 Guo-qing Song
Guo-qing Song Xue Han
Xue Han- Plant Biotechnology Resource and Outreach Center, Department of Horticulture, Michigan State University, East Lansing, MI, United States
MADS-box genes are considered as the foundation of all agronomic traits because they play essential roles in almost every aspect of plant reproductive development. Keratin-like (K) domain is a conserved protein domain of tens of MIKC-type MADS-box genes in plants. K-domain technology constitutively expresses a K-domain to mimic expression of the K-domains of other MADS-box genes simultaneously and thus to generate new opportunities for yield enhancement, because the increased K-domains can likely prevent MADS-domain proteins from binding to target DNA. In this study, we evaluated utilizing the K-domain technology to increase maize yield. The K-domain of a blueberry’s SUPPRESSOR of CONSTITUTIVE EXPRESSION OF CONSTANS 1 (VcSOC1K) has similarities to five MADS-box genes in maize. Transgenic maize plants expressing the VcSOC1K showed 13–100% of more grain per plant than the nontransgenic plants in all five experiments conducted under different experimental conditions. Transcriptome comparisons revealed 982 differentially expressed genes (DEGs) in the leaves from 83-day old plants, supporting that the K-domain technology were powerful and multiple functional. The results demonstrated that constitutive expression of the VcSOC1K was very effective to enhance maize grain production. With the potential of mimicking the K-domains of multiple MADS-box genes, the K-domain technology opens a new approach to increase crop yield.
Introduction
Plant-specific MIKC proteins have conserved MADS (M-), intervening (I-), keratin-like (K-), and C-terminal (C-) domains (Theissen et al., 1996; Gramzow and Theissen, 2010). These MIKC proteins consist of MIKC∗ and MIKCc (classical MIKC) subgroups and are key regulators in plant reproductive processes (Verelst et al., 2007; Adamczyk and Fernandez, 2009; Smaczniak et al., 2012; Liu et al., 2013; Dreni and Kater, 2014; Gramzow and Theissen, 2015; Dreni and Zhang, 2016). For example, six MIKC∗-type genes (AGAMOUS-LIKE 30 (AGL30), AGL65, AGL66, AGL67, AGL94, and AGL104) play a significant role in regulating pollen development in Arabidopsis thaliana (Verelst et al., 2007; Kwantes et al., 2012; Liu et al., 2013). The MIKCc genes play specific roles in the ABC model of floral development and in timing plant flowering (Amasino, 2010; Lee and Lee, 2010; Wellmer and Riechmann, 2010; Heijmans et al., 2012; Smaczniak et al., 2012).
MADS-box genes were frequent targets of selection during maize domestication and improvement (Zhao et al., 2011; Schilling et al., 2018). They play essential roles in every aspect of plant reproductive development and were considered as the jack of all traits (Heuer et al., 2001; Schilling et al., 2018). For MIKC proteins, the I-domain defines specificity in the formation of DNA binding dimers (Masiero et al., 2011); the K-domain contributes to specificity in protein-protein interactions (Kaufmann et al., 2005; Rumpler et al., 2015). Both the K and C domains function in the formation of higher-order protein complexes, and the C domain also determines the specificity of interactions of MADS-box proteins (van Dijk et al., 2010; Liu et al., 2013). Of the MIKCc gene clades, SUPPRESSOR of CONSTITUTIVE EXPRESSION OF CONSTANS 1 (SOC1) is a positive regulator of the downstream MADS-box genes such as APETALA1 (AP1) and FRUITFUL (FUL)/AGAMOUS-like 8 (AGL8) (Lee and Lee, 2010; Alter et al., 2016). Due to their regulatory roles, many of the MIKCc genes have a potential to change agronomic traits (Cacharron et al., 2000; Takatsuji and Kapoor, 2002; Lee et al., 2004; Podila et al., 2005; Ryu et al., 2009; Bae et al., 2011; Giovannoni et al., 2013; Alter et al., 2016). For example, a maize ZMM28 gene (patent application # WO2008148872A1), which is a homolog of the AGL8, has been successfully applied to enhance grain yield by its constitutive expression (Munster et al., 2002; Anderson et al., 2019a,b; Catron, 2019).
Being part of the K domain of the blueberry’s (Vaccinium corymbosum L.) SOC1 gene (VcSOC1K), overexpression of the VcSOC1K was found effective in promoting flowering, reducing plant height, enhancing abiotic tolerance, and increasing blueberry yield potential through its broad impact the expression of numerous genes (Song et al., 2013; Song and Chen, 2018). This laid a foundation of the K-domain technology, which utilizes a constitutively expressing K-domain to mimic or affect the expression of multiple MADS-box genes simultaneously. In this study, we evaluated the K-domain technology for maize yield increase. We provide the phenotypic data of transgenic maize plants containing a constitutively expressed VcSOC1K (VcSOC1K-CX) from five experiments conducted under different conditions. We show the data of transcriptome comparison to reveal a broad effect of VcSOC1K-CX on plant development at transcript levels. The K-domain technology opens a new approach to enhance crop yield potential by mimicking expression of the K-domains of multiple MADS-box genes.
Materials and Methods
Constructs and Plant Transformation
Maize SOC1 gene (ZmSOC1 or ZmMADS1) was cloned from the cDNA of maize inbred line B104. The protein sequence of the cloned 696-bp ZmSOC1 is identical to that derived from the HQ858775.1 in the GenBank. The ZmSOC1 and VcSOC1K protein sequences were aligned using Clustal Omega at EBI with default parameters1.
The VcSOC1K was previously cloned into the T-DNA region of the binary vector pBI121 between the CaMV 35S promoter and the Nos terminator for constitutive expression (Song and Chen, 2018). The CaMV 35S-VcSOC1K-Ocs expression cassette in the PBI121 vector was released by a digestion using Hind III and EcoR I, purified from gel, and then ligated to the T-DNA region of the Hind III- and EcoR I-digested binary vector pTF101.1. The pTF101.1 contains the bialaphos resistance (bar) gene under the CaMV 35S promoter for selection of transformed plant cells using glufosinate (GS) herbicide. The resulting pTF101.1-VcSOC1K was verified by sequencing the VcSOC1K and was then transformed into Agrobacterium tumefaciens strain EHA101.
Transformation of the pTF101.1-VcSOC1K into maize cultivar Hi-II (A188 × B73) calluses was conducted at the Plant Transformation Facility of Iowa State University. The first generation (T0) of transgenic (TR) Hi-II plants were backcrossed with nontransgenic inbred line B73 to produce first generation of backcross (BC1) seeds, which have about 75% of the B73 genetic background. T0 plants from separate callus clusters were defined as independent transgenic lines. BC1 seeds from 18 transgenic lines were obtained. Transgenic BC1 plants that showed PCR-positive for both the bar gene and the VcSOC1K were crossed with inbred line B73 to produce BC2 seeds, which have about 87.5% of the B73 genetic background.
