 Chaorui Liu1,2
Chaorui Liu1,2- 1State Key Laboratory of Crop Biology, Shandong Agricultural University, Taian, China
- 2College of Forestry, Shandong Agricultural University, Taian, China
To reveal whether the response of mulberry to phytoplasma infection is associated with genome-wide DNA methylation changes, the methylome and transcriptome patterns of mulberry in response to phytoplasma infection were explored. Though the average methylation level of the infected leaves showed no significant difference from that of healthy leaves, there were 1,253 differentially methylated genes (DMGs) and 1,168 differentially expressed genes (DEGs) in the infected leaves, and 51 genes were found simultaneously to be differently methylated and expressed. It was found that the expression of G-type lectin S-receptor-like serine/threonine protein kinase gene (Mu-GsSRK) was increased, but its methylation level was decreased in the pathogen-infected or salicylic acid (SA)-treated leaves. Overexpression of Mu-GsSRK in Arabidopsis and in the hairy roots of mulberry enhanced transgenic plant resistance to the phytoplasma. Moreover, overexpression of Mu-GsSRK enhanced the expressions of pathogenesis-related protein 1, plant defensin, and cytochrome P450 protein CYP82C2 genes in transgenic plants inoculated with pathogens, which may contribute to the enhanced disease resistance against various pathogens. Finally, the DNA methylation dynamic patterns and functions of the differentially expressed and methylated genes were discussed. The results suggested that DNA methylation has important roles in mulberry responses to phytoplasma infection.
Introduction
Phytoplasmas are cell wall-less plant pathogens of the class, Mollicutes (Sugio et al., 2011), and they are associated with hundreds of diseases in more than 1,000 plants in the world and cause serious losses in vegetables, fruit crops, and ornamental plants (Bertaccini and Duduk, 2009; Cao et al., 2019). Phytoplasmas colonize in the host phloem tissue, where they secrete a variety of effector molecules to affect the expression of host genes, resulting in the metabolism disorder of hosts which shows various symptoms (Christensen et al., 2005; Gai et al., 2018). Though some virulence factors of phytoplasmas have been described, how these pathogens manipulate the physiological functions of plant hosts remains unclear (Namba, 2019).
As an important epigenetic mechanism, DNA methylation is associated with many biological processes, including transcriptional silencing, gene regulation, and genomic imprinting (Ji et al., 2018; Huang et al., 2019). It was reported that DNA methylation plays important roles in regulating responses to biotic stresses in plants, and evidence showed that DNA methylation levels were altered in potato (Solanum tuberosum), Arabidopsis thaliana, and tomato (S. lycopersicum) plants infected by pathogenic fungi, bacteria, or virus (Pavet et al., 2006; Torchetti et al., 2016; Zhu et al., 2016; De Palma et al., 2019). Some studies also showed that phytoplasmas were involved in the methylation changes of host genes. In Candidatus Phytoplasma aurantifolia-infected periwinkle (Catharanthus roseus) and tomato plants, sterol-C-methyltransferase and some methylase and demethylase genes were downregulated (Jagoueix-Eveillard et al., 2001), and it was found that DNA methylation was involved in the epigenetic regulation of SlDEFICIENS (Ahmad et al., 2012). In addition, it was shown that demethylation of some genes associated with floral development were inhibited in tomato plants infected by phytoplasmas (Pracros et al., 2006). It was also revealed that there were different DNA methylation marks between spontaneous and cultivar-dependent recovery and healthy leaves, occurring within the promoters of genes involved in secondary metabolism and photosynthesis (Pagliarani et al., 2020). Moreover, it has been shown that the global DNA methylation level was reduced in the phytoplasma-infected Paulownia seedlings (Cao et al., 2014). Taken together, these studies suggested that DNA methylation may play an important role in regulating gene expression in phytoplasma-infected plants.
Mulberry trees are planted all over the world and have been used to feed silkworms for about 5,000 years. Mulberry is not only the sole feed tree for silkworms, but it is also a nutritionally and economically important fruit tree, and mulberry fruits have long been used as edible fruits and traditional medicines. Mulberry plants are susceptible to dwarf disease associated with the presence of phytoplasma belonging to the subgroup, 16SrI-B (aster yellows) (Gai et al., 2014), and when faced with phytoplasma infection, they will activate radical changes of gene expression (Gai et al., 2018). Phytoplasmas are introduced into mulberry phloem by vector insects, parasitize in the sieve elements of phloem, and directly contact the cytoplasm of sieve elements and companion cells (Sugio et al., 2011). In the process of mulberry–phytoplasma interaction, the sieve elements and companion cells can not only perceive phytoplasma invasion and initiate local defense responses but can also transmit the infection signal over long distances and activate the response at the whole-plant level (Henry et al., 2013; Gai et al., 2018). Previous studies have shown that phytoplasma infection can cause differential expression of some genes involved in signal transduction in plants (Hren et al., 2009; Gai et al., 2018; Liu et al., 2019). Since gene transcriptional reprogramming may be controlled by epigenetic modifications triggered by pathogen challenges, the infection of phytoplasma may cause the differential expression of some genes involved in signal transduction mediated by DNA methylation in plants. However, the gene expression changes associated with phytoplasma infection and the molecular mechanism at DNA methylation level are still not well understood. In this study, methylation-dependent restriction site-associated DNA sequencing (MethylRAD-Seq) which allows for de novo (reference-free) methylation analysis can be applied to both model plants and non-eschatological plants, and it is performed to assess genome-wide DNA methylation patterns and to identify the differentially methylated genes (DMGs) involved in response to phytoplasma infection. The information provided will facilitate the elucidation of the epigenetic mechanisms underlying the responses of mulberry to phytoplasma infection, and also provide important clues for further studying the disease resistance genes for mulberry breeding.
Materials and Methods
Plant Materials and Growth Conditions
Mulberry (Morus multicaulis) cutting seedlings were inoculated with phytoplasmas by being grafted with the scions collected from phytoplasma-infected mulberry trees Gai et al. (2018), and the cutting seedlings derived from the same mother tree were grafted with healthy scions and used as controls. The grafted mulberry seedlings were cultivated in a greenhouse under light/dark regime (16 h light/8 h dark) at 24°C with 50–60% humidity. The grafted mulberry seedlings showing Witches' broom disease symptoms were used to detect phytoplasma by PCR amplification of the 16S rRNA gene of phytoplasma using a universal primer set (Forward primer: 5′ TAAAAGACCTAGCAATAGG 3′; Reverse primer: 5′ CAATCCGAACTGAGACTGT 3′) (Ji et al., 2009). The sixth leaves from the top of the grafted healthy and phytoplasma-infected shoots were used for subsequent experiments, and three phytoplasma-infected and healthy plants were used as independent biological replicates for RNA sequencing (RNAseq) and MethylRAD sequencing analyses. A. thaliana (Col-0) and Nicotiana benthamiana seedlings were cultivated in the greenhouse under light/dark regime (16 h light/8 h dark) at 22 and 24°C, respectively, with 50–60% humidity.
Transcriptome Analysis
Total RNA was extracted using a TRIzol reagent (Invitrogen Corporation, CA, United States), and the poly-A-containing messenger RNAs (mRNAs) were purified using beads with Oligo (dT). The purified mRNAs were used as templates to synthesize complementary DNAs (cDNAs) which were then added to a single “A” base to their ends. These cDNA fragments were subsequently connected with sequencing adapters and then were purified and underwent PCR amplification to create cDNA libraries. Transcriptome sequencing was conducted on the Illumina HiSeq™ 2000S platform, Illumina, San Diego, CA, United States. Raw reads were cleaned and assembled into unigenes and then the assembled unigenes were annotated by BLASTn searches against the M. notabilis genome (https://www.ncbi.nlm.nih.gov/genome/?term=Morus). The expressions of genes were calculated using the reads per kilobase million (RPKM), and if the expression value of a gene changed more than two times (P ≤ 0.05), and the false discovery rate (FDR) was <0.01 between infected and healthy samples, it was designated as a significantly differentially expressed gene (DEG). The identification of DEGs was performed using DESeq software (Zenoni et al., 2010). Three biological replicate experiments for healthy and infected samples and three technical replicates per biological experiment were performed.
MethylRAD Sequencing and Relative Quantification of DNA Methylation Levels
Genomic DNA was extracted from the infected and healthy mulberry leaves using the cetyltrimethyl ammonium bromide method (Clarke, 2009) and then was digested with FspEI. The digestion productions were added with adaptor primers and amplified through PCR to create the MethylRAD libraries which were subjected to sequencing on an Illumina HiSeq2000 sequencer, Illumina, San Diego, CA, United States. Input sequencing data were cleaned and then subjected to Pair-End sequencing on a HiSeq X Ten platform. After mapping the paired-end sequencing reads, the MethylRAD sequencing data were cleaned to obtain high quality reads which were mapped to the reference CCGG/CCWGG sites built with the FspEI sites extracted from the M. notabilis genome using the SOAP program. A site with the sequencing depth of not <3 was determined as a reliable methylation site, and the RPKM was used to determine the relative DNA methylation levels of each site. If the methylation level of a methylation site changed more than two times (P ≤ 0.05) between the infected and healthy samples, it was designated as a significantly differentially methylated site. Then the methylation levels of sites that were localized in the gene regions were summed to evaluate the DNA methylation levels of the genes using the R package edge R (Robinson et al., 2010), and the thresholds of statistically significant difference were found to have a p < 0.05 and log2FC > 1.
Gene Ontology Analysis
BlastN searches against the reference M. notabilis database were performed with online tools (https://morus.swu.edu.cn/morusdb/blast) to provide gene ID for the target genes. Gene ontology (GO) analysis was performed with online tools (https://morus.swu.edu.cn/morusdb/searchgo) based on the gene IDs obtained.
Quantitative Real-Time PCR Analysis
Total RNA was extracted using a TRIzol reagent (Invitrogen Corporation, CA, United States) following the instructions of the manufacturer and then it was digested with DNase I. The cDNA was synthesized from 1 μg of total RNA extracted using oligo (dT)18 primers (Invitrogen Corporation, CA, United States) with reverse transcriptase M-MLV (Promega Corporation, Madison, WI, United States) in 20 ml reactions. The qRT-PCR was performed on the CFX96TM Real-time System (Bio-Rad Laboratories, CA, United States) according to the protocol of the kit (SYBR Premix Ex Taq™) with 20 μl of PCR mix. The PCR mix contains 2 μl of synthesized cDNA, 10 μl of 2 × SYBR Premix Ex Taq, 200 nmol l−1 of gene-specific primers, and 0.4 μl of 50 × ROX Reference Dye II. The PCR mix was preheated to 95°C for 30 s followed by 39 cycles of 95°C for 10 s and 58°C for 30 s. Melting curve analysis was conducted from 65°C up to 95°C followed by a 0.5°C incremental ramp to validate product specificity. The two genes, EF1-a and ACTIN, were used as endogenous controls to quantify the gene expression levels by the 2−ΔΔCT method (Livak and Schmittgen, 2001). All samples were assayed in at least three biological replicates, and three technical replicates were run for each of them. The primers used for quantitative real-time PCR (qRT-PCR) are given in Supplementary Table 1.
DNA Methylation Analysis by Semi-Quantitative PCR
Since the restriction endonuclease, FspEI can recognize 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) in the CmC and mCDS sites in genomic DNA, the genomic DNA double-strand can be cleaved by FspEI at CCGG and CCWGG methylated sites. The extracted genomic DNA was digested with FspEI, and then the digested DNA was used as a template to perform 28 cycles of PCR amplification. The genomic DNA without being digested was also used as a template to perform 28 cycles of PCR amplification using the same primers and PCR conditions. All the PCR experiments were repeated at least three times. The primers used for semi-quantitative PCR are listed in Supplementary Table 1.
