 Peilei Chen
Peilei Chen Yanqing Zhou
Yanqing Zhou Hongying Duan
Hongying Duan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 August 2021
Sec. Plant Metabolism and Chemodiversity
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.693758
This article is part of the Research Topic Synthesis and Bioactivities of Plant-Derived Biomolecules View all 8 articles
Rehmannia glutinosa has important medicinal value; terpenoid is one of the main active components in R. glutinosa. In this study, iTRAQ technique was used to analyze the relative abundance of proteins in roots of R. glutinosa, and 6,752 reliable proteins were quantified. GO enrichment results indicated that most proteins were involved in metabolic process or cellular process, 57.63% proteins had catalytic activity, and 65.80% proteins were enriched in membrane-bounded organelle. In roots of R. glutinosa, there were 38 KEGG enrichments with significance, more DEPs were found in some pathways, especially the proteasome pathway and TCA cycle with 15.0% DEPs between elongation stage and expansion stage of roots. Furthermore, five KEGG pathways of terpenoid synthesis were found. Most prenyltransferases belong to FPP/GGPP synthase family, involved in terpenoid backbone biosynthesis, and all interacted with biotin carboxylase CAC2. Compared with that at the elongation stage, many prenyltransferases exhibited higher expression at the expansion stage or maturation stage of roots. In addition, eight FPP/GGPP synthase encoding genes were cloned from R. glutinosa, namely FPPS, FPPS1, GGPS, GGPS3, GGPS4, GGPS5, GPPS and GPPS2, introns were also found in FPPS, FPPS1, GGPS5 and GGPS2, and FPP/GPP synthases were more conservative in organisms, especially in viridiplantae, in which the co-occurrence of GPPS or GPPS2 was significantly higher in plants. Further analysis found that FPP/GGPP synthases of R. glutinosa were divided into three kinds, GGPS, GPPS and FPPS, and their gene expression was significantly diverse in different varieties, growth periods, or tissues of R. glutinosa. Compared with that of GGPS, the expression of GPPS and FPPS was much higher in R. glutinosa, especially at the expansion stage and maturation stage. Thus, the synthesis of terpenoids in roots of R. glutinosa is intricately regulated and needs to be further studied.
Terpenoids are important secondary metabolites in plants and have important economic value, not only because of their ability to resist diseases and fungus (Keeling and Bohlmann, 2006; Gershenzon and Dudareva, 2007), but also because of their various physiological activities, such as being anti-tumor, anti-inflammatory, and anti-oxidizing (Bohlmann and Zerbe, 2012). In plants, there are two different biosynthetic pathways of terpenoids: MVA pathway in the cytoplasm and MEP pathway in the plastid (Vranová et al., 2013; Henry et al., 2018). The biosynthetic pathway of terpenoids is mainly composed of three stages in plants: the first stage is the formation of intermediates, IPP and DMAPP, which are common precursors of terpenoids and could be interchangeably used in cytoplasm and plastid (Bochar et al., 1999; Lichtenthaler, 1999); the second stage is the synthesis of three direct precursors, GPP (C10), FPP (C15), and GGPP (C20) (Dewick, 2002); and the last stage leads to the formation of terpenoids with different structures and functions by the modification of various enzymes, such as methylation, hydroxylation, glycosylation, and so on (Liu, 2017).
Prenyltransferase is a key enzyme in the biosynthetic pathways of terpenoids and could catalyze the formation of the precursors for monoterpenes, sesquiterpenes, or diterpenes by IPP and DMAPP (Koyama and Ogura, 1999). At present, prenyltransferase encoding genes have been cloned and studied in some plants (Takei et al., 2001; Miyawaki et al., 2004; Brugière et al., 2008), and it was found that prenyltransferase mainly includes FPPS, GGPPS, and GPPS. Under the catalysis of GPPS, GPP is synthesized from one IPP molecule and one DMAPP molecule, and would lead to the synthesis of monoterpene (Wise and Croteau, 1999). Two IPP molecules and one DMAPP molecule are catalyzed to form FPP by FPPS, which would lead to the synthesis of sesquiterpenes and triterpenes (Cane, 1999). Three IPP molecules and one DMAPP molecule are catalyzed to form GGPP by GGPPS, which would lead to the synthesis of diterpenes and tetraterpenes (Martin et al., 2004).
R. glutinosa belongs to Scrophulariaceae Rehmannia and is rich in sugars, glycosides, alcohols, terpenoids, and other important components; more than 30 kinds of terpenoids have been discovered so far, such as catalpol, motherwort, arachidin, dihydroanthraquinone, glucoside A, D, and so on (Liu et al., 2012). Catalpol is a higher content of terpenoids in R. glutinosa, is one of the criteria evaluating medicinal effects of R. glutinosa (Wang et al., 2019), and has obvious pharmacological effects on osteoporosis, nervous system, the cardiovascular-cerebrovascular system, and the immune system, as well as having the effect of lowering blood glucose and regulating blood lipid (Huang et al., 2010; Shieh et al., 2011), anti-ionizing radiation (Chen et al., 2013), and anti-inflammation (Han et al., 2012).
At present, multiple omics techniques have been applied in R. glutinosa to better understand the formation and development of roots (Li et al., 2015; Sun et al., 2015; Zhou et al., 2018), and to explore the biosynthesis of bioactive components in roots (Sun et al., 2012; Zhou et al., 2016). In order to explore the biosynthetic pathway of terpenoids in R. glutinosa, iTRAQ was used for proteomic study on the roots of R. glutinosa, furthermore, some prenyltransferase encoding genes were identified and analyzed in R. glutinosa.
In this experiment, R. glutinosa Jinjiu and R. glutinosa 85-5 were used. R. glutinosa Jinjiu is a new breed recently bred, its main advantages are high yield, stable yield, high quality, and multiple resistances. R. glutinosa 85-5 also has high yield and high quality, but is not resistant to water stain and the degradation of some characteristics are more serious. The tuberous roots of R. glutinosa were kindly provided by Yongkang Liu and Cuihong Lu in Agricultural Research Institute of Wenxian County, Henan, China.
As described by Duan et al. (2018), tuberous roots of R. glutinosa were grown and cultured in test field of Henan Normal University, Xinxiang City, Henan, China. The type of planting soil was loam, planting density of R. glutinosa was 30×30 cm, and R. glutinosa was managed by conventional field management. Along with the growth and development of R. glutinosa, plant experiment materials were taken and studied, especially in the following three growth stages of R. glutinosa: the elongate stage (the root is fleshy and cylindrical), expansion stage (the root displays expansion), and the mature stage (the root is spindle-shaped). Roots, stems, and leaves of R. glutinosa were taken and stored at −80°C after quick-freezing with liquid nitrogen.
According to the extraction method of phenylic acid (Isaacson et al., 2006) and some improvements, proteins in roots of R. glutinosa Jinjiu were extracted. The concentration of protein extracts was determined by BCA method (Smith et al., 1985), protein extracts were detected by 12% SDS-PAGE electrophoresis, and the SDS-PAGE gel was visualized by CBB stain according to Candiano's protocol (Giovanni et al., 2004).
The shotgun comparative proteomic analysis of protein extracts from roots of R. glutinosa Jinjiu was studied by the iTRAQ 8-plex experiment. In this study, protein reduce, cysteine block, and digest were performed by FASP method (Jacek et al., 2009). iTRAQ labeling of protein sample was made according to the instruction of iTRAQ 8-plex kit (AB Sciex). After 50 μl tryptic digest (100 μg peptide) and iTRAQ reagent, the extracts were incubated for 2 h at room temperature, the labeled samples were pooled and collected, and then were dried in the vacuum freeze dryer for iTRAQ analysis.
The separation of peptides was performed by RPLC using the following method. The dry sample was re-suspended with 100 μl buffer A and the RPLC was employed on the Agilent 1200 HPLC System. The parameter of HPLC column (Agilent) was: Analytical Guard Column 4.6×12.5 mm 5-Micron, Narrow-Bore 2.1×150 mm 5 μm with 215 and 280 nm UV detection. The separation was performed at 0.3 ml/min. Elution of peptides was made with a mobile phase B of 2–90% over 8 min, each segment with 1min interval for 8-52 min was collected into one tube, four or five tubes were mixed with a total of 10 segments, and every segment was dried in a vacuum freeze dryer. Then the freeze-dried polypeptide samples were re-suspended with Nano-RPLC buffer A for RPLC-MS/MS analysis.
The online Nano-RPLC was employed on the Eksigent nanoLC-Ultra™ 2D System (AB SCIEX). The dissolved samples were loaded on C18 nanoLC trap column (100 μm×3 cm, C18, 3μm, 150Å) and washed by Nano-RPLC Buffer A (0.1% FA, 2% ACN) at 2 μl/min for 10 min. An elution gradient of 5-35% acetonitrile (0.1% formic acid) in 70 min gradient was used on an analytical ChromXP C18 column (75 μm × 15 cm, C18, 3 μm 120 Å, ChromXP Eksigent) with spray tip. Data acquisition was performed with a Triple TOF 5600 System (AB SCIEX, USA) fitted with a Nanospray III source (AB SCIEX, USA) and a pulled quartz tip as the emitter (New Objectives, USA). For information dependent acquisition, survey scans were acquired in 250 ms and as many as 35 product ion scans were collected if they exceeded a threshold of 150 counts per second (counts/s) with a 2+-5+ charge-state. The total cycle time was fixed to 2.5 s, a rolling collision energy setting was applied to all precursor ions for CID, and dynamic exclusion was set for ½ of peak width (18 s).
Against transcriptome data of R. glutinosa, raw data (.wiff, Sciex) was processed by Protein Pilot Software v. 5.0 (AB SCIEX, USA) using the Paragon algorithm (Shilov et al., 2007).
Furthermore, the experimental data from tandem MS was used to match the theory data to obtain results of protein identification, and protein identification was performed with the search option: emphasis on biological modifications.
In this study, the ID of reliable proteins was nonstandard protein ID; it was necessary to extract sequences from the transcriptome data of R. glutinosa (Sun et al., 2012). Based on sequence similarity and the sequence of retrieved transcriptome data of R. glutinosa, biological information functions of reliable proteins were analyzed using Arabidopsis thaliana as background population.