Sequence Analysis of Maize MADS-Box Genes
The amino acid sequence of the VcSOC1K was used to search the sequence database “all gene model protein sequences” at Maize Genetics and Genomics Database (MaizeGDB)2 using BLAST program blastp. The BLAST hits with E-value cutoff of 1e-4 were retained and annotated by BLAST against the database at GenBank. Protein sequence alignment was conducted using CLC Sequence Viewer 8.0. Phylogenetic tree analysis was performed using the Maximum Likelihood method conducted in MEGA X (Jones et al., 1992; Kumar et al., 2018; Stecher et al., 2020).
Plant Phenotyping
To collect phenotypic data of the BC1 plants, three experiments started on May 17th, June 11th, and June 25th were conducted in 2018, and one experiment started on may 11th was performed in 2019. Plants of four transgenic lines were evaluated in all four experiments, and five additional transgenic lines were also evaluated in one or two experiments. For all of the four experiments, BC1 seeds were germinated in water-soaked Suremix Perlite planting medium (Michigan Grower Products Inc., Galesburg, MI) in 4-inch plastic pots (8.9 cm width × 12.7 cm height). Individual BC1 plant was transplanted to a 4-gallon pot (top diameter 30 cm, bottom diameter 24 cm, depth 27 cm) and the plants were grown in a secured courtyard under natural environmental conditions at Michigan State University, East Lansing, Michigan. All of the plants were irrigated and fertilized as needed. During the summer time, plants were watered every other day and fertilized weekly using 20-20-20 fertilizer. Young leaves of 30 to 40-day old plants, 0.5 g per plant, were collected for each plant, frozen in liquid nitrogen, and stored in a freezer at −80°C for DNA isolation. To avoid biases in phenotypic data collection, verification of the transgenic plants through polymerase chain reaction (PCR) was conducted after phenotypic data collection.
To collect phenotypic data of the BC2 plants, one field test started on May 26th was conducted in 2020. One of the four transgenic lines evaluated in the BC1 generation was tested in six field plots. A total of 30 plants in three lanes were randomly grown in each of the six plots, including three plots for a high planting density of 40,000 plants/acre and another three for a low planting density of 32,000 plants/acre. Two extra lanes of B73 plants for each plot were used as protection lanes (Supplementary Figure 1). A drip irrigation system was installed in the field for plant irrigation as needed.
Phenotypic data collections included plant height, seed germination date, date of tassel and silk appearance, the total number of stem nodes and leaves, the number of cobs, dry weight of aerial parts without ears, dry weight of ear(s) excluding husk(s), and dry wright of grains. Plant heights measured during plant growth refer to stalk heights from the soil surface to the node of the highest leaf. The final heights of the maize plants refer to stalk heights from the soil surface to the base of the first branch of tassels at the harvest time. All of the plants for each experiment were harvested at a same time after they reached full physiological maturity in late Octobers. The ear(s) of each plant were collected in a paper bag and dried at 25°C for over two months in the lab prior to weighing the dry weights of cob(s) and grains. Grain quality from BC2 transgenic and nontransgenic null segregate (NT) and B73 plants was measured using a Grain Analyser (Infratec 1241, FOSS Analytical AB, Denmark).
Transgene Detection
DNA was isolated from about 200 mg of leaf tissues for each sample using the cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). Two pairs of primers, bar-F and bar-R for the bar gene, 35S-F (3′ portion of the CaMV 35S promoter) and SOK for the VcSOC1K gene (Supplementary Table 1), were used to detect the presence of transgenes in each sample. PCR reaction conditions for all primer pairs started with an initial denaturation for two min at 94°C, 30 cycles of 45 s at 94°C, 60 s at 58°C and 90 s at 72°C, and a final extension for 10 min at 72°C. All amplified PCR and RT-PCR products were separated on 1.0% agarose gel containing ethidium bromide and visualized and photographed under UV light.
RNA Sequencing and Transcriptome Analysis
The third leaf from the top of 83-day old plants at the Blister (R2) stage, which is a reproductive growth stage occurs 10–14 days after silking, were harvested, frozen immediately in liquid nitrogen, and stored at −80°C in a freezer for RNA isoation. Three transgenic and three NT plants from one field plot were used as individual biological replicates. Total RNA of each sample was isolated from about 500 mg young leaf tissues using a separate CTAB method (Zamboni et al., 2008) and was purified using RNeasy Mini Kit (Qiagen, Valencia, CA, United States). On-Column DNase digestion with the RNase-free DNase Set was used to remove DNA in the RNA samples (Qiagen, Valencia, CA, United States). RNA quality was determined using the High Sensitivity RNA ScreenTape system (Agilent technologies, Santa Clara, CA, United States). All of the RNA samples used for RNA sequencing had an RNA integrity number (RIN) equivalent score greater than 5.0.
The RNA samples were sequenced (150 bp-paired end reads) using the Illumina HiSeq4000. All sequencing was performed at the Research Technology Support Facility at Michigan State University (East Lansing, Michigan, United States). The FastQC program3 was used to assess the quality of sequencing reads for the per base quality scores. A total of 7.3–11.0 million pair-reads (MR) for each of the six biological samples with average scores ranging from 38.4 to 39.4 were obtained for transcriptome analysis. A transcriptome reference of BC1 plants (ZmTrinity) assembled from about 100 MR of multiple NT and transgenic lines using Trinity/2.8.5 was used to conduct differential expression analysis (Haas et al., 2013). The differentially expressed transcripts (DETs) with the false discovery rate (FDR) value below 0.05 were used for further analyses of different pathway genes. The transcriptome reference ZmTrinity was annotated using Trinotate (Bryant et al., 2017).
Pathway genes of nine phytohormones in Arabidopsis, including auxin, cytokinin, ABA, ethylene, gibberellin, brassinosteroid, jasmonic acid, salicylic acid, and strigolactones, were retrieved from RIKEN Plant Hormone Research Network4. Similarly, pathway genes of sugar in Arabidopsis were identified. These Arabidopsis hormone, MADS-box, and sugar genes were used as queries to blast against the transcriptome reference ZmTrinity and the isoforms showing e-values less than −20 were identified and used for transcriptome comparisons. Flowering pathway genes in Arabidopsis and cereals (Walworth et al., 2016) were used to analyze flowering-related DETs identified in this study. Cytoscape 3.8.2 was used to construct gene networks of overrepresented gene ontology (GO) terms for the selected DETs under BiNGO’s default parameters with selected ontology file “GOSlim_Plants” and selected organism “A. thaliana” (Shannon et al., 2003; Maere et al., 2005).