Gene Cloning and Phylogenetic Analysis
The isolated RNA was used to synthesize cDNA using the reverse transcriptase, M-MLV (Promega). The specific primers (Supplementary Table 2) used for PCR amplifications were designed based on the nucleotide sequence of the gene available from the M. notabilis genome (https://www.ncbi.nlm.nih.gov/genome/?term=Morus) database; the PCR products were separated by electrophoresis and the target DNA fragment was recovered and subcloned into the pMD18-T vector (Invitrogen). After transformation into DH5α, the positive clones were identified and selected for further sequencing. The multiple alignments of the deduced amino acid sequences with the sequences from other plants were conducted using the DNAMAN program. SignalP-5.0 Server (http://www.cbs.dtu.dk/services/SignalP/) was used with the default parameters to predict signal peptides, and TargetP-2.0 Server (http://www.cbs.dtu.dk/services/TargetP/) was used to predict the subcellular localization of proteins. Putative conserved domains were detected using the online NCBI program (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). A phylogenetic tree was generated using the MEGA program by the neighbor-joining method, and bootstrapping was run 1,000 times. The three-dimensional (3D) structure of the protein was generated by the SWISS-MODEL pipeline.
Subcellular Localization
In order to elucidate its subcellular localization, the fusion gene of Mu-GsSRK (GenBank: MN364943.1) and the green fluorescent protein gene (GFP) under the control of 35S was generated and cloned into the binary vector, pBI121 to produce the 35S::Mu-GsSRK-GFP expression vector. The N. benthamiana leaf epidermal cells were infiltrated with Agrobacteria, containing the vector with an optical density of 0.4. After filtration for about 48–72 h, small sections of the infiltrated leaves were excised and mounted in water. Confocal imaging was performed using a Bio-Rad MRC1024 confocal laser scanning microscope (Bio-Rad Microscience Ltd., Manchester, United Kingdom).
Promoter Activity Analysis
The promoter of Mu-GsSRK (designed as pMu-GsSRK) was obtained with specific primers (Supplementary Table 2), and designed based on the nucleotide sequence of the gene available from the M. notabilis genome and used to create the promoter expression vector, pMu-GsSRK::GUS by replacing the 35S promoter and fusing it with the GUS gene. The pMu-GsSRK::GUS vector was then introduced into the Agrobacterium tumefaciens strain, GV3101 which was used to infiltrate N. benthamiana leaves as described previously (Arpat et al., 2012), and β-glucuronidase (GUS) expression in the infiltrated tobacco leaves was assessed by histochemical staining (Jefferson et al., 1987).
Production of Transgenic Arabidopsis Lines
The coding region of the Mu-GsSRK gene was amplified and ligated into binary plasmid vector, pBI121 under the control of the 35S promoter. Then, the constructed transgenic plant expression vectors were introduced into the A. tumefaciens strain, GV3101 which was used to transform the wild-type (WT) Arabidopsis plants with the floral dip method (Harrison et al., 2006). Transgenic seeds were selected on selection plates [Murashige and Skoog (MS) media supplemented with 50 mg l−1 of kanamycin], and the T3 generation seeds from independent transgenic lines were used for further functional studies.
Plant Treatment
Jasmonate (JA) and salicylic acid (SA) treatments were conducted by spraying 100 μmol l−1 of JA or 5 mmol l−1 of SA solution onto the adaxial surface of mulberry leaves. Pseudomonas syringae pv. mori inoculation was performed by spraying the bacterial suspension (108 CFU ml−1) onto the adaxial surface of young leaves of mulberry. As for Colletotrichum dematium inoculation, it was conducted by spraying the conidial suspension (2.5 × 106 ml−1 of conidia) onto the adaxial surface of mulberry leaves. The leaves sprayed with sterilized water were used as controls. All of the inoculated and control mulberry seedlings were incubated in a glass chamber for 48 h to maintain sufficient humidity. P. syringae pv. tomato DC3000 (Pst DC3000) inoculation was performed by injecting 50 μl of Pst DC3000 (105 CFU ml−1) bacterial suspensions into the rosette leaves of 4-week-old Arabidopsis plants. The rosette leaves were detached and inoculated with Botrytis cinerea or Phytophthora capsici using 2-mm-diameter mycelium plugs taken from the actively growing strain colonies. The inoculated leaves were placed in covered Petri dishes to maintain high humidity, and disease symptoms were evaluated by determining the diameter of the lesions 1 week after inoculation. Three biological replicates per treatment were assayed and three technical replicates were performed for each sample.
Detection of Colony-Forming Units
The leaves inoculated with Pst DC3000 were sampled 72 h after inoculation and ground in sterile water, and the suspension was continuously diluted 10 times with sterile water and spread-plated onto King's B medium. Colonies were counted after 48 h of incubation. All the experiments were conducted at least three times.
Production of Hairy Root Transgenic Mulberry Plants
The Mu-GsSRK gene was cloned into the vector, pROK2 under the control of the 35S promoter to produce 35S::Mu-GsSRK. The pROK2 vector containing 35S::Mu-GsSRK was introduced into A. rhizogenes strains, K599 that are used for the transformation of mulberry seedlings. Gui sang You 62 mulberry seeds were sown in plastic pots containing vermiculite medium and watered with MS medium and grown in an artificial growth chamber at 25°C with a photoperiod of 16 h of light and 8 h of darkness. About 2 weeks after germination, the seedlings with the first two fully expanded true leaves were selected for agro infiltration. The A. rhizogenes strains, K599 harboring vectors, pROK2 containing 35S::Mu-GsSRK were cultured in yeast extract peptone (YEP) liquid medium containing rifampicin (20 mg l−1) and kanamycin (50 mg l−1), and then were centrifuged when its OD600 value reached 0.4 followed by re-suspension in 2-N-morpholino ethanesulfonic acid (MES) buffer (10 mmol l−1 MgCl2, 10 mmol l−1 MES-KOH and 100 μmol l−1 acetosyringone, pH 5.2). About 0.1 ml of A. rhizogenes harboring vectors pROK2 containing 35S::Mu-GsSRK suspension was injected into the junction of true leaves and cotyledons as described previously (Meng et al., 2019). At the same time, the plant injected with A. rhizogenes strains K599 harboring empty vectors, pROK2 were used as the control group. About 1 month later, when the hairy roots developed well, they were detected by PCR and the original roots were cut off.
Phytoplasma Inoculation and Determination
To obtain mulberry yellow dwarf phytoplasma (16SrI-B)-infected leafhoppers (Hishmonus sellatus), the leafhoppers were transferred to phytoplasma–infected mulberry plants for 2 weeks. A. thaliana and mulberry seedlings were inoculated with mulberry phytoplasma by the sap-feeding method with phytoplasma-infected leafhoppers (Sugio et al., 2011), and the seedlings challenged with uninfected leafhoppers were used as controls. Each of the A. thaliana or mulberry seedling was challenged with six adult leafhoppers for 6 days. All the seedlings were incubated in a growth chamber at 26°C, 60% humidity, and under 12 h of light. Four weeks after inoculation, the DNA was extracted following the method described previously (Prince, 1993), and phytoplasma concentration in the plants was determined using real-time PCR described previously (Christensen et al., 2004). The primers and TaqMan probes used to amplify the 16S rRNA gene of phytoplasma, Arabidopsis, and mulberry are listed in Supplementary Table 2. Three biological replicates per treatment were assayed and three technical replicates were performed for each sample.
Results
Differential Transcriptome Analysis Between Phytoplasma-Infected and Healthy Mulberry Leaves
Using RNA-seq, a total of 48.9 and 48.7 million clean reads were obtained in the healthy and phytoplasma-infected mulberry (M. multicaulis Perr.) leaf mRNA libraries, respectively. The expression level of genes identified was calculated, and a total of 1,168 genes were found to be differentially expressed between phytoplasma-infected and healthy leaves, among which 769 genes were upregulated and 399 genes were downregulated (Figure 1A). All the detected DEGs are shown in Supplementary Table 1 and the top 40 up and downregulated ones are listed in Table 1. The GO analysis of the DEGs classified them into 11 functional categories (Figure 1B). The first category of gene was involved in stress and environment response, and the genes associated with catabolic process belonged to the second category, and the third category included the genes involved in transcription and post-transcription regulation. The other DEGs belonged to categories, such as secondary metabolite biosynthesis, signaling pathway, growth and development, cell component, and cycle process. The detailed functional GO terms of the DEGs are given in Supplementary Table 3. Interestingly, 6% of the DEGs were found to be associated with signal transduction pathways, indicating that diverse signal transduction pathways were involved in the response of mulberry to phytoplasma infection. Therefore, the regulatory networks involved in response to phytoplasma infection in mulberry plants are found to be complex.
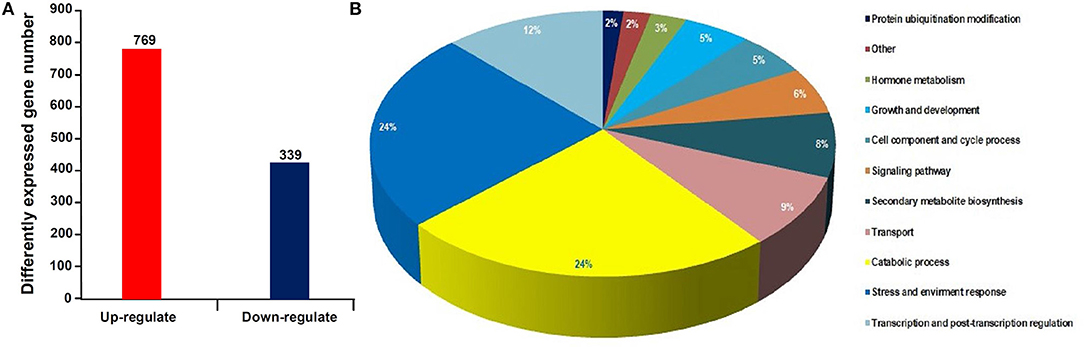
Figure 1. Statistical histogram of the differentially expressed genes (DEGs) and distribution percentage of them in various categories. (A) Number of DEGs between healthy and infected leaves. (B) Distribution percentage of the DEGs in various categories.