GO analysis was performed by the mainstream database David6.7 (http://david.abcc.ncifcrf.gov/) and QuickGO (http://www.ebi.ac.uk/QuickGO/) to describe GO classification annotation and enrichment analysis of reliable proteins. KEGG Pathway analysis and enrichment analysis results were obtained by mapping protein information to KEGG database. Furthermore, the analysis of PPI is based on string database (http://string.embl.de/) and cytoscape software (http://www.cytoscape.org/).
CTAB method was used to extract genomic DNA from roots and leaves of R. glutinosa and was performed as described by Duan et al. (2018). The yield and purity of genomic DNA were respectively determined with spectrophotometry at 260 nm, and the integrity of genomic DNA was detected by 0.8% agarose gel electrophoresis.
According to the instruction of RNAiso Plus (TaKaRa, Japan), total RNA was extracted from roots, stems, and leaves of R. glutinosa. In this experiment, DNase treatment and phenol-chloroform extraction were used to remove DNase, furthermore total RNA was dissolved in RNase-free dH2O. As described in Duan et al. (2019), the integrity of total RNA was detected by 1.0% agarose gel electrophoresis and the yield and purity of total RNA was estimated according to the absorbance at 260 and 280 nm by UV spectrophotometer. Furthermore, the first-strand cDNA of total RNA from roots, stems, or leaves of R. glutinosa was synthesized by reverse transcription referring to the instruction of TaKaRa kit (TaKaRa, Japan).
Based on transcriptome sequencing results of R. glutinosa, the full-length cDNA sequences of target genes were obtained by electronic cloning. According to UTR regions of target genes, primers were designed, and their sequences were listed in Supplementary Table 1. In this study, cDNA and genomic DNA of R. glutinosa were respectively used as templates to obtain full-length cDNA or DNA sequence of target genes by PCR amplification. PCR amplification reaction was performed in a volume of 20.0 μl, which was composed of 2.0 μl template, 1.0 μl each primer (10 μM), 10.0 μl 2×Taq plus, and 6.0 μl ddH2O. PCR amplification conditions were as follow: pre-denaturation for 5 min at 95°C, followed by 35 cycles (denaturation for 30 s at 94°C, annealing for 30 s and extension for 1 min at 72°C), and the final extension for 10 min at 72°C. Furthermore, the annealing temperatures of FPP/GGP synthase encoding genes were between 51 and 60°C in PCR amplification reaction.
Bioinformatics analysis of target genes were performed using the following method: ORF was analyzed with ORF Finder program (http://www.ncbi.nlm.nih.gov/gorf/gorf.html); amino acid sequence, MW, and other physicochemical properties of putative proteins were predicted by ExPASy proteomics server (http://www.expasy.ch/tools/protparam.html); and functional domain analysis of putative proteins were performed with NCBI-CDS (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). In addition, the signal peptide and transmembrane region in putative proteins were detected with Signal P 4.1 (http://www.cbs.dtu.dk/services/SignalP/) or TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), similar sequences of target genes were analyzed by BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi), amino acid sequences of target genes were compared with DNAMAN, the phylogenetic tree of target genes was constructed by MEGA, and the conservative analysis of target genes was performed based on multiple alignments of homologs using string genome (https://string-db.org/).
In this experiment, gene expression of target genes in R. glutinosa was detected by qRT-PCR which was conducted by LightCycler 96 Real-time PCR reaction. The cDNA template of qRT-PCR was prepared from total RNA of R. glutinosa by PrimeScript RT reagent Kit with gDNA Eraser (Perfect Real Time: TaKaRa, Japan), and the internal reference gene was TIP41. Primers used in qRT-PCR were listed in Supplementary Table 1, and were synthesized by YingjieJi Trade Co., Ltd., (Shanghai, China).
The total volume of qRT-PCR reaction system was 20 μl, composed of 2.0 μl cDNA template, 1.0 μl each primer (10 μM), 10 μL SYBR® Green Master Mix, and 6.0 μl ddH2O. The reaction conditions of qRT-PCR were as follows: predenaturation for 5 min at 95°C, followed by 45 cycles of denaturation for 5 s at 95°C, and annealing for 20 s at 60°C. The dissolution conditions were 95°C for 15 s, 60°C for 1 min, and 65°C for 15 s, with continuous detection of signals. In addition, the relative expression levels of target genes were normalized and analyzed by the comparative Ct (2−ΔΔct) method (Duan et al., 2019).
In this study, iTRAQ quantitative proteomic analysis was performed to study the relative abundance of proteins in roots of R. glutinosa. Peptides of six or more amino acids in length and with a maximum of two missed cleavages were exclusively considered for the analysis; the original data were obtained according to search results of protein mass spectrometry (ProteomeXchange identifier: PXD025914). After the database search, 8,929 proteins were detected, and 6,752 reliable proteins were obtained according to the screening criteria of reliable protein.
GO enrichment of all proteins were performed. The significant number of BP, CC, and MF was respectively 1704, 360, or 592 (P < 0.05) (Supplementary Figure 1). GO function and enrichment analysis were shown in Figure 1, most proteins were involved in metabolic process or cellular process (80% or so), about 42% proteins were in response to stimulus, about 22% proteins were involved in developmental process, and 4 and 3% proteins in root development or root morphogenesis, respectively (Figure 1A). Furthermore, there were some metabolic processes with higher significance (P < 0.01), such as small molecule metabolic process, nucleotide metabolic process, and carboxylic acid metabolic process, the biological process response to cadmium ion was also significant, and catabolic process had higher significance (Figure 1B).
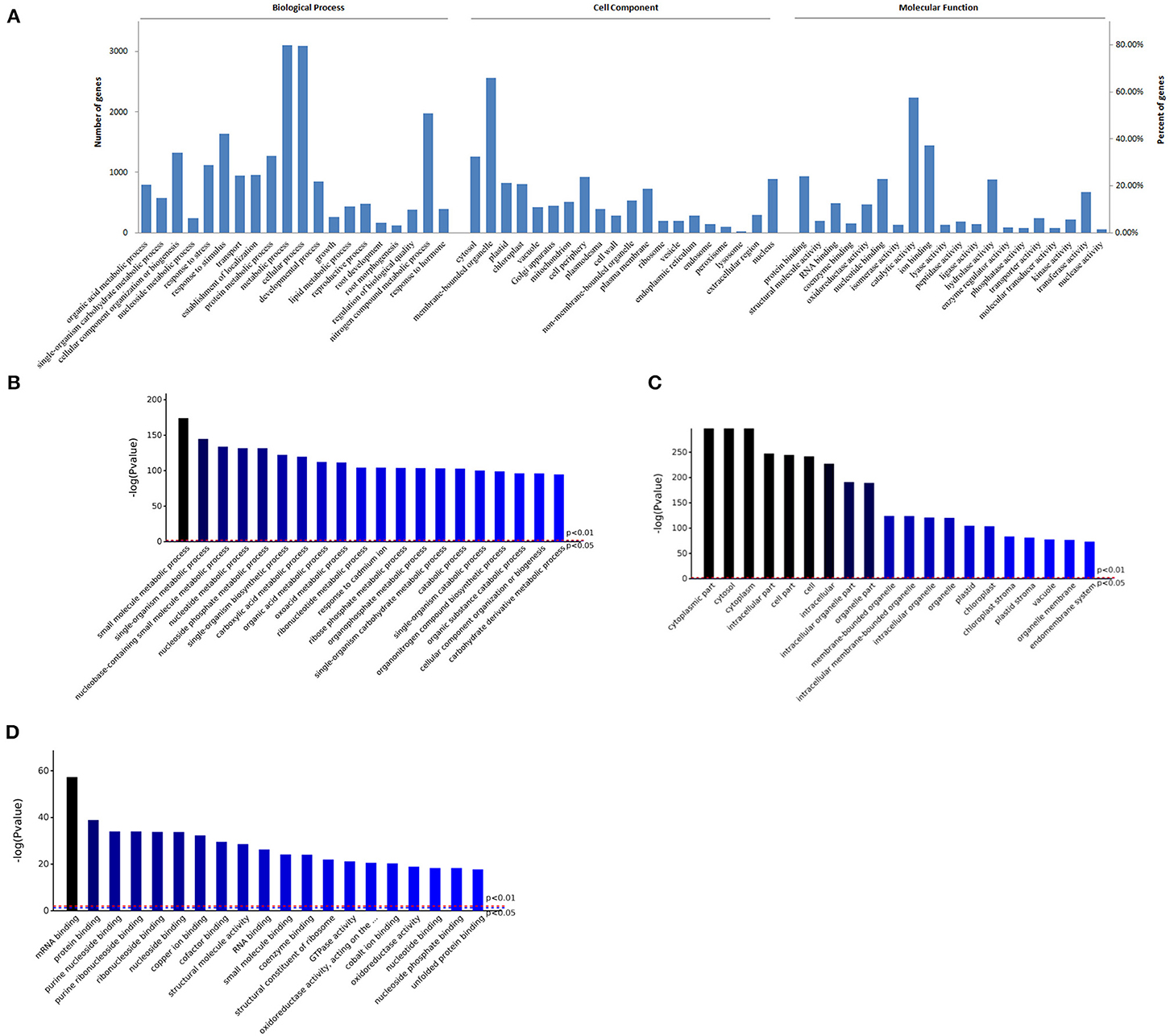
Figure 1. GO enrichment analysis of proteins in roots of R. glutinosa. (A) GO distribution of proteins, the abscissa represents the entries in each category (Biological Process, Cell Component, Molecular Function), the left and right ordinate represent the number or percent of proteins which are included in each entry. (B–D) Represents the top 20 entries in BP, CC, or MF enrichment results, respectively, the abscissa represents the entries, P value of red line and blue line is respectively 0.01 or 0.05.
In addition, almost every cellular compartment was enriched. Compared to cytosol, about 66% proteins were enriched in membrane-bounded organelle, such as nucleus, chloroplast, mitochondrion, vacuole, ribosome, endoplasmic and reticulum (Figure 1A), and cytosol, membrane-bounded organelle, and chloroplast had higher fold enrichment (P < 0.01) (Figure 1C). As shown in Figure 1A, about 58% proteins had catalytic activity, followed by hydrolase activity, transferase activity, or oxidoreductase activity. Furthermore, most proteins belong to ion binding, protein binding, or nucleotide binding, and protein binding or nucleotide binding had higher fold enrichment (P < 0.01) (Figure 1D).