Quantitative reverse transcript PCR (RT-qPCR) using SYBR Green system (LifeTechnologies, Carlsbad, CA, United States) was conducted to check the selected transcripts. The primers were designed according to the RNA-seq data, ZmActin1 was used to normalize the RT-qPCR results (Supplementary Table 1). RT-qPCR was performed on a Roche LightCycler 480 Instrument II (Roche). The reaction conditions for RT-qPCR were 95°C for 5 min, 45 cycles of 30 s at 95°C, 45 s at 62°C and 30 s at 72°C. Transcript levels within samples were normalized to Actin. Fold changes were calculated using 2–ΔΔCt, where ΔΔCt = (CtTARGET − CtNOM)transgenic − (CtTARGET − CtNOM)nontransgenic. Three biological samples and three technical replicates were used for the analysis of each transgenic and nontransgenic line.
Statistical Analysis
Statistical analysis of the phenotypic data was conducted using ANOVA and TukeyHSD in RStudio (Version 1.3.1093).
Results and Discussion
VcSOC1K Sequence Has Similarities to the Proteins of Multiple Maize MADS-Box Genes
The VcSOC1K, lacking the MADS box domain, had 36.9% of identity to the maize SOC1 gene (ZmSOC1/ZmMADS1) (Figures 1A,B). It contained both the K-, I-, and C-domains (Figure 1B). A total of five MADS-box genes on five chromosomes in the maize B73 representative genome showed similarities to the VcSOC1K (Figure 1C). This was the foundation of our hypothesis of utilizing constitutive expression of the VcSOC1K to mimic expression of the K-domains of multiple MADS-box genes and thus to regulate plant development for grain yield increase in this study.
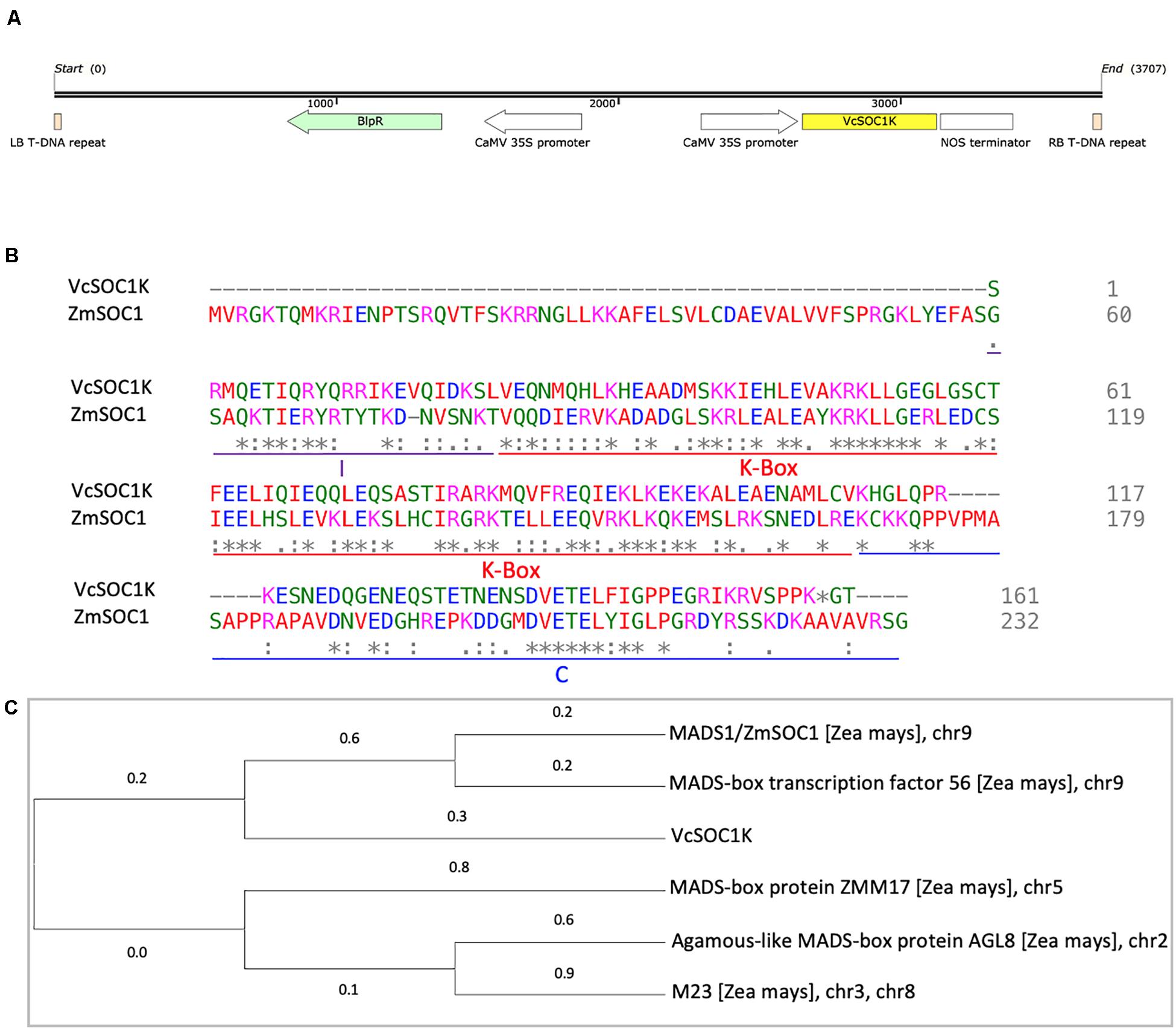
Figure 1. Comparison of VcSOC1K and the selected maize MADS-box genes. (A) Diagram of the T-DNA region of the pTF101.1-ZmSOC1 vector. LB: left border. RB: right border. (B) Protein sequence alignment of the VcSOC1K and ZmSOC1 (GenBank #: HQ858775.1). (C) Phylogenetic analysis of maize MADS-box proteins that showed similarities to the VcSOC1K using the Maximum Likelihood method conducted in MEGA X. There were a total of 172 positions in the final dataset. The tree with the highest log likelihood (–1672.77) is shown. Chromosome numbers refer to the positions in the sequence database Zm-B73-REFERENCE-NAM-5.0 (the maize representative genome, B73, version 5) at Maize Genetics and Genomics Database (MaizeGDB).
VcSOC1-OX Enhanced Grain Production
Five experiments were conducted to evaluate the phenotypic changes in VcSOC1K-CX plants under five environmental conditions in three years. Pot-growing VcSOC1K-CX BC1 plants from nine transgenic lines in four experiments and field-growing VcSOC1K-CX BC2 plants at two planting densities from one transgenic line were compared with the NT plants (Figure 2A and Supplementary Figure 1). Of the nine agronomic traits investigated, the VcSOC1K-CX plants had a higher grain production per plant than the NT plants in all five experiments (Figure 2B and Supplementary Table 2). The increases for VcSOC1K-CX BC1 plants ranged from 13 to 27%. Incredibly, for the 180 BC2 plants tested in the field in 2020, the average dry grain weight for the VcSOC1K-CX BC2 (81 g/plant) was two-fold as many as that for the NT plants (40 g/plant) (Figure 2B). This difference for the BC2 plants, compared to field-growing BC1 plants, was due likely to the increased abiotic and biotic stresses caused by the field conditions. All of the other traits (e.g., flowering time, leaf number, and plant height) showed no significant changes (Figure 2C and Supplementary Table 2). Quality of the grains from the BC2 plants was measured. The grains from the VcSOC1K-CX plants showed no significant differences from those of the NT plants (Supplementary Table 3).