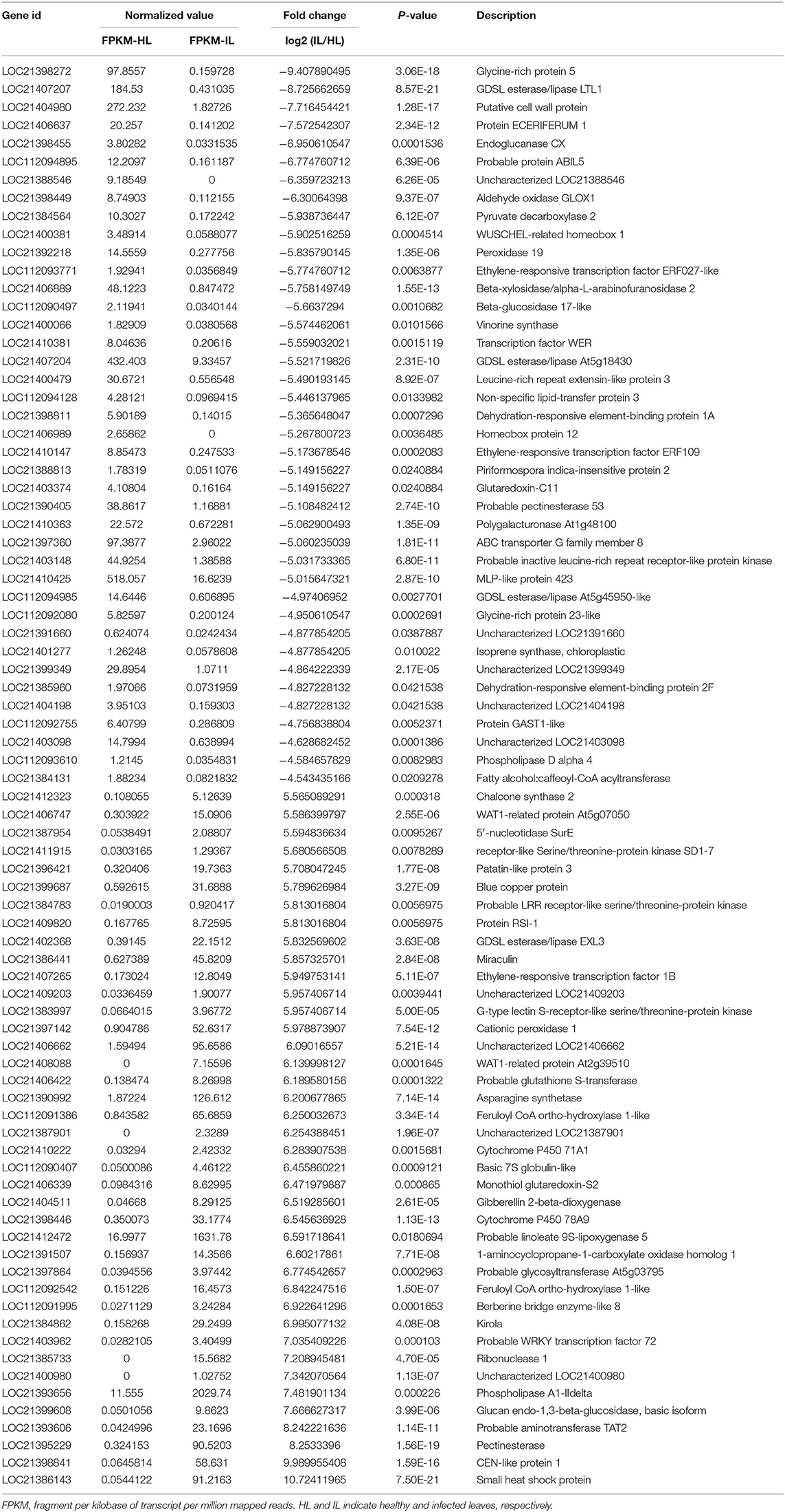
Table 1. The top 40 up and downregulated genes in the infected mulberry leaves.
To validate the expression profiles obtained by RNA-seq, a qRT-PCR analysis was performed for 10 genes including the upregulated and downregulated ones in response to phytoplasma infection (Figure 2). The data showed that all the selected genes exhibited similar changes in expression levels between qRT-PCR and RNA-seq results, indicating that the differential expression profiles of these genes obtained by RNA-seq are reliable.
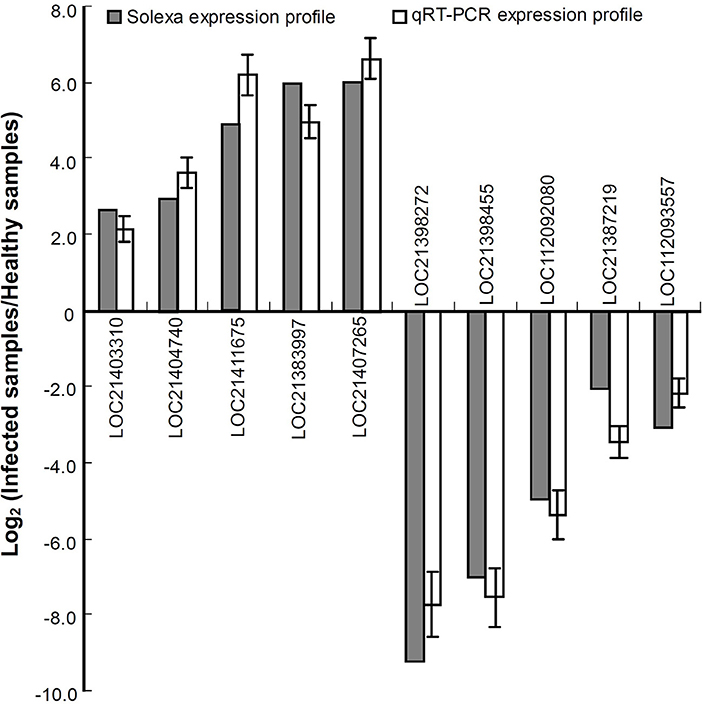
Figure 2. Relative expression levels of the genes analyzed by quantitative real time PCR (qRT-PCR). The relative expression levels of the genes were evaluated using the 2−ΔΔCt method with Mul-ACTIN and Mul-EF1-α as reference genes. The column indicates the log2 ratio of phytoplasma-infected leaves/healthy leaves. Statistical values are expressed as the mean ± SD, n = 3 in each group.
DNA Methylation Site Distributions
There were 23,056 CCGG and 8,520 CCWGG DNA methylation sites detected in the phytoplasma-infected leaves, and a total of 22,733 CCGG and 7,856 CCWGG DNA methylation sites were found in healthy leaves. When the MethyIRAD reads were aligned onto a unique locus in different regions of the genome, the genome-wide methylation pattern was obtained, and the results showed that the distributions of methylation sites in different components of the genome were differential. However, it was found that the distribution patterns of methylation sites at different elements of genomes were similar in the phytoplasma-infected and healthy mulberry leaves (Figures 3A–F). Most CCGG DNA methylation sites were mainly enriched in the exon regions followed by the intergenic, intron, and upstream regions (Figure 3G). As for the CCWGG DNA methylation sites, the major sites were mainly enriched in the intergenic regions followed by the exon, upstream, and intron regions (Figure 3H).
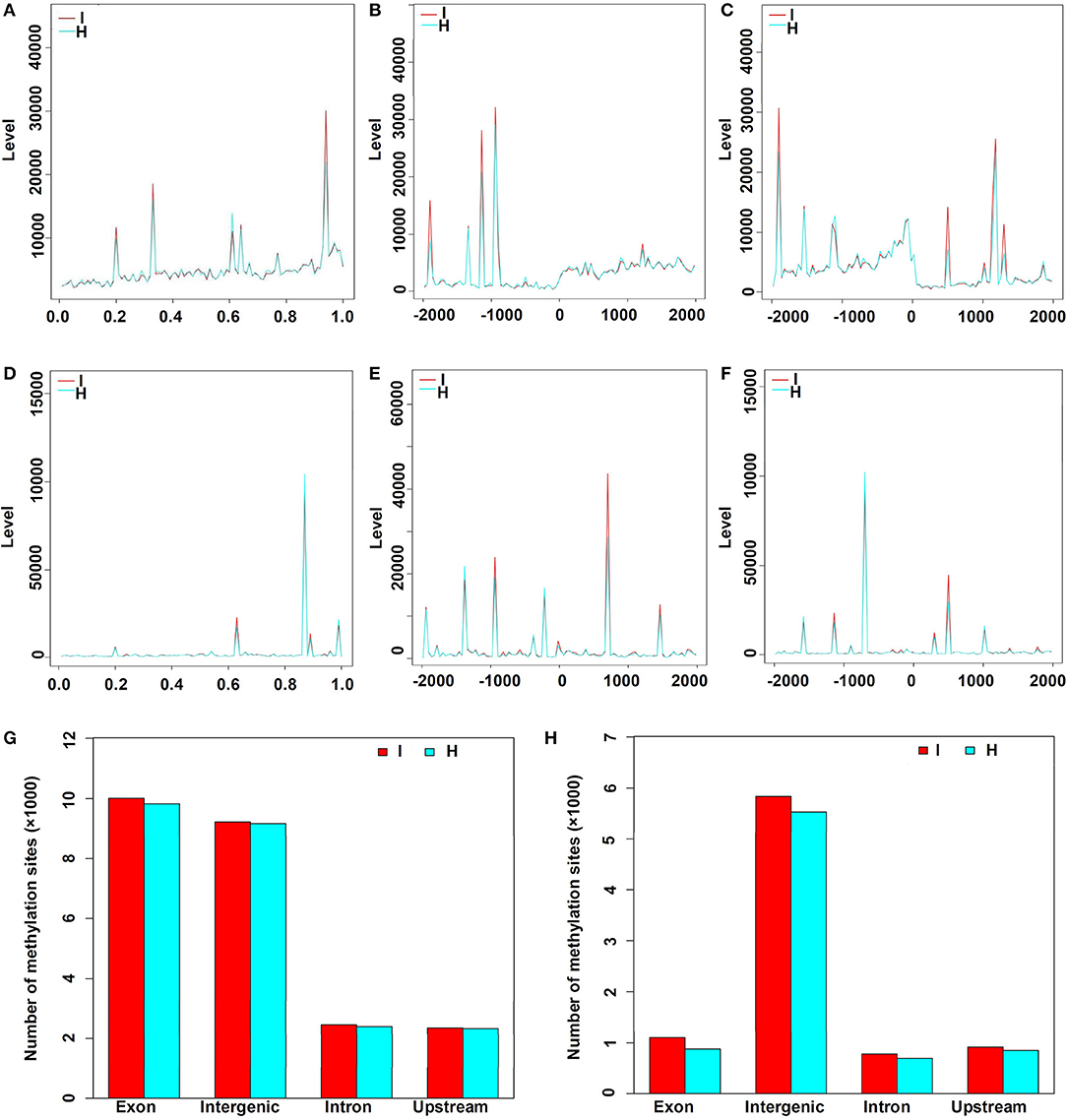
Figure 3. Distribution of methylation sites in different gene regions and elements. (A) Distribution of CCGG methylation sites in the genes. X-axis represents the relative position of the locus on the gene. (B) Distribution of CCGG methylation sites 2 kb upstream or downstream of the transcriptional start sites (TSS). (C) Distribution of CCGG methylation sites 2 kb upstream or downstream of the transcriptional termination sites (TTS). (D) Distribution of CCWGG methylation sites in genes. X-axis represents the relative position of the locus on the gene. (E) Distribution of CCWGG methylation sites 2 kb upstream or downstream of the TSS. (F) Distribution of CCWGG methylation sites 2 kb upstream or downstream of the TTS. (G) CCGG methylation sites are shown. (H) CCWGG methylation sites are shown. I, infected leaves; H, healthy leaves. Exon indicates the exon regions; Intergenic indicates the intergenic regions; Intron indicates the intron regions; Upstream indicates 2 kb upstream of TSS.
DNA Methylation Levels
In this study, a total of 3,676 and 1,366 differentially methylated sites in CCGG and CCWGG, respectively, were found between phytoplasma-infected and healthy mulberry leaves. Most of the differentially methylated sites were found in the intergenic and exon regions, and only about 10% of the differentially methylated sites were found in the upstream and intron regions, respectively (Figure 4). The genes with differentially methylated sites in phytoplasma-infected and healthy mulberry leaves were screened and termed as DMGs. In total, 935 and 347 DMGs were identified in CCGG (Supplementary Table 4) and CCWGG sites (Supplementary Table 5), respectively. The results revealed that 511 DMGs in CCGG sites as well as 220 DMGs in CCWGG sites showed upregulation and 424 DMGs in CCGG sites as well as 127 DMGs in CCWGG sites showed downregulation in the phytoplasma-infected leaves compared to healthy leaves. Interestingly, there were 29 genes which were differentially methylated at both CCGG and CCWGG sites. These results suggested that phytoplasma infection induced the changes of methylation level of some genes.
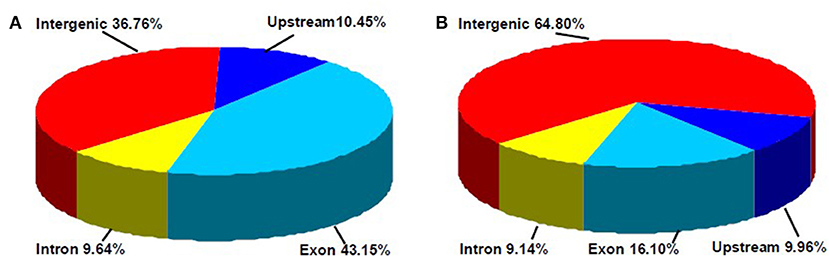
Figure 4. Distribution percentage of differential DNA methylation sites on different gene function components. (A) CCGG methylation sites are shown; (B) CCWGG methylation sites are shown. Exon, the regions of exon; Upstream, the upstream 2,000 bp of the transcriptional start sites (TSS); Intron, the whole introns of genes; Intergenic, the intergenic regions.
