
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 23 June 2021
Sec. Plant Systematics and Evolution
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.679047
This article is part of the Research TopicEvolution and Biodiversity of Wild PolyploidsView all 11 articles
 J. Lucas Boatwright1*
J. Lucas Boatwright1* Cheng-Ting Yeh2Heng-Cheng Hu2,3
Cheng-Ting Yeh2Heng-Cheng Hu2,3 Alfonso Susanna4Douglas E. Soltis5,6,7,8,9
Alfonso Susanna4Douglas E. Soltis5,6,7,8,9 Pamela S. Soltis6,7,8,9
Pamela S. Soltis6,7,8,9 Patrick S. Schnable2
Patrick S. Schnable2 William B. Barbazuk5
William B. Barbazuk5Polyploidization can have a significant ecological and evolutionary impact by providing substantially more genetic material that may result in novel phenotypes upon which selection may act. While the effects of polyploidization are broadly reviewed across the plant tree of life, the reproducibility of these effects within naturally occurring, independently formed polyploids is poorly characterized. The flowering plant genus Tragopogon (Asteraceae) offers a rare glimpse into the intricacies of repeated allopolyploid formation with both nascent (< 90 years old) and more ancient (mesopolyploids) formations. Neo- and mesopolyploids in Tragopogon have formed repeatedly and have extant diploid progenitors that facilitate the comparison of genome evolution after polyploidization across a broad span of evolutionary time. Here, we examine four independently formed lineages of the mesopolyploid Tragopogon castellanus for homoeolog expression changes and fractionation after polyploidization. We show that expression changes are remarkably similar among these independently formed polyploid populations with large convergence among expressed loci, moderate convergence among loci lost, and stochastic silencing. We further compare and contrast these results for T. castellanus with two nascent Tragopogon allopolyploids. While homoeolog expression bias was balanced in both nascent polyploids and T. castellanus, the degree of additive expression was significantly different, with the mesopolyploid populations demonstrating more non-additive expression. We suggest that gene dosage and expression noise minimization may play a prominent role in regulating gene expression patterns immediately after allopolyploidization as well as deeper into time, and these patterns are conserved across independent polyploid lineages.
The consequences of plant polyploidization have been a subject of intense interest for several decades (reviewed in Wendel, 2000, 2015; Doyle et al., 2008; Leitch and Leitch, 2008; Van de Peer et al., 2009; Barker et al., 2012; Soltis et al., 2016). Polyploidization results in broad-scale genomic changes that serve as potentially novel avenues upon which evolution may act (reviewed in Otto and Whitton, 2000; Flagel and Wendel, 2009). Many changes occur in the generations immediately after polyploidization including changes in genome size (reviewed in Soltis et al., 2003; Leitch et al., 2008; Leitch and Leitch, 2013) spanning the extremes in both gain (e.g., Paris japonica Pellicer et al., 2010) and loss (e.g., Utricularia gibba Ibarra-Laclette et al., 2013), expression (Chen and Pikaard, 1997; reviewed in Adams and Wendel, 2005b; Chaudhary et al., 2009; Hu et al., 2015), epigenetic modifications (Shaked et al., 2001; Salmon et al., 2005; reviewed in Chen, 2007; Madlung and Wendel, 2013; Cheng et al., 2016), transposon activity (reviewed in Woodhouse et al., 2014; Vicient and Casacuberta, 2017; Wendel et al., 2018) as well as changes in protein folding and dosage (reviewed in Birchler and Veitia, 2010, 2012; Pires and Conant, 2016). These changes are variable across lineages (Anssour and Baldwin, 2010; reviewed in Soltis et al., 2016) and may occur in repeated cycles (Soltis and Soltis, 1999; Buggs et al., 2012; reviewed in Wendel, 2015; Soltis et al., 2016). In some paleopolyploids, these changes appear to largely converge over time, at least within closely related lineages (Blanc and Wolfe, 2004; reviewed in Barker et al., 2008; Edger and Pires, 2009; Freeling, 2009).
Polyploids are categorized as either autopolyploids, which are formed from a whole-genome duplication within a single species (reviewed in Otto and Whitton, 2000; Spoelhof et al., 2017), or allopolyploids, which are generated by the combination of entire genomes from two different species (Kihara and Ono, 1926). However, these definitions represent an oversimplification of the dynamic range of variability that polyploids may cover (reviewed in Stebbins, 1947; Ramsey and Schemske, 1998) and the various mechanisms by which they are formed (reviewed in Mason and Pires, 2015). Allopolyploid formation results in duplicated gene copies originating from each parent known as homoeologs. Immediately after polyploidization, homoeologs are expected to be functionally redundant and as such, one copy may be altered without deleterious effect or conserved in duplicate (reviewed in Conant et al., 2014; Pires and Conant, 2016). Whole-genome duplication in an organism can impose unfavorable dosage effects upon cellular functions unless gene balance is maintained (Freeling, 2009; Birchler and Veitia, 2010, 2012). These dosage effects likely represent one aspect of a larger framework that directs genome evolution after polyploidization (Conant et al., 2014). As such, duplicate loci in allopolyploids may experience a number of possible fates. Genomes may experience silencing or loss of one homoeologous copy via fractionation over time. Homoeolog functions may diverge from the parentally inherited state such that functions are partitioned between homoeologs (subfunctionalization), or copies may develop novel functionality (neofunctionalization) (reviewed in Edger and Pires, 2009; Freeling, 2009). Homoeologs may also interact via convergent evolution, homoeologous recombination or gene conversion (Langham et al., 2004; Doyle et al., 2008).
Expression patterns may also vary in the polyploid such that loci demonstrate spatiotemporally divergent expression from the progenitors (Pires et al., 2004b; Wang et al., 2006a; Buggs et al., 2010b; Baldauf et al., 2016), homoeolog-specific expression (HSE) (Buggs et al., 2010a; reviewed in Grover et al., 2012; Yoo et al., 2013; Woodhouse et al., 2014) or additive expression (Guo et al., 2006; Stupar and Springer, 2006; Wang et al., 2006b; reviewed in Yoo et al., 2014). HSE occurs when the polyploid expresses one parental homoeolog over the other (Woodhouse et al., 2014; Boatwright et al., 2018). HSE is similar to allele-specific expression in that both refer to expression differences that are caused by cis- and trans-regulatory variation (Bell et al., 2013), and each has been a topic of interest in hybrid and polyploid studies (Wright et al., 1998; Adams and Wendel, 2005a; Aguilar-Rangel et al., 2017). HSE differs from allele-specific expression in that HSE examines expression across homoeologous chromosomes in contrast to allele-specific expression, which examines expression between homologous chromosomes. Homoeolog expression may also diverge in an additive manner where expression in the polyploid is the arithmetic mean of the two diploid progenitors (reviewed in Yoo et al., 2014).
It is worth noting that the degree of similarity/dissimilarity in expression between parents of a polyploid and the polyploid itself, also known as parental legacy (Buggs et al., 2014), may have a significant effect upon the fate of homoeolog expression in the polyploid (Conant et al., 2014). Similarly, differences among polyploids and their diploid progenitors may derive from numerous processes such as divergent evolution of the lineages after polyploidization, effects of whole-genome duplication (i.e., larger cells and stomata, higher photosynthetic rates and gas exchange, and different stress tolerance) (Hegarty et al., 2006; Sémon and Wolfe, 2007; De Smet and Van de Peer, 2012), or hybridization (resulting in heterosis, increased genetic variation and additive expression) (Mallet, 2007; Bell et al., 2013; Soltis et al., 2016). While the fates of homoeologs after polyploidization are convergent within some lineages (Blanc and Wolfe, 2004; reviewed in Edger and Pires, 2009; and Freeling, 2009), establishing a paradigm has proved elusive (reviewed in Soltis et al., 2016).
The evolutionary model organism Tragopogon serves as a prominent example of repeated, naturally occurring allopolyploidization. The Tragopogon system includes synthetic lines, nascent (< 90 years) and meso- (~2.6 million years) polyploids (Mavrodiev et al., 2015; Soltis et al., 2016). While most species of Tragopogon have chromosome numbers of 2n = 12, there are several well-studied allopolyploids (2n = 24), Tragopogon mirus, T. miscellus, and T. castellanus. Both T. mirus and T. miscellus represent neoallotetraploids that formed recently in the northwestern United States after their three diploid progenitors (T. dubius-T. porrifolius and T. dubius-T. pratensis, respectively) were introduced from Europe in the early 1900s (Ownbey, 1950; Soltis et al., 2004). These allopolyploids never formed in Europe due to their geographic isolation but have formed repeatedly in the USA since the diploids were brought into close proximity. These polyploids are estimated to be approximately 45 generations old (80–90 years for these biennials) (Soltis et al., 1995; Symonds et al., 2010).
Similarly, T. castellanus has formed multiple times from independent allopolyploidization events (Mavrodiev et al., 2015). Tragopogon castellanus is endemic to Spain and occurs only along the northern half of the Iberian Peninsula (Blanca and de la Guardia, 1996). Morphological, cytological, and molecular phylogenetic analyses support T. lamottei and T. crocifolius as putative parents. Tragopogon castellanus is morphologically variable and somewhat similar to parental T. crocifolius; as a result T. castellanus was once considered a subspecies of T. crocifolius (Willkomm, 1893; de la Guardia Guerrero and López, 1989). The parentage of T. castellanus was validated using phylogenetic analyses of external transcribed spacers, internal transcribed spacers, Adh, and plastid datasets, fluorescence in situ hybridization, and genome in situ hybridization (Mavrodiev et al., 2008, 2015). Tragopogon castellanus may have formed before the last glacial maximum that would date the formation of this polyploid species to perhaps as long ago as 2.6 million years (Mavrodiev et al., 2015). As such, the multiple, independent occurrences of Tragopogon allopolyploid formation in young US species and the older T. castellanus permits the assessment of the fate of homoeologs in both neo- and mesopolyploids.
