
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 08 March 2021
Sec. Plant Systematics and Evolution
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.643548
This article is part of the Research TopicMolecular Organization, Evolution, and Function of Ribosomal DNAView all 15 articles
 Radka Vozárová1,2
Radka Vozárová1,2 Veit Herklotz3
Veit Herklotz3 Aleš Kovařík1
Aleš Kovařík1 Yuri O. Tynkevich4
Yuri O. Tynkevich4 Roman A. Volkov4
Roman A. Volkov4 Christiane M. Ritz3,5
Christiane M. Ritz3,5 Jana Lunerová1*
Jana Lunerová1*The genus Rosa comprises more than 100 woody species characterized by intensive hybridization, introgression, and an overall complex evolutionary history. Besides many diploid species (2n = 2x = 14) polyploids ranging from 3x to 10x are frequently found. Here we analyzed 5S ribosomal DNA in 19 species covering two subgenera and the major sections within subg. Rosa. In addition to diploids and polyploids with regular meiosis, we focused on 5x dogroses (Rosa sect. Caninae), which exhibit an asymmetric meiosis differentiating between bivalent- and univalent-forming chromosomes. Using genomic resources, we reconstructed 5S rDNA units to reveal their phylogenetic relationships. Additionally, we designed locus-specific probes derived from intergenic spacers (IGSs) and determined the position and number of 5S rDNA families on chromosomes. Two major 5S rDNA families (termed 5S_A and 5S_B, respectively) were found at variable ratios in both diploid and polyploid species including members of the early diverging subgenera, Rosa persica and Rosa minutifolia. Within subg. Rosa species of sect. Rosa amplified the 5S_A variant only, while taxa of other sections contained both variants at variable ratios. The 5S_B family was often co-localized with 35S rDNA at the nucleolar organizer regions (NOR) chromosomes, whereas the co-localization of the 5S_A family with NOR was only exceptionally observed. The allo-pentaploid dogroses showed a distinct distribution of 5S rDNA families between bivalent- and univalent-forming chromosomes. In conclusion, two divergent 5S rDNA families dominate rose genomes. Both gene families apparently arose in the early history of the genus, already 30 myrs ago, and apparently survived numerous speciation events thereafter. These observations are consistent with a relatively slow genome turnover in the Rosa genus.
Ribosomal RNA genes (rDNA) encoding 5S, 5.8S, 18S, and 26S ribosomal RNA are ubiquitous in plants and are organized into arrays containing hundreds to thousands of tandem units at one or more genomic loci (Hemleben et al., 1988; Nieto Feliner and Rossello, 2012; Roa and Guerra, 2012). Each unit consists of an evolutionary conserved coding region of 120 bp and a variable intergenic spacer (IGS) (Long and Dawid, 1980). The units within the 5S arrays retain a high degree of identity due to homogenizing forces referred to as concerted evolution (Dover, 1982; Eickbush and Eickbush, 2007) where unequal crossing-over and gene conversion are major forces driving the process. Regardless of the mechanism, numerous factors such as the number of arrays, their mutation rate, formation of new variants, the intensity of natural selection, or efficient population size can affect homogenization of repeats (Dover, 1982; Ohta, 1984; Nagylaki, 1990). In plant hybrids and allopolyploids, homogenization of 5S rDNA arrays may not always occur as efficiently as that of 35S rDNA (Pedrosa-Harand et al., 2006; Weiss-Schneeweiss et al., 2008; Garcia et al., 2016; Amosova et al., 2019). As a consequence, two or more variants differing in the length and nucleotide sequence may simultaneously exist per genome (Cronn et al., 1996; Volkov et al., 2001, 2017; Fulnecek et al., 2002; Pastova et al., 2019; Benson et al., 2020).