To validate the results obtained with MethylRAD-seq data, six genes were selected for analysis by PCR. The genomic DNA digested with FspEI was used as the template for semi-quantitative PCR, and there were more amplification products obtained from the samples, in which the genomic DNAs were lower methylated than those from the samples in which the genomic DNAs were highly methylated at CCGG or CCWGG sites (Figure 5). The PCR results showed a high degree of consistency with the MethylRAD-seq data, indicating that the genomic DNA methylation results obtained by MethylRAD are reliable.
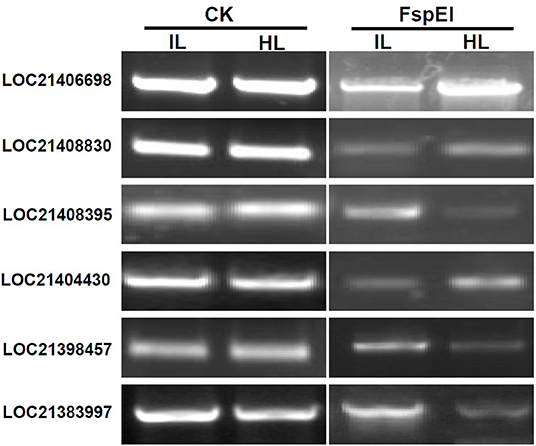
Figure 5. Validation of methylation levels of differentially methylated genes (DMGs) by PCR. CK, the genomic DNA without being digested with FspEI used as the template for PCR; FspEI, the genomic DNA digested with FspEI used as the template for PCR; IL, the infected leaves; HL, the healthy leaves.
Association Analysis of MethylRAD and Transcriptome Sequencing
Based on the RNA-Seq and MethylRAD sequencing data, the methylation and expression levels of the genes in the infected and healthy leaves were compared. Though a lot of genes were methylated or expressed differently, only 51 genes were differentially methylated and expressed between the infected and healthy leaves. Among these differentially expressed and methylated genes (DEMGs), there were 40 and 11 genes with CCGG and CCWGG differential methylation sites, respectively (Tables 2, 3). The correlation between DNA methylation and gene expression showed that the expression levels of 33 genes were negatively correlated with their methylation levels, while the expression levels of 18 genes were positively correlated with their methylation levels. Moreover, it was found that most of the DEMGs were differentially methylated in the exon regions, and only 5 DEMGs were differentially methylated in the intron regions and one was differentially methylated in the intergenic region (Supplementary Tables 6, 7).
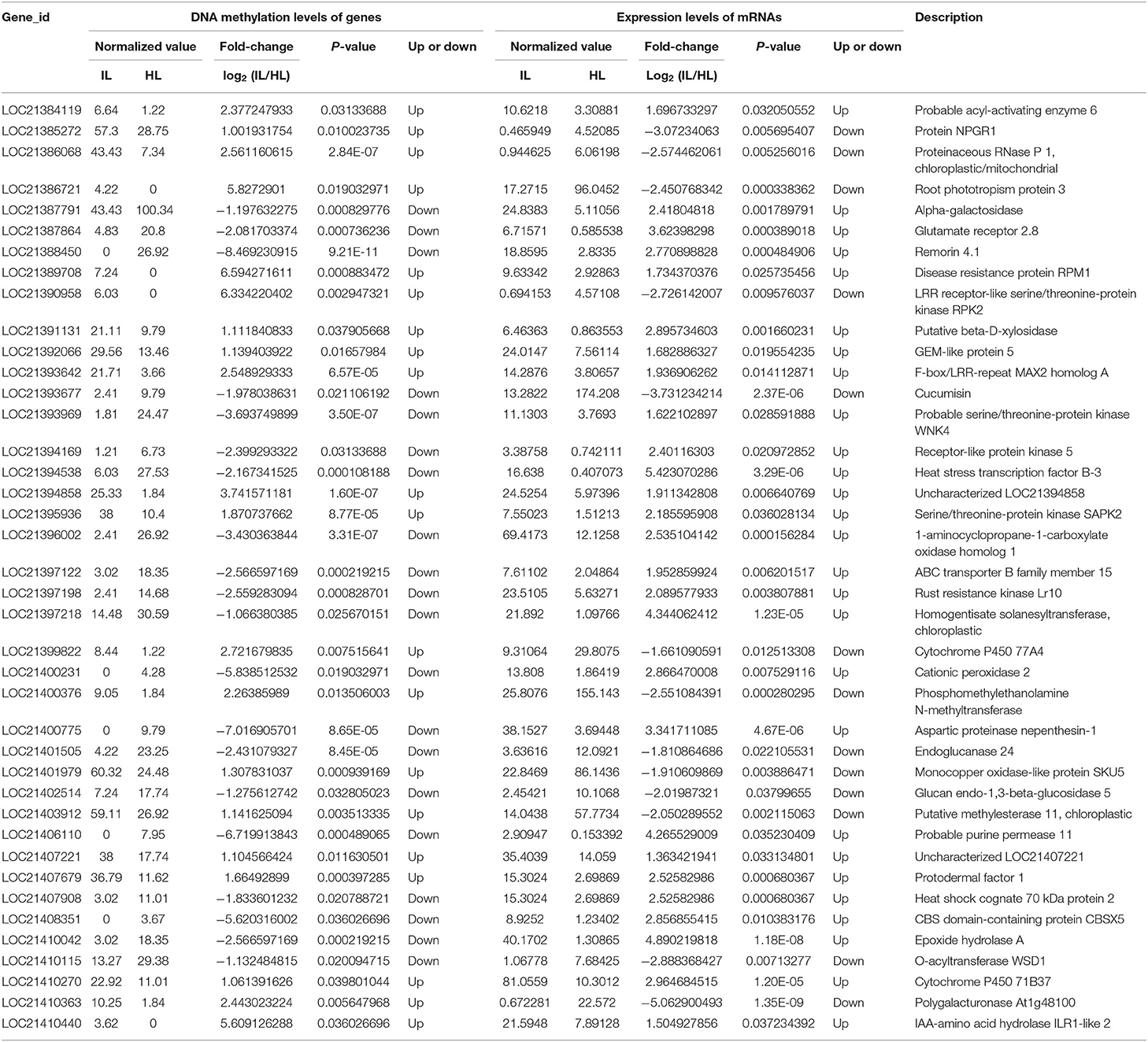
Table 2. Association analysis of the DNA methylation levels in CCGG sites and expression levels of genes in the healthy and infected mulberry leaves.
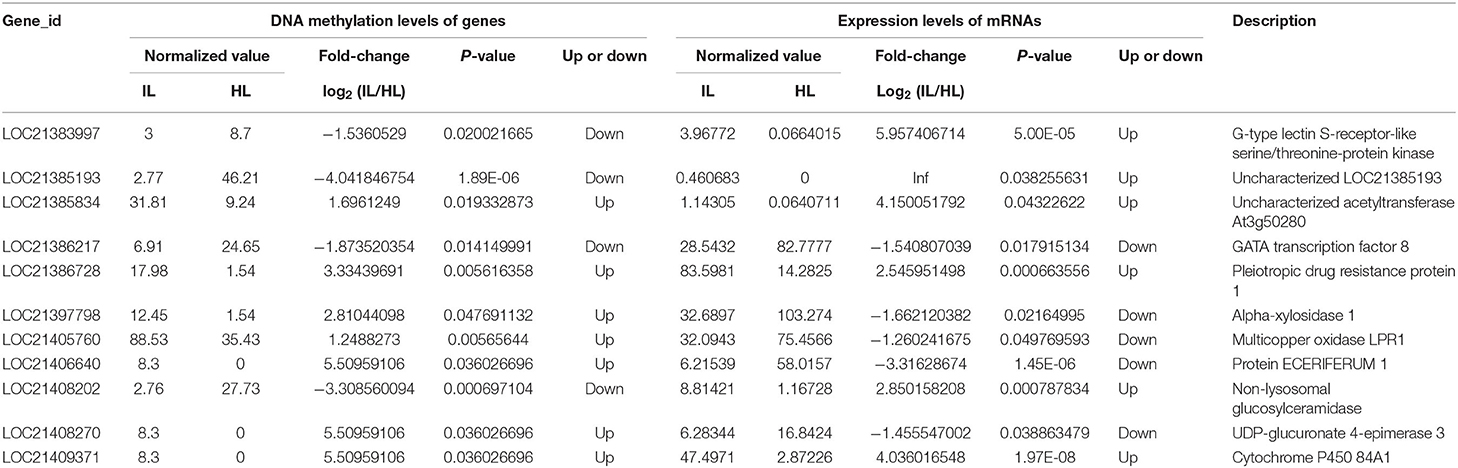
Table 3. Association analysis of the DNA methylation levels in CCWGG sites and expression levels of genes in the healthy and infected mulberry leaves.
The GO analysis of these DEMGs indicated that these genes were classified into nine functional categories (Figure 6). The first category of genes was involved in metabolism followed by the second category of genes which was associated with stress response, and the third category of genes was involved in signal transduction. The other genes were classified into transportion, RNA processing, regulation of transcription, development, and unknown categories.
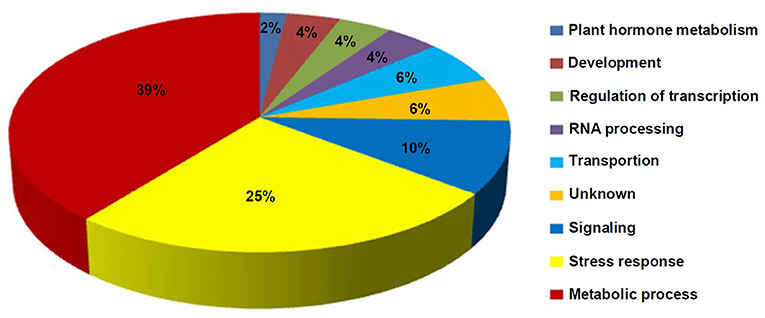
Figure 6. Distribution percentage of the differential DNA methylation and expressed genes in various categories.
Characterization of the Phytoplasma-Responsive G-Type Lectin S-Receptor-Like Serine/Threonine Protein Kinase
Integrated analysis showed that the expression of LOC21383997, which was annotated as Mu-GsSRK was increased, but its methylation level was decreased in the phytoplasma-infected leaves, and these results were confirmed by qRT-PCR (Figure 2) and semi-quantitative PCR analyses (Figure 5). The protein encoded by the Mu-GsSRK gene contains 781 amino acids, and its predicted molecular weight (MW) and isoelectric point (pI) were found to be 87.6 kDa and 6.28, respectively. Multiple sequence alignments revealed the protein-shared homology regions with GsSRKs in various species (Figure 7A). Similar to other typical G-type lectin receptor-like kinases (LecRLKs), Mu-GsSRK protein has some putative conserved domains, such as protein kinase active sites, ATP binding sites, substrate binding sites, and two activation loops. Besides these conserved kinase domains, the protein also contains a B lectin (bulb-type mannose-specific lectin) region and an S-locus-glycoprotein region at the N terminus. In addition, a plant PAN/APPLE-like domain associated with various biological functions by mediating protein–carbohydrate or protein–protein interactions was detected in the Mu-GsSRK protein (Figure 7B).
