 Adam Chin-Fatt
Adam Chin-Fatt Reza Saberianfar
Reza Saberianfar Rima Menassa
Rima Menassa
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 14 April 2021
Sec. Plant Biotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.651262
This article is part of the Research TopicPlant-Production Platforms for Veterinary BiopharmaceuticalsView all 8 articles
We previously isolated a single domain antibody (VHH) that binds Enterohemorrhagic Escherichia coli (EHEC) with the end-goal being the enteromucosal passive immunization of cattle herds. To improve the yield of a chimeric fusion of the VHH with an IgA Fc, we employed two rational design strategies, supercharging and introducing de novo disulfide bonds, on the bovine IgA Fc component of the chimera. After mutagenizing the Fc, we screened for accumulation levels after transient transformation in Nicotiana benthamiana leaves. We identified and characterized five supercharging and one disulfide mutant, termed ‘(5 + 1)Fc’, that improve accumulation in comparison to the native Fc. Combining all these mutations is associated with a 32-fold increase of accumulation for the Fc alone, from 23.9 mg/kg fresh weight (FW) to 599.5 mg/kg FW, as well as a twenty-fold increase when fused to a VHH that binds EHEC, from 12.5 mg/kg FW tissue to 236.2 mg/kg FW. Co-expression of native or mutated VHH-Fc with bovine joining chain (JC) and bovine secretory component (SC) followed by co-immunoprecipitation suggests that the stabilizing mutations do not interfere with the capacity of VHH-Fc to assemble with JC and FC into a secretory IgA. Both the native and the mutated VHH-Fc similarly neutralized the ability of four of the seven most prevalent EHEC strains (O157:H7, O26:H11, O111:Hnm, O145:Hnm, O45:H2, O121:H19 and O103:H2), to adhere to HEp-2 cells as visualized by immunofluorescence microscopy and quantified by fluorometry. These results collectively suggest that supercharging and disulfide bond tethering on a Fc chain can effectively improve accumulation of a VHH-Fc fusion without impacting VHH functionality.
Fragment crystallizable (Fc) fusion proteins represent 15% of all recently approved biologics by the United States Food and Drug Administration (FDA) (2011-2016), with significant market increases forecasted for 2016 to 2025 (Lagasse et al., 2017). Fc-fusion proteins, also known as Fc chimeric proteins or Fc-tagged proteins, comprise the Fc domain of an immunoglobulin (Ig) that has been genetically linked to a protein of interest. The appeal of the Fc as a fusion partner is afforded by its structural modularity allowing independent folding of its fusion partner while still providing structural stability (Czajkowsky et al., 2012). Additionally, the Fc domain homodimerizes with itself via an inter-chain disulfide bond allowing an increase in avidity for a fused partner (Bastian et al., 1992) and enabling effector functions (Park et al., 2016).
Although the market consists predominantly of IgG subtype Fc fusions, there has been much interest in developing an alternative IgA-based Fc scaffold for the control of mucosal pathogens in applications such as oral passive immunization (Bakema et al., 2011; Bakema and Van Egmond, 2011; Lohse et al., 2011; De Greve et al., 2020). Unlike the IgG Fc, the IgA Fc assembles with two other subunits, a 15 kDa joining chain (JC) and a 70 kDa secretory component (SC), to form a secretory complex (sIgA) (Supplementary Figure 1). Despite the therapeutic value of IgA, transition to clinical testing has been hampered by technical difficulties in producing and purifying the full-size IgA in conventional microbial and mammalian platforms because of native IgA’s large size (380 kDa; compared to 150 kDa for IgG) (Reinhart and Kunert, 2015). There are several reports of the production of IgA in plants and in yeast intended for oral passive enteromucosal immunization of animals (Virdi et al., 2013, 2019; Vanmarsenille et al., 2018; Saberianfar et al., 2019). While accumulation of IgA in some of those reports has met the 1% TSP/100 mg/kg FW/100 mg/L culture medium rule of thumb benchmark for commercial viability (Rybicki, 2009; Schillberg et al., 2019), an improvement in accumulation levels by way of stabilization would reduce the cost of production. According to a techno-economic analysis for monoclonal antibodies (mAbs) in a plant platform, an increase in accumulation level from 0.25 g/kg FW to 1 g/kg FW cuts production costs by almost 60% from ∼$290/g to ∼$120/g (Nandi et al., 2016). Therefore, there is value in exploring design strategies that improve IgA accumulation in a plant platform.
Structurally, IgA is a modular glycoprotein with an antigen binding fragment (Fab) linked to an Fc scaffold which consists of two dimerized constant domains, CH2 and CH3, that form interchain disulfides with the SC and JC respectively to enable assembly of the secretory complex (Supplementary Figure 1). To develop a stabilized IgA Fc chain that could improve yield of its fusion partner without impairing function, we investigated two rational design strategies, supercharging and introducing de novo disulfide bonds, on a bovine IgA Fc chain. The underlying idea behind both of these two designs is based on a number of previous proteomic observations that suggest that heterologous protein stability and solubility strongly correlate with protein abundance (Walther et al., 2015; Leuenberger et al., 2017). Conceptually, supercharging involves the addition of charged amino acids to the protein surface. Because these additional charges exhibit small charge-charge repulsive forces, protein accumulation can be enhanced by reducing the non-specific protein aggregation that typically occurs in the macromolecular crowded environment when producing recombinant proteins (Lawrence et al., 2007). Since such aggregates can be either refolded by chaperones, terminally sequestered or targeted for degradation via the proteasome (Mogk et al., 2018; McLoughlin et al., 2019), reducing the occurrence of non-specific aggregation may increase yield. The introduction of novel disulfides at strategic locations can enhance accumulation by preventing exposure of the reactive hydrophobic interior normally susceptible to proteolytic enzymes or non-specific aggregation to adjacent proteins (Zabetakis et al., 2014). The use of protease inhibitors to prevent degradation from proteolytic enzymes has been shown to improve heterologous mAb yield in plants (Jutras et al., 2016).
Enterohemorrhagic Escherichia coli (EHEC) has consistently been one of the foremost foodborne pathogen threats worldwide, conservatively estimated in causing 2.8 million acute illnesses annually (Majowicz et al., 2014). It primarily colonizes the bovine digestive tract as a reservoir from which it can be transmitted via fecal shedding or during slaughter to ultimately compromise food and water safety (Montenegro et al., 1990; Beutin et al., 1993). Collectively, seven EHEC strains (O157:H7, O26:H11, O111:Hnm, O145:Hnm, O45:H2, O121:H19 and O103:H2), account for over 95% of all EHEC-related illnesses and have been the main focus of North American food surveillance efforts for the past decade (Mathusa et al., 2010; Public Health Agency of Canada, 2015). Towards addressing this problem, we previously isolated camelid-derived single-domain antibodies (VHHs) specific for intimin, an EHEC adhesin required for colonization, fused them to a bovine IgA Fc and demonstrated that these chimeric VHH-Fc fusions could bind and neutralize the adherence of three of the most prevalent EHEC serotypes: O157, O26, and O111 (Saberianfar et al., 2019). We subsequently showed that these chimeric fusions neutralize a 4th EHEC serotype, O145 (Chin-Fatt, unpublished). Simplified chimeric VHH-IgA Fc, which involve replacing the Fab region with a VHH, are much easier to produce and are encoded by a single gene, rather than four required for full sized sIgA (De Greve et al., 2020).
In this study, we identified a stabilized rationally designed bovine IgA Fc chain with five mutations predicted to supercharge the protein, and one mutation predicted to introduce a novel disulfide respectively. After transient transformation into Nicotiana benthamiana leaves, these mutations, when combined, improved in planta accumulation of both the Fc chain alone and of the Fc fused to an anti-EHEC VHH partner. Co-expression of the engineered VHH-Fc fusion with a bovine joining chain and secretory component and subsequent co-immunoprecipitation suggest that the mutations do not impair correct folding and assembly of components to the secretory IgA form. We also demonstrate that the engineered Fc fusion does not compromise the VHH’s function of binding and neutralizing EHEC strains O26:H11, O111:Hnm, O145:Hnm and O157:H7. This study is notable because it provides proof of concept that an Fc chain may be rationally designed for improved IgA yield in a plant platform without compromising functionality or its ability to assemble into its secretory form.
We chose to work with the bovine Fc for neutralizing the pathogen by immune exclusion for several reasons. In the context of passive immunization there may be some activation of the host immune system that requires the bovine Fc. There is some evidence, though not conclusive, that secretory IgAs bound to pathogens in the gut associated lymphoid tissue (GALT) can pass through the epithelium and activate a subset of dendritic cells via the FcαR to upregulate CD86 costimulatory molecules, class II major histocompatibility complex (MHC) expression, and increased allostimulatory activity (Geissmann et al., 2001). We have intentionally not mutated the residues involved in this interaction. As well, using a bovine sequence reduces the chance of an allergic reaction by the host and may be more acceptable to regulatory approval than a non-bovine Fc.
Although the crystal structure of human IgA Fc has been determined and is publicly available (pdb: 1IGA) (Boehm et al., 1999), bovine IgA Fc, which is 70% similar in sequence, has not yet been documented (Figure 1A). Because the structure of the Fc is generally well conserved across species, we hypothesized that we could use the human Fc for homology-based structural prediction of the bovine Fc rather than modeling it ab initio (Figure 1B). Therefore, we used the I-TASSER online program (Zhang, 2008) to predict the structure of bovine IgA Fc using the human IgA Fc as a threading template (Wu and Zhang, 2008; Zhang, 2008). Because the resulting predicted structure had a high confidence score of 1.35 (given a range of -5 to 2), it may be sufficient for rational design of mutations. The predicted secondary structure of the bovine IgA Fc consists of a characteristic β sandwich of seven anti-parallel β strands for both its CH2 and CH3 domains. Both domains exhibit Greek key connectivity, a topological signature of β sandwiches, (ABED CFG) forming two distinct β sheets that fold over each other (Figure 1C). For both domains, an intra-chain disulfide in the center of the β sheet stabilizes the tertiary structure. There are also inter-chain disulfides adjacent to the hinge area that enable dimerization of the Fc.
