 Eva C. Henningsen1
Eva C. Henningsen1 Vahid Omidvar1Rafael Della Coletta2Jean-Michel Michno3
Vahid Omidvar1Rafael Della Coletta2Jean-Michel Michno3 Erin Gilbert1
Erin Gilbert1 Feng Li1Marisa E. Miller1†
Feng Li1Marisa E. Miller1† Chad L. Myers3,4Sean P. Gordon5†John P. Vogel5,6
Chad L. Myers3,4Sean P. Gordon5†John P. Vogel5,6 Brian J. Steffenson1Shahryar F. Kianian1,7
Brian J. Steffenson1Shahryar F. Kianian1,7 Cory D. Hirsch1*
Cory D. Hirsch1* Melania Figueroa8*
Melania Figueroa8*- 1Department of Plant Pathology, University of Minnesota, St. Paul, MN, United States
- 2Department of Agronomy and Plant Genetics, University of Minnesota, St. Paul, MN, United States
- 3Bioinformatics and Computational Biology Graduate Program, University of Minnesota, Minneapolis, MN, United States
- 4Department of Computer Science and Engineering, University of Minnesota, Minneapolis, MN, United States
- 5Joint Genome Institute, Walnut Creek, CA, United States
- 6Department of Plant and Microbial Biology, University of California, Berkeley, Berkeley, CA, United States
- 7USDA-ARS Cereal Disease Laboratory, St. Paul, MN, United States
- 8Commonwealth Scientific and Industrial Research Organisation, Agriculture and Food, Canberra, ACT, Australia
Wheat stem rust disease caused by Puccinia graminis f. sp. tritici (Pgt) is a global threat to wheat production. Fast evolving populations of Pgt limit the efficacy of plant genetic resistance and constrain disease management strategies. Understanding molecular mechanisms that lead to rust infection and disease susceptibility could deliver novel strategies to deploy crop resistance through genetic loss of disease susceptibility. We used comparative transcriptome-based and orthology-guided approaches to characterize gene expression changes associated with Pgt infection in susceptible and resistant Triticum aestivum genotypes as well as the non-host Brachypodium distachyon. We targeted our analysis to genes with differential expression in T. aestivum and genes suppressed or not affected in B. distachyon and report several processes potentially linked to susceptibility to Pgt, such as cell death suppression and impairment of photosynthesis. We complemented our approach with a gene co-expression network analysis to identify wheat targets to deliver resistance to Pgt through removal or modification of putative susceptibility genes.
Introduction
Stem rust caused by Puccinia graminis f. sp. tritici (Pgt) is one of the most devastating foliar diseases of wheat (Triticum aestivum) and barley (Hordeum vulgare). The economic relevance of this pathogen to food security is demonstrated by the impact of historical and recent epidemics (Pretorius et al., 2000; Peterson, 2001; Olivera et al., 2015; Singh et al., 2015; Bhattacharya, 2017; Steffenson et al., 2017). Consistent with its biotrophic lifestyle, Pgt develops an intricate relationship with its host in order to acquire nutrients and survive. Early stages of infection involve the germination of urediniospores (asexual spores) and host penetration through the formation of appressoria over stomata (Staples and Macko, 1984). As the fungus reaches the mesophyll cavity of the plant, it develops infection hyphae which penetrate plant cell walls and differentiate into specialized feeding structures, known as haustoria. Haustorial development takes place during the first 24 h post-infection and is critical for colony establishment and sporulation that re-initiates the infection cycle (Harder and Chong, 1984). Similar to other plant pathogens, cereal rust infections involve the translocation of effectors to the plant cell as a mechanism to shut down basal defenses activated by PAMP triggered immunity (PTI) and manipulate host metabolism (Dodds and Rathjen, 2010; Couto and Zipfel, 2016). In rust fungi, the haustorium mediates the secretion of effectors, although the underlying molecular mechanism that facilitates this process is not known (Garnica et al., 2014; Petre et al., 2014). The plant targets of effectors and other plant genes that mediate compatibility and facilitate pathogen infection are often regarded as susceptibility (S) genes (Lapin and van den Ackerveken, 2013; van Schie and Takken, 2014; Lo Presti et al., 2015).
To avoid infection by adapted pathogens, plants employ effector-triggered immunity (ETI) which is mediated by the recognition of effectors by nucleotide-binding domain leucine-rich repeat (NLR) receptors (Flor, 1971; Dodds and Rathjen, 2010; Garnica et al., 2014; Petre et al., 2014). These specific recognition events often induce localized cell death at infection sites (hypersensitive response, HR) which restrict pathogen growth. In wheat-rust interactions, ETI is manifested by the reduction or absence of fungal growth and sporulation (Periyannan et al., 2017). The use of NLR genes to provide crop protection was a critical component of the Green Revolution which diminished the impact of stem rust epidemics (Ellis et al., 2014). While this approach still contributes to the development of wheat cultivars with genetic resistance to stem rust, the durability of such resistant cultivars is hampered by the evolution of rust populations to avoid recognition by NLRs. Given the economic and environmental advantages of genetic disease control over chemical applications, the identification of alternative genetic sources of resistance are a priority for securing future wheat production. In this context, the discovery of S genes could have important translational applications for agriculture and potential durable disease control. Mutations in S genes, although often recessive, could shift a genotype to a non-suitable host due to alterations in initial recognition stages or loss of pathogen establishment requirements (van Schie and Takken, 2014; Lo Presti et al., 2015).
The genetic factors that contribute to wheat susceptibility to biotrophic pathogens such as rust fungi remain largely unknown. Numerous structural and physiological alterations have been observed in wheat-rust compatible interactions. At early infection stages, 4–6 days post-inoculation (dpi), the cytoplasm of infected mesophyll cells increases in volume and an extensive network of the endoplasmic reticulum is built near the haustorium (Bushnell, 1984). The nucleus of infected cells also increases in size and migrates toward the haustorium, and in some cases both structures appear in proximity. These observations suggest that plant cells undergo a massive transcriptional reprogramming to either accommodate rust colonization or initiate a cascade of plant defenses to prevent infection. In addition, many biotrophs are known to increase the ploidy of host cell nuclei near infection sites (Wildermuth, 2010). Advances in next generation sequencing and data mining bring new opportunities to deepen our understanding of plant-pathogen interactions and the relationship between plant metabolism and disease resistance or susceptibility. Several transcriptome profiling studies comparing compatible and incompatible wheat-rust interactions provide strong evidence for the complexity of these interactions (Bozkurt et al., 2010; Zhang et al., 2014; Chandra et al., 2016; Dobon et al., 2016; Yadav et al., 2016). Although S genes in rust pathosystems are largely unknown, several susceptibility factors to other plant pathogenic fungi have been identified in Arabidopsis thaliana, H. vulgare (barley), and solanaceous plants (van Schie and Takken, 2014; Zaidi et al., 2018).
To expand our knowledge of wheat-rust interactions and identify candidate S genes to direct future functional studies, we conducted a comparative RNA-seq analysis of the molecular responses to Pgt in compatible and incompatible interactions. We included a susceptible genotype (W2691) of Triticum aestivum (bread wheat) and the same genotype containing the resistance gene Sr9b, which confers race-specific responses to various Pgt isolates (McIntosh et al., 1995). We also included the related grass species Brachypodium distachyon, which is recognized as a non-host to various cereal rust species (Kellogg, 2001; Ayliffe et al., 2013; Figueroa et al., 2013, 2015; Bettgenhaeuser et al., 2014, 2018; Gilbert et al., 2018; Omidvar et al., 2018). As part of our analysis, we examined the expression profiles of T. aestivum and B. distachyon orthologs of several known S genes in Arabidopsis thaliana, H. vulgare, as well as other characterized S genes in T. aestivum, and identified groups of genes co-regulated with these S gene candidates. In conclusion, this study provides an overview of global expression changes associated with failure or progression of Pgt infection in T. aestivum and B. distachyon and insights into the molecular processes that define disease incompatibility.
Materials and Methods
Plant and Fungal Materials
Two near-isogenic lines of T. aestivum, W2691 (Luig and Watson, 1972) and W2691 carrying the Sr9b gene (referred to onward as W2691+Sr9b, U.S. National Plant Germplasm System Accession Identifier: CI 17386) and the B. distachyon Bd21-3 inbred line (Vogel and Hill, 2008) were used in this study. T. aestivum and B. distachyon seeds were received from the USDA-ARS Cereal Disease Laboratory (CDL) St. Paul, MN, United States and the USDA-ARS Plant Science Unit, St. Paul, MN, United States, respectively. The fungal isolate P. graminis f. sp. tritici (Pgt; isolate # CDL 75-36-700-3 race SCCL; Duplessis et al., 2011) was obtained from the USDA-ARS CDL.
Pgt Infection of T. aestivum and B. distachyon Genotypes
Brachypodium distachyon seeds were placed in petri dishes with wet grade 413 filter paper (VWR International) at 4°C for 5 days and germinated at room temperature for 3 days before sowing to synchronize growth with wheat plants which did not require stratification. Seeds of both wheat and B. distachyon were sown in Fafard® Germination Mix soil (Sun Gro Horticulture, Agawam, MA, United States). All plants were grown in growth chambers with a 18/6 h light/dark cycle at 21/18°C light/dark and 50% relative humidity. Urediniospores of Pgt were activated by heat-shock treatment at 45°C for 15 min and suspended in Isopar M oil (ExxonMobil) at 10 mg/mL concentration. Inoculation treatments consisted of 50 μl of spore suspension per plant, whereas mock treatments consisted of 50 μl of oil per plant. Fungal and mock inoculations were conducted on 7-day old wheat plants (first-leaf stage) and 12-day old B. distachyon plants (three-leaf stage). After inoculations, plants were kept for 12 h in mist chambers with repeated misting for 2 min every 30 min and returned to growth chambers under the previously described conditions.
Analysis of Fungal Colonization and Growth
At 2, 4, and 6 dpi T. aestivum and B. distachyon leaves were sampled and cut into 1 cm sections before staining with Wheat Germ Agglutinin Alex Fluor® 488 conjugate (WGA-FITC; ThermoFisher Scientific) following previously described procedures (Omidvar et al., 2018). Time points to represent stages of Pgt infection were selected based on previous characterization (Figueroa et al., 2013, 2015). To determine the level of fungal colonization, the percentage of urediniospores that germinated (GS), formed an appressorium (AP), established a colony (C), and differentiated a sporulating colony (SC) were visualized using a fluorescence microscope (Leica model DMLB; 450–490 nM excitation). The progression of fungal growth was recorded for 100 infection sites for each of the three biological replicates. Genomic DNA was extracted from T. aestivum (three infected primary leaves) and B. distachyon (three infected secondary leaves) using the DNeasy Plant Mini Kit (Qiagen) and were standardized to a 10 ng/μl concentration. The ITS regions were amplified by qPCR using ITS-specific primers provided by the FemtoTM Fungal DNA Quantification Kit (Zymo Research) to quantify the relative abundance of fungal DNA following the manufacturer’s recommendations for the three biological replicates. The GAPDH housekeeping gene from each species was used as an internal control to normalize fungal DNA quantities (Omidvar et al., 2018).
RNA Isolation, Purification, and Sequencing
Infected and mock treated primary leaves from W2691 and W2691+Sr9b and secondary leaves from Bd21-3 were collected at 2, 4, and 6 dpi. For each of the three biological replicates, three infected leaves were pooled for RNA extraction using the RNeasy Plant Mini Kit (Qiagen). Subsequently, stranded-RNA libraries were constructed, and 125 bp paired-end reads were sequenced on an Illumina HiSeqTM 2500 instrument at the University of Minnesota Genomics Center. On average, more than 10 million reads were generated per time point in each of the previously listed plant-rust interactions (Supplementary Table 1).
Alignment of Reads to the T. aestivum and B. distachyon Reference Genomes
Short reads and low-quality bases were trimmed using cutadapt v1.18 (Martin, 2011) with the following parameters: minimum-length 40, quality-cutoff 30, and quality-base = 33. Subsequently, W2691 and W2691+Sr9b reads were mapped to the T. aestivum cv. Chinese Spring reference genome IWGSC RefSeq v1.0 (Alaux et al., 2018) and Bd21-3 reads were mapped to the Bd21-3 reference genome from the Joint Genome Institute (B. distachyon Bd21-3 v1.1 DOE-JGI, http://phytozome.jgi.doe.gov/). Read mapping was conducted using STAR v2.5.3 (Dobin et al., 2012) set for two-pass mapping mode with the following parameters: twopassMode Basic and outSAMmapqUnique 20.
Expression Profiling and Identification of Differentially Expressed Genes
Reads were mapped to T. aestivum and B. distachyon gene features using htseq v.0.11.0 to obtain count values (Anders et al., 2015). Normalized read counts and differential expression (DE) analysis were performed with DESeq2 v1.28.1 (Love et al., 2014). Genes with a | log2 fold change| ≥1.5 and a p-value < 0.05 were identified as differentially expressed genes (DEGs).