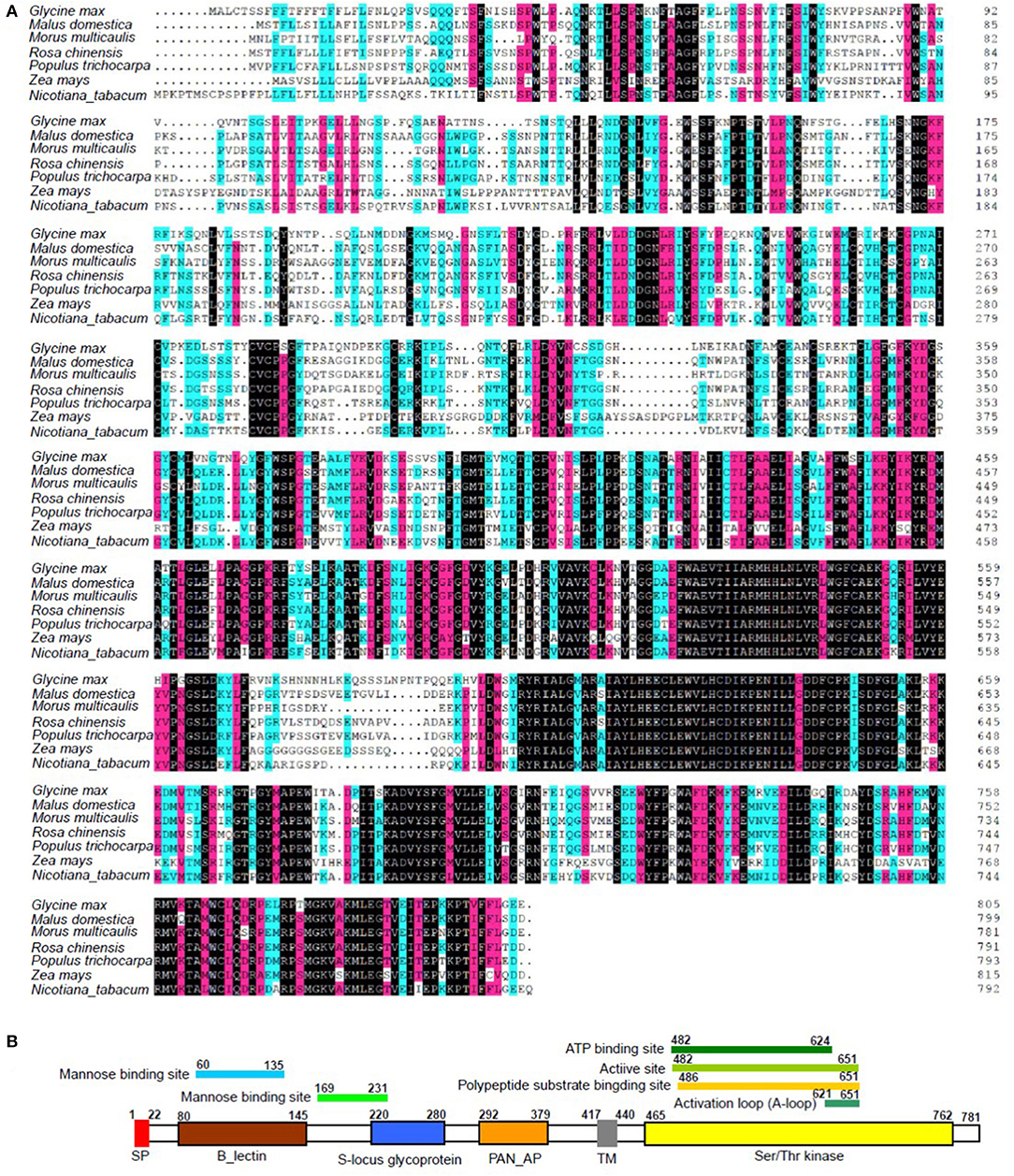
Figure 7. Multiple sequence alignment of Mu-GsSRK with other GsSRK proteins (A) and its putative conserved domains (B). Amino acid residues black and red or blue-shaded are conserved and similar amino acid residues, respectively. The aligned sequences included those GsSRK proteins from Glycine max (XP_003529230.1), Malus domestica (XP_028951539.1), Rosa chinensis (XP_024193513.1), Populus trichocarpa (XP_002314767.3), Zea mays (PWZ52786.1), and Nicotiana tabacum (XP_016458326.1).
Phylogenetic analysis of Mu-GsSRK and GsSRKs from other plants showed that there was closest homology between Mu-GsSRK and GsSRK in M. notabilis (Figure 8A). The structural properties of the Mu-GsSRK protein was predicted using SWISS–MODEL, and the result showed that the protein contained 54.93% random coil, 23.30% alpha helix, and 21.77% extended strands and had a β-barrel structure composed of a number of strands in their N-terminal lectin domains (Figure 8B). The prediction of N-terminal extension suggested that Mu-GsSRK contained an obvious signal peptide, but no obvious sublocalization sequence was detected. In order to elucidate its subcellular localization, Mu-GsSRK was fused to a green fluorescent protein gene and introduced into N. benthamiana leaves. The green fluorescent signal was detected within the plasma membrane suggesting the localization of Mu-GsSRK in the plasma membrane (Figure 8C). Therefore, as a protein kinase, Mu-GsSRK may play a role in sensing and transmitting signals arising from different biotic stress conditions.
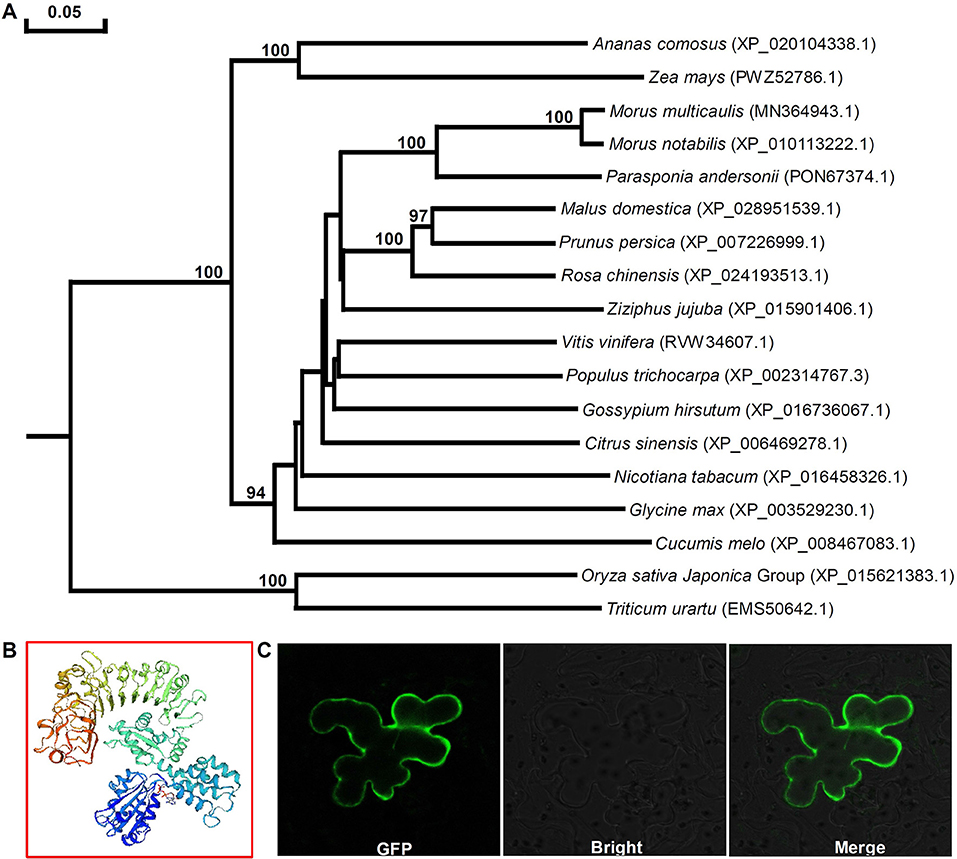
Figure 8. Phylogenetic analyses of GsSRK proteins from different plants and predicted three-dimensional structure and subcellular localization of Mu-GsSRK. (A) Phylogenetic tree was generalized using the neighbor-joining method. The numbers given on the nodes are bootstrap values, and the scale indicates genetic distance. GenBank accession numbers of the proteins are shown in the brackets. (B) Three-dimensional structure of Mu-GsSRK proteins was established by SWISS-MODEL. (C) Subcellular localization of Mu-GsSRK-GFP in N. benthamiana leaf epidermal cells. Mu-GsSRK-GFP fusion protein was transiently expressed in N. benthamiana leaf epidermal cells and visualized with a confocal laser scanning microscope (Zeiss LSM880, Zeiss, Jena, Germany). The left image shows the cell with GFP signal, and the bright-field view of the same cells is shown in the middle image. The right image indicates the overlays of the fluorescent and bright images.
Expression Profile of Mu-GsSRK
To explore the function of the Mu-GsSRK gene, its expression pattern in different tissues and organs was analyzed by qRT-PCR. The results showed that Mu-GsSRK was expressed ubiquitously in the investigated parts of mulberry plants, but its expression level was higher in the leaves than that in other parts (Figure 9A). Moreover, the induced expression pattern of Mu-GsSRK was explored by challenging the mulberry seedlings with P. syringae pv. mori and C. dematium and by treating the seedlings with JA and SA, respectively. The results indicated that the expression level of Mu-GsSRK was increased in the leaves challenged by P. syringae pv. mori or C. dematium. Furthermore, it showed that the exogenous application of JA or SA enhanced the expression of Mu-GsSRK in the leaves (Figure 9B). In addition, pMu-GsSRK was fused with the GUS gene and then was transiently expressed in tobacco leaves. The results of GUS staining showed that GUS activity was induced in the leaves infected by B. cinerea or Pst DC3000, as well as in the leaves upon inoculation with JA or SA (Figure 9C). These data indicated that Mu-GsSRK may be associated with stress resistance, and JA and SA may modulate its expression in mulberry.
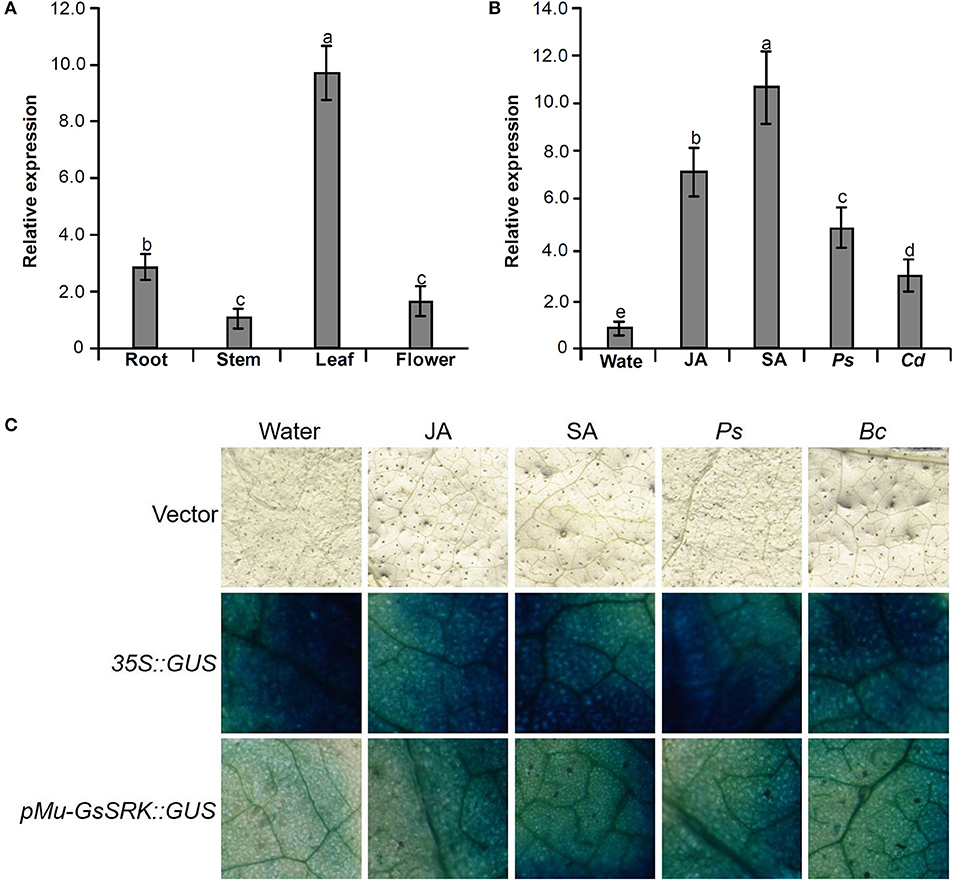
Figure 9. Expression pattern of the Mu-GsSRK gene. (A) Tissue expression pattern of Mu-GsSRK. (B) Induced expression pattern of Mu-GsSRK. Ps and Cd indicate P. syringae pv. mori and C. dematium, respectively. The relative expression levels of the genes were evaluated using the 2−ΔΔCt method with Mul-actin and Mul-EF1-α as reference genes. Data represent the mean values of triplicate samples ± SD. Columns with different letters above indicate significant differences at P < 0.05 according to Duncan's multiple range test. (C) Transient expression of pMu-GsSRK::GUS fusion in N. benthamiana leaves. Tobacco leaves infiltrated with transformed Agrobacterium were sampled at 36 and 72 h after Pst DC3000 and B. cinerea inoculation, respectively. The leaves infiltrated were sampled at 6 h after salicylic acid (SA) or jasmonic acid (JA) treatments.