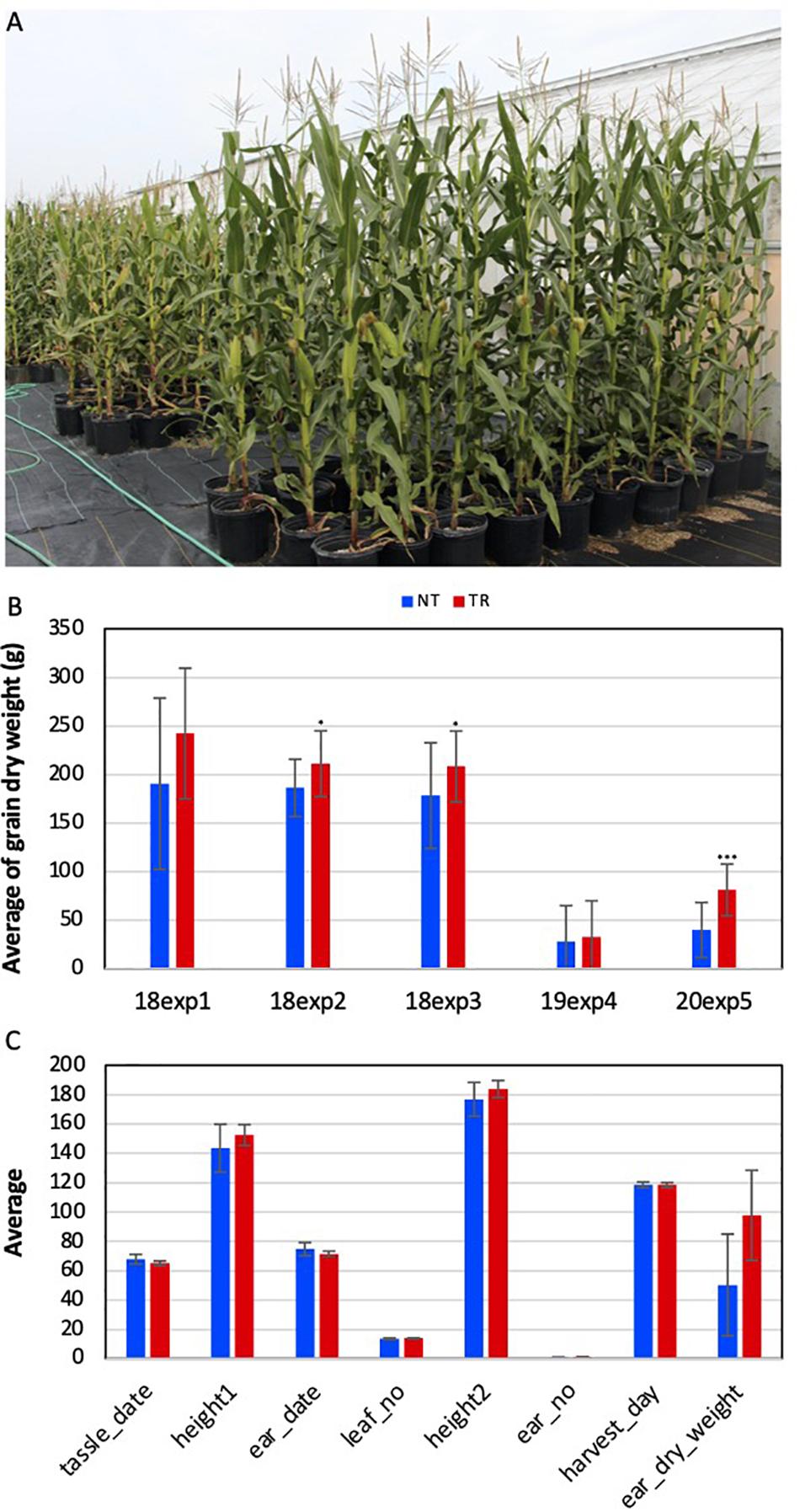
Figure 2. Phenotype assessment of transgenic VcSOC1K-CX (TR) and nontransgenic null segregant (NT) maize plants of BC1 and BC2 lines. The BC1 plants were evaluated in three experiments in 2018 [i.e., 18exp1 (No. of NT = 7, No. of TR = 10), 18exp2 (No. of NT = 25, No. of TR = 16), and 18exp3 (No. of NT = 38, No. of TR = 15)] and one experiment (19exp4, No. of NT = 44, No. of TR = 40) in 2019. The BC2 plants were evaluated in one field test (20exp5, No. of NT = 87, No. of TR = 86) in 2020. (A) BC1 plants randomly grown for phenotyping. (B) Averages of grain dry weight per plant. (C) Phenotypic data of all BC2 plants. Tassle date: Days from sowing to the appearance of tassle. Height1: Plant height at the time of tassel appearance. Ear date: Days from sowing to the appearance of silk. Leaf no: Total number of leaves of a mature plant. Height2: Plant height at the time of harvest. Ear no: Total number of ears of a mature plant. Harvest day: Days from sowing to harvest. Ear dry weight: Dry weight of the ears from each plant. Bars indicate standard deviation. Significance codes: “***” < 0.001, “**” < 0.01, and “*” < 0.05.
With the MADS box of MIKC-type SOC1 protein removal, constitutive expression of the truncated SOC1 genes resulted in SOC1-deficient phenotypes in model plants, petunia, Arabidopsis, and Brachypodium (Ferrario et al., 2004; Seo et al., 2012). Overexpression of a truncated petunia SOC1 gene containing the K-domain and C-region delayed flowering in transgenic petunia plants (Ferrario et al., 2004). Similarly, in both Arabidopsis and Brachypodium, expression of the truncated SOC1 genes containing either the K-domain only or the K-domain and I-region inactivated the SOC1 and caused delay of flowering (Seo et al., 2012). In contrast, unlike the other truncated SOC1 genes reported (Ferrario et al., 2004; Seo et al., 2012), when the truncated VcSOC1K lacking only the MADS box were constitutively expressed, both VcSOC1K-CX tobacco or VcSOC1K-OX blueberry plants had SOC1-OX phenotypes (Song et al., 2013; Song and Chen, 2018). Surprisingly, the VcSOC1K-CX maize plants in this study showed neither SOC1-deficient phenotype in delayed flowering nor obvious SOC1-OX phenotypes of promoted flowering and plant dwarfing (Alter et al., 2016).
