 2019; Wang et al., 2019). Many recent studies aiming to identify those key genes involved in wood development in many plant species were carried out to investigate xylem evolution at the transcriptome level (Cronk and Forest, 2017; He and Groover, 2017; Sundell et al., 2017; Tuskan et al., 2018; Roodt et al., 2019). The expanding genomic resources of tree species are invaluable for exploring secondary xylem formation and its evolution. Yet woody gymnosperm species typically have large genome sizes and high heterozygosity, so their transcriptome analyses remain limited by the poor quality of assembled genomes.
Alternative splicing (AS) is an important model of post-transcriptional regulation that can increase transcriptional and proteomic diversity in eukaryotic organisms (Chen and Manley, 2009). AS that combines different transcript splice junctions results in transcripts with shuffled exons, alternative 5′ or 3′ splicing sites, retained introns, and different transcript termini. In plants, 33−60% of the mRNAs are alternatively spliced (Shen et al., 2014), of which more than 60% are in the form of retained introns (Zhang et al., 2010; Syed et al., 2012). Transcriptome analysis has revealed that approximately 36% of wood-expressed genes undergo AS in the xylem of P. trichocarpa (Bao et al., 2013), and that 28.3% and 20.7% of the highly expressed transcripts in developing xylem tissue undergo AS events in Populus and Eucalyptus, respectively (Xu et al., 2014). Interestingly, most of the key TFs in the first layer of SCW regulatory network also undergo AS. In Populus, PtrSND1-A2IR, a splice variant of stem-differentiating xylem (SDX)- SND1, acts as a dominant negative regulator of the SND1 transcriptional network (Li et al., 2012). PtrSND1-A2IR is derived from PtrSND1-A2 (also named PtrWND1B/PtrVNS11), having lost its DNA binding and activation domain but retaining its dimerization capability; it represses the transcription of PtrSND1 members and their target gene PtrMYB021 by translocating into the nucleus exclusively as a heterodimeric partner with full-size PtrSND1s (Li et al., 2012). This is the first time to report about the TF family's auto-repression by its own splice variant found in plants (Zhang et al., 2018; Camargo et al., 2019). Other research has shown that overexpression of PtrSND1-A2 enhances fiber cell wall thickening, while overexpression of PtrSND1-A2IR (PtrWND1B-l) inhibits the fiber cell wall-thickening process (Zhao et al., 2014). VND6, another key TF in the first layer of the SCW regulatory network, was also confirmed to undergo alternative splicing during wood formation in poplar (Lin et al., 2017). Its splice variant, PtrVND6-C1IR derived from PtrVND6-C1 (PtVNS01/ PtrWND5A), suppresses the protein functioning of all PtrVND6 and PtrSND1 family members, whereas PtrVND6-C1IR is unable to suppress PtrSND1-A2; further, PtrSND1-A2IR has no effect on PtrVND6-C1IR. Both PtrVND6-C1IR and PtrSND1-A2IR function together in the reciprocal cross-regulation of VND and SND members to maintain homeostasis during xylem differentiation and plant development. Recently, for a gymnosperm species, LaSCL6, a member of the GRAS transcription factor family, was determined to have two variants which are differentially expressed during the growth and development in L. kaempferi (Zang et al., 2019). Our knowledge of AS of other genes, especially the key TFs in SCW regulatory network, is still vastly limited.
Although some molecular players are distinguished by their conserved functions during wood formation in gymnosperms and angiosperms, wood formation between gymnosperms and angiosperms is clearly different (i.e., xylem cell type and wood composition, among others). This suggests their gene regulatory networks are also not the same and that certain genes may have distinct functions in the two tree species above. Here, we employed next-generation sequencing and Iso-Seq technology to explore the gene expression model across wood-forming tissues in the angiosperm P. alba × P. glandulosa and the gymnosperm L. kaempferi. By screening xylem specific-expressed genes, associating these genes with wood-formation related pathways, building a co-expression network and analyzing the AS of common genes among the two tree species, differential mechanisms of wood formation between angiosperm and gymnosperm tree species were investigated and preliminarily discussed. Our research provides theoretical support for searching for the differing wood formation mechanisms between angiosperm and gymnosperm species.
Samples were collected from 10-year-old P. alba × P. glandulosa trees in the Beiwu garden (39°59′N, 116°15′E; Beijing, China) and 10-year-old L. kaempferi tress in the Dagujia seed orchard (42°22′N, 124°51′E; Liaoning Province, China) as describe in He et al. (2020). Briefly, the bark and phloem were peeled off at breast height, the immature xylem and phloem were collected from each species by scratching with single-end razors. These, along with leaf samples, were immediately frozen in liquid nitrogen. Each sample was collected from three trees per species as three biological replicates. Total RNAs were extracted by using the improved CTAB method (Lorenz et al., 2010). The RNA was precipitated by ethanol, and then dissolved in RNase-free water. After digesting it with DNaseI, we used the Agilent 2100 Bioanalyzer and NanoDrop to measure the RNA concentration, RIN value, 28s/18s value, and size of the fragment, to confirm the integrity and purity of RNA in the samples.
To correct the Hi-Seq results and analyze AS events in the two tree species, we also constructed PacBio libraries. By taking 1 μg RNA subsamples, first-strand cDNA was synthesized using the Clontech SMARTer PCR cDNA synthesis Kit (cat. no. 634926, http://www.clontech.com/), with an anchored olig (dT) 30 as the primer. Then the double-strand cDNA was amplified by carrying out a long PCR (LD-PCT) using the Advantage 2 PCT kit (Clonetech, cat. No.639206). The 1–2 kb, 2–3 kb, and 3–6 kb cDNA fragments were generated by the BluePinppin size-selection system (Sage Science, http://www.sagescience.com/). For those transcripts whose size was more than 3 kb, we generated the BluePippin selection again. The PacBio libraries were then constructed with a Pacific Biosciences SMARTbell template Prep Kit 1.0 (part 100-259-100, http://www.pacb.com/), following the manufacture's protocol. These libraries were later sequenced on a PacBio RSII real-time (RT) platform, using the SMRT Cell 8 Pac v3 (part100-171-800) with a total of 8 SMRT cells: that is, the 1–2 k and 2–3 k libraries were each sequenced with 3 SMRT cells, while the 3–6 k libraries were sequenced with 2 SMRT cells.
We used magnetic beads with Oli (dT) to enrich the mRNA and broke it into pieces by applying a fragmentation buffer. Employing these mRNAs as the template, random hexamers were used to synthesize the first-strand cDNA. Next, together with buffer, the dNTPs, RNaseH, and DNA polymerase I synthesized the second-strand cDNA. Each cDNA library was purified and to it a joint end with A was added. Following their PCR amplification, the ensuing PCR products were sequenced by the Illumina HiSeq 2500 platform to generate 100-bp paired sequence reads.
We used the SMRT Analysis Server (v2.3) to handle the Raw SMRT sequencing reads in order to obtain the Full-Length non-chimeric Read (FLNC). The first step in doing this was to properly deal with the inserted anti-molecular sequence and assess the length of cDNAs loaded into SMRT cell using these parameters “minFullPasses” = 0, “minPredictedAccruacy” = 80, “numThreads” = 12. Reads-of-Insert can be obtained from a single molecular inserted sequence. After removing the cDNA primer and polyA, we classified the Reads-of-Insert into full-length or non-full-length, chimeric or non-chimeric, fragments with these two parameter settings: “min_seq_len” = 300, “cpus” = 12. The full-length and non-chimeric transcripts were corrected, using the Interactive Clustering and Erro Correction (ICE) algorithm, to obtain the corresponding sequences. Then the cover rating for these corresponding sequences was predicted, after which the non-full length and non-chimeric sequences were corrected by the Quiver algorithm.
Completing these steps left us with high- and low-quality full-length transcripts. The LSC software was then used to correct the FLNC by referencing short reads of HiSeq (Au et al., 2012). For each corrected FLNC, we used the CD-HIT-EST to reduce the redundant highly similar transcripts (Li and Godzik, 2006); these transcripts were merged by Cogent software to obtain the unigenes. For their annotation, unigenes were BLAST-searched to NR (ftp://ftp.ncbi.nlm.nih.gov/blast/db), NT (ftp://ftp.ncbi.nlm.nih.gov/blast/db), COG (http://www.ncbi.nlm.nih.gov/COG), KEGG (http://www.genome.jp/kegg) and Swiss-Prot (http://ftp.ebi.ac.uk/pub/databases/swissprot) databases. According to those annotations of the NR database, Blast2GO (https://www.blast2go.com) (Conesa et al., 2005) was used to obtain the annotations from the GO database. According to the latter, the CDs of best-matched unigenes were selected for further analysis. For those unigenes that could not be annotated by any database, ESTscan software was relied upon to build a model and predict the CDs for them (Iseli et al., 1999).
Raw data were filtered using the NGS QC Toolkit to obtain clean reads. These were mapped onto the genome or PacBio Isoform transcripts via Hierarchical Indexing for Spliced Alignment of Transcripts (HISAT) (Kim et al., 2015). The mapped output was processed by Cufflinks to obtain the Fragments per Kilobase Million (FPKM) for all genes in each sample, for which correlations among different replicate samples were determined by calculating Pearson correlation. Using the DEseq2 software (Love et al., 2014), we obtained the DEGs among the types of plant tissue. The DEGs in the two species were filtered according to these criteria: the DEGs that had a log2 (X vs. P or L) ≥ 1 (Q-value ≤ 0.05) were designated as up-regulated genes in xylem compared to phloem and leaf parts; those DEGs that met log2 (X vs. P or L) ≤ −1 (Q-value ≤ 0.05) were considered down-regulated genes compared with the other two tissue types. The intersection of X vs. P and X vs. L correspond to the xylem-specifically expressed genes.
For all DEGs, their Gene Ontology (GO) enrichment analysis was conducted using GOseq and topGO (Young et al., 2010). To do this, we first mapped all the detected genes in the two tree species to the GO database (http://geneontology.org/), to obtain their GO annotations. According to these results, we enriched the xylem-specifically expressed genes by comparing them to the reference gene background by applying the hypergeometric test. The gene numbers for each term were then calculated, among which the significantly enriched GO terms were determined. We also mapped all detected genes to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/), to obtain their corresponding KEGG annotations. According to the KEGG annotations, we enriched the xylem-specifically expressed genes in the main biochemical and signal transduction pathways and identified those significantly enriched metabolic pathways or signal transduction pathways. The “phyper” package for the R computing platform was used to calculate the p values, as described in Li et al. (2020).