Methylation Profile of Mu-GsSRK
To explore whether DNA methylation was involved in the expression change of the Mu-GsSRK gene, genomic DNA digested with FspEI was used as the template for PCR analysis. The semi-quantitative PCR results indicated that the Mu-GsSRK gene was methylated at CCGG or CCWGG sites, but the methylation level of Mu-GsSRK did not differ significantly among the different parts of mulberry plants (Figure 10A). Therefore, the expression difference of Mu-GsSRK in the different parts is not caused by DNA methylation. However, the PCR results indicated that the methylation level of the Mu-GsSRK gene was reduced significantly in the leaves inoculated with P. syringae pv. mori or C. dematium or treated with SA, but its DNA methylation level may not be affected by JA treatment. So, the expression change of Mu-GsSRK in the leaves inoculated with Pst DC3000 or C. dematium or treated with SA may be also caused by its methylation level change (Figure 10B).
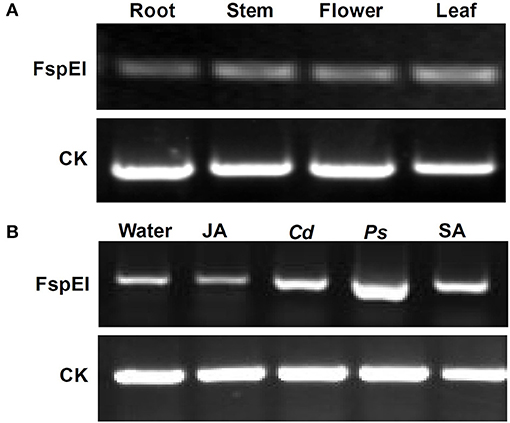
Figure 10. Methylation profile of Mu-GsSRK analyzed by PCR. Methylation level of the Mu-GsSRK gene in different organs detected by PCR (A). Methylation level of the Mu-GsSRK gene in the leaves inoculated with P. syringae pv. mori or C. dematium and treated with SA or JA detected by PCR (B). Ps and Cd indicate P. syringae pv. mori and C. dematium, respectively.
Overexpression of Mu-GsSRK in A. thaliana Enhances Disease Resistance
To explore the defensive role of Mu-GsSRK, wild-type (WT) and transgenic Mu-GsSRK plants were challenged by Pst DC3000. Three days post-inoculation (DPI), the WT plants showed severe disease symptoms. In contrast, there were no evident disease symptoms observed in the leaves of transgenic plants. Meanwhile, the transgenic Mu-GsSRK A. thaliana plants were challenged by B. cinerea or Phytophthora capsici to explore the possible role of Mu-GsSRK in defense against fungal pathogens. Four DPI, B. cinerea or P. capsici were successfully colonized on the inoculation leaf surface of WT plants, and obvious necrotic lesions were observed on the leaves. On the contrary, mild disease symptoms were observed on the inoculated leaf surface of Mu-GsSRK-overexpressing plants (Figure 11A). In addition, the bacterial populations of Pst DC3000 strains in the inoculated leaves were determined, and the result revealed that the population of the strains in the leaves of WT plants was significantly higher than that in the ones of Mu-GsSRK-overexpressing plants at 3 DPI (Figure 11B). Moreover, the lesion areas in the leaves of WT plants inoculated with B. cinerea and P. capsici were larger than those in the leaves of Mu-GsSRK-overexpressing plants (Figure 11C). These results indicate that the overexpression of Mu-GsSRK in Arabidopsis confer enhanced resistance to Pst DC 3000, B. cinerea, and P. capsici, and the Mu-GsSRK gene may play an important role in disease resistance.
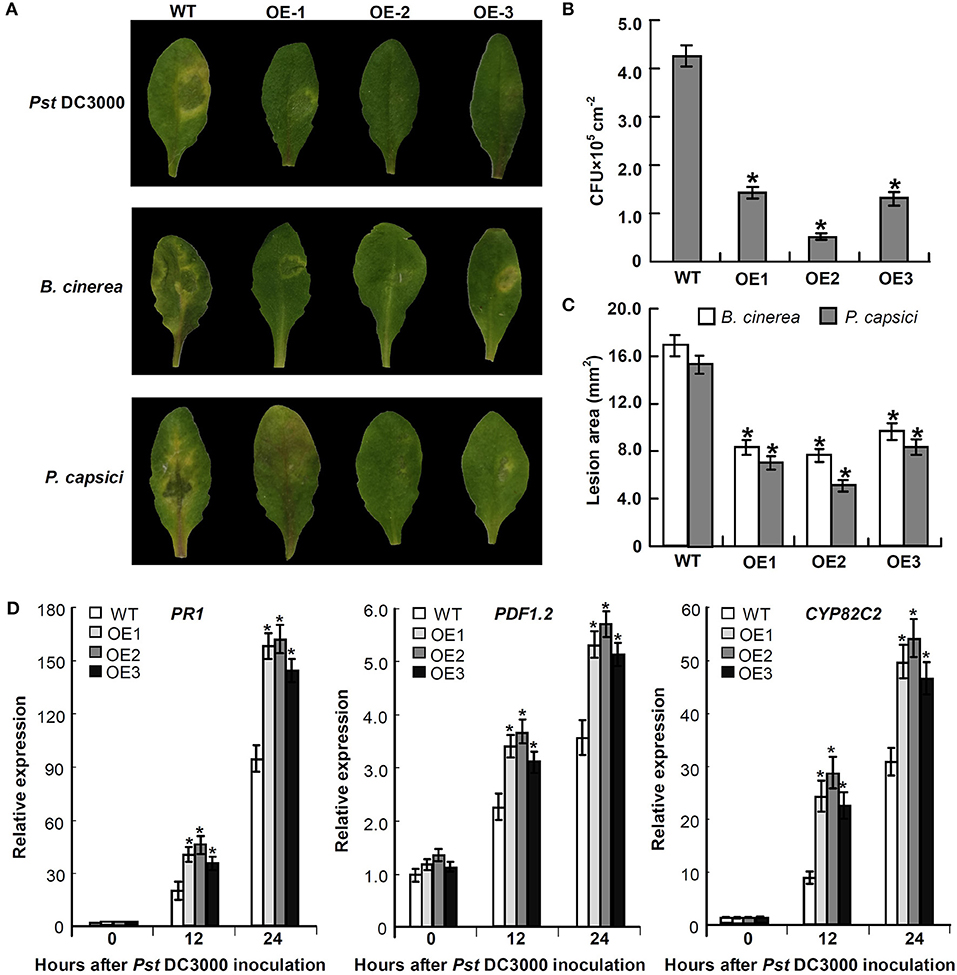
Figure 11. Transgenic Mu-GsSRK gene A. thaliana plants that are resistant to Pst DC 3000, B. cinerea, and P. capsici. (A) Disease symptoms observed on the leaves at 3 days post-inoculation (DPI) with Pst DC 3000 and 4 DPI with B. cinerea or P. capsici. (B) Bacterial populations of Pst DC 3000 strains in the inoculated leaves at 3 DPI. Values represent the mean and SD of three leaves for three independent plants. The asterisk indicates significant difference at P < 0.05 between WT and OE according to Student's t-test. (C) Lesion area in the leaves of WT and OE plants inoculated with B. cinerea and P. capsici. Lesion areas were measured 7 days after inoculation by determining the average lesion diameter on three leaves per line. Values represent the mean and SD of three leaves for three independent plants. The asterisk indicates significant difference at P < 0.05 between WT and OE according to Student's t-test. (D) Expression of PR-1, PDF1.2, and CYP82C2 in Mu-GsSRK-overexpressing A. thaliana plants. The relative expression levels of the genes were evaluated using the 2−ΔΔCt method with Ath-actin and Ath-EF1-α as reference genes. Data represent the mean values of triplicate samples ± SD. The relative expressions of the genes were compared with their expressions in WT without treatment, and the asterisk indicates significant difference at P < 0.05 between WT and OE according to Student's t-test. WT indicates wild-type plants and OE indicates Mu-GsSRK-overexpressing plants.
To examine whether the expressions of some defense-related genes were elevated in the Mu-GsSRK-overexpressing A. thaliana plants, the expression levels of the pathogenesis-related protein 1 (PR-1) gene, plant defensin gene (PDF1.2), and cytochrome P450 protein CYP82C2 gene in the transgenic Mu-GsSRK plants were evaluated. The data showed that the expression levels of PR-1, PDF1.2, and CYP82C2 genes were all very low both in the WT and transgenic Mu-GsSRK plants in the absence of Pst DC3000 inoculation, indicating that the constitutive overexpression of the Mu-GsSRK gene might not affect the basal expression levels of these defense-related genes. However, after Pst DC3000 treatment, the expression levels of PR-1, PDF1.2, and CYP82C2 genes were higher in the Mu-GsSRK-overexpressing plants than in WT plants at 12 and 24 h post-inoculation (Figure 11D). These results demonstrated that Mu-GsSRK may have a positive role in the regulation of defense gene expressions, but pathogen induction is a necessary step for defense gene expression.
Overexpression of Mu-GsSRK in Mulberry Enhances Resistance to Phytoplasma
Since the efficient genetic transformation and regeneration system has not yet been established in mulberry trees, in order to examine the role of Mu-GsSRK in the defense response to phytoplasma, transgenic Mu-GsSRK mulberry plants with hairy roots were generated (Supplementary Figure 1A). qRT-PCR showed that the Mu-GsSRK gene was constitutively highly expressed in the transgenic hairy roots (Supplementary Figure 1B). After the confirmation of transgenic Mu-GsSRK hairy roots, the original roots were cut off and the seedlings carrying the transgenic hairy roots were used in the follow-up of phytoplasma challenge experiments using the sap-feeding method. Four weeks post-challenge, the mulberry seedlings carrying the non-transgenic hairy roots showed severe symptoms of Witches' broom disease, such as short internodes, leaf curling, and axillary bud sprouting. Contrarily, the mulberry seedlings carrying the transgenic Mu-GsSRK hairy roots showed mild dwarfism symptoms and enhanced resistance to phytoplasma (Figures 12A,B). Quantitative PCR analysis showed that the concentrations of phytoplasmas in the aboveground parts and roots of the seedlings carrying the non-transgenic hairy roots were about 22.5 × 105 and 1.3 × 105 cells μg−1 plant DNA, respectively. While the concentrations of phytoplasmas in the aboveground parts and roots of the seedlings carrying the transgenic Mu-GsSRK hairy roots were about 6.0 × 105 and 0.4 × 105 cells μg−1 plant DNA, respectively, there was no phytoplasma detected in the plants challenged by healthy leafhoppers (Figure 12C). Therefore, the concentrations of phytoplasmas in the aboveground parts and roots of the seedlings carrying the non-transgenic hairy roots were higher than those in the seedlings carrying the transgenic Mu-GsSRK hairy roots, and the overexpression of the Mu-GsSRK gene in the hairy roots partially inhibited the growth of phytoplasmas and led to enhanced resistance to phytoplasma.