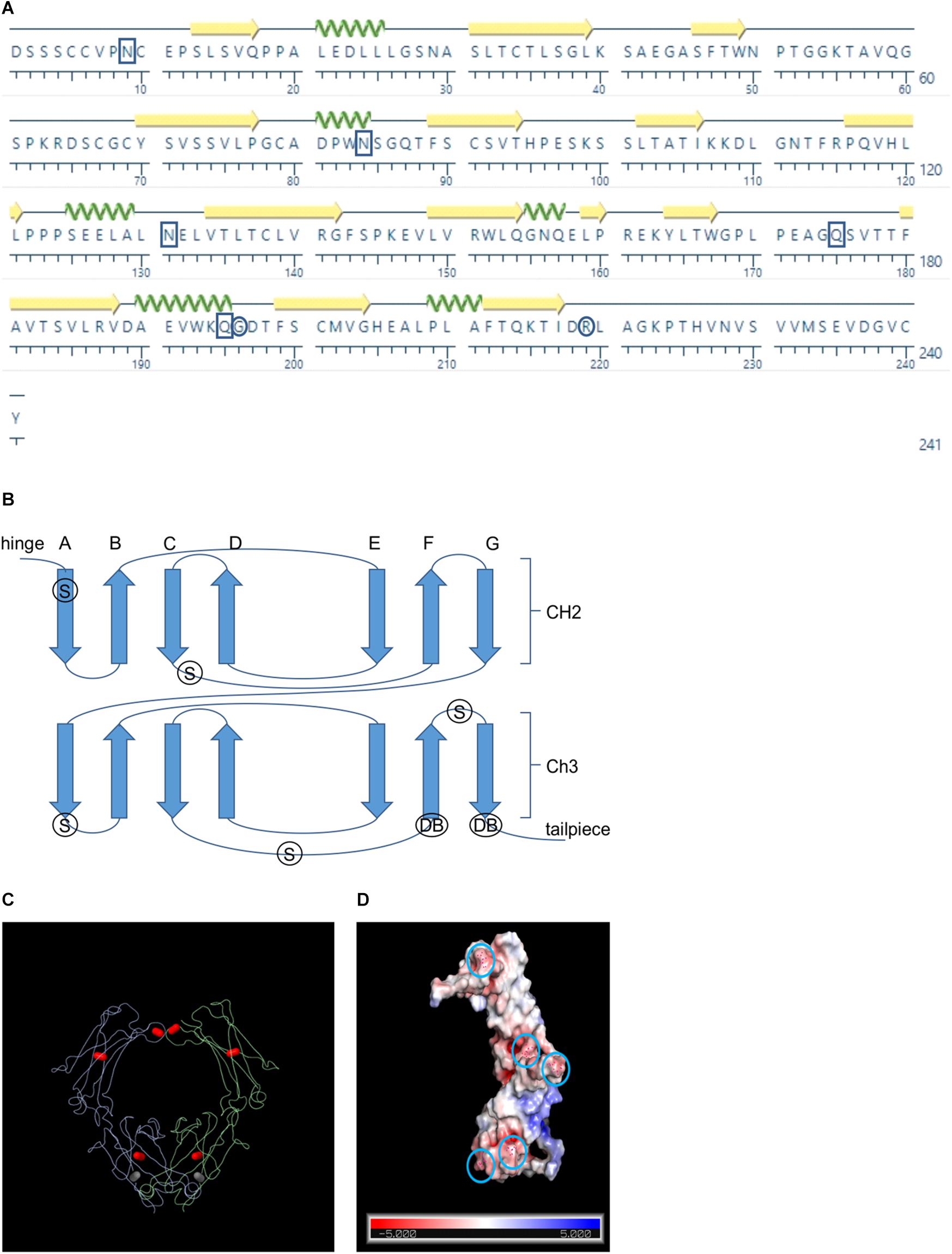
Figure 1. Schematics showing the positions of the rational design candidates. (A) Amino acid sequence of the bovine IgA Fc sequence. Boxes indicate positions of the candidates for supercharging and circles indicate the positions of the candidates for de novo disulfide bonds (B) Schematic showing the Greek key connectivity of the Fc’s β barrel structure. Arrows indicate β strands, S indicates the positions of supercharging candidates and DB indicates the positions of de novo disulfide bond candidates (C) Wire diagram of a dimerized Fc with native intra- and inter- chain disulfides colored in red and the de novo disulfide colored in gray (D) Surface representation of the bovine Fc chain with predicted charge colored on a scale of red indicating more electronegative to blue indicating more electropositive. Circles indicate the positions of the supercharging candidates
To develop a more stable Fc chain that could act as a stabilization partner when fused to a VHH, we tested two different rational design strategies: (1) changing key surface residues to give a more electronegative net charge, and (2) introducing de novo intra-chain disulfide bonds to stabilize the internal structure (Figures 1A–C). A total of 12 mutants were assessed of which five supercharging (N9D, N84D, N131D, Q175E, Q195E) and one disulfide (G196C/R219C) mutants were advanced for further characterization (Supplementary Table 1).
Supercharging residue candidates were determined by modeling a predicted bovine IgA Fc structure using the I-TASSER and PyMol programs (Schrodinger, 2010; Yang et al., 2015) and then selecting the most surface-exposed asparagine and glutamine residues for mutating to their conservative but negatively charged counterparts: aspartic acid and glutamic acid respectively. Aspartic acid and glutamic acid are structurally similar to asparagine and glutamine, differing only in their exposed side chains, and these substitutions are usually well tolerated in maintaining tertiary structure. The asparagines and glutamines were also selected based on non-involvement in native glycosylation (residues 29, 228) or Fc α receptor (FcαR) binding (residues 26, 117, 151, 153, 156, 158, 202, 205, 214). The asparagines that were mutated are unlikely to be involved in N-glycosylation considering that the adjacent residues do not match the standard glycosylation motif (N-X-S/T where X is a non-proline residue) and crystallization studies of human IgA have not suggested otherwise.
For the selection of de novo intra-chain disulfide bonds, the predicted Fc structure was modeled and the disulfide candidates chosen either by manual inspection of the molecule in PyMol or by ranked selection based on stability recommendations using the Disulfide by Design 2.0 software (Supplementary Table 2) (Craig and Dombkowski, 2013). The G196C/R219C disulfide candidate was manually chosen based on neighboring proximity with the intent of tethering the C-terminal end of strand G to the N terminal end of strand F (Figure 1B). The tailpiece leading out from strand G is unstructured and contains a free S-H that natively forms a disulfide bond with the J chain in dimeric IgA. With the G196C/R219C mutation, we hypothesized that tethering the tailpiece to strand F could potentially stabilize the molecule by hiding any vulnerable hydrophobic regions that may become exposed as the strand becomes unstructured, and in so doing, prevent access by proteolytic enzymes. In particular, free floating unstructured tails are known to be preferentially loaded into the 26S proteasome apparatus for degradation (Inobe and Matouschek, 2014). Thus, tethering the tailpiece by use of G196C/R219C could potentially prevent proteasome access.
The rationally designed candidates were generated by site-directed mutagenesis of the native bovine IgA Fc and confirmed by sequencing. All candidates were then cloned into a pCaMGate plant expression vector for ER targeting and retrieval (Pereira et al., 2014). We chose to target the constructs to the ER because of the requirement for disulfide formation for correct folding and assembly and also because VHH-Fc accumulation has been previously shown to be robust (220 mg/kg FW) when targeted to the ER using the pEAQ plant expression vector (Sainsbury et al., 2009; Saberianfar et al., 2019). Screening of ER-targeted wild type and mutant Fc (Supplementary Table 1) was done by agroinfiltration of N. benthamiana leaves and semi-quantitative western blotting at four, six and eight days post-infiltration (dpi). Compared to accumulation of native Fc (42.30 ± 10.83 mg/kg), by day eight, each of the supercharged Fc mutants showed between a two- to four-fold improvement (between N84D at 112.39 ± 17.11 mg/kg to Q195E at 175.17 ± 20.22 mg/kg) that persisted across the time course post infiltration (Figure 2A, Supplementary Table 3). Of the six disulfide pairs tested (Supplementary Table 1), two (K40C/pre-existing cysteine at position 10, and G196C/R219C) improved yield by two- and 10-fold (35.91 mg/kg for K40C and 132.02 mg/kg for G196C/R219C) (Figure 2B). However, upon further examination of the predicted model of the bovine IgA Fc, we decided to abandon K40C because of the potential for distorting correct inter-chain disulfide formation at the hinge. To test if these mutations could be combined to further improve accumulation, we combined the mutations step-wise and measured accumulation in transformed leaf extract by western blot. Combining the mutations gave an incremental increase in accumulation after transient expression. The Fc construct containing all five supercharging residues showed a seven-fold improvement (296.18 ± 18.47 mg/kg) in accumulation compared to native (Figure 2C). Adding the de novo disulfide to these five supercharging residues further improved accumulation to an approximately 32-fold improvement (599.47 ± 25.40 mg/kg) (Figure 2C). To test if these Fc mutants could also enhance accumulation as an Fc fusion protein, we fused each to a VHH that was identified from panning a phage display library with intimin of O157:H7 (denoted as VHH9 in Saberianfar et al., 2019). Similar to the comparison using Fc alone, each of the mutant VHH-Fc fusions showed between a three to six-fold improvement (between 26.42 ± 4.96 mg/kg for N9D to 52.38 ± 1.39 mg/kg for G196C/R219C) in accumulation when compared to native (7.57 ± 0.89 mg/kg) by day eight (Figure 2D). When fused to the VHH, combining the mutations on the Fc incrementally improved accumulation; mutants with all five supercharging mutations plus the disulfide showed a twenty-fold improvement (236.19 ± 41.32 mg/kg) compared to native (12.45 ± 5.86 mg/kg) on day eight (Figure 2E). For consistency, day 8 post-infiltration was selected for extraction towards binding and neutralization assays. Because the engineered construct containing all supercharging and disulfide mutations (5 + 1 Fc) showed the most promise, we focused on it for further characterization.
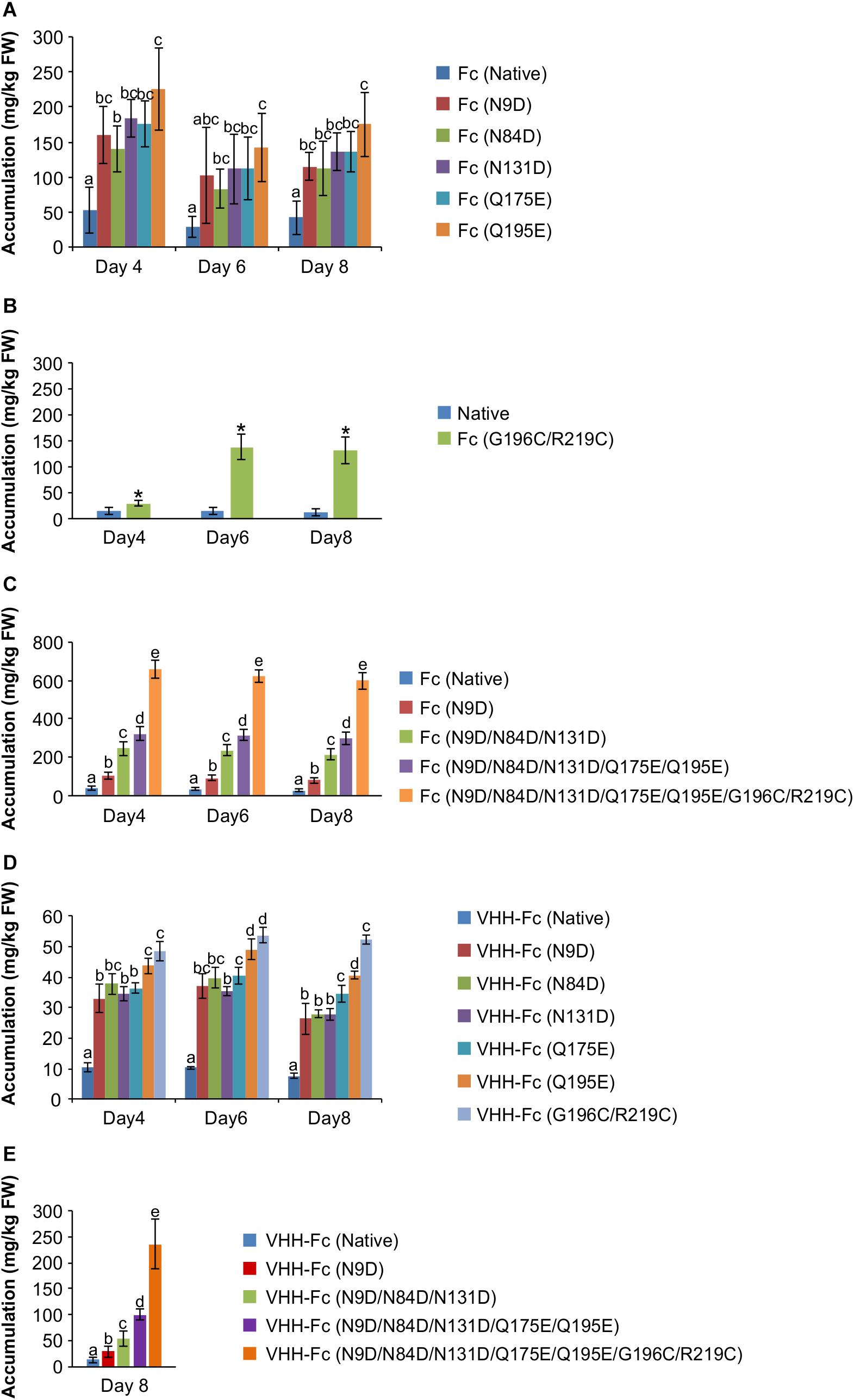
Figure 2. Time courses showing accumulation after transient transformation of N. benthamiana by agroinfiltration. Shown are (A) Supercharging mutations on the Fc chain. (B) Novel disulfides on the Fc chain (C) Combining of mutations on the Fc (D) Fc with individual mutations fused to a VHH (E) Fc with combined mutations fused to a VHH on 8 dpi. * represents statistically significant difference from native as determined by a T-test. Letters denote significantly different treatments as determined by one way ANOVA and post hoc Tukey HSD test. P < 0.05, n = 3-5 biological replicates. Error bars shown are standard error of the mean.