Previous studies have demonstrated that duplicate gene fates after polyploidization are non-random within the newly formed allopolyploid species of Tragopogon. That is, many gene loss and expression changes were repeated across polyploid populations of independent origins (Buggs et al., 2012; Soltis et al., 2012). However, these studies were primarily small-scale and the fates of duplicated gene copies do not generalize across all polyploid plants (reviewed in Soltis et al., 2016). Here, we examine multiple, independently formed allopolyploid T. castellanus lineages estimated to have formed as long as 2.6 mya (Mavrodiev et al., 2015). We show that not only are expression patterns similar, but duplicate loss is largely convergent across independent lineages of T. castellanus. We further compare duplicate fates in populations of this mesopolyploid from Spain to the neopolyploids from the US, T. mirus and T. miscellus (based on earlier studies; Buggs et al., 2010a,b, 2012; Boatwright et al., 2018) in which identical methods were used so that we may examine changes due to natural allopolyploidization over a large evolutionary time scale of perhaps several million years.
Leaf tissue was collected from plants grown in controlled conditions as described by Tate et al. (2006), and RNA was extracted as described in Tate et al. (2006). Three individuals of the diploid T. crocifolius were sampled from the P-B lineage along with five individuals of diploid T. lamottei composed of two and three individuals/lineages from lineage P-I and P-II lineages, respectively (Mavrodiev et al., 2015). Both parental species are phylogenetically distinct and appeared as members of two distinct clades based on ITS phylogeny as estimated in Mavrodiev et al. (2005); namely, clade Majores s. l. [incl. clade Hebecarpus] (T. crocifolius) and clade Tragopogon (T. lamottei) (Mavrodiev et al., 2008). Sample localities and voucher information for all samples are given in Supplementary Table 1. Additional information is provided in Mavrodiev et al. (2015) and vouchers are deposited at the University of Florida herbarium (FLAS). We sequenced 12 allotetraploid T. castellanus individuals representing three bioreplicates for four independent polyploidization events (Supplementary Figure 1 and Supplementary Table 1). RNA-Seq samples were bar-coded and processed using the Illumina TruSeq kit.
Samples were sequenced using the Illumina MiSeq sequencing platform resulting in 2 X 300 paired-end reads (Supplementary Table 2). Adapters were removed using CutAdapt (Martin, 2011), and sequences were trimmed using Trimmomatic (Bolger et al., 2014). RNA reads were pooled from all individuals of each diploid species and assembled using the Trinity de novo assembler (Grabherr et al., 2011), resulting in one assembly per species. Redundant isoforms were removed from our assemblies using a previously described pipeline (Boatwright et al., 2018). The final assemblies were annotated using Trinotate (Altschul et al., 1990; Ashburner et al., 2000; Krogh et al., 2001; Lagesen et al., 2007; Finn et al., 2011; Grabherr et al., 2011; Kanehisa et al., 2011; Petersen et al., 2011; Powell et al., 2011; Punta et al., 2011) with default parameters (https://github.com/jlboat/Tragopogon_castellanus).
Putative orthologs were identified between the T. crocifolius and T. lamottei assemblies using a reciprocal best-hit approach (Moreno-Hagelsieb and Latimer, 2008) as described in Boatwright et al. (2018). Common Orthologous REgions (COREs) were identified between orthologous pairs using the local alignment provided by WU-BLAST (Gish, 2005) and a custom CPython script (https://github.com/BBarbazukLab/papers/). This resulted in BED files containing COREs that were used to filter BAM files after aligning reads to complete assemblies (Boatwright et al., 2018).
As in Boatwright et al. (2018), parental RNA-Seq reads were mapped to both complete, diploid references independently using Bowtie v0.12.9, -m1,-v 3] (Langmead et al., 2009) and Last [v531, -l 25] (Frith et al., 2010; Graze et al., 2012; Munger et al., 2014). The BED files containing COREs were used to filter the resulting SAM files for respective references. Parental reads that mapped uniquely within COREs were isolated, and the reads were subsequently identified as mapping equally well to both references or better to one of the two parents. A Bayesian Poisson-Gamma model (León-Novelo et al., 2014), which provides conservative estimates of the type I error (Fear et al., 2016), was used to identify COREs biasedly mapping reads from the alternative parent. COREs demonstrated expression bias if the credible interval did not overlap 0.5 for all priors (0.4, 0.5, 0.6). Polyploid reads were mapped following the same procedure, and the biased COREs—as determined by diploid read mapping—were filtered out after processing, leaving the remaining set of unbiased COREs for inference. Within the set of unbiased COREs, remaining expression bias corresponded to loci demonstrating HSE. Overlapping gene sets were visually displayed using UpSet plots generated using R (Team, 2014) and the UpSetR package (Lex et al., 2014).
Reads mapping to both references within COREs were used to generate an expression matrix for diploids and all independent polyploids. Loci were filtered from the expression matrix that did not contain at least 10 counts-per-million based upon the average library size in 11 samples. Differentially expressed genes were identified in R (Team, 2014) using the empirical Bayesian analysis pipeline within the limma package (Ritchie et al., 2015) after using voom (Law et al., 2014) to apply precision weights to account for the mean-variance trend. Loci were considered differentially expressed at a false discovery rate of 0.05 (Benjamini and Hochberg, 1995). Contrasts were performed between T. lamottei and T. crocifolius to determine when parental expression was the same or different. To test for additivity, contrasts were performed between each population of the polyploid T. castellanus and its two parents where polyploid expression is expected to be the arithmetic mean of the two parental expression levels. Overlapping gene sets were visually displayed using UpSet plots generated using R (Team, 2014) and the UpSetR package (Lex et al., 2014).
Orthologs were used to design probes for NimbleGen sequence capture to isolate genomic reads from allopolyploid T. castellanus individuals. Each probe was designed to target unique regions of each contig with 1-3 probes along each contig. These probes were used to isolate genomic DNA corresponding to expressed transcripts (Supplementary Table 3). Polyploid DNA reads obtained from sequence capture were aligned to diploid references in the same manner as polyploid RNA reads, and homoeolog loss and silencing were assessed within COREs using the unbiased homoeolog set. Homoeologs with mapped DNA and mapped RNA reads represent genes that are both present and expressed. Homoeologs with DNA reads and no RNA reads represent putative silencing events. Homoeologs with RNA reads but no DNA reads likely represent a failed capture or mismapped reads. Homoeologs with neither DNA nor RNA reads represent putative loss. Overlapping gene sets were visually displayed using UpSet plots generated using R (Team, 2014) and the UpSetR package (Lex et al., 2014).
Loci common to all independently formed polyploid populations that were lost, exhibited additive or non-additive expression as well as loci that demonstrated HSE were individually tested for interaction enrichment. Arabidopsis thaliana orthologs were identified for Tragopogon contigs using WU-BLAST blastx with an A. thaliana protein database. The E-value cutoff was set at 1E-75, and the high-scoring segment pair had to represent 70% of either the total query or subject length. The resulting lists of A. thaliana genes were used to construct functional protein association networks using STRING10 (Szklarczyk et al., 2014). The resulting networks used only high-confidence, experimentally validated protein-protein interactions with disconnected nodes in the networks hidden, and the edge thickness represented confidence of data supporting interaction. Protein-protein interaction enrichment p-values were FDR corrected (Benjamini and Hochberg, 1995). All gene sets were further checked for GO enrichment using GOSeq (Young et al., 2012). The background for HSE and lost loci was the set of unbiased COREs. The background for additively expressed genes included all loci tested for additivity. Functional network details and GO annotations are available at (https://github.com/jlboat/Tragopogon_castellanus).
The assemblies of the diploids, T. crocifolius and T. lamottei, contained 113,865 and 155,600 contigs, respectively. Assemblies were annotated using Trinotate, and putative orthologs and domains were identified. For each of the diploid species, over 7,000 entries hit Arabidopsis thaliana sequences, and approximately 500 of the remainder hit Oryza sativa ssp. japonica using NCBI-BLAST against the SwissProt database. We also identified approximately 4,800 unique eggNOG hits for each diploid, where eggNOG hits represent hierarchical orthologous groups and provide functional annotations for homologous sequences. We identified 14,388 orthologs between the diploid genomes and delimited COREs for downstream processing (Gish, 2005; Moreno-Hagelsieb and Latimer, 2008). COREs were assessed for similarity in both length and GC content (Supplementary Figures 1, 2) and were found to be highly similar between the two species, with length differences never exceeding 16 bases and GC content differences primarily falling under 2%.
Additivity was assessed by first performing a contrast between diploid parents to identify loci where parental expression deviated or was the same (Table 1 and Supplementary Figure 4). The matrix of counts used to estimate additive expression was subjected to multi-dimensional scaling, where samples that lie in close proximity exhibit more similar expression patterns, and plotted. The clustering of T. castellanus individuals is consistent with the known lineages as samples for Cast_2 and Cast_10 both come from lineage I and cluster together (Figures 1, 2, and Supplementary Figure 1). Similarly, the T. lamottei individuals Lam_1 and Lam_2 come from the same lineage, P-I, and are adjacent. Of the 5,806 loci remaining after filtering, parental expression was the same at 4,533 loci and different at 1,273. We found that polyploid expression is primarily non-additive where additivity was examined with respect to parental expression (Table 1), and overlap of each additive/non-additive category was assessed (Figure 3). Approximately 65% (2,155) of the loci were not additive in all four of the independent polyploids, whereas only 43% (909) of the loci consistently exhibited additivity over the four polyploids. There was no significant (FDR < 0.05) GO enrichment for shared additively expressed loci.
