 Harsh Raman
Harsh Raman Rosy Raman
Rosy Raman Yu Qiu
Yu Qiu Yuanyuan Zhang
Yuanyuan Zhang Jacqueline Batley
Jacqueline Batley Shengyi Liu
Shengyi Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 12 May 2021
Sec. Plant Breeding
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.654604
Canola exhibits an extensive genetic variation for resistance to blackleg disease, caused by the fungal pathogen Leptosphaeria maculans. Despite the identification of several Avr effectors and R (race-specific) genes, specific interactions between Avr-R genes are not yet fully understood in the Brassica napus–L. maculans pathosystem. In this study, we investigated the genetic basis of resistance in an F2:3 population derived from Australian canola varieties CB-Telfer (Rlm4)/ATR-Cobbler (Rlm4) using a single-spore isolate of L. maculans, PHW1223. A genetic linkage map of the CB-Telfer/ATR-Cobbler population was constructed using 7,932 genotyping-by-sequencing-based DArTseq markers and subsequently utilized for linkage and haplotype analyses. Genetic linkage between DArTseq markers and resistance to PHW1223 isolate was also validated using the B. napus 60K Illumina Infinium array. Our results revealed that a major locus for resistance, designated as Rlm13, maps on chromosome C03. To date, no R gene for resistance to blackleg has been reported on the C subgenome in B. napus. Twenty-four candidate R genes were predicted to reside within the quantitative trait locus (QTL) region. We further resequenced both the parental lines of the mapping population (CB-Telfer and ATR-Cobbler, > 80 × coverage) and identified several structural sequence variants in the form of single-nucleotide polymorphisms (SNPs), insertions/deletions (InDels), and presence/absence variations (PAVs) near Rlm13. Comparative mapping revealed that Rlm13 is located within the homoeologous A03/C03 region in ancestral karyotype block “R” of Brassicaceae. Our results provide a “target” for further understanding the Avr–Rlm13 gene interaction as well as a valuable tool for increasing resistance to blackleg in canola germplasm.
Blackleg, caused by the hemibiotrophic fungal pathogen Leptosphaeria maculans (Desmaz.) Ces. et de Not., is one of the highly widespread and devastating diseases of canola (Brassica napus L) and its relatives (Howlett, 2004; Rouxel and Balesdent, 2017). It continues to be a major threat to the sustainable production of canola across many parts of the world, particularly Australia, Europe, and North America (West et al., 2001; Fitt et al., 2006). Host resistance, qualitative (race-specific resistance mediated by R genes) and quantitative resistance [non-race-specific resistance mediated by quantitative trait loci (QTL)], is considered as the most effective and environmentally safe method of disease management. Since the 1970s, concerted efforts are being made in developing improved varieties of canola with resistance to L. maculans by accessing genetic variation from diploid and amphidiploid members of the Brassicaceae (Roy, 1984; Chèvre et al., 1997; Saal and Struss, 2005; Yu et al., 2012).
Qualitative resistance largely relies on the classical gene-to-gene hypothesis: avirulence/effector (Avr)–R gene recognition interaction (Flor, 1971), also called elicitor-triggered immunity (ETI, Jones and Dangl, 2006). In Brassica species, at least 18 genes, conferring “complete” resistance to blackleg, have been identified on the Ar, An, Bn/Bj, and Co subgenomes of Brassica (Delourme et al., 2011; Raman et al., 2013, 2016a; Ferdous et al., 2019), while in L. maculans, at least eight Avr genes have been cloned (Gout et al., 2006; Fudal et al., 2007; Parlange et al., 2009; Ghanbarnia et al., 2012, 2018; Balesdent et al., 2013; Van de Wouw et al., 2014; Ghanbarnia et al., 2015; Plissonneau et al., 2016, 2018), which correspond to at least nine of the R genes (Ghanbarnia et al., 2018). To date, none of the blackleg R genes have been reported on the Cn subgenome of B. napus.
Both linkage and genome-wide association mapping approaches have been employed to identify loci having major and minor allelic effects for resistance to blackleg. Although R genes do not provide long-term disease control due to the adaptive nature of L. maculans (Rouxel et al., 2003; Li et al., 2006; Sprague et al., 2006), they are easy to manipulate in the breeding programs due to their simple (predominantly monogenic) inheritance and high additive genetic variance. Therefore, new sources of resistance are highly sought after by canola breeding programs to increase diversity in the resistance gene pool for commercial deployment into new varieties.
Understanding the genetic network involved in Avr–R gene interactions is crucial for the deployment of effective blackleg management strategies. However, investigating the genetic effects for race-specific R genes in field conditions, as well as in the ascospore shower test (with stubble collected from the field) under controlled conditions, is challenging due to the highly heterogenous nature of L. maculans populations, reliance on the environment (timing of spore release and weather conditions), method of evaluation, and genotype × environment interactions (Hayden et al., 2007). The L. maculans–B. napus system has also revealed several interesting R–Avr interactions, such as AvrLm4-7/Rlm4 and Rlm7 (Parlange et al., 2009) and AvrLm1/Rlm1 and LepR3 (Larkan et al., 2013). Recently, epistatic R–Avr interactions for AvrLm3/Rlm3/AvrLm4-7 (Plissonneau et al., 2016) and AvrLm5-9/Rlm9/AvrLm4-7 (Ghanbarnia et al., 2018) have also been reported. To gain an understanding of “novel” genes involved in resistance, we analyzed an intercross population derived from the cross, CB-Telfer (Rlm4)/ATR-Cobbler (Rlm4).
In this study, we identify a major locus, designated Rlm13, on chromosome C03 that accounts for genetic variation in resistance to L. maculans in an Australian canola population derived from the CB-Telfer/ATR-Cobbler. We further resequenced both the parental lines of the mapping population (CB-Telfer and ATR-Cobbler) and identified several structural sequence variants in the form of single-nucleotide polymorphisms (SNPs), insertions/deletions (InDels), and presence/absence variations (PAVs) near the Rlm13. Comparative mapping revealed that the Rlm13 region is localized within the “R” ancestral block of Brassicaceae. Our results provide a novel “target” for further understanding Avr–R gene interactions, as well as a valuable tool for combining resistance loci in new varieties.
For linkage mapping, we developed an F2 population from a single F1 plant derived from the cross CB-Telfer/ATR-Cobbler. CB-Telfer is a current, homozygous Australian commercial variety, developed by Canola Breeders Western Australia Pty Ltd (CBWA, now NPZA, South Perth, Western Australia), while ATR-Cobbler (NMT040) is a current commercial canola variety developed by Nuseed Pty Ltd, Horsham, Victoria, Australia from the cross of ATR-Eyre/Ag-Emblem1. Both parental lines carry R genes for resistance to blackleg: CB-Telfer (Rlm4) and ATR-Cobbler [Rlm4 and Rlm9 (H; segregating for resistance)] (Marcroft et al., 2012).
To confirm specific Avr–R interaction between the parental lines of the mapping population, we utilized a differential set of single-spore isolates of L. maculans, which were previously characterized for avirulence (Marcroft et al., 2012). Isolates were procured from Marcroft Grains Pathology Pty Ltd, Horsham, Australia, and then multiplied at the Wagga Wagga Agricultural Institute (WWAI), NSW, Australia. Pycnidiospores were used for the cotyledon inoculation tests. The PHW1223 (AvrLm5-6-8-9-S) was the only isolate that showed a differential response between the parental lines (Supplementary Table 1); we used this isolate to phenotype F2 and F2:3 derived progenies from the CB-Telfer/ATR-Cobbler.
A total of 464 F2 lines, two parental lines, and five control cultivars [Westar (no R gene), AV-Garnet (Rlm1 and Rlm9), Thunder TT (Rlm4), Charlton (Rlm4), and Caiman (Rlm7)] were evaluated for resistance to L. maculans under glasshouse conditions. The seedlings were grown in plastic trays (7 × 8 cells) in a controlled glasshouse at WWAI, Australia, and maintained at 20 ± 2°C. Each tray had a row of eight genotypes, representing either F2/parental or control cultivars. The cotyledon lobes of each plant were inoculated with the PHW1223 isolate, at a concentration of 107 spores ml–1 as described previously (Raman et al., 2012). Briefly, 10-days-old plants were inoculated and then placed in a humidity chamber for 48 h at 100% relative humidity in the dark at 18°C to allow spore germination and further penetration into canola cells. Subsequently, inoculated plants were transferred to the glasshouse and maintained at 20 ± 2°C for 3–4 weeks. Disease symptoms on cotyledons were assessed at 18 days post inoculation, using a scale described previously (Koch et al., 1991): 0 (resistant response/no disease development) to 9 (highly susceptible/and profuse sporulation). Quantitative disease scores (0–9) were used to identify significant trait–marker associations for resistance. After phenotyping F2 lines for resistance, each line was transplanted individually into a small pot (70 mm square tubes, Garden City Plastics, Australia) and subsequently raised under glasshouse conditions.
Both parental lines, as well as each individual F2 plant from the CB-Telfer/ATR-Cobbler cross, were bagged at the flower bud initiation stage to generate selfed F2:3 families. To confirm the disease scores of F2 plants, 96 F2:3 families were randomly selected and assessed for response to infection using a cotyledon assay with the PHW1223 isolate. Eight seedlings per F2:3 derived family were grown in plastic trays (7 × 8 cells), accommodating seven accessions in each tray. Phenotyping and assessment for resistance to L. maculans were carried-out as described above.
DNA was isolated from young leaves from the same parental and F2 lines that were evaluated for resistance to PHW1223 using a standard cetyltrimethylammonium bromide (CTAB) method. DNA samples were genotyped with DArTseq (Raman et al., 2014) and 60K Illumina Infinium SNP markers (Clarke et al., 2016). SNP genotypes generated from the 60K Illumina Infinium were scored using the Genotyping Module of Genome Studio Data Analysis software (Illumina Inc., San Diego, United States). Polymorphic DArTseq SNPs and presence/absence markers were determined by aligning the Illumina reads with the sequenced genomes of B. napus cv. Darmor-bzh version 4.1 (Chalhoub et al., 2014), after trimming the barcodes and using Bowtie version 0.12 as described previously (Raman et al., 2016b). Markers having ≥ 90% call rate and < 10% missing data were used for linkage analysis.