The genus Rosa L. (Rosaceae) comprises about 150 species widely distributed across the northern hemisphere. Taxonomy is considered to be challenging because frequent polyploidy in app. 50% of the species (Yokoya et al., 2000; Roberts et al., 2009) and recurrent hybridization events may blur species boundaries (Ritz et al., 2005; Joly and Bruneau, 2006; Koopman et al., 2008). The existence of multiple cytotypes and variable degree of retention of progenitor alleles leading to incomplete lineage sorting complicating taxonomic classifications. In addition, species identification is generally hampered because most species are characterized rather by combinations of morphological characters than by single discriminating traits (Christ, 1873; Wissemann, 2003). Moreover, roses are one of oldest ornamentals (Wang, 2007) and their complex history of cultivation and breeding may generate another uncertainty in phylogenetic studies. Several attempts have been made to reconstruct the phylogeny of the genus (Millan et al., 1996; Matsumoto et al., 1998; Wissemann and Ritz, 2005; Bruneau et al., 2007; Zhang et al., 2013; Fougere-Danezan et al., 2015; Liu et al., 2015). Currently, the system comprises four subgenera: Hulthemia (one species), Hesperhodos (two species), Platyrhodon (one species), and Rosa, the latter consisting of 10 sections and comprising the vast majority of species (Wissemann, 2003). The most recent phylogenies detected Rosa persica (subg. Hulthemia) and Rosa minutifolia (subg. Hesperhodos) as early diverging lineages, and a major split of the genus into two large clades: the Synstylae and allies clade consisting of sect. Synstylae, Indicae, Caninae, Bracteatae, Laevigatae, and Gallicanae and the Rosa and allies clade comprising sect. Rosa [=Cinnamomeae] and Pimpinellifoliae (Wissemann and Ritz, 2005; Bruneau et al., 2007; Fougere-Danezan et al., 2015; Zhu et al., 2015; Debray et al., 2019).
The exclusively polyploid members of a large section Caninae (DC.) Ser. (dogroses), originated by multiple hybridization events (Ritz et al., 2005; Herklotz et al., 2018) represent a remarkable evolutionary lineage because they exhibit a peculiar unbalanced mode of sexual reproduction also known as Canina meiosis (Täckholm, 1920; Blackburn and Harrison, 1921). Canina meiosis results in a strongly matroclinal inheritance of genetic information since two pairing genomes form bivalents, while the remaining genomes remain unpaired as univalents and are transmitted by the female germ line only. Thus, at least hemisexual reproduction is ensured in the mostly pentaploid (2n = 5x = 35) species but tetraploids, hexaploids, and heptaploids also occur and their meiosis just differs by the number univalents (Wissemann, 2003; Roberts et al., 2009; Pachl, 2011). Amazingly, in plastid phylogenies, sect. Caninae appeared to be polyphyletic since species with fragrant glands (subsect. Rubigineae and Vestitae) were separated from the remaining species (subsect. Caninae) by Rosa gallica and Rosa arvensis which perform regular meiosis (Wissemann and Ritz, 2005; Fougere-Danezan et al., 2015). Thus, Canina meiosis has been probably evolved twice, which is supported by fluorescence in situ hybridization (FISH) analyses of meiotic chromosomes (Herklotz et al., 2018; Lunerova et al., 2020).
Ribosomal DNA loci have been studied in several diploid and polyploid species of Rosa so far. Ma et al. (1997) found one 35S rDNA locus per genome, located terminally on the short arms of small submetacentric chromosomes in five diploid species and one tetraploid cultivar of Rosa. Fernandez-Romero et al. (2001) found one 35S rDNA locus per genome at terminal locations on submetacentric chromosomes in five diploid species. These studies indicate the presence of a single nucleolar organizer regions (NOR) chromosome per haploid set of x = 7. In pentaploid dogroses, four to five 35S loci were reported implying the occasional loss of one locus (Lim et al., 2005; Herklotz et al., 2018). The 5S locus has been less commonly studied, while there is evidence for more than one 5S locus per haploid set. Two loci were found in the diploid Rosa lucieae [=Rosa wichurana] (Kirov et al., 2016), and some pentaploid dogroses may contain more than five sites (Lim et al., 2005; Herklotz et al., 2018) indicating a variable number of 5S loci per haploid set. The analysis of 5S rDNA clones from diploid Rosa rugosa revealed a conserved bipartite polymerase III promoter and non-coding IGS region (Tynkevich and Volkov, 2014b) evidencing that organization at the unit level is similar to most other plants. Analysis of 5S rDNA clones from four distantly related diploid species of Rosa (R. nitida, R. rugosa, R. sericea, and R. lucieae) showed a high level of intragenomic homogeneity. In contrast, the level of IGS similarity between R. lucieae and three other diploid species appeared to be unusually low (less than 58%) arguing for interspecies diversity in Rosa (Tynkevich and Volkov, 2014a, b).