Based on the KEGG results, we located the enriched xylem-specifically expressed genes in two pathways, the phenylpropanoid biosynthesis pathway, and the starch and sucrose pathway, to derive and display a putative gene expression model. These pathways were visualized in Chewdraw software (version Professional 15.0). The heatmaps were drawn with TBtools 0.6669 (Chen et al., 2020).
We used “WGNCA” package in R software to carry out a co-expression network analysis for xylem-specifically expressed genes in the two tree species. Then, for each module per species, we also conducted GO and KEGG analyses. After combining these GO and KEGG results, we selected the modules in which wood formation-related genes were evidently enriched.
To discern the possible relationships among wood formation-related genes, we chose the top 50,000 gene relation pairs in the selected edge file of WGCNA and imported these pairs into Cytoscape software (Kohl et al., 2011). According to the GO and KEGG analyses and BLASTn results in the selected module of each species, we built co-expression networks of wood formation-related genes in Cytoscape software to depict the co-expression relationships of the two tree species.
To classify the AS events, Cogent software was used to reconstruct the PacBio transcripts and this yielded the UniTransModels. After blasting the non-redundant transcripts to UniTransModels, using GMAP software, the output results from GMAP were inputted into SUPPA software to detect the AS events (Li et al., 2017).
To validate the RNA-Seq and AS events experimentally, PCRs were run for eight pairs of homologous genes that underwent different AS types in the two tree species. For each sample, 1 mg of total RNA was reverse-transcribed into first-strand cDNA, by using the PrimeScript RT reagent Kit gDNA Eraser (Takara, Dalian, China). The primers were designed based on the consensus gene sequences of the two tree species, and these primer sequences are listed in Supplementary Tables 1A,B. For validation of RNA-Seq results, we chose several genes from both P. alba × P. glandulosa and L. kaempferi to perform the qRT-PCR, by following the instructions for SYBR® Premix Ex Taq™ II (Takara), for the xylem, phloem, and leaf tissues, respectively. Linear fitting between RNA-Seq and qRT-PCR data was done in Origin 2016 software. To validate the AS events, the PCR was run in a 25-ml reaction system, using the High-Fidelity PCR Master Mix (NEB), whose procedure went as follows: initial denaturation at 98°C for 1 min, 98°C for 10 s, 60°C for 30 s, and 72°C for 1–3 min (35 cycles) and a final extension at 72°C for 5 min. All the PCR products were visualized by 1.5% agarose gel electrophoresis analysis.
We conducted the RNA-Seq of xylem, phloem, and leaf tissues of two tree species, with three biological replicates per tissue. Before the formal data analysis, we tested for correlations (using Pearson's r) among the different replicates and tissue types (Figure 1). The tests confirmed that the three replicates of the same tissue in each of the species were strongly correlated, having r values > 0.8 (Figures 1A,B), thus showing that their transcriptome data was suitable for further analysis. We also derived key statistics for gene expression levels in different tissue of the two tree species. InP. alba × P. glandulosa, two high proportions of expressed genes focus at 0.1 and 1.0, respectively. By contrast, there was only one high proportion of expressed genes focus at 0.1 in L. kaempferi. The median gene expression levels in the three tissues of P. alba × P. glandulosa were about 0.8–0.9, and higher than those of L. kaempferi (Figure 1C). These phenomena reflected different gene expression distributions between angiosperm and gymnosperm tree species.
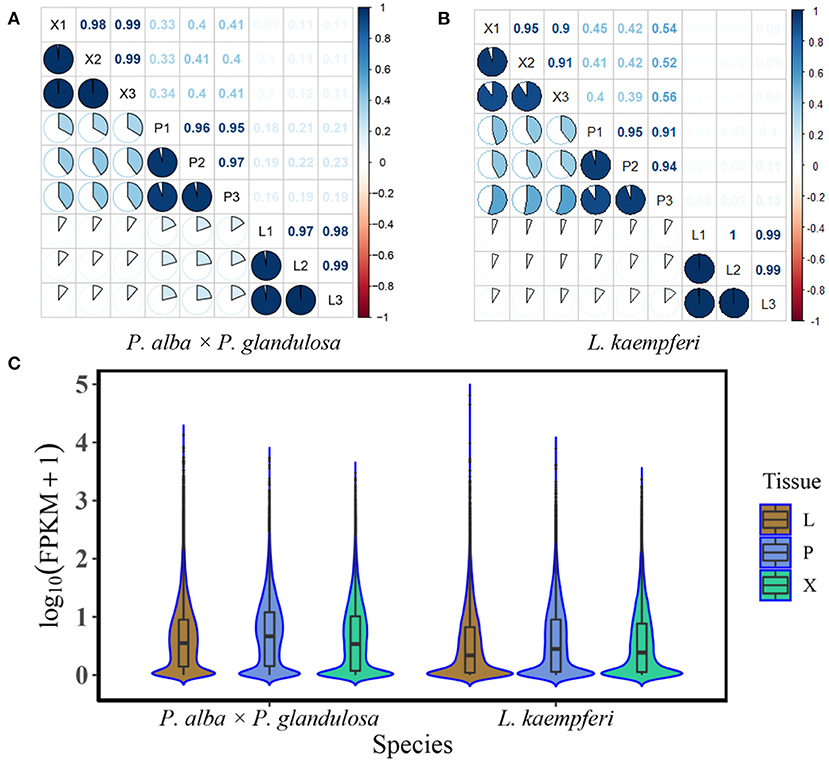
Figure 1. Correlation analysis and the gene expression distributions of different tissues in the two tree species. (A) Correlations among different tissues and replicates of the angiosperm Populus alba × P. glandulosa clone 84K (P. alba × P. glandulosa). (B) Correlations among different tissues and replicates of the gymnosperm Larix kaempferi (Lamb.) Carr (L. kaempferi). (C) Gene expression distribution of different tissues (Xylem: X; Phloem: P; Leaf: L) in two tree species. Box plots appear in the center of the violin graphs. The top edge, middle edge, and bottom edge represent third quartile, median, and first quartile, respectively. The expression levels of the genes were standardized by the log10 (FPKM) algorithm. FPKM represents Fragments per Kilobase Million.
We next profiled the RNA transcriptome for three tissues (xylem, phloem and leaves) of the two tree species. In P. alba × P. glandulosa, we obtained 50,995,164, 56,113,145, and 50,995,164 clean reads in the xylem, phloem, and leaves, respectively; in L. kaempferi, the corresponding counts were 66,463,409, 77,166,823, and 63,449,442 clean reads. The reads from P. alba × P. glandulosa were mapped to the reference genome (https://bigd.big.ac.cn/), while those reads from L. kaempferi were mapped to the SMRT library which had corrected by RNA-Seq data. The mapping rates for xylem, phloem, and leaf tissue were 95.39 95.41 and 95.60 for P. alba × P. glandulosa, and 46.79 41.41 and 54.95 for L. kaempferi, respectively. Eventually, we obtained 44,561 genes in xylem, 47,465 in phloem, and 48,091 in leaves of P. alba × P. glandulosa, and likewise, 21,535 genes in xylem, 22,986 in phloem and 23,233 in leaves of L. kaempferi (Table 1).
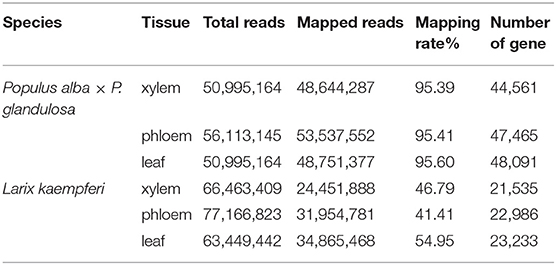
Table 1. Summary information of the RNA-Seq results for the two tree species.
To understand the expression discrepancy of RNAs in three different tissues of the two tree species, we conducted a DEG analysis for P. alba × P. glandulosa, we obtained 9,025 up-regulated DEGs and 12,024 down-regulated DEGs in the X vs. P group, and 12,086 up-regulated DEGs and 19,241 down-regulated DEGs in the X vs. L group (Figure 2A; Supplementary Tables 2A,B). For L. kaempferi, we obtained 2,746 up-regulated DEGs and 2,080 down-regulated DEGs in the X vs. P group, and 3,150 up-regulated DEGs and 3,063 down-regulated DEGs in the X vs. L group (Figure 2B; Supplementary Tables 2C,D). By taking the overlapping intersection of X vs. P and X vs. L, finally, we finally obtained 5,954 up-regulated DEGs and 7,953 down-regulated DEGs in the xylem of P. alba × P. glandulosa (Figure 2A), and 1,648 up-regulated and 948 down-regulated DEGs in the xylem of L. kaempferi (Figure 2B).
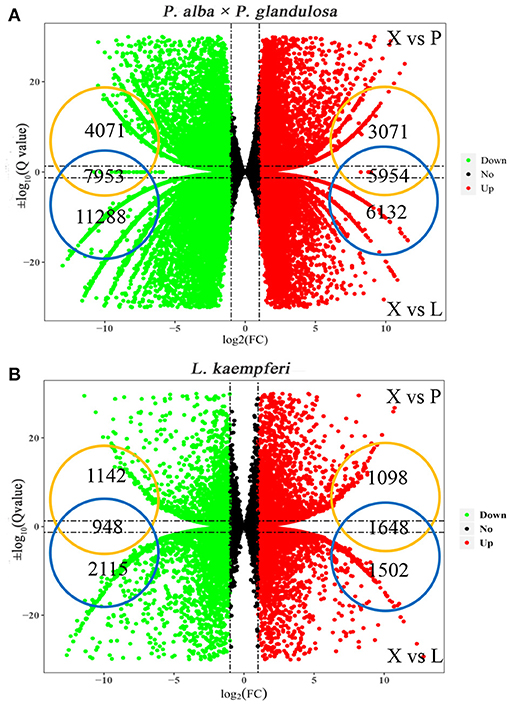
Figure 2. Distributions of up- and down-regulated DEGs in the two tree species. (A) Up and down-regulated DEGs in P. alba × P. glandulosa. (B) Up and down-regulated DEGs in L. kaempferi. In both panels, X vs. P represents the differential expression analysis between the xylem and phloem; likewise, X vs. L represents the differential expressed analysis between the xylem and leaf. The vertical dotted lines correspond to the log2 (fold- change) = ±1, while the horizontal dotted lines are positioned at log10 (Q-value) = ±1.3 (Q-value = 0.05). The genes at the intersection of two groups are the up- and down-regulated genes in xylem tissue.