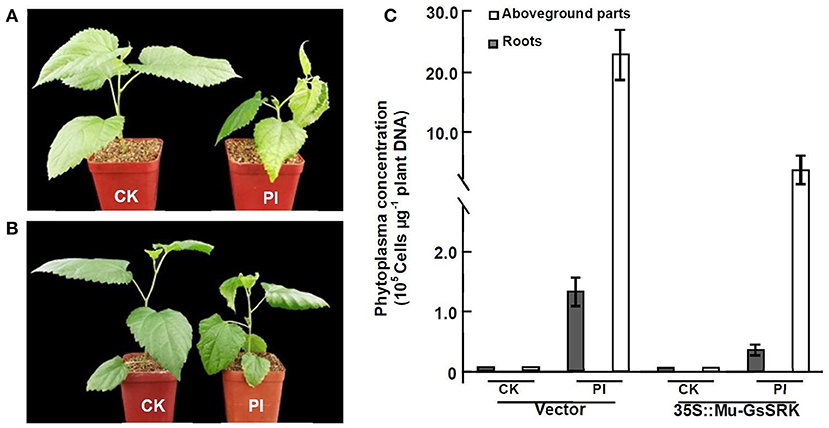
Figure 12. Transgenic Mu-GsSRK gene mulberry seedlings were resistant to phytoplasma. (A) Phenotypes of mulberry seedlings carrying the transgenic empty vector hairy roots challenged by phytoplasma. (B) Phenotypes of mulberry seedlings carrying the transgenic Mu-GsSRK hairy roots challenged by phytoplasma. (C) Phytoplasma concentration in the plants detected by real-time PCR amplification of phytoplasma 16S rDNA. Statistics values are expressed as the mean ± SD, n = 3 in each group. CK, plants challenged by uninfected leafhoppers. PI, plants challenged by phytoplasma-infected leafhoppers. Vector, mulberry seedlings carrying the transgenic empty vector hairy roots. 35S::Mu-GsSRK, mulberry seedlings carrying the transgenic Mu-GsSRK hairy roots.
Discussion
DNA Dynamic Methylation Levels Were Associated With Expression Changes of the Genes Involved in Response to Phytoplasma Infection
As an important and conserved epigenetic modification, DNA methylation is associated with many important biological processes, and has been extensively studied in recent years. However, few studies have focused on mulberry, and this is the first report about the genome-wide DNA methylation profiles in mulberry. Previous reports showed that DNA methylation is not randomly distributed in genomes. In plants, the gene body regions are often highly methylated, while the transcriptional start sites (TSS) and transcriptional termination sites (TTS) mostly lack DNA methylation. Moreover, many intergenic regions also show hypermethylation; on the contrary, most promoter regions are hypomethylated (Zhang et al., 2006). The results presented here showed that DNA methylation is enriched in intergenic and gene body regions, whereas the promoter regions are also hypomethylated in mulberry genomes. In addition, it was also observed that the methylation level in exons was higher than that in introns and CCGG methylation sites (Figure 4). These results indicated that DNA methylation profiles of mulberry are analogous to those of other plants, and the methylation patterns among different plant species may be conservative. It is widely accepted that DNA methylation is a major transcription silencing pathway, which is negatively correlated with gene expression in plants (Zhang et al., 2006). However, it has also been reported that DNA methylation is positively correlated with gene expression (Lou et al., 2014). Analysis of gene expression differences between the healthy and infected leaves indicated that DNA hypermethylation is associated with activation of several genes during phytoplasma infection (Tables 2, 3). Therefore, the mechanism of gene regulation mediated by DNA methylation is very complicated and further study is necessary to elucidate it in detail.
Previous studies have shown that DNA methylation patterns can be changed when plants are infected with pathogens, and it was reported that the DNA methylation levels decreased in Paulownia plantlets infected with phytoplasma (Cao et al., 2014). However, no significant difference in the average DNA methylation level between healthy and phytoplasma-infected mulberry samples was detected (Figure 3). The transcriptome data also indicated that phytoplasma infection did not lead to significant changes in the expression of methyltransferase and demethylase in the infected samples. Although the average DNA methylation level was not changed, the methylation levels of more than 1,253 genes were changed significantly in the infected samples (Supplementary Tables 4, 5). So the methylation levels of certain genes may be dynamically regulated to control plant resistance against phytoplasma infection, but the genome was closely monitored to maintain stability. However, whether there is a link between loci-specific methylation and phytoplasma infection remains to be established.
It has been reported that DNA methylation is associated with gene expression changes in response to phytoplasma infection (Jagoueix-Eveillard et al., 2001; Pracros et al., 2006; Ahmad et al., 2012; Cao et al., 2014; Pagliarani et al., 2020). In this study, 51 DEMGs were identified in the infected leaves. Among these, there were some receptor kinase genes, such as LRR receptor-like serine/threonine-protein kinase, Mu-GsSRK, and serine/threonine-protein kinase SAPK2e (Tables 2, 3), which play a central role in signaling during the pathogen recognition in plants (Bouwmeester and Govers, 2009; Wang and Bouwmeester, 2017). By adjusting the methylation levels of these genes, plants can regulate their expression levels to enhance the perception of phytoplasma infection. On the other hand, phytoplasma may change the methylation level of these genes to affect their expression, thus avoiding plant perception (Zhai et al., 2010; Cao et al., 2014). In addition, there were 22 DEMGs involved in metabolic, growth and developmental processes were found in the infected leaves; the changes of these genes may disturb the normal metabolic growth and developmental processes leading to various symptoms. Although these symptoms may hinder the normal growth and development of mulberry plants, they may promote parasite multiplication of phytoplasma in the infected plants. Moreover, among these DEMGs, there were 13 genes associated with stress response (Tables 2, 3). This suggests that mulberry plants can change the expression of some disease resistance genes by altering their methylation status, so as to improve their resistance to phytoplasma. Therefore, our evidence supported that the DNA dynamic methylation levels were associated with expression changes of the genes involved in response to phytoplasma infection. Although there were many genes which were simultaneously methylated and expressed differently in the infected and healthy leaves, there is a wide variety of mechanisms that control gene expression, and DNA methylation may be the reason for the change of gene expression, but the change may be affected by other factors rather than DNA methylation.
DNA Methylation Plays an Important Role in Regulating Mu-GsSRK Gene Expression in Response to Phytoplasma Infection
The lectin receptor-like kinases (LecRLKs) are categorized into three sub-classes: G-, L-, and C-type depending on the features of their N-terminal lectin domains (Vaid et al., 2012), and the Mu-GsSRK protein has some domains conserved in G-type LecRLKs (Figure 7B). So it might have similar biological functions with other G-type LecRLKs. Due to the resemblance of the extracellular domain with the lectin protein, known to bind to fungal and bacterial cell wall components, LecRLKs are hypothesized to predominantly participate in biotic stress tolerance (Wang and Bouwmeester, 2017). There were many G-type LecRLKs that have been found to be associated with the responses to pathogen infections, and some LecRLKs have been reported to confer resistance to a variety of pathogens (Chen et al., 2006; Sanabria et al., 2012; Vaid et al., 2013; Ranf et al., 2015). However, to the best of our knowledge, the current study is the first to report that G-type LecRLK genes are associated with the response to phytoplasma infection.
It was reported that some members of the LecRLK family are located on the plasma membrane and can sense signals arising from different biotic stress conditions and transmit the stress signal to the nucleus (Vaid et al., 2013). So some LecRLK family genes were proposed to play a role in mediating and strengthening the cell wall–plasma membrane (CW–PM) links and continuum which is essential for defense against pathogens (André et al., 2005; Bouwmeester et al., 2011; Singh et al., 2012; Ranf et al., 2015). Our results showed that the Mu-GsSRK protein is localized on the plasma membrane (Figure 8C), and it can significantly increase the expressions of disease resistance-related genes upon pathogenic infection (Figure 11C) and enhanced resistance to the pathogens (Figure 11A). Moreover, our results showed that the overexpression of Mu-GsSRK in the roots not only enhanced the roots but also the resistance of the aboveground parts to phytoplasma (Figure 12). Therefore, the Mu-GsSRK protein may be able to sense the signals arising from phytoplasma infection and transmit the signals to the nucleus and regulate the expressions of disease resistance-related genes, and the signal might be transmitted a long distance and stimulate the immune response of the whole plant.
It was reported that there was an increase in endogenous SA in the phytoplasma-infected plants (Dermastia, 2019). In addition, our results showed that the methylation level of the Mu-GsSRK gene was reduced significantly in the leaves treated with SA (Figure 10B). Therefore, the endogenous SA may be increased in the phytoplasma-infected mulberry leaves, which may reduce the methylation level and enhance the expression of Mu-GsSRK. Moreover, our results showed that the activity of the Mu-GsSRK promoter was induced by SA (Figure 9C). So, the activities of the Mu-GsSRK promoter may be induced by the increase of endogenous SA caused by phytoplasma infection and enhance the expression of the Mu-GsSRK gene. In response to phytoplasma infection, SA may be an important factor in the regulation of methylation and the expression level of Mu-GsSRK in mulberry; further study is required to elucidate the precise mechanism behind this process.
Conclusion
In conclusion, a large number of genes with different methylation and expression levels were identified, which laid a foundation for further study on the regulation mechanism of gene expression during the response to phytoplasma infection in mulberry. Our results proved that phytoplasma infection induced changes both in the methylation and expression of Mu-GsSRK gene which positively regulates the resistance of plant disease. The information provided here is particularly useful to better understand the interactions between mulberry and phytoplasma.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: PRJNA718973 (https://www.ncbi.nlm.nih.gov/sra/PRJNA718973).
Author Contributions
XJ conceived and designed the experiments. CL, XD, YX, YW, and QD performed the experiments and analyzed the data. YG wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (2018YFD1000602), the National Natural Science Foundation of China (31670600), the Natural Science Foundation of Shandong Province (ZR2019MC017), and the Modern Agricultural Technology System of Shandong Province (No. SDAIT-18-04).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.697702/full#supplementary-material
Supplementary Table 1. The primers used for gene cloning.
Supplementary Table 2. The primers used for qRT-PCR and semi-quantitative PCR.
Supplementary Table 3. Differentially expressed genes (DEGs) between the healthy and infected mulberry leaves.
Supplementary Table 4. Differentially methylated genes (DMGs) in CCGG sites between the healthy and infected mulberry leaves.
Supplementary Table 5. Differentially methylated genes (DMGs) in CCWGG sites between the healthy and infected mulberry leaves.
Supplementary Table 6. Differentially methylated levels in CCGG sites in the gene regions of differentially methylated and expressed genes in the healthy and infected mulberry leaves.
Supplementary Table 7. Differentially methylated levels in CCWGG sites in the gene regions of differential methylated and expressed genes in the healthy and infected mulberry leaves.
Supplementary Figure 1. Regeneration of transgenic Mu-GsSRK hairy roots in mulberry seedling. (A) The hairy root phenotype of mulberry seedling. The arrow points to the hairy root. (B) Quantitative analysis of the expression levels of the Mu-GsSRK genes in the transgenic hairy roots. The relative expression levels of the genes were evaluated using the 2−ΔΔCt method with the Mul-actin and Mul-EF1-α as reference genes. Data represent the mean values of triplicate samples ± SD. Asterisk indicates significant difference at P < 0.05 between WT and OE plants according to Student's t-test. WT, wild type plants; CK, the transgenic empty vector hairy roots; OE, the transgenic Mu-GsSRK hairy roots.