In this study, based on genomic and cytogenetic approaches, we aim to map the evolutionary history of 5S rDNA loci across the genus Rosa. Based on available phylogenies of the genus, we selected 11 diploid and eight polyploid species representing the genus’ diversity (Table 1). Bioinformatic methods were used to determine the abundance and homogeneity of 5S rDNA in the genomes. Using locus-specific probes derived from 5S IGSs, we identified the two major 5S rDNA loci on the chromosomes by FISH.
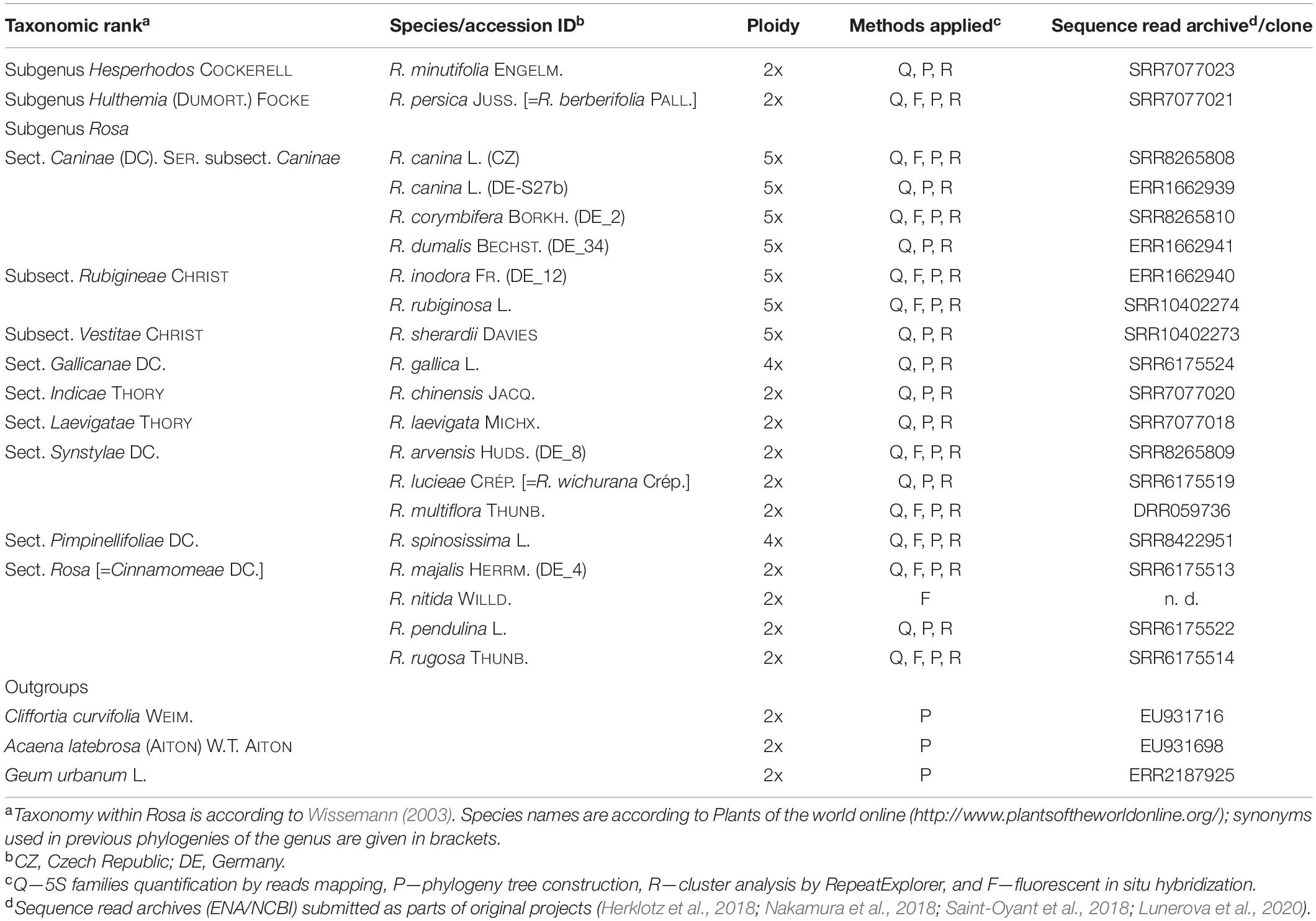
Table 1. List of Rosaceae species used in this study, ploidy, source, and read archive accessions and the analyses employed.