To detect and check the robustness of our RNA-Seq results, we selected four genes in P. alba × P. glandulosa: Pop_G14G045130 (PagBXL2), Pop_G16G060785 (PagIRX9), Pop_G06G002976 (PagPME2), and Pop_A14G029833 (PagARA12), and similarly four genes in L. kaempferi: Lkgene4125 (LkIRX9H), Lkgene760 (LkMSR2), Lkgene166 (LkC4H), and Lkgene11869 (LkSUS4). These were used to perform qRT-PCR in xylem, phloem, and leaf tissues (Figure 3). In P. alba × P. glandulosa, except for Pop_G06G002976, the other three genes were significantly up-regulated or down-regulated in xylem when compared to the other tissues. In L. kaempferi, all four selected genes were significantly up-regulated or down-regulated in xylem (Figure 3). Hence, the qRT-PCR results agreed with the RNA-Seq analysis. The linear relationship between the RNA-Seq and qRT-PCR results was fitted in the Origin software; this showed their data were significantly correlated (p value = 1.47E-14). The slope of the fitted line was 20.15, indicating a positive relationship between RNA-Seq and qRT-PCR results, and the goodness-of-fit was R2 = 0.553 (Figure 3). Taken together, these results indicated our RNA-Seq results were reliable and useful for further analysis.
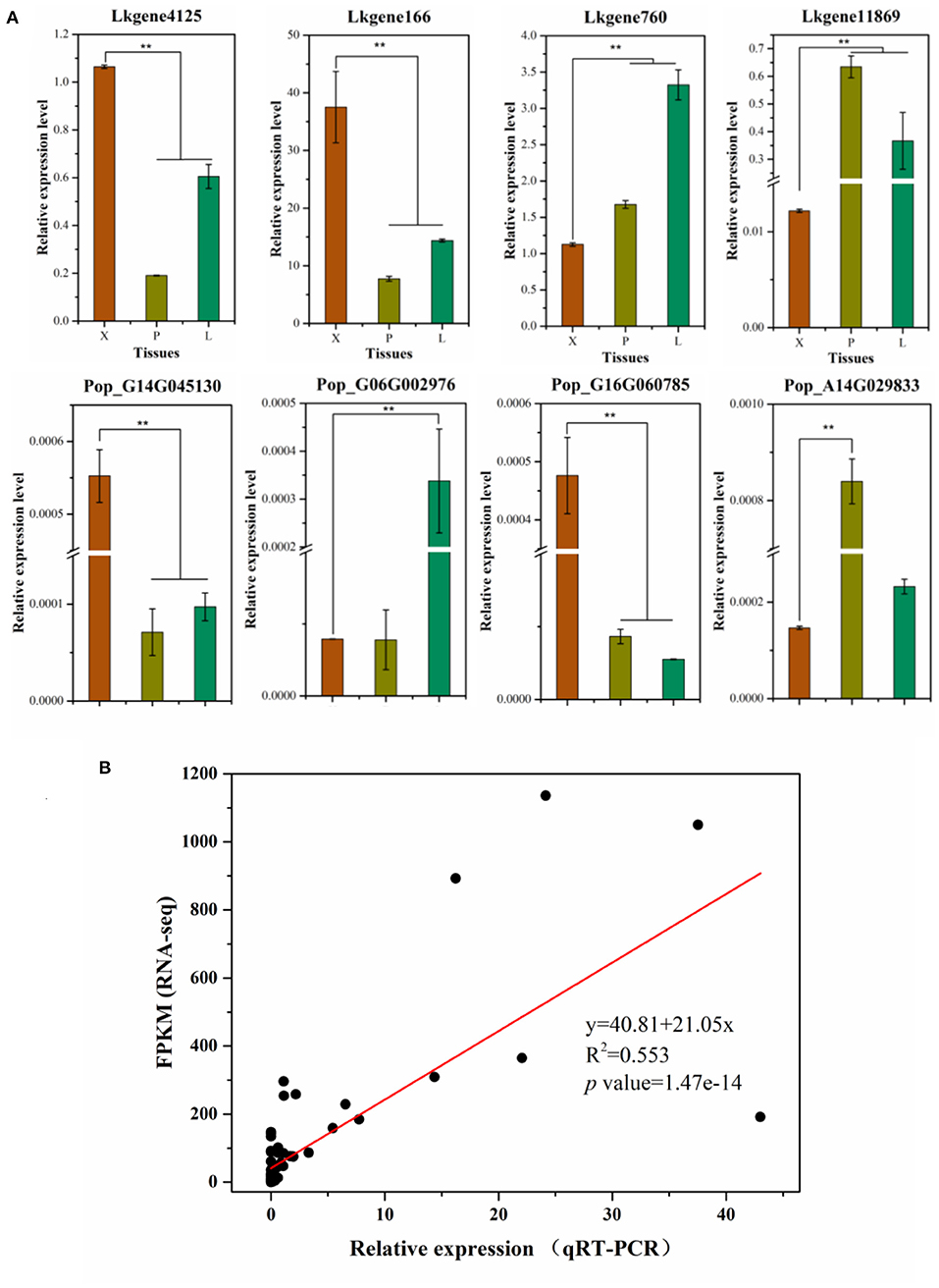
Figure 3. Validation of RNA-Seq results by qRT-PCR. (A) The qRT-PCR results for selected genes in the two tree species. (B) Linear regression for the RNA-Seq and qRT-PCR results. In this graph, gene expression levels in RNA-Seq are standardized by FPKM, the expression levels in qRT-PCR were standardized by the 2−Δct algorithm. In (A), the relative expression levels in the three tissue types are shown as the mean ± sd; the ** indicates a significant difference at P < 0.01 according to a one-way ANOVA.
By filtering the DEGs, we obtained 13,907 xylem-specifically expressed genes (5,954 up-regulated, 7,953 down-regulated) in P. alba × P. glandulosa, and 2,596 xylem-specifically expressed genes (1,648 up-regulated, 948 down-regulated) in L. kaempferi. To assess and compare the functional differences of these genes between the two species, we implemented GO and KEGG analyses. Three main ontologies—biological process (BP), cellular component (CC), and molecular function (MF)—were considered here. To fully display the GO terms, we reordered them according to the Q-values of the GO results and selected only the top 10 GO terms in each ontology category for the visualization.
In P. alba × P. glandulosa, 13,907 xylem-specifically expressed genes were clustered in 50 GO terms. Under BP, genes were significantly enriched in single organism process (GO: 0044699, Q-value = 1.4E-07), localization (GO: 0051179, Q-value = 5.45E-05), and growth (GO: 0040007, Q-value = 0.017); under MF, genes were significantly enriched in transporter activity (GO: 0005215, Q-value = 2.20E-14) and electron carrier activity (GO: 0009055, Q-value = 1.1E-4); finally, under CC, genes were significantly enriched in membrane (GO: 0016020, Q-value = 3.16E-58), membrane part (GO: 0044425, Q-value = 3.16E-58), supramolecular fiber (GO: 0099512, Q-value = 1E-5), extracellular region (GO: 0005576, Q-value = 1.1E-3), cell junction (GO: 0030054, Q-value = 2.37E-2), and symplast (GO: 0055044, Q-value = 2.37E-2) (Supplementary Table 3A). In L. kaempferi, 2,596 xylem-specifically expressed genes were enriched in 43 GO terms. Under BP, genes were significantly enriched only in single organism process (GO: 0044699, Q-value = 6.84E-09); under MF, genes were significantly enriched in catalytic activity (GO: 0005576, Q-value = 1.903E-33), nucleic acid binding transcription factor activity (GO: 0099512, Q-value = 6.0E-08), and structural molecule activity (GO: 0044425, Q-value = 9.05E-3); under CC, genes were significantly enriched in membrane part (GO: 0003824, Q-value = 3.486E-11), membrane (GO: 0001071, Q-value = 2.39E-2), and nucleoid (GO: 0005198, Q-value = 2.7E-2) (Supplementary Table 3C). In Supplementary Figures 1A,C, more detailed GO terms are shown for the two species. According to the GO results, genes in P. alba × P. glandulosa tend to increase plant growth and enable the directed movement of substances within cells; in contrast, genes in L. kaempferi involved in the structural integrity of a complex or its assembly within or outside a cell, catalyzed reactions process. In sum, our GO analysis results indicated different functions between the angiosperm and gymnosperm trees.
For the KEGG analysis, we reordered the results according to the Rich factor and chose the top 30 pathways for visualization purposes. In P. alba × P. glandulosa, 13,907 xylem-specifically expressed genes were enriched in 132 pathways. The top three significantly enriched pathways were photosynthesis (ko00195), metabolic pathways (ko01100), and photosynthesis-antenna proteins (ko00196) (Supplementary Figure 1B; Supplementary Table 3B) with corresponding enriched gene numbers of 99, 1,605, and 34 respectively. In L. kaempferi, 2,596 xylem-specifically expressed genes were clustered in 112 pathways; its top three significantly enriched pathways were starch and sucrose metabolism (ko00500), flavonoid biosynthesis (ko00941), and metabolic pathways (ko01100). The enriched gene numbers respectively were 196, 59, and 655 (Supplementary Figure 1D; Supplementary Table 3D). Hence, these KEGG results also uncovered different gene functions during the metabolism process between the two tree species.