Analysis and enrichment results of KEGG pathway in roots of R. glutinosa were listed in Supplementary File 1, there were 38 KEGG enrichments with significance (P < 0.05) (Supplementary Figure 1), such as metabolic pathways, carbon metabolism, biosynthesis of amino acids, and TCA cycle. The significant top ranking 10 entries (P < 0.01) were shown in Figure 2A, the P value was very high in carbon metabolism or biosynthesis of amino acids, but was very low in arginine biosynthesis. Furthermore, many proteins were enriched in carbon metabolism, biosynthesis of amino acids, metabolic pathway, spliceosome, and protein processing in endoplasmic reticulum, there were significantly more in the metabolic pathway with 795 proteins (Supplementary File 1). However, less proteins were enriched in proteasome, glycolysis, pyruvate metabolism, TCA cycle, and arginine biosynthesis; only 26 proteins were enriched in arginine biosynthesis (Supplementary File 1).
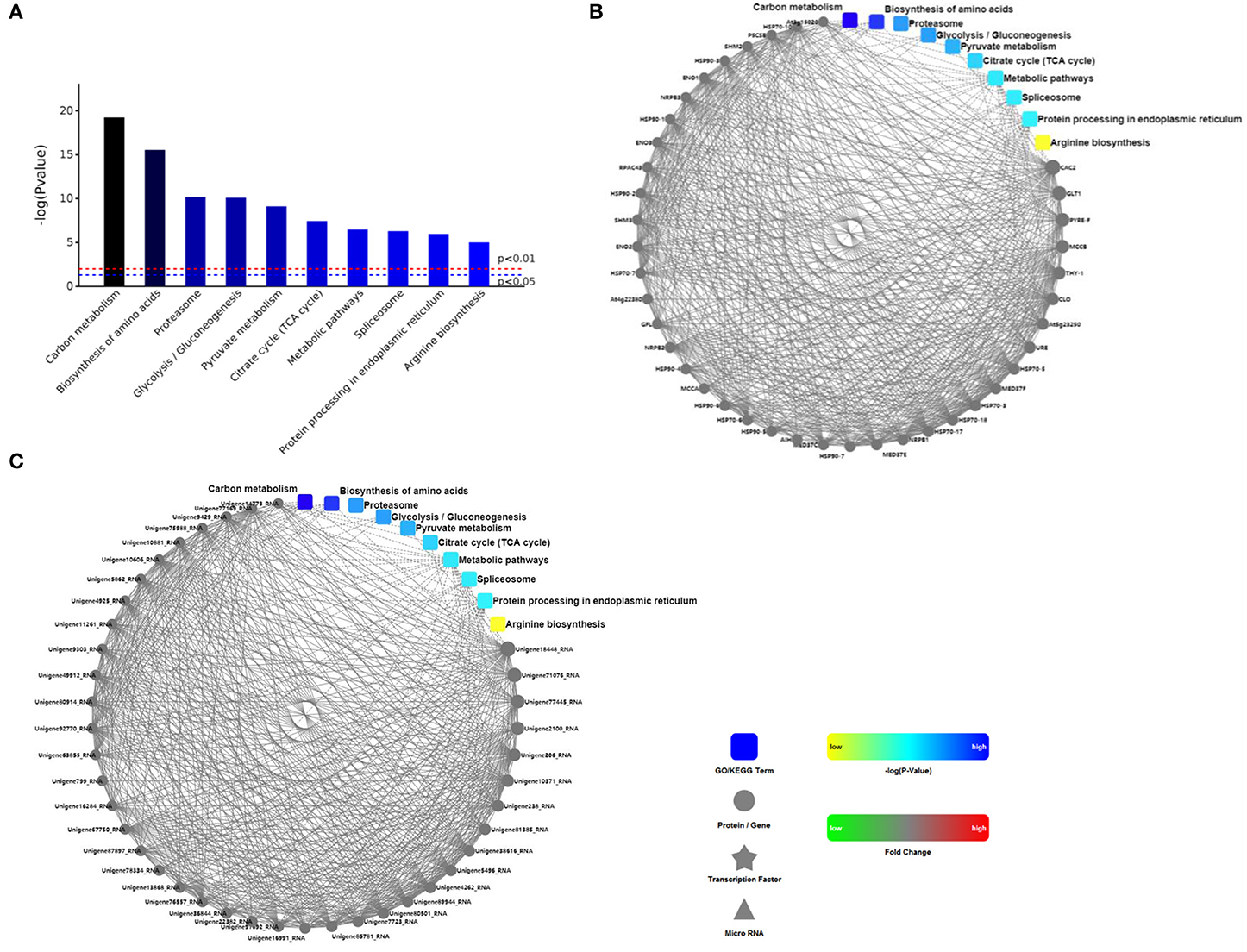
Figure 2. The analysis of KEGG and PPI proteins in roots of R. glutinosa. (A) The significant top ranking 10 KEGG enrichments, (B, C) represents the interaction between the 10 regulatory pathways and the interaction with the protein, is respectively ppigene (B) and ppiquery (C) of interaction network. P value of red line and blue line in (A) is respectively 0.01 or 0.05, only when the top of the column is higher than the blue line or the red line, the regulatory pathway is significant (P < 0.05) or very significant (P < 0.01). Interactive mapping (B) or (C) is based on dots (gene or protein), polygon (metabolic product), triangle (miRNA), five angle (transcription factor) and rounded rectangle (biological process, cellular localization, molecular functional or signaling pathway) connecting lines, which show the molecular interaction networks and connection mechanism model. The dashed line is without the experimental verification, the solid line is already the relevant verification reports. Furthermore, the dot color change is that red sketch expression is up-regulated, green sketch expression is down-regulated, the rounded rectangle color is that yellow and blue gradient said is from low to high.
According to string database and Cytoscape software, PPI in roots of R. glutinosa was analyzed (Supplementary File 2), PPI network of proteins in the top ranking 10 KEGG pathways was shown in Figures 2B,C. There were more PPIs in carbon metabolism, metabolic pathway, and protein processing in endoplasmic reticulum, followed by biosynthesis of amino acids and spliceosome, yet PPIs were less in glycolysis, TCA cycle, or arginine biosynthesis, and were not found in proteasome (Figures 2B,C).
During the growth and development of R. glutinosa, some proteins were differentially expressed, and DEPs in KEGG enrichments of Figure 2 were further analyzed (Table 1). In some KEGG pathways, there were more DEPs between elongation stage and expansion stage, such as carbon metabolism, biosynthesis of amino acids, proteasome, pyruvate metabolism, and TCA cycle pathway, especially the pathway of proteasome and TCA cycle with 15.0% DEPs (Table 1). More DEPs were also found between other stages, such as proteasome between elongation stage and maturation stage or the pathway of pyruvate metabolism and arginine biosynthesis between expansion stage and maturation stage. However, as listed in Table 1, the percentage of DEPs was relatively lower in some KEGG pathways during the growth and development of R. glutinosa, especially spliceosome.
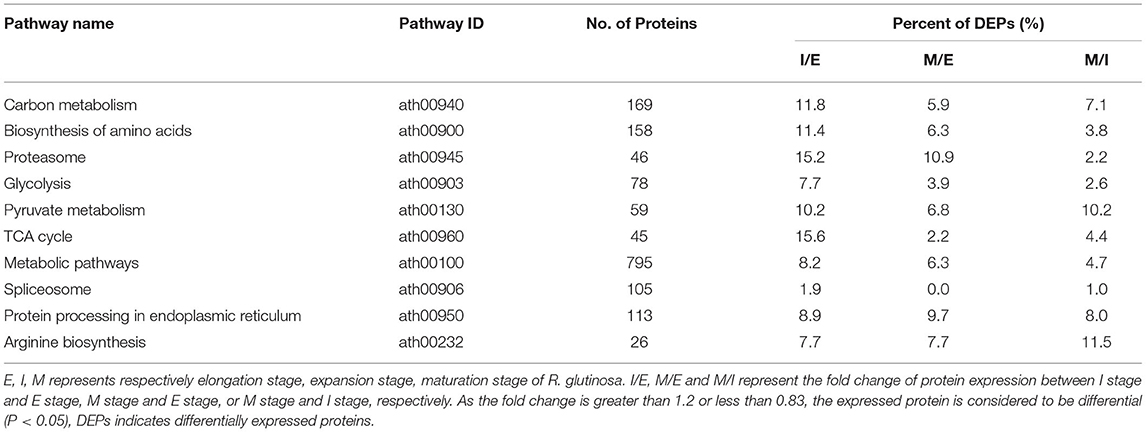
Table 1. KEGG enrichment analysis of differentially expressed proteins in roots of R. glutinosa.
As shown in Supplementary File 1, about 457 proteins were enriched to Biosynthesis of secondary metabolites (ath01110). Some KEGG pathways about the active ingredients of R. glutinosa were also found, such as phenylpropanoid, terpene, stilbenoid, steroid, alkaloid, flavonoid, and so on (Table 2). In R. glutinosa, there were five KEGG regulatory pathways of terpene synthesis: terpenoid backbone biosynthesis pathway, ubiquinone and other terpenoid-quinone biosynthesis pathway, monoterpenoid biosynthesis pathway, diterpenoid biosynthesis pathway, and sesquiterpenoid and triterpenoid biosynthesis pathway (Table 2). Furthermore, 27 and 15 proteins, respectively, were enriched in terpenoid backbone biosynthesis or other terpenoid-quinone biosynthesis.