Although the VHH-Fc fusion lacks the light chains and CH1 domains found in native mammalian sIgA, assembly to the JC and SC is directed specifically via disulfide bond formation with the Fc (Supplementary Figure 1). JC forms a disulfide bond with the tailpiece in the CH3 domain from opposite Fc chains orienting them end-to-end, and SC forms a disulfide bond with opposite CH2 domains from each Fc. To determine if engineering of the VHH-Fc affected its ability to assemble with the SC and JC subunits, we co-expressed all three subunits for immunoprecipitation experiments. The JC and SC sequences had previously been cloned into pEAQ-DEST-1 expression vectors (Sainsbury et al., 2009) with ER targeting and retrieval sequences added (Saberianfar et al., 2019). Each subunit is also linked to a different tag (VHH-Fc-c-myc; SC-Flag; JC-HA). Agrobacterium cultures containing each of the VHH-Fc, SC and JC constructs were resuspended in infiltration media, pooled in a 4:1:1 ratio, and co-infiltrated into leaf tissue. Crude extracts were immunoprecipitated with the anti-FLAG antibody specific to the SC subunit, then separated and detected on a western blot probing for either anti-cmyc (VHH-Fc subunit), or anti-HA (JC subunit). Bands matching the predicted 44 kDa size of VHH-Fc were detected with anti-c-myc antibody in crude extract transformed with VHH-Fc, VHH-(5 + 1) Fc, co-expressed VHH-Fc/SC/JC and co-expressed VHH-(5 + 1)Fc/SC/JC, but no bands were detected in crude extract expressing only JC or SC (Figure 3A). After co-IP, ∼44 kDa bands were seen only in extracts co-expressing VHH-Fc/SC/JC and VHH-(5 + 1)Fc/SC/JC (Figure 3B). This suggests that both SC and VHH-Fc or SC and VHH-(5 + 1)Fc interact, and that the mutations in Fc did not hinder this interaction. The size mismatch between the observed band of ∼50 kDa and the predicted size of ∼44 kDa may possibly either be due to a gel shift from the effect of SDS on the tertiary structure (Rath et al., 2009). A laddering effect showing several bands of higher molecular weight may suggest the presence of folding intermediates of the complex or alternatively may be a product of aggregation. Similarly, detection with anti-HA indicated bands of ∼20 kDa, matching the predicted size of JC, in crude extract transformed with JC, VHH-Fc/SC/JC and VHH-(5 + 1)Fc/SC/JC (Figure 3C). After Co-IP, ∼20 kDa bands were seen only for the co-expressed VHH-Fc/SC/JC and VHH-(5 + 1)Fc/SC/JC, indicating that SC and JC are present in the same complex (Figure 3D).
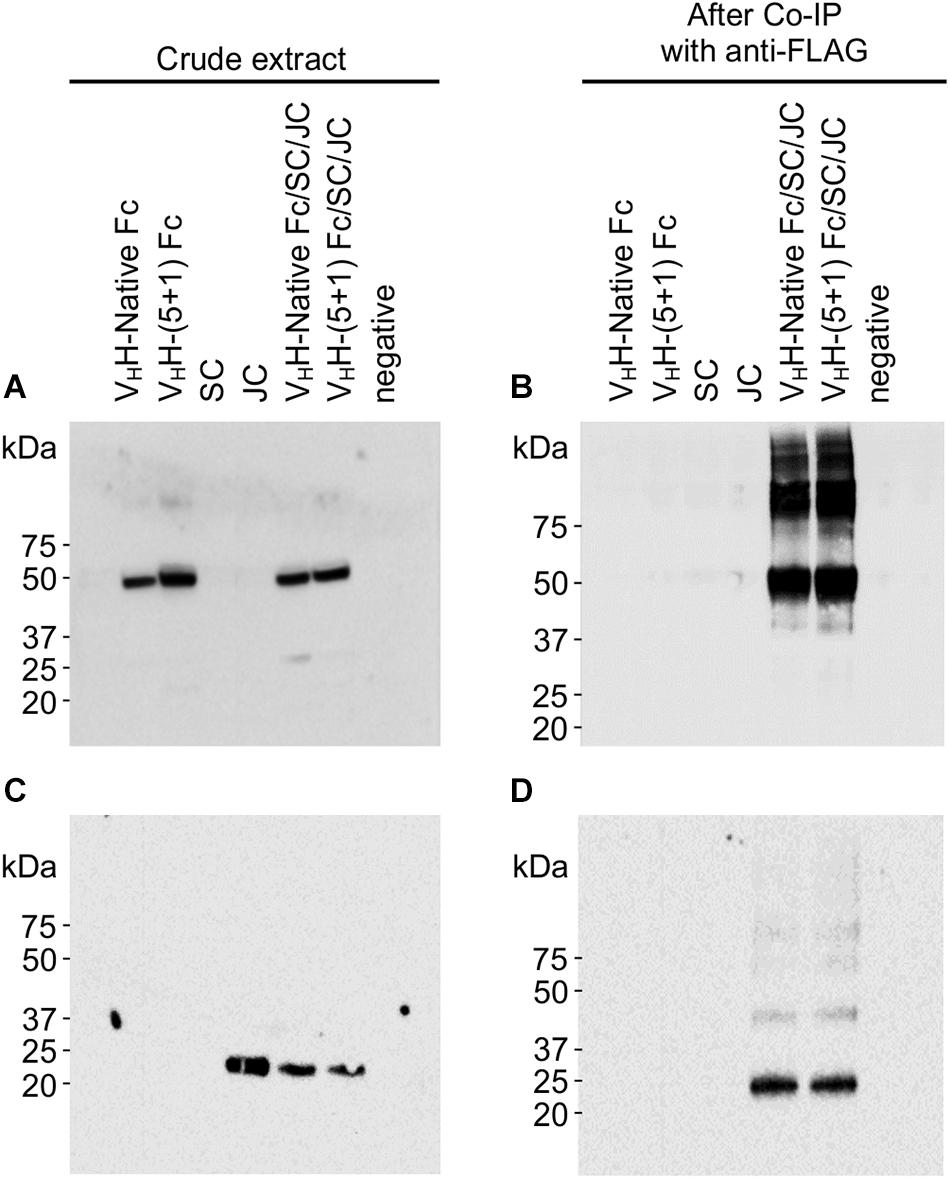
Figure 3. VHH-(5 + 1) Fc retains the ability to assemble with other secretory IgA subunits SC and JC. Shown are Western blots probed with either anti-cmyc (A,B) or anti-HA (C,D), which correspond to differently tagged subunits VHH-Fc and JC respectively. Leaf issue was transformed with constructs of each subunit individually and also with combinations of VHH-Fc/SC/JC and VHH-(5 + 1)Fc/SC/JC for intended co-expression and assembly. Detection was done for both diluted crude leaf extract (A,C) and for the eluent after the extract had been co-immunoprecipitated using an anti-FLAG column (B,D).
To determine if the rationally designed mutations impact VHH-Fc’s pattern of cross-serotype binding to EHEC, we used a FITC conjugated secondary antibody to track the binding of VHH-Fc and VHH-(5 + 1)Fc to bacterial cell surfaces. We saw consistent co-localization of FITC signal with strains O26:H11, O145:Hnm, O111:Hnm and O157:H7 cells for both VHH-Fc and VHH-(5 + 1)Fc (Figure 4). Both VHH-Fc and VHH-(5 + 1)Fc do not seem to bind O45:H2, O121:H19 and O103:H2. We have previously shown by a sequence alignment of the extracellular residues of intimin that this pattern of binding may be explained by greater sequence similarity across strains O26:H11, O145:Hnm, O111:Hnm and O157:H7 and conversely, greater divergence for strains O45:H2, O121:H19 and O103:H2, presumably at antigenic sites to which the VHH binds (Saberianfar et al., 2019). As a negative control, EHEC cells were also treated with PBS instead of antibodies and similarly stained but did not show fluorescence under FITC-related imaging conditions (480 nm excitation and 520–540 nm detection).