Here, we selected two main pathways related to wood formation: phenylpropanoid biosynthesis, and starch and sucrose metabolism (Figure 4), and went on to associate each with those xylem-specifically expressed genes according to the KEGG results. Evidently, in both pathways, most of those genes were active in the same step during lignin and cellulose synthesis processes in the two species. Yet differences did exist between these tree species. For instance, in the phenylpropanoid biosynthesis pathway, during the transformation from ferulic acid to 5-hydroxyl ferulic acid, coniferyl-aldehyde to 5-hydroxy-coniferaldehyde, and coniferyl-alcohol to 5-hydroy-coniferyl alcohol, we found that six highly expressed genes (homologous to FAH1) participated in these steps in P. alba × P. glandulosa. These genes encode enzymes that catalyze the hydroxylation of coniferyl alcohol and coniferaldehyde during syringyl lignin formation (Wu et al., 2018). In stark contrast, such genes were apparently absent in L. kaempferi (Figure 4A; Supplementary Tables 4A,B).
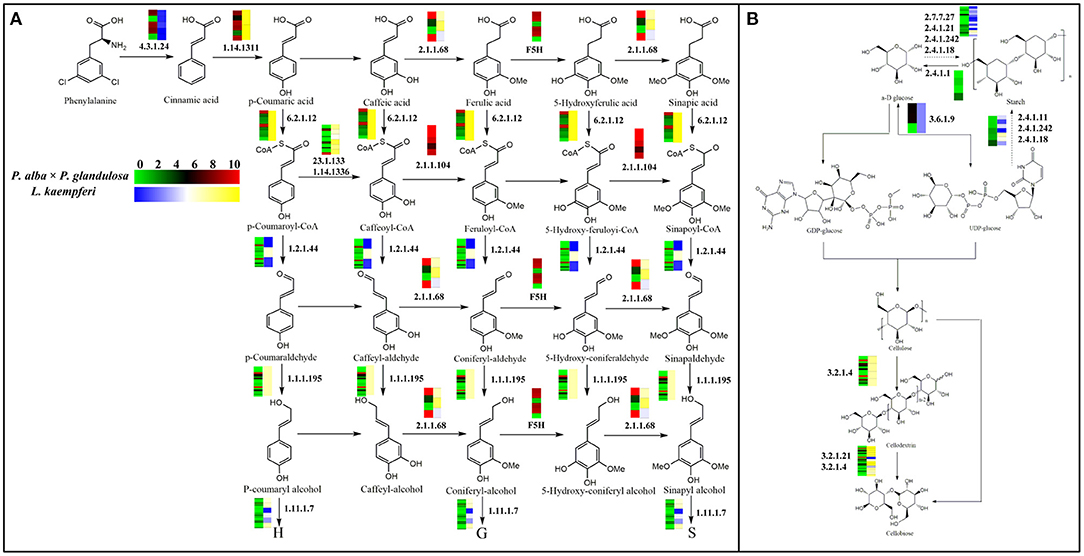
Figure 4. Gene expression model for the lignin and cellulose biosynthesis pathway. (A) Branch of phenylpropanoid biosynthesis pathway that is related to lignin biosynthesis. (B) Branch of starch and sucrose metabolism pathway that is related to cellulose biosynthesis. The heatmaps in each pathway are visualized according to gene expression levels. In the color bar, red indicates a high expression level and green a low expression level in P. alba × P. glandulosa; yellow corresponds to high expression level and blue a low expression level in L. kaempferi. The dotted arrows in the pathways denote an indirect linkage. The solid arrows refer to a direct relationship during compound synthesis. Numbers in the graph are EC identifier of corresponding enzyme.
During the biosynthesis process of cinnamic acid from phenylalanine, most genes in P. alba × P. glandulosa were highly expressed, whereas most genes in L. kaempferi were expressed at low levels. In the synthesis process converting cafferyl-CoA to feruloyl-CoA, and 5-hydroxy-feruloyl CoA to sinapoyl-CoA, we identified five active genes (homologous to CCoAOMT1 and CCOAMT) in P. alba × P. glandulosa (Figure 4A; Supplementary Tables 4A,B), known to related to lignin content and composition (Xie et al., 2019). In the starch and sucrose metabolism pathway, four genes (homologous toPHS1 and DPE2) were expressed in P. alba × P. glandulosa, these participating in the transformation from starch to α-D glucose (Figure 4B; Supplementary Tables 4A,B). These divergent results may explain why the compound synthesis mechanism differs between gymnosperm and angiosperm species during their wood formation.
Although both tree species also harbor homologous genes involved in the same steps, their respective expression patterning was not totally identical, with certain discrepancies among them. For example, during the formation of an alcohol-derived compound from an aldehyde-derived compound, we distinguished 21 genes—homologous to CAD4, 6, 7, 9, K9L2.15,0.18,0.19,0.20, MEE23, T17H7.1, and FOX2—expressed in these steps in P. alba × P. glandulosa. Among them, CAD (cinnamyl alcohol dehydrogenase) family members are involved in lignin biosynthesis and catalyze the final step specific for the production of lignin monomers (Kim et al., 2004). A member of the BBE-like family, T17H7.1, mediates the oxidation of cinnamyl alcohol and of p-hydroxylated derivatives of cinnamyl alcohol during the monolignin metabolism process (Daniel et al., 2015). Yet only Lkgene2978 (homologous to F19I3.4) was expressed in L. kaempferi (Figure 4A, Supplementary Tables 4A,B). This disparity between species suggests that P. alba × P. glandulosa relies on a more complicated mechanism than the gymnosperm species during these steps of wood formation. Nevertheless, despite belong to different phyla, P. alba × P. glandulosa and L. kaempferi have expression models with notable similarities. For example, some genes found (homologous to T16L4.190) participated in the reciprocal transformation between α-D glucose and UDP-glucose in both P. alba × P. glandulosa and L. kaempferi (Figure 4B; Supplementary Table 4B). This emphasizes that different species within the same phyla could be similar in some aspects.
To better understand the relationships among those xylem-specifically expressed genes, we performed a WGCNA analysis (Lukens and Downs, 2012) (Supplementary Figure 2). For P. alba × P. glandulosa, its 13,907 xylem-specifically expressed genes could be divided into 20 dynamic modules. After merging them, eventually we obtained four modules: a dark magenta module, an orange module, a saddle brown module, and a white module (Supplementary Figure 2A). For L. kaempferi, its 2,596 xylem-specifically expressed genes were divided into 22 dynamic modules. After merging those, three modules were obtained: a dark green module, a green module, and a midnight blue module (Supplementary Figure 2B).
After doing the WGCNA analysis for each selected module per species, the top 50,000 gene pairs ordered by the edge weight coefficient were chosen to build a gene co-expression network (Figure 5; Supplementary Table 5). A follow-up GO analysis was then performed for each module, from which only the top five GO numbers in BP are shown graphically (Figure 5). For P. alba × P. glandulosa, the GO analysis showed that more of its wood formation-related genes were clustered in the orange module. Among these top five GO terms, GO: 0009808 had a relationship with lignin biosynthesis (Figure 5; Supplementary Table 5C). In L. kaempferi, the genes were significantly enriched in starch metabolic process (GO: 0005982) and starch biosynthetic process (GO: 0019252) in the dark green module (Figure 5; Supplementary Table 5I), as were the phenylpropanoid catabolic process (GO: 0046271), lignin catabolic process (GO: 0046274), lignin metabolic process (GO: 0009808), and phenylpropanoid metabolic process (GO: 0009698) in the midnight blue module (Figure 5; Supplementary Table 5M).
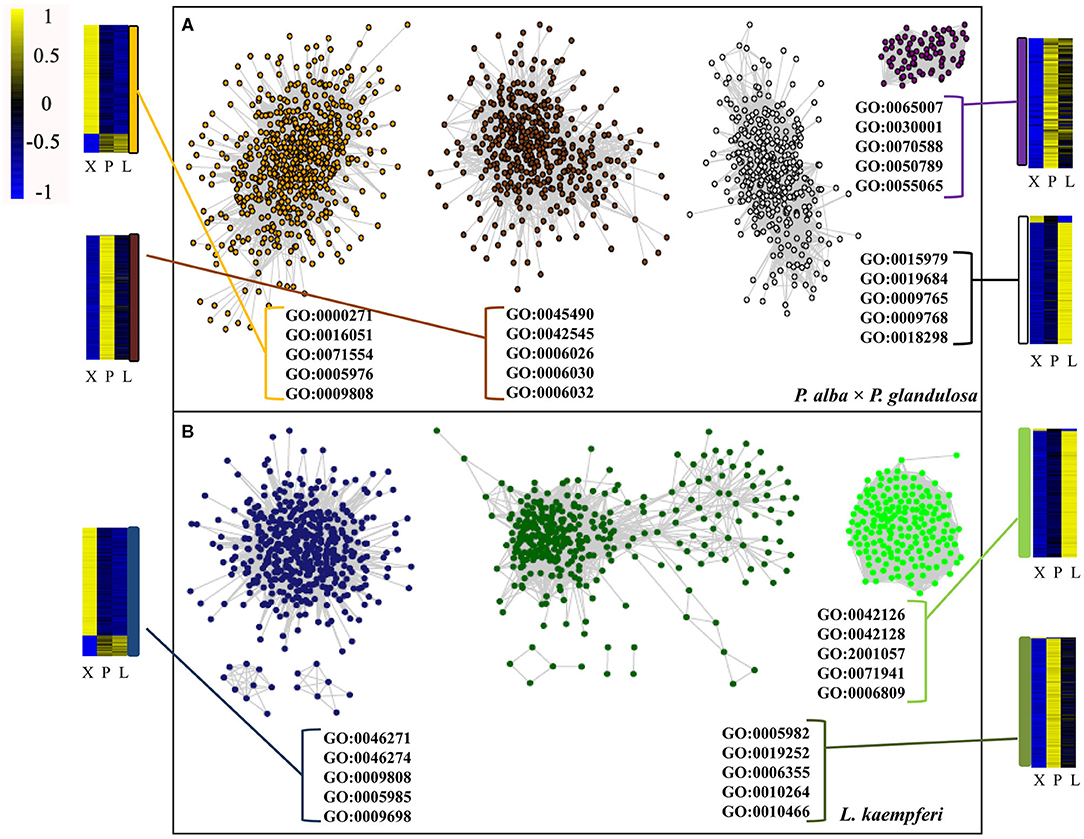
Figure 5. Co-expression network and GO analysis for each module in the two tree species. (A) Co-expression network in P. alba × P. glandulosa. (B) Co-expression network in L. kaempferi. In these networks, only the top 50 000 gene co-expression pairs are displayed. Only the top five GO terms under the biological process category appear in each module. The heatmaps are for gene expression levels in the different modules.