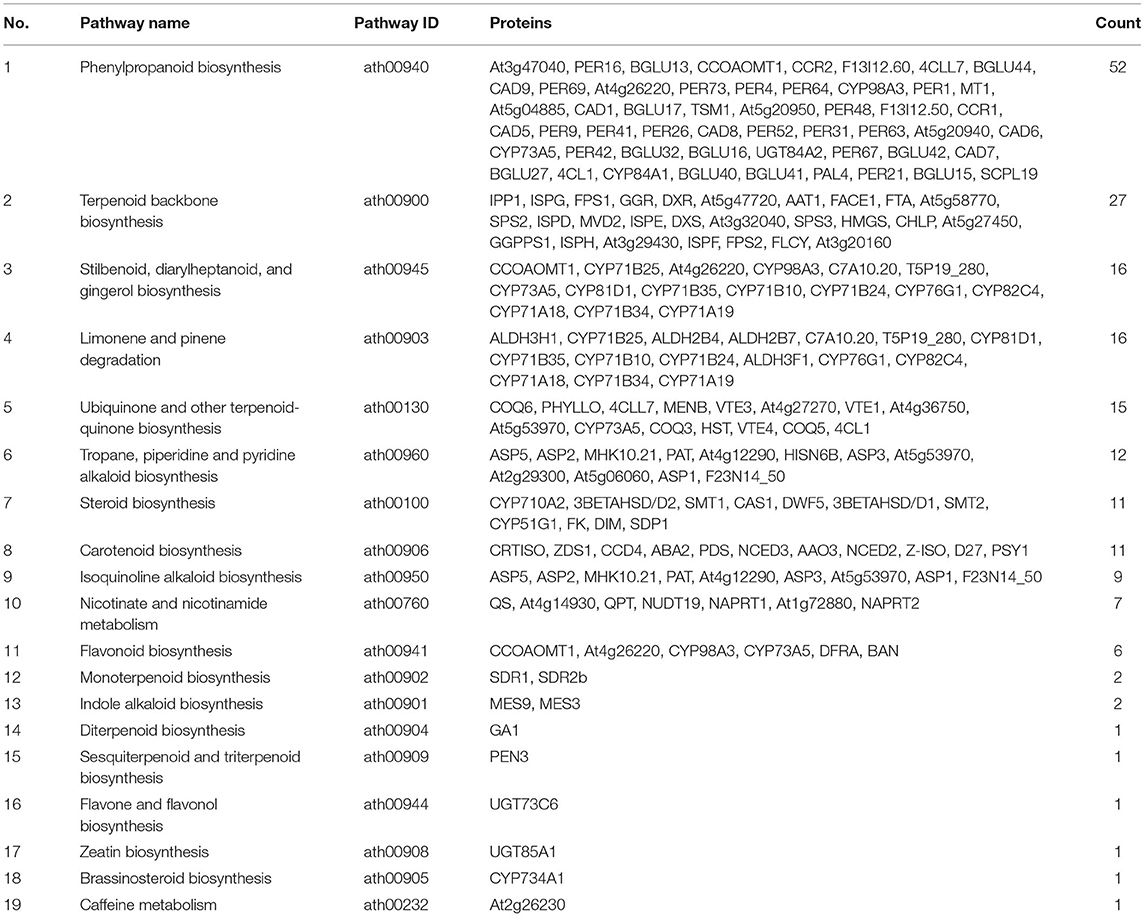
Table 2. KEGG pathway on some secondary metabolites in roots of R. glutinosa.
Based on string database, PPI analysis of proteins associated with terpenoid synthesis was performed (Supplementary Table 2). Except for AAT1 and TEGA10, most proteins could interact with each other, especially At5G27450, HMGS, ISPD, HST, IPP1, ISPF, and SPS2 (Figure 3A). Further analysis indicated that these terpenoid synthesis proteins could simultaneously interact with other proteins, in which most proteins interacted with CAC2, followed by GLU1 and MPA1, and there was also more interaction with ALDH10A8, PPC3, and ASP5 (Figure 3B).
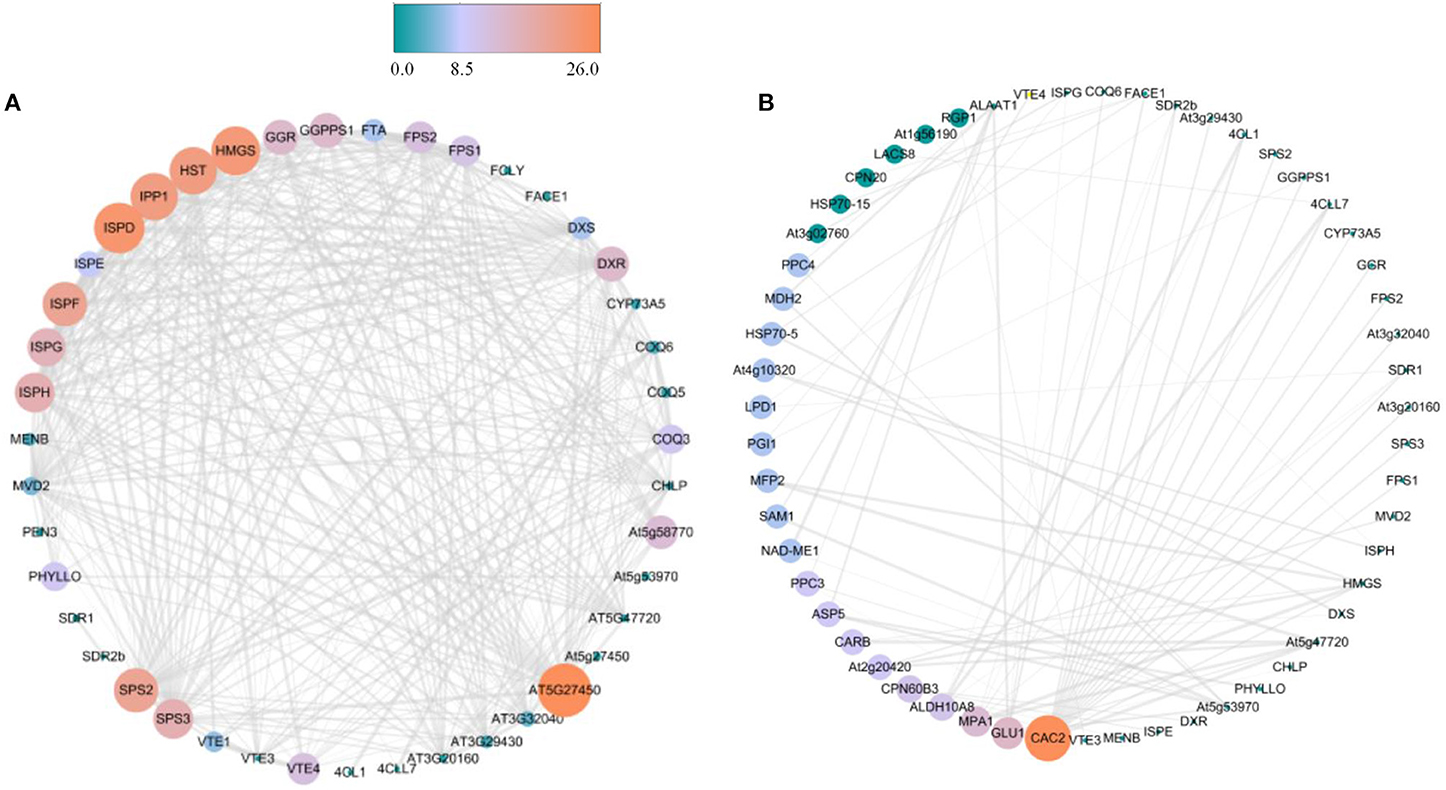
Figure 3. PPI analysis of proteins associated with terpenoid synthesis in R. glutinosa. (A) PPI analysis between proteins of terpenoid synthesis in R. glutinosa, (B) PPI analysis between proteins of terpenoid synthesis and others in R. glutinosa. The size and color of the circle represents the indegree of PPI, atrovirens circle is low values, but jacinth circle is high values.
During the growth and development of R. glutinosa, most proteins associated with terpenoid synthesis can be found differently expressed (P < 0.05) (Supplementary Table 3 and Figure 4A). Compared with that at elongation stage, some proteins were up-regulated at expansion stage and maturation stage, such as CHLPA, DXR, AAT1, ISPG, ISPE, PEN3, FPS2, and At3g20160, especially at expansion stag (P < 0.05) (Figure 4A). However, the expression of about 26% proteins at elongation stage was significantly higher than that at expansion stage (P < 0.05), in which some proteins were also up-regulated at maturation stage, such as 4CLL7, FTA, TGA10, COQ5, FACE1, At4g27270, and ISPH, and the expression of 4CLL7, FTA, and TGA10 at the maturation stage was significantly higher than that at elongation stage (P < 0.05) (Figure 4A).
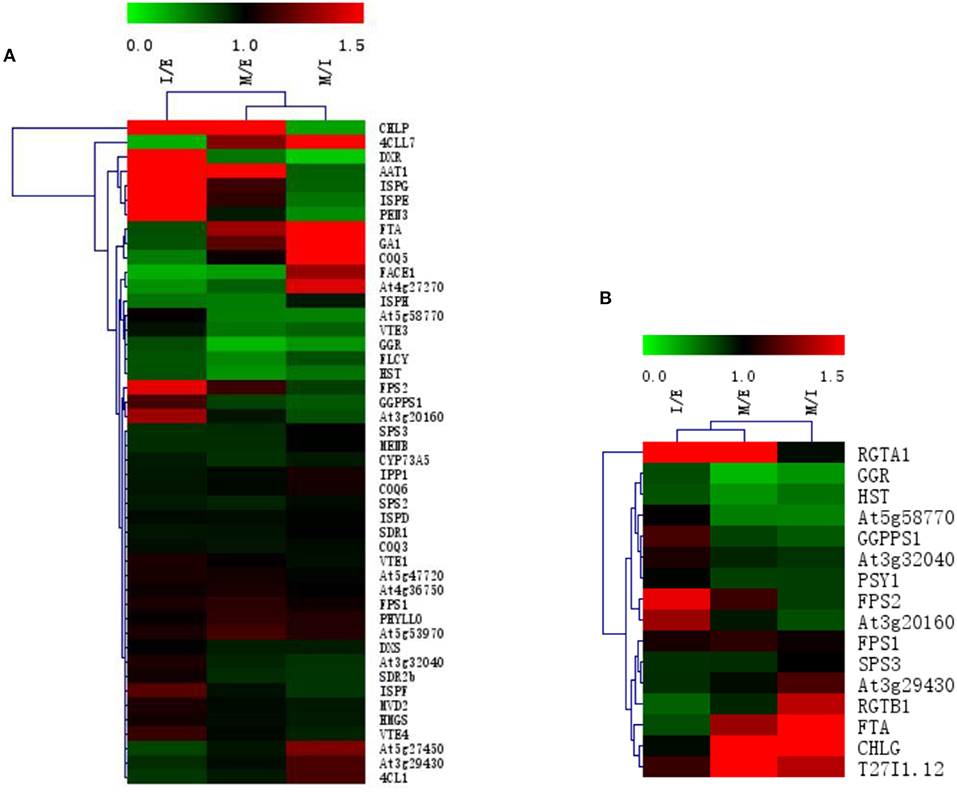
Figure 4. The expression clustering heatmap of some proteins in roots of R. glutinosa. (A) The expression of proteins associated with terpenoid synthesis in R. glutinosa; (B) The expression of prenyltransferase in R. glutinosa. I/E represents the fold change of protein expression between I stage and E stage of R. glutinosa, M/E represents the fold change of protein expression between M stage and E stage of R. glutinosa, M/I represents the fold change of protein expression between M stage and I stage of R. glutinosa. Green and red color refers to the down or up expression of proteins, respectively. Each line represents the expression of each protein in different groups. Each column represents the expression of all proteins in each group.