Gene Ontology Analysis
Gene ontology (GO) terms were obtained from GOMAP track data for T. aestivum (Alaux et al., 2018) and previously published data for B. distachyon (Brachypodium distachyon Bd21-3 v1.2 DOE-JGI, http://phytozome.jgi.doe.gov/) annotation files. GO terms in wheat and B distachyon were mapped to the GOslim plant subset using OWLTools with the command owltools –map2slim1. GO enrichment analysis for DEGs was performed using the topGO R package using the “weight01” algorithm and fisher test statistic (Alexa and Rahnenfuhrer, 2020). Enriched terms were considered significant with a Fisher test p-value < 0.01 (Supplementary Table 2). Enrichment analyses using the GOslim subset were performed on all differentially expressed wheat and B. distachyon genes, as well as on genes within the S-gene orthologs clusters. Enrichment analysis with the full GO set was only performed on the differentially expressed T. aestivum and B. distachyon genes using the same methods described above.
Orthology Analysis
Protein sequences from S genes of interest (Supplementary Table 3) as cited in original publications as reviewed by van Schie and Takken (2014) were cross-checked using gene name and synonym information and the Basic Local Alignment Search Tool (BLAST) functions in the TAIR gene search database2, EnsemblPlants3, UniPro4, and the IPK blast server5. OrthoFinder version 2.4.0 (Emms and Kelly, 2019) was used to identify orthologs between A. thaliana6, H. vulgare7, T. aestivum (annotation version 1.1 https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.1/iwgsc_refseqv1.1_genes_2017July06.zip), and B. distachyon (annotation version 1.2 https://phytozome-next.jgi.doe.gov/info/BdistachyonBd21_3_v1_2) proteins. For genes in A. thaliana, H. vulgare, and T. aestivum with multiple isoforms, perl scripts for each species were used to retain only the longest representative transcript for use in the orthology analysis8. The longest transcript file for B. distachyon (BdistachyonBd21_3_537_v1.2.protein_primaryTranscriptOnly. fa) was obtained from Phytozome. The default settings of OrthoFinder were used, and orthologs of the four species were obtained in a single run. The URGI BLAST tool9 was used to identify candidates for missing subgenome representatives.
Protein Sequence Phylogenetic Analysis
Using the longest protein sequence from known S genes in A. thaliana (Lamesch et al., 2011) and H. vulgare (Howe et al., 2019), as well as the longest protein sequences from the orthologous candidate S genes in T. aestivum and B. distachyon (Supplementary Table 3), phylogenetic trees were constructed to examine the relationship of ortholog families using the web-based tool NGPhylogeny (Lemoine et al., 2019). Default parameters for the FastME one-click workflow were used for MAFFT alignment, BMGE curation, and FASTME tree inference10. A R script using the packages ggplot2, ggtree, and ape was used to generate visualizations of the generated phylogenetic trees (Wickham, 2016; Yu et al., 2017; Paradis and Schliep, 2018).
Gene Co-expression Network Analysis
Individual gene co-expression networks (GCNs) were constructed and analyzed for T. aestivum W2691, W2691+Sr9b, and B. distachyon Bd21-3 genotypes using the python package Camoco (Schaefer et al., 2018). To build each network, all three independent RNA-seq replicates from all three time points (2, 4, and 6 dpi) of infected and mock-inoculated treatments were used. HTSeq read counts were converted to FPKM values for Camoco compatibility, and subjected to inverse hyperbolic sine transformation normalized against median FPKMs across all samples. Genes with coefficient of variation <0.1 across all samples or without a single sample having an expression above 0.5 FPKM were removed from analysis. Additionally, genes with a FPKM value > 0.001 across 60% of samples were included in network analyses. Pearson correlation metrics between all gene pairs were calculated and subjected to Fisher transformation to generate Z-scores with a cutoff of Z > = 3 to allow comparisons between networks (Huttenhower et al., 2006). Finally, correlation metrics were used to build weighted GCNs. Clusters containing susceptibility gene orthologs were visualized using ggplot2 (Wickham, 2016), ggnetwork (Briatte, 2020), sna (Butts, 2019), and network (Butts, 2015) R packages.
Data Availability
Sequence data was deposited in NCBI under BioProject PRJNA483957 (Supplementary Table 1). Unless specified otherwise, Supplementary Tables, scripts and files for analysis and visualizations are available at https://github.com/henni164/stem_rust_susceptibility. Due to large file sizes Supplementary Tables 8, 9 can only be found the github page mentioned above.
Results
T. aestivum and B. distachyon Differ in Susceptibility to Pgt
We compared the infection and colonization of Pgt in two T. aestivum isogenic lines that were susceptible (W2691) or resistant (W2691+Sr9b) to Pgt as well as in the non-host B. distachyon Bd21-3. The Bd21-3 line was selected to ease future adoption of reverse genetics approaches as an extensive collection of T-DNA insertional mutants is available in the same background (Bragg et al., 2012). Symptom development upon infection was consistent with previous observations reporting susceptibility of W2691 and W2691+Sr9b mediated resistance (intermediate) to race (pathotype) SCCL (Figures 1A,B; Zambino et al., 2000). Susceptibility was manifested by formation of large sporulating pustules in W2691, while small pustules surrounded by a chlorotic halo were characteristic of Sr9b mediated-resistance at 6 days post-inoculation (dpi). Susceptibility differences between W2691 and W2691+Sr9b were evident at 6 dpi as formation of fungal colonies was present in both genotypes, but colony sizes were larger in W2691 than W2691+Sr9b (Figure 1). B. distachyon supports the formation of colonies that are smaller than those in the resistant T. aestivum line W2691+Sr9b with no visible macroscopic symptoms observed at 6 dpi (Figure 1C). To monitor the progression of fungal growth and colonization, we quantified the percentage of GS, and interaction sites displaying the formation of AP, colony formation (C), and colony sporulation (SC) at 2, 4, and 6 dpi using microscopy (Figure 1D). The germination frequency (∼95%) was similar between all three genotypes tested (ANOVA test, p > 0.05). The percentage of interaction sites showing appressorium formation (AP) was higher in wheat than in B. distachyon at 4 dpi (ANOVA test, p ≤ 0.035). The genotype W2691 displayed the highest percentage of interaction sites showing colony formation at 4 and 6 dpi (ANOVA test, p ≤ 0.002), and sporulation at 6 dpi (ANOVA test, p ≤ 0.0015). In contrast, a smaller number of rust colonies formed in B. distachyon, and these colonies did not show signs of sporulation. To estimate rust colonization levels on T. aestivum and B. distachyon, we quantified the abundance of fungal DNA in infected leaves at 2, 4, and 6 dpi (Figure 1E). Fungal DNA abundance (rust colonization) among all genotypes was not significantly different at 2 dpi (ANOVA test, p > 0.05); however, there was a trend at 4 and 6 dpi for higher rust colonization in W2691 than in W2691+Sr9b and B. distachyon (ANOVA test, p > 0.05).
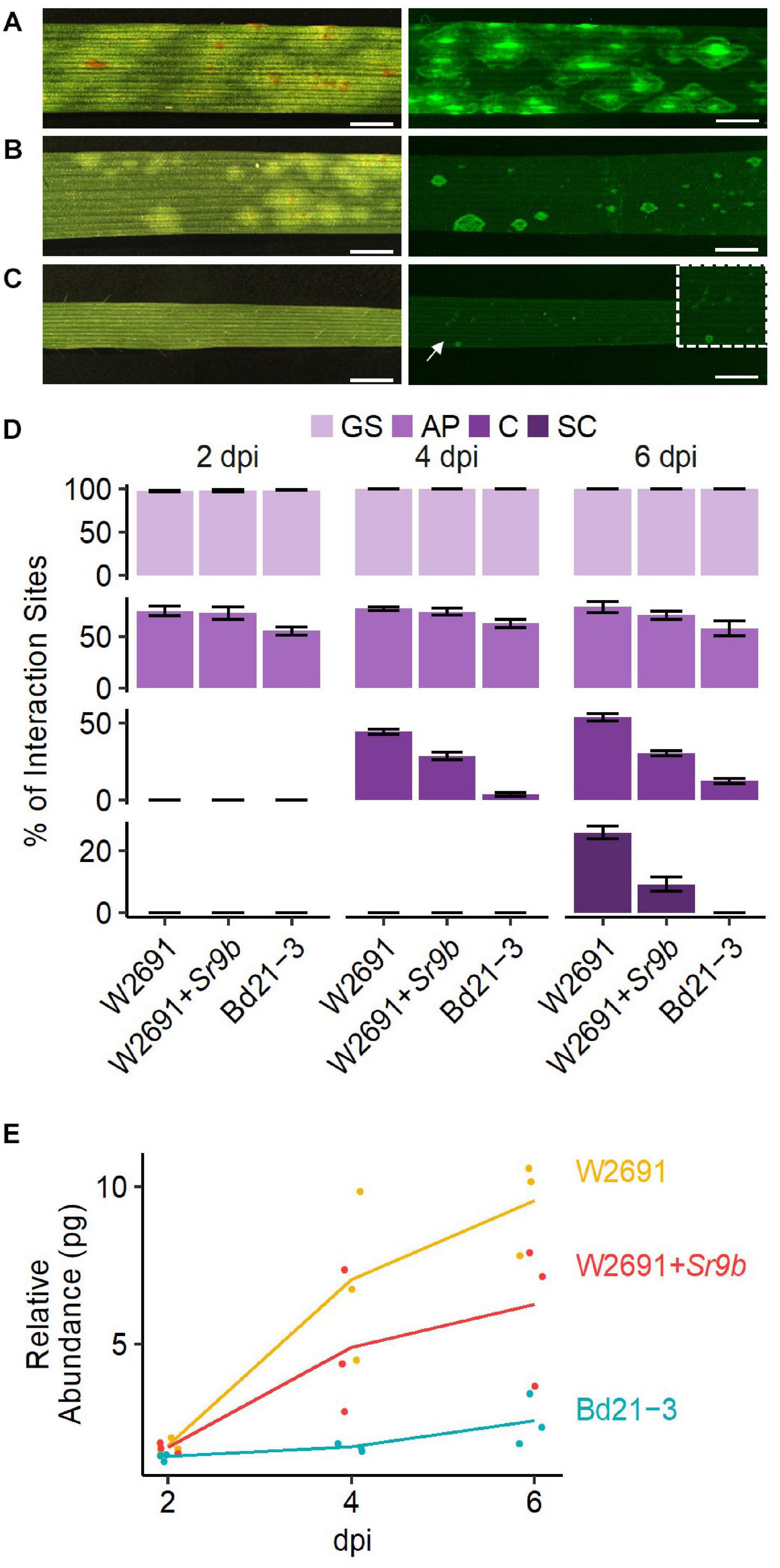
Figure 1. Infection of T. aestivum and B. distachyon genotypes with P. graminis f. sp. tritici race SCCL. (A–C) Development of disease symptoms (left) and fungal colonization (right) at 6 dpi. (A) W2691 (susceptible wheat line). (B) W2691+Sr9b (intermediate resistant wheat line). (C) B. distachyon Bd21-3 line (non-host). The white arrow and the white box indicate the area which was enlarged for better visualization of colonies. Scale bars indicate 2 mm. (D) Percentage of fungal infection sites which showed germinated urediniospores (GS), appressorium formation (AP), colony establishment (C), and sporulating colony (SC). Error bars represent the standard error of three independent biological replicates. (E) Fungal DNA abundance in infected W2691, W2691+Sr9b, and Bd21-3 genotypes as measured using qPCR. The points show the sample values and the lines represents the mean of the samples.
Putative Biological Processes Associated With in planta Responses to Pgt
The transcriptome profiles of T. aestivum (W2691 and W2691+Sr9b) and B. distachyon (Bd21-3) in response to Pgt infection at 2, 4, and 6 dpi were examined using RNA-seq expression profiling (Supplementary Table 1). DE analysis was used to compare responses to rust infection relative to the baseline mock treatments. Overall, the number of DEGs increased in W2691, W2691+Sr9b, and Bd21-3 over the course of infection (Table 1). Between 11 and 12.9% of T. aestivum genes were differentially expressed at 6 dpi, whereas in Bd21-3 only 6.2% were differentially expressed. We conducted a GOslim enrichment analysis on up- and down-regulated DEGs for each interaction at the infection time points (Figure 2). At 2 dpi, W2691 and W2691+Sr9b had only a few GOslim terms enriched in either up- or down-regulated DEGs. At 4 dpi, greater similarities between the T. aestivum genotypes emerged with very similar enrichment patterns in GOslim terms. The similarity of GOslim term enrichment continued at 6 dpi, with W2691 and W2691+Sr9b having nearly identical enrichment patterns. W2691+Sr9b had one additional term enriched in both up-regulated (cytoplasm, GO:0005737) and down-regulated (chromatin binding, GO:0003682) genes. Compared to the two T. aestivum genotypes, Bd21-3 had fewer terms enriched across all three timepoints and only a few terms were in common with W2691 and W2691+Sr9b, i.e., extracellular region (GO:0005576), DNA-binding transcription factor activity (GO:0003700). Bd21-3 had several unique terms in both up- and down-regulated categories, among them mitochondrion (GO:0005739), transporter activity (GO:0005215), catalytic activity (GO:0003824), and DNA binding (GO:0003677) were up-regulated, while intracellular (GO:0005622), DNA-binding transcription factor activity (GO:0003700), catalytic activity (GO:0003824), and DNA binding (GO:0003677) were down-regulated. The full GO set also demonstrated clear differences between the T. aestivum genotypes and Bd21-3. Photosynthesis-related terms such as chloroplast photosystem I and II (GO:0030093 and GO:0030095), photosystem II antenna complex (GO:0009783), and PSII associated light-harvesting complex II (GO:0009517) were overrepresented at 4 and 6 dpi in W2691 and W2691+Sr9b, but not in Bd21-3 (Supplementary Table 2). In addition, Bd21-3 only had enrichment in 11 terms across the cellular component (CC), biological process (BP), and molecular function (MF) categories compared to the terms enriched 741 across the three categories in W2691 and W2691+Sr9b (Supplementary Table 2). Overall, this analysis highlights how the molecular and genetic responses of Bd21-3 to Pgt differ from those in W2691 and W2691+Sr9b over the course of the experiment.