In order to confirm the presence of Rlm9 in the population, all individuals were genotyped using Rlm9-specific primers (Rlm9F1: 5′-TCGTATAGTTCTTATCGCCTGCC-3′, Rlm9R1: 5′-TCCGTAAGTCAGGCTATAGTGT-3′). Amplification was performed using DreamTaq Green PCR Master Mix (2X, Thermo Fisher Scientific) with the reaction mixture prepared following the manufacturer’s protocol. The cycling conditions were initial denaturation at 95°C for 3 min, 35 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s, with a final elongation of 5 min. PCR products were visualized on an agarose gel.
A set of 7,932 DArTseq markers that showed segregation among DH lines were selected for genetic linkage map construction, as detailed by Raman et al. (2016b). Map distance in centimorgan (cM) was calculated using the Kosambi mapping function. A genome scan was performed to identify any associations for resistance to blackleg using the linear marker analysis algorithm, implemented in the SVS package2. The allele frequency of SNP markers and their association with mean disease scores (from F2 lines) were determined by using linear marker regression, F-test, and correlation/trend test, implemented in SVS. For reducing Type 1 and Type II errors, multiple testing corrections for Bonferroni adjustment (on SNPs), false discovery rate, and full genome scan permutations (5,000 iterations) were conducted in the SVS package. We further performed haplotype association analysis using the expectation/maximization (EM) method following 1,000 iterations, a dynamic window size of 5 kb, and an EM convergence tolerance value of 0.0001. Trait–marker association was graphically represented using the MapChart software (Voorrips, 2002).
To determine the position of the Rlm13 locus in relation to the previously reported QTL for resistance to blackleg (Raman et al., 2016a, 2018), we performed comparative mapping of marker sequences underlying QTL using the Darmor-bzh reference genome of B. napus version 4.1 (Chalhoub et al., 2014). If the physical positions of the genomic region (QTL interval) and Rlm13 overlapped (within 50 kb) on C03, the genomic region was considered to be the same. Since the LepR4 locus for resistance to blackleg is mapped on chromosome A06 (Yu et al., 2013) and the latter showed synteny with the homoeologous chromosomes A03/C03, we searched the sequences of marker intervals encompassing LepR4.
The proximity of candidate genes to identified associations was inferred based on the functional annotation of the Arabidopsis thaliana genome and implemented in the reference sequenced genome of “Darmor-bzh.” Sequences were searched for their identities with the disease resistance NLR-R genes (Chalhoub et al., 2014; Alamery et al., 2018; Tirnaz et al., 2020) using the RGAugury function using the B. napus genome3 to obtain their physical map positions.
Whole-genome resequencing (2 × 150 bp) of both parental lines, CB-Telfer and ATR-Cobbler, was performed at the Novogene facility (Novogene Co., Ltd, Hong Kong) using the Illumina HiSeq 2000 sequencing platform. The coverage of the parental lines ranged from 83.7 × (94.6 Gb, ATR-Cobbler) to 90.1 × (102.6 Gb, CB-Telfer). Reads were mapped onto the “Darmor-bzh” reference assembly (version 4.1) using BWA version 0.7.8 (Li and Durbin, 2010). SNP and InDel calling based on the short-read alignment data were performed using the GATK haplotype caller (version 3.5).
Genomic structural variation (SV) detection for two parental lines was performed using novoBreak (Chong et al., 2017) compared against the Darmor-bzh reference sequence (version 4.1). SVs inferred by no less than 10 reads were further filtered with the following conditions: (1) more than five supporting split reads or (2) no fewer than three discordant read pairs. We detected deletions, duplications, and inversions with sizes between 100 bp and 1 Mb. Two adjacent SVs were identified as the same SV if their start and end positions varied no more than 1 kb, and the overlapping region was more than 50% of the total size of that two SVs.
To identify the HE events between An and Cn subgenomes in each of the two parental lines, we followed a standard protocol, described earlier with minor parameter optimization (Chalhoub et al., 2014). First, the resequenced reads for each line were aligned to the Darmor-bzh reference genome version 4.1 by using BWA with default parameters (Li and Durbin, 2010). Second, successive 10-kb windows are scanned to calculate the average depth on the whole genome. If the coverage depth is more than 1.5-fold of the average coverage of the whole genome, this was defined as a double coverage. By contrast, if the coverage depth is less than half of the average coverage of the whole genome, it would be defined as a deletion coverage. Adjacent windows with depths greater than the threshold and that were at most 10 windows distant were linked together. Only the regions spanning more than 10 windows (100 kb total) were retained as HE regions. Finally, the read coverage depth was used to indicate HE, where regions with double-read coverage were considered duplicated while regions with low or no coverage in its corresponding syntenic region of homoeologous subgenomes were considered to be deleted/replaced.
We further sought to identify whether marker intervals associated with Rlm13 in CB-Telfer/ATR-Cobbler are located within the same duplicated regions on chromosome A03/C03 for resistance to L. maculans that were detected previously under field and glasshouse conditions (Fomeju et al., 2014; Fopa Fomeju et al., 2015; Raman et al., 2016a, 2018; Kumar et al., 2018). The marker sequences associated with Rlm13 were searched for their identities using the reference B. napus cv. Darmor-bzh and ancestral karyotype blocks of A. thaliana (TAIR104), which is considered as the pre-triplication ancestor of B. napus. The co-location between NLR gene/marker sequences underlying QTL/synteny (AK) and physical positions of Rlm13 were analyzed using Microsoft Excel.
To confirm the specific Avr–R interactions in parental lines of a mapping population, CB-Telfer (Rlm4) and ATR-Cobbler (Rlm4, heterogenous), we utilized a differential set of L. maculans isolates. The PHW1223 (AvrLm5-6-8-9-S) was the only isolate that revealed a contrasting disease response between parents (Supplementary Table 1). Inferred R gene profiles from a differential set of isolates suggested that ATR-Cobbler likely possesses Rlm9, consistent with earlier findings (Marcroft et al 2012). Recently, the Rlm9 gene (BnaA07g20220D), encoding the wall-associated kinase-like (WAKL) protein, has been cloned in B. napus (Larkan et al., 2020); we resequenced this gene from both the parental lines CB-Telfer and ATR-Cobbler and compared it with the reference Darmor-bzh genome sequence version 4.1 (Supplementary Table 2). There were three identical SNPs in exon 1 at the coordinates 15,912,895 bp (C > T), 15,912,904 bp (T > C), and 15,912,927 bp (C > A) in both parental lines compared to the Darmor, which harbors Rlm9 (Delourme et al., 2004). Sequence analysis suggests that the Rlm9 allele present in the parental lines does not contribute to variation in resistance in this mapping population. Both parents contained a susceptible allele, along with all progeny when tested with Rlm9 gene-specific primers.
To understand the genetic basis underlying the differential disease expression response to the PHW1223 isolate (Supplementary Table 1), we phenotyped a total of 464 F2 lines from CB-Telfer/ATR-Cobbler. Upon infection, ATR-Cobbler displayed a low cotyledon lesion score (1) for resistance, whereas the CB-Telfer had a high disease lesion score of (8) for susceptibility (Figure 1). The cotyledons of resistant genotypes exhibited a classical hypersensitive response with limited necrosis but no sporulation, while the susceptible parental lines showed extensive cell death and sporulation. Phenotypic evaluation of 464 F2 plants exhibited a bimodal distribution of disease scores to isolate PHW1223 (Figure 2). A Chi-square test (χ2 = 0.74; P0.05 = 0.39) confirmed that a single locus controls resistance in this F2 population, as the allelic ratio conforms to a 3 (340 resistant plants)-to-1 (116 susceptible plants) segregation. The observed response is consistent with the highly specific interaction of a pathogen effector (Avr protein) and host resistance (R) protein, for the gene-for-gene complementarity hypothesis (Flor, 1971; Keen, 1990; Jones and Dangl, 2006). In the F2 population, homozygous resistant genotypes could not be discriminated conclusively, based on the extent of cotyledon infection, from heterozygous resistant ones, while those which lacked a resistance allele developed large lesions on the inoculated cotyledons. To verify the disease scores of each individual F2 line, we randomly selected 96 lines from 464 F3 families. Of these, 30 were homozygous resistant, 42 were heterozygous (segregation for both resistance and susceptibility), and 24 were homozygous susceptible. No segregation was observed within susceptible F3 families, suggesting that phenotypic conditions for disease expression were optimal. The data on F2:3 families fitted in a codominant monogenic segregation ratio (χ2 = 2.25, P0.05 = 0.32), indicating that a single gene controls variation in resistance to isolate PHW1223.

Figure 1. Infection response of parental lines CB-Telfer (maternal, Rlm4) and ATR-Cobbler (paternal, Rlm4 and Rlm9) to the L. maculans isolate PHW1223 (AvrLm5-6-8-9-S). The visible symptoms of blackleg commenced after 7–10 days of inoculation. Resistant parental and intercross lines displayed a limited cotyledon lesion compared to susceptible lines, which displayed extensive cotyledon lesions, characterized by discoloration and pyncindia sporulation (after 21 days of inoculation).
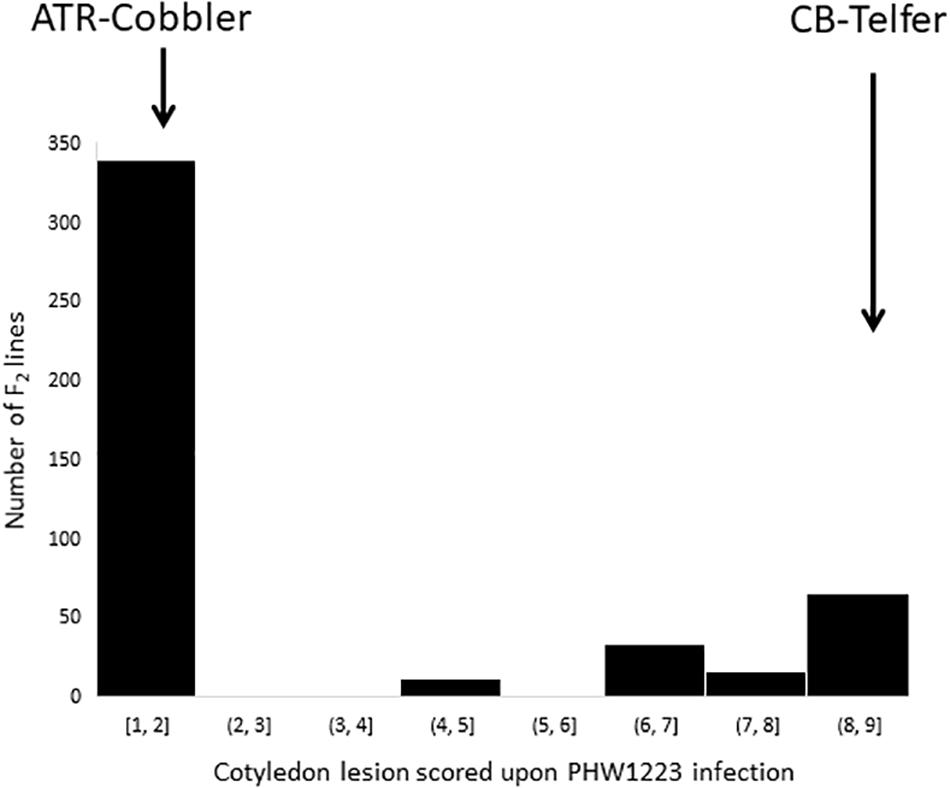
Figure 2. Frequency distributions of the cotyledon lesion scores in an F2 population derived from the CB-Telfer/ATR-Cobbler. Plants were infected with a single-spore isolate, PHW1223, at the cotyledon stage. Parental means are shown with arrows.