Combining the GO and KEGG results and the BLASTn to Arabidopsis, we then screened the wood formation-related genes among all xylem-specifically expressed genes and built the former's co-expression network in the two tree species (Figure 6). In the resulting co-expression network, 36 co-expressed genes were specific to P. alba × P. glandulosa (Supplementary Table 6A), 34 co-expressed genes were specific to L. kaempferi (Supplementary Table 6B), and 27 genes were shared by the two species (Supplementary Table 6C). Of the 36 specifically expressed genes in P. alba × P. glandulosa, four NAC members (VND1, VND5, SND2, SND3), two MYB members (MYB26, MYB52), two laccase members (LAC2, LAC4), and two cysteine protease members (XCP1, XCP2) were obtained. Among these, SND2 is known to play a crucial role in the biosynthesis of cellulose, mannan, and xylan, in addition to cell wall modification and lignin polymerization, but not so in monolignol biosynthesis. The SND2 promotes the up-regulation of several TF genes, such as MYB103 and SND1, and it occupies a subordinate position in the transcriptional regulatory network (Hussey et al., 2011). SND3 is directly activated by SND1/NST1 and VND6/VND7, yet there is still no evidence to prove SND3 is a direct target of VND6 or VND7 (Hussey et al., 2011). Both VND1 and VND5 are specifically expressed in vessels, where they activate the expression of secondary wall biosynthetic genes for cellulose, xylan, and lignin and concomitantly induce the ectopic deposition of secondary walls (Zhou et al., 2014).
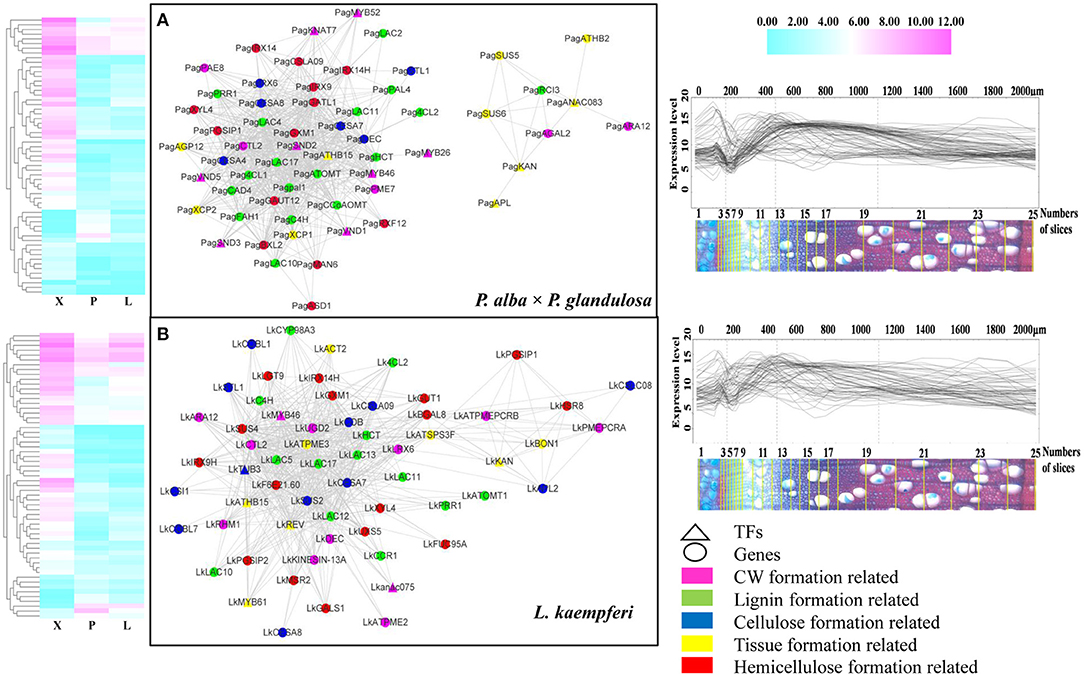
Figure 6. Co-expression network of wood formation-related genes in the two tree species. (A) Network for wood formation-related genes in P. alba × P. glandulosa. (B) Network for wood formation-related genes in L. kaempferi. The heatmaps (left) and gene expression abundance curves (right) depict the corresponding genes in each co-expression network. The scales above the expression curves indicate the slice position; the scales under expression curves indicate the number of slices. Gene expression abundance curves were derived and drawn using an online tool (http://aspwood.popgenie.org).
We also obtained two notable cysteine protease gene family members, XCP1 and XCP2, in the co-expression network of P. alba × P. glandulosa. This gene family is involved in other PCD (programmed cell death) events that contribute to plant organ senescence, plant defense responses, and nutrient mobilization. Both XCP1 and XCP2 are frequently used as markers for xylogenesis, and prominently positioned in models of TEs' (tracheary elements) PCD to carry out their autolysis. They are dispersed in the cytoplasmic compartment and imported into the vacuole (Funk et al., 2002; Avci et al., 2008; Zhang et al., 2014). In our results, however, the absence of XCP1 and XCP2 in L. kaempferi (Supplementary Table 6B) may have led to a TE PCD process unlike that which characterizes P. alba × P. glandulosa.
In 34 specifically expressed genes in L. kaempferi (Supplementary Table 6B), one NAC member (ANAC075), three laccase members (LAC5, LAC12, LAC13), and one MYB member (MYB61) were obtained. Among them, we know that ANAC075 promotes the expression of the secondary wall-associated TF called MYB46 (Endo et al., 2015) and functions upstream of NAC030, VND7, NAC101, VND6, LBD30, and ASL19 (Sakamoto and Mitsuda, 2015). We then verified the co-expression relation for all the genes forming the network via AspWood web resources (http://aspwood.popgenie.org). We used BLASTn to find the homologous genes in both tree species (Supplementary Tables 6D,E). As Figure 6 shows, for P. alba × P. glandulosa, 59 out of 63 genes in its network were co-expressed in xylem (Figure 6A; Supplementary Table 6D). Similarly, in L. kaempferi, 57 out of 64 genes in its network were co-expressed in xylem (Figure 6B, Supplementary Table 6E). For either tree species, the genes in the co-expression network corresponded to actual co-expression relationships present across xylem tissue (≥ 300 μm). This suggests our co-expression networks were robust and valid for inference.
Seven AS forms were detected, namely the alternative 3′ splice site (A3SS), alternative 5′ splice site (A5SS), alternative first exon, alternative last exon, mutually exclusive exon, retained intron (RI), and skipped exon (Figure 7A). We derived an AS model of genes expressed in xylem of P. alba × P. glandulosa and L. Kaempferi (Figure 7B). This showed similar proportions for different tree species, in that A3SS, A5SS, and RI are the main splicing models. Further investigation of the common wood formation-related genes for both species showed that CSLA9, UGD2, and DEC featured AS. The verification of AS in both tree species was pursued and these results confirmed that CSLA9 in L. kaempferi accorded with the putative AS results presented in Figure 8. Although our bioinformatic analysis indicated the gene CSLA9 also can undergo AS in P. alba × P. glandulosa, our RT-PCR result failed to detect it. We also confirmed AS variants of UGD2 in L. kaempferi, and those of DEC in P. alba × P. glandulosa (Figure 8). Collectively, these results demonstrated there is differential post-transcriptional regulation in P. alba × P. glandulosa and L. kaempferi, which could point to the possible causes for the differing wood structure in these two kinds of trees.
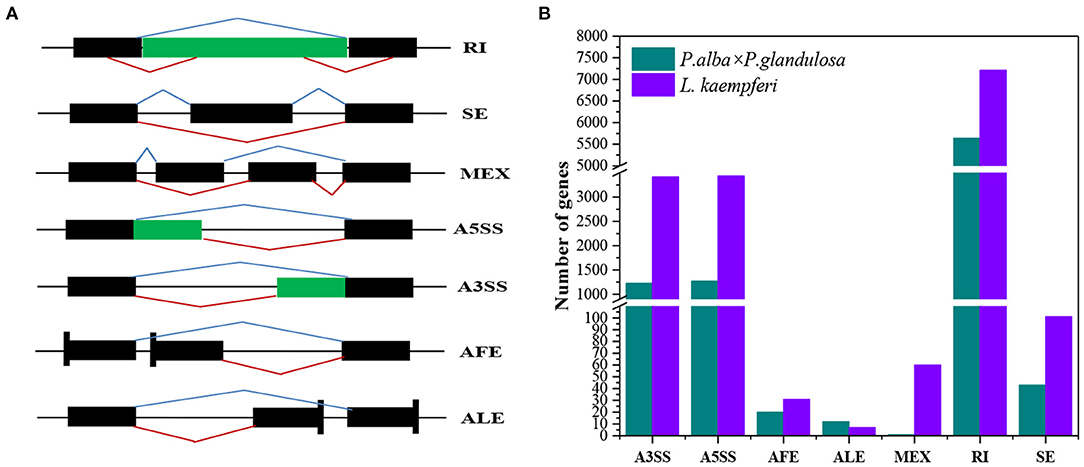
Figure 7. Comparison of different alternative splicing events between the two tree species. (A) Splicing patterns of seven kinds of alternative splicing. (B) Distributions of seven kinds of alternative splicing in each of the two tree species. A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; AFE, alternative first exon; ALE, alternative last exon; MEX, mutually exclusive exon; RI, retained intron; SE, skipped exon. Black blocks are exons, while the green blocks and black lines represent introns. Blue broken lines above the block(s) represent normal transcripts; red broken lines are the alternative splicing events.