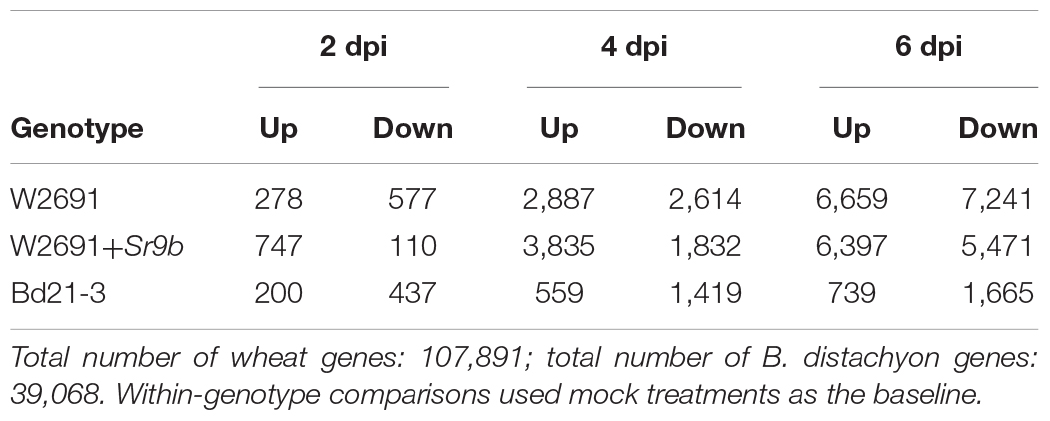
Table 1. Differentially expressed genes in T. aestivum and B. distachyon in response to P. graminis f. sp. tritici infection.
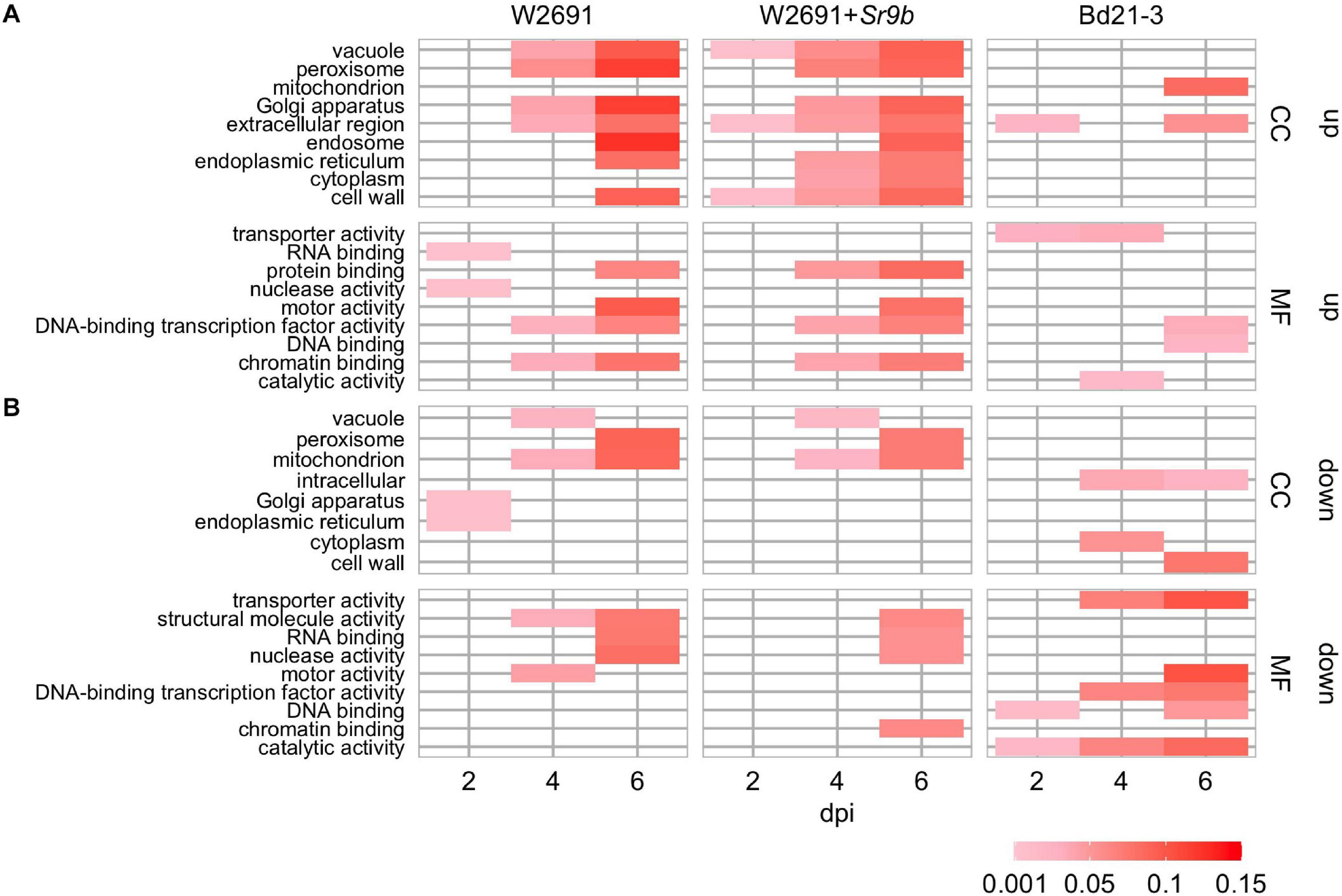
Figure 2. GOslim enrichment analysis of differentially expressed (DE) genes in mock vs inoculated T. aestivum (W2691 and W2691+Sr9b) and B. distachyon (Bd21-3) genotypes across three time points (bottom x-axis) upon infection with P. graminis f. sp. tritici. (A) Enrichment of plant GOslim terms of up-regulated (up) DE genes and (B) down-regulated (down) DE genes. The y-axis shows plant GO slim terms separated by category: cellular component (CC) and molecular function (MF). The scale represents the proportion of genes annotated with each GO term to all the genes tested.
Differential Regulation of Candidate Orthologous Susceptibility (S) Genes in T. aestivum and B. distachyon Upon Pgt Infection
Various S genes have been previously characterized or postulated in several species, including A. thaliana and H. vulgare (Büschges et al., 1997; Chen et al., 2007, 2010; Low et al., 2020), and this knowledge has allowed us to further understand molecular plant-microbe interactions. With an interest in identifying potential S genes in T. aestivum as well as creating resources to enable future studies, we designed an experimental workflow based on the identification of known S gene orthologs, gene expression comparisons and co-expression network analysis (Figure 3). A curated set of previously characterized or postulated S genes as summarized by van Schie and Takken (2014) was narrowed down by selecting genes in A. thaliana, and H. vulgare, and eliminating S genes that were discovered or characterized for viruses or necrotrophic fungi, leaving 112 potential candidate S genes to examine (Supplementary Table 3). We then conducted an orthology analysis using all H. vulgare, A. thaliana, B. distachyon, and T. aestivum transcripts to identify orthogroups of longest transcript of all genes. Orthogroups were constructed from 211,973 genes across these species (Supplementary Table 3). A total of 182,206 genes were assigned to 29,420 orthogroups, the largest of which (OG0000000) contained 211 genes. Of the total genes, 92,913 (86%) wheat, 31,334 B. distachyon (80%), 34,075 barley (91%), and 23,883 A. thaliana (87%) genes were assigned to orthogroups. We identified 91 of the reported S genes from A. thaliana and H. vulgare across 70 orthogroups, that also consisted of at least one T. aestivum gene and one B. distachyon gene (Supplementary Table 4). These genes from T. aestivum and B. distachyon were selected as S gene orthologs. A total of 29,767 genes (orthogroup OG0029421 to OG0059187) were assigned groups with only one member (singleton orthogroups; Supplementary Table 5).
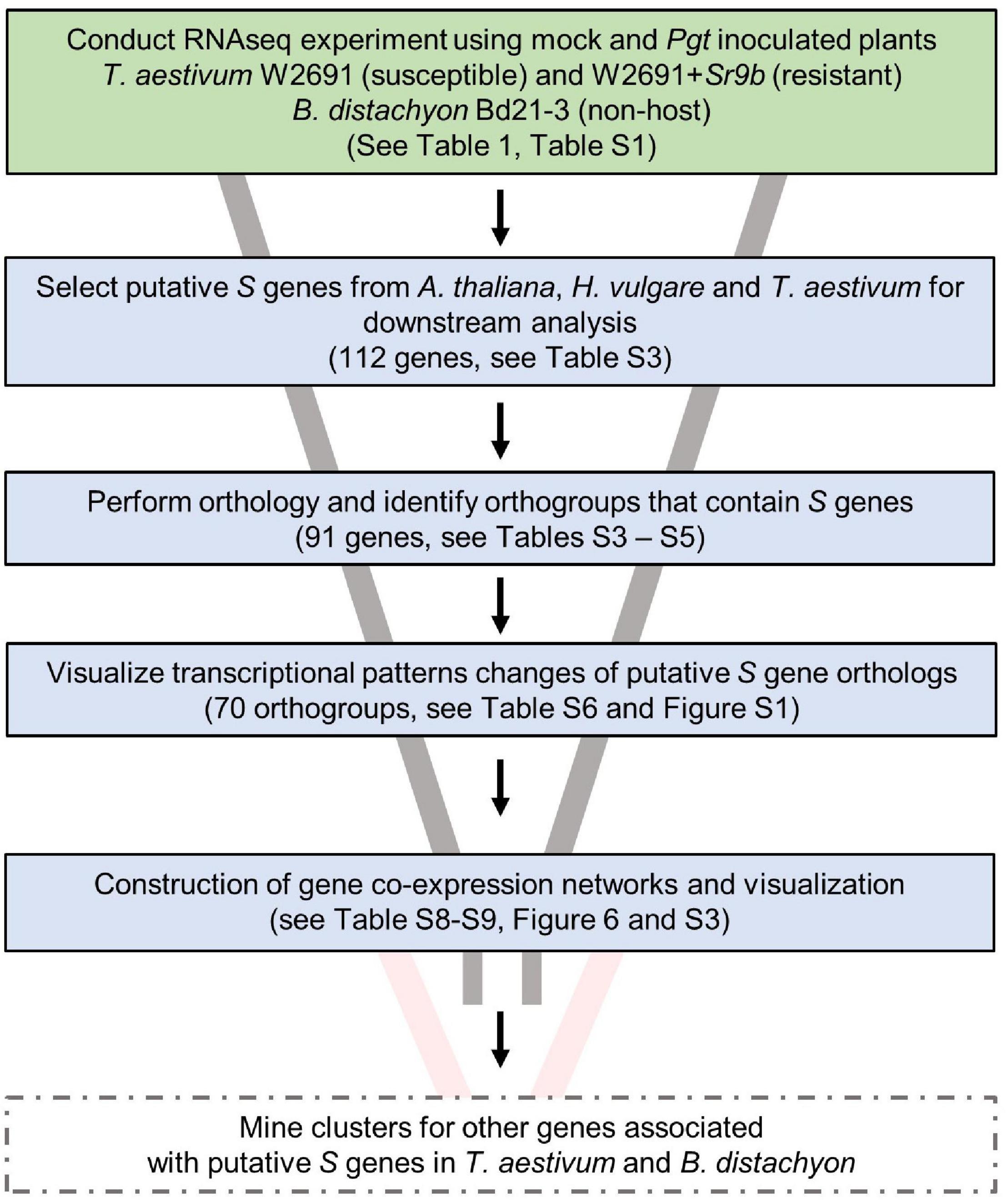
Figure 3. Experimental workflow used to identify candidates of S genes that contribute to infection of T. aestivum by P. graminis f. sp. tritici. Solid box outlines indicate work completed in this publication. Future work is indicated by dashed box outlines.