VcSOC1K-CX Affected the Expression of Numerous Genes
Kernel Blister Stage (growth stage R2) is a reproductive growth stage occurs 10–14 days after silking. The R2 stage is of importance for the determination of grain yield. At this stage, we found the leaves of the VcSOC1K-CX BC1 plants were often greener leaves than the NT plants. Thus, we conducted transcriptome comparisons between the VcSOC1K-CX and the NT BC2 plants. The comparison revealed 2,247 DETs, which were annotated to 982 differentially expressed genes (DEGs) with E-value cutoff of 1e-19 (Supplementary Table 4). RT-qPCR analysis of seven selected DEGs were consistent with those from RNA-sequencing data, suggesting that the RNA-seq data were reliable (Supplementary Figure 2).
Of the 982 DEGs, we further identified 21 DEGs in the flowering pathway and 41 DEGs related to phytohormones, including abscisic acid (9 DEGs), auxin (9), brassinosteroid (8), cytokinin (5), ethylene (1), gibberellin (8), and Jasmonate (1). Additionally, there were 3 DEGs of MADS-box genes, 18 DEGs related to sucrose synthesis, and 28 DEGS in the family of mitogen-activated protein kinase (MAPK) related to plant resistance to abiotic and biotic stresses (Bigeard and Hirt, 2018; Krysan and Colcombet, 2018; He et al., 2020). Remarkably, of these essential DEGs, greater than 100-fold changes occurred for 10 repressed and 40 up-regulated DEGs (Table 1). The examples of these DEGs indicated that VcSOC1K-CX could affect grain production, at least, through flowering, phytohormones, MAPK-mediated signaling (Bigeard and Hirt, 2018), or photosynthetic sucrose synthesis (Stein and Granot, 2019), although these DEGs only represented the changes in a specific tissue at a specific developmental stage. For instance, the upregulated ZEAXANTHIN EPOXIDASE Gene (ZEP_ORYSJ) could increase plant resistance to osmotic and drought stresses, seed development and dormancy (Agrawal et al., 2001; Cao et al., 2018). The increased expression of the floral homeotic protein APETALA 2 (AP2_ARATH) could play a broad role in flower and seed development by controlling the expression of other floral organ identity genes (Jofuku et al., 1994; Krogan et al., 2012). The upregulated Mitogen-activated protein kinase kinase kinase YODA (YODA_ARATH) could enhance the regulation of florescence architecture due to its role in promoting extra-embryogenic fate (Lukowitz et al., 2004; Meng et al., 2012). Alpha, alpha-trehalose-phosphate synthase [UDP-forming] 1 (TPS1_ARATH) plays a critical role in vegetative growth and transition to flowering, embryo development and growth, and starch and sucrose degradation (Blazquez et al., 1998; van Dijken et al., 2004; Avonce et al., 2005; Gomez et al., 2006). It was likely that the increased expression of TPS10_ARATH enhanced grain yield (Table 1). More studies at protein levels are still needed to find out how and why the expression of the truncated VcSOC1K with the MADS box removal had such a broad impact on gene expressions.
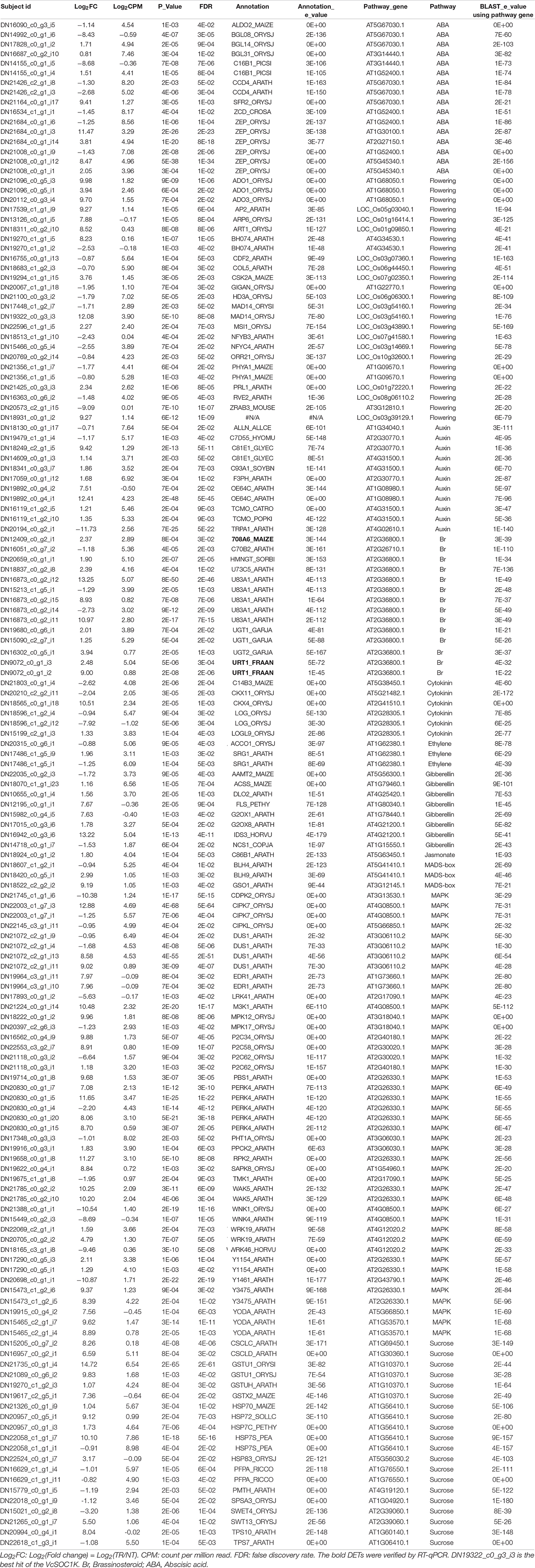
Table 1. Differentially expressed transcripts (DETs) of flowering pathway, hormone genes, MADS-box genes, and mitogen-activated protein kinase (MAPK) genes in maize new leaves from 83-day old plants.
Gene Networks of the DETs
As a quantitative trait, since there are no convincing molecular criteria available to define different crop yield potentials, we visualized the overall impact of VcSOC1K-CX by further analyses of the DETs using the ontology file of GOSlim_Plants in BiNGO to identify overrepresented GO terms. Thirty-nine overrepresented GO terms were revealed in the gene networks, including 13 in “biological process,” nine in “molecular function,” and 17 in “cellular component” (Figure 3). These overrepresented GO terms indicated a broad impact of the VcSOC1K-CX at different levels, which provided alternative evidence to support that the VcSOC1K-CX worked effectively in regulating plant development.