References
Ahmad, J. N., Garcion, C., Teyssier, E., Hernould, M., Gallusci, P., Pracros, P., et al. (2012). Effects of stolbur phytoplasma infection on DNA methylation processes in tomato plants. Plant Pathol. 62, 205–216. doi: 10.1111/j.1365-3059.2012.02605.x
André, S., Siebert, H. C., Nishiguchi, M., Tazaki, K., and Gabius, H. J. (2005). Evidence for lectin activity of a plant receptor-like protein kinase by application of neoglycoproteins and bioinformatic algorithms. Biochim. Biophys. Acta 1725, 222–232. doi: 10.1016/j.bbagen.2005.04.004
Arpat, A. B., Magliano, P., Wege, S., Rouached, H., Stefanovic, A., and Poirier, Y. (2012). Functional expression of PHO1 to the Golgi and trans-Golgi network and its role in export of inorganic phosphate. Plant J. 71, 479–491. doi: 10.1111/j.1365-313X.2012.05004.x
Bertaccini, A., and Duduk, B. (2009). Phytoplasma and phytoplasma diseases: a review of recent research. Phytopathol. Mediterr. 48, 355–378. doi: 10.14601/Phytopathol_Mediterr-3300
Bouwmeester, K., de Sain, M., Weide, R., Gouget, A., Klamer, S., Canut, H., et al. (2011). The lectin receptor kinase lecRK-I.9 is a novel phytophthora resistance component and a potential host target for a RXLR effector. PLoS Pathog. 7:e1001327. doi: 10.1371/journal.ppat.1001327
Bouwmeester, K., and Govers, F. (2009). Arabidopsis L-type lectin receptor kinases: phylogeny, classification, and expression profiles. J. Exp. Bot. 60, 4383–4396. doi: 10.1093/jxb/erp277
Cao, X., Fan, G., Deng, M., Zhao, Z., and Dong, Y. (2014). Identification of genes related to Paulownia witches' broom by AFLP and MSAP. Int. J. Mol. Sci. 15, 14669–14683. doi: 10.3390/ijms150814669
Cao, Y., Fan, G., Wang, Z., and Gu, Z. (2019). Phytoplasma-induced changes in the acetylome and succinylome of Paulownia tomentosa provide evidence for involvement of acetylated proteins in witches' broom disease. Mol. Cell. Proteomics 18, 1210–1226. doi: 10.1074/mcp.RA118.001104
Chen, X., Shang, J., Chen, D., Lei, C., Zou, Y., Zhai, W., et al. (2006). A B-lectin receptor kinase gene conferring rice blast resistance. Plant J. 46, 794–804. doi: 10.1111/j.1365-313X.2006.02739.x
Christensen, N. M., Axelsen, K. B., Nicolaisen, M., and Schulz, A. (2005). Phytoplasmas and their interactions with hosts. Trends Plant Sci. 10, 526–535. doi: 10.1016/j.tplants.2005.09.008
Christensen, N. M., Nicolaisen, M., Hansen, M., and Schulz, A. (2004). Distribution of phytoplasmas in infected plants as revealed by real-time PCR and bioimaging. Mol. Plant Microbe Interact. 17, 1175–1184. doi: 10.1094/MPMI.2004.17.11.1175
Clarke, J. D. (2009). Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harb. Protoc. 4, 1–2. doi: 10.1101/pdb.prot5177
De Palma, M., Salzano, M., Villano, C., Aversano, R., Lorito, M., Ruocco, M., et al. (2019). Transcriptome reprogramming, epigenetic modifications and alternative splicing orchestrate the tomato root response to the beneficial fungus Trichoderma harzianum. Hortic. Res. 6:5. doi: 10.1038/s41438-018-0079-1
Dermastia, M. (2019). Plant hormones in phytoplasma infected plants. Front. Plant Sci. 10:477. doi: 10.3389/fpls.2019.00477
Gai, Y. P., Han, X. J., Li, Y. Q., Yuan, C. Z., Mo, Y. Y., Guo, F. Y., et al. (2014). Metabolomic analysis reveals the potential metabolites and pathogenesis involved in mulberry yellow dwarf disease. Plant Cell Environ. 37, 1474–1490. doi: 10.1111/pce.12255
Gai, Y. P., Yuan, S. S., Liu, Z. Y., Zhao, H. N., Liu, Q., Qin, R. L., et al. (2018). Integrated phloem sap mRNA and protein expression analysis reveals phytoplasma-infection responses in mulberry. Mol. Cell. Proteomics 17, 1702–1719. doi: 10.1074/mcp.RA118.000670
Harrison, S. J., Mott, E. K., Parsley, K., Aspinall, S., Gray, J. C., and Cottage, A. (2006). A rapid and robust method of identifying transformed Arabidopsis thaliana seedlings following floral dip transformation. Plant Methods 2:19. doi: 10.1186/1746-4811-2-19
Henry, E., Yadeta, K. A., and Coaker, G. (2013). Recognition of bacterial plant pathogens: local, systemic and transgene rational immunity. New Phytol. 199, 908–915. doi: 10.1111/nph.12214
Hren, M., Nikolić, P., Rotter, A., Blejec, A., Terrier, N., Ravnikar, M., et al. (2009). 'Bois noir' phytoplasma induces significant reprogramming of the leaf transcriptome in the field grown grapevine. BMC Genomics 10:460. doi: 10.1186/1471-2164-10-460
Huang, H., Liu, R., Niu, Q., Tang, K., Zhang, B., Zhang, H., et al. (2019). Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. U.S.A. 116, 1430–1436. doi: 10.1073/pnas.1815441116
Jagoueix-Eveillard, S., Tarendeau, F., Guolter, K., Danet, J. L., Bové, J. M., and Garnier, M. (2001). Catharanthus roseus genes regulated differentially by mollicute infections. Mol. Plant Microbe Interact. 14, 225–233. doi: 10.1094/MPMI.2001.14.2.225
Jefferson, R. A., Kavanagh, T. A., and Bevan, M. W. (1987). GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x
Ji, L., Jordan, W. T., Shi, X., Hu, L., He, C., and Schmitz, R. J. (2018). TET-mediated epimutagenesis of the Arabidopsis thaliana methylome. Nat. Commun. 9:895. doi: 10.1038/s41467-018-03289-7
Ji, X., Gai, Y., Zheng, C., and Mu, Z. (2009). Comparative proteomic analysis provides new insights into mulberry dwarf responses in mulberry (Morus alba L.). Proteomics 9, 5328–5339. doi: 10.1002/pmic.200900012
Liu, Z. G., Zhao, Z. H., Xue, C. L., Wang, L. X., Wang, L. L., Feng, C. F., et al. (2019). Three main genes in the MAPK cascade involved in the Chinese jujube-phytoplasma interaction. Forests 10:392. doi: 10.3390/f10050392
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lou, S., Lee, H. M., Qin, H., Li, J. W., Gao, Z., Liu, X., et al. (2014). Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 15:408. doi: 10.1186/s13059-014-0408-0
Meng, D., Yang, Q., Dong, B., Song, Z., Niu, L., Wang, L., et al. (2019). Development of an efficient root transgenic system for pigeon pea and its application to other important economically plants. Plant Biotechnol. J. 17, 1804–1813. doi: 10.1111/pbi.13101
Namba, S. (2019). Molecular and biological properties of phytoplasmas. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 95, 401–418. doi: 10.2183/pjab.95.028
Pagliarani, C., Gambino, G., Ferrandino, A., Chitarra, W., Vrhovsek, U., Cantu, D., et al. (2020). Molecular memory of Flavescence dorée phytoplasma in recovering grapevines. Hortic. Res. 7:126. doi: 10.1038/s41438-020-00348-3
Pavet, V., Quintero, C., Cecchini, N. M., Rosa, A. L., and Alvarez, M. E. (2006). Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol. Plant Microbe Interact. 19, 577–587. doi: 10.1094/MPMI-19-0577
Pracros, P., Renaudin, J., Eveillard, S., Mouras, A., and Hernould, M. (2006). Tomato flower abnormalities induced by stolbur phytoplasma infection are associated with changes of expression of floral development genes. Mol. Plant Microbe Interact. 19, 62–68. doi: 10.1094/MPMI-19-0062
Prince, J. P. (1993). Molecular detection of diverse mycoplasma like organisms (MLOs) associated with grapevine yellows and their classification with aster yellows, X-disease, and elm yellows MLOs. Phytopathology 83, 1130–1137. doi: 10.1094/Phyto-83-1130
Ranf, S., Gisch, N., Schäffer, M., Illig, T., Westphal, L., Knirel, Y. A., et al. (2015). A lectin S-domain receptor kinase mediates lipopolysaccharide sensing in Arabidopsis thaliana. Nat. Immunol. 16, 426–433. doi: 10.1038/ni.3124
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Sanabria, N. M., van Heerden, H., and Dubery, I. A. (2012). Molecular characterisation and regulation of a Nicotiana tabacum S-domain receptor-like kinase gene induced during an early rapid response to lipopolysaccharides. Gene 501, 39–48. doi: 10.1016/j.gene.2012.03.073
Singh, P., Kuo, Y. C., Mishra, S., Tsai, C. H., Chien, C. C., Chen, C. W., et al. (2012). The lectin receptor kinase-VI.2 is required for priming and positively regulates Arabidopsis pattern-triggered immunity. Plant Cell 24, 1256–1270. doi: 10.1105/tpc.112.095778
Sugio, A., Kingdom, H. N., MacLean, A. M., Grieve, V. M., and Hogenhout, S.A. (2011). Phytoplasma protein effector SAP11 enhances insect vector reproduction by manipulating plant development and defense hormone biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 19111–19112. doi: 10.1073/pnas.1105664108
Torchetti, E. M., Pegoraro, M., Navarro, B., Catoni, M., and Noris, E. (2016). A nuclear-replicating viroid antagonizes infectivity and accumulation of a geminivirus by upregulating methylation-related genes and inducing hypermethylation of viral DNA. Sci. Rep. 6:35101. doi: 10.1038/srep35101
Vaid, N., Macovei, A., and Tuteja, N. (2013). Knights in action: lectin receptor-like kinases in plant development and stress responses. Mol. Plant. 6, 1405–1418. doi: 10.1093/mp/sst033
Vaid, N., Pandey, P. K., and Tuteja, N. (2012). Genome-wide analysis of lectin receptor-like kinase family from Arabidopsis and rice. Plant Mol. Biol. 80, 365–388. doi: 10.1007/s11103-012-9952-8
Wang, Y., and Bouwmeester, K. (2017). L-type lectin receptor kinases: new forces in plant immunity. PLoS Pathog. 13:e1006433. doi: 10.1371/journal.ppat.1006433
Zenoni, S., Ferrarini, A., Giacomelli, E., Xumerle, L., Fasoli, M., Malerba, G., et al. (2010). Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 152, 1787–1795. doi: 10.1104/pp.109.149716
Zhai, X. Q., Cao, X. B., and Fan, G. Q. (2010). Growth of paulownia witches' broom seedlings treated with methyl methane sulphonate and SSR analysis. Sci. Silva Sin. 46, 176–181.
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006). Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201. doi: 10.1016/j.cell.2006.08.003
Keywords: mulberry, phytoplasma, DNA methylation, transcriptome, G-type lectin S-receptor-like serine/threonine protein kinase
Citation: Liu C, Dong X, Xu Y, Dong Q, Wang Y, Gai Y and Ji X (2021) Transcriptome and DNA Methylome Reveal Insights Into Phytoplasma Infection Responses in Mulberry (Morus multicaulis Perr.). Front. Plant Sci. 12:697702. doi: 10.3389/fpls.2021.697702
Received: 20 April 2021; Accepted: 05 July 2021;
Published: 03 August 2021.
Edited by:
Dirk Albert Balmer, Syngenta, SwitzerlandReviewed by:
Du Seok Choi, LG Chem, South KoreaKhayalethu Ntushelo, University of South Africa, South Africa
Copyright © 2021 Liu, Dong, Xu, Dong, Wang, Gai and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingping Gai, gyp@sdau.edu.cn; Xianling Ji, xlji@sdau.edu.cn