To locate the locus for race-specific resistance in ATR-Cobbler, we constructed a linkage map of the genetic population derived from CB-Telfer/ATR-Cobbler. A total of 7,932 polymorphic DArTseq markers that showed segregation in the F2 population were utilized for map construction. These markers were grouped into 21 linkage groups, representing the 19 haploid canola chromosomes and unassigned ChrA_random and ChrC_random linkage groups (Table 1). The total length of the genetic map was 2,252.22 cM, with an average coverage of one marker per 0.28 cM. Of the 19 chromosomes of B. napus, A06 had the highest marker density (4.42/cM), while chromosome C01 had the lowest marker coverage and C05 had the lowest marker density (2.37/cM). The majority of the DArTseq markers (75%) could be anchored on the reference “Darmor-bzh” genome (Supplementary Table 3).
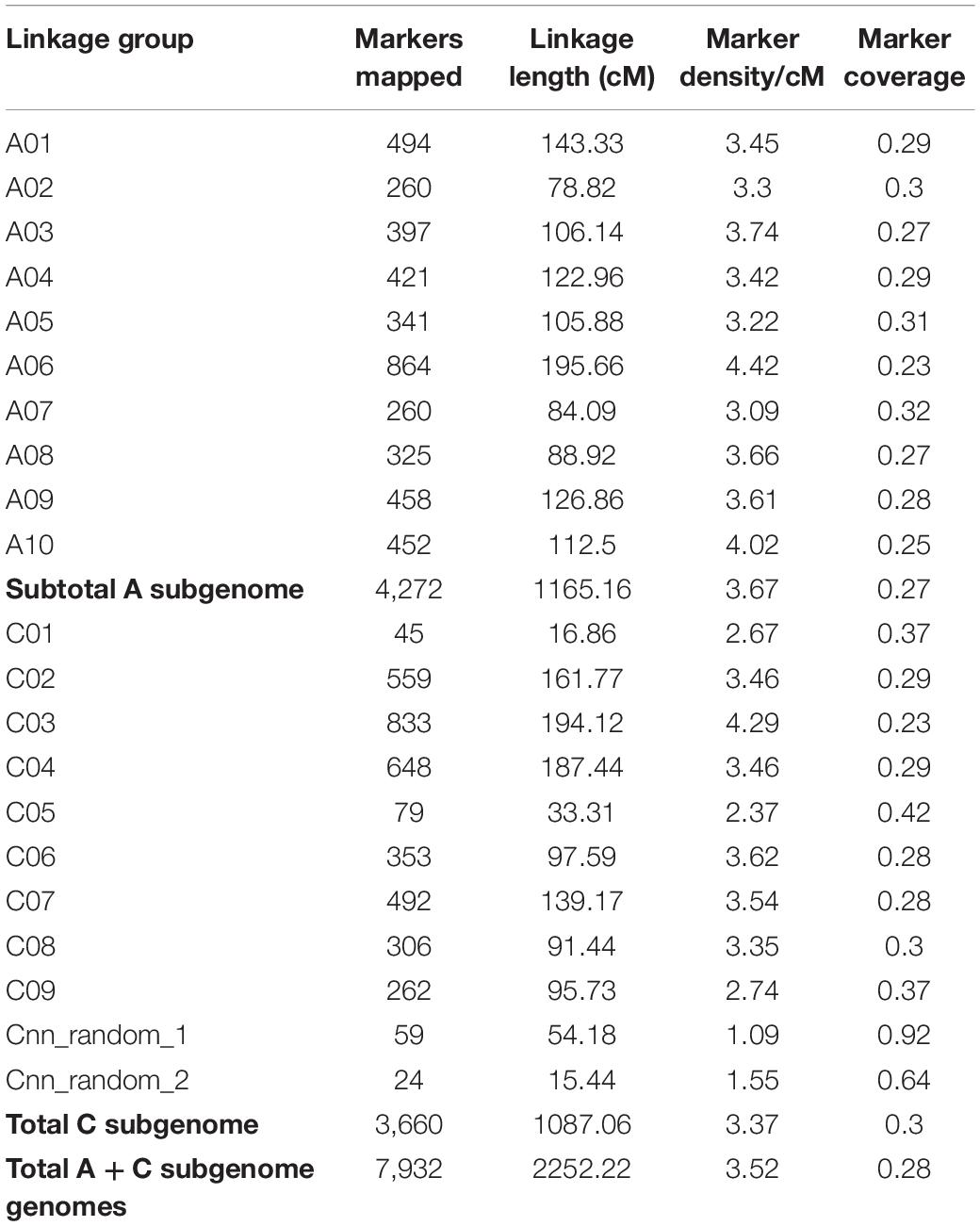
Table 1. Linkage map of the CB-Telfer/Ag-Cobbler population constructed using DArTseq markers.
Utilizing the linkage map and cotyledon lesion score data, we scanned the genome for association with resistance to the PHW1223 isolate using multiple association tests (Table 2). We dropped markers with a call rate < 0.9 and selected a subset of 6,849 markers for trait–marker analyses. On chromosome C03, a major QTL for resistance was identified (Figure 3 and Table 2). Regression analysis showed that the paternal parent, ATR-Cobbler, contributed the allele for resistance, consistent with the response to PHW1223. We further confirmed the linkage between DArTseq markers and resistance to PHW1223 using 60K Illumina SNP markers, genotyped on selected F2 plants exhibiting resistance and susceptibility to the PHW1223 isolate. A genome scan using linear marker regression revealed that an Illumina SNP marker, Bn-scaff_15877_1-p719944, on chromosome C03 showed the most significant association (−log10 P = 20.22), followed by Bn-scaff_21778_1-p75811 (−log10 P = 13.35) (Supplementary Table 4). The allelic effect confirmed that resistance was contributed by the resistant parent, ATR-Cobbler, as observed with DArTseq marker analysis. Haplotyping-based association analysis also identified significant association with resistance to PHW1223 (−log10 P = 17.79) with the marker haplotype “3094566-3087181-4334770” mapped at 4.87 Mb on the Darmor-bzh genome. Since all the associated markers on chromosome C03 had high logarithm of the odds (LOD) scores in multiple tests (Table 2 and Supplementary Table 4), we binned the disease scores into discrete categories (resistance and susceptibility). Linkage analysis showed that the resistance to isolate PHW1223 indeed maps on chromosome C03 and was 1.3 cM from the Bn-scaff_15877_1-p719944 Illumina SNP marker (Figure 4). So far, no race-specific gene for resistance (R)/Avr interaction on C03 chromosome of B. napus has been reported in the literature; as such, we designated this locus for race-specific resistance to L. maculans as a novel locus, Rlm13.
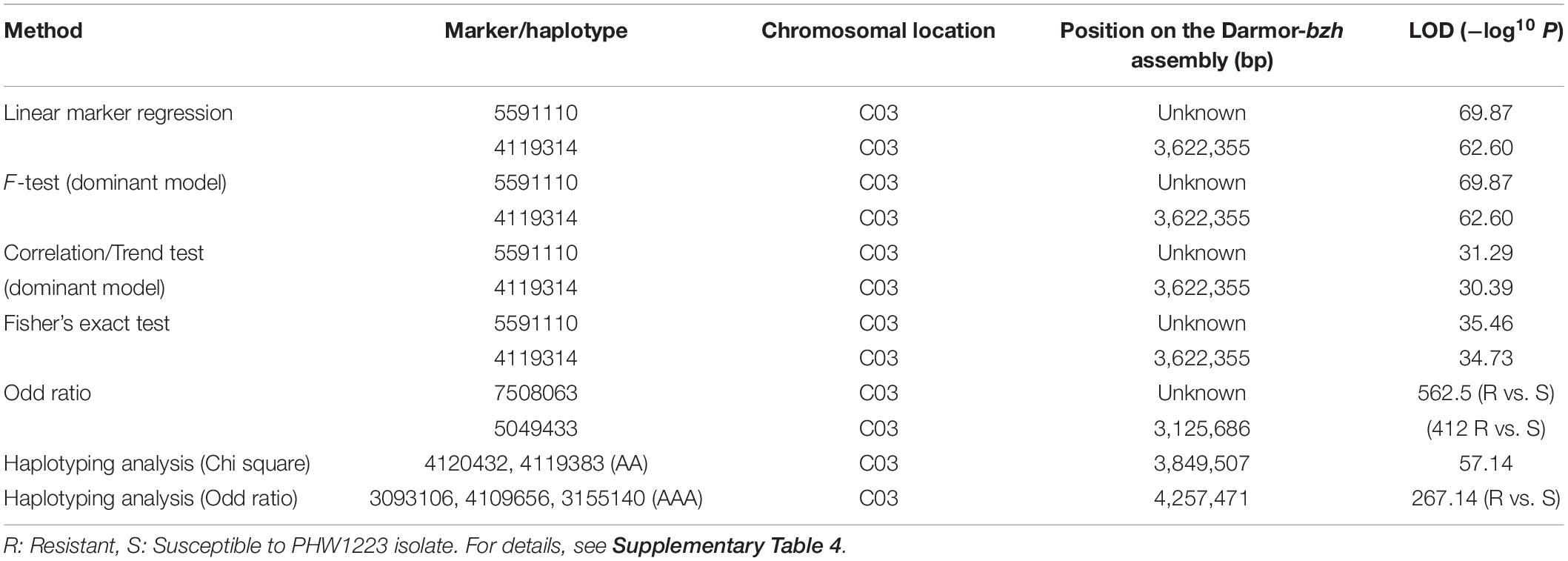
Table 2. Genetic association between GBS-based DArTSeq markers and resistance to L. maculans. Only highly significantly associated markers are shown herein.