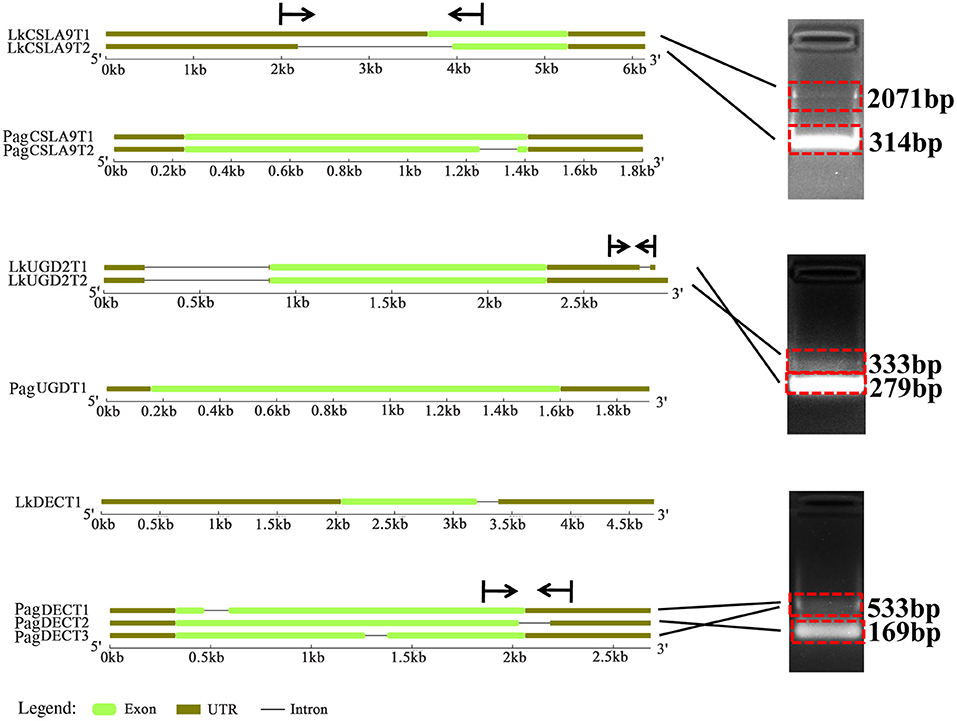
Figure 8. Comparison of the gene structure for three common genes and their variants in the two tree species. Vertical lines and arrows indicate the position of designed primers and amplification direction of fragments, respectively.
In comparison to gymnosperm species, angiosperms underwent massive adaptive radiation to supplant the gymnosperms as the world's dominant vascular plant group (Sanderson and Donoghue, 1994; Pavy et al., 2012). During evolutionary process, angiosperm wood form their own structure characters which is mostly composed of vessels, rays, fibers, and parenchyma cells and sharply contrast to the relatively simple gymnosperms' wood which mainly consists of tracheid and rays (Zhang et al., 2017). Besides these wood structural differences, gymnosperms and angiosperms also differ in their HD-Zip III genes and hemicelluloses ingredients (Côté et al., 2010); in the binding of multiple xylan chains to adjacent planes of the cellulose fibril (Marta et al., 2016); metabolically, in the formation of syringyl lignin (Nakamura et al., 1974), and; in their cell wall-associated peroxidases involved in xylem lignification (Mcdougall, 2001).
Gene co-expression represents interactive relationships among genes to a certain extent (Mitsuda and Ohme-Takagi, 2009). For TFs, their co-expression is indicative of coding function-related or protein–protein interactions (Mitsuda and Ohme-Takagi, 2009), and more generally, co-expressed genes may have up- or down-stream relationships in the transcriptional cascade (Hirai et al., 2007). In the constructed co-expression network, we detected similar and different co-expressed genes between the two tree species (Supplementary Tables 6A–C). Although both species do share some common genes, these genes still had some differences in their structure. For example, in their investigation of the structure of HD-Zip III family between angiosperms and gymnosperms, Côté et al. (2010) found that gymnosperm sequences derived from lineages that diverged earlier than angiosperm sequences, and that some sequences were lost in angiosperms indicates the full-length cDNA is longer in gymnosperms than angiosperms. Consistent with the above research (Côté et al., 2010), our gene structure analysis showed that UGD2, CSLA9, and DEC in L. kaempferi are longer than in P. alba × P. glandulosa, especially in the UTR region, suggesting lost sequences in P. alba × P. glandulosa.
Both FAH1 and CYP98A3 are members of P450 (Cytochrome P450). We found that FAH1, also named F5H, was expressed specifically in the xylem of P. alba × P. glandulosa (Figure 4A, Supplementary Table 6A). In contrast, CYP98A3 was expressed only in the xylem of L. kaempferi (Supplementary Table 6B) where it could catalyze the hydroxylation of shikimic acid and quinic acid to form monolignin. Its substrate mainly includes p-coumarate, p-coumaraldehyde, and p-coumaroyl methyl ester, which differs from FAH1. Other studies also found that CYP98A3 could participate in the biosynthesis process of the coumarins scopoletin and scopolin (Schoch et al., 2001; Abdulrazzak et al., 2006). Previous research indicated that FAH1 is associated with S-lignin monomer formation, in that the expression of FAH1 could increase the ratio of S/G (Boerjan et al., 2003). In our study, FAH1 was only present in P. alba × P. glandulosa, indicating a high syringyl lignin content and high proportion of S/G in this species. This result is consistent with work by Boerjan et al. (2003), which revealed that most dicotyledonous angiosperms (hardwood) are rich in G and S lignin monomers yet poor in the H lignin monomer. In gymnosperms (softwood), the G lignin monomer is the main lignin monomer and S and H lignin monomers' content is low (Boerjan et al., 2003). This implies a pronounced difference in the lignin monomer formation process between angiosperm and gymnosperm plants. We found SND2 was specific to P. alba × P. glandulosa whereas ANAC075 was specific to L. kaempferi. Both SND2 and ANAC075 can influence glucose and xylose and lignin contents, but ANAC075 has a greater transcriptional activation ability than does SND2, suggesting a different wood formation mechanism between angiosperm and gymnosperm species (Sakamoto and Mitsuda, 2015).
Although MYB family members were found in the studied two tree species, MYB26 and MYB52 were specifically expressed in the xylem of P. alba × P. glandulosa (Supplementary Table 6A). MYB26 localizes to the nucleus and regulates endothelial development and the secondary wall- thickening process (Yang et al., 2007). MYB52 is involved in both the ABA response and cell wall biosynthesis, and its overexpression in Arabidopsis improves this plant's drought tolerance and salt-sensitivity (Park et al., 2011). Even so, MYB52, SND2, and SND3 together with KAN7 can also influence the secondary cell wall-thickening process in fiber cells under the control of SND1 (Zhong et al., 2008). MYB61 mainly participates in xylem formation, by inducing qualitative changes to the xylem cell structure and lateral root development (Romano et al., 2012). The characteristic expression model in P. alba × P. glandulosa illustrated a distinctive wood formation process when compared with that of gymnosperm species. Laccase genes were reportedly active in the late stage of lignin formation and could promote lignification of the secondary xylem cell walls (Brown et al., 2005) and their different subcellular localization implies different functions among the laccase members. For instance, some research indicates that laccase can play additional roles in plants that go beyond the lignification process (Cai et al., 2006). In our results, we found co-expression of laccase members LAC2 and LAC4 in P. alba × P. glandulosa, and likewise that of LAC5, LAC12, and LAC13 in L. kaempferi, which suggests laccase genes may be the key factors influencing wood structure. Fine-scale distinctions among these laccase genes' functioning deserve further investigation.
Alternative splicing (AS) is a criticalpost-regulation process for the expression of genes. In this way, different transcripts could encode different proteins to perform various functions and this contributes to the variety of transcripts and proteins available for use (Chen and Manley, 2009; Reddy and Yamile, 2013). Researchers have since discovered that approximately 30 to 60% of mRNAs have variants (Zhang et al., 2010; Shen et al., 2014). According to our results, AS events characterized about 14.5% of the genes (8,207 of 56,711) in P. alba × P. glandulosa, but this was threefold greater, at 46.7% (14,259 of 30,726 genes), in L. kaempferi. Compared with other research (Zhang et al., 2010; Shen et al., 2014), our results uncovered a lower proportion of AS events in P. alba × P. glandulosa, perhaps due to the quality of its references. A previous study also showed that the retained intron is the main AS form, constituting approximately 60% of all AS events in plants (Syed et al., 2012). Similarly, in our study, the chief form of AS encountered was the retained intron, occurring in about 68.7% of genes (5,639 out of 8,207) in P. alba × P. glandulosa and 50.6% of those (7,209 out of 14,259) in L. kaempferi.
In this study, we compared the xylem-specifically expressed genes between P. alba × P. glandulosa and L. kaempferi trees on the transcriptional level and post-transcriptional level. Our results showed that differences exist in the enriched GO terms and KEGG pathways, gene expression models of the lignin and cellulose biosynthesis-related pathway, co-expression relationships, and alternative splicing forms, which together indicates different wood formation processes between P. alba × P. glandulosa and L. kaempferi. Our research provides a timely foundation for the further discovery and elucidation of differing wood formation mechanisms between angiosperm and gymnosperm species.
The original contributions presented in the study are publicly available. This data can be found at: NCBI repository, accession number: PRJNA723483.
HL and XD conceived and designed the study. QW and XD collected the plant materials. XD, QW, and HL extracted the RNA. HP, XD, and HL conducted the validation of RNA-Seq and alternative splicing events. HL and GC performed the bioinformatics analyses and carried out the data visualization. HL and XD wrote the manuscript. All of the authors revised and approved the manuscript.