As listed in Table 3, these proteins with prenyltransferase activity in R. glutinosa were divided into five families, namely FPP/GGPP synthase family, UPP synthase family, protein prenyltransferase family, phytoene/squalene synthase family and UbiA prenyltransferase family, and were enriched in some KEGG pathways, except for T27I1.12, RGTA1, and RGTB1 which belong to protein prenyltransferase subunit family. Further analysis found that most prenyltransferases in R. glutinosa belong to FPP/GGPP synthase family, such as FPS1, FPS2, At3g32040, At3g20160, GGPPS1, GGR, and SPS3, which were involved in terpenoid backbone biosynthesis and interacted with biotin carboxylase CAC2. GGPPS1 also interacted with Glucose-6-phosphate isomerase PGI1 (Supplementary Table 3). Furthermore, UPP synthase At5g58770 and protein prenyltransferase subunit alpha FTA were also involved in terpenoid backbone biosynthesis of R. glutinosa (Table 3).
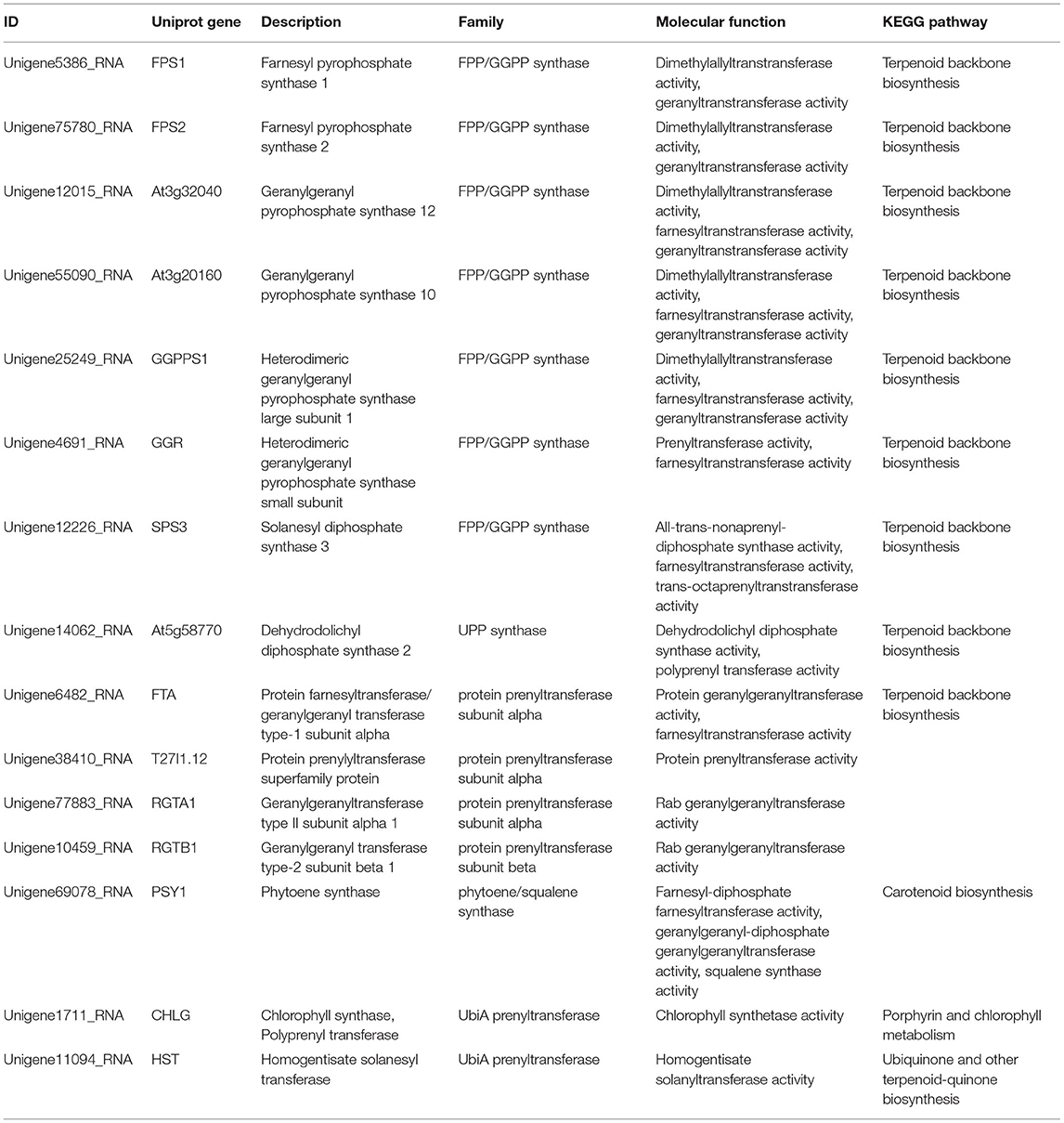
Table 3. The analysis of prenyltransferase in R. glutinosa.
In addition, protein expression of prenyltransferase in R. glutinosa (shown in Supplementary Table 3 and Figure 4B) showed that the expression of FPS2 and At3g20160 was higher at expansion stage (P < 0.05). Compared with that at elongation stage, RGTA1 was continuously up-regulated at expansion stage and maturation stage (P < 0.05). FTA, CHLG, and T27I1.12 all exhibited higher expression at maturation stage, but the expressions of GGR and HST continued to decrease at expansion stage and maturation stage, and GGPPS1, At5g58770, and PSY1 were down-regulated at maturation stage (P < 0.05).
In this study, some FPP/GGPP synthase encoding genes were cloned from R. glutinosa and their cDNA sequences were submitted to GenBank (Supplementary Figure 2), such as FPPS (MG770217), FPPS1 (MT680921), GGPS (MG770218), GGPS3 (MT680922), GGPS4 (MT680923), GGPS5 (MW298275), GPPS (MG770219), and GPPS2 (MW656184). Their full-length CDS sequences were, respectively, 1029, 1050, 1119, 987, 1119, 990, 1272, and 1263 bp (Table 4). Furthermore, intron sequence was found in some FPP/GGPP synthase encoding genes, such as FPPS, FPPS1, GGPS5, and GGPS2; their DNA sequences were shown in Supplementary Figure 3. The full-length DNA sequence of FPPS gene was 3572 bp and was composed of 11 exons and 10 introns, the longest intron was 1073 bp, the shortest intron was 86 bp (Supplementary Figure 3a). As shown in Supplementary Figure 3b, DNA sequence of FPPS1 gene was 4767 bp with 10 introns; the longest intron was 2015 bp, the shortest intron was 83 bp. DNA sequence of GPPS2 gene was 3193 bp and was composed of 6 exons and 5 introns, the longest intron was 625 bp and the shortest intron was 81 bp (Supplementary Figure 3d), yet only one intron with 1259 bp was found in GGPS5 gene (Supplementary Figure 3c).
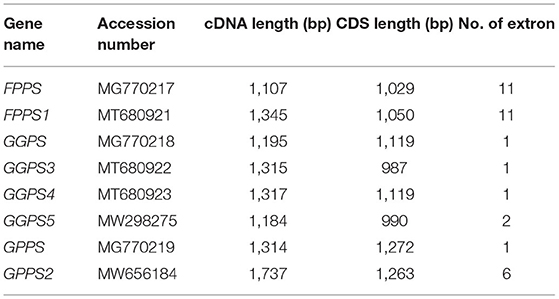
Table 4. Sequence information of FPP/GGPP synthase encoding genes in R. glutinosa.
CDD analysis results showed that FPP/GGPP synthase of R. glutinosa is all composed of three functional domains, namely polyprenyl-synt, IspA, and Trans-IPPS-HT, and belongs to Isoprenoid_biosyn_C1 superfamily which share the same isoprenoid synthase fold. The physical and chemical properties of FPP/GGPP synthase in R. glutinosa were listed in Table 5. PI was 5.5–6.1 and MW was 40 kD or so. MW of GGPS3 and GPPS5 was about 35 kD, but MW of GPPS and GPPS2 was 46 kD or so. FPP/GGPP synthases were all hydrophilic; only one transmembrane region was detected in FPPS (at 180th−220th) and the subcellular locations of FPP/GGPP synthases were different, for example, FPPS and FPPS1 were located in cytoplasm, GGPS4 and GPPS were located in mitochondrion, and others were located in chloroplast. The secondary structures of FPP/GGPP synthase were generally composed of four parts, namely alpha helix, extended strand, beta turn, and random coil. The proportion of alpha helix was the most (50–65%), followed by random coils (25–33%); the proportion of beta turn was the least (Table 5).
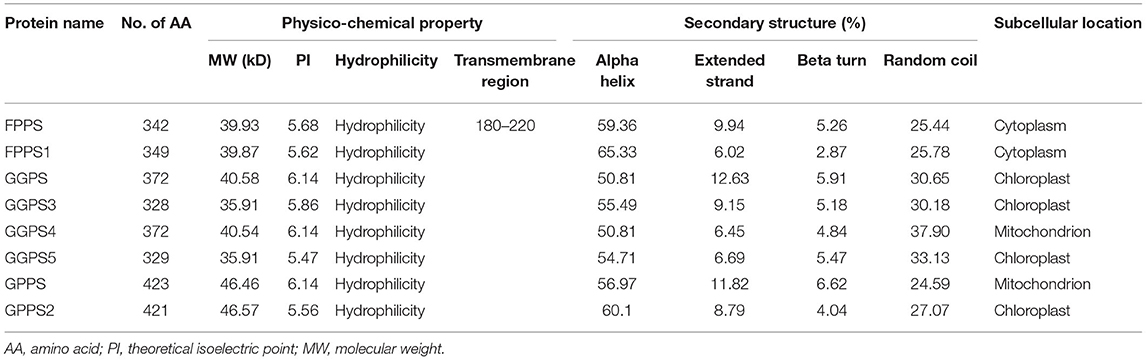
Table 5. The analysis of FPP/GGPP synthase in R. glutinosa.