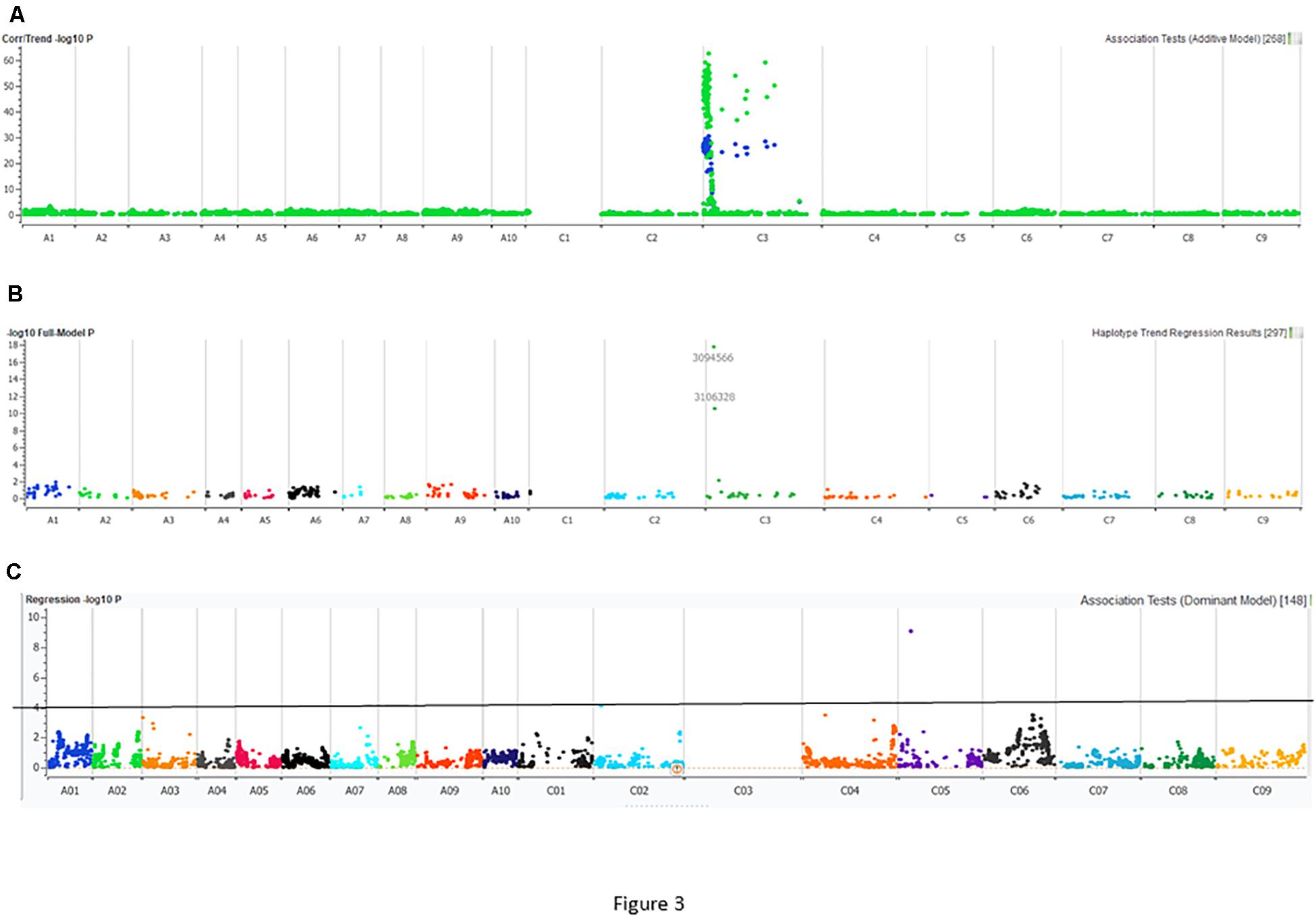
Figure 3. Manhattan plots describing marker associations for resistance to the PHW1223 isolate of L. maculans in an intercross population derived from CB-Telfer/ATR-Cobbler. Resistance to L. maculans was assessed at the cotyledon stage under glasshouse conditions. (A) Markers highlighted in green represent associations detected for resistance to the PHW1223 isolate using linear marker regression (additive model), and those in blue represent associations detected for resistance to the PHW1223 isolate using correlation/trend (additive model). (B) Marker associations identified for resistance using haplotype trend regression. (C) Marker association identified for resistance to PHW1223 isolates (without markers mapped on chromosome C03). Different colors represent chromosomes of B. napus (A01–A10 and C01–C09). Significant associations with a −log10 P ≤ 3 are shown with a solid horizontal line (in black).

Figure 4. Genetic and physical mapping of the Rlm13 locus in relation to DArTseq and Illumina SNP-based markers. The order of markers was determined using the Record program. The marker order is shown on the right-hand side, while the genetic and physical positions of markers and candidate genes (labeled in Red color) are on the left-hand side. Positions of common markers across genetic and physical maps are in bold. For clarity, physical distances are given in 0.1 Mb.
To determine the physical location of Rlm13, we compared the sequence identities between the significant (DArTseq and Illumina SNP) marker sequences and the Darmor-bzh reference sequence. All the top 50 DArTseq markers that were significantly associated with Rlm13 were located on chromosome C03, with the majority of them located within the 3.12–6.8 Mb interval (Figure 4 and Supplementary Table 4). Several Illumina SNP markers that had significant association with resistance were also mapped within the 4.41–5.7 Mb interval (Supplementary Table 4). Of these, the Bn-scaff_15877_1-p719944 marker, located on chromosome C03 at 4,417,287 bp (Clarke et al., 2016), had the highest LOD score (20.2). A further six Illumina SNP markers having a −log10 P score ≤ 13 were also located within 0.5 Mb from the Bn-scaff_15877_1-p719944 marker (Figure 4 and Supplementary Table 3). Mapping of multiple markers that revealed significant association with resistance to isolate PHW1223 within a small genomic region on C03 suggests that the Rlm13 is indeed localized in this region.
We compared the physical positions of significantly associated markers with Rlm13 and previously identified QTL for resistance, which was assessed previously under (i) glasshouse conditions with isolate PHW1223 (used in this study) and five isolates representing pathogenicity group 4, (ii) an ascospore shower test in diverse Australian B. napus germplasm (Raman et al., 2016a), and (iii) natural field conditions in diverse European and Australian B. napus germplasms (Fomeju et al., 2014; Fopa Fomeju et al., 2015; Jestin et al., 2015; Rahman et al., 2016; Kumar et al., 2018) on chromosomes A03 and C03. This analysis showed that the Rlm13 region does not overlap with QTL intervals for quantitative resistance on C03 described in a diverse panel of accessions, representing winter, spring, semi-winter, and rutabaga types (Fopa Fomeju et al., 2015; Rahman et al., 2016). However, Rlm13 is located within the QTL regions for quantitative resistance, detected on C03 in the Darmor/Bristol DH population and genome-wide association study (GWAS) panel of B. napus (Fomeju et al., 2014; Jestin et al., 2015; Raman et al., 2016a; Kumar et al., 2018). The Rlm13 was located within the QTL region, spanning 3.62–4.25 Mb of the Darmor-bzh genome for resistance identified previously in a GWAS panel of B. napus accessions to six single-spore isolates of L. maculans, D1, D2, D6, D9, D10 (PHW1223), and 04MGPS021 (Raman et al., 2016a), suggesting that it may be conditioning resistance response to L. maculans across multiple isolates; some of the associations were detected within 4 kb (Supplementary Table 5).
Earlier studies have shown that the race-specific resistance locus LepR4 maps on chromosome A06 in B. napus (Yu et al., 2013), which shares homoeology with group 3 chromosomes (Chalhoub et al., 2014). Therefore, we determined whether the genomic regions associated with the Rlm13 and LepR4 resistance are present on the same homoeologous genomic regions. First, we investigated the syntenic relationship among marker intervals, collinear with Brassica rapa genes on A06, flanking LepR4 (Bra017959, Bra018037, Bra018198, and Bra024309, Yu et al., 2013), Brassica oleracea, and B. napus genomes. These B. rapa genes on A06 showed synteny with B. oleracea genes Bo3g132890 and Bo3g101190 located on chromosome O3 and Bo8g078980 and Bo8g077270 on O8 and B. napus genes BnaA06g15600D and BnaA06g23540D on A06, BnaC03g56070D on C03, and BnaC08g20450D and BnaC08g21330D on C08 (Supplementary Table 6). Of these, BnaC03g56070D was the only one mapped on C03 of the Darmor-bzh genome, but at 45,361,849–45,362,475 bp, suggesting that the LepR4 region does not map near Rlm13.
We further aligned the genomic sequences of both parental lines with > 80 × Illumina read coverage and interrogated the syntenic regions on the An and Cn subgenomes of B. napus, corresponding to LepR4. As expected, chromosome C03 exhibited extensive synteny to genomic regions on chromosomes A03, A06, and A08 (Figure 5). Our results reconfirmed that the 3.62–4.25 Mb genomic region encompassing Rlm13 is not syntenic to the LepR4 region on A06. These findings indicate that Rlm13 indeed represents a new locus for resistance and is different to LepR4 derived from B. rapa ssp. sylvestris (Yu et al., 2013).