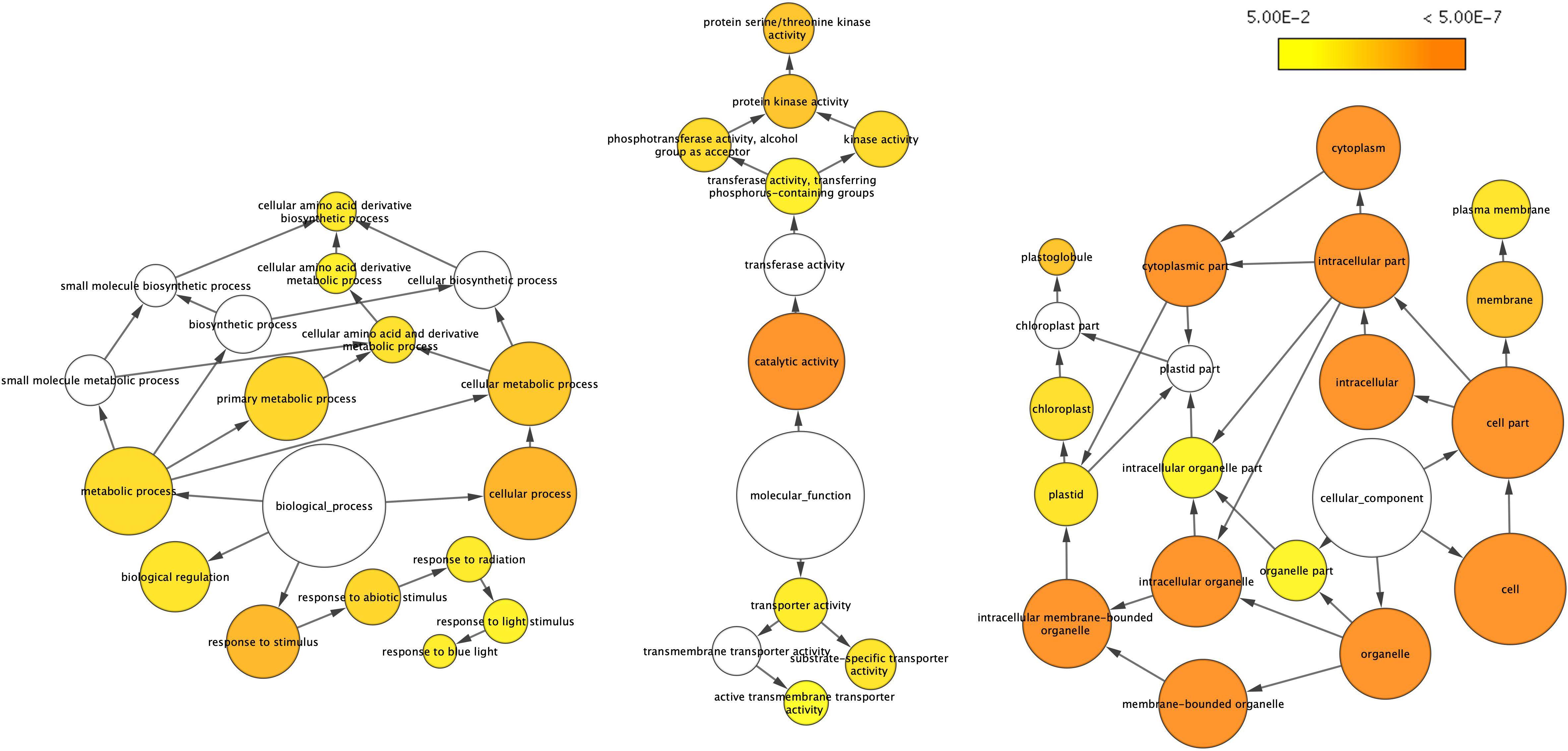
Figure 3. Gene networks of differentially expressed transcripts (DETs) identified from the leaves of the 83-day old plants. The ontology file of GOSlim_Plants in BiNGO was used to identify overrepresented GO terms (p < 0.05). Bubble size and color indicate the frequency of the GO term and the P-value, respectively.
Of the 13 overrepresented GO terms “biological process,” five were related to abiotic factors (Figure 3). In this study, the field-grown BC2 plants were likely exposed to more abiotic stresses than the pot-grown BC1 plants tested under more controlled conditions (i.e., water and fertilizing). The higher grain yield increase by 100% for the BC2 TR (vs BC2 NT) plants (100%), compared to the grain yield increase by 13 to 27% observed in four experiments for BC1 TR (vs BC1 NT) plants, could be attributed to enhanced abiotic tolerance in transgenic plants that could be more obvious under stress conditions. In the previous report, VcSOC1K-OX resulted in high pH tolerance in blueberry plants (Song and Chen, 2018).
Conclusion
K-domain technology utilizes expression of the VcSOC1K to regulate plant growth. In maize, the VcSOC1K showed similarities to five MADS-box genes. VcSOC1K-CX resulted in grain yield increase by 13 to 100% in all five experiments conducted under different experimental conditions. Transcriptome comparisons revealed 982 DEGs in the leaves from the growth stage R2 plants, supporting that the K-domain technology were multiple functional. The K-domain technology opens a new approach to increase crop yield by its potential of mimicking the K-domains of multiple MADS-box genes.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI repository, accession number PRJNA701291.
Author Contributions
G-qS conceived, supervised the study, analyzed data, and wrote the manuscript. XH and G-qS conducted the experiments. Both authors read and approved the final manuscript.
Funding
The work was supported was supported under the MTRAC Program by the State of Michigan 21st Century Jobs Fund received through the Michigan Strategic Fund and administered by the Michigan Economic Development Corporation, and it was also supported partially by AgBioResearch of Michigan State University (http://www.canr.msu.edu/research/agbioresearch/).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.664983/full#supplementary-material
Supplementary Figure 1 | 80-day old BC2 plants growing for phenotyping in the field test in 2020 (20exp5).
Supplementary Figure 2 | Comparison of the RT-qPCR analysis result and the RNA-seq data of the selected DETs (Table 1 and Supplementary Table 4). −ΔΔCt is an average of three biological and three technical replicates for each DET. ZmActin1 (SAC1_ARATH) was used to normalize the RT-qPCR results. Bars indicate standard deviation.
Supplementary Table 1 | PCR primers.
Supplementary Table 2 | Summary of phenotype assessment of BC1 plants.
Supplementary Table 3 | Effect of VcSOC1K-CX on seed quality of field grown plants. The mean values (±STDEV) were for 3 transgenic, 5 non-transgenic, and 15 wild type B73 plants.
Supplementary Table 4 | Differentially expressed transcripts (DETs) in maize new leaves from 83-day old plants. Log2FC: Log2(Fold change) = Log2(TR/NT). CPM, count per million read; FDR, false discovery rate; #N/A, no annotation.