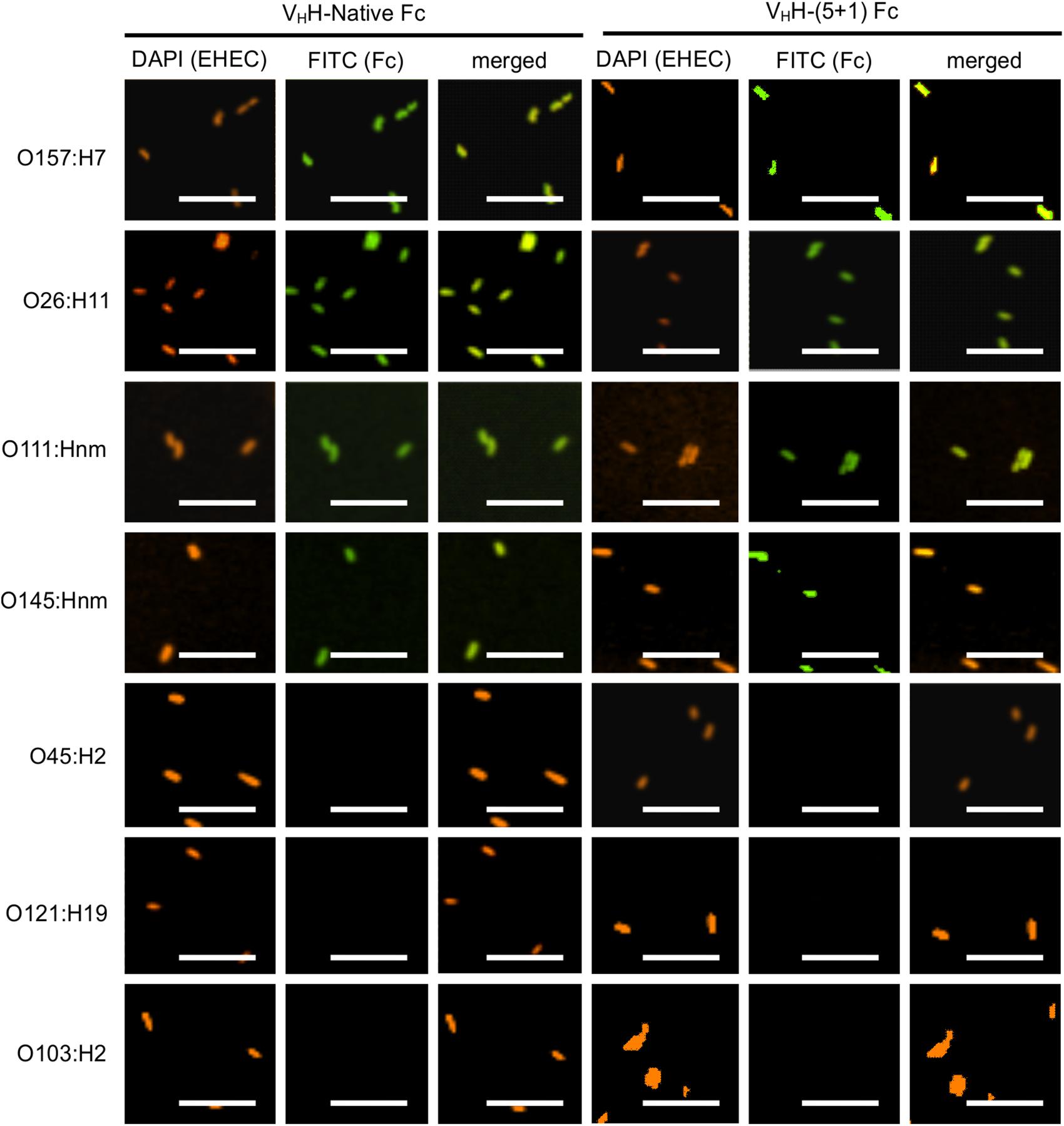
Figure 4. VHH-(5 + 1) Fc retains the same binding pattern across EHEC strains as VHH-Native Fc. Shown are confocal images of the seven most prevalent EHEC strains incubated with either VHH-Native Fc or VHH-(5 + 1) Fc. DAPI was used to visualize EHEC cells (orange) and a FITC-conjugated antibody (green) was used to immunolabel the Fc specifically. Size bar = 10 μm
Intimin, the antigenic target of the VHH-Fc, mediates the intimate attachment of EHEC to epithelial cells. We previously showed that VHH-sIgA neutralizes EHEC’s ability to intimately adhere to epithelial cells in vitro (Saberianfar et al., 2019). Here, we investigated if the VHH-Fc alone is sufficient to neutralize EHEC’s ability to adhere to human epithelial type 2 (HEp-2) cells, and if the rationally designed mutations impacted the VHH-Fc’s pattern of cross-serotype protection. HEp-2 cells have previously been shown to be an appropriate model system for intimin-based adherence and colonization (McKee et al., 1995). Compared to the respective positive controls of no VHH-Fc (+ PBS treatment), the addition of either VHH-Fc or VHH-(5 + 1)Fc seemed to abrogate the adhesion of EHEC strains O26:H11, O111:Hnm, O145:Hnm and O157:H7 to HEp-2 cells (Figure 5). Similarly to the binding assay, both VHH-Fc and VHH-(5 + 1)Fc showed no observable effect on inhibiting adhesion for strains O45:H2, O103:H2 and O121:H19. As a control to show that neutralization is specifically mediated by the VHH, the Fc without an attached VHH was incubated with O157:H7 and HEp-2 cells and showed a similar degree of adherent bacteria, based on microscopy and relative fluorescence, as did incubation without antibody (Figure 5). To quantify the relative neutralization capacity of the VHH-Fc compared to the VHH-(5 + 1)Fc, we adapted the adhesion assay for fluorometry and measured the relative fluorescence of HEp-2 cells incubated with a culture of each of the seven EHEC strains with and without either VHH-Fc or VHH-(5 + 1)Fc. The addition of either antibody showed the same pattern of reducing the relative fluorescence caused by adherent bacteria for strains O26:H11, O111:Hnm, O145:Hnm and O157:H7 to background levels (Figure 6). Similar to the binding assay, strains O45:H2, O121:H19 and O103:H2 showed no neutralization.
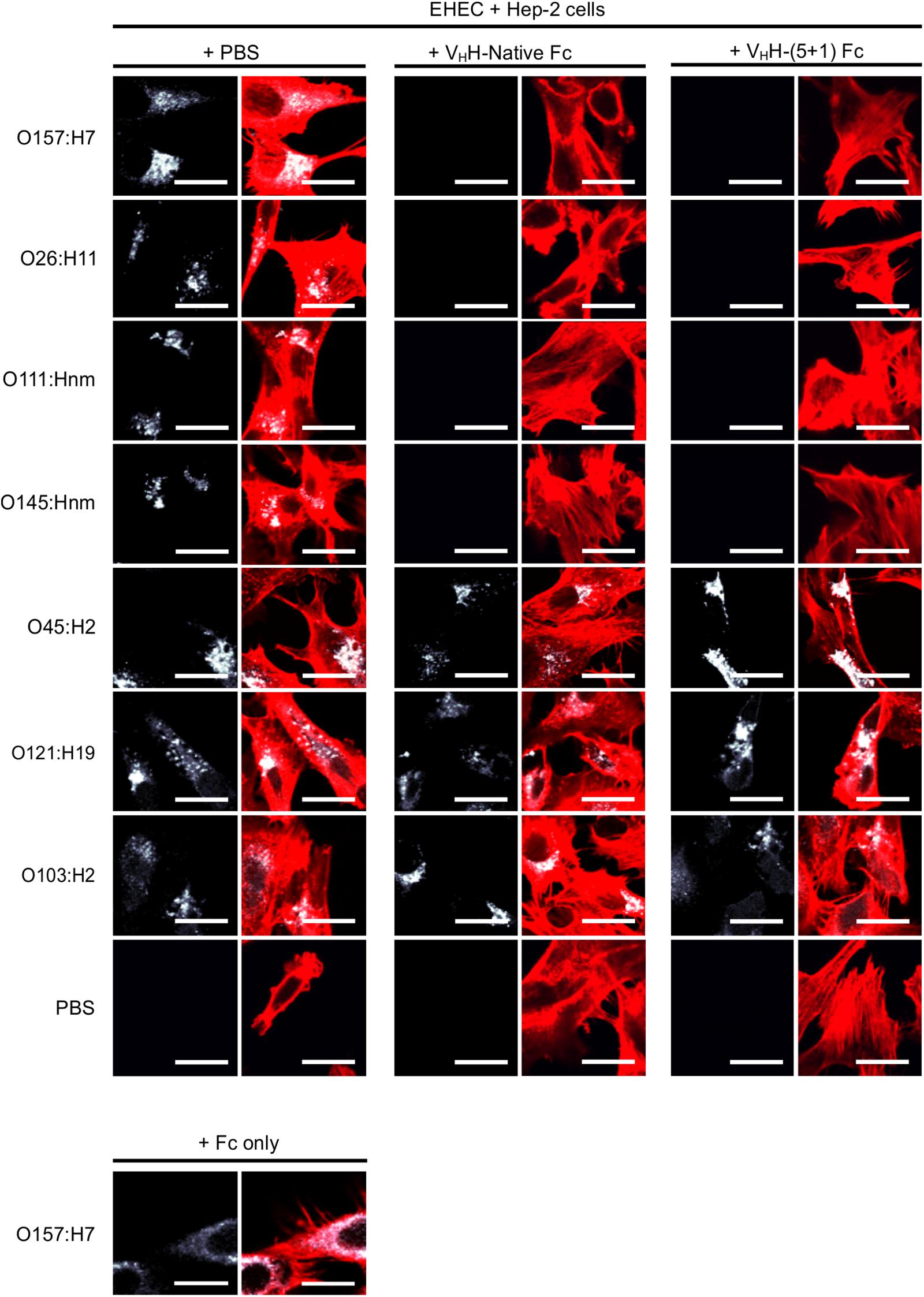
Figure 5. VHH-(5 + 1)Fc retains the VHH-Native Fc’s pattern of cross-serotype protection by neutralizing bacterial levels of O26:H11, O111:Hnm, O145:Hnm and O157:H7. Shown are confocal images of the seven most prevalent EHEC strains (white) that have been incubated with HEp-2 cells (red) in the presence of either 1x PBS as a control, with VHH-Native Fc or with VHH-(5 + 1)Fc. As a control against non-specific Fc binding, O157:H7 was incubated with Fc only to confirm that neutralization was mediated through the VHH. Size bar = 20 μm
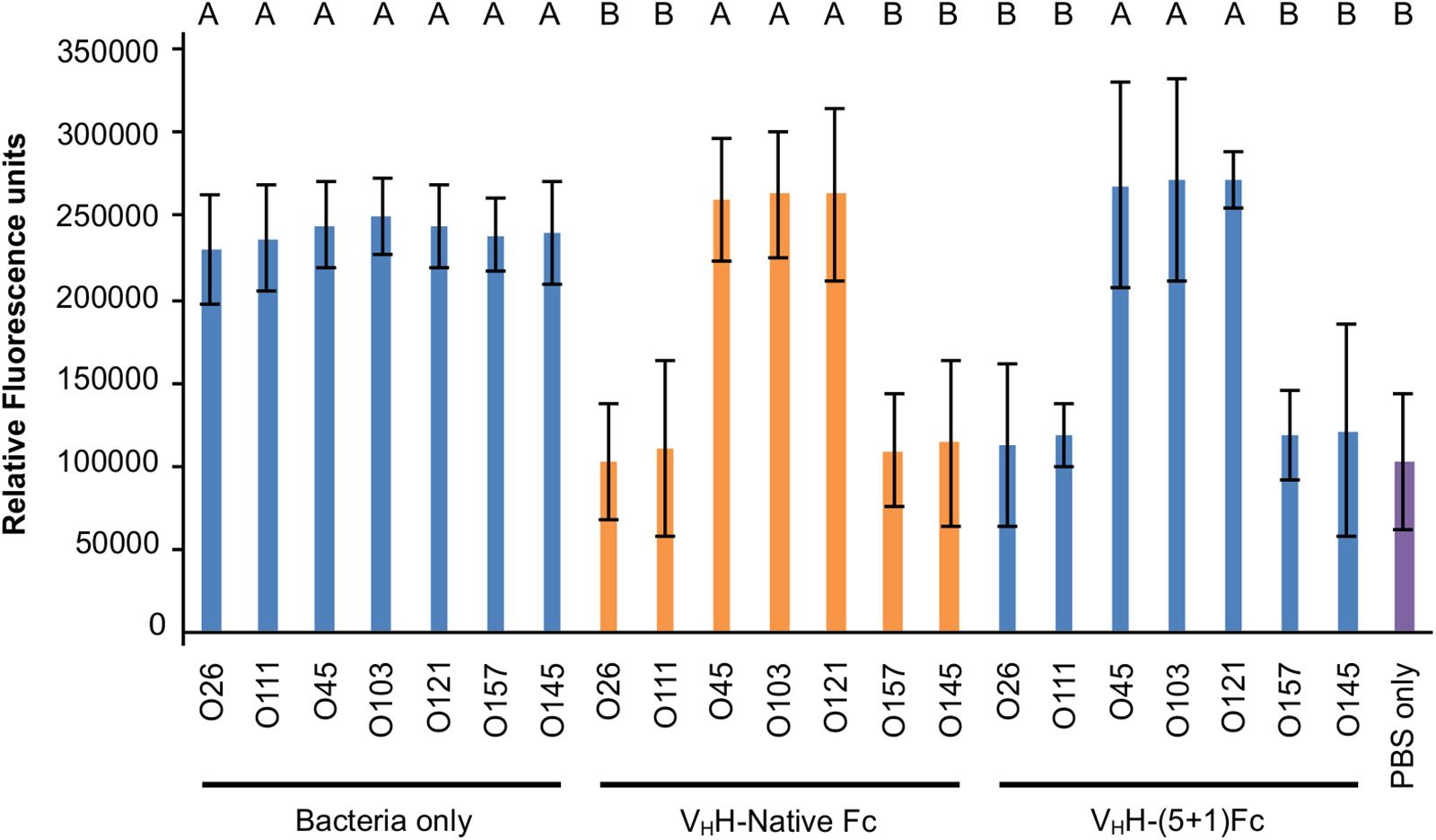
Figure 6. VHH-(5 + 1)Fc neutralizes bacterial levels of O26:H11, O111:Hnm, O145:Hnm and O157:H7 to baseline levels similarly to VHH-Native Fc. Shown is the relative fluorescence of seven EHEC strains that were immunolabelled, incubated on HEp-2 cells and treated with either VHH-Native Fc, VHH-(5 + 1)Fc or with 1x PBS as a positive control. As a negative control, HEp-2 cells were incubated with 1x PBS without addition of a bacterial strain or antibody. Letters indicate a significant difference of the amount of immunolabelled adherent bacteria as determined by a one-way ANOVA with a post hoc Tukey HSD test (p < 0.05, N = 3 biological replicates). Error bars indicate standard error of the mean.