The gene expression patterns of S gene orthologs in T. aestivum and B. distachyon were used to identify which orthologs may act as susceptibility factors (Figure 3 and Supplementary Table 6). The selection criterion was applied to include DEGs that showed a progressive increase in log2 fold change (mock vs infected, | log2 fold change| ≥1.5 and a p-value < 0.05) in W2691 or in both W2691 and W2691+Sr9b, but the corresponding orthologs in B. distachyon and/or W2691+Sr9b showed a decrease or no change, as observed in various systems (Chen et al., 2010; Pessina et al., 2014). The assumption is that S genes will be up-regulated during infection when the pathogen reaches the sporulation stage (e.g., in a susceptible or intermediate resistant host represented by W2691 and W2691+Sr9b, respectively) but with a low or no regulatory change in a non-host (Bd21-3). Expression data for all genes can be found in Supplementary Table 6 in association with orthogroup number. Most genes in the 70 orthogroups did not demonstrate major changes in expression over the course of the experiment (Supplementary Figure 1), including the orthogroup OG0001703, which contains the Mlo (Mildew locus O) alleles and orthologous sequences. Eight orthogroups that demonstrated these expression patterns were chosen for further analysis; these included ortholog genes for AGD2 (aberrant growth and death 2), BI-1 (BAX inhibitor-1), DMR6 (downy mildew resistance 6), DND1 (defense, no death), FAH1 (fatty acid hydroxylase 1), IBR3 (IBA response 3), VAD1 (vascular associated death 1), and WRKY25 (WRKY DNA binding protein 25; Figure 4, Table 2 and Supplementary Table 7). Among the eight susceptibility orthogroups, T. aestivum orthologs of BI-1, DMR6, and WRKY25 showed the greatest increase in fold change (Supplementary Table 7) in either W2691 or W2691+Sr9b, particularly at 6 dpi (Figure 4). The gene ortholog of DND1 displayed a higher fold change in W2691 than in W2691+Sr9b.
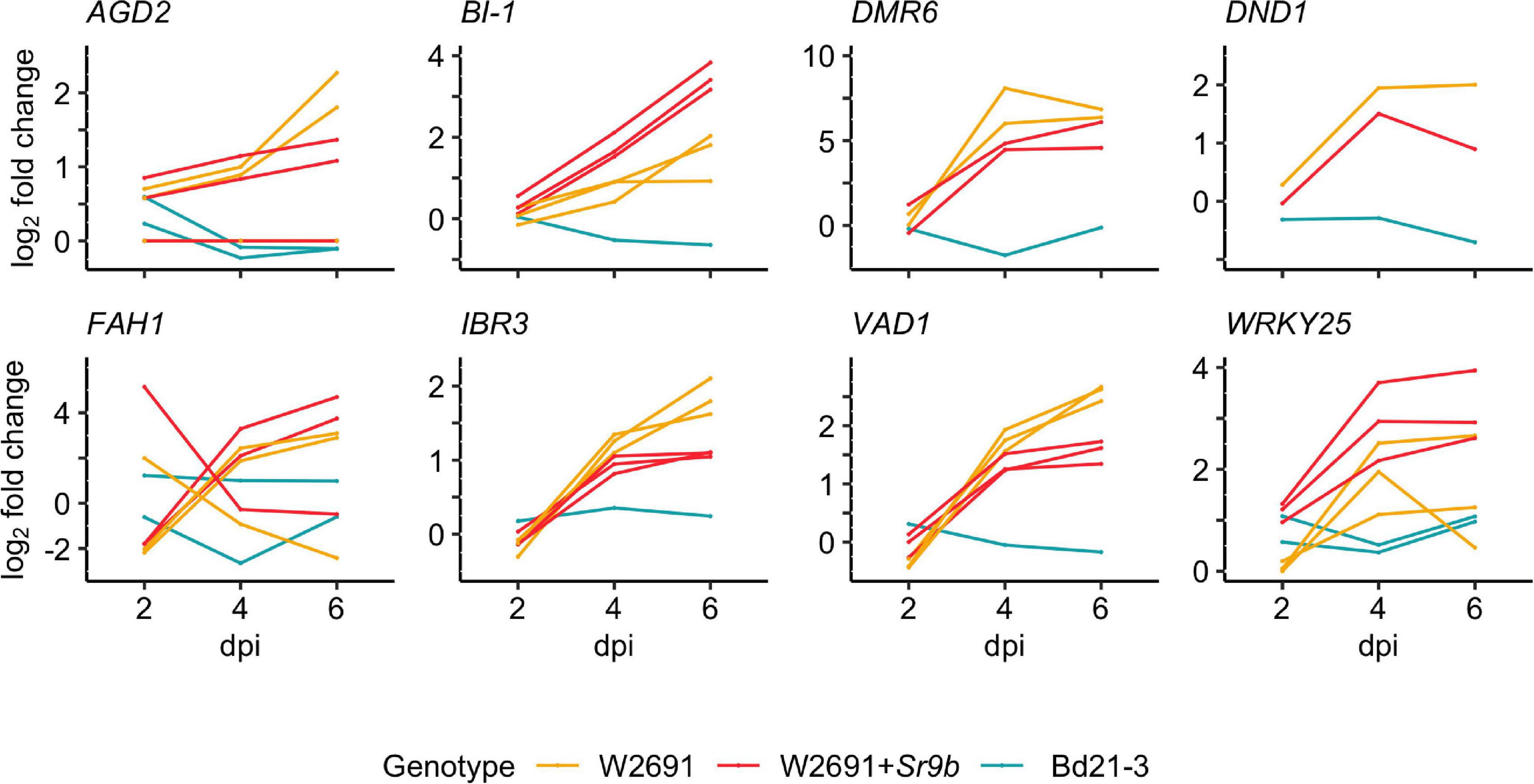
Figure 4. RNAseq expression profile patterns of selected orthogroups containing candidate S genes in T. aestivum (W2691 and W2691+Sr9b) and B. distachyon (Bd21-3) genotypes throughout infection with P. graminis f. sp. tritici. Log2 fold change values for all gene orthologs are presented for each infected genotype compared to the mock treatment per sampling time point. Gene IDs, average FPKM values, orthogroup, and co-expression cluster identifiers are presented in Supplementary Table 7.
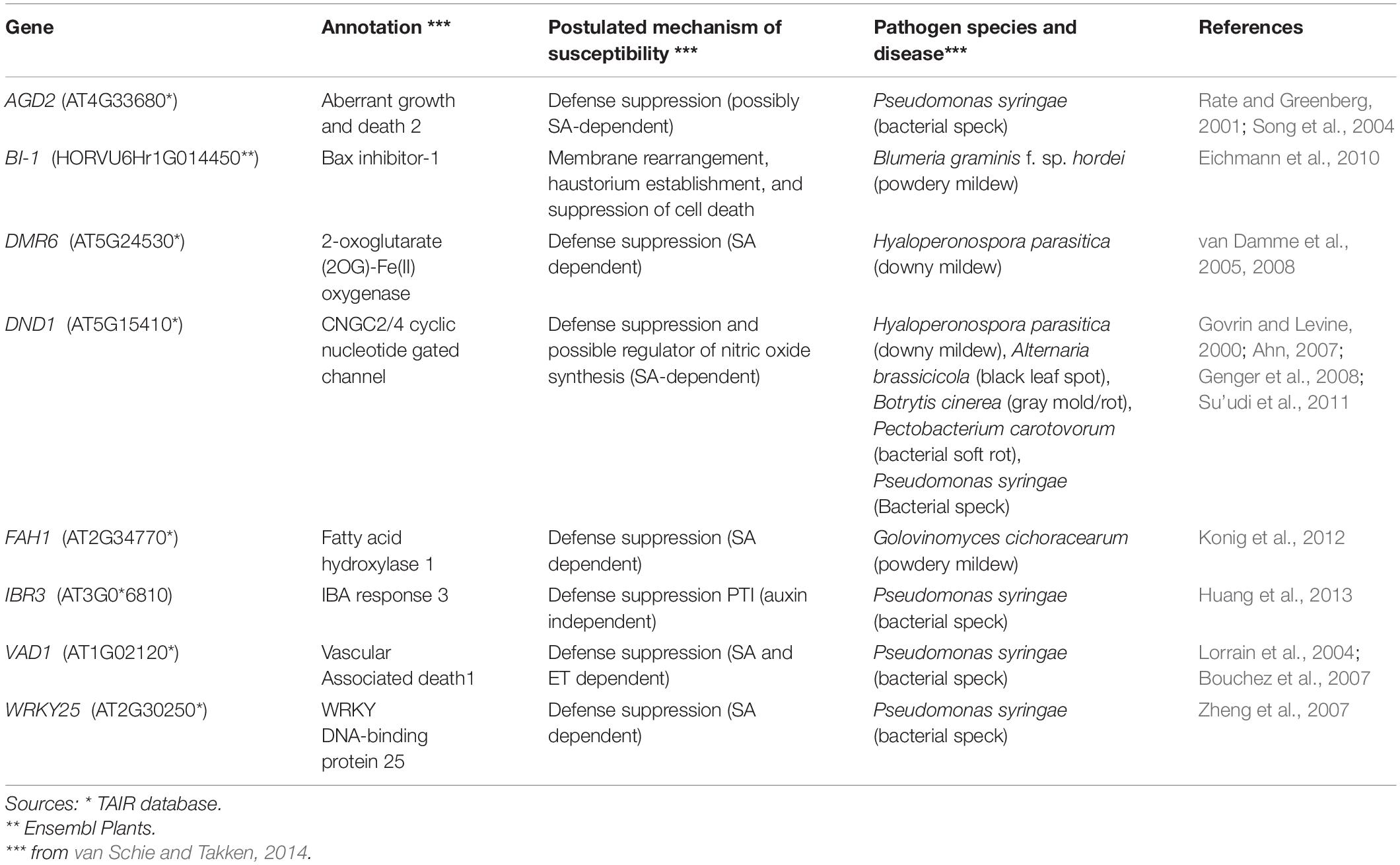
Table 2. List of S genes explored through the gene expression analysis.
The phylogenetic relationships of the orthogroups to known S genes were confirmed using NGphylogeny (Supplementary Figure 2). A phylogenetic tree for DND1 was not generated since the orthogroup (OG0018857) only contains three genes (TraesCS5D01G404600, BdiBd21-3.1G0110600, and AT5G15410). Complete sets of T. aestivum homeologs from the three subgenomes were found in four out of the eight examined orthogroups. There were only two of three expected T. aestivum homeologs in the DMR6 orthogroup, with TraesCS4B02G346900 and TraesCS4D02G341800 representing the B and D subgenomes, respectively. A tblastn of these sequences to chromosome 4A revealed TraesCS4A02G319100, a partial match of 30–31% identity (1e-42 to 1e-44). This gene has low expression and is found in orthogroup OG0006808, which contains two other T. aestivum genes, one B. distachyon gene, two A. thaliana genes, and one H. vulgare gene (Supplementary Tables 4, 6). Despite the low sequence similarity, TraesCS4A02G319100 and TraesCS4B02G346900 are at more similar positions (4A:608043459 and 4B:640532917, respectively) to each other than to TraesCS4D02G341800 (4D:498572979). A tblastn to the entire genome revealed 20 other matches for the two DMR6 orthologs with 31–74% identity. Thus, it does not seem that the T. aestivum genome reference (Chinese Spring) contains a homeolog of DMR6 in the A genome. To further examine if a DMR6 ortholog is present in the A subgenome, the B and D subgenome DMR6 homeologss were BLASTed to the wheat pangenome CDS sequences11. Similar results were obtained; there were 30 total hits ranging from 36.765 to 39.564% identity (1.07e-61 to 1.99e-69) on chromosome 4A of the 10 genomes. Based on the low sequence identity, it is likely that there is not an ortholog of DMR6 on chromosome 4A. However, hits with high identity (97.619–98.214, e = 0) were found on chromosome 5A in all 10 genomes.
Another S gene orthogroup without full subgenome representation was OG0018857 which contained DND1. This orthogroup only has one T. aestivum gene, TraesCS5D02G404600 from subgenome D. A tblastn to chromosomes 5A and 5B resulted in matches with high identity on both 5A (TraesCS5A02G395300, 94%, and 6e-159) and 5B (89%, 1e-176). TraesCS5A02G395300 is present in orthogroup OG0048986 as a singleton with low expression in W2691 (FPKM = 2.61) and W2691+Sr9b (FPKM = 1.65), and TraesCS5B02G400100 is included in orthogroup OG0048844 as a singleton as well with notable expression at 6 dpi in infected W2691 (FPKM = 4.28) and low expression in W2691+Sr9b (FPKM = 1.19; Supplementary Tables 5, 6). A tblastn to the entire genome identified 19 other candidates with identity 33–97%. The most notable matches with high identity were TraesCS7B02G161600 (97%, 4e-152) and TraesCS3B02G306700 (97%, 2e-147), which are the only two genes together in orthogroup OG0027858. Both top matches had essentially no expression in either T. aestivum genotype (FPKM = 0 to 0.07; Supplementary Table 6). A third genomic region on chromosome 2B also has 97% identity, but is annotated as a nested repeat.