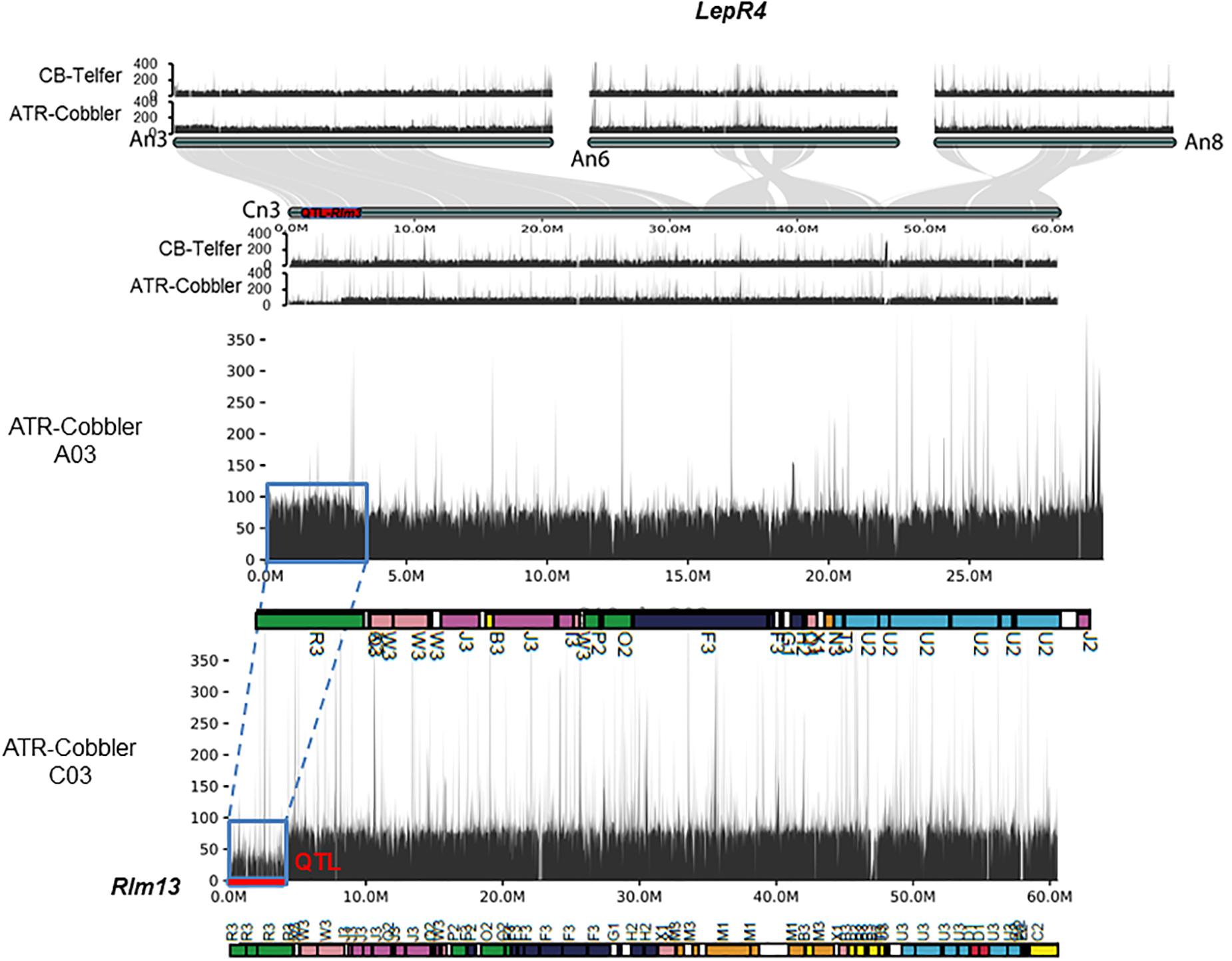
Figure 5. Syntenic relationships between LepR4 (on A06) and homoeologous group 3 chromosomes, encompassing the Rlm13 region on chromosome C03. The gene syntenies between A03, A06, A08, and C08 are shown with gray connections. The Rlm13 region is marked in red. The coverage depth of homoeologous chromosomes was calculated after mapping resequenced reads to the Darmor-bzh genome assembly version 4.1. In ATR-Cobbler, the coverage depth of the Rlm13 region on chromosome C03 is less than 50 (1×), while in A03, the coverage depth is double (∼100), indicating the occurrence of HE between A03 and C03. The syntenic region, subjected to HE, is marked with dotted/solid lines (in blue). Gene sequences which were located in different ancestral blocks are also marked in different colors. Localization of marker sequences to ancestral blocks was carried out using the method described previously (Zou et al., 2016).
Using the resequenced data of CB-Telfer and ATR-Cobbler, we identified several SNPs, InDels, and SVs in the form of transversion, deletion, duplication, and inversions, which were polymorphic between the parental lines, CB-Telfer and ATR-Cobbler, of a mapping population. To understand whether the Rlm13 region is subjected to HE, we used the read coverage depth of resequenced genomes of CB-Telfer and ATR-Cobbler. HE analysis revealed that the A03 region corresponding to Rlm13 on chromosome C03 is in fact subjected to a non-reciprocal duplication (Figure 5). We also found that chromosome A07 is also duplicated, as the corresponding read coverage depth in the C06 region is almost half (Figure 6). However, this duplicated region on chromosome A07 does not represent the Rlm9 genomic region (15.9 Mb, Raman et al., 2018; Larkan et al., 2020), which was anticipated to map in the intercross population from CB-Telfer (Rlm4)/ATR-Cobbler (Rlm4 and Rlm9). Both these non-reciprocal HEs (C03 to A03 and C06 to A07) occurred from the Cn subgenome to An subgenome in the CB-Telfer/ATR-Cobbler. Interestingly, A03/C03 HE occurred at the start of the chromosomes, while the A07/C06 occurred at the end of the homoeologous chromosomes (Figures 5, 6). The SVs are known to occur in B. napus and play an important role in phenotypic and functional diversity in various plants which have undergone genome duplication and fractionation events (Chalhoub et al., 2014; Clark and Donoghue, 2018; Hurgobin et al., 2018). Research to establish the role of HE and PAVs in resistance to L. maculans is in progress.
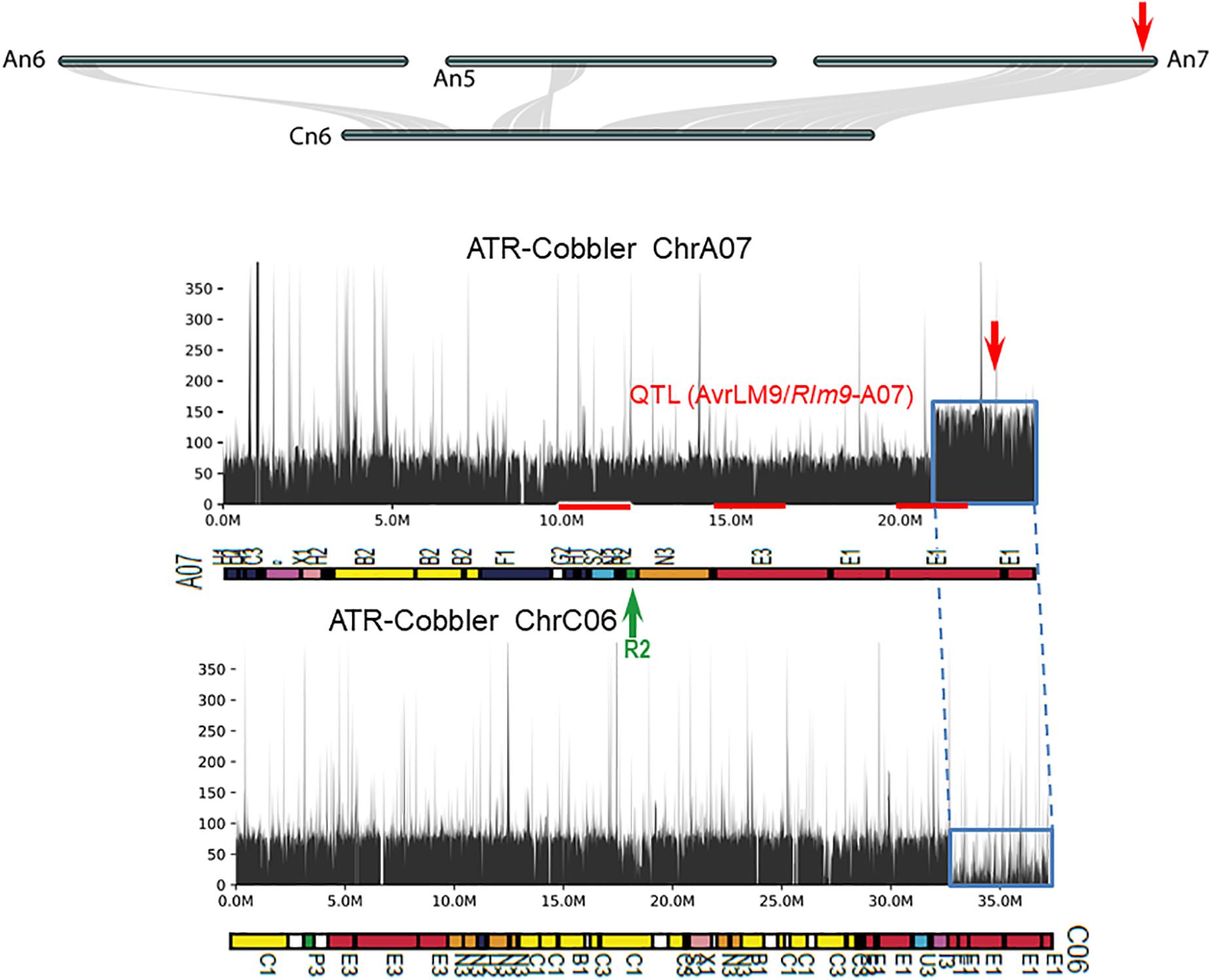
Figure 6. Syntenic relationships between homoeologous chromosome harboring LepR4 and Rlm9 genes for resistance to L. maculans. The gene syntenies between A05, A06, A07, and C06 are shown with gray connections. The Rlm9 region maps to 15.1 Mb of the Darmor-bzh sequence and is marked in red. The coverage depth of homoeologous chromosomes was calculated after mapping resequenced reads to the Darmor-bzh genome assembly version 4.1. Gene syntenies show that in ATR-Cobbler, the coverage depth of the C06 region is almost nil, while in A07, the coverage depth is more than double, illustrating the occurrence of HE between A07 and C06. The syntenic region, subjected to HE, is marked with dotted/solid lines (in blue). Gene sequences which were located in different ancestral blocks are also marked in different colors. Localization of marker sequences to ancestral blocks was carried out using the method described previously (Zou et al., 2016).
To investigate whether Rlm13 is located in the duplicated homoeologous regions involved in resistance to blackleg, we searched the synteny between significantly associated markers with Rlm13 and 22 ancestral blocks of Brassicaceae, as investigated in earlier studies (Schranz et al., 2006; Delourme et al., 2013; Fomeju et al., 2014; Parkin et al., 2014; Fopa Fomeju et al., 2015). Comparative mapping showed that the Rlm13 region is collinear to sequences of A02, A03, A10, C02, and C09, as shown previously (Fopa Fomeju et al., 2015). The syntenic region of Rlm13 corresponded to the most fractionated subgenome MF2 of the R block, delimited with coordinates of the Darmor-bzh, 3,138,662–5,148,532 bp on C03 (Fopa Fomeju et al., 2015). However, this study did not detect any significant SNP association for resistance to blackleg in R and W blocks of chromosome C03 (Supplementary Table 7).