Material of polyploid dogroses was sampled in wild populations in Germany and the Czechia (Supplementary Table S1). Diploid species and tetraploid species with regular meiosis were obtained from various Botanical Gardens (Supplementary Table S1). In addition, we retrieved sequence information from published work stored in the ENA database for bioinformatics analyses (Supplementary Table S1).
Total genomic DNA of Rosa canina was extracted from fresh leaves applying the standard protocol (Rogers and Bendich, 1985). The 5S rDNA repeats were amplified using the primers pr5S-14 and pr5S-15 (Tynkevich and Volkov, 2014b) with the 5’-extensions containing restriction endonuclease NotI recognition site. The PCR products were treated by NotI, ligated into the Eco52I recognition site of the pLitmus 38 plasmid, and used for transformation of Escherichia coli XL_blue line by electroporation method. Selected recombinant clones were sequenced using a BigDye Terminator Cycle Sequencing Kit (Thermofisher Scientific, United States). Clones containing inserts of A and B variants of 5S rDNA were identified by sequence analysis. The sequences were submitted to GenBank under the accession numbers MW349696 and MW349697.
The inserts of cloned 5S rDNA sequences contained genic regions and IGS. In order to increase the specificity of probe hybridization, we amplified the IGS sequences using specific primers annealing to 5S_A and 5S_B variants. The oligonucleotide primers’ sequences for the 5S_A IGS were as follows: A_for: 5′-CCTCTTTTTTCTGTTTCGGT-3′; A_rev: 5′-ATAAACTCCATTCGCTCAG-3′. Primers for the 5S_B variants were: B_for: 5′-ACCCCTCTTTTTGCCTTT-3′; B_rev: 5′-GCTTCGTCTCACTCCTCT-3’. The 25 μl PCR reaction contained 0.1 ng of plasmid DNA as the template, 4 pmol of each primer, 2.4 nmol of each dNTP, and 0.4 units of Kapa Taq DNA polymerase I (Kapa Biosystems). Cycling conditions were as follows: initial denaturation step (94°C, 180 s); 35 cycles (94°C, 20 s; 57°C, 30 s; and 72°C, 30 s). The length of amplified products was 373 nt for the A variant and 394 nt for the B variant. Purified PCR products were labeled by fluorescent dyes (as below) and used in FISH.
For bioinformatic analyses, the whole genomic sequencing data for 19 Rosa accessions were used (Supplementary Table S2). The genome proportion of 5S rDNA families was determined using the total Illumina reads trimmed for quality (Phred score ≥ 30 over ≥ 95% read length). Trimmed reads (typically > 7 million) were mapped to corresponding 5S_A and 5S_B reference sequences (IGS subregion between the primers, Figure 1) using the following parameters: insertion and deletion costs_3, lengths fraction_0.5, similarity fraction_0.8, and deletion cost_2 (Qiagen, Germany). The distribution of SNPs across the 5S rDNA sequences was recorded when the distribution exceeded a threshold of at least 20 identical SNPs over at least 200 reads that covered the variant position and occurred at ≥ 10% frequency. For the alignment, minimal sequence length coverage was 50% and minimum sequence similarity was 90%. For the more distantly related species, Rosa spinosissima and R. persica, similarity threshold parameter was decreased to 80% (for the 5S_B variant). The genome abundance and copy number was calculated from genome proportions according to the formula stated in (Lunerova et al., 2020).