For FAH1, three T. aestivum orthologs are present in the orthogroup OG0006155, but one is from subgenome A (TraesCS5A02G019200) while the other two are from subgenome D (TraesCS5D02G024600 and TraesCS5D02G424200). A tblastn of all three sequences to chromosome 5B revealed two matches, TraesCS5B02G016700 (87%, 3e-49) and TraesCS5B02G418800 (62/87%, 2e-64/2e-49), while a tblastn to the entire genome uncovered a partial match on 5A (TraesCS5A02G416500, 46–62%, and 7e-68-1e-104) and a partial match on 3D which was not annotated (68–89%, 6e-34-3e-43). All three annotated genes are in singleton orthogroups (TraesCS5B02G016700, OG0056228; TraesCS5B02G418800, OG0055841; and TraesCS5A02G416500, OG0057903) and have low expression in both W2691 and W2691+Sr9b (FPKM = 0.05 to 1.8; Supplementary Tables 5, 6). Orthogroup OG0005265 for AGD2 is similar to the orthorgoup for FAH1, having one A subgenome representative (TraesCS4A02G116000) and two D subgenome representatives (TraesCS4D02G189600 and TraesCS7D02G452900). The tblastn of these sequences to chromosome 4B revealed one possible match with two annotations in the same location (61–62%, 9e-108-1e-113), TraesCS4B02G264500 on the – strand and TraesCS4B02G264400 on the + strand. The former is a singleton in orthogroup OG0047603 with low expression in W2691 (FPKM = 0.09) and high expression in infected W2691+Sr9b at 6 dpi (FPKM = 4.64), while the latter is in OG0015484 with several other genes and is not highly expressed in either T. aestivum genotype (FPKM = 1.3 to 1.7; Supplementary Table 6). The tblastn to the entire genome revealed several hits of identity varying between 22 and 97%.
Gene Co-expression Network Analysis
To further explore potential processes and novel genes linked to stem rust susceptibility, a gene co-expression network for B. distachyon and each T. aestivum genotype using the mock and infected RNA-seq data at each timepoint was constructed (Supplementary Table 8, see github page). The complete Bd21-3 network has 572,179 edges that connected 21,746 nodes (55.7% of protein-coding genes), while the W2691 and W2691+Sr9b networks are larger (W2691: 3,433,279 edges, 49,082 nodes, 45.6% of protein-coding genes; W2691+Sr9b: 3,817,404 edges, 49,000 nodes, 45.5% of protein-coding genes). The B. distachyon network was expected to be smaller as it represents a diploid species with fewer annotated genes (39,068), while the hexaploid wheat contains more gene annotations (107,891). There are 189 clusters with more than 10 genes in Bd21-3, 258 in W2691, and 391 in W2691+Sr9b. Thus, more genes have similar expression patterns in W2691+Sr9b than in W2691, and Bd21-3 has the lowest number of genes with similar patterns. The eight S gene orthogroups of interest are represented by 14 clusters in W2691 (cluster IDs: 0, 3, 4, 5, 8, 11, 13, 60, 110, 178, 11114, 11235, 12377, and 20128), 11 in W2691+Sr9b (cluster IDs: 0, 2, 4, 112, 1139, 1916, 2729, 2772, 3133, 10229, and 11079), and 11 in Bd21-3 (cluster IDs: 3, 4, 35, 51, 272, 513, 652, 1359, 1662, 1848, and 3087; Supplementary Table 9, see github page). Some orthogroups are represented across multiple clusters, while others are only represented in singleton clusters. The ortholog clusters in B. distachyon contain fewer genes than the corresponding W2691 and W2691+Sr9b ortholog clusters.
Gene ontology enrichment tests using GOslim annotations were conducted on the clusters to investigate functional processes. Across all eight S gene orthogroups, at least one gene from each is in a cluster with GO enrichment in at least one genotype (Figure 5). DMR6, FAH1, and WRKY25 are the only candidates to have enrichment in all three genotypes, AGD2 and DND1 only has enrichment in W2691, and BI-1, IBR3, and VAD1 have enrichment in both W2691 and W2691+Sr9b. Terms commonly enriched in the T. aestivum genotypes include the Golgi apparatus (GO:0005794), endosome (GO:0005768), endoplasmic reticulum (GO:0005783), protein binding (GO:0005515), transporter activity (GO:0005215), vacuole (GO:0005773), and peroxisome (GO:0005777; Figure 5). Only one GO term, catalytic activity (GO:0003824) is unique to Bd21-3, with other terms like DNA-binding transcription factor activity (GO:0003700) being enriched in the Bd21-3 and T. aestivum genotypes.

Figure 5. GOslim term enrichment for all genes in co-expression gene clusters containing S gene orthologs in T. aestivum and B. distachyon. The y-axis shows GOslim terms separated into categories: cellular component (CC) and molecular function (MF).
For each genotype a cluster containing one or more orthologs of DND1, VAD1, and DMR6 was selected as examples for presentation (Figure 6). Selection criteria for these examples included (1) higher expression in infected than in mock treatments in T. aestivum and (2) varied cluster sizes across genotypes. DND1 is represented by TraesCS5D02G404600 within cluster 4 in the W2691 genotype (557 genes), by TraesCS5D02G404600 within cluster 122 in the W2691+Sr9b genotype (21 genes) and by BdiBd21-3.1G0110600 within cluster 652 in the Bd21-3 genotype (4 genes; Figure 6A and Supplementary Table 8). VAD1 represents a mid-point between DND1 and DMR6, with the large cluster 0 (TraesCS2D02G236800) representing VAD1 for the W2691 genotype (4527 genes), a singleton cluster (cluster 3087) for the Bd21-3 genotype (1 gene, BdiBd21-3.1G0357000), and the large cluster 0 (TraesCS2D02G236800) for the W2691+Sr9b genotype (3400 genes; Figure 6B and Supplementary Table 8). DMR6 is also represented by cluster 0 (TraesCS4B02G346900 and TraesCS4D02G341800) for both W2691 and W2691+Sr9b; however, cluster 4 representing DMR6 in Bd21-3 (BdiBd21-3.1G1026800) is larger than in the previous examples (443 genes; Figure 6C and Supplementary Table 8). In all cases, the S gene candidates are not the most differentially-expressed genes at 6 dpi among the T. aestivum genotype clusters; the most DEG at 6 dpi in cluster 0 is TraesCS7A02G157400 (not functionally annotated) in W2691 (log2FC = 13.64) and TraesCS1A02G266000 (IPR002921:Fungal lipase-like domain IPR029058:Alpha/Beta hydrolase fold IPR033556:Phospholipase A1-II) in W2691+Sr9b (log2FC = 14.61). For cluster 4 in W2691, TraesCS4D02G120200 (IPR001471:AP2/ERF domain IPR016177:DNA-binding domain superfamily IPR036955:AP2/ERF domain superfamily) is the most DEG at 6 dpi (log2FC = 12.71), while for cluster 122 in W2691+Sr9b it is TraesCS1A02G276800 (IPR013087:Zinc finger C2H2-type IPR036236:Zinc finger C2H2 superfamily; log2FC = 3.83). In B. distachyon, BdiBd21-3.1G0110600, which is the Bd21-3 ortholog to A. thaliana DND1, is most differentially expressed in the cluster representing DND1 and was highly down-regulated in infected tissue at 6 dpi (log2FC = −0.70). By necessity the most DEG in the network representing VAD1 in B. distachyon is the ortholog of VAD1, as Bd21-3 cluster 3087 is a singleton cluster. The most differentially expressed Bd21-3 gene in cluster 4 representing DMR6 is BdiBd21-3.2G0466100 (log2FC = 0.28). This gene is annotated as a Leucine-rich repeat protein kinase family protein due to homology with the A. thaliana gene AT1G79620, though the orthology analysis places these genes in different clusters (OG0019394 and OG0010938, respectively). All clusters representing the eight S gene candidates are shown in Supplementary Figure 3.
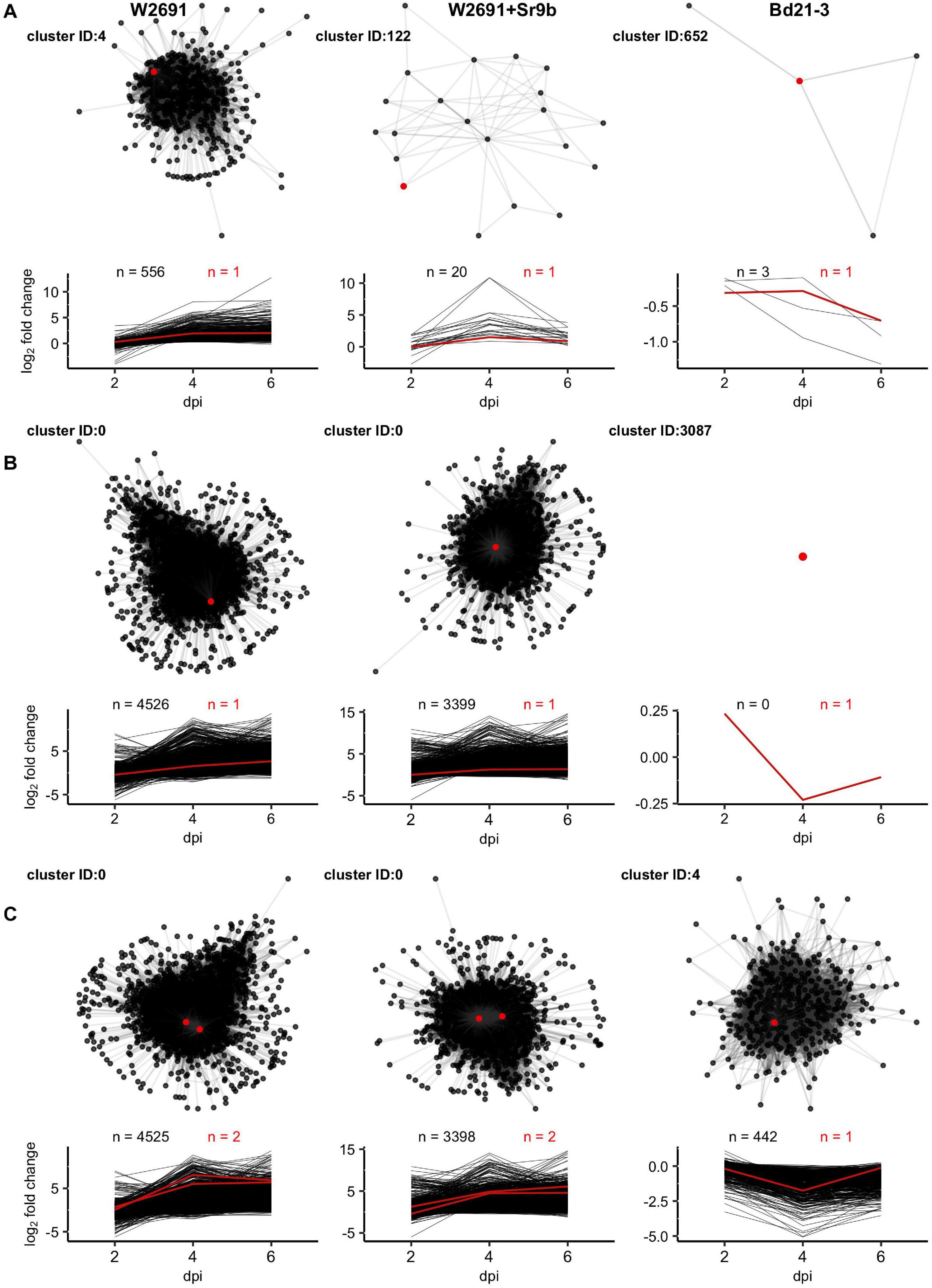
Figure 6. Network diagrams for clusters containing orthologs of (A) DND1, (B) VAD1, and (C) DMR6 with corresponding plots showing log2 fold change of all nodes across 2, 4, and 6 dpi. Only connections with Z > = 3 are shown. Red lines, points, and counts represent T. aestivum and B. distachyon orthologs of S genes. Cluster identifiers (IDs) and gene names presented, left to right: DND1: 4 (TraesCS5D02G404600), 122 (TraesCS5D02G404600), and 652 (BdiBd21-3.1G0110600); VAD1: 0 (TraesCS2D02G236800), 0 (TraesCS2D02G236800), and 3085 (BdiBd21-3.1G0357000); and DMR6: 0 (TraesCS4B02G346900), 0 (TraesCS4D02G341800), and 4 (BdiBd21-3.1G1026800).
Discussion
Susceptibility (S) genes are an essential component of compatible plant pathogen interactions (Engelhardt et al., 2018). The opportunity to genetically manipulate such genes to engineer disease resistance in important crops such as T. aestivum has captured significant scientific interest in recent years. However, our understanding of the genetic basis of disease susceptibility in cereals is limited to a few examples (van Schie and Takken, 2014; Engelhardt et al., 2018). Thus, important questions regarding the biological functions of these genes and their activation remain to be answered. As a first step to uncover putative stem rust S genes, we conducted a comparative RNA-seq experiment coupled with gene co-expression network analysis to determine transcriptional responses in T. aestivum genotypes and B. distachyon Bd21-3. We compared a compatible interaction (W2691) with an incompatible interaction controlled by the race-specific resistance gene Sr9b in the same genetic background (W2691+Sr9b). Sr9b restricts pathogen growth; however, it also allows the development of small sporulating colonies of a Pgt isolate which belongs to the race SCCL (Zambino et al., 2000). A more stringent incompatibility scenario is given by Bd21-3 genotype of B. distachyon, which allows restricted colony formation of Pgt without sporulation. These observations were consistent with previous descriptions of B. distachyon as a non-host to rust pathogens (Figueroa et al., 2013, 2015, Omidvar et al., 2018). Thus, a strength of this study is the survey of molecular responses associated with increasing levels of susceptibility.