In this study, we engineered a bovine IgA Fc to improve its accumulation alone, and when fused to a VHH, by a combination of five supercharging residues and a disulfide pair to tether an unstructured domain at the C-terminal. Our initial motivation for engineering the Fc was to explore if it could be a practical solution for the problem of heterologous mAb yield in plant platforms, which has been a primary limiting factor for advancing many therapeutics toward market. Conceptually, engineering for protein supercharging relies on an inverse relationship that exists between protein charge extent and aggregation propensity (Carballo-Amador et al., 2019). The concept of ‘resurfacing’ a protein has recently been of interest in the context of biobetter development because substitutions to protein surfaces are usually better tolerated than buried side chains and folding is driven primarily by the loss of solvation of the core hydrophobic residues (Dill et al., 2008; Chapman and McNaughton, 2016). Indeed, because surface side chains do not become buried and retain a similar environment in both a folded versus unfolded state, solvent-exposed amino acids are thought to contribute less energy toward stabilizing the folded state compared to buried core residues (Tokuriki et al., 2007). Our results indicate that each of these supercharging substitutions, along with disulfide mutations, can individually improve in planta accumulation of either the Fc or VHH-Fc by as much as four- and six-fold respectively (Fc: 175.17 ± 20.22 mg/kg FW; VHH-Fc: 52.38 ± 1.40 mg/kg FW), and then be combined for an incremental increase [(5 + 1)Fc: 599.47 ± 25.40; VHH-(5 + 1)Fc: 236.19 ± 41.32]. Regarding market accessibility, these mutations permit overcoming of the rule of thumb benchmark of 100 mg/kg FW (Rybicki, 2009; Schillberg et al., 2019). We have previously found the yield of the native VHH-Fc construct to reach as high as 220 mg/kg FW when expressed using the pEAQ-DEST vector (Saberianfar et al., 2019) whereas in this study, we obtained only 12.5 mg/kg FW for the same construct when expressed using the pCaMGate vector. There are several differences in vector features that may possibly account for this difference in accumulation. In particular, the pEAQ vector contains modified 5′ and 3′-untranslated regions from cowpea mosaic virus (CPMV) RNA-2 that were shown to enhance the level of expression of the gene they flank (Sainsbury and Lomonossoff, 2008). As well, the pEAQ vector permits multi-gene expression of both the native VHH-Fc and p19, a suppressor of post-transcriptional gene silencing (Silhavy et al., 2002). A multi-gene vector may allow for a more thorough infiltration of both constructs into plant cells compared to co-infiltrating Agrobacteria containing different vectors, as done in the current study, and thus the lower accumulation may reflect a greater degree of post-transcriptional gene silencing (Westerhof et al., 2016). Although the absolute accumulation levels are lower with the pCaMGate vector, the relative yield increase from engineering the Fc is nonetheless meaningful as a yield improvement technique and we expect the mutations we have introduced in the bovine IgA Fc to lead to higher accumulation than reported by Saberianfar et al. (2019) when using the pEAQ vector. Previous studies using IgG Fc in bacterial systems have found similar success in promoting mAb stabilization by supercharging, particularly substitutions using aspartic and glutamic acid (Dudgeon et al., 2012; Buchanan et al., 2013; Lee et al., 2013; Courtois et al., 2015). This study is the first report demonstrating that this strategy can be successfully implemented for an IgA Fc in a plant platform.
Of the six supercharging candidates tested, five improved yield. The failure of the sixth to improve yield may be due to an incorrect prediction of surface exposure of that residue. If that residue was buried then its mutation to aspartic acid would not be expected to have an effect on surface charge and thus an effect on aggregation. Regarding the appropriateness of using a predicted structure for rational design, a previous validation study of I-TASSER on de novo protein design of 87 proteins showed that I-TASSER could accurately fold the designed sequences with an average root mean square deviation (RMSD) of 2.1Å (Mitra et al., 2013). Previous studies have also used I-TASSER for rationally designing stability into a variety of proteins (Wan et al., 2019; Guo et al., 2019; Ashfaq et al., 2021). Still, the resolution of the model is a key limitation on the success rate of the designed screen whereby inaccurately estimated surface exposure of asparagine and glutamine residues or the Cα distance of neighboring cysteines directly affects the prediction of supercharging or de novo disulfide mutations respectively. In another related project, we performed a similar screen on an EHEC subunit vaccine and found that using two supercharging candidates for an EHEC subunit vaccine improved yield by ∼20% when produced in the chloroplast (Chin-Fatt, unpublished). Notably, this protein was both intrinsically disordered and a membrane protein. Thus, this rational design approach may potentially be broadly applicable for other plant produced recombinant proteins.
We also designed a novel disulfide bridge in the CH3 domain of the Fc to fasten the C-terminal end of strand G to the N terminal end of strand F. In the disulfide screen, only two of the six disulfide candidates showed improved yield. Incorrect disulfide mutants are known to be strongly destabilizing because free thiol groups of unpaired cysteines are highly reactive when folding in the ER leading to intra-molecular disulfide scrambling, covalent oligomerization and consequently degradation (Trivedi et al., 2009). However, in a similar study, a de novo disulfide bond that tethered two β strands in two opposite β sheets of a variable heavy chain domain was proposed as a general strategy to increase thermal and conformational stability in nanobodies without compromising functionality (Saerens et al., 2008). Because nanobodies also tend to preserve the Ig-like fold, this may suggest a broader applicability of disulfide based domain tethering for stabilization of Greek key proteins. Interestingly, the study’s novel cysteine pairs were also not able to be predicted by algorithms, including Disulfide by Design, and were instead, like for G196C/R219C, identified based on manual inspection of the Cα distance in crystal structures and side chain orientation.
Another protein engineering approach for overcoming low yields of plant-made antibodies was based on exchanging sequences in the variable heavy chain domains of separate antibodies that showed differential accumulation (Zischewski et al., 2016). After transient expression in leaves of N. benthamiana, yield increased for several chimeras ranging from ∼250 mg/kg FW to as high as 2,000 mg/kg FW. Their analysis of the transcript and protein levels of the native and chimeric antibodies also suggested that the main events limiting yield were either translational or post-translational in nature. So, to address low yields of plant-made antibodies, the main research focus should arguably be to stabilize the protein folded state, such as by the rational design of supercharging and de novo disulfides. Another approach for stabilizing a murine IgG in Nicotiana tabacum has been to mutate protease-sensitive sites in the antibody light and heavy chains to increase proteolytic resistance (Hehle et al., 2016). Alteration of these sites was shown to enrich for the full-size antibody and to reduce the accumulation of fragments from proteolytic degradation. The negative impact of endogenous proteases on the yields of plant recombinant proteins targeted to the secretory pathway in N. benthamiana is well documented (Benchabane et al., 2010; Robert et al., 2013; Jutras et al., 2020). Although targeting sequence specific cleavage sites is important, our results suggest that degradation by proteolysis at unstructured regions may not only be substantial but also may be mitigated by appropriate disulfide tethering.
Co-immunoprecipitation of the differentially tagged subunits that had been co-expressed indicated that engineering of the Fc did not impact its capacity to assemble with the JC and SC subunits to form the secretory complex. Although the JC can double the avidity, and thus the potency, and the SC can protect the complex from proteolytic attack, full assembly comes at the cost of yield (Saberianfar et al., 2019). We had previously characterized the binding and neutralization of a separate anti-EHEC VHH-sIgA because it was previously thought that the entire complex was needed for protection as the secretory form is the predominant functional unit in mammals (Saberianfar et al., 2019). Although these VHH-sIgA’s are effective, accumulation is apparent as multiple assembly intermediates and, if selecting for only the final multi-subunit assembly, yield is diminished. Other groups have observed such a reduction for murine, chicken and human sIgA in plants with a significant proportion of monomeric IgA and assembly intermediates occurring more so than fully assembled sIgA (Ma et al., 1995; Wieland et al., 2006; Juarez et al., 2013; Paul et al., 2014). A possible reason for this is a cryptic sequence in the tailpiece targeting the Fc to the vacuole before disulfide formation with the JC can be accomplished (Frigerio et al., 2000; Hadlington et al., 2003). Indeed, joining chain incorporation has been shown to be a major limiting step for assembly (Westerhof et al., 2016). Also, the presence of assembly intermediates may be due to sporadic transformation of plant cells such that expression of the fully assembled sIgA may be limited to occurring only in the cells receiving all transgenes. Indeed, expression of the fully assembled sIgA has been shown to be enriched using a multi-gene vector (Westerhof et al., 2016). Recently, an anti-F4 VHH-Fc monomer was shown to be effective in protecting piglets against F4 ETEC challenge suggesting that the assembled secretory format may not be necessary for protection (Virdi et al., 2019). The use of a single transgene, rather than three, simplifies production, improves yield and is still protective in animal models. So, with yield as a priority, we opted to focus on the simplified monomer to characterize VHH functionality.