As can be seen in Figures 5A,B, the similarity was lower among FPP/GGPP synthases in R. glutinosa, but the identity between their domains was about 37.8%. Further analysis showed that the identity of GGPS and GGPS4 was as high as 98.4% and was also higher (81.6%) between GGPS3 and GGPS5, but the identity of GGPS with GGPS3 or GGPS5 was relatively low, at 38% or so. The similarity of FPPS and FPPS1 was also higher (81.0% identity), yet was 31.4% between GPPS and GPPS2. The identity among other FPP/GGPP synthases was also relatively low. In addition, the Phylogenetic tree of FPP/GGPP synthases in R. glutinosa was divided into two branches, FPPS and FPSS1 were clustered into one branch, other synthases were clustered together, in which GGPPS synthases were clustered into one sub-branch, such as GPPS, GPPS3, GPPS4, and GPPS5, while the other sub-branch was composed of GPPS and GPPS2 (Figure 5C).

Figure 5. Sequence comparison of FPP/GGPP synthases in R. glutinosa. (A) Sequence alignment of FPP/GGPP synthase in R. glutinosa, (B) Homology matrix of FPP/GGPP synthase in R. glutinosa, (C) Phylogenetic analysis of FPP/GGPP synthase in R. glutinosa.
Based on string genome, gene co-occurrence on FPP/GPP synthase of R. glutinosa was analyzed. It was found that FPP/GPP synthase was conservative in organisms and occurred in Archaea, Eukaryota, and Bacteria, in which FPPS, FPPS1, GGPS4, GGPS5, GPPS, or GPPS2 of R. glutinosa was more conservative, especially in viridiplantae (Figure 6A). Many genes in viridiplantae appeared to match FPP/GPP synthase of R. glutinosa (Table 6), gene co-occurrence for all FPP/GGPP synthases of R. glutinosa were all found in 39 species of plants, such as Arabidopsis thaliana, Brassica rapa, Erythranthe guttata, Glycine max, Gossypium raimondii, Nicotiana tomentosiformis, and Vitis vinifera. 5–7 FPP/GPP synthases were respectively matched in 14 species of plants, in which GGPS and GGPS4 had less co-occurrence. Only 1–2 synthases in 14 species of plants had co-occurrence, and mainly belong to GPPS. In addition, the co-occurrence of FPP/GPP synthase in plants was higher, especially GPPS and GPPS2 (85% co-occurrence), yet the co-occurrence of GGPS, GGPS4 in plants was 57% or so (Table 6).
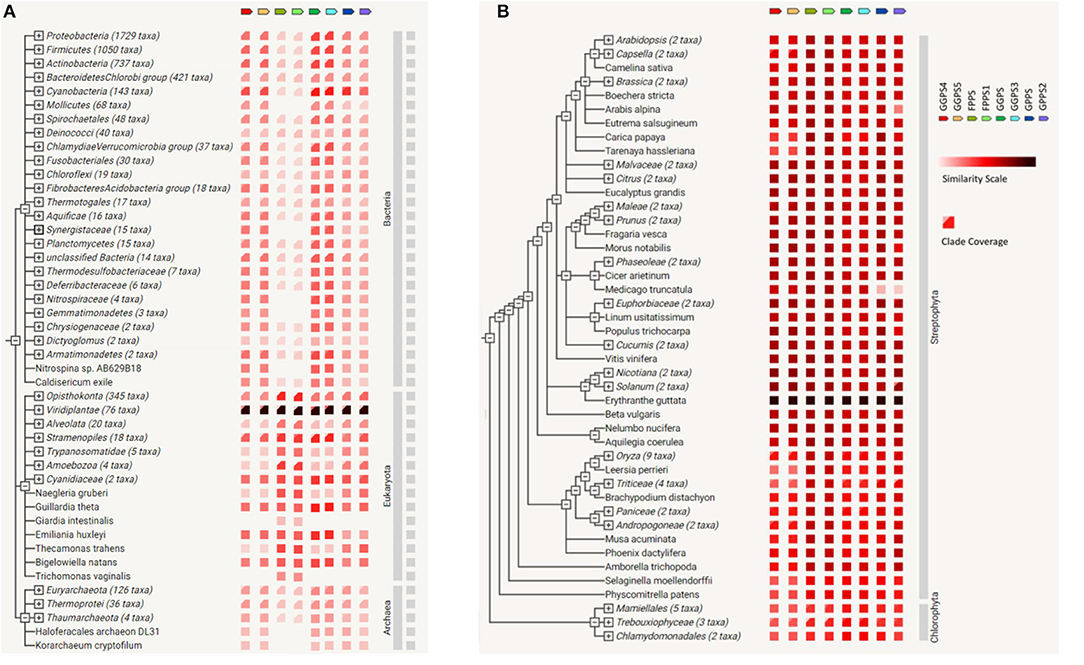
Figure 6. The co-occurrence on FPP/GPP synthase family of R. glutinosa. (A) Gene co-occurrence of FPP/GPP synthase in the organisom, (B) The co-occurrence of FPP/GPP synthase in plants. Similarity Scale: the color denotes, for each gene of FPP/GPP synthase, the similarity of its best hit in a given string genome. Clade Coverage: for groups of genomes that are collapsed in the phylogenetic tree, two distinct colors indicate the lowest and highest similarity observed within that clade.

Table 6. The gene co-occurrence on FPP/GPP synthase family of R. glutinosa.
Compared with other FPP/GPP synthases of R. glutinosa, GGPS and GGPS3 had higher similarity with their relative homologs in Bacteria, the similarity of FPPS or FPPS1 with the relative homologs was also higher in Eukaryota, but was lower in Bacteria and Archaea (Figure 6A). Further analysis showed that all FPP/GGPP synthases of R. glutinosa had higher similarity with their homologs in streptophyta, especially in Erythranthe guttata, followed by Nicotiana sylvestris, Nicotiana tomentosiformis, Vitis vinifera, Beta vulgaris, Solanum tuberosum, Beta vulgaris, and Arabidopsis thaliana (Figure 6B; Supplementary Table 4). In addition, FPPS or FPPS1 of R. glutinosa had higher similarity with their relative homologs in most plants compared to other synthases (Figure 6B).
As shown in Figure 7A, along with the growth and development of R. glutinosa 85-5, the expression of FPPS gene in root, stem, and leaf increased, and was higher at M stage (P < 0.05), especially in leaf. Similarly, the expression of FPPS gene in R. glutinosa Jinjiu also increased continuously and was higher in root and stem at M stage (P < 0.05). Furthermore, the expression profile of FPPS was obviously varied between R. glutinosa 85-5 and R. glutinosa Jinjiu, except for that in leaf at M stage, the expression of FPPS was generally higher in R. glutinosa Jinjiu, and was especially significant in root and stem (Figure 7A).
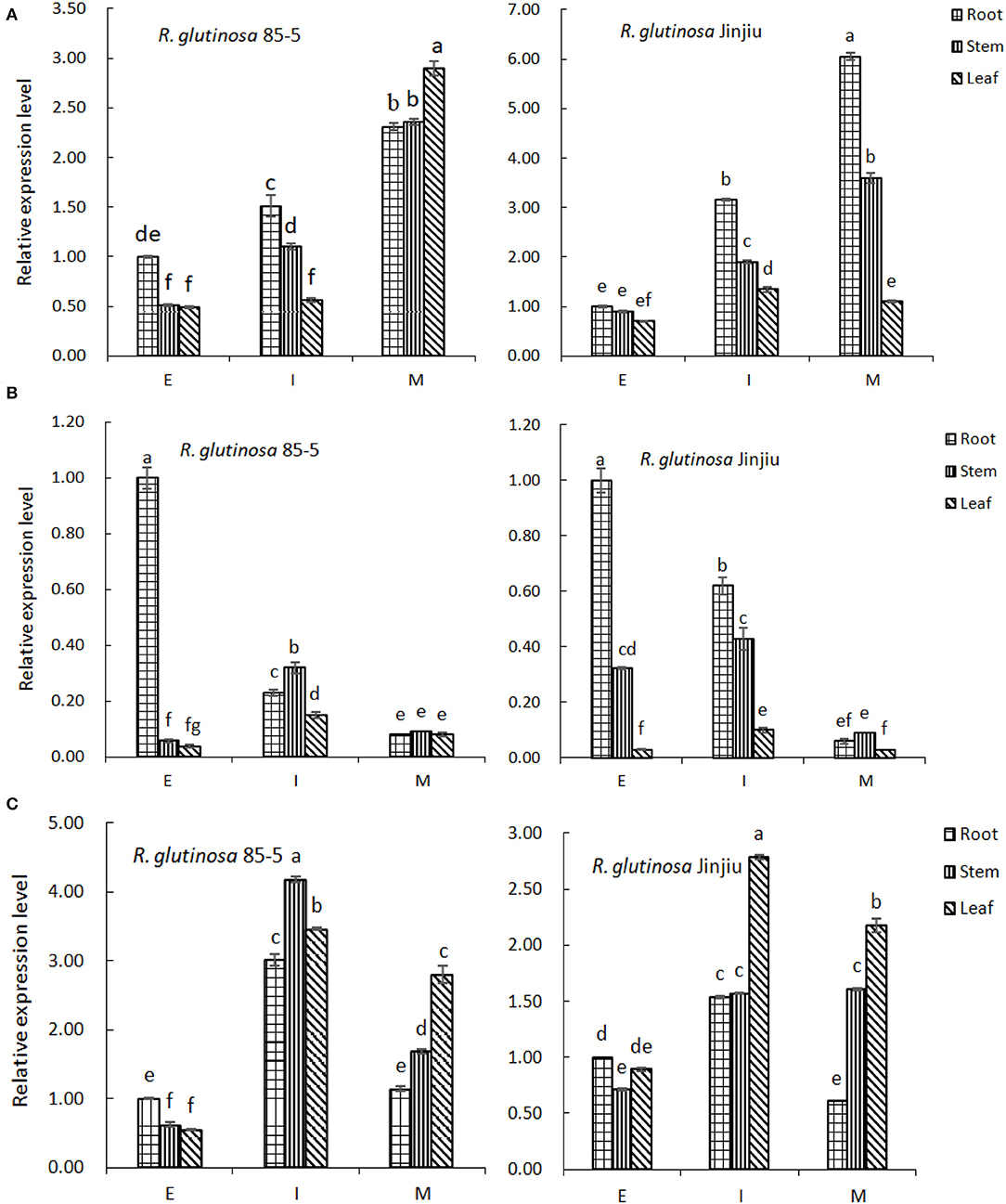
Figure 7. The expression pattern of FPP/GGPP gene in R. glutinosa. (A–C) represents the relative expression of FPPS, GGPS, or GPPS in R. glutinosa 85-5 and R. glutinosa Jinjiu, E, I, and M respectively represents elongation stage, expansion stage, and maturation stage of R. glutinosa. The error bar is standard error of mean, and the lower letter above the bar indicated the significant difference in root, stem, or leaf among the different growth stages of R. glutinosa (P < 0.05).