Consistent with findings from other transcriptomic studies of wheat-rust interactions (Manickavelu et al., 2010; Zhang et al., 2014; Chandra et al., 2016; Dobon et al., 2016; Yadav et al., 2016), major transcriptional changes were detected in response to infection in both T. aestivum and B. distachyon, which reflect the complexity of these plant-microbe interactions. A significantly higher number of up- or down-regulated genes were found in T. aestivum than B. distachyon. The greater fungal colonization of T. aestivum as indicated by in planta fungal growth assays of Pgt is likely a result of the pathogen’s failure to effectively manipulate the metabolism of B. distachyon. GOslim term analyses indicated an enrichment for Golgi apparatus, peroxisome, vacuole, and cell wall related functions in up-regulated genes in T. aestivum. These results are not surprising as a large proportion of immune receptors and plant defense signaling components play a role in plant-microbe interactions (Dodds and Rathjen, 2010; Couto and Zipfel, 2016). The plant Golgi apparatus and peroxisomes have been reported as targets of effectors from various pathogenic filamentous fungi (Robin et al., 2018). The enrichment of these GO terms in up-regulated genes in T. aestivum suggests that these CCs may be direct or indirect targets for effectors derived from Pgt. Analyses with the full GO term set revealed many enriched terms among downregulated genes related to photosynthesis in W2691 and W2691+Sr9b; a decrease in chlorophyll and photosynthetic activity has been previously reported in wheat infected with Pgt (Berghaus and Reisener, 1984; Moerschbacher et al., 1994).
Several S genes to diverse pathogens have been identified or postulated in various plant species (van Schie and Takken, 2014; Engelhardt et al., 2018). While this area of research for cereal rust pathogens is in its infancy, positive results from other pathosystems make a strong case to consider the modification of S genes as an approach to deliver durable and broad-spectrum disease resistance. So far, only a few host-delivered avirulence effectors, AvrSr50 (Chen et al., 2017), AvrSr35 (Salcedo et al., 2017), and AvrSr27 (Upadhyaya et al., in press) from any cereal rust fungi have been isolated. These were identified in Pgt and how these effectors disrupt defense responses in compatible interactions remains unknown. Future research seeking to identify which plant proteins these effectors target will help elucidating S genes or processes required for stem rust susceptibility.
Here, expression patterns of gene orthologs in T. aestivum and B. distachyon corresponding to previously characterized S genes in H. vulgare and A. thaliana were examined to develop a framework to study S genes in wheat. A key focus of this study was to develop a workflow to extract orthologs with high expression in stem rust susceptible T. aestivum, but low expression in either T. aestivum with intermediate resistance, or B. distachyon. We note that differential gene expression between T. aestivum genotypes (W2691 and W2691+Sr9b) can also provide an opportunity to discovery S genes since rust infection in both genotypes differs. To link these candidate S genes with the biological pathways in T. aestivum and B. distachyon, we constructed GCNs, which can be explored to determine the role of components of these pathways and the complex interplay toward regulation of susceptibility in Pgt-T. aestivum interactions.
The biological functions of S genes in compatible-plant microbe interactions are diverse, as these genes play roles in a wide array of events that are critical for pathogen accommodation and survival (Engelhardt et al., 2018). Some of these susceptibility genes can act as negative regulators of immune responses, such as PTI, cell death, and phytohormone-related defense. Our study determined that T. aestivum orthologs of the BAX inhibitor-1 (BI-1) gene in H. vulgare are candidate S genes, as these were up-regulated in W2691 (6 dpi) and W2691+Sr9b (4–6 dpi) whereas their expression in B. distachyon was not affected. BI-1 is an endoplasmic reticulum membrane-localized cell death suppressor in A. thaliana, and its wheat ortholog TaBI-1 (accession GR305011) is proposed to contribute to susceptibility in T. aestivum to the biotrophic pathogen Puccinia striiformis f. sp. tritici (Wang et al., 2012). Interestingly, the highest upregulation of the BI-1 was detected in the W2691+Sr9b genotype where it is necessary to regulate a HR upon Pgt recognition. Given this result it should be examined if BI-1 may be a conserved plant S factor to wheat rust fungi. Various orthologs of FAH1, which encodes a ferulate 5-hydroxylase in A. thaliana, were up-regulated in the T. aestivum genotypes upon Pgt infection (Mitchell and Martin, 1997). According to studies in A. thaliana FAH1 plays a role in BI-1-mediated cell death suppression through interaction with cytochrome b5 and biosynthesis of very-long-chain fatty acids (Nagano et al., 2012). Additional findings further suggest that Pgt can also interfere with cell death signaling by altering VAD1 expression. The VAD1 gene encodes a putative membrane-associated protein with lipid binding properties and it is proposed to act as negative regulator of cell death (Lorrain et al., 2004; Khafif et al., 2017). Transcriptional activation of VAD1 has been shown to occur in advanced stages in plant pathogen interactions (Bouchez et al., 2007). We detected an upregulation of VAD1 orthologs in T. aestivum at 6 dpi, which is considered a late infection stage in the establishment of rust colonies.
Salicylic acid (SA) is a key phytohormone required to orchestrate responses to many pathogens (Ding and Ding, 2020). Similar to VAD1 whose function as a S factor is SA-dependent, we also uncovered other up-regulated genes that may also participate in defense suppression. The orthologs of the DMR6 are highly up-regulated in T. aestivum at 4 and 6 dpi in both compatible and incompatible interactions. As characterized in A. thaliana, DMR6 encodes a putative 2OG−Fe(II) oxygenase that is defense−associated and required for susceptibility to downy mildew through regulation of the SA pathway (van Damme et al., 2008; Zhang et al., 2017). The role of DMR6 in disease susceptibility holds significant promise to control diverse pathogens. For instance, mutations in DMR6 confer resistance to hemibiotrophic pathogens Pseudomonas syringae and Phytophthora capsici (Zeilmaker et al., 2015) and silencing of DMR6 in potato increases resistance to the potato blight causal agent, P. infestans (Sun et al., 2016). It has also been shown that the H. vulgare ortholog genetically complements DMR6 knock-out A. thaliana lines and restores susceptibility to Fusarium graminearum (Low et al., 2020). Gene orthologs of DND1 were also identified as up-regulated in both T. aestivum genotypes. The gene DND1 encodes a cyclic nucleotide-gated ion channel and its activity is also related to SA regulation (Clough et al., 2000). Mutations in A. thaliana DND1 display enhanced resistance to viruses, bacteria and fungal pathogens (Yu et al., 2000; Jurkowski et al., 2004; Genger et al., 2008; Sun et al., 2017). We also noted that several wheat orthologs of the A. thaliana gene IBR3 also increased in expression as Pgt infection advanced. The role of IBR3 in susceptibility to P. syringae in A. thaliana has been confirmed by mutations and overexpression approaches (Huang et al., 2013). Consistent with our results, IBR3 is up-regulated in A. thaliana upon infection by P. syringae.
Plant transcriptional reprogramming triggered by pathogen perception is often mediated by WRKY transcription factors through activation of the MAP kinase pathways (Eulgem and Somssich, 2007; Rushton et al., 2010). Here, we detected an upregulation of the expression of WRKY25 orthologs that was most prominent at 6 dpi in the W2691+Sr9b genotype. The Arabidopsis gene AtWRKY25 is induced in response to the bacterial pathogen Pseudomonas syringae and the SA-dependent activity of AtWRKY25 is also linked to defense suppression (Zheng et al., 2007). According to results from this study, the contribution of orthologs of AGD2 to stem rust susceptibility in T. aestivum should also be examined. AGD2 encodes an aminotransferase and participates in lysine biosynthesis at the chloroplast (Song et al., 2004). Given that several oomycete and fungal effectors target the chloroplast (Kretschmer et al., 2020), effector research in cereal rust pathogens will be crucial to determine if these pathogens also target this organelle.
A classic example of S genes in barley is given by the Mlo gene (Jørgensen, 1992) in which a recessive mutation results in broad spectrum resistance to Blumeria graminis f. sp. hordei, the causal agent of powdery mildew. The Mlo gene family is highly conserved across monocot and dicot plants and gene editing of Mlo homeologs in wheat confers resistance to powdery mildew (Acevedo-Garcia et al., 2017). Interestingly, Mlo genes in T. aestivum have not been reported to provide protection against cereal rust diseases. Consistent with this, this study did not detect a significant change in the expression of Mlo alleles in T. aestivum genotypes (W2691 and W2691+Sr9b) over the course of the experiment.
One caveat of this study is that some S genes in T. aestivum for Pgt may not be found in model species like A. thaliana or detected using other pathogens. However, this is a first step to identify candidates to guide functional studies. While in this study we focused on orthologous S genes, the GCNs presented here are excellent resources to identify additional candidate S factors. It is possible that some of the genes included in clusters of these networks are part of the regulatory process that control expression of S genes or are part of essential pathways although their function may not be characterized yet in other systems. Future functional studies are required to validate the function of these genes in T. aestivum as S factors for rust infection and determine if these can be exploited for agricultural practice. A key aspect for the success of these novel approaches is the absence of plant developmental defects resulting from mutations of S genes. In some cases, the loss-of-function of negative regulators leads to constitutive activation of plant defense responses that manifest as poor growth or lesion-mimic phenotypes among other pleiotropic effects (Büschges et al., 1997; Ge et al., 2016). VIGS-mediated transient gene silencing (Lee et al., 2015), RNAi-mediated silencing (Helliwell and Waterhouse, 2003; Waterhouse and Helliwell, 2003; Sun et al., 2016), TILLING populations include some of the approaches to explore the potential use of these S gene candidates. Gene editing technologies through Zinc Finger nucleases, TALENs, CRISPR/Cas9 systems also offer options to generate transgene free plants (Urnov et al., 2010; Gaj et al., 2013; Wang et al., 2014; Jia et al., 2017; Nekrasov et al., 2017; Kim et al., 2018; Luo et al., 2019). In conclusion, as the demands for multi-pathogen durable disease resistance rise, our ability to target S genes may serve as a sound approach to harness genetic diversity and maximize the resources to meet critical these grand challenges.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, Bioproject PRJNA483957.
Author Contributions
MF, CH, CM, and SK conceived and designed the study. VO, MM, FL, and MF conducted the experiments. EH, EG, J-MM, RD, CH, SG, JV, and MM contributed to data analysis. EH, BS, SK, CH, and MF interpreted results. EH, VO, CH, and MF wrote the manuscript. All authors contributed to manuscript editing, revisions and approved the submitted version.
Funding
This work was supported by a seed grant from the Microbial Plant Genome Institute at The University of Minnesota, USDA-NIFA grant #2018-67013-27819, University of Minnesota Experimental Station USDA-NIFA Hatch project MIN-22-086, as well as the USDA-ARS/ The University of Minnesota Standard Cooperative Agreement (3002-11031-00053115) between SK and MF. EH is supported by a scholarship from the College of Food, Agriculture, and Natural Resource Science at the University of Minnesota. The work conducted by the United States DOE Joint Genome Institute is supported by the Office of Science of the United States Department of Energy under Contract no. DE-AC02-05CH11231.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank staff at the Minnesota Supercomputing Institute at the University of Minnesota for technical assistance.
Supplementary Material
Supplementary Tables 1–7 and Supplementary Figures 1–3 for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.657796/full#supplementary-material. Further supplementary material, including Supplementary Tables 8 and 9, scripts and files for analysis and visualizations, are available at https://github.com/henni164/stem_rust_susceptibility.
Footnotes
- ^ https://github.com/owlcollab/owltools
- ^ https://www.arabidopsis.org/index.jsp
- ^ https://plants.ensembl.org/index.html
- ^ https://www.uniprot.org/
- ^ https://webblast.ipk-gatersleben.de/barley_ibsc/
- ^ https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Athaliana
- ^ http://floresta.eead.csic.es/rsat/data/genomes/Hordeum_vulgare.IBSCv2.36/genome/Hordeum_vulgare.IBSCv2.36.pep.all.fa
- ^ https://github.com/henni164/stem_rust_susceptibility/longest_transcript/perl
- ^ https://wheat-urgi.versailles.inra.fr/Seq-Repository/BLAST
- ^ https://ngphylogeny.fr/documentation
- ^ https://galaxy-web.ipk-gatersleben.de/
References
Acevedo-Garcia, J., Spencer, D., Thieron, H., Reinstädler, A., Hammond-Kosack, K., Phillips, A. L., et al. (2017). mlo-based powdery mildew resistance in hexaploid bread wheat generated by a non-transgenic TILLING approach. Plant Biotechnol. J. 15, 367–378. doi: 10.1111/pbi.12631
Ahn, I. P. (2007). Disturbance of the Ca(2+)/calmodulin-dependent signaling pathway is responsible for the resistance of Arabidopsis dnd1 against Pectobacterium carotovorum infection. Mol. Plant Pathol. 8, 747–759. doi: 10.1111/j.1364-3703.2007.00428.x
Alaux, M., Rogers, J., Letellier, T., Flores, R., Alfama, F., Pommier, C., et al. (2018). Linking the international wheat genome sequencing consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 19:111.