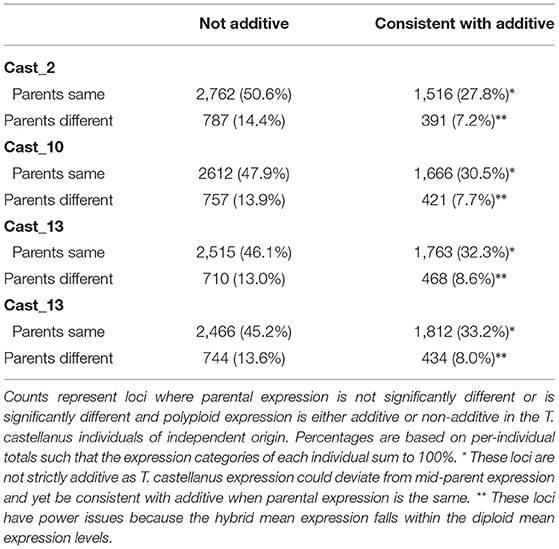
Table 1. Test for additivity in polyploid expression.
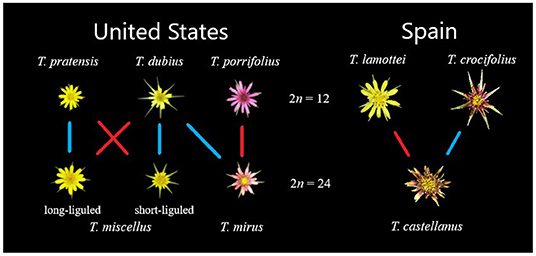
Figure 1. Relationships among the Tragopogon diploids and polyploids. US species are left of the chromosome counts and Spanish species are on the right. Diploids are aligned along the top row and polyploid offspring are along the bottom row. Colored lines indicate whether the diploid serves as the maternal or paternal parent for the corresponding polyploid, where blue is paternal and red is maternal.
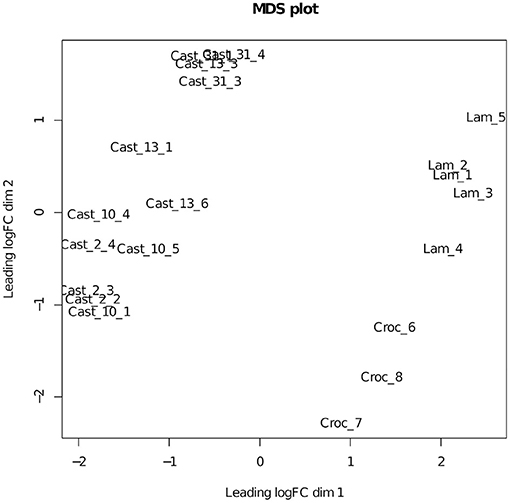
Figure 2. MDS plot of the additive expression matrix for T. castellanus and its diploid progenitors. Lam represents T. lamottei, Croc represents T. crocifolius, and Cast represents T. castellanus.
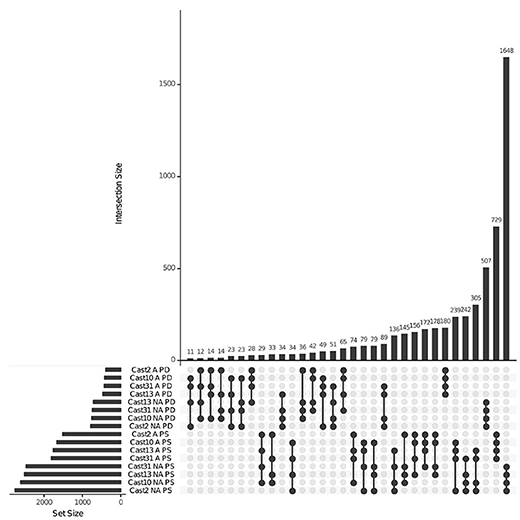
Figure 3. Additive and non-additive expression overlap across T. castellanus individuals. Set IDs represent samples that are additive (A) or non-additive (NA) where parents are different (PD) or the same (PS). Sample sets with common loci are indicated by filled circles with connecting lines, and the number of loci within that intersection may be seen directly above in the bar chart with corresponding size over each bar. The total sample sizes are found in the left bar chart and correspond to the adjacent sample.
HSE was assessed using the PG model and, similar to the additivity assessment, was examined in light of parental expression using unbiased COREs (Table 2). The number of polyploid loci exhibiting homoeolog expression bias toward each parent was similar, with a moderate, consistent bias toward T. crocifolius of about 50 loci, which accounts for ~7% of loci in which parental gene expression is the same but ~23% of loci which exhibit significantly non-equal expression in the parents. The percent of loci overlapping between independent polyploids was ~60% when parental expression was the same and ~64% when parental expression differed (Figure 4). There was no significant (FDR < 0.05) difference in GO enrichment for common loci demonstrating HSE.
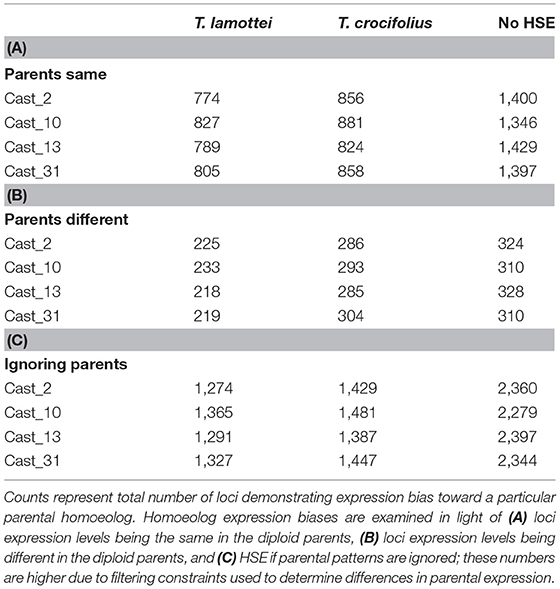
Table 2. Homoeolog-Specific Expression.
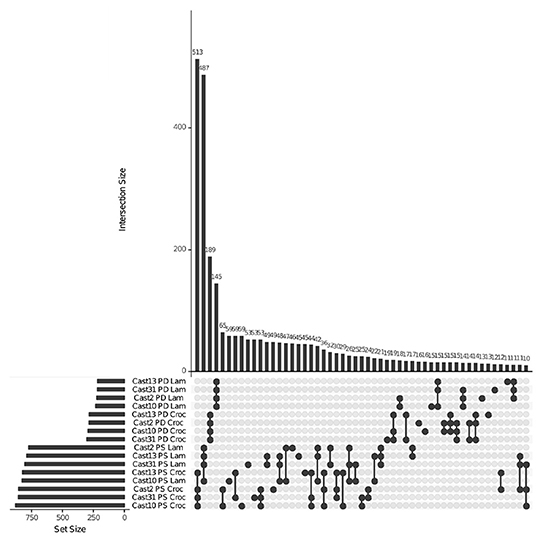
Figure 4. Homoeolog-specific expression overlap across T. castellanus. Set IDs represent samples where parents are different (PD) or the same (PS) and the direction of homoeolog-specific expression (i.e. toward higher expression of the T. lamottei (Lam) or T. crocifolius (Croc) homoeolog). Overlapping sets are indicated by filled circles, and the number of loci within that intersection may be seen directly above in the bar chart with corresponding size over each bar. The total sample sizes are found in the left bar chart and correspond to the adjacent sample.
After orthologs were identified between diploid assemblies, exon-capture probes were designed so that genomic data could be used to distinguish between loci lost vs. silenced after polyploidization. As seen with both additivity and HSE, the number of loci expressed, silenced or lost is highly consistent across all polyploids of independent origin (Table 3). However, the degree of overlap varies among expressed, silenced or lost loci. For expressed loci, approximately 95% of the same parentally derived homoeologs (4,113 for T. lamottei and 4,054 for T. crocifolius) overlap among the four polyploids (Figure 5). Of those few homoeologs demonstrating loss, approximately 66% overlap, again, for both T. lamottei (92) and T. crocifolius (99) homoeologs, independently (Figure 6).
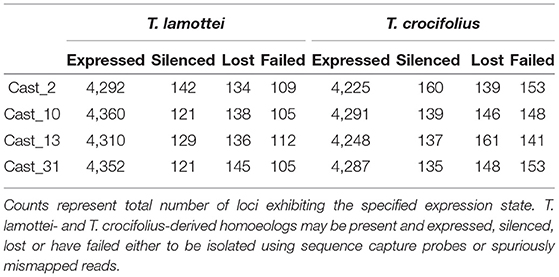
Table 3. Expression states of homoeologs derived from T. lamottei or T. crocifolius.
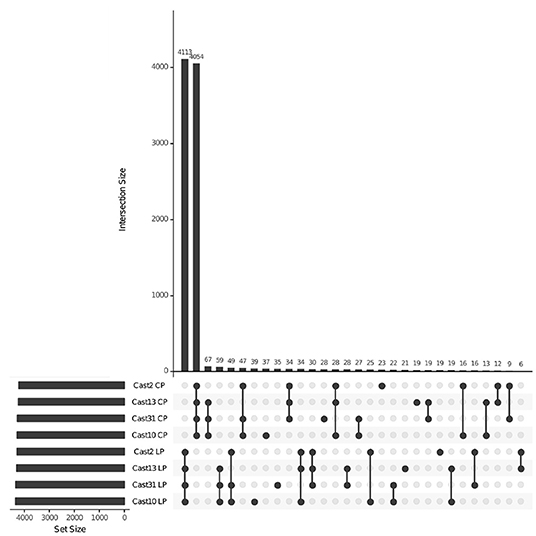
Figure 5. UpSet plot showing homoeologs mapping both DNA and RNA reads across T. castellanus individuals. Set IDs represent samples where homoeologs from T. crocifolius (C) or T. lamottei (L) are present (P) based upon both DNA and RNA alignment. Overlapping sets are indicated by filled circles, and the number of loci within that intersection may be seen directly above in the bar chart with corresponding size over each bar. The total sample sizes are found in the left bar chart and correspond to the adjacent sample. As expected, there should be no overlap across T. crocifolius and T. lamottei homoeologs.