We further searched candidate genes within a 0.5-Mb window either side of the physical location of the Rlm13 region. Plausible candidates for disease resistance were selected, which are annotated in the Darmor-bzh genome (Chalhoub et al., 2014; Alamery et al., 2018; Tirnaz et al., 2020). At least 24 R genes were identified in the Rlm13 region (Figure 4 and Table 3). Of these, 10 were RLKs, four were RLPs, seven were NBS-LRRs, and three were TM-CC. So far, three R genes (two RLP-type genes: LepR3 and Rlm2 and one RLK gene: Rlm9) were identified for resistance to L. maculans (Larkan et al., 2013, 2015, 2020). The candidates for Rlm13 genes were located within 0.4 Mb of the Darmor-bzh genome and were localized in the R ancestral block in the most fractionated (MF2) subgenome on chromosome C03 (Supplementary Table 7). The BnaC03g12220D (DANGEROUS MIX2H gene) was located in the W block in the subgenome MF2. Of the plausible candidate genes, at least nine of them showed InDel (11), SNP (163), and SVs (21) in the form of transversion, inversion, duplication, and deletion between CB-Telfer and ATR-Cobbler (Supplementary Table 2), which remains to be validated for their association to Rlm13-mediated resistance.
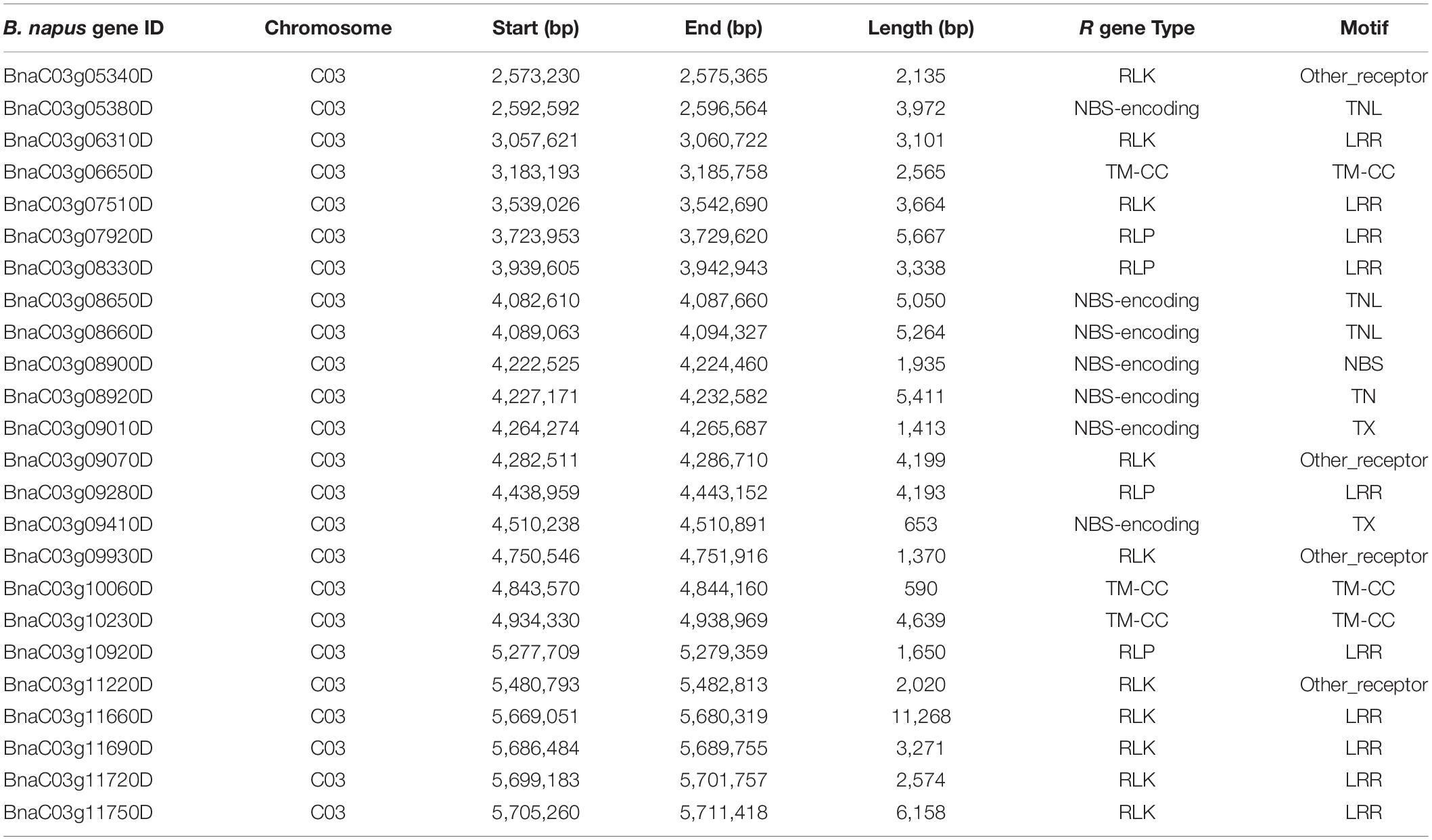
Table 3. Candidate genes identified in the vicinity of the Rlm13 genomic region associated with resistance to L. maculans, causing blackleg disease in canola (B. napus L.).
In this study, we identified a new locus for resistance, Rlm13, on chromosome C03 that confers resistance to the PHW1223 isolate of L. maculans in an Australian canola population derived from CB-Telfer/ATR-Cobbler. Previously, several genes for qualitative resistance to L. maculans were mapped in B. napus, such as Rlm1, Rlm3, Rlm4, Rlm7, and Rlm9 on A07 (Dion et al., 1995; Mayerhofer et al., 1997; Delourme et al., 2004; Tollenaere et al., 2012; Raman et al., 2013); Rlm2, BlmR2, and LepR3 on chromosome A10 (Delourme et al., 2004; Long et al., 2011; Larkan et al., 2013, 2015); Rlm12 on chromosome A01 (Raman et al., 2016a); LepR1 and LepR2 on A02 (Yu et al., 2005); and LepR4 on A06 (Yu et al., 2013). To date, no R gene for resistance on chromosome C03 has been reported in B. napus and its ancestral species (A03 in B. rapa and C03 in B. oleracea).
PHW1223 has AvrLm5-6-8-9-S genes for avirulence, which interact with the corresponding R genes in a host (Delourme et al., 2004; Raman et al., 2018); however, we could not identify any association on chromosomes A02, A07, A06, and A10 for resistance with known R genes (described above). We further checked whether identification of the Rlm13 locus is due to the masking effect of Rlm1, Rlm3, Rlm4, Rlm7, and Rlm9 genes; this was discounted by “hiding” markers on chromosome C03. Interestingly, we did not detect any significant association with resistance to the PHW1223 isolate on chromosome A07 (Figure 2). These results implicate that identification of Rlm13 (on C03) is not a result of the masking effect of A07 Rlm genes (Rlm1, Rlm3, Rlm4, Rlm7, and Rlm9). However, it is possible that another gene network is masking the allelic effect of Rlm9, as reported for the AvrLm4/9-Rlm4/Rlm9 interaction (Ghanbarnia et al., 2018). In the present study, we could exclude this possibility, as both parental lines of a mapping population possess the Rlm4 gene for resistance (Marcroft et al., 2012; Supplementary Table 1). Another possibility is that PHW1223 may possess an uncharacterized Avr gene corresponding to the Rlm13 gene in B. napus.
With Illumina SNP and DArTseq markers, comparisons of physical locations of Rlm13 and QTL detected in DH populations and diverse winter and spring GWAS panels were made in this study. We showed that the Rlm13 is collocated with some of the QTLs for quantitative resistance that were mapped on the homoeologous regions on A03 and C03 chromosomes (Fomeju et al., 2014; Fopa Fomeju et al., 2015; Kumar et al., 2018). For example, Rlm13 was mapped to the same genomic region associated with resistance in a diverse Australian panel of B. napus accessions that was assessed against multiple isolates of the L. maculans (D1, D2, D6, D9, and 04MGPS021), including PHW1223, used in this study (Raman et al., 2016a). It is not established whether QTL effects for resistance at the adult plant stage in described populations is either due to Rlm13 and/or due to tight linkage between R and quantitative resistance locus, as suggested previously for Rlm2 and QTL for the adult plant stage in the Darmor/Samourai population (Pilet et al., 2001; Delourme et al., 2011).
Further research is required to establish the allelic effects of Rlm13 in diverse winter and spring ecotypes. The parents of ATR-Cobbler (ATR-Eyre and ATR-Emblem and their ancestral sources) have been extensively used in Australian canola breeding programs; as a result, several canola cultivars have been released for commercial cultivars. Our results also hint that Rlm13 may represent a non-race-specific resistance gene that is effective against multiple isolates (described above). Similar types of resistance against multiple races of stripe rust, blast, and powdery mildew have been reported in wheat barley and rice (Buschges et al., 1997; Fu et al., 2009; Fukuoka et al., 2009). Non-race-specific resistance genes are also shown to be more durable under field conditions (Lagudah, 2011). Detection of the same consistent region for resistance to multiple isolates in the CB-Telfer/ATR-Cobbler (this study) and GWAS panel (Raman et al., 2016a) also suggests that the Rlm13 gene is prevalent in diverse B. napus accessions and confers resistance to various isolates/pathotypes of L. maculans existing under the Australian conditions. Given that L. maculans is highly diverse and reproduces both sexually and asexually under field conditions, it is likely that deployment of Rlm13 alone may not provide long-term durable resistance. Therefore, it may be deployed in different combinations with other effective R and QR genes to increase the level of resistance in new varieties and its durability, as demonstrated in previous studies (Brun et al., 2010). Closely linked molecular markers with Rlm13 provide an excellent tool for various applications in Brassica improvement programs.