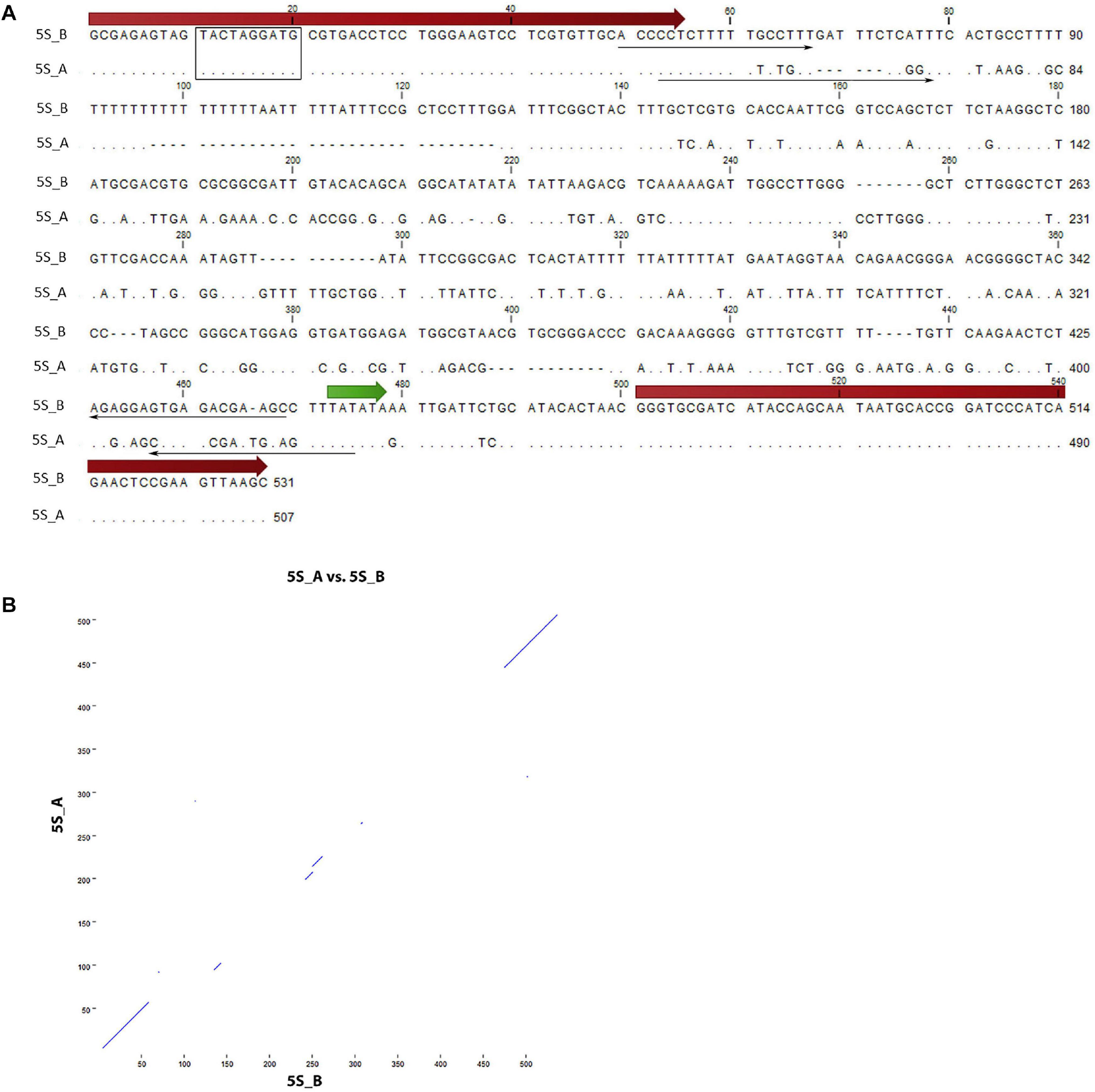
Figure 1. Sequence analysis of 5S rDNA clones from Rosa canina. (A) Alignment of the 5S_A and 5S_B clones. The position of 5S rRNA coding regions (thick arrows), primers (thin arrows), and regulatory regions (TATA box—in green, Box-C—rectangle) are highlighted. (B) Dot plot of pairwise comparisons clones. Note that only coding regions showed significant similarity.