This research was supported by the Fundamental Research Funds for the Central Non-profit Research Institution of CAF (CAFYBB2018ZY001-8 and CAFYBB2017ZY001) and the Youth Program of National Natural Science Foundation of China (31800267).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.698602/full#supplementary-material
Abdulrazzak, N., Pollet, B., Ehlting, J., Larsen, K., Asnaghi, C., Ronseau, S., et al. (2006). A coumaroyl-ester-3-hydroxylase insertion mutant reveals the existence of nonredundant meta-hydroxylation pathways and essential roles for phenolic precursors in cell expansion and plant growth. Plant Physiol. 140, 30–48. doi: 10.1104/pp.105.069690
Aspeborg, H., Schrader, J., Coutinho, P. M., Stam, M., Kallas, Å., Djerbi, S., et al. (2005). Carbohydrate-active enzymes involved in the secondary cell wall biogenesis in hybrid aspen. Plant Physiol. 137, 983–997. doi: 10.1104/pp.104.055087
Au, K. F., Underwood, J. G., Lee, L., and Wong, W. H. (2012). Improving PacBio long read accuracy by short read alignment. PLoS ONE. 7:e46679. doi: 10.1371/journal.pone.0046679
Avci, U., Petzold, H. E., Ismail, I. O., Beers, E. P., and Haigler, C. H. (2008). Cysteine proteases XCP1 and XCP2 aid micro-autolysis within the intact central vacuole during xylogenesis in Arabidopsis roots. Plant J. 56, 303–315. doi: 10.1111/j.1365-313X.2008.03592.x
Bao, H., Li, E., Mansfield, S. D., Cronk, Q. C., El-Kassaby, Y. A., and Douglas, C. J. (2013). The developing xylem transcriptome and genome-wide analysis of alternative splicing in Populus trichocarpa (black cottonwood) populations. BMC Genomics 14:359. doi: 10.1186/1471-2164-14-359
Bell, C. D., Soltis, D. E., and Soltis, P. S. (2010). The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303. doi: 10.3732/ajb.0900346
Boerjan, W., Ralph, J., and Baucher, M. (2003). Lignin biosynthesis. Annu. Rev. Plant Biol. 54, 519–546. doi: 10.1146/annurev.arplant.54.031902.134938
Brown, D. M., Zeef, L. A., Ellis, J., Goodacre, R., and Turner, S. R. (2005). Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell 17, 2281–2295. doi: 10.1105/tpc.105.031542
Cai, X., Davis, E. J., Ballif, J., Liang, M., Bushman, E., Haroldsen, V., et al. (2006). Mutant identification and characterization of the laccase gene family in Arabidopsis. J. Exp. Bot. 57, 2563–2569. doi: 10.1093/jxb/erl022
Camargo, E. L., Ployet, R., Cassan-Wang, H., Mounet, F., and Grima-Pettenati, J. (2019). Digging in wood: new insights in the regulation of wood formation in tree species. Adv. Bot. Res. 89, 201–233. doi: 10.1016/bs.abr.2018.11.007
Carvalho, A., Paiva, J., Louzada, J., and Lima-Brito, J. (2013). The transcriptomics of secondary growth and wood formation in conifers. Mol. Biol. Int. 2013, 1–12. doi: 10.1155/2013/974324
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. MOL PLANT, 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, M., and Manley, J. L. (2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741–754. doi: 10.1038/nrm2777
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Côté, C. L., Boileau, F., Roy, V., Ouellet, M., Levasseur, C., Morency, M.-J., et al. (2010). Gene family structure, expression and functional analysis of HD-Zip III genes in angiosperm and gymnosperm forest trees. BMC Plant Biol. 10, 1–17. doi: 10.1186/1471-2229-10-273
Courtois-Moreau, C. L., Pesquet, E., Sjödin, A., Muñiz, L., Bollhöner, B., Kaneda, M., et al. (2009). A unique program for cell death in xylem fibers of Populus stem. Plant J. 58, 260–274. doi: 10.1111/j.1365-313X.2008.03777.x
Cronk, Q. C. B., and Forest, F. (2017). “The evolution of angiosperm trees: from palaeobotany to genomics,” in Comparative and Evolutionary Genomics of Angiosperm Trees. Plant Genetics and Genomics: Crops and Models, eds A. Groover and Q. Cronk (Cham: Springer), 1–17. doi: 10.1007/7397_2016_31
Daniel, B., Pavkov-Keller, T., Steiner, B., Dordic, A., Gutmann, A., Nidetzky, B., et al. (2015). Oxidation of monolignols by members of the berberine bridge enzyme family suggests a role in plant cell wall metabolism. J. Bio. Chem. 290, 18770–18781. doi: 10.1074/jbc.M115.659631
Dieset, A. (2011). Genetic variation of xylem formation in Norway spruce (Picea abies (L.) Karst.) clones with contrasting growth rhytm (Master thesis). Norwegian University of Life Sciences, Ås, Norway.
Du, J., Gerttula, S., Li, Z., Zhao, S. T., Liu, Y. L., Liu, Y., et al. (2019). Brassinosteroid regulation of wood formation in poplar. New Phytol. 225, 1516–1530. doi: 10.1111/nph.15936
Endo, H., Yamaguchi, M., Tamura, T., Nakano, Y., Nishikubo, N., Yoneda, A., et al. (2015). Multiple classes of transcription factors regulate the expression of vascular-related NAC-DOMAIN7, a master switch of xylem vessel differentiation. Plant Cell Physiol. 56, 242–254. doi: 10.1093/pcp/pcu134
Funk, V., Kositsup, B., Zhao, C., and Beers, E. P. (2002). The Arabidopsis xylem peptidase XCP1 is a tracheary element vacuolar protein that may be a papain ortholog. Plant Physiol. 128, 84–94. doi: 10.1104/pp.010514
He, S., Xie, Y., Sun, X., and Zhang, S. (2020). Comparative transcriptome analyses reveal candidate genes regulating wood quality in Japanese larch (Larix kaempferi). J. For. Res. 31, 65–73. doi: 10.1007/s11676-019-00997-8
He, X., and Groover, A. T. (2017). “The genomics of wood formation in angiosperm trees,” in Comparative and Evolutionary Genomics of Angiosperm Trees. Plant Genetics and Genomics: Crops and Models, eds E. Groover A. and E. Cronk Q. (Cham: Springer), 205–225. doi: 10.1007/7397_2016_17
Higuchi, T., Shimada, M., Nakatsubo, F., and Tanahashi, M. (1977). Differences in biosyntheses of guaiacyl and syringyl lignins in woods. Wood Sci. Technol. 11, 153–167.
Hirai, M. Y., Sugiyama, K., Sawada, Y., Tohge, T., Obayashi, T., Suzuki, A., et al. (2007). Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Sci. U S A. 104, 6478–6483. doi: 10.1073/pnas.0611629104
Hussey, S. G., Mizrachi, E., Spokevicius, A. V., Bossinger, G., Berger, D. K., and Myburg, A. A. (2011). SND2, a NAC transcription factor gene, regulates genes involved in secondary cell wall development in Arabidopsis fibres and increases fibre cell area in Eucalyptus. BMC Plant Biol. 11, 1–17. doi: 10.1186/1471-2229-11-173
Iseli, C., Jongeneel, C. V., and Bucher, P. (1999). “ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences,” in Proceedings of the International Conference on Intelligent Systems for Molecular Biology (Heidelberg), 138–148.
Jokipii-Lukkari, S., Delhomme, N., Schiffthaler, B., Mannapperuma, C., Prestele, J., Nilsson, O., et al. (2018). Transcriptional roadmap to seasonal variation in wood formation of Norway Spruce. Plant Physiol. 176, 2851–2870. doi: 10.1104/pp.17.01590
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 357–360. doi: 10.1038/nmeth.3317
Kim, M.-H., Cho, J.-S., Jeon, H.-W., Sangsawang, K., Shim, D., Choi, Y.-I., et al. (2019). Wood transcriptome profiling identifies critical pathway genes of secondary wall biosynthesis and novel regulators for vascular cambium development in Populus. Genes. 10:690. doi: 10.3390/genes10090690
Kim, S.-J., Kim, M.-R., Bedgar, D. L., Moinuddin, S. G., Cardenas, C. L., Davin, L. B., et al. (2004). Functional reclassification of the putative cinnamyl alcohol dehydrogenase multigene family in Arabidopsis. Proc. Natl. Sci. U S A. 101, 1455–1460. doi: 10.1073/pnas.0307987100
Kohl, M., Wiese, S., and Warscheid, B. (2011). Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 696, 291–303. doi: 10.1007/978-1-60761-987-1_18
Li, H., Huang, X., Li, W., Lu, Y., and Li, Q. (2020). MicroRNA comparison between poplar and larch provides insight into the different mechanism of wood formation. Plant Cell Rep. 39, 1199–1217. doi: 10.1007/s00299-020-02559-3
Li, J., Harata-Lee, Y., Denton, M. D., Feng, Q., Rathjen, J. R., Qu, Z., et al. (2017). Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 3, 1–13. doi: 10.1038/celldisc.2017.31
Li, Q., Lin, Y.-C., Sun, Y.-H., Song, J., Chen, H., Zhang, X.-H., et al. (2012). Splice variant of the SND1 transcription factor is a dominant negative of SND1 members and their regulation in Populus trichocarpa. Proc. Natl. Sci. U S A. 109, 14699–14704. doi: 10.1073/pnas.1212977109
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, X., Wu, H. X., and Southerton, S. G. (2010). Comparative genomics reveals conservative evolution of the xylem transcriptome in vascular plants. BMC Evol. Biol. 10, 1–14. doi: 10.1186/1471-2148-10-190
Lin, Y.-C. J., Chen, H., Li, Q., Li, W., Wang, J. P., Shi, R., et al. (2017). Reciprocal cross-regulation of VND and SND multigene TF families for wood formation in Populus trichocarpa. Proc. Natl. Sci. U S A. 114, E9722–E9729. doi: 10.1073/pnas.1714422114
Lorenz, W. W., Yu, Y.-S., and Dean, J. F. (2010). An improved method of RNA isolation from loblolly pine (P. taeda L.) and other conifer species. J. Vis. Exp. 36:e1751. doi: 10.3791/1751
Love, M., Anders, S., and Huber, W. (2014). Differential analysis of count data–the DESeq2 package. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, S., Li, Q., Wei, H., Chang, M.-J., Tunlaya-Anukit, S., Kim, H., et al. (2013). Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Sci. U S A. 110, 10848–10853. doi: 10.1073/pnas.1308936110
Lukens, L., and Downs, G. (2012). “Bioinformatics techniques for understanding and analyzing tree gene expression data,” in Genomics of Tree Crops, eds E. Schnell R.J. and E. Priyadarshan P.M. (New York, NY: Springer), 17–38. doi: 10.1007/978-1-4614-0920-5_2
Marta, Busse-Wicher, Li, A., Silveira Rodrigo, L., Caroline, S., et al. (2016). Evolution of xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant physiol. 171, 2418–2431. doi: 10.1104/pp.16.00539
Mcdougall, G. J. (2001). Cell-wall-associated peroxidases from the lignifying xylem of angiosperms and gymnosperms: monolignol oxidation. Holzforschung 55, 246–249. doi: 10.1515/HF.2001.040
Mitsuda, N., and Ohme-Takagi, M. (2009). Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiol. 50, 1232–1248. doi: 10.1093/pcp/pcp075
Nakamura, Y., Fushiki, H., and Higuchi, T. (1974). Metabolic differences between gymnosperms and angiosperms in the formation of syringyl lignin. Phytochemistry 13, 1777–1784. doi: 10.1016/0031-9422(74)85088-0
Nawawi, D. S., Syafii, W., Akiyama, T., and Matsumoto, Y. (2016). Characteristics of guaiacyl-syringyl lignin in reaction wood in the gymnosperm Gnetum gnemon L. Holzforschung 70, 593–602. doi: 10.1515/hf-2015-0107
Park, M. Y., Kang, J. Y., and Kim, S. Y. (2011). Overexpression of AtMYB52 confers ABA hypersensitivity and drought tolerance. Mol Cells. 31, 447–454. doi: 10.1007/s10059-011-0300-7
Pavy, N., Pelgas, B., Laroche, J., Rigault, P., Isabel, N., and Bousquet, J. (2012). A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 10, 1–19. doi: 10.1186/1741-7007-10-84
Reddy Anireddy, S. N.Yamile, Marquez, et al. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. doi: 10.1105/tpc.113.117523
Romano, J. M., Dubos, C., Prouse, M. B., Wilkins, O., and Campbell, M. M. (2012). AtMYB61, an R2R3-MYB transcription factor, functions as a pleiotropic regulator via a small gene network. New Phytol. 195, 774–786. doi: 10.1111/j.1469-8137.2012.04201.x
Roodt, D., Li, Z., Van de Peer, Y., and Mizrachi, E. (2019). Loss of wood formation genes in monocot genomes. Genome Biol Evol. 11, 1986–1996. doi: 10.1093/gbe/evz115
Sakamoto, S., and Mitsuda, N. (2015). Reconstitution of a secondary cell wall in a secondary cell wall-deficient Arabidopsis mutant. Plant Cell Physiol. 56, 299–310. doi: 10.1093/pcp/pcu208
Sanderson, M. J., and Donoghue, M. J. (1994). Shifts in diversification rate with the origin of angiosperms. Science. 264, 1590–1593. doi: 10.1126/science.264.5165.1590
Sarkanen, K. (1971). “Classification and distribution,” in Lignins Occurrence, Formation, Structure and Reactions, eds E. Sarkanen, K.V. and Ludwig, C.H. (New York: Wiley-Interscience). 43–94.