Along with the growth and development of R. glutinosa, the expression of GGPS gene in roots decreased continuously (P < 0.05), but increased initially and then decreased in stem and leaf, and was higher at I stage (Figure 7B). Compared with that in stem and leaf, the expression of GGPS was higher in root of R. glutinosa 85-5 at E stage, and was also higher in root of R. glutinosa Jinjiu at E–I stage (P < 0.05). Although the expression trend of GGPS gene was similar between R. glutinosa 85-5 and R. glutinosa Jinjiu and was both lower at M stage, the expression of GGPS was higher at E-I stage of R. glutinosa Jinjiu compared with that of R. glutinosa 85-5 (Figure 7B).
As can be seen from Figure 7C, the expression of GPPS gene in R. glutinosa 85-5 first increased and then decreased along with the growth and development of R. glutinosa, and was higher at I stage (P < 0.05). Compared with that in root, the expression of GPPS was significantly higher except in E stage (P < 0.05). Similarly, the expression of GPPS gene in R. glutinosa Jinjiu also first increased and then decreased, reached the highest peak at I stage, and the expression of GPPS gene in leaf was higher (P < 0.05) (Figure 7C). In addition, the expression of GGPS gene was much lower compared with that of FPPS and GPPS, especially at I–M stage of R. glutinosa (P < 0.05).
R. glutinosa is a perennial herb, its tuberous root is commonly used in Chinese herbal medicines (Duan et al., 2018), can be divided into dried rehamannia root, prepared rehamannia root, and fresh rehamannia root (Lian, 2016). Along with the development of biological technology, omics technique provides a theoretical basis for further study on the molecular mechanism of quality and yield of R. glutinosa (Li et al., 2015; Zhou et al., 2018). In this study, iTRAQ quantitative proteomic analysis was used to analyze the relative abundance of proteins in roots of R. glutinosa; 8,929 proteins were identified and 6,752 proteins were quantified, but only about 50% reliable proteins in R. glutinosa could be further analyzed using Arabidopsis thaliana as background population. GO enrichment results indicated that most proteins in roots of R. glutinosa were involved in metabolic process or cellular process, 42.26% proteins were in response to stimulus, about 22% proteins were involved in developmental process, and 4.33% were involved in root development. Furthermore, 57.63% proteins had catalytic activity, and 65.80% proteins were enriched in membrane-bounded organelle, especially in nucleus and chloroplast. On account of its high sensitivity, iTRAQ technology is also used to detect proteins in some plants, such as Beta vulgaris (Wang, 2017), Rice (Qian et al., 2015), Camelina sativa (Alvarez et al., 2015), and Tobacco (He et al., 2020).
It is well known that roots of R. glutinosa are rich in bioactive compounds, such as glycosides, alcohols, terpenoids, and so on (Zhou et al., 2018). In this study, there were 38 KEGG enrichments with significance, such as metabolic pathway, carbon metabolism, biosynthesis of amino acids, and TCA cycle, and more PPIs were found in these pathways. During the growth and development of R. glutinosa, more DEPs were found in some KEGG pathways, especially the pathway of proteasome and TCA cycle with 15.0% DEPs between elongation stage and expansion stage of R. glutinosa. In addition, some proteins in roots of R. glutinosa were also enriched in the secondary metabolism pathway, such as phenylpropanoid biosynthesis, terpenoid backbone biosynthesis, limonene and pinene degradation, and so on. Terpenoids are important secondary metabolites in plants and have a wide range of pharmacological effects and important economic value (Keeling and Bohlmann, 2006; Gershenzon and Dudareva, 2007; Bohlmann and Zerbe, 2012). In R. glutinosa, five KEGG terpenoid synthesis pathways were found, many proteins associated with terpenoid synthesis were mainly enriched in terpenoid backbone biosynthesis pathway, but were less in the downstream pathways of terpenoid synthesis, such as monoterpenoid and diterpenoid. Furthermore, most proteins associated with terpenoid synthesis were differently expressed along with the growth and development of R. glutinosa and could interact with each other or with CAC2, GLU1, MPA1, and other proteins, suggesting they might participate in the complex metabolic networks.
Prenyltransferase plays an important role in terpenoid synthesis and could catalyze the formation of precursors for monoterpenes, sesquiterpenes, diterpenoids, or other terpenes by IPP and DMAPP (Liu, 2017). In R. glutinosa, most prenyltransferases belong to FPP/GGPP synthase family, such as FPS1, FPS2, At3g32040, At3g20160, GGPPS1, GGR, and SPS3, and are involved in terpenoid backbone biosynthesis. All interacted with CAC2. Compared with that at elongation stage, many prenyltransferases exhibited higher expression in roots at expansion stage or maturation stage of R. glutinosa. In addition, eight FPP/GGPP synthase encoding genes were cloned from R. glutinosa, and introns were found in FPPS, FPPS1, GGPS5, and GGPS2, in which FPPS and FPPS1 had 10 introns, 5 introns were found in GPPS2 gene, and only one intron with 1259 bp was found in GGPS5 gene. Further analysis found that FPP/GPP synthases of R. glutinosa were more conservative in organisms, especially in viridiplantae, and the co-occurrence of GPPS or GPPS2 was significantly higher in plants. Furthermore, FPP/GPP synthases of R. glutinosa had higher similarity with their homologs in streptophyta, especially in Erythranthe guttata.
Although the physical and chemical properties of FPP/GGPP synthases were diverse in R. glutinosa, their structures were similarly composed, and these FPP/GGPP synthases of R. glutinosa were further divided into three kinds: GGPS, GPPS, and FPPS. As is well known, GGPS is a key enzyme in MEP pathway and is involved in the synthesis of diterpenes and tetraterpenes (Martin et al., 2004), yet FPPS is a key enzyme in MVA pathway and is involved in the synthesis of sesquiterpene and triterpenoids (Cane, 1999). GPPS is mainly involved in MEP pathway, and is related to the synthesis of monoterpene (Wise and Croteau, 1999). Catalpol belongs to monoterpene and is a higher content of terpenoids in R. glutinosa (Wang et al., 2019). Compared with that of GPPS and FPPS, the expression of GGPS gene was much lower in R. glutinosa, especially at expansion stage and maturation stage, the expression of GPPS gene was also higher than that of FPPS gene at these stages, which is consistent with the accumulation of catalpol in R. glutinosa. Further analysis found that the expression of FPPS and GGPS in root and stem was higher at expansion stage or maturation stage of R. glutinosa Jinjiu, yet GPPS gene had high expression in R. glutinosa 85-5, which may be related to the characteristics of R. glutinosa 85-5 and R. glutinosa Jinjiu, which also indicates that gene expression of FPP/GGPP synthase is significantly different in different varieties, growth periods, and tissues of R. glutinosa. Similar phenomena were also found in other research (Miyawaki et al., 2004; Fang et al., 2017; Liu, 2017; Duan et al., 2019). In conclusion, prenyltransferase, as a key enzyme in the synthesis pathway of terpenoids, regulates the synthesis direction of terpenoids, but the anabolism network is complex and is regulated by various enzymes. Therefore, the biosynthesis and metabolic regulation mechanism of terpenoids in R. glutinosa are complicated and need to be further studied.
The datasets presented in this study can be found in online repositories (ProteomeXchange, the accession number is PXD025914).
HD and PC conceived this experiment. XW, WJ, and HW obtained experiment data. PC and QQ analyzed experiment data and wrote this paper. MZ, PC, and YZ participated in text editing. YZ and HD revised this manuscript. All authors read and approved the final manuscript.
This research was completed under the financial aid of National Science Foundation of China (Nos. 31870312 and 31500262), Fund of Henan Normal University (No. 2019JQ01), and Key Project of Henan Province (No. 212102110405) in China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.693758/full#supplementary-material
BCA, bicinchonininc acid; BP, biological process; CAC2, Biotin carboxylase; CC, cell component; CID, collision induced dissociation; CTAB, cetyltriethyl ammonium bromide; DMAPP, dimethylallyl diphosphate; DEPs, differentially expressed proteins; MEP, 2-methyl erythritose-4-phosphate; E stage, elongation stage; FASP, filter aided sample preparation; FPP, farnesyl pyrophosphate; FPPS, farnesyl pyrophosphate synthase; I stage, expansion stage; GGPP, geranylgeranyl pyrophosphate; GGPPS, geranylgeranyl pyrophosphate synthase; GO, Gene ontology; GPP, geranyl pyrophosphate; GPPS, geranyl pyrophosphate synthase; ID, identification number; IPP, isopentenyl pyrophosphate; iTRAQ, isobaric tags for relative and absolute quantitation; KEGG, kyoto encyclopedia of genes and genomes; M stage, maturation stage; MF, molecular function; MS, mass spectrometry; MVA, mevalonate; MW, molecular weight; ORF, open reading frame; PGI1, Glucose-6-phosphate isomerase 1; PI, isoelectric point; PPI, protein-protein interaction; qRT-PCR, quantitative real-time PCR; R. glutinosa, Rehmannia glutinosa; TCA cycle, citrate cycle; UTR, untranslated regions.
Alvarez, S., Choudhury, S. R., Sivagnanam, K., Hicks, L. M., and Pandey, S. (2015). Quantitative proteomics analysis of camelina sativa seeds overexpressing the agg3 gene to identify the proteomic basis of increased yield and stress tolerance. J. Proteome Res. 14, 2606–2616. doi: 10.1021/acs.jproteome.5b00150
Bochar, D. A., Friesen, J. A., Stauffacher, C. V., and Rodwell, V. W. (1999). “Biosynthesis of mevalonic acid from acetyl-CoA. In: Cane DE, ed. Isoprenoids, including carotenoids and steroids, Vol. 2,” in Comprehensive Natural Products Chemistry (London: Elsevier), 15–44.