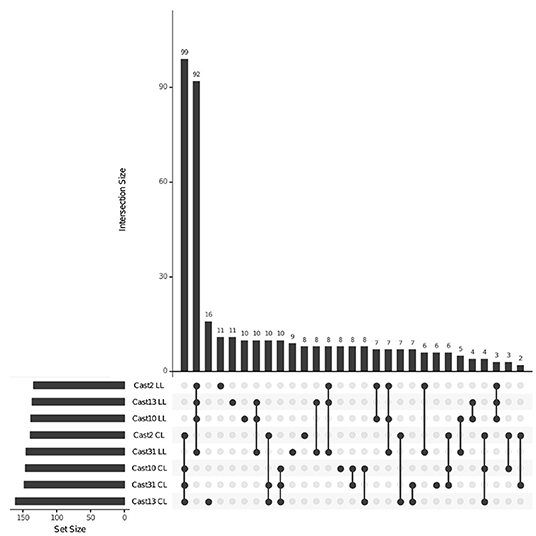
Figure 6. Loci lost across T. castellanus individuals. Set IDs represent samples where either T. crocifolius (C) or T. lamottei (L) homoeologs are lost (L). Overlapping sets are indicated by filled circles, and the number of loci within that intersection may be seen directly above in the bar chart with corresponding size over each bar. The total sample sizes are found in the left bar chart and correspond to the adjacent sample. As expected, there should be no overlap across T. crocifolius and T. lamottei homoeologs.
Silenced homoeologs showed the most variability even though a similar number of loss events occurred across all independently formed polyploids. Only 14 T. lamottei-derived homoeologs (~10% of all T. lamottei homoeolog silencing events) and 35 T. crocifolius-derived homoeologs (~25% of all T. crocifolius homoeolog silencing events) were silenced in all four polyploids. In fact, the majority of silencing events were unique to each polyploid, suggesting that silencing is likely a much more stochastic process than homoeolog loss (Figures 7, 8). There was no significant (FDR < 0.05) GO enrichment for loci expressed, lost or silenced.
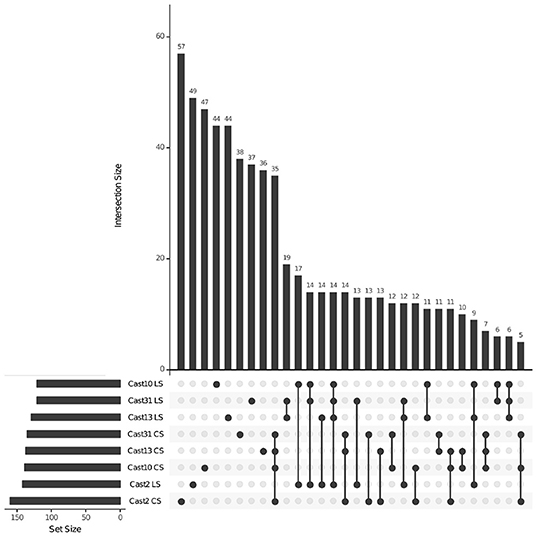
Figure 7. Loci silenced across T. castellanus individuals. Set IDs represent samples where either T. crocifolius (C) or T. lamottei (L) homoeologs are silenced (S). Overlapping sets are indicated by filled circles, and the number of loci within that intersection may be seen directly above in the bar chart with corresponding size over each bar. The total sample sizes are found in the left bar chart and correspond to the adjacent sample. As expected, there should be no overlap across T. crocifolius and T. lamottei homoeologs.
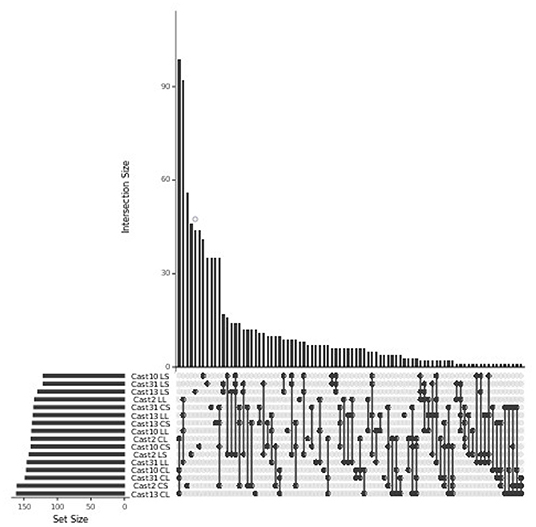
Figure 8. Loci lost or silenced across T. castellanus individuals. Set IDs represent samples where either T. crocifolius (C) or T. lamottei (L) homoeologs are lost (L) or silenced (S). Overlapping sets are indicated by filled circles, and the number of loci within that intersection may be seen directly above in the bar chart. The total sample sizes are found in the left bar chart and correspond to the adjacent sample. As expected, there should be no overlap across T. crocifolius and T. lamottei homoeologs.
The only group of loci that demonstrated significantly more interactions than expected based on chance included the additively expressed genes common to all independently formed allopolyploids (Supplementary Figure 5). The resulting network included 787 nodes with 185 edges with an expected edge count of 101. The average node degree was 0.47 with an average local clustering coefficient of 0.132. The FDR-corrected q-value was 2.75e-13.
Transcript assembly sizes were similar in these Tragopogon diploids from Spain (113,865 and 155,600 contigs for T. crocifolius and T. lamottei, respectively) to those seen in the US diploid parental species (105,282, 116,777, and 122,024 for T. dubius, T. porrifolius, and T. pratensis, respectively) (Boatwright et al., 2018). The number of orthologous pairs identified between diploid progenitors was also similar, with 14,389 pairs identified in this study between T. lamottei and T. crocifolius, while US species were represented by 15,493 pairs between T. dubius-T. pratensis and 15,587 between T. dubius-T. porrifolius. Differences in CORE lengths were similar between studies with no difference over 16 bp, while the %GC difference never exceeded 5% (Supplementary Figures 2, 3). The number of hits against the SwissProt database was nearly identical for all diploid assemblies (~7,000 to A. thaliana and ~500 to O. sativa for both US and Spanish species) as were the numbers of unique eggNOG hits (~4,800). These metrics are significant in that they demonstrate that these studies contain large, similarly sized and comparable data. Thus, differences between the studies should largely be due to biological differences and not methodological differences.
Additive and non-additive gene expression patterns are commonly studied in hybrid and polyploid plants (Guo et al., 2006; Stupar and Springer, 2006; Swanson-Wagner et al., 2006, 2009; Wang et al., 2006a,b; Baldauf et al., 2016). Synthetic Brassica napus exhibits proteome additivity where differential regulation was not related to protein function (Albertin et al., 2007). Additive protein expression has been previously described in the neopolyploid Tragopogon mirus (Koh et al., 2012), and additive gene expression in both neopolyploids T. mirus and T. miscellus (Boatwright et al., 2018).
However, expression in the diploid parents of T. castellanus is significantly different from that seen in the parents of the nascent polyploids. For the neopolyploids, the expression of the diploids T. dubius and T. porrifolius was the same for 5,806 loci and different for 4,706 loci; T. dubius and T. pratensis expression was the same for 5,026 and different for 5,121 loci (Boatwright et al., 2018). While expression was different between diploid parents for the neopolyploids about 50% of the time, the values seen here for T. crocifolius and T. lamottei indicate that parental expression is primarily the same. Similarly, whereas the homoeolog expression was consistent with additivity for the majority of loci within the neopolyploids (Boatwright et al., 2018), plants of the mesopolyploid T. castellanus exhibit more non-additive expression.
A recent publication in Spartina (Giraud et al., 2021) has demonstrated that a high degree transcriptome repatterning (52% of genes deviated from parental additivity) occurs following neopolyploidy (within the last 170 years), and long-term, divergent transcriptome evolution is evident between the mesohexaploid parents that diverged 2-3 MYA (with 36% genes deviating from parental additivity).
One potential reason for this difference in additive expression may be that neopolyploid survival is dependent upon reduction in gene expression noise, as expression noise can have negative impacts upon fitness (Barkai and Leibler, 2000; Rao et al., 2002; Fraser et al., 2004; Pires and Conant, 2016). Thus, shrinkage toward mean parental expression within neopolyploids may alleviate the effects of transcriptomic shock, especially for polyploids with sub-genome trans-acting factors that are largely interchangeable. Over longer periods of time, mutation and selection may then optimize expression of genes, resulting in more non-additive expression. Both noise reduction and gene dosage are expected to play a large role after polyploidization (reviewed in Conant et al., 2014; Pires and Conant, 2016). Interestingly, dosage effects are seen in numerous additively expressed genes within polyploids (Guo et al., 1996; Chen, 2007). These dosage effects are expected to primarily affect genes that function in protein complexes or biological pathways (reviewed in Freeling, 2009; Birchler and Veitia, 2010, 2012). This explanation appears to be the case for additively expressed genes conserved across these independently formed polyploids in that our functional protein association network was significantly enriched for protein-protein interactions.