We also showed that engineering of the Fc did not impair the VHH’s pattern of cross serotype binding and neutralization for strains O26:H11, O111:Hnm, O145:Hnm and O157:H7. Collectively, these strains account for ∼72% of all EHEC-related illnesses, affecting an estimated 1,158 per 100,000 individuals in the U.S. (CDC, 2017). Testing for cross-serotype protection is important because the vast majority of EHEC surveillance and therapeutic development has historically focused on O157:H7 only, despite the growing importance of non-O157 strains. Based on surveillance of agricultural sites across Canada, the prevalence of non-O157:H7 strains now exceeds that of O157:H7 by about 7% (Public Health Agency of Canada, 2015). Currently, there are no EHEC diagnostics or antibody therapeutics in the market or in the pipeline that have been shown to detect O157:H7 as well as non-O157:H7 strains.
In this study we demonstrated that rationally designing an IgA Fc for supercharged residues and de novo disulfides can impart a significant improvement of in planta accumulation without sacrificing the binding efficacy of the VHH or the Fc’s ability to structurally assemble with other secretory subunits. Given that combining these mutations did not seem to destabilize the protein, it is tempting to speculate on identifying both the upper limit of mutation that can be accommodated as well as an exhaustive list of all relevant mutations in order to fully optimize accumulation. Based on a stability estimating algorithm across globular proteins, about 70% of amino acid mutations are neutral and 20% are significantly destabilizing (Tokuriki et al., 2007). Within the scope of all possible amino acid variations of the Fc being 1.15 × 1048, isolating stabilizing mutants by systematic or random mutation is not practical. Therefore, relevant techniques to identify stabilizing mutations are required. With regards to combining mutations, supercharging has been used to alter the net charge of GFP by as much as 55 charge units without compromising protein folding or function and enabling significant resistance to aggregation (Lawrence et al., 2007). Also, various separate rational design strategies have been combined for improving protein stability without compromising function (Courtois et al., 2016; Fei et al., 2013). Given that we were able to combine supercharging and disulfide mutations without noticeably compromising function, this raises questions of whether this strategy can be combined with other strategies to further improve yield or if there is a theoretical mutational load that can be tolerated within the context of the plant cell as a heterologous environment. Because it folds independently from the VHH, this engineered IgA Fc scaffold may potentially be useful as a modular tool for improving accumulation of other VHHs as well. Future experiments will seek to test how universally applicable (5 + 1)Fc is as a scaffold. Although plant-based IgA production is still an emerging field, we are optimistic that such a tool could be of value in overcoming the yield hurdles that have thus far hindered transition of plant-based antibody therapeutics to market.
Estimating of negatively supercharged Fc candidates was performed computationally by first ranking residues for solvent accessibility by their average number of neighboring atoms (within 10 Å) per side-chain atom (AvNAPSA) and then identifying highly polar solvent-exposed Asn and Gln residues for mutation to their negatively charged counterparts, Asp and Glu respectively (Schrodinger, 2010). For visualizing the multi-subunit complex, the VHH-Fc sequence was submitted as a dimer to the SPRING server which uses a template-based threading algorithm across the PDB library to predict structure and assembly (Guerler et al., 2013)
Disulfide candidates were selected by manual inspection of the model in PyMol (Schrodinger, 2010) based on residue proximity (under 5 Å) or by ranked selection of ∑B-factor, a measure of dynamic mobility for each atom, in the DisulfidebyDesign 2.0 software (Craig and Dombkowski, 2013). To retain the functionality of the native Fc, native disulfide sites were avoided (inter-chain: 16-18, 205-207, 199-201, 13-15; intra-chain: 100-102, 271-273, 412-414, 601-603; tailpiece to JC: 718-720; 235-237; free S-H: 235-237).
The bovine Fc, JC and SC sequences were obtained from the NCBI public database (ANN46383, NP_786967 and NP_776568 respectively) and synthesized by Bio Basic Inc. (Markham, ON, Canada). Rational design mutations were individually made to the native bovine IgA Fc sequence using an in vitro single primer site-directed mutagenesis method (Huang and Zhang, 2017). Combination of mutations was done using a multi-site-directed mutagenesis method (Liang et al., 2012). Genetic fusion to an anti-EHEC VHH (denoted as VHH9 identified previously in Saberianfar et al., 2019) was done using a sequence and ligation independent cloning (SLIC) method (Li and Elledge, 2007). All cloning was confirmed by sequencing.
To enable expression in leaf tissue, each Fc and VHH-Fc construct was cloned into an in-house pCaMGate plant expression vector (Pereira et al., 2014) using the Gateway® cloning kit (Thermo-Fisher Scientific Inc., Waltham, MA, United States). The pCaMGate vector attaches a PR1b tobacco signal peptide for targeting the protein to the secretory pathway, an N-terminal Xpress tag for protein stability, a C-terminal c-myc tag for detection and purification as well as a C-terminal KDEL tag for retrieval to the ER of the plant cell. The JC and SC sequences were cloned into pEAQ-DEST-1 expression vectors (Sainsbury et al., 2009) along with C-terminal HA and FLAG tags respectively (Saberianfar et al., 2019). Vectors were then transformed into Agrobacterium tumefaciens (EHA105) and transformed bacteria were selected, using kanamycin and rifampicin antibiotics. Cultures were resuspended into infiltration media consisting of Gamborg’s B-5 medium solution (3.2 g/L Gamborg’s B5 salts with vitamins, 20 g/L sucrose, 10 mM MES, pH 5.6, 200 μM acetosyringone). Each construct was co-infiltrated with a vector carrying p19, a suppressor of post-transcriptional gene silencing from Cymbidium ringspot virus (Silhavy et al., 2002), each at a final optical density (OD600) of 0.3. Transient expression was performed by syringe infiltration into leaf tissue of N. benthamiana plants. Plants were grown in a growth chamber at 22°C with a 16 h photoperiod at a light intensity of 110 μmol m–2 s–1 for 7 weeks and fertilized with water soluble N:P:K (20:8:20) at 0.25 g/L (Plant products, Brampton, ON, Canada).
Pre-weighed leaf samples were frozen in liquid nitrogen and homogenized with silica beads (Bio Spec Products Inc., Bartlesville, OK, United States) for 2 min using a TissueLyser II (Retsch Inc., Newton, PA, United States). One mL of either a denaturing extraction buffer (1x PBS, pH 7.5, 4% SDS, 2% PVPP) or a native buffer (1x PBS, pH 7.5, 0.1% Tween-20, 1 mM EDTA, 2% PVPP, 100 mM sodium ascorbate, 8 M sucrose, 1 μg/mL leupeptin, 1 mM PMSF, 1 μg/mL pepstatin A) was added per approximately one hundred mg of sample. All samples were vortexed on high speed for 30 s and centrifuged at 20,000 x g for 10 min to remove insoluble debris. Extracted proteins were combined with 1/5th volume of 5 x reducing loading buffer (0.3 M Tris-HCl pH 8.0, 5% SDS, 10% glycerol, 100 mM DTT, 0.05% Phenol Red) heated at 90°C for 10 min, then loaded onto Express Plus 4-20% gradient polyacrylamide gels (Genscript Inc., Piscataway, NJ, United States). Gels were run at 100 V for 100 min, then transferred to polyvinylidine difluoride (PVDF) membrane using the Trans-Blot Turbo transfer system (Bio-Rad Laboratories Inc., Hercules, CA, United States). Blots were blocked overnight with 5% skimmed milk in tris-buffered saline, pH 7.5, and proteins of interest were probed with a mouse anti-c-myc antibody (diluted 1:1,000; Genscript Inc., Piscataway, NJ, United States) and the One-Hour Basic western kit for mouse primary antibody (Genscript Inc., Piscataway, NJ, United States). Detection was performed using Amersham ECL western blot detection reagents (GE Healthcare, Mississauga, ON, Canada) or Enhanced Chemiluminescent detection solution (Biorad Laboratories Inc., Hercules, CA, United States) and a MicroChemi 4.2 imaging system with GelCapture acquisition software (DNA Bio-Imaging Systems Ltd., Jerusalem, Israel). For staining, membranes were rinsed in methanol followed by ultrapure water, stained using GelCode Blue (Thermo-Fisher Scientific Inc., Waltham, MA, United States) for 15 min, and destained in 50% methanol 1% acetic acid for 15 min. Quantification of accumulation was done by densitometry using a calibrated standard curve of an in-house-produced purified protein. Statistical significance for accumulation in tissue expressing the native and mutant Fc was determined using a one-way ANOVA with three to five biological replicates. Post hoc comparisons were then performed on the accumulation means using the Tukey HSD test.
Plant extracts were prepared under native conditions as described above. All samples were vortexed on high speed for 30 s and centrifuged twice at 20,000 × g for 10 min to remove insoluble debris. Purification on the supernatant was performed by affinity chromatography using an anti-c-myc mild purification kit (MBL International Corp., Woburn, MA, United States) according to the manufacturers’ protocol. Briefly, 100 μL anti-c-Myc tag bead suspension was added to 3 mL of clarified extract and incubated at 4°C for one hour using an end-over-end shaker. The extract was then added to a spin column and centrifuged for 10 s. The beads were then washed three times and the protein eluted using a c-Myc tag peptide in 1x PBS.
Enterohemorrhagic E. coli strains O26:H11, O45:H2, O103:H2, O121:H19, O111:Hnm and O157:H7 were obtained from Dr. Michael Mulvey at the Public Health Agency of Canada, National Microbiology Laboratory, E. coli Unit, Enteric Diseases Program, Winnipeg, MB. EHEC strain O145 (C625) was obtained from the American Type Culture Collection supplied by Cedarlane Labs in Burlington, ON. Strains were stored at −80°C in a level 2 containment laboratory. All experiments with EHEC were conducted in a level 2 containment laboratory. For propagation of live cultures, single colonies were picked and inoculated in Luria-Bertani (LB) medium overnight and then subculture into pre-warmed Dulbecco’s Modified Eagle Medium (DMEM) (Thermo-Fisher Scientific, Cat. No. 10566016) at a 1: 50 dilution and incubated at 37°C in 5% CO2 for 2 h without shaking. Binding and neutralization assays were performed as previously described (Saberianfar et al., 2019).
To assess binding, we incubated either VHH-Fc or VHH-(5 + 1)Fc with the seven EHEC strains recognized as food adulterants, EHEC O26:H11, O45:H2, O103:H2, O145:Hnm, O121:H19, O111:Hnm or O157:H7. After washing with PBS and fixing with 2.5% paraformaldehyde, we visualized bacteria with DAPI and VHH-Fc binding using a secondary fluorescent antibody (rabbit anti-bovine-FITC) (1:40 dilution, Thermo-Fisher Scientific, Cat. No. SA1-36043) that binds Fc. The cells were then dried onto poly-L-lysine coated coverslips (Millipore Sigma, Cat, No. S1815) and mounted onto glass slides with Aqua-Poly/Mount (Polyscience Inc., Warrington, PA, United States, Cat. No. 18606). To visualize co-localization, FITC and DAPI sequential imaging was performed with an Olympus LSM FV 1200 using a 100x oil objective lens. FITC fluorescence was excited using a 480 nm laser and detected at 520–540 nm. DAPI fluorescence was excited at 350 nm and detected at 455–465 nm.