Bohlmann, J., and Zerbe, P. (2012). Pine terpenoid defences in the mountain pine beetle epidemic and in other conifer pest interactions: specialized enemies are eating holes into a diverse, dynamic and durable defence system. Tree Physiol. 32, 943–994. doi: 10.1016/j.tibtech.2015.04.006
Brugière, N., Humbert, S., Rizzo, N., Bohn, J., and Habben, J. E. (2008). A member of the maize isopentenyl transferase gene family, zea mays isopentenyl transferase 2 (ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Plant Mol. Biol. 67, 215–229. doi: 10.1007/s11103-008-9312-x
Cane, D. E. (1999). “Sesquiterpene biosynthesis: cyclization mechanisms,” in Isoprenoids, including carotenoids and steroids, Vol. 2. Comprehensive natural products chemistry, Cane DE, eds (London, UK: Elsevier), 155–200
Chen, C., Zhe, C., Fang, X., Chao, Z., Fang, F., Shu, S., et al. (2013). Radio-protective effect of catalpol in cultured cells and mice. J. Radiat. Res. 54, 76–82. doi: 10.1093/jrr/rrs080
Dewick, P. M. (2002). The biosynthesis of C5–C25 terpenoid compounds. Nat. Prod. Rep. 19, 181–222. doi: 10.1039/b002685i
Duan, H. Y., Li, J. J., Wang, H. N., Chen, F., and Zhou, Y. Q. (2018). Effects of DNA methylation on growth and development of Rehmannia glutinosa. Int. J. Agric. Biol. 20, 2161–2168. doi: 10.17957/IJAB/15.0745
Duan, H. Y., Liu, W. X., Zeng, Y. P., Jia, W. J., Wang, H. H., and Zhou, Y. Q. (2019). Expression analysis of key enzymes involved in the accumulation of iridoid in Rehmannia glutinosa. Plant Omics. 12, 102–108. doi: 10.21475/POJ.12.02.19.p2221
Fang, J., Li, C. F., Ma, C. L., and Chen, L. (2017). Molecular cloning, bioinformatics and expression analysisof GGPS gene family in Tea plant. J. Tea Sci. 37, 130–138. doi: 10.13305/j.cnki.jts.2017.02.002
Gershenzon, J., and Dudareva, N. (2007). The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414. doi: 10.1038/nchembio.2007.5
Giovanni, C., Bruschi, M., Musante, L., Santucci, L., Ghiggeri, G. M., Carnemolla, B., et al. (2004). Blue silver: a very sensitive colloidal coomassie g-250 staining for proteome analysis. Electrophoresis 25, 1327–1333. doi: 10.1002/elps.200305844
Han, Y., Jung, H. W., Lee, J. Y., Kim, J. S., Kang, S. S., Kim, Y. S., et al. (2012). 2,5-dihydroxyacetophenone isolated from rehmanniae radix preparata inhibits inflammatory responses in lipopolysaccharide-stimulated raw264.7 macrophages. J. Med. Food. 15, 505–510. doi: 10.1089/jmf.2011.1940
He, C., Zheng, Z., Chen, Y., Zhao, G., He, C., Hu, B., et al. (2020). Screening differentially expressed proteins in response to senescence of tobacco leaves based on iTRAQ. Acta Tabacaria Sin. 26, 98–107. doi: 10.16472/j.chinatobacco.2019.413
Henry, L. K., Thomas, S. T., Widhalm, J. R., Lynch, J. H., Davis, T. C., Kessler, S. A., et al. (2018). Contribution of isopentenyl phosphate to plant terpenoid metabolism. Nat. Plants 4, 721–729. doi: 10.1038/s41477-018-0220-z
Huang, W. J., Niu, H. S., Lin, M. H., Cheng, J. T., and Hsu, F. L. (2010). Antihyperglycemic effect of catalpol in streptozotocin-induced diabetic rats. J. Nat. Prod. 73, 1170–1172. doi: 10.1021/np9008317
Isaacson, T., Damasceno, C., Saravanan, R. S., He, Y. C, Catalá, M., et al. (2006). Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 1, 769–774. doi: 10.1038/nprot.2006.102
Jacek, R. W., Alexandre, Z., Nagarjuna, N., and Matthias, M. (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. doi: 10.1038/nmeth.1322
Keeling, C. I., and Bohlmann, J. (2006). Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens. New Phytol. 170, 657–675. doi: 10.1111/j.1469-8137.2006.01716.x
Koyama, T., and Ogura, K. (1999). “Isopentenyl diphosphate isomerase and prenyltransferases,” in Isoprenoids, including carotenoids and steroids, Vol. 2. Comprehensive natural products chemistry, eds D. E. Cane (London: Elsevier), 69–96.
Li, M. J., Yang, Y. H., Li, X. Y., Gu, L., Wang, F., Feng, F., et al. (2015). Analysis of integrated multiple ‘omics’ datasets reveals the mechanisms of initiation and determination in the formation of tuberous roots in Rehmannia glutinosa. J. Exp. Bot. 66, 5837–5851. doi: 10.1093/jxb/erv288
Lian, J. H. (2016). Characteristic agriculture development research of Jiaozuo Four Huaiqing Chinese medicines. Zhengzhou: Henan University of Technology
Lichtenthaler, H. K. (1999). The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 47–65 doi: 10.1146/annurev.arplant.50.1.47
Liu, W. X. (2017). Study on Total Iridoid Glycosides and Related Enzyme' Gene of Rehmannia glutinosa. XinXiang: Henan Nomal University, 12–16.
Liu, Y. F., Liang, D., Luo, H., Hao, Z. Y., and Yu, D. Q. (2012). Hepatoprotective iridoid glycosides from the roots of Rehmannia glutinosa. J. Nat. Prod. 75, 1625–1631. doi: 10.1021/np300509z
Martin, D. M., Fäldt, J., and Bohlmann, J. (2004). Functional characterization of nine Norway Spruce TPS genes and evolution of gymnosperm terpene synthases of the TPS-d subfamily. Plant Physiol. 135, 1908–1927. doi: 10.1104/pp.104.042028
Miyawaki, K., Matsumoto, M., and Kakimoto, T. (2004). Expression of cytokinin biosynthetic isopentenyltransferase genes in arabidopsis: tissue specificity and regulation by auxin, cytokinin, and nitrate. Plant J. 37, 128–138. doi: 10.1046/j.1365-313X.2003.01945.x
Qian, D., Tian, L., and Qu, L. (2015). Proteomic analysis of endoplasmic reticulum stress responses in rice seeds. Sci. Rep. 5, 14255. doi: 10.1038/srep14255
Shieh, J. P., Cheng, K. C., Chung, H. H., Kerh, Y. F., Yeh, C. H., and Cheng, J. T. (2011). Plasma glucose lowering mechanisms of catalpol, an active principle from roots of Rehmannia glutinosa, in streptozotocin-induced diabetic rats. J. Agric. Food Chem. 59, 3747–3753. doi: 10.1021/jf200069t
Shilov, I. V., Seymour, S. L., Patel, A. A., Loboda, A., Tang, W. H., Keating, S. P., et al. (2007). The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655. doi: 10.1074/mcp.T600050-MCP200
Smith, P. K., Krohn, R. I., and Hermanson, G. T. (1985). Measurement of protein using bicinchoninic acid. Anal. Biochem.. 163, 76–85. doi: 10.1016/0003-2697(85)90442-7
Sun, P., Song, S., Zhou, L., Zhang, B., Qi, J., and Li, X. (2012). Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int. J. Mol. Sci. 13, 13748–13763. doi: 10.3390/ijms131013748
Sun, P., Xiao, X., Duan, L., Guo, Y., Qi, J., Liao, D., et al. (2015). Dynamic transcriptional profiling provides insights into tuberous root development in Rehmannia glutinosa. Front. Plant Sci. 6:396. doi: 10.3389/fpls.2015.00396
Takei, K., Sakakibara, H., and Sugiyama, T. (2001). Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J. Biol. Chem. 276, 26405–26410. doi: 10.1074/jbc.M102130200
Vranová, E., Coman, D., and Gruissem, W. (2013). Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu. Rev. Plant Biol. 64, 665–700. doi: 10.1146/annurev-arplant-050312-120116
Wang, H., Bingyi, L. I., Chen, F., Zhou, Y., and Jiaojiao, L. I. (2019). Analysis on phenotype, catalpol accumulation and methylation of Rehmannia glutinosa. Pak. J. Bot. 51, 461–467. doi: 10.30848/PJB2019-2(13)
Wang, X. F. (2017). Study on proteomics of beet tuber enlargement based on iTRAQ technology. Hohhot: Inner Mongolia Agricultural University
Wise, M. L., and Croteau, R. (1999). “Monoterpene biosynthesis,” in Isoprenoids, Including Carotenoids and Steroids, Vol. 2. Comprehensive Natural Products Chemistry, ed D. E. Cane (London: Elsevier), 97–153.
Zhou, Y., Yang, K., Zhang, D., Duan, H., Liu, Y., and Guo, M. (2018). Metabolite accumulation and metabolic network in developing roots of Rehmannia glutinosa reveals its root developmental mechanism and quality. Sci. Rep. 8:14127. doi: 10.1038/s41598-018-32447-6
Keywords: Rehmannia glutinosa, iTRAQ, terpenoids, prenyltransferase, qRT-PCR
Citation: Chen P, Wei X, Qi Q, Jia W, Zhao M, Wang H, Zhou Y and Duan H (2021) Study of Terpenoid Synthesis and Prenyltransferase in Roots of Rehmannia glutinosa Based on iTRAQ Quantitative Proteomics. Front. Plant Sci. 12:693758. doi: 10.3389/fpls.2021.693758
Received: 12 April 2021; Accepted: 24 May 2021;
Published: 04 August 2021.
Edited by:
Zeng-Yu Wang, Qingdao Agricultural University, ChinaReviewed by:
Dongming Ma, Guangzhou University of Chinese Medicine, ChinaCopyright © 2021 Chen, Wei, Qi, Jia, Zhao, Wang, Zhou and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqing Zhou, eXF6aG91QGh0dS5jbg==; Hongying Duan, ZHhkaHlAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.