There is evidence that members of protein complexes within yeast, fruit flies, and humans all exhibit reduced expression noise (Ohno, 1970; Lemos et al., 2004; Schuster-Böckler et al., 2010; reviewed in Pires and Conant, 2016). As such, finding protein-protein interaction enrichment among additively expressed genes may be further evidence that noise reduction and dosage play a significant role in expression changes after allopolyploidization. The degree of dissimilarity between parental expression may also significantly affect homoeolog expression fate between neo- and mesopolyploids (Conant et al., 2014). Environmental differences may also select for different expression patterns over time (Otto and Whitton, 2000). As such, there is likely a complex interplay among the processes governing expression patterns after polyploidization.
HSE, also sometimes called homoeolog expression bias, has been observed in neopolyploids such as Senecio (Hegarty et al., 2012), mesopolyploids such as Gossypium (Adams et al., 2004; Chaudhary et al., 2009; Yoo et al., 2013), and even more broadly across polyploid plants (Buggs et al., 2010b; Schnable et al., 2011; Grover et al., 2012; Woodhouse et al., 2014; Yang et al., 2016). Notably, we observed numerous loci demonstrating HSE, but overall, we see a similar proportion of loci exhibiting homoeolog expression bias toward each parent (Grover et al., 2012). This balanced proportion of HSE in Tragopogon is interesting in that numerous other allopolyploid plants have exhibited substantial subgenome expression bias (Chen and Pikaard, 1997; Wang et al., 2006b; Flagel et al., 2008; Chaudhary et al., 2009; Akhunova et al., 2010; Schnable and Freeling, 2011; Schnable et al., 2011). However, both neoallopolyploid Tragopogon (Boatwright et al., 2018) and the mesoallopolyploid T. castellanus demonstrate similar proportions of homoeolog expression bias toward their corresponding parents. HSE in resynthesized Brassica neoallopolyploids is established soon after the initial genome merger and allopolyploidization (Yang et al., 2016). So, HSE is potentially yet another ameliorative response to whole-genome duplication and/or hybridization (Pires and Conant, 2016). The cause of these expression patterns in Tragopogon is unclear, but numerous genetic and epigenetic mechanisms have been proposed to affect expression in polyploids (Chen, 2007).
The maintenance of dosage balance is not likely to occur indefinitely after whole-genome duplication (Conant et al., 2014; McGrath et al., 2014). HSE is believed to allow duplicated copies to undergo subfunctionalization, neofunctionalization or fractionation, but it is possible that recurrent gene conversion between duplicated copies may maintain sequence identity between them (Pires and Conant, 2016). Biased sub-genome expression dominance has been observed following whole-genome duplication in maize where biased expression occurs within neofunctionalized regulatory genes, and non-regulatory neofunctionalized genes incrementally acquire sub-genome dominance during development (Hughes et al., 2014). Epigenetic regulation has been shown to facilitate sub-genome dominance after whole-genome triplication in B. rapa where a biased distribution of transposable elements among sub-genomes as well as small targeting RNAs are responsible for expression dominance at a sub-genome scale (Cheng et al., 2016). It is also possible that HSE reconciles problems arising from heterologous protein complexes for proteins that function more efficiently as homopolymers or require precise binding affinities, stoichiometry or product ratios (Birchler and Veitia, 2010, 2012; Boatwright et al., 2018).
The process of genome evolution after polyploidization is characterized by alterations in methylation, transposable element activity, expression and function changes as well as genome rearrangement and downsizing (reviewed in Van de Peer et al., 2009; Wendel, 2015; Soltis et al., 2016; Wendel et al., 2018). While these changes have been observed in mesopolyploids (Wang et al., 2011) and paleopolyploids (Schnable et al., 2011), they also occur in neopolyploids where a wide spectrum of genomic changes may occur soon after genome merger and duplication (Madlung and Wendel, 2013), indicating that neopolyploid genomes are not necessarily additive or static (Leitch et al., 2008). Stochastic silencing has been proposed to play an important role in the formation of new species and diploidization after polyploidization. Polyploid species are notable in their tendency to preserve duplicate gene copies, which could be a result of gene dosage effects (Lynch and Conery, 2000; Conant et al., 2014). Dynamic silencing likely serves as a damage-control mechanism to temper potentially adverse effects of polyploidization on gene dosage to improve chances of establishment and adaptation of nascent polyploids (Wendel, 2000; Chaudhary et al., 2009; Buggs et al., 2011). In this study, the silencing of specific homoeologs was more inconsistent across independent polyploids than were loss events or expressed genes. In fact, the majority of silencing events were unique to each polyploid, which seems to support the role of stochastic silencing in polyploid plants. However, it is notable that even though silencing appeared to be stochastic, the homoeologs that were lost were more consistent. This may suggest that the mechanisms governing fractionation are more systematic.
Tragopogon castellanus was previously shown to exhibit a nearly additive genome size of its parents, and the degree of loss seen here (~3% of loci examined) is consistent with that finding (Mavrodiev et al., 2015). Neopolyploid Tragopogon species from the US also exhibited very little putative gene loss (Boatwright et al., 2018) and exhibit an additive genome size (Pires et al., 2004a). Long-term gene loss and retention after whole-genome duplication has demonstrated what appears to be a non-random progression in previous studies (Barker et al., 2008; Freeling, 2009; Birchler and Veitia, 2010; Schnable et al., 2011; Severin et al., 2011; De Smet et al., 2013; Soltis et al., 2016). These observations may also be consistent with the biased fractionation hypothesis, where genome dominance is expected when the subgenomes are highly diverged but not when the subgenomes are similar (Garsmeur et al., 2014; Zhao et al., 2017). While the exact divergence between for diploid parents of T. castellanus has not been thoroughly investigated, the P-derived parental genetic divergence index, the ratio between parental divergence and the average genetic divergence in the respective genus, is 1.14 (Paun et al., 2009), indicating that the balanced expression may be justified by the low parental divergence. This biased fractionation theory is also supported by the contrasting case of recently formed Mimulus peregrinus allopolyploids (Edger et al., 2017) where subgenome expression dominance occurs immediately following the hybridization of divergent genomes and increases significantly over subsequent generations and results from Ephedra allotetraploids whose subgenomes are approximately 8MY diverged, where it has been shown that the rapid formation of large genomes could be attributed to even and slow fractionation following polyploidization (Wu et al., 2021).
Tragopogon seems to be yet another case of convergent homoeolog loss after multiple, independent polyploidization events similar to recent results from Capsella allotetraploids have demonstrated predictable patterns of gene retention and loss following polyploidization (Douglas et al., 2015). We further checked for gene ontology enrichment within our retained, lost and silenced genes but found no significant enrichment. Differential regulation of proteome additivity was not related to protein function in Brassica napus allotetraploids (Albertin et al., 2007). So, a lack of enrichment within additively expressed genes may be expected. While the lack of enrichment within lost genes contrasts with studies that found binding proteins, protein kinases, transcription factors, and transferases are usually retained in duplicate (Jiao et al., 2011), and photosynthesis and cell cycle genes typically drop to singleton status (De Smet et al., 2013), it is the same result found in the neopolyploid Tragopogon species (Boatwright et al., 2018). It may be that loss is not predominantly determined by functional category but rather by some other genetic or epigenetic characteristic such as noise reduction or dosage, at least within Tragopogon.
The short- and long-term effects of cis- and trans-acting interactions are sure to have a significant, if not dynamically different, effect on duplicate gene fate within allopolyploid species. Studies of these processes lack duplication but are certain to identify broader physiological, ecological, and evolutionary implications of polyploidization (Soltis et al., 2016). Here, we compared both homoeolog fate convergence within independently formed mesoallopolyploid populations (T. castellanus) and how those compare to neoallopolyploids within the same genus using the same methodology. While homoeolog expression bias was balanced in both the two neopolyploids and in the mesopolyploid, the degree of additive expression was significantly different, with populations of the mesopolyploid demonstrating more non-additive expression. We found that homoeologs that are retained or lost seem to be strongly convergent across independently formed allopolyploids, while silencing tends to occur stochastically. Further, this non-random trend in long-term homoeolog retention and loss is not unique to Tragopogon but may be selectively advantageous for polyploid speciation and survival (Barker et al., 2008; Freeling, 2009; Birchler and Veitia, 2010; Severin et al., 2011; Schnable et al., 2012; De Smet et al., 2013; Soltis et al., 2016). While there was no GO enrichment among the studied gene sets, additively expressed genes demonstrated enrichment for protein-protein interactions within a functional network. It may be that gene dosage and noise minimization play leading roles in regulating gene expression patterns after allopolyploidization, and these patterns are conserved across independent lineages.
The data presented in this study are deposited in the NCBI's Sequence Read Archive (SRA) under BioProject PRJNA728143. Scripts used to run the analyses are available on GitHub at https://github.com/jlboat/Tragopogon_castellanus.
JB, DS, PSo, PSc, and WB designed the experiments. H-CH and AS generated data. JB and C-TY performed analyses. JB, WB, PSo, and DS wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Department of Biological Sciences at the University of Florida, the University of Florida College of Liberal Arts and Sciences, and the Florida Genetics Institute and National Science Foundation grant IOS-1146065 (DS, PSo, WB, PSc). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NSF.