For phylogenetic reconstructions, the consensus 5S_A and 5S_B rDNA sequences were extracted from mapped reads using CLC genomic workbench. Additionally, we added the partial sequence of 5S ribosomal RNA genes from Acaena latebrosa (EU931698.1) and Cliffortia curvifolia (EU931716.1). Paired 250 bp Illumina reads of Geum urbanum (SRA accession ERR2187925) were mapped in a first round to the C. curvifolia (EU931716.1) sequence. Mapping was done with Bowtie2 (Langmead and Salzberg, 2012) implemented in Geneious® 10.0.91 with the lowest sensitivity pre-set. This resulted in two reads out of 22.8 million hitting to a 39 bp conserved region. The two reads were aligned and served in a second round with same parameters as mapping scaffold of 334 bp length. In the second mapping, 50 reads out of 22.8 million were assembled and its consensus was added to the alignment including all rose sequences and the two species from the Acaena clade. Alignments were done with MAFFT v7.450 algorithm (Katoh and Standley, 2013) implemented in Geneious using default parameters. Model test implemented in MEGA X v. 10.1.8 (Kumar et al., 2018) revealed the Tamura–parameter substitution model with invariant sites as most appropriate for the data based on Akaike information criterion (Tamura, 1992). Based on this model, we computed a maximum-likelihood tree in MEGA X whose branch support was evaluated by 1000 bootstrap replicates. Rooted with G. urbanum, this tree was used for the dating approach conducted with MEGA X. Therefore, we used two calibration points along the tree taken from the respective fossils given in Xiang et al. (2017) Rosa germerensis, 48.6 Mya (Edelman, 1975) and Acaena sp., 37.2 Mya (Zetter et al., 1999). A timetree inferred by applying the RelTime method (Tamura et al., 2018) was computed using two fixed calibration constraints. All positions containing gaps and missing data were eliminated (complete deletion option). A neighbor joining tree was constructed with the Seaview software (Gouy et al., 2010).
The fastq reads were initially filtered for quality and trimmed to uniform length using the pre-processing and QC tools in RepeatExplorer2 (Novak et al., 2013). Read length ranged between 100 and 150 bp, depending on sequencing library and the Illumina sequencing platform. After the fastq > fasta conversion and trimming to uniform length, reads were analyzed with the RepeatExplorer2 clustering program using default parameters. We used 1 million paired-end reads, or 1 million single-end reads as inputs for RepeatExplorer2 clustering. This bioinformatic pipeline runs a graph-based clustering algorithm (Novak et al., 2013) that assembles groups of frequently overlapping reads into clusters of reads, representing a repetitive element or part of a repetitive element with a higher order genome structure. The similarity and structure-based repeat identification tools in RepeatExplorer2 aid in identification of the repeats. RepeatExplorer2 uses a BLAST threshold of 90% similarity across 55% of the read to assign reads to clusters (minimum overlap = 55, cluster threshold = 0.01%, minimum overlap for assembly = 40), and the clusters are identified based on the principle of maximum modularity. We also used the SeqGrapheR (Novak et al., 2010) software in virtual space of Ubuntu 18.04 to visualize the specific reads corresponding to the 5S_A and 5S_B variants.
For slide preparations, we used young anthers from flower buds of about 0.5 cm in length, which were harvested during spring 2019. Male meiosis was studied at prophase I (diplotene/diakinesis) where the bivalents and univalents could be easily distinguished from each other. Fresh flower buds were fixed using Carnoy solution (acetone:acetic acid, 2:1 or ethanol:acetic acid, 3:1 in some cases), and stored in 70% ethanol at −20°C. Before slide preparation, anthers were pre-treated by 0.5% PVP and 2% Triton-X100 (Sigma–Aldrich, United States) for 2–5 min followed by enzyme digestion overnight at 10°C in 1% cellulase, 0.2% pectolyase Y23, 0.5% hemi-cellulase, and 0.5%, macerozym R10 (Sigma–Aldrich, United States; Duchefa Biochemie, Netherlands) dissolved in citric buffer (0.04 M citric acid and 0.06 M sodium citrate). FISH followed the procedures described in Herklotz et al. (2018). Anthers were separated and squashed on slides in a drop of 70% acetic acid and fixed in liquid nitrogen.
For FISH, we used two probes derived from the 5S_A and 5S_B clones of the IGS region, respectively, and in addition, an 18S rDNA probe that was a 1.7-kb fragment of the 18S rRNA gene of Solanum lycopersicum (GenBank # X51576.1). The 5S rDNA genic region originated from Artemisia tridentata S4 clone, GenBank # JX101915.1. The probes were labeled by nick translation using Spectrum green dUTPs (Abbott, United States) for 5S_A rDNA, and Cy3-dUTPs (Roche, Switzerland) for 5S_B and 18S rDNA; 5S rDNA was labeled by Atto647N (Jena Bioscience, Germany). Slide preparation and hybridization followed standard protocols (Schwarzacher and Heslop-Harrison, 2000). Chromosomes were counterstained with 1 μg ml–1 DAPI (4’, 6-diamidine-2’-phenylindole dihydrochloride) diluted in mounting medium for fluorescence (Vectashield, Vector Laboratories, United Kingdom). The slides were scanned using epifluorescence microscopes (Olympus Provis AX70, with cold cube camera, Metasystems, Germany). Imaging software was ISIS (MetaSystems, Germany), and images were optimized for contrast and brightness with Adobe Photoshop CS6 and PS2020.