In conclusion, we identified a new player, the Rlm13 gene, that is associated with resistance to blackleg in Australian canola. Based on the current and previous findings, it appears that there are two groups of qualitative R genes for resistance to blackleg in canola. The first group includes the race-specific resistance genes, which are effective for specific isolates of L. maculans such as Rlm1, Rlm2, Rlm3, Rlm4, Rlm7, Rlm9, and Rlm10 and LepR1-4. The second group represents genes that provide non-race-specific resistance to multiple isolates such as Rlm13; however, it differs from quantitative resistance, which is described as polygenic, having small additive allelic effects and being stable over a longer evolutionary time frame (Delourme et al., 2006; Niks et al., 2015). Further research is required to verify this type of resistance in a wider Brassica germplasm. Nevertheless, our results provide a “target” for further understanding Avr–R gene interactions as well as provide a valuable tool for genomic-assisted breeding in canola improvement programs.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
HR conceived the experiments, supervised the research, and drafted the manuscript. RR developed a mapping population. HR and RR conducted phenotyping experiments and genetic analysis. YQ assisted with inoculation, DNA isolation, and management of plants. JB genotyped a subset of F2 lines with Illumina SNP markers. YZ and SL conducted comparative mapping. All authors reviewed and approved this manuscript.
This research was supported by the New South Wales Department of Primary Industries (NSW DPI). The F2 population was developed under the project DAN00208 supported by the Grains Research and Development Corporation, Australia, and NSW DPI.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank DArT P/L for providing genotyping service and Novogene for resequencing the parental lines.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.654604/full#supplementary-material
Alamery, S., Tirnaz, S., Bayer, P., Tollenaere, R., Chaloub, B., Edwards, D., et al. (2018). Genome-wide identification and comparative analysis of NBS-LRR resistance genes in Brassica napus. Crop Pasture Sci. 69, 72–93. doi: 10.1071/cp17214
Balesdent, M.-H., Fudal, I., Ollivier, B., Bally, P., Grandaubert, J., Eber, F., et al. (2013). The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa. New Phytol. 198, 887–898. doi: 10.1111/nph.12178
Brun, H., Chevre, A. M., Fitt, B. D. L., Powers, S., Besnard, A. L., Ermel, M., et al. (2010). Quantitative resistance increases the durability of qualitative resistance to Leptosphaeria maculans in Brassica napus. New Phytol. 185, 285–299. doi: 10.1111/j.1469-8137.2009.03049.x
Buschges, R., Hollricher, K., Panstruga, R., Simons, G., Wolter, M., Frijters, A., et al. (1997). The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 88, 695–705. doi: 10.1016/s0092-8674(00)81912-1
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953.
Chèvre, A. M., Barret, P., Eber, F., Dupuy, P., Brun, H., and Tanguy, X. (1997). Selection of stable Brassica napus-B. juncea recombinant lines resistant to blackleg (Leptosphaeria maculans). 1. Identification of molecular markers, chromosomal and genomic origin of the introgression. Theor. Appl. Genet. 95, 1104–1111. doi: 10.1007/s001220050669
Chong, Z., Ruan, J., Gao, M., Zhou, W., Chen, T., Fan, X., et al. (2017). novoBreak: local assembly for breakpoint detection in cancer genomes. Nat. Methods 14, 65–67. doi: 10.1038/nmeth.4084
Clark, J. W., and Donoghue, P. C. J. (2018). Whole-genome duplication and plant macroevolution. Trends Plant Sci. 23, 933–945. doi: 10.1016/j.tplants.2018.07.006
Clarke, W. E., Higgins, E. E., Plieske, J., Wieseke, R., Sidebottom, C., Khedikar, Y., et al. (2016). A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor. Appl. Genet. 129, 1887–1899. doi: 10.1007/s00122-016-2746-7
Delourme, R., Barbetti, M. J., Snowdon, R., Zhao, J., and Manzanares-Dauleux, M. J. (2011). Genetics and Genomics of Disease Resistance USA: Science Publishers. Boca Raton, FL: CRC Press.
Delourme, R., Chevre, A. M., Brun, H., Rouxel, T., Balesdent, M. H., Dias, J. S., et al. (2006). Major gene and polygenic resistance to Leptosphaeria maculans in oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 41–52. doi: 10.1007/1-4020-4525-5_4
Delourme, R., Falentin, C., Fomeju, B., Boillot, M., Lassalle, G., Andre, I., et al. (2013). High-density SNP-based genetic map development and linkage disequilibrium assessment in Brassica napus L. BMC Genomics 14:120. doi: 10.1186/1471-2164-14-120
Delourme, R., Pilet-Nayel, M. L., Archipiano, M., Horvais, R., Tanguy, X., Rouxel, T., et al. (2004). A cluster of major specific resistance genes to Leptosphaeria maculans in Brassica napus. Phytopathology 94, 578–583. doi: 10.1094/phyto.2004.94.6.578
Dion, Y., Gugel, R. K., Rakow, G. F. W., Séguin-Swartz, G., and Landry, B. S. (1995). RFLP mapping of resistance to the blackleg disease [causal agent, Leptosphaeria maculans (Desm.) Ces. et de Not.] in canola (Brassica napus L.). Theor. Appl. Genet. 91, 1190–1194. doi: 10.1007/bf00220928
Ferdous, M. J., Hossain, M. R., Park, J.-I., Robin, A. H. K., Jesse, D. M. I., Jung, H.-J., et al. (2019). Inheritance pattern and molecular markers for resistance to blackleg disease in cabbage. Plants 8:583. doi: 10.3390/plants8120583
Fitt, B., Brun, H., Barbetti, M., and Rimmer, S. (2006). World-wide importance of Phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 3–15. doi: 10.1007/1-4020-4525-5_1
Flor, H. H. (1971). The current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. doi: 10.1146/annurev.py.09.090171.001423
Fomeju, B. F., Falentin, C., Lassalle, G., Manzanares-Dauleux, M. J., and Delourme, R. (2014). Homoeologous duplicated regions are involved in quantitative resistance of Brassica napus to stem canker. BMC Genomics 15:498. doi: 10.1186/1471-2164-15-498
Fopa Fomeju, B., Falentin, C., Lassalle, G., Manzanares-Dauleux, M. J., and Delourme, R. (2015). Comparative genomic analysis of duplicated homoeologous regions involved in the resistance of Brassica napus to stem canker. Front. Plant Sci. 6:772. doi: 10.3389/fpls.2015.00772
Fu, D., Uauy, C., Distelfeld, A., Blechl, A., Epstein, L., Chen, X., et al. (2009). A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science 323, 1357–1360. doi: 10.1126/science.1166289
Fudal, I., Ross, S., Gout, L., Blaise, F., Kuhn, M. L., Eckert, M. R., et al. (2007). Heterochromatin-like regions as ecological niches for avirulence genes in the Leptosphaeria maculans genome: map-based cloning of AvrLm6. Mol. Plant Microbe Interact. 20, 459–470. doi: 10.1094/mpmi-20-4-0459
Fukuoka, S., Saka, N., Koga, H., Ono, K., Shimizu, T., Ebana, K., et al. (2009). Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325, 998–1001. doi: 10.1126/science.1175550
Ghanbarnia, K., Fudal, I., Larkan, N. J., Links, M. G., Balesdent, M. H., Profotova, B., et al. (2015). Rapid identification of the Leptosphaeria maculans avirulence gene AvrLm2 using an intraspecific comparative genomics approach. Mol. Plant Pathol. 16, 699–709. doi: 10.1111/mpp.12228
Ghanbarnia, K., Lydiate, D., Rimmer, S. R., Li, G., Kutcher, H. R., Larkan, N., et al. (2012). Genetic mapping of the Leptosphaeria maculans avirulence gene corresponding to the LepR1 resistance gene of Brassica napus. Theor. Appl. Genet. 124, 505–513. doi: 10.1007/s00122-011-1724-3
Ghanbarnia, K., Ma, L., Larkan, N. J., Haddadi, P., Fernando, W. G. D., and Borhan, M. H. (2018). Leptosphaeria maculans AvrLm9: a new player in the game of hide and seek with AvrLm4-7. Mol. Plant Pathol. 19, 1754–1764. doi: 10.1111/mpp.12658
Gout, L., Fudal, I., Kuhn, M. L., Blaise, F., Eckert, M., Cattolico, L., et al. (2006). Lost in the middle of nowhere: the AvrLm1 avirulence gene of the Dothideomycete Leptosphaeria maculans. Mol. Microbiol. 60, 67–80. doi: 10.1111/j.1365-2958.2006.05076.x
Hayden, H. L., Cozijnsen, A. J., and Howlett, B. J. (2007). Microsatellite and minisatellite analysis of Leptosphaeria maculans in Australia reveals regional genetic differentiation. Phytopathology 97, 879–887. doi: 10.1094/phyto-97-7-0879
Howlett, B. J. (2004). Current knowledge of the interaction between Brassica napus and Leptosphaeria maculans. Can. J. Plant Pathol. 26, 245–252.
Hurgobin, B., Golicz, A. A., Bayer, P. E., Chan, C.-K. K., Tirnaz, S., Dolatabadian, A., et al. (2018). Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 16, 1265–1274. doi: 10.1111/pbi.12867
Jestin, C., Bardol, N., Lodé, M., Duffé, P., Domin, C., Vallée, P., et al. (2015). Connected populations for detecting quantitative resistance factors to phoma stem canker in oilseed rape (Brassica napus L.). Mol. Breeding 35:167.