PSc is a Changjiang Scholar at China Agriculture University, and co-founder and managing partner of Data2Bio, LLC; Dryland Genetics, LLC; and EnGeniousAg, LLC. He is a member of the scientific advisory board and a shareholder of Hi-Fidelity Genetics, Inc. and a member of the scientific advisory boards of Kemin Industries and Centro de Tecnologia Canavieira. H-CH was employed by the company Covance Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the University of Florida's High-Performance Computing Center for providing computational resources and support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.679047/full#supplementary-material
Adams, K. L., Percifield, R., and Wendel, J. F. (2004). Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics 168, 2217–2226. doi: 10.1534/genetics.104.033522
Adams, K. L., and Wendel, J. F. (2005a). Allele-specific, bidirectional silencing of an alcohol dehydrogenase gene in different organs of interspecific diploid cotton hybrids. Genetics 171, 2139–2142. doi: 10.1534/genetics.105.047357
Adams, K. L., and Wendel, J. F. (2005b). Novel patterns of gene expression in polyploid plants. Trends Genet. 21, 539–543. doi: 10.1016/j.tig.2005.07.009
Aguilar-Rangel, M. R., Montes, R. A. C., González-Segovia, E., Ross-Ibarra, J., Simpson, J., and Sawers, R. J. (2017). Allele specific expression analysis identifies regulatory variation associated with stress-related genes in the mexican highland maize landrace palomero toluqueño. bioRxiv 152397. doi: 10.7717/peerj.3737
Akhunova, A. R., Matniyazov, R. T., Liang, H., and Akhunov, E. D. (2010). Homoeolog-specific transcriptional bias in allopolyploid wheat. BMC Genomics 11:505. doi: 10.1186/1471-2164-11-505
Albertin, W., Alix, K., Balliau, T., Brabant, P., Davanture, M., Malosse, C., et al. (2007). Differential regulation of gene products in newly synthesized brassica napus allotetraploids is not related to protein function nor subcellular localization. BMC Genomics 8:56. doi: 10.1186/1471-2164-8-56
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anssour, S., and Baldwin, I. T. (2010). Variation in antiherbivore defense responses in synthetic nicotiana allopolyploids correlates with changes in uniparental patterns of gene expression. Plant Physiol. 153, 1907–1918. doi: 10.1104/pp.110.156786
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Baldauf, J., Marcon, C., Paschold, A., and Hochholdinger, F. (2016). Nonsyntenic genes drive tissue-specific dynamics of differential, nonadditive and allelic expression patterns in maize hybrids. Plant Physiol. 171, 1144–1155. doi: 10.1104/pp.16.00262
Barkai, N., and Leibler, S. (2000). Biological rhythms: Circadian clocks limited by noise. Nature 403, 267–268. doi: 10.1038/35002258
Barker, M. S., Baute, G. J., and Liu, S. L. (2012). “Duplications and turnover in plant genomes,” in Plant Genome Diversity Volume 1 (Vienna: Springer), 155–169.
Barker, M. S., Kane, N. C., Matvienko, M., Kozik, A., Michelmore, R. W., Knapp, S. J., et al. (2008). Multiple paleopolyploidizations during the evolution of the compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol. Biol. Evol. 25, 2445–2455. doi: 10.1093/molbev/msn187
Bell, G. D., Kane, N. C., Rieseberg, L. H., and Adams, K. L. (2013). Rna-seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations. Genome Biol. Evol. 5, 1309–1323. doi: 10.1093/gbe/evt072
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 67, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Birchler, J. A., and Veitia, R. A. (2010). The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol. 186, 54–62. doi: 10.1111/j.1469-8137.2009.03087.x
Birchler, J. A., and Veitia, R. A. (2012). Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U.S.A. 109, 14746–14753. doi: 10.1073/pnas.1207726109
Blanc, G., and Wolfe, K. H. (2004). Functional divergence of duplicated genes formed by polyploidy during arabidopsis evolution. Plant Cell 16, 1679–1691. doi: 10.1105/tpc.021410
Blanca, G., and de la Guardia, C. D. (1996). Sinopsis del género Tragopogon L. (Asteraceae) en la Península Ibérica. Anales del Jardin Botanico de Madrid, Vol. 54, 358–363
Boatwright, J. L., McIntyre, L. M., Morse, A. M., Chen, S., Yoo, M.-J., Koh, J., et al. (2018). A robust methodology for assessing differential homeolog contributions to the transcriptomes of allopolyploids. Genetics 210, 883–894. doi: 10.1534/genetics.118.301564
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buggs, R. J., Chamala, S., Wu, W., Gao, L., May, G. D., Schnable, P. S., et al. (2010a). Characterization of duplicate gene evolution in the recent natural allopolyploid tragopogon miscellus by next-generation sequencing and sequenom iplex massarray genotyping. Mol. Ecol. 19, 132–146. doi: 10.1111/j.1365-294X.2009.04469.x
Buggs, R. J., Chamala, S., Wu, W., Tate, J. A., Schnable, P. S., Soltis, D. E., et al. (2012). Rapid, repeated, and clustered loss of duplicate genes in allopolyploid plant populations of independent origin. Curr. Biol. 22, 248–252. doi: 10.1016/j.cub.2011.12.027
Buggs, R. J., Elliott, N. M., Zhang, L., Koh, J., Viccini, L. F., Soltis, D. E., et al. (2010b). Tissue-specific silencing of homoeologs in natural populations of the recent allopolyploid tragopogon mirus. New Phytol. 186, 175–183. doi: 10.1111/j.1469-8137.2010.03205.x
Buggs, R. J., Wendel, J. F., Doyle, J. J., Soltis, D. E., Soltis, P. S., and Coate, J. E. (2014). The legacy of diploid progenitors in allopolyploid gene expression patterns. Philos. Trans. R. Soc. B Biol. Sci. 369, 20130354. doi: 10.1098/rstb.2013.0354
Buggs, R. J., Zhang, L., Miles, N., Tate, J. A., Gao, L., Wei, W., et al. (2011). Transcriptomic shock generates evolutionary novelty in a newly formed, natural allopolyploid plant. Curr. Biol. 21, 551–556. doi: 10.1016/j.cub.2011.02.016
Chaudhary, B., Flagel, L., Stupar, R. M., Udall, J. A., Verma, N., Springer, N. M., et al. (2009). Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (gossypium). Genetics 182, 503–517. doi: 10.1534/genetics.109.102608
Chen, Z. J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Ann. Rev. Plant Biol. 58:377. doi: 10.1146/annurev.arplant.58.032806.103835
Chen, Z. J., and Pikaard, C. S. (1997). Transcriptional analysis of nucleolar dominance in polyploid plants: biased expression/silencing of progenitor rrna genes is developmentally regulated in brassica. Proc. Natl. Acad. Sci. U.S.A. 94, 3442–3447. doi: 10.1073/pnas.94.7.3442
Cheng, F., Sun, C., Wu, J., Schnable, J., Woodhouse, M. R., Liang, J., et al. (2016). Epigenetic regulation of subgenome dominance following whole genome triplication in brassica rapa. New Phytol. 211, 288–299. doi: 10.1111/nph.13884
Conant, G. C., Birchler, J. A., and Pires, J. C. (2014). Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 19, 91–98. doi: 10.1016/j.pbi.2014.05.008
de la Guardia Guerrero, C. D., and López, G. B. (1989). “Tragopogon castellanus” Levier = “T. crocifolius” subsp. “badalii” Willk. Anales del Jardin Botanico de Madrid, Vol. 47, 253–256.
De Smet, R., Adams, K. L., Vandepoele, K., Van Montagu, M. C., Maere, S., and Van de Peer, Y. (2013). Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl. Acad. Sci. U.S.A. 110, 2898–2903. doi: 10.1073/pnas.1300127110
De Smet, R., and Van de Peer, Y. (2012). Redundancy and rewiring of genetic networks following genome-wide duplication events. Curr. Opin. Plant Biol. 15, 168–176. doi: 10.1016/j.pbi.2012.01.003
Douglas, G. M., Gos, G., Steige, K. A., Salcedo, A., Holm, K., Josephs, E. B., et al. (2015). Hybrid origins and the earliest stages of diploidization in the highly successful recent polyploid capsella bursa-pastoris. Proc. Natl. Acad. Sci. U.S.A. 112, 2806–2811. doi: 10.1073/pnas.1412277112
Doyle, J. J., Flagel, L. E., Paterson, A. H., Rapp, R. A., Soltis, D. E., Soltis, P. S., et al. (2008). Evolutionary genetics of genome merger and doubling in plants. Ann. Rev. Genet. 42, 443–461. doi: 10.1146/annurev.genet.42.110807.091524
Edger, P. P., and Pires, J. C. (2009). Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17, 699–717. doi: 10.1007/s10577-009-9055-9
Edger, P. P., Smith, R., McKain, M. R., Cooley, A. M., Vallejo-Marin, M., Yuan, Y., et al. (2017). Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29, 2150–2167. doi: 10.1105/tpc.17.00010
Fear, J. M., Leon-Novelo, L. G., Morse, A. M., Gerken, A. R., Van Lehman, K., Tower, J., et al. (2016). Buffering of genetic regulatory networks in drosophila melanogaster. Genetics 203, 1177–1190. doi: 10.1534/genetics.116.188797
Finn, R. D., Clements, J., and Eddy, S. R. (2011). Hmmer web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Flagel, L., Udall, J., Nettleton, D., and Wendel, J. (2008). Duplicate gene expression in allopolyploid gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 6:16. doi: 10.1186/1741-7007-6-16
Flagel, L. E., and Wendel, J. F. (2009). Gene duplication and evolutionary novelty in plants. New Phytol. 183, 557–564. doi: 10.1111/j.1469-8137.2009.02923.x
Fraser, H. B., Hirsh, A. E., Giaever, G., Kumm, J., and Eisen, M. B. (2004). Noise minimization in eukaryotic gene expression. PLoS Biol. 2:e137. doi: 10.1371/journal.pbio.0020137
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Ann. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Frith, M. C., Wan, R., and Horton, P. (2010). Incorporating sequence quality data into alignment improves dna read mapping. Nucleic Acids Res. 38, e100–e100. doi: 10.1093/nar/gkq010
Garsmeur, O., Schnable, J. C., Almeida, A., Jourda, C., D'Hont, A., and Freeling, M. (2014). Two evolutionarily distinct classes of paleopolyploidy. Mol. Biol. Evol. 31, 448–454. doi: 10.1093/molbev/mst230
Giraud, D., Lima, O., Rousseau-Gueutin, M., Salmon, A., and Aïnouche, M. (2021). Gene and transposable element expression evolution following recent and past polyploidy events in spartina (poaceae). Front. Genet. 12:589160. doi: 10.3389/fgene.2021.589160
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Graze, R., Novelo, L., Amin, V., Fear, J., Casella, G., Nuzhdin, S., and McIntyre, L. (2012). Allelic imbalance in drosophila hybrid heads: exons, isoforms, and evolution. Mol. Biol. Evol. 29, 1521–1532. doi: 10.1093/molbev/msr318
Grover, C., Gallagher, J., Szadkowski, E., Yoo, M., Flagel, L., and Wendel, J. (2012). Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 196, 966–971. doi: 10.1111/j.1469-8137.2012.04365.x
Guo, M., Davis, D., and Birchler, J. A. (1996). Dosage effects on gene expression in a maize ploidy series. Genetics 142, 1349–1355. doi: 10.1093/genetics/142.4.1349
Guo, M., Rupe, M. A., Yang, X., Crasta, O., Zinselmeier, C., Smith, O. S., et al. (2006). Genome-wide transcript analysis of maize hybrids: allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 113, 831–845. doi: 10.1007/s00122-006-0335-x
Hegarty, M. J., Abbott, R. J., and Hiscock, S. J. (2012). “Allopolyploid speciation in action: the origins and evolution of Senecio cambrensis,” in Polyploidy and Genome Evolution (Berlin; Heidelberg: Springer), 245–270.