Footnotes
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo/
- ^ https://www.maizegdb.org/assembly
- ^ www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://hormones.psc.riken.jp/
References
Adamczyk, B. J., and Fernandez, D. E. (2009). MIKC∗ MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 149, 1713–1723. doi: 10.1104/pp.109.135806
Agrawal, G. K., Yamazaki, M., Kobayashi, M., Hirochika, R., Miyao, A., and Hirochika, H. (2001). Screening of the rice viviparous mutants generated by endogenous retrotransposon Tos17 insertion. tagging of a zeaxanthin epoxidase gene and a novel ostatc gene. Plant Physiol. 125, 1248–1257. doi: 10.1104/pp.125.3.1248
Alter, P., Bircheneder, S., Zhou, L. Z., Schluter, U., Gahrtz, M., Sonnewald, U., et al. (2016). Flowering time-regulated genes in maize include the transcription factor ZmMADS1. Plant Physiol. 172, 389–404. doi: 10.1104/pp.16.00285
Amasino, R. (2010). Seasonal and developmental timing of flowering. Plant J. 61, 1001–1013. doi: 10.1111/j.1365-313X.2010.04148.x
Anderson, J. A., Brustkern, S., Cong, B., Deege, L., Delaney, B., Hong, B. N., et al. (2019a). Evaluation of the history of safe use of the maize ZMM28 protein. J. Agricult. Food Chem. 67, 7466–7474. doi: 10.1021/acs.jafc.9b00391
Anderson, J. A., Hong, B., Moellring, E., TeRonde, S., Walker, C., Wang, Y., et al. (2019b). Composition of forage and grain from genetically modified DP202216 maize is equivalent to non- modified conventional maize (Zea mays L.). GM Crops Food 10:13. doi: 10.1080/21645698.2019.1609849
Avonce, N., Leyman, B., Thevelein, J., and Iturriaga, G. (2005). Trehalose metabolism and glucose sensing in plants. Biochem. Soc. Trans. 33, 276–279. doi: 10.1042/Bst0330276
Bae, J. M., Noh, S. A., Kwak, M. S., Shin, J. S., and Lee, H. S. (2011). Sweetpotato Mads-Box Promoter Directing High Level Expression in Plant Storage Root. Google Patents.
Bigeard, J., and Hirt, H. (2018). Nuclear signaling of plant MAPKs. Front. Plant Sci. 9:469. doi: 10.3389/fpls.2018.00469
Blazquez, M. A., Santos, E., Flores, C. L., Martinez-Zapater, J. M., Salinas, J., and Gancedo, C. (1998). Isolation and molecular characterization of the Arabidopsis TPS1 gene, encoding trehalose-6-phosphate synthase. Plant J. 13, 685–689. doi: 10.1046/j.1365-313X.1998.00063.x
Bryant, D. M., Johnson, K., DiTommaso, T., Tickle, T., Couger, M. B., Payzin-Dogru, D., et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776. doi: 10.1016/j.celrep.2016.12.063
Cacharron, J., Theissen, G., Deleu, W., and Saedler, H. (2000). Mads-Box Genes and Uses Thereof. Google Patents.
Cao, Y., Zhang, Z., Zhang, T., You, Z., Geng, J., Wang, Y., et al. (2018). Overexpression of zeaxanthin epoxidase gene from Medicago sativa enhances the tolerance to low light in transgenic tobacco. Acta Biochim. Pol. 65, 431–435. doi: 10.18388/abp.2018_2551
Catron, S. A. (2019). Petition for Determination of Nonregulated Status for Enhanced Grain Yield Potential and Glufosinate-ammonium Resistant DP202216 Maize [Online]. Available online at: https://www.aphis.usda.gov/brs/aphisdocs/19-10101p.pdf (accessed December 2, 2019).
Doyle, J. J., and Doyle, J. L. (1987). A rapid dna isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19:5.
Dreni, L., and Kater, M. M. (2014). MADS reloaded: evolution of the AGAMOUS subfamily genes. New Phytol. 201, 717–732. doi: 10.1111/nph.12555
Dreni, L., and Zhang, D. (2016). Flower development: the evolutionary history and functions of the AGL6 subfamily MADS-box genes. J. Exp. Bot. 67, 1625–1638. doi: 10.1093/jxb/erw046
Ferrario, S., Busscher, J., Franken, J., Gerats, T., Vandenbussche, M., Angenent, G. C., et al. (2004). Ectopic expression of the petunia MADS box gene UNSHAVEN accelerates flowering and confers leaf-like characteristics to floral organs in a dominant-negative manner. Plant Cell 16, 1490–1505. doi: 10.1105/tpc.019679
Giovannoni, J. J., Friedman, H., Vrebalov, J., and Elitzer, T. (2013). Banana Mads-Box Genes for Banana Ripening Control. Google Patents.
Gomez, L. D., Baud, S., Gilday, A., Li, Y., and Graham, I. A. (2006). Delayed embryo development in the Arabidopsis trehalose-6-phosphate synthase 1 mutant is associated with altered cell wall structure, decreased cell division and starch accumulation. Plant J. 46, 69–84. doi: 10.1111/j.1365-313X.2006.02662.x
Gramzow, L., and Theissen, G. (2010). A hitchhiker’s guide to the MADS world of plants. Geno. Biol. 11:214. doi: 10.1186/gb-2010-11-6-214
Gramzow, L., and Theissen, G. (2015). Phylogenomics reveals surprising sets of essential and dispensable clades of MIKC(c)-group MADS-box genes in flowering plants. J. Exp. Zool. B Mol. Dev. Evol. 324, 353–362. doi: 10.1002/jez.b.22598
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protocols 8, 1494–1512. doi: 10.1038/Nprot.2013.084
He, X. W., Wang, C. Z., Wang, H. B., Li, L. G., and Wang, C. (2020). The function of MAPK cascades in response to various stresses in horticultural plants. Front. Plant Sci. 11:952. doi: 10.3389/fpls.2020.00952
Heijmans, K., Morel, P., and Vandenbussche, M. (2012). MADS-box genes and floral development: the dark side. J. Exp. Bot. 63, 5397–5404. doi: 10.1093/jxb/ers233
Heuer, S., Hansen, S., Bantin, J., Brettschneider, R., Kranz, E., Lorz, H., et al. (2001). The maize MADS box gene ZmMADS3 affects node number and spikelet development and is co-expressed with ZmMADS1 during flower development, in egg cells, and early embryogenesis. Plant Physiol. 127, 33–45. doi: 10.1104/pp.127.1.33
Jofuku, K. D., den Boer, B. G., Van Montagu, M., and Okamuro, J. K. (1994). Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6, 1211–1225. doi: 10.1105/tpc.6.9.1211
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kaufmann, K., Melzer, R., and Theissen, G. (2005). MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347, 183–198. doi: 10.1016/j.gene.2004.12.014
Krogan, N. T., Hogan, K., and Long, J. A. (2012). APETALA2 negatively regulates multiple floral organ identity genes in Arabidopsis by recruiting the co-repressor TOPLESS and the histone deacetylase HDA19. Development 139, 4180–4190. doi: 10.1242/dev.085407
Krysan, P. J., and Colcombet, J. (2018). Cellular complexity in MAPK signaling in plants: questions and emerging tools to answer them. Front. Plant Sci. 9:1674. doi: 10.3389/fpls.2018.01674
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA x: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kwantes, M., Liebsch, D., and Verelst, W. (2012). How MIKC∗ MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol. Biol. Evol. 29, 293–302. doi: 10.1093/molbev/msr200
Lee, J., and Lee, I. (2010). Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 61, 2247–2254. doi: 10.1093/jxb/erq098
Lee, S., Kim, J., Han, J. J., Han, M. J., and An, G. (2004). Functional analyses of the flowering time gene OsMADS50, the putative suppressor of overexpression Of CO 1/agamous-like 20 (SOC1/AGL20) ortholog in rice. Plant J. 38, 754–764. doi: 10.1111/j.1365-313X.2004.02082.x
Liu, Y., Cui, S., Wu, F., Yan, S., Lin, X., Du, X., et al. (2013). Functional conservation of MIKC∗-type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25, 1288–1303. doi: 10.1105/tpc.113.110049
Lukowitz, W., Roeder, A., Parmenter, D., and Somerville, C. (2004). A MAPKK kinase gene regulates extra-embryonic cell fate in Arabidopsis. Cell 116, 109–119. doi: 10.1016/s0092-8674(03)01067-5
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A., and Kater, M. M. (2011). The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872. doi: 10.1105/tpc.110.081737
Meng, X., Wang, H., He, Y., Liu, Y., Walker, J. C., Torii, K. U., et al. (2012). A MAPK cascade downstream of ERECTA receptor-like protein kinase regulates Arabidopsis inflorescence architecture by promoting localized cell proliferation. Plant Cell 24, 4948–4960. doi: 10.1105/tpc.112.104695
Munster, T., Deleu, W., Wingen, L. U., Ouzunova, M., Cacharron, J., Faigl, W., et al. (2002). Maize MADS-box genes galore. Maydica 47, 287–301.