Keen, N. T. (1990). Gene-for-gene complementarity in plant-pathogen interactions. Annu. Rev. Genet. 24, 447–463. doi: 10.1146/annurev.ge.24.120190.002311
Koch, E., Song, K., Osborn, T. C., and Williams, P. H. (1991). Relationship between pathogenicity based on restriction fragment length polymorphism in Leptosphaeria maculans. Mol. Plant Microbe Interact. 4, 341–349. doi: 10.1094/mpmi-4-341
Kumar, V., Paillard, S., Fopa-Fomeju, B., Falentin, C., Deniot, G., Baron, C., et al. (2018). Multi-year linkage and association mapping confirm the high number of genomic regions involved in oilseed rape quantitative resistance to blackleg. Theor. Appl. Genet. 131, 1627–1643. doi: 10.1007/s00122-018-3103-9
Lagudah, E. S. (2011). Molecular genetics of race non-specific rust resistance in wheat. Euphytica 179, 81–91. doi: 10.1007/s10681-010-0336-3
Larkan, N. J., Lydiate, D. J., Parkin, I. A. P., Nelson, M. N., Epp, D. J., Cowling, W. A., et al. (2013). The Brassica napus blackleg resistance gene LepR3 encodes a receptor-like protein triggered by the Leptosphaeria maculans effector AvrLM1. New Phytol. 197, 595–605. doi: 10.1111/nph.12043
Larkan, N. J., Ma, L., and Borhan, M. H. (2015). The Brassica napus receptor-like protein RLM2 is encoded by a second allele of the LepR3/Rlm2 blackleg resistance locus. Plant Biotechnol. J. 13, 983–992. doi: 10.1111/pbi.12341
Larkan, N. J., Ma, L., Haddadi, P., Buchwaldt, M., Parkin, I. A. P., Djavaheri, M., et al. (2020). The Brassica napus wall-associated kinase-like (WAKL) gene Rlm9 provides race-specific blackleg resistance. Plant J. 104, 892–900. doi: 10.1111/tpj.14966
Li, H., Barbetti, M. J., and Sivasithamparam, K. (2006). Concomitant inoculation of an avirulent strain of Leptosphaeria maculans prevents break-down of a single dominant gene-based resistance in Brassica napus cv. Surpass 400 by a virulent strain. Field Crops Res. 95, 206–211. doi: 10.1016/j.fcr.2005.02.010
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Long, Y., Wang, Z., Sun, Z., Fernando, D. W., Mcvetty, P. B., and Li, G. (2011). Identification of two blackleg resistance genes and fine mapping of one of these two genes in a Brassica napus canola cultivar ‘Surpass 400’. Theor. Appl. Genet. 122, 1223–1231. doi: 10.1007/s00122-010-1526-z
Marcroft, S. J., Elliott, V. L., Cozijnsen, A. J., Salisbury, P. A., Howlett, B. J., and Van De Wouw, A. P. (2012). Identifying resistance genes to Leptosphaeria maculans in Australian Brassica napus cultivars based on reactions to isolates with known avirulence genotypes. Crop Pasture Sci. 63, 338–350. doi: 10.1071/cp11341
Mayerhofer, R., Bansal, V. K., Thiagarajah, M. R., Stringam, G. R., and Good, A. G. (1997). Molecular mapping of resistance to Leptosphaeria maculans in Australian cultivars of Brassica napus. Genome 40, 294–301. doi: 10.1139/g97-041
Niks, R. E., Qi, X., and Marcel, T. C. (2015). Quantitative resistance to biotrophic filamentous plant pathogens: concepts, misconceptions, and mechanisms. Annu. Rev. Phytopathol. 53, 445–470. doi: 10.1146/annurev-phyto-080614-115928
Parkin, I., Koh, C., Tang, H., Robinson, S., Kagale, S., Clarke, W., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15:R77.
Parlange, F., Daverdin, G., Fudal, I., Kuhn, M.-L., Balesdent, M.-H., Blaise, F., et al. (2009). Leptosphaeria maculans avirulence gene AvrLm4-7 confers a dual recognition specificity by the Rlm4 and Rlm7 resistance genes of oilseed rape, and circumvents Rlm4-mediated recognition through a single amino acid change. Mol. Microbiol. 71, 851–863. doi: 10.1111/j.1365-2958.2008.06547.x
Pilet, M. L., Duplan, G., Archipiano, M., Barret, P., Baron, C., Horvais, R., et al. (2001). Stability of QTL for field resistance to blackleg across two genetic backgrounds in oilseed rape. Crop Sci. 41, 197–205. doi: 10.2135/cropsci2001.411197x
Plissonneau, C., Daverdin, G., Ollivier, B., Blaise, F., Degrave, A., Fudal, I., et al. (2016). A game of hide and seek between avirulence genes AvrLm4-7 and AvrLm3 in Leptosphaeria maculans. New Phytol. 209, 1613–1624. doi: 10.1111/nph.13736
Plissonneau, C., Rouxel, T., Chevre, A. M., Van De Wouw, A. P., and Balesdent, M. H. (2018). One gene-one name: the AvrLmJ1 avirulence gene of Leptosphaeria maculans is AvrLm5. Mol. Plant Pathol. 19, 1012–1016. doi: 10.1111/mpp.12574
Rahman, M., Mamidi, S., Del Rio, L., Ross, A., Kadir, M. M., Rahaman, M. M., et al. (2016). Association mapping in Brassica napus (L.) accessions identifies a major QTL for blackleg disease resistance on chromosome A01. Mol. Breeding 36:90.
Raman, H., Raman, R., Coombes, N., Song, J., Diffey, S., Kilian, A., et al. (2016a). Genome-wide association study identifies new loci for resistance to Leptosphaeria maculans in canola. Front. Plant Sci. 7:1513. doi: 10.3389/fpls.2016.01513
Raman, H., Raman, R., Diffey, S., Qiu, Y., Mcvittie, B., Barbulescu, D. M., et al. (2018). Stable quantitative resistance loci to blackleg disease in canola (Brassica napus L.) over continents. Front. Plant Sci. 9:1622. doi: 10.3389/fpls.2018.01622
Raman, H., Raman, R., Kilian, A., Detering, F., Carling, J., Coombes, N., et al. (2014). Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS One 9:e101673. doi: 10.1371/journal.pone.0101673
CrossRef Full Text 9, e101673 | PubMed Abstract | Google Scholar
Raman, H., Raman, R., and Larkan, N. (2013). Genetic Dissection of Blackleg Resistance Loci in Rapeseed (Brassica napus L.). Plant Breeding from Laboratories to Fields (Ed. Prof. Sven Bode Andersen). Available online at: http://www.intechopen.com/books/export/citation/EndNote/plant-breeding-from-laboratories-to-fields/genetic-dissection-of-blackleg-resistance-loci-in-rapeseed-brassica-napus-l- (accessed 2013-05-22).
Raman, R., Diffey, S., Barbulescu, D. M., Coombes, N., Luckett, D., Salisbury, P., et al. (2020). Genetic and physical mapping of loci for resistance to blackleg disease in canola (Brassica napus L.). Sci. Rep. 10:4416.
Raman, R., Diffey, S., Carling, J., Cowley, R., Kilian, A., Luckett, D., et al. (2016b). Quantitative genetic analysis of yield in an Australian Brassica napus doubled haploid population. Crop Pasture Sci. 67, 298–307. doi: 10.1071/cp15283
Raman, R., Taylor, B., Marcroft, S., Stiller, J., Eckermann, P., Coombes, N., et al. (2012). Molecular mapping of qualitative and quantitative loci for resistance to Leptosphaeria maculans; causing blackleg disease in canola (Brassica napus L.). Theor. Appl. Genet. 125, 405–418. doi: 10.1007/s00122-012-1842-6
Rouxel, T., and Balesdent, M.-H. (2017). Life, death and rebirth of avirulence effectors in a fungal pathogen of Brassica crops, Leptosphaeria maculans. New Phytol. 214, 526–532. doi: 10.1111/nph.14411
Rouxel, T., Penaud, A., Pinochet, X., Brun, H., Gout, L., Delourme, R., et al. (2003). A 10-year survey of populations of Leptosphaeria maculans in France indicates a rapid adaptation towards the Rlm1 resistance gene of oilseed rape. Eur. J. Plant Pathol. 109, 871–881.
Roy, N. N. (1984). Interspecific transfer of Brassica juncea-type high blackleg resistance to Brassica napus. Euphytica 33, 295–303. doi: 10.1007/bf00021125
Saal, B., and Struss, D. (2005). RGA- and RAPD-derived SCAR markers for a Brassica B-genome introgression conferring resistance to blackleg in oilseed rape. Theor. Appl. Genet. 111, 281–290. doi: 10.1007/s00122-005-2022-8
Schranz, M., Lysak, M., and Mitchell-Olds, T. (2006). The ABC’s of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11, 535–542. doi: 10.1016/j.tplants.2006.09.002
Sprague, S. J., Marcroft, S. J., Hayden, H. L., and Howlett, B. J. (2006). Major gene resistance to blackleg in Brassica napus overcome within three years of commercial production in Southeastern Australia. Plant Dis. 90, 190–198. doi: 10.1094/pd-90-0190
Tirnaz, S., Bayer, P. E., Inturrisi, F., Zhang, F., Yang, H., Dolatabadian, A., et al. (2020). Resistance gene analogs in the brassicaceae: identification, characterization, distribution, and evolution. Plant Physiol. 184, 909–922. doi: 10.1104/pp.20.00835
Tollenaere, R., Hayward, A., Dalton-Morgan, J., Campbell, E., Lee, J. R. M., Lorenc, M., et al. (2012). Identification and characterization of candidate Rlm4 blackleg resistance genes in Brassica napus using next-generation sequencing. Plant Biotechnol. J. 10, 709–715. doi: 10.1111/j.1467-7652.2012.00716.x
Van de Wouw, A. P., Lowe, R. G., Elliott, C. E., Dubois, D. J., and Howlett, B. J. (2014). An avirulence gene, AvrLmJ1, from the blackleg fungus, Leptosphaeria maculans, confers avirulence to Brassica juncea cultivars. Mol. Plant Pathol. 15, 523–530. doi: 10.1111/mpp.12105
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
West, J. S., Kharbanda, P. D., Barbetti, M. J., and Fitt, B. D. L. (2001). Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada and Europe. Plant Pathol. 50, 10–27. doi: 10.1046/j.1365-3059.2001.00546.x
Yu, F., Lydiate, D. J., Gugel, R. K., Sharpe, A. G., and Rimmer, S. R. (2012). Introgression of Brassica rapa subsp. sylvestris blackleg resistance into B. napus. Mol. Breeding 30, 1495–1506 doi: 10.1007/s11032-012-9735-6
Yu, F., Lydiate, D. J., and Rimmer, S. R. (2005). Identification of two novel genes for blackleg resistance in Brassica napus. Theor. Appl. Genet. 110, 969–979. doi: 10.1007/s00122-004-1919-y
Yu, F. Q., Gugel, R. K., Kutcher, H. R., Peng, G., and Rimmer, S. R. (2013). Identification and mapping of a novel blackleg resistance locus LepR4 in the progenies from Brassica napus x B. rapa subsp sylvestris. Theor. Appl. Genet. 126, 307–315. doi: 10.1007/s00122-012-1919-2
Keywords: natural variation, resistance to L. maculans, canola, genome-wide analysis (GWA), linkage map construction
Citation: Raman H, Raman R, Qiu Y, Zhang Y, Batley J and Liu S (2021) The Rlm13 Gene, a New Player of Brassica napus–Leptosphaeria maculans Interaction Maps on Chromosome C03 in Canola. Front. Plant Sci. 12:654604. doi: 10.3389/fpls.2021.654604
Received: 16 January 2021; Accepted: 25 March 2021;
Published: 12 May 2021.
Edited by:
Ryo Fujimoto, Kobe University, JapanReviewed by:
Mukhlesur Rahman, North Dakota State University, United StatesCopyright © 2021 Raman, Raman, Qiu, Zhang, Batley and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harsh Raman, aGFyc2gucmFtYW5AZHBpLm5zdy5nb3YuYXU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.