Hegarty, M. J., Barker, G. L., Wilson, I. D., Abbott, R. J., Edwards, K. J., and Hiscock, S. J. (2006). Transcriptome shock after interspecific hybridization in senecio is ameliorated by genome duplication. Curr. Biol. 16, 1652–1659. doi: 10.1016/j.cub.2006.06.071
Hu, G., Koh, J., Yoo, M.-J., Chen, S., and Wendel, J. F. (2015). Gene-expression novelty in allopolyploid cotton: a proteomic perspective. Genetics 200, 91–104. doi: 10.1534/genetics.115.174367
Hughes, T. E., Langdale, J. A., and Kelly, S. (2014). The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 24, 1348–1355. doi: 10.1101/gr.172684.114
Ibarra-Laclette, E., Lyons, E., Hernández-Guzmán, G., Pérez-Torres, C. A., Carretero-Paulet, L., Chang, T.-H., et al. (2013). Architecture and evolution of a minute plant genome. Nature 498, 94. doi: 10.1038/nature12132
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. doi: 10.1038/nature09916
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2011). Kegg for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi: 10.1093/nar/gkr988
Kihara, H., and Ono, T. (1926). Chromosomenzahlen und systematische gruppierung der rumex-arten. Z. Zellforsch. Mikroskopisc. Anatom. 4, 475–481. doi: 10.1007/BF00391215
Koh, J., Chen, S., Zhu, N., Yu, F., Soltis, P. S., and Soltis, D. E. (2012). Comparative proteomics of the recently and recurrently formed natural allopolyploid tragopogon mirus (asteraceae) and its parents. New Phytologist 196, 292–305. doi: 10.1111/j.1469-8137.2012.04251.x
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Lagesen, K., Hallin, P., Rødland, E., Stærfeldt, H., Rognes, T., and Ussery, D. (2007). Rnammer: consistent annotation of rrna genes in genomic sequences. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langham, R. J., Walsh, J., Dunn, M., Ko, C., Goff, S. A., and Freeling, M. (2004). Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166, 935–945. doi: 10.1093/genetics/166.2.935
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short dna sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Law, C. W., Chen, Y., Shi, W., and Smyth, G. K. (2014). voom: Precision weights unlock linear model analysis tools for rna-seq read counts. Genome Biol. 15:R29. doi: 10.1186/gb-2014-15-2-r29
Leitch, A., and Leitch, I. (2008). Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483. doi: 10.1126/science.1153585
Leitch, I., Hanson, L., Lim, K., Kovarik, A., Chase, M., Clarkson, J., et al. (2008). The ups and downs of genome size evolution in polyploid species of nicotiana (solanaceae). Ann. Bot. 101, 805–814. doi: 10.1093/aob/mcm326
Leitch, I. J., and Leitch, A. R. (2013). Genome Size Diversity and Evolution in Land Plants (Springer), 307–322.
Lemos, B., Meiklejohn, C. D., and Hartl, D. L. (2004). Regulatory evolution across the protein interaction network. Nat. Genet. 36, 1059–1060. doi: 10.1038/ng1427
León-Novelo, L. G., McIntyre, L. M., Fear, J. M., and Graze, R. M. (2014). A flexible bayesian method for detecting allelic imbalance in RNA-seq data. BMC Genomics 15:920. doi: 10.1186/1471-2164-15-920
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R., and Pfister, H. (2014). Upset: visualization of intersecting sets. IEEE Trans. Visual. Comput. Graph. 20, 1983–1992. doi: 10.1109/TVCG.2014.2346248
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Madlung, A., and Wendel, J. F. (2013). Genetic and epigenetic aspects of polyploid evolution in plants. Cytogenet Genome Res. 140, 270–285. doi: 10.1159/000351430
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mason, A. S., and Pires, J. C. (2015). Unreduced gametes: meiotic mishap or evolutionary mechanism? Trends Genet. 31, 5–10. doi: 10.1016/j.tig.2014.09.011
Mavrodiev, E. V., Chester, M., Suárez-Santiago, V. N., Visger, C. J., Rodriguez, R., Susanna, A., et al. (2015). Multiple origins and chromosomal novelty in the allotetraploid tragopogon castellanus (asteraceae). New Phytol. 206, 1172–1183. doi: 10.1111/nph.13227
Mavrodiev, E. V., Soltis, P. S., and Soltis, D. E. (2008). Putative parentage of six old world polyploids in tragopogon l. (asteraceae: Scorzonerinae) based on its, ets, and plastid sequence data. Taxon 57, 1215–1222E. doi: 10.1002/tax.574014
Mavrodiev, E. V., Tancig, M., Sherwood, A. M., Gitzendanner, M. A., Rocca, J., Soltis, P. S., et al. (2005). Phylogeny of tragopogon l. (asteraceae) based on internal and external transcribed spacer sequence data. Int. J. Plant Sci. 166, 117–133. doi: 10.1086/425206
McGrath, C. L., Gout, J.-F., Doak, T. G., Yanagi, A., and Lynch, M. (2014). Insights into three whole-genome duplications gleaned from the paramecium caudatum genome sequence. Genetics 197, 1417–1428. doi: 10.1534/genetics.114.163287
Moreno-Hagelsieb, G., and Latimer, K. (2008). Choosing blast options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324. doi: 10.1093/bioinformatics/btm585
Munger, S. C., Raghupathy, N., Choi, K., Simons, A. K., Gatti, D. M., Hinerfeld, D. A., et al. (2014). Rna-seq alignment to individualized genomes improves transcript abundance estimates in multiparent populations. Genetics 198, 59–73. doi: 10.1534/genetics.114.165886
Otto, S. P., and Whitton, J. (2000). Polyploid incidence and evolution. Annu. Rev. Genet. 34, 401–437. doi: 10.1146/annurev.genet.34.1.401
Ownbey, M. (1950). Natural hybridization and amphiploidy in the genus tragopogon. Am. J. Bot. 37, 487–499. doi: 10.1002/j.1537-2197.1950.tb11033.x
Paun, O., Forest, F., Fay, M. F., and Chase, M. W. (2009). Hybrid speciation in angiosperms: parental divergence drives ploidy. New Phytol. 182, 507–518. doi: 10.1111/j.1469-8137.2009.02767.x
Pellicer, J., Fay, M. F., and Leitch, I. J. (2010). The largest eukaryotic genome of them all? Bot. J. Linnean Soc. 164, 10–15. doi: 10.1111/j.1095-8339.2010.01072.x
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). Signalp 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pires, J. C., and Conant, G. C. (2016). Robust yet fragile: expression noise, protein misfolding, and gene dosage in the evolution of genomes. Ann. Rev. Genet. 50, 113–131. doi: 10.1146/annurev-genet-120215-035400
Pires, J. C., Lim, K. Y., Kovarík, A., Matyásek, R., Boyd, A., Leitch, A. R., et al. (2004a). Molecular cytogenetic analysis of recently evolved tragopogon (asteraceae) allopolyploids reveal a karyotype that is additive of the diploid progenitors. Am. J. Bot. 91, 1022–1035. doi: 10.3732/ajb.91.7.1022
Pires, J. C., Zhao, J., Schranz, M., Leon, E. J., Quijada, P. A., Lukens, L. N., et al. (2004b). Flowering time divergence and genomic rearrangements in resynthesized brassica polyploids (brassicaceae). Biol. J. Linnean Soc. 82, 675–688. doi: 10.1111/j.1095-8312.2004.00350.x
Powell, S., Szklarczyk, D., Trachana, K., Roth, A., Kuhn, M., Muller, J., et al. (2011). eggnog v3. 0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 40, D284–D289. doi: 10.1093/nar/gkr1060
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2011). The pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Ramsey, J., and Schemske, D. W. (1998). Pathways, mechanisms, and rates of polyploid formation in flowering plants. Ann. Rev. Ecol. System. 29, 467–501. doi: 10.1146/annurev.ecolsys.29.1.467
Rao, C. V., Wolf, D. M., and Arkin, A. P. (2002). Control, exploitation and tolerance of intracellular noise. Nature 420, 231–237. doi: 10.1038/nature01258
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Salmon, A., Ainouche, M. L., and Wendel, J. F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in spartina (poaceae). Mol. Ecol. 14, 1163–1175. doi: 10.1111/j.1365-294X.2005.02488.x
Schnable, J. C., and Freeling, M. (2011). Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6:e17855. doi: 10.1371/journal.pone.0017855
Schnable, J. C., Freeling, M., and Lyons, E. (2012). Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol. Evol., 4, 265–277. doi: 10.1093/gbe/evs009
Schnable, J. C., Springer, N. M., and Freeling, M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074. doi: 10.1073/pnas.1101368108
Schuster-Böckler, B., Conrad, D., and Bateman, A. (2010). Dosage sensitivity shapes the evolution of copy-number varied regions. PLoS ONE 5:e9474. doi: 10.1371/journal.pone.0009474
Sémon, M., and Wolfe, K. H. (2007). Consequences of genome duplication. Curr. Opin. Genet. Dev. 17, 505–512. doi: 10.1016/j.gde.2007.09.007
Severin, A. J., Cannon, S. B., Graham, M. M., Grant, D., and Shoemaker, R. C. (2011). Changes in twelve homoeologous genomic regions in soybean following three rounds of polyploidy. Plant Cell 23, 3129–3136. doi: 10.1105/tpc.111.089573
Shaked, H., Kashkush, K., Ozkan, H., Feldman, M., and Levy, A. A. (2001). Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13, 1749–1759. doi: 10.1105/TPC.010083
Soltis, D. E., Buggs, R. J., Barbazuk, W. B., Chamala, S., Chester, M., Gallagher, J. P., et al. (2012). The Early Stages of Polyploidy: Rapid and Repeated Evolution in Tragopogon (Springer), 271–292.