Two 5S rDNA clones (5S_A and 5S_B) were isolated from R. canina IGS. Sequence analysis revealed some conserved regulatory elements: Box-C within the coding region, the TATA box at –29 (both clones), and T-rich terminators downstream of the coding sequences (Figure 1A). Box-A could not be unambiguously determined due to primer overlap. The 5S_B clone had a long (20 nt) T-tract which appears to be missing or was much shorter in clone 5S_A. By analogy with other 5S rRNA transcripts, the putative transcription started at the first G within the GGG motif following the C at −1 (Tynkevich and Volkov, 2014b). Intragenomic homogeneity was high, and no significant SNPs were revealed in mapping experiments (not shown). Pairwise alignment revealed conserved coding regions, while most of the IGS was dissimilar between both sequences (Figure 1B). We took advantage of considerable sequence divergence between both clones and amplified the locus-specific IGS subregions from plasmids. The resulting 373 bp (5S_A family) and 394 bp (5S_B family) PCR products were subsequently used in FISH.
To determine the abundance of individual 5S rRNA gene families in Rosa genomes, we used available genomic resources (Table 1). The genome proportion of the 5S_A family was in average twice of that of the 5S_B family (Supplementary Tables S2, S3). The copy number per somatic cell (2C) ranged from 80–8000 (5S_A family) and 0–2400 (5S_B family). Both families appeared to be equally homogeneous containing a relatively low number of SNPs consistent with our previous findings obtained by comparison of individual 5S rDNA clones (Tynkevich and Volkov, 2014a, b). The contribution of each family to total 5S rDNA was expressed for each species by pie charts and visualized in a phylogenetic context (Figure 2). The diploid species from sect. Synstylae, all polyploid species, and R. persica (subg. Hulthemia) carried both families. Rosa laevigata contained the 5S_A family in low copy (c. 80 copies/2C), while its 5S rDNA was dominated by the 5S_B family (980 copies/2C) (Supplementary Table S3). Species from sect. Rosa and R. minutifolia (subg. Hesperhodos) carried the 5S_A rDNA family only. Blast searches failed to reveal significant hits of 5S_A and 5S_B sequences in genomic reads from Prunus, Rubus, Fragaria, Cliffortia, Acaena, and Sanguisorba (all Rosaceae) even at relaxed (e = 0.1) stringencies (not shown).