Podila, G. K., Cseke, L. J., Sen, B., and Karnosky, D. F. (2005). Application of Aspen Mads-Box Genes to Alter Reproduction and Development in Trees. Google Patents.
Rumpler, F., Gramzow, L., Theissen, G., and Melzer, R. (2015). Did convergent protein evolution enable phytoplasmas to generate ‘zombie plants’? Trends Plant Sci. 20, 798–806. doi: 10.1016/j.tplants.2015.08.004
Ryu, C. H., Lee, S., Cho, L. H., Kim, S. L., Lee, Y. S., Choi, S. C., et al. (2009). OsMADS50 and OsMADS56 function antagonistically in regulating long day (LD)-dependent flowering in rice. Plant Cell. Environ. 32, 1412–1427. doi: 10.1111/j.1365-3040.2009.02008.x
Schilling, S., Pan, S., Kennedy, A., and Melzer, R. (2018). MADS-box genes and crop domestication: the jack of all traits. J. Exp. Bot. 69, 1447–1469. doi: 10.1093/jxb/erx479
Seo, P. J., Hong, S. Y., Ryu, J. Y., Jeong, E. Y., Kim, S. G., Baldwin, I. T., et al. (2012). Targeted inactivation of transcription factors by overexpression of their truncated forms in plants. Plant J. 72, 162–172. doi: 10.1111/j.1365-313X.2012.05069.x
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Geno. Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Smaczniak, C., Immink, R. G., Angenent, G. C., and Kaufmann, K. (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098. doi: 10.1242/dev.074674
Song, G.-Q., and Chen, Q. (2018). Overexpression of the MADS-box gene K-domain increases the yield potential of blueberry. Plant Sci. 276:10. doi: 10.1016/j.plantsci.2018.07.018
Song, G. Q., Walworth, A., Zhao, D. Y., Hildebrandt, B., and Leasia, M. (2013). Constitutive expression of the K-domain of a Vaccinium corymbosum SOC1-like (VcSOC1-K) MADS-box gene is sufficient to promote flowering in tobacco. Plant Cell Rep. 32, 1819–1826. doi: 10.1007/S00299-013-1495-1
Stecher, G., Tamura, K., and Kumar, S. (2020). Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 37, 1237–1239. doi: 10.1093/molbev/msz312
Stein, O., and Granot, D. (2019). An overview of sucrose synthases in plants. Front. Plant Sci. 10:95. doi: 10.3389/fpls.2019.00095
Takatsuji, H., and Kapoor, M. (2002). Improvement of Plant Flower Type Targeting Mads Box Gene. Google Patents.
Theissen, G., Kim, J. T., and Saedler, H. (1996). Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43, 484–516.
van Dijk, A. D., Morabito, G., Fiers, M., van Ham, R. C., Angenent, G. C., and Immink, R. G. (2010). Sequence motifs in MADS transcription factors responsible for specificity and diversification of protein-protein interaction. PLoS Comput. Biol. 6:e1001017. doi: 10.1371/journal.pcbi.1001017
van Dijken, A. J. H., Schluepmann, H., and Smeekens, S. C. M. (2004). Arabidopsis trehalose-6-phosphate synthase 1 is essential for normal vegetative growth and transition to flowering. Plant Physiol. 135, 969–977. doi: 10.1104/pp.104.039743
Verelst, W., Saedler, H., and Munster, T. (2007). MIKC∗ MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiol. 143, 447–460. doi: 10.1104/pp.106.089805
Walworth, A. E., Chai, B., and Song, G. Q. (2016). Transcript profile of flowering regulatory genes in VcFT-overexpressing blueberry plants. PLoS One 11:e0156993. doi: 10.1371/journal.pone.0156993
Wellmer, F., and Riechmann, J. L. (2010). Gene networks controlling the initiation of flower development. Trends Genet. 26, 519–527. doi: 10.1016/j.tig.2010.09.001
Zamboni, A., Pierantoni, L., and De Franceschi, P. (2008). Total RNA extraction from strawberry tree (Arbutus unedo) and several other woody plants. Iforest Biogeosci. Forestry 1, 122–125. doi: 10.3832/Ifor0465-0010122
Keywords: flowering mechanism, MADS-box genes, MIKC-type MADS-box protein, SOC1, yield increase, Zea mays
Citation: Song G-q and Han X (2021) K-Domain Technology: Constitutive Expression of a Blueberry Keratin-Like Domain Mimics Expression of Multiple MADS-Box Genes in Enhancing Maize Grain Yield. Front. Plant Sci. 12:664983. doi: 10.3389/fpls.2021.664983
Received: 06 February 2021; Accepted: 15 April 2021;
Published: 07 May 2021.
Edited by:
Marcos Egea-Cortines, Universidad Politécnica de Cartagena, SpainReviewed by:
Rainer Melzer, University College Dublin, IrelandYaroslav B. Blume, Institute of Food Biotechnology and Genomics, National Academy of Sciences of Ukraine (NAN Ukraine), Ukraine
Copyright © 2021 Song and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-qing Song, c29uZ2dAbXN1LmVkdQ==