To assess neutralization, eight-well chamber slides were seeded with ∼2 × 105 human epithelial-2 (HEp-2) cells (American Type Culture Collection) in DMEM supplemented with 10% fetal bovine serum (FBS) and incubated overnight at 37°C in 5% CO2. For the assay, the DMEM was removed from the culture and replaced with 225 μL fresh DMEM. HEp2 cells were then incubated with 25 μL of an overnight starter culture of one of seven EHEC strains (O26:H11, O45:H2, O103:H2, O145:Hnm, O121:H19, O111:Hnm and O157:H7) in the presence or absence of either the VHH-Fc or VHH-(5 + 1)Fc. Cells were then washed in 1x PBS to remove any non-adherent bacteria, fixed in 2.5% paraformaldehyde and then visualized using fluorescent actin staining with Alexa 647 rhodamine phalloidin (red) (Thermo-Fisher Scientific, Cat. No. A22287) on HEp-2 cells and a donkey anti-rabbit alexa 350 secondary antibody (blue) (Thermo-Fisher Scientific, Cat. No. A10039) that specifically hybridizes to EHEC cells. Cells were then washed in 1x PBS and mounted using AquaPoly/Mount (PolyScience Inc., Warrington, PA, United States, Cat. No. 18606). To visualize adherence to HEp-2 cells, sequential imaging was done using an Olympus LSM FV 1200 and a 64x water objective lens. Alexa 647 phalloidin was excited at 650 nm and detected at 660 nm-680 nm. The donkey anti-rabbit Alexa 350 antibody was excited at 350 nm and detected at 455-465 nm. To quantify HEp-2 adherence inhibition by fluorometry, the assay was adapted by growing the HEp-2 cells in 96-well black fluorometry plates that had been previously coated with polyD-lysine, which enables cell adherence. Incubation, washing and immunolabelling was done similarly as above. Relative fluorescence was then measured using a Synergy2 plate reader (Biotek, Winooski, United States) using the Gen5 v1.10 software (Biotek). Wells were measured at 37°C with 5 s intermediate shaking with the same excitation and detection conditions as above.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
AC-F and RM conceived the study. AC-F performed the experiments and wrote the manuscript. AC-F, RS, and RM edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by Agriculture and Agri-Food Canada A-base project 1258 to RM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Hong Zhu and Angelo Kaldis at Agriculture and Agri-Food Canada for providing technical support, Andrew Scott for help with E.coli and HEp-2 cell culture, and Alex Molnar for assistance with preparation of figures.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.651262/full#supplementary-material
Ashfaq, U. A., Saleem, S., Masoud, M. S., Ahmad, M., Nahid, N., Bhatti, R., et al. (2021). Rational design of multi epitope-based subunit vaccine by exploring MERS-COV proteome: reverse vaccinology and molecular docking approach. PLoS One 16:e0245072. doi: 10.1371/journal.pone.0245072
Bakema, J. E., and Van Egmond, M. (2011). Immunoglobulin A: a next generation of therapeutic antibodies? MAbs 3, 352–361. doi: 10.4161/mabs.3.4.16092
Bakema, J. E., Ganzevles, S. H., Fluitsma, D. M., Schilham, M. W., Beelen, R. H., Valerius, T., et al. (2011). Targeting FcalphaRI on polymorphonuclear cells induces tumor cell killing through autophagy. J. Immunol. 187, 726–732. doi: 10.4049/jimmunol.1002581
Bastian, A., Kratzin, H., Eckart, K., and Hilschmann, N. (1992). Intra- and interchain disulfide bridges of the human J chain in secretory immunoglobulin A. Biol. Chem. Hoppe Seyler 373, 1255–1263. doi: 10.1515/bchm3.1992.373.2.1255
Benchabane, M., Schlüter, U., Vorster, J., Goulet, M. C., and Michaud, D. (2010). Plant cystatins. Biochimie 92, 1657–1666. doi: 10.1016/j.biochi.2010.06.006
Beutin, L., Geier, D., Steinruck, H., Zimmermann, S., and Scheutz, F. (1993). Prevalence and some properties of verotoxin (Shiga-like toxin)-producing Escherichia coli in seven different species of healthy domestic animals. J. Clin. Microbiol. 31, 2483–2488. doi: 10.1128/jcm.31.9.2483-2488.1993
Boehm, M. K., Woof, J. M., Kerr, M. A., and Perkins, S. J. (1999). The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 286, 1421–1447. doi: 10.1006/jmbi.1998.2556
Buchanan, A., Clementel, V., Woods, R., Harn, N., Bowen, M. A., Mo, W., et al. (2013). Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. MAbs 5, 255–262. doi: 10.4161/mabs.23392
Carballo-Amador, M. A., Mckenzie, E. A., Dickson, A. J., and Warwicker, J. (2019). Surface patches on recombinant erythropoietin predict protein solubility: engineering proteins to minimise aggregation. BMC Biotechnol. 19:26. doi: 10.1186/s12896-019-0520-z
CDC (2017). Foodborne Diseases Active Surveillance Network (FoodNet): FoodNet 2015 Surveillance Report (Final Data). Atlanta, GA: U.S. Department of Health and Human Services.
Chapman, A. M., and McNaughton, B. R. (2016). Scratching the surface: resurfacing proteins to endow new properties and function. Cell Chem. Biol. 23, 543–553. doi: 10.1016/j.chembiol.2016.04.010
Courtois, F., Agrawal, N. J., Lauer, T. M., and Trout, B. L. (2016). Rational design of therapeutic mAbs against aggregation through protein engineering and incorporation of glycosylation motifs applied to bevacizumab. MAbs 8, 99–112. doi: 10.1080/19420862.2015.1112477
Courtois, F., Schneider, C. P., Agrawal, N. J., and Trout, B. L. (2015). Rational design of biobetters with enhanced stability. J. Pharm. Sci. 104, 2433–2440. doi: 10.1002/jps.24520
Craig, D. B., and Dombkowski, A. A. (2013). Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinform. 14, 1–7. doi: 10.1186/1471-2105-14-346
Czajkowsky, D. M., Hu, J., Shao, Z., and Pleass, R. J. (2012). Fc-fusion proteins: new developments and future perspectives. EMBO Mol. Med. 4, 1015–1028. doi: 10.1002/emmm.201201379
De Greve, H., Virdi, V., Bakshi, S., and Depicker, A. (2020). Simplified monomeric VHH-Fc antibodies provide new opportunities for passive immunization. Curr. Opin. Biotechnol. 61, 96–101. doi: 10.1016/j.copbio.2019.11.006
Dill, K. A., Ozkan, S. B., Shell, M. S., and Weikl, T. R. (2008). The protein folding problem. Annu. Rev. Biophys. 37, 289–316.
Dudgeon, K., Rouet, R., Kokmeijer, I., Schofield, P., Stolp, J., Langley, D., et al. (2012). General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 10879–10884. doi: 10.1073/pnas.1202866109
Fei, B., Xu, H., Cao, Y., Ma, S., Guo, H., Song, T., et al. (2013). A multi-factors rational design strategy for enhancing the thermostability of Escherichia coli AppA phytase. J. Ind. Microbiol. Biotechnol. 40, 457–464. doi: 10.1007/s10295-013-1260-z
Frigerio, L., Vine, N. D., Pedrazzini, E., Hein, M. B., Wang, F., Ma, J. K.-C., et al. (2000). Assembly, secretion, and vacuolar delivery of a hybrid immunoglobulin in plants. Plant Physiol. 123, 1483–1494. doi: 10.1104/pp.123.4.1483
Geissmann, F., Launay, P., Pasquier, B., Lepelletier, Y., Leborgne, M., Lehuen, A., et al. (2001). A subset of human dendritic cells expresses IgA Fc receptor (CD89), which mediates internalization and activation upon cross-linking by IgA complexes. J. Immunol. 166, 346–352. doi: 10.4049/jimmunol.166.1.346
Guerler, A., Govindarajoo, B., and Zhang, Y. (2013). Mapping monomeric threading to protein–protein structure prediction. J. Chem. Inf. Model. 53, 717–725. doi: 10.1021/ci300579r
Guo, C., Wang, X., Zhang, H., Zhi, L., Lv, T., Li, M., et al. (2019). Structure-based rational design of a novel chimeric PD1-NKG2D receptor for natural killer cells. Mol. Immunol. 114, 108–113. doi: 10.1016/j.molimm.2019.07.009
Hadlington, J. L., Santoro, A., Nuttall, J., Denecke, J., Ma, J. K.-C., Vitale, A., et al. (2003). The C-terminal extension of a hybrid immunoglobulin A/G heavy chain is responsible for its Golgi-mediated sorting to the vacuole. Mol. Biol. Cell 14, 2592–2602. doi: 10.1091/mbc.e02-11-0771
Hehle, V. K., Paul, M. J., Roberts, V. A., van Dolleweerd, C. J., and Ma, J. K. (2016). Site-targeted mutagenesis for stabilization of recombinant monoclonal antibody expressed in tobacco (Nicotiana tabacum) plants. FASEB J. 30, 1590–1598. doi: 10.1096/fj.15-283226
Huang, Y., and Zhang, L. (2017). An in vitro single-primer site-directed mutagenesis method for use in biotechnology. Methods Mol. Biol. 1498, 375–383. doi: 10.1007/978-1-4939-6472-7_26
Inobe, T., and Matouschek, A. (2014). Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 24, 156–164. doi: 10.1016/j.sbi.2014.02.002
Juarez, P., Huet-Trujillo, E., Sarrion-Perdigones, A., Falconi, E. E., Granell, A., and Orzaez, D. (2013). Combinatorial analysis of secretory immunoglobulin A (sIgA) expression in plants. Int. J. Mol. Sci. 14, 6205–6222. doi: 10.3390/ijms14036205
Jutras, P. V., Dodds, I., and van der Hoorn, R. A. (2020). Proteases of Nicotiana benthamiana: an emerging battle for molecular farming. Curr. Opin. Biotechnol. 61, 60–65. doi: 10.1016/j.copbio.2019.10.006
Jutras, P. V., Marusic, C., Lonoce, C., Deflers, C., Goulet, M. C., Benvenuto, E., et al. (2016). An accessory protease inhibitor to increase the yield and quality of a tumour-targeting mAb in Nicotiana benthamiana leaves. PLoS One 11:e0167086. doi: 10.1371/journal.pone.0167086
Lagasse, H. A., Alexaki, A., Simhadri, V. L., Katagiri, N. H., Jankowski, W., Sauna, Z. E., et al. (2017). Recent advances in (therapeutic protein) drug development. F1000Res. 6:113. doi: 10.12688/f1000research.9970.1
Lawrence, M. S., Phillips, K. J., and Liu, D. R. (2007). Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 129, 10110–10112. doi: 10.1021/ja071641y
Lee, C. C., Perchiacca, J. M., and Tessier, P. M. (2013). Toward aggregation-resistant antibodies by design. Trends Biotechnol. 31, 612–620. doi: 10.1016/j.tibtech.2013.07.002
Leuenberger, P., Ganscha, S., Kahraman, A., Cappelletti, V., Boersema, P. J., Von Mering, C., et al. (2017). Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 355:eaai7825. doi: 10.1126/science.aai7825
Li, M. Z., and Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256. doi: 10.1038/nmeth1010
Liang, X., Peng, L., Li, K., Peterson, T., and Katzen, F. (2012). A method for multi-site-directed mutagenesis based on homologous recombination. Anal. Biochem. 427, 99–101. doi: 10.1016/j.ab.2012.05.002
Lohse, S., Derer, S., Beyer, T., Klausz, K., Peipp, M., Leusen, J. H., et al. (2011). Recombinant dimeric IgA antibodies against the epidermal growth factor receptor mediate effective tumor cell killing. J. Immunol. 186, 3770–3778. doi: 10.4049/jimmunol.1003082
Ma, J. K.-C., Hiatt, A., Hein, M., Vine, N. D., Wang, F., Stabila, P., et al. (1995). Generation and assembly of secretory antibodies in plants. Science 268, 716–719. doi: 10.1126/science.7732380
Majowicz, S. E., Scallan, E., Jones-Bitton, A., Sargeant, J. M., Stapleton, J., Angulo, F. J., et al. (2014). Global incidence of human Shiga toxin-producing Escherichia coli infections and deaths: a systematic review and knowledge synthesis. Foodborne Pathog. Dis. 11, 447–455.