Alexa, A., and Rahnenfuhrer, J. (2020). topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.40.0.
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Ayliffe, M., Singh, D., Park, R., Moscou, M., and Pryor, T. (2013). Infection of Brachypodium distachyon with selected grass rust pathogens. Mol. Plant Microbe Interact. 26, 946–957. doi: 10.1094/mpmi-01-13-0017-r
Berghaus, R., and Reisener, H. J. (1984). Changes in photosynthesis of wheat plants infected with wheat stem rust (Puccinia graminis f. sp. tritici). Phytopathology 112, 165–172. doi: 10.1111/j.1439-0434.1985.tb04825.x
Bettgenhaeuser, J., Gardiner, M., Spanner, R., Green, P., Hernández-Pinzón, I., Hubbard, A., et al. (2018). The genetic architecture of colonization resistance in Brachypodium distachyon to non-adapted stripe rust (Puccinia striiformis) isolates. PLoS Genet. 14:e1007637. doi: 10.1371/journal.pgen.1007637
Bettgenhaeuser, J., Gilbert, B., Ayliffe, M., and Moscou, M. J. (2014). Nonhost resistance to rust pathogens- a continuation of continua. Front. Plant Sci. 5:664. doi: 10.3389/fpls.2014.00664
Bhattacharya, S. (2017). Deady new wheat disease threatens Europe’s crops. Nature 542, 145–146. doi: 10.1038/nature.2017.21424
Bouchez, O., Huard, C., Lorrain, S., Roby, D., and Balague, C. (2007). Ethylene is one of the key elements for cell death and defense response control in the Arabidopsis lesion mimic mutant vad1. Plant Physiol. 145, 465–477. doi: 10.1104/pp.107.106302
Bozkurt, T. O., McGrann, G. R., MacCormack, R., Boyd, L. A., and Akkaya, M. S. (2010). Cellular and transcriptional responses of wheat during compatible and incompatible race-specific interactions with Puccinia striiformis f. sp. tritici. Mol. Plant Pathol. 11, 625–640.
Bragg, J. N., Wu, J., Gordon, S. P., Guttman, M. E., Thilmony, R., Lazo, G. R., et al. (2012). Generation and characterization of the Western Regional Research Center Brachypodium T-DNA insertional mutant collection. PLoS One 7:e41916. doi: 10.1371/journal.pone.0041916
Büschges, R., Hollricher, K., Panstruga, R., Simons, G., Wolter, M., Frijters, A., et al. (1997). The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 88, 695–705. doi: 10.1016/s0092-8674(00)81912-1
Bushnell, W. R. (1984). “15 - Structural and physiological alterations in susceptible host tissue,” in The Cereal Rusts, eds W. R. Bushnell and A. P. Roelfs (Orlando, FL: Academic Press), 477–507. doi: 10.1016/b978-0-12-148401-9.50021-2
Chandra, S., Singh, D., Pathak, J., Kumari, S., Kumar, M., Poddar, R., et al. (2016). De novo assembled wheat transcriptomes delineate differentially expressed host genes in response to leaf rust. PLoS One 11:e0148453. doi: 10.1371/journal.pone.0148453
Chen, B., Jiang, J., and Zhou, X. (2007). A TOM1 homologue is required for multiplication of Tobacco mosaic virus in Nicotiana benthamiana. J. Zhejiang Univ. Sci. B 8, 256–259. doi: 10.1631/jzus.2007.b0256
Chen, J., Upadhyaya, N. M., Ortiz, D., Sperschneider, J., Li, F., Bouton, C., et al. (2017). Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 358, 1607–1610. doi: 10.1126/science.aao4810
Chen, L.-Q., Hou, B.-H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532.
Clough, S. J., Fengler, K. A., Yu, I. C., Lippok, B., Smith, R. K. Jr., and Bent, A. F. (2000). The Arabidopsis dnd1 “defense, no death” gene encodes a mutated cyclic nucleotide-gated ion channel. Proc. Natl. Acad. Sci. U.S.A. 97, 9323–9328. doi: 10.1073/pnas.150005697
Couto, D., and Zipfel, C. (2016). Regulation of pattern recognition receptor signaling in plants. Nat. Rev. Immunol. 16, 537–552. doi: 10.1038/nri.2016.77
Ding, P., and Ding, Y. (2020). Stories of salicylic acid: a plant defense hormone. Trends Plant Sci. 25, 549–565. doi: 10.1016/j.tplants.2020.01.004
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dobon, A., Bunting, D. C., Cabrera-Quio, L. E., Uauy, C., and Saunders, D. G. (2016). The host-pathogen interaction between wheat and yellow rust induces temporally coordinated waves of gene expression. BMC Genomics 17:380. doi: 10.1186/s12864-016-2684-4
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Duplessis, S., Cuomo, C. A., Lin, Y. C., Aerts, A., Tisserant, E., Veneault-Fourrey, C., et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. U.S.A. 108, 9166–9171. doi: 10.1073/pnas.1019315108
Eichmann, R., Bischof, M., Weis, C., Shaw, J., Lacomme, C., Schweizer, P., et al. (2010). BAX INHIBITOR-1 is required for full susceptibility of barley to powdery mildew. Mol. Plant Microbe Interact. 23, 1217–1227. doi: 10.1094/mpmi-23-9-1217
Ellis, J. G., Lagudah, E. S., Spielmeyer, W., and Dodds, P. N. (2014). The past, present and future of breeding rust resistant wheat. Front. Plant Sci. 5:641. doi: 10.3389/fpls.2014.00641
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238.
Engelhardt, S., Stam, R., and Hückelhoven, R. (2018). Good riddance? Breaking disease susceptibility in the era of new breeding technologies. Agronomy 8:114. doi: 10.3390/agronomy8070114
Eulgem, T., and Somssich, I. E. (2007). Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 10, 366–371. doi: 10.1016/j.pbi.2007.04.020
Figueroa, M., Alderman, S., Garvin, D. F., and Pfender, W. F. (2013). Infection of Brachypodium distachyon by formae speciales of Puccinia graminis: early infection events and host-pathogen incompatibility. PLoS One 8:e56857. doi: 10.1371/journal.pone.0056857
Figueroa, M., Castell-Miller, C. V., Li, F., Hulbert, S. H., and Bradeen, J. M. (2015). Pushing the boundaries of resistance: insights from Brachypodium-rust interactions. Front. Plant Sci. 6:558. doi: 10.3389/fpls.2015.00558
Flor, H. H. (1971). Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. doi: 10.1146/annurev.py.09.090171.001423
Gaj, T., Gersbach, C. A., and Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Garnica, D. P., Nemri, A., Upadhyaya, N. M., Rathjen, J. P., and Dodds, P. N. (2014). The ins and outs of rust haustoria. PLoS Pathog. 10:e1004329. doi: 10.1371/journal.ppat.1004329
Ge, X. T., Deng, W. W., Lee, Z. Z., Lopez-Ruiz, F. J., Schweizer, P., and Ellwood, S. R. (2016). Tempered mlo broad-spectrum resistance to barley powdery mildew in an Ethiopian landrace. Sci. Rep. 7:29558.
Genger, R. K., Jurkowski, G. I., McDowell, J. M., Lu, H., Jung, H. W., Greenberg, J. T., et al. (2008). Signaling pathways that regulate the enhanced disease resistance of Arabidopsis “defense, no death” mutants. Mol. Plant Microbe Interact. 21, 1285–1296. doi: 10.1094/mpmi-21-10-1285
Gilbert, B., Bettgenhaeuser, J., Upadhyaya, N., Soliveres, M., Singh, D., Park, R. F., et al. (2018). Components of Brachypodium distachyon resistance to nonadapted wheat stripe rust pathogens are simply inherited. PLoS Genet. 14:e1007636. doi: 10.1371/journal.pgen.1007636
Govrin, E. M., and Levine, A. (2000). The hypersensitive response facilitates plant infection by the necrotrophic pathogen Botrytis cinerea. Curr. Biol. 10, 751–757. doi: 10.1016/s0960-9822(00)00560-1
Harder, D. E., and Chong, J. (1984). “Structure and physiology of haustoria,” in The Cereal Rusts, Vol. 1, eds W. Bushnell and A. P. Roelfs (Orlando, FL: Academic Press), 416–460.
Helliwell, C., and Waterhouse, P. (2003). Constructs and methods for high-throughput gene silencing in plants. Methods 30, 289–295. doi: 10.1016/s1046-2023(03)00036-7
Howe, K. L., Contreras-Moreira, B., de Silva, N., Maslen, G., Akanni, W., Allen, J., et al. (2019). Ensembl genomes 2020—enabling non-vertebrate genomic research. Nucleic Acids Res. 48, D689–D695.
Huang, T. Y., Desclos-Theveniau, M., Chien, C. T., and Zimmerli, L. (2013). Arabidopsis thaliana transgenics overexpressing IBR3 show enhanced susceptibility to the bacterium Pseudomonas syringae. Plant Biol. (Stuttg) 15, 832–840.
Huttenhower, C., Hibbs, M., Myers, C., and Troyanskaya, O. G. (2006). A scalable method for integration and functional analysis of multiple microarray datasets. Bioinformatics 22, 2890–2897. doi: 10.1093/bioinformatics/btl492
Jia, H. G., Zhang, Y. Z., Orbovic, V., Xu, J., White, F. F., Jones, J. B., et al. (2017). Genome editing of the disease susceptibility gene CsLOB1 in citrus confers resistance to citrus canker. Plant Biotechnol. J. 15, 817–823. doi: 10.1111/pbi.12677
Jørgensen, J. H. (1992). Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. Euphytica 63, 141–152. doi: 10.1007/bf00023919
Jurkowski, G. I., Smith, R. K. Jr., Yu, I., Ham, J. H., Sharma, S. B., Klessig, D. F., et al. (2004). Arabidopsis DND2, a second cyclic nucleotide-gated ion channel gene for which mutation causes the “defense, no death” phenotype. Mol. Plant Microbe Interact. 17, 511–520. doi: 10.1094/mpmi.2004.17.5.511
Kellogg, E. A. (2001). Evolutionary history of the grasses. Plant Physiol. 125, 1198–1205. doi: 10.1104/pp.125.3.1198
Khafif, M., Balagué, C., Huard-Chauveau, C., and Roby, D. (2017). An essential role for the VASt domain of the Arabidopsis VAD1 protein in the regulation of defense and cell death in response to pathogens. PLoS One 12:e0179782. doi: 10.1371/journal.pone.0179782
Kim, D., Alptekin, B., and Budak, H. (2018). CRISPR/Cas9 genome editing in wheat. Funct. Integr. Genomics 18, 31–41. doi: 10.1007/s10142-017-0572-x
Konig, S., Feussner, K., Schwarz, M., Kaever, A., Iven, T., Landesfeind, M., et al. (2012). Arabidopsis mutants of sphingolipid fatty acid alpha-hydroxylases accumulate ceramides and salicylates. New Phytol. 196, 1086–1097. doi: 10.1111/j.1469-8137.2012.04351.x
Kretschmer, M., Damoo, D., Djamei, A., and Kronstad, J. (2020). Chloroplasts and plant immunity: where are the fungal effectors? Pathogens 9:19. doi: 10.3390/pathogens9010019
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., et al. (2011). The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210.
Lapin, D., and van den Ackerveken, G. (2013). Susceptibility to plant disease: more than a failure of host immunity. Trends Plant Sci. 18, 546–554. doi: 10.1016/j.tplants.2013.05.005
Lee, W. S., Rudd, J. J., and Kanyuka, K. (2015). Virus induced gene silencing (VIGS) for functional analysis of wheat genes involved in Zymoseptoria tritici susceptibility and resistance. Fungal Genet. Biol. 79, 84–88. doi: 10.1016/j.fgb.2015.04.006
Lemoine, F., Correia, D., Lefort, V., Doppelt-Azeroual, O., Mareuil, F., Cohen-Boulakia, S., et al. (2019). NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265.
Lo Presti, L., Lanver, D., Schweizer, G., Reissman, S., and Kahmann, R. (2015). Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 66, 513–545. doi: 10.1146/annurev-arplant-043014-114623
Lorrain, S., Lin, B., Auriac, M. C., Kroj, T., Saindrenan, P., Nicole, M., et al. (2004). Vascular associated death1, a novel GRAM domain-containing protein, is a regulator of cell death and defense responses in vascular tissues. Plant Cell 16, 2217–2232. doi: 10.1105/tpc.104.022038
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Low, Y. C., Lawton, M. A., and Di, R. (2020). Validation of barley 2OGO gene as a functional orthologue of Arabidopsis DMR6 gene in Fusarium head blight susceptibility. Sci. Rep. 10:9935.