Soltis, D. E., and Soltis, P. S. (1999). Polyploidy: recurrent formation and genome evolution. Trends Ecol. Evol. 14, 348–352. doi: 10.1016/S0169-5347(99)01638-9
Soltis, D. E., Soltis, P. S., Bennett, M. D., and Leitch, I. J. (2003). Evolution of genome size in the angiosperms. Am. J. Bot. 90, 1596–1603. doi: 10.3732/ajb.90.11.1596
Soltis, D. E., Soltis, P. S., Pires, J. C., Kovarik, A., Tate, J. A., and Mavrodiev, E. (2004). Recent and recurrent polyploidy in tragopogon (asteraceae): cytogenetic, genomic and genetic comparisons. Biol. J. Linnean Soc. 82, 485–501. doi: 10.1111/j.1095-8312.2004.00335.x
Soltis, D. E., Visger, C. J., Marchant, D. B., and Soltis, P. S. (2016). Polyploidy: pitfalls and paths to a paradigm. Am. J. Bot. 103, 1146–1166. doi: 10.3732/ajb.1500501
Soltis, P. S., Plunkett, G. M., Novak, S. J., and Soltis, D. E. (1995). Genetic variation in tragopogon species: additional origins of the allotetraploids t. mirus and t. miscellus (compositae). Am. J. Bot. 82, 1329–1341. doi: 10.1002/j.1537-2197.1995.tb12666.x
Spoelhof, J. P., Soltis, P. S., and Soltis, D. E. (2017). Pure polyploidy: closing the gaps in autopolyploid research. J. Syst. Evol. 55, 340–352. doi: 10.1111/jse.12253
Stebbins Jr, G. L. (1947). Types of polyploids: their classification and significance. Adv. Genetics 1, 403–429.
Stupar, R. M., and Springer, N. M. (2006). Cis-transcriptional variation in maize inbred lines b73 and mo17 leads to additive expression patterns in the f1 hybrid. Genetics 173, 2199–2210. doi: 10.1534/genetics.106.060699
Swanson-Wagner, R. A., DeCook, R., Jia, Y., Bancroft, T., Ji, T., Zhao, X., et al. (2009). Paternal dominance of trans-eqtl influences gene expression patterns in maize hybrids. Science 326, 1118–1120. doi: 10.1126/science.1178294
Swanson-Wagner, R. A., Jia, Y., DeCook, R., Borsuk, L. A., Nettleton, D., and Schnable, P. S. (2006). All possible modes of gene action are observed in a global comparison of gene expression in a maize f1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. U.S.A. 103, 6805–6810. doi: 10.1073/pnas.0510430103
Symonds, V. V., Soltis, P. S., and Soltis, D. E. (2010). Dynamics of polyploid formation in tragopogon (asteraceae): recurrent formation, gene flow, and population structure. Evol. Int. J. Org. Evol. 64, 1984–2003. doi: 10.1111/j.1558-5646.2010.00978.x
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2014). String v10: proteinprotein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Tate, J. A., Ni, Z., Scheen, A.-C., Koh, J., Gilbert, C. A., Lefkowitz, D., et al. (2006). Evolution and expression of homeologous loci in tragopogon miscellus (asteraceae), a recent and reciprocally formed allopolyploid. Genetics 173, 1599–1611. doi: 10.1534/genetics.106.057646
Team, R. C. (2014). R: a Language and Environment for Statistical Computing. Vienna: R foundation for statistical computing 2012.
Van de Peer, Y., Maere, S., and Meyer, A. (2009). The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10:725. doi: 10.1038/nrg2600
Vicient, C. M., and Casacuberta, J. M. (2017). Impact of transposable elements on polyploid plant genomes. Ann. Bot. 120, 195–207. doi: 10.1093/aob/mcx078
Wang, J., Tian, L., Lee, H.-S., and Chen, Z. J. (2006a). Nonadditive regulation of fri and flc loci mediates flowering-time variation in arabidopsis allopolyploids. Genetics 173, 965–974. doi: 10.1534/genetics.106.056580
Wang, J., Tian, L., Lee, H.-S., Wei, N. E., Jiang, H., Watson, B., et al. (2006b). Genomewide nonadditive gene regulation in arabidopsis allotetraploids. Genetics 172, 507–517. doi: 10.1534/genetics.105.047894
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Wendel, J. F. (2015). The wondrous cycles of polyploidy in plants. Am. J. Bot. 102, 1753–1756. doi: 10.3732/ajb.1500320
Wendel, J. F., Lisch, D., Hu, G., and Mason, A. S. (2018). The long and short of doubling down: polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. opin. Genet. Dev. 49, 1–7. doi: 10.1016/j.gde.2018.01.004
Willkomm, M. (1893). Supplementum prodromi florae hispanicae: sive, Enumeratio el'descriptio omnium plantarum inde ab anno 1862 usque ad annum 1893 in Hispania detectarum quae innotuerunt auctori, adjectis locis novis specierum jam notarum. E. Schweizerbart.
Woodhouse, M. R., Cheng, F., Pires, J. C., Lisch, D., Freeling, M., and Wang, X. (2014). Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proc. Natl. Acad. Sci. U.S.A. 111, 5283–5288. doi: 10.1073/pnas.1402475111
Wright, R. J., Thaxton, P. M., El-Zik, K. M., and Paterson, A. H. (1998). D-subgenome bias of xcm resistance genes in tetraploid gossypium (cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 149, 1987–1996. doi: 10.1093/genetics/149.4.1987
Wu, H., Yu, Q., Ran, J.-H., and Wang, X.-Q. (2021). Unbiased subgenome evolution in allotetraploid species of ephedra and its implications for the evolution of large genomes in gymnosperms. Genome Biol. Evol. 13:evaa236. doi: 10.1093/gbe/evaa236
Yang, J., Liu, D., Wang, X., Ji, C., Cheng, F., Liu, B., et al. (2016). The genome sequence of allopolyploid brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. doi: 10.1038/ng.3657
Yoo, M., Szadkowski, E., and Wendel, J. (2013). Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180. doi: 10.1038/hdy.2012.94
Yoo, M.-J., Liu, X., Pires, J. C., Soltis, P. S., and Soltis, D. E. (2014). Nonadditive gene expression in polyploids. Ann. Rev. Genet. 48, 485–517. doi: 10.1146/annurev-genet-120213-092159
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2012). goseq: gene ontology testing for RNA-seq datasets. R Bioconductor 8, 1–25.
Zhao, T., Holmer, R., de Bruijn, S., Angenent, G. C., van den Burg, H. A., and Schranz, M. E. (2017). Phylogenomic synteny network analysis of mads-box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell 29, 1278–1292. doi: 10.1105/tpc.17.00312
Keywords: Tragopogon, allopolyploid, additive expression, homoeologs, expression bias, homoeolog-specific expression, non-model system, RNA-Seq
Citation: Boatwright JL, Yeh C-T, Hu H-C, Susanna A, Soltis DE, Soltis PS, Schnable PS and Barbazuk WB (2021) Trajectories of Homoeolog-Specific Expression in Allotetraploid Tragopogon castellanus Populations of Independent Origins. Front. Plant Sci. 12:679047. doi: 10.3389/fpls.2021.679047
Received: 10 March 2021; Accepted: 20 May 2021;
Published: 23 June 2021.
Edited by:
Elvira Hörandl, University of Göttingen, GermanyReviewed by:
Malika Ainouche, University of Rennes 1, FranceCopyright © 2021 Boatwright, Yeh, Hu, Susanna, Soltis, Soltis, Schnable and Barbazuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Lucas Boatwright, amJvYXR3MkBjbGVtc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.