Mathusa, E. C., Chen, Y., Enache, E., and Hontz, L. (2010). Non-O157 Shiga toxin–producing Escherichia coli in foods. J. Food Prot. 73, 1721–1736. doi: 10.4315/0362-028x-73.9.1721
McKee, M. L., Melton-Celsa, A. R., Moxley, R. A., Francis, D. H., and O’Brien, A. D. (1995). Enterohemorrhagic Escherichia coli O157: H7 requires intimin to colonize the gnotobiotic pig intestine and to adhere to HEp-2 cells. Infect. Immun. 63, 3739–3744. doi: 10.1128/iai.63.9.3739-3744.1995
McLoughlin, F., Kim, M., Marshall, R. S., Vierstra, R. D., and Vierling, E. (2019). HSP101 interacts with the proteasome and promotes the clearance of ubiquitylated protein aggregates. Plant Physiol. 180, 1829–1847. doi: 10.1104/pp.19.00263
Mitra, P., Shultis, D., Brender, J. R., Czajka, J., Marsh, D., Gray, F., et al. (2013). An evolution-based approach to de novo protein design and case study on Mycobacterium tuberculosis. PLoS Comput. Biol. 9:e1003298. doi: 10.1371/journal.pcbi.1003298
Mogk, A., Bukau, B., and Kampinga, H. H. (2018). Cellular handling of protein aggregates by disaggregation machines. Mol. Cell 69, 214–226. doi: 10.1016/j.molcel.2018.01.004
Montenegro, M. A., Bulte, M., Trumpf, T., Aleksic, S., Reuter, G., Bulling, E., et al. (1990). Detection and characterization of fecal verotoxin-producing Escherichia coli from healthy cattle. J. Clin. Microbiol. 28, 1417–1421. doi: 10.1128/jcm.28.6.1417-1421.1990
Nandi, S., Kwong, A. T., Holtz, B. R., Erwin, R. L., Marcel, S., and Mcdonald, K. A. (2016). Techno-economic analysis of a transient plant-based platform for monoclonal antibody production. MAbs 8, 1456–1466. doi: 10.1080/19420862.2016.1227901
Park, H. I., Yoon, H. W., and Jung, S. T. (2016). The highly evolvable antibody Fc domain. Trends Biotechnol. 34, 895–908. doi: 10.1016/j.tibtech.2016.04.005
Paul, M., Reljic, R., Klein, K., Drake, P. M., van Dolleweerd, C., Pabst, M., et al. (2014). Characterization of a plant-produced recombinant human secretory IgA with broad neutralizing activity against HIV. MAbs 6, 1585–1597. doi: 10.4161/mabs.36336
Pereira, E. O., Kolotilin, I., Conley, A. J., and Menassa, R. (2014). Production and characterization of in planta transiently produced polygalacturanase from Aspergillus niger and its fusions with hydrophobin or ELP tags. BMC Biotechnol. 14:59. doi: 10.1186/1472-6750-14-59
Public Health Agency of Canada (2015). Foodnet Canada Short Report 2015. Ottawa, ON: Public Health Agency of Canada.
Rath, A., Glibowicka, M., Nadeau, V. G., Chen, G., and Deber, C. M. (2009). Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Nat. Acad. Sci. U.S.A. 106, 1760–1765. doi: 10.1073/pnas.0813167106
Reinhart, D., and Kunert, R. J. (2015). Upstream and downstream processing of recombinant IgA. Biotechnol. Lett. 37, 241–251. doi: 10.1007/s10529-014-1686-z
Robert, S., Khalf, M., Goulet, M. C., D’Aoust, M. A., Sainsbury, F., and Michaud, D. (2013). Protection of recombinant mammalian antibodies from development-dependent proteolysis in leaves of Nicotiana benthamiana. PLoS One 8:e70203. doi: 10.1371/journal.pone.0070203
Rybicki, E. P. (2009). Plant-produced vaccines: promise and reality. Drug Discov. Today 14, 16–24. doi: 10.1016/j.drudis.2008.10.002
Saberianfar, R., Chin-Fatt, A., Scott, A., Henry, K. A., Topp, E., and Menassa, R. (2019). Plant-produced chimeric VHH-sIgA against enterohemorrhagic E. coli intimin shows cross-serotype inhibition of bacterial adhesion to epithelial cells. Front. Plant Sci. 10:270. doi: 10.3389/fpls.2019.00270
Saerens, D., Conrath, K., Govaert, J., and Muyldermans, S. (2008). Disulfide bond introduction for general stabilization of immunoglobulin heavy-chain variable domains. J. Mol. Bol. 377, 478–488. doi: 10.1016/j.jmb.2008.01.022
Sainsbury, F., and Lomonossoff, G. P. (2008). Extremely high-level and rapid transient protein production in plants without the use of viral replication. Plant Physiol. 148, 1212–1218. doi: 10.1104/pp.108.126284
Sainsbury, F., Thuenemann, E. C., and Lomonossoff, G. P. (2009). pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J. 7, 682–693. doi: 10.1111/j.1467-7652.2009.00434.x
Schillberg, S., Raven, N., Spiegel, H., Rasche, S., and Buntru, M. (2019). Critical analysis of the commercial potential of plants for the production of recombinant proteins. Front. Plant Sci. 10:720. doi: 10.3389/fpls.2019.00720
Silhavy, D., Molnar, A., Lucioli, A., Szittya, G., Hornyik, C., Tavazza, M., et al. (2002). A viral protein suppresses RNA silencing and binds silencing-generated, 21- to 25-nucleotide double-stranded RNAs. EMBO J. 21, 3070–3080. doi: 10.1093/emboj/cdf312
Tokuriki, N., Stricher, F., Schymkowitz, J., Serrano, L., and Tawfik, D. S. (2007). The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 369, 1318–1332. doi: 10.1016/j.jmb.2007.03.069
Trivedi, M. V., Laurence, J. S., and Siahaan, T. J. (2009). The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 10, 614–625. doi: 10.2174/138920309789630534
Vanmarsenille, C., Elseviers, J., Yvanoff, C., Hassanzadeh-Ghassabeh, G., Garcia Rodriguez, G., Martens, E., et al. (2018). In planta expression of nanobody-based designer chicken antibodies targeting Campylobacter. PLoS One 13:e0204222. doi: 10.1371/journal.pone.0204222
Virdi, V., Coddens, A., De Buck, S., Millet, S., Goddeeris, B. M., Cox, E., et al. (2013). Orally fed seeds producing designer IgAs protect weaned piglets against enterotoxigenic Escherichia coli infection. Proc. Natl. Acad. Sci. U.S.A. 110, 11809–11814. doi: 10.1073/pnas.1301975110
Virdi, V., Palaci, J., Laukens, B., Ryckaert, S., Cox, E., Vanderbeke, E., et al. (2019). Yeast-secreted, dried and food-admixed monomeric IgA prevents gastrointestinal infection in a piglet model. Nat. Biotechnol. 37, 527–530. doi: 10.1038/s41587-019-0070-x
Walther, D. M., Kasturi, P., Zheng, M., Pinkert, S., Vecchi, G., Ciryam, P., et al. (2015). Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932. doi: 10.1016/j.cell.2015.03.032
Wan, C., Zhang, J., Zhao, L., Cheng, X., Gao, C., Wang, Y., et al. (2019). Rational design of a chimeric derivative of PcrV as a subunit vaccine against Pseudomonas aeruginosa. Front. Immunol. 10:781. doi: 10.3389/fimmu.2019.00781
Westerhof, L. B., Wilbers, R. H., van Raaij, D. R., van Wijk, C. Z., Goverse, A., Bakker, J., et al. (2016). Transient expression of secretory IgA in planta is optimal using a multi-gene vector and may be further enhanced by improving joining chain incorporation. Front. Plant Sci. 6:1200. doi: 10.3389/fpls.2015.01200
Wieland, W. H., Lammers, A., Schots, A., and Orzáez, D. V. (2006). Plant expression of chicken secretory antibodies derived from combinatorial libraries. J. Biotechnol. 122, 382–391. doi: 10.1016/j.jbiotec.2005.12.020
Wu, S., and Zhang, Y. (2008). MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins 72, 547–556. doi: 10.1002/prot.21945
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Zabetakis, D., Olson, M. A., Anderson, G. P., Legler, P. M., and Goldman, E. R. (2014). Evaluation of disulfide bond position to enhance the thermal stability of a highly stable single domain antibody. PLoS One 9:e115405. doi: 10.1371/journal.pone.0115405
Zhang, Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40
Keywords: enterohemorrhagic E. coli-EHEC, IgA, VHH antibody fragment, single domain antibody (sdAb), Fc fusion, plant-made antibodies, transient expression, rational design antibody engineering
Citation: Chin-Fatt A, Saberianfar R and Menassa R (2021) A Rationally Designed Bovine IgA Fc Scaffold Enhances in planta Accumulation of a VHH-Fc Fusion Without Compromising Binding to Enterohemorrhagic E. coli. Front. Plant Sci. 12:651262. doi: 10.3389/fpls.2021.651262
Received: 09 January 2021; Accepted: 22 March 2021;
Published: 14 April 2021.
Edited by:
Inge Broer, University of Rostock, GermanyReviewed by:
Eva Stoger, University of Natural Resources and Life Sciences Vienna, AustriaCopyright © 2021 Chin-Fatt, Saberianfar and Menassa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rima Menassa, cmltYS5tZW5hc3NhQENhbmFkYS5jYQ==
†Present address: Reza Saberianfar, Toronto Recombinant Antibody Centre, University of Toronto, Toronto, ON, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.