Luig, N. H., and Watson, I. A. (1972). The role of wild and cultivated grasses in the hybridization of formae speciales of Puccinia graminis. Aust. J. Biol. Sci. 25, 335–342. doi: 10.1071/bi9720335
Luo, M., Li, H., Chakraborty, S., Morbitzer, R., Rinaldo, A., Upadhyaya, N., et al. (2019). Efficient TALEN-mediated gene editing in wheat. Plant Biotechnol. J. 17, 2026–2028. doi: 10.1111/pbi.13169
Manickavelu, A., Kawaura, K., Oishi, K., Shin-I, T., Kohara, Y., and Yahiaoui, N. (2010). Comparative gene expression analysis of susceptible and resistant near-isogenic lines in common wheat infected by Puccinia triticina. DNA Res. 17, 211–222. doi: 10.1093/dnares/dsq009
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
McIntosh, R., Wellings, C., and Park, R. (1995). Wheat Rusts: An Atlast of Resistance Genes. Canberra, ACT: CSIRO.
Mitchell, A. G., and Martin, C. E. (1997). Fah1p, a Saccharomyces cerevisiae cytochrome b5 fusion protein, and its Arabidopsis thaliana homolog that lacks the cytochrome b5 domain both function in the α−hydroxylation of sphingolipid-associated very long chain fatty acids. J. Biol. Chem. 272, 28281–28288. doi: 10.1074/jbc.272.45.28281
Moerschbacher, B. M., Vander, P., Springer, C., Noll, U., and Schmittmann, G. (1994). Photosynthesis in stem rust-infected, resistant and susceptible near-isogenic wheat leaves. Can. J. Bot. 72, 990–997. doi: 10.1139/b94-124
Nagano, M., Takahara, K., Fujimoto, M., Tsutsumi, N., Uchimiya, H., and Kawai-Yamada, M. (2012). Arabidopsis sphingolipid fatty acid 2-hydroxylases (AtFAH1 and AtFAH2) are functionally differentiated in fatty acid 2-hydroxylation and stress responses. Plant Physiol. 159, 1138–1148. doi: 10.1104/pp.112.199547
Nekrasov, V., Wang, C., Win, J., Lanz, C., Weigel, D., and Kamoun, S. (2017). Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci. Rep. 7:482.
Olivera, P., Newcomb, M., Szabo, L. J., Rouse, M., Johnson, J., Gale, S., et al. (2015). Phenotypic and genotypic characterization of race TKTTF of Puccinia graminis f. sp. tritici that caused a wheat stem rust epidemic in Southern Ethiopia in 2013–14. Phytopathology 105, 917–928. doi: 10.1094/phyto-11-14-0302-fi
Omidvar, V., Dugyala, S., Li, F., Rottschaefer, S. M., Miller, M. E., Ayliffe, M., et al. (2018). Detection of race-specific resistance against Puccinia coronata f. sp. avenae in Brachypodium species. Phytopathology 108, 1443–1454. doi: 10.1094/phyto-03-18-0084-r
Paradis, E., and Schliep, K. (2018). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Periyannan, S., Milne, R. J., Figueroa, M., Lagudah, E. S., and Dodds, P. N. (2017). An overview of genetic rust resistance: from broad to specific mechanisms. PLoS Pathog. 13:e1006380. doi: 10.1371/journal.ppat.1006380
Pessina, S., Pavan, S., Catalano, D., Gallotta, A., Visser, R. G. F., Bai, Y., et al. (2014). Characterization of the MLO gene family in Rosaceae and gene expression analysis in Malus domestica. BMC Genomics 15:618. doi: 10.1186/1471-2164-15-618
Peterson, P. D. (2001). Stem Rust of Wheat: From Ancient Enemy to Modern Foe. St. Paul, MN: American Phytopathological Society.
Petre, B., Joly, D. L., and Duplessis, S. (2014). Effector proteins of rust fungi. Front. Plant Sci. 5:416. doi: 10.3389/fpls.2014.00416
Pretorius, Z. A., Singh, R. P., Wagoire, W. W., and Payne, T. S. (2000). Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis f. sp. tritici in Uganda. Plant Dis. 84:203. doi: 10.1094/pdis.2000.84.2.203b
Rate, D. N., and Greenberg, J. T. (2001). The Arabidopsis aberrant growth and death2 mutant shows resistance to Pseudomonas syringae and reveals a role for NPR1 in suppressing hypersensitive cell death. Plant J. 27, 203–211. doi: 10.1046/j.0960-7412.2001.1075umedoc.x
Robin, G. P., Kleemann, J., Neumann, U., Cabre, L., Dallery, J. F., Lapalu, N., et al. (2018). Subcellular localization screening of Colletotrichum higginsianum effector candidates identifies fungal proteins targeted to plant peroxisomes, Golgi bodies, and microtubules. Front. Plant Sci. 9:562. doi: 10.3389/fpls.2018.00562
Rushton, P. J., Somssich, I. E., Ringler, P., and Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15, 247–258.
Salcedo, A., Rutter, W., Wang, S., Akhunova, A., Bolus, S., Chao, S., et al. (2017). Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 358, 1604–1606. doi: 10.1126/science.aao7294
Schaefer, R. J., Michno, J. M., Jeffers, J., Hoekenga, O., Dilkes, B., Baxter, I., et al. (2018). Integrating coexpression networks with GWAS to prioritize causal genes in maize. Plant Cell 30, 2922–2942. doi: 10.1105/tpc.18.00299
Singh, R. P., Hodson, D. P., Jin, Y., Lagudah, E. S., Ayliffe, M. A., Bhavani, S., et al. (2015). Emergence and spread of new races of wheat stem rust fungus: continued threat to food security and prospects of genetic control. Phytopathology 105, 872–884. doi: 10.1094/phyto-01-15-0030-fi
Song, J. T., Lu, H., and Greenberg, J. T. (2004). Divergent roles in Arabidopsis thaliana development and defense of two homologous genes, aberrant growth and death2 and AGD2-LIKE DEFENSE RESPONSE PROTEIN1, encoding novel aminotransferases. Plant Cell 16, 353–366. doi: 10.1105/tpc.019372
Staples, R., and Macko, V. (1984). “Germination of urediospores and differentiation of infection structures,” in The Cereal Rusts, eds W. Bushnell and A. P. Roelfs (New York, NY: Academic Press), 255–289. doi: 10.1016/b978-0-12-148401-9.50015-7
Steffenson, B. J., Case, A. J., Pretorius, Z. A., Coetzee, V., Kloppers, F. J., Zhou, H., et al. (2017). Vulnerability of barley to African pathotypes of Puccinia graminis f. sp. tritici and sources of resistance. Phytopathology 107, 950–962. doi: 10.1094/phyto-11-16-0400-r
Sun, K., van Tuinen, A., van Kan, J. A. L., Wolters, A.-M. A., Jacobsen, E., Visser, R. G. F., et al. (2017). Silencing of DND1 in potato and tomato impedes conidial germination, attachment and hyphal growth of Botrytis cinerea. BMC Plant Biol. 17:235. doi: 10.1186/s12870-017-1184-2
Sun, K., Wolters, A.-M. A., Vossen, J. H., Rouwet, M. E., Loonen, A. E. H. M., Jacobsen, E., et al. (2016). Silencing of six susceptibility genes results in potato late blight resistance. Transgenic Res. 25, 731–742. doi: 10.1007/s11248-016-9964-2
Su’udi, M., Kim, M. G., Park, S. R., Hwang, D. J., Bae, S. C., and Ahn, I. P. (2011). Arabidopsis cell death in compatible and incompatible interactions with Alternaria brassicicola. Mol. Cells 31, 593–601. doi: 10.1007/s10059-011-2203-z
Upadhyaya, N. M., Mago, R., Panwar, V., Hewitt, T., Luo, M., and Chen, J., et al. (in press). Genomics accelerated isolation of a new stem rust avirulence gene - wheat resistance gene pair. Nat. Plants
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., and Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646. doi: 10.1038/nrg2842
van Damme, M., Andel, A., Huibers, R. P., Panstruga, R., Weisbeek, P. J., and Van den Ackerveken, G. (2005). Identification of Arabidopsis loci required for susceptibility to the downy mildew pathogen Hyaloperonospora parasitica. Mol. Plant Microbe Interact. 18, 583–592. doi: 10.1094/mpmi-18-0583
van Damme, M., Huibers, R. P., Elberse, J., and Van den Ackerveken, G. (2008). Arabidopsis DMR6 encodes a putative 2OG-Fe(II) oxygenase that is defense-associated but required for susceptibility to downy mildew. Plant J. 54, 785–793. doi: 10.1111/j.1365-313x.2008.03427.x
van Schie, C. C., and Takken, F. L. (2014). Susceptibility genes 101: how to be a good host. Annu. Rev. Phytopathol. 52, 551–581. doi: 10.1146/annurev-phyto-102313-045854
Vogel, J., and Hill, T. (2008). High-efficiency Agrobacterium-mediated transformation of Brachypodium distachyon inbred line Bd21-3. Plant Cell Rep. 27, 471–478. doi: 10.1007/s00299-007-0472-y
Wang, X., Tang, C., Huang, X., Li, F., Chen, X., Zhang, G., et al. (2012). Wheat BAX inhibitor-1 contributes to wheat resistance to Puccinia striiformis. J. Exp. Bot. 63, 4571–4584. doi: 10.1093/jxb/ers140
Wang, Y., Cheng, X., Shan, Q., Zhang, Y., Liu, J., Gao, C., et al. (2014). Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 32, 947–951. doi: 10.1038/nbt.2969
Waterhouse, P., and Helliwell, C. (2003). Exploring plant genomes by RNA-induced gene silencing. Nat. Rev. Genet. 4, 29–38. doi: 10.1038/nrg982
Wildermuth, M. C. (2010). Modulation of host nuclear ploidy: a common plant biotroph mechanism. Curr. Opin. Plant Biol. 13, 449–458. doi: 10.1016/j.pbi.2010.05.005
Yadav, I. S., Sharma, A., Kaur, S., Nahar, N., Bhardwaj, S. C., Sharma, T. R., et al. (2016). Comparative temporal transcriptome profiling of wheat near isogenic line carrying Lr57 under compatible and incompatible interactions. Front. Plant Sci. 7:1943. doi: 10.3389/fpls.2016.01943
Yu, G., Smith, D. K., Zhu, H., Guan, Y., and Lam, T. T.-Y. (2017). ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210x.12628
Yu, I., Fengler, K. A., Clough, S. J., and Bent, A. F. (2000). Identification of Arabidopsis mutants exhibiting an altered hypersensitive response in gene-for-gene disease resistance. Mol. Plant Microbe Interact. 13, 277–286. doi: 10.1094/mpmi.2000.13.3.277
Zaidi, S. S., Mukhtar, M. S., and Mansoor, S. (2018). Genome editing: targeting susceptibility genes for plant disease resistance. Trends Biotechnol. 36, 898–906. doi: 10.1016/j.tibtech.2018.04.005
Zambino, P. J., Kubelik, A. R., and Szabo, L. J. (2000). Gene action and linkage of avirulence genes to DNA markers in the rust fungus Puccinia graminis. Phytopathology 90, 819–826. doi: 10.1094/phyto.2000.90.8.819
Zeilmaker, T., Ludwig, N. R., Elberse, J., Seidl, M. F., Berke, L., Van Doorn, A., et al. (2015). DOWNY MILDEW RESISTANT 6 and DMR6-LIKE OXYGENASE 1 are partially redundant but distinct suppressors of immunity in Arabidopsis. Plant J. 81, 210–222. doi: 10.1111/tpj.12719
Zhang, H., Yang, Y., Wang, C., Liu, M., Li, H., Fu, Y., et al. (2014). Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics 15:898. doi: 10.1186/1471-2164-15-898
Zhang, Y., Zhao, L., Zhao, J., Li, Y., Wang, J., Guo, R., et al. (2017). S5H/DMR6 encodes a salicylic acid 5-hydroxylase that fine-tunes salicylic acid homeostasis. Plant Physiol. 175, 1082–1093. doi: 10.1104/pp.17.00695
Keywords: susceptibility, rust (disease), wheat, transcription, co-expression, non-host
Citation: Henningsen EC, Omidvar V, Della Coletta R, Michno J-M, Gilbert E, Li F, Miller ME, Myers CL, Gordon SP, Vogel JP, Steffenson BJ, Kianian SF, Hirsch CD and Figueroa M (2021) Identification of Candidate Susceptibility Genes to Puccinia graminis f. sp. tritici in Wheat. Front. Plant Sci. 12:657796. doi: 10.3389/fpls.2021.657796
Received: 24 January 2021; Accepted: 22 March 2021;
Published: 21 April 2021.
Edited by:
Zuhua He, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Urmil Bansal, The University of Sydney, AustraliaJin-Ying Gou, Fudan University, China
Copyright © 2021 Henningsen, Omidvar, Della Coletta, Michno, Gilbert, Li, Miller, Myers, Gordon, Vogel, Steffenson, Kianian, Hirsch and Figueroa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cory D. Hirsch, Y2RoaXJzY2hAdW1uLmVkdQ==; Melania Figueroa, bWVsYW5pYS5maWd1ZXJvYUBjc2lyby5hdQ==
†Present address: Marisa E. Miller, Pairwise Plants, Durham, NC, United States Sean P. Gordon, Roche, Albany, CA, United States