Schoch, G., Goepfert, S., Morant, M., Hehn, A., Meyer, D., Ullmann, P., et al. (2001). CYP98A3 from Arabidopsis thaliana is a 3′-hydroxylase of phenolic esters, a missing link in the phenylpropanoid pathway. J. Biol. Chem. 276, 36566–36574. doi: 10.1074/jbc.M104047200
Shen, Y., Zhou, Z., Wang, Z., Li, W., Fang, C., Wu, M., et al. (2014). Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell. 26, 996–1008. doi: 10.1105/tpc.114.122739
Shi, R., Sun, Y.-H., Li, Q., Heber, S., Sederoff, R., and Chiang, V. L. (2009). Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 51, 144–163. doi: 10.1093/pcp/pcp175
Sperry, J. S., Hacke, U. G., and Pittermann, J. (2006). Size and function in conifer tracheids and angiosperm vessels. Am. J. Bot. 93, 1490–1500. doi: 10.3732/ajb.93.10.1490
Sundell, D., Street, N. R., Kumar, M., Mellerowicz, E. J., Kucukoglu, M., Johnsson, C., et al. (2017). AspWood: high-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell. 29, 1585–1604. doi: 10.1105/tpc.17.00153
Suzuki, S., Li, L., Sun, Y.-H., and Chiang, V. L. (2006). The cellulose synthase gene superfamily and biochemical functions of xylem-specific cellulose synthase-like genes in Populus trichocarpa. Plant physiol. 142, 1233–1245. doi: 10.1104/pp.106.086678
Syed, N. H., Kalyna, M., Barta, B., et al. (2012). Alternative splicing in plants - coming of age. Trends Plant Sci. 17, 616–623. doi: 10.1016/j.tplants.2012.06.001
Tuskan, G. A., Groover, A. T., Schmutz, J., DiFazio, S. P., Myburg, A., Grattapaglia, D., et al. (2018). Hardwood tree genomics: unlocking woody plant biology. Front. Plant Sci. 9, 1799–1799. doi: 10.3389/fpls.2018.01799
Vanholme, R., Demedts, B., Morreel, K., Ralph, J., and Boerjan, W. (2010). Lignin biosynthesis and structure. Plant Physiol. 153, 895–905. doi: 10.1104/pp.110.155119
Wang, J. P., Matthews, M. L., Naik, P. P., Williams, C. M., Ducoste, J. J., Sederoff, R. R., et al. (2019). Flux modeling for monolignol biosynthesis. Curr. Opin. Biotechnol. 56, 187–192. doi: 10.1016/j.copbio.2018.12.003
Weng, J.-K., Li, X., Stout, J., and Chapple, C. (2008). Independent origins of syringyl lignin in vascular plants. Proc. Natl. Sci. U S A. 105, 7887–7892. doi: 10.1073/pnas.0801696105
Wu, Z., Wang, N., Hisano, H., Cao, Y., and Fu, C. (2018). Simultaneous regulation of F5H in COMT-RNAi transgenic switchgrass alters effects of COMT suppression on syringyl lignin biosynthesis. Plant Biotechnol. J. 17, 836–845. doi: 10.1111/pbi.13019
Xie, H., Engle, N. L., Venketachalam, S., Yoo, C. G., Barros, J., Lecoultre, M., et al. (2019). Combining loss of function of Folylpolyglutamate synthetase1 and caffeoyl-coa 3-o-methyltransferase1 for lignin reduction and improved saccharification efficiency in Arabidopsis thaliana. Biotechnol. Biofuels. 12, 1–20. doi: 10.1186/s13068-019-1446-3
Xu, P., Kong, Y., Song, D., Huang, C., Li, X., and Li, L. (2014). Conservation and functional influence of alternative splicing in wood formation of Populus and Eucalyptus. BMC Genom. 15, 1–12. doi: 10.1186/1471-2164-15-780
Yamamoto, R., Demura, T., and Fukuda, H. (1997). Brassinosteroids induce entry into the final stage of tracheary element differentiation in cultured Zinnia cells. Plant Cell Physiol. 38, 980–983. doi: 10.1093/oxfordjournals.pcp.a029262
Yang, C., Xu, Z., Song, J., Conner, K., Barrena, G. V., and Wilson, Z. A. (2007). Arabidopsis MYB26/MALE STERILE35 regulates secondary thickening in the endothecium and is essential for anther dehiscence. Plant Cell. 19, 534–548. doi: 10.1105/tpc.106.046391
Ye, Z.-H., and Zhong, R. (2015). Molecular control of wood formation in trees. J. Exp. Bot. 66, 4119–4131. doi: 10.1093/jxb/erv081
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, 1–12. doi: 10.1186/gb-2010-11-2-r14
Yuan, Y., Teng, Q., Zhong, R., and Ye, Z.-H. (2014). Identification and biochemical characterization of four wood-associated glucuronoxylan methyltransferases in Populus. PLoS ONE. 9:e87370. doi: 10.1371/journal.pone.0087370
Zang, Q.-L., Li, W.-F., and Qi, L.-W. (2019). Regulation of LaSCL6 expression by genomic structure, alternative splicing, and microRNA in Larix kaempferi. Tree Genet. Genomes 15, 1–7. doi: 10.1007/s11295-019-1362-5
Zeng, L., Zhang, Q., Sun, R., Kong, H., Zhang, N., and Ma, H. (2014). Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 5, 1–12. doi: 10.1038/ncomms5956
Zhang, B., Tremousaygue, D., Denance, N., van Esse, H. P., Horger, A. C., Dabos, P., et al. (2014). PIRIN2 stabilizes cysteine protease XCP2 and increases susceptibility to the vascular pathogen Ralstonia solanacearum in Arabidopsis. Plant J. 79, 1009–1019. doi: 10.1111/tpj.12602
Zhang, G., Guo, G., Hu, X., Zhang, Y., Li, Q., Li, R., et al. (2010). Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 20, 646–654. doi: 10.1101/gr.100677.109
Zhang, J., Xie, M., Tuskan, G. A., Muchero, W., and Chen, J.-G. (2018). Recent advances in the transcriptional regulation of secondary cell wall biosynthesis in the woody plants. Front. Plant Sci. 9:1535. doi: 10.3389/fpls.2018.01535
Zhang, M., Ji, C., Zhu, J., et al. (2017). Comparison of wood physical and mechanical traits between major gymnosperm and angiosperm tree species in China. Wood Sci. Technol. 51, 1405–1419. doi: 10.1007/s00226-017-0954-1
Zhao, Y., Sun, J., Xu, P., Zhang, R., and Li, L. (2014). Intron-mediated alternative splicing of PtrWND1B regulates cell wall thickening during fiber development in Populus. Plant Physiol. 164, 765–776. doi: 10.1104/pp.113.231134
Zhong, R., Lee, C., Zhou, J., McCarthy, R. L., and Ye, Z.-H. (2008). A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 20, 2763–2782. doi: 10.1105/tpc.108.061325
Zhong, R., and Ye, Z.-H. (2014). Complexity of the transcriptional network controlling secondary wall biosynthesis. Plant Sci. 229, 193–207. doi: 10.1016/j.plantsci.2014.09.009
Keywords: angiosperm, gymnosperm, transcriptome, co-expression network, alternative splicing, wood formation
Citation: Li H, Chen G, Pang H, Wang Q and Dai X (2021) Investigation Into Different Wood Formation Mechanisms Between Angiosperm and Gymnosperm Tree Species at the Transcriptional and Post-transcriptional Level. Front. Plant Sci. 12:698602. doi: 10.3389/fpls.2021.698602
Received: 21 April 2021; Accepted: 01 June 2021;
Published: 02 July 2021.
Edited by:
Wei Li, Northeast Forestry University, ChinaReviewed by:
Qingzhang Du, Beijing Forestry University, ChinaCopyright © 2021 Li, Chen, Pang, Wang and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinren Dai, eGlucmVuZEBjYWYuYWMuY24=
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.