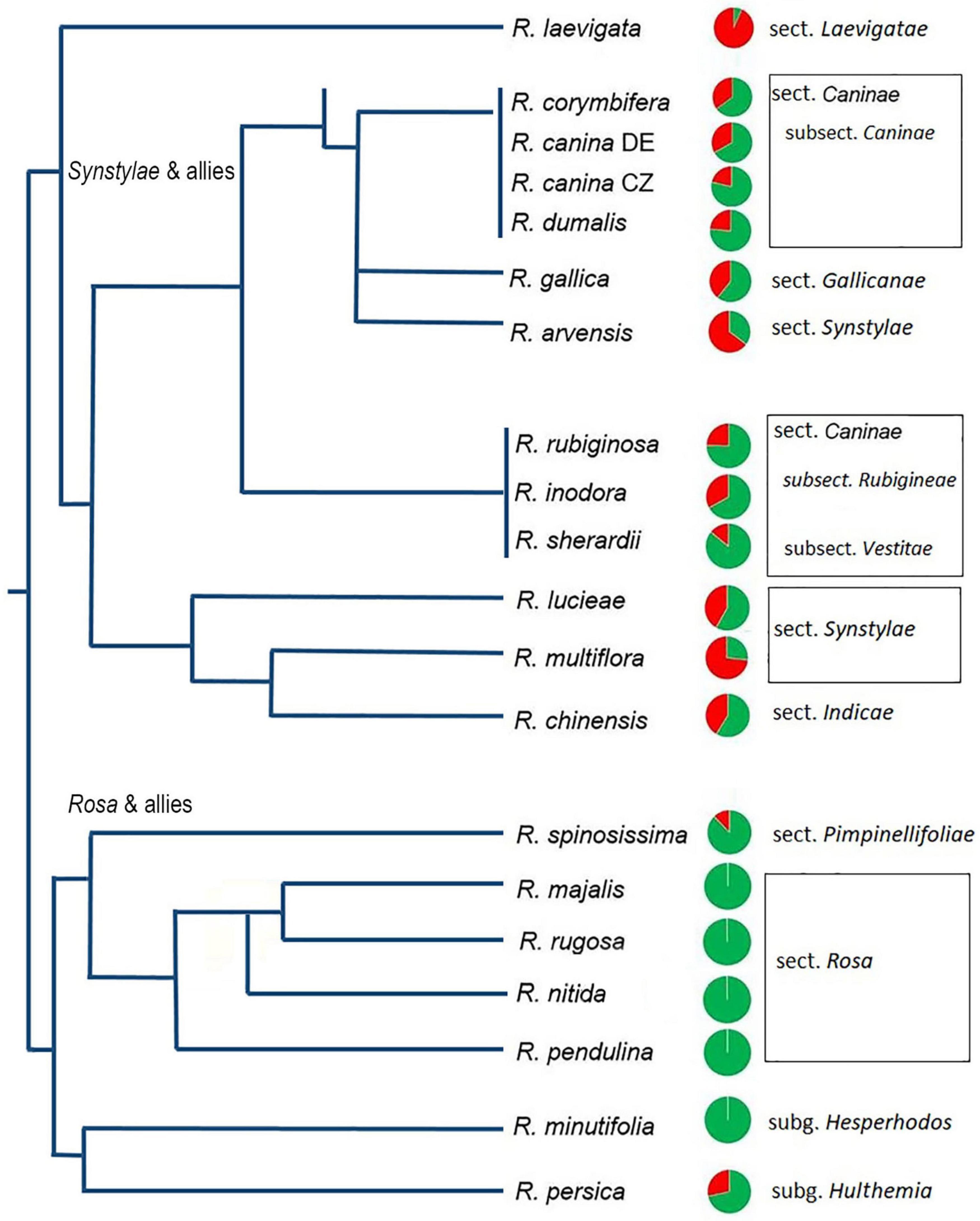
Figure 2. Quantitative relationships between 5S_A and 5S_B rDNA families in Rosa genomes. Genomic proportions of families calculated from high-throughput reads are shown as pie charts next to the species names. Green—5S_A family, red—5S_B family. Data are in Supplementary Table S3; data for R. nitida are taken from FISH (Supplementary Figure S5). A simplified phylogenetic tree is redrawn according to published phylogenies based on plastid sequences (Fougere-Danezan et al., 2015).
Cloning experiments cannot address the question about the distribution of gene families in the genome. Thus, in order to determine the number and genomic representation of individual 5S rRNA gene families, we applied clustering analysis (Figure 3). The cluster graph shapes provide information about the number and type of 5S gene families revealing potential hybridization and introgression (Garcia et al., 2020). It visualizes divergent IGS families as loops emanating from the bridge region which contains reads derived from a conserved coding region. Any loop can be considered as a separate gene family. The subregions in the graph can be annotated based on the read alignment against the 5S_A and 5S_B reference clones. Visual exploration indicates that there is no other 5S family amplified except of 5S_A and 5S_B types. The cluster graphs obtained from different Rosa genomes were categorized based on their structure into three groups (Figure 3). Group 1 comprised a single species R. laevigata (sect. Laevigatae) with predominant 5S_B type family representation. Group 2 comprised R. persica (subg. Hulthemia) and the majority of diploid species (sect. Synstylae and Indicae) showing a typical two-loop structure representing relatively balanced ratios of both families. Group 3 contained the diploid species R. minutifolia (subg. Hesperhodos) and species of sect. Rosa harboring a single 5S rDNA family (A). All polyploid species (both, those with regular meiosis and those with Canina meiosis) showed a Group 2 profile indicating the presence of both A and B families (Supplementary Figure S1). In sum, quantitative relationships between both 5S rDNA families were confirmed. Moreover, cluster analysis showed that the maximum number of 5S rDNA families in the Rosa genomes is always two, irrespective of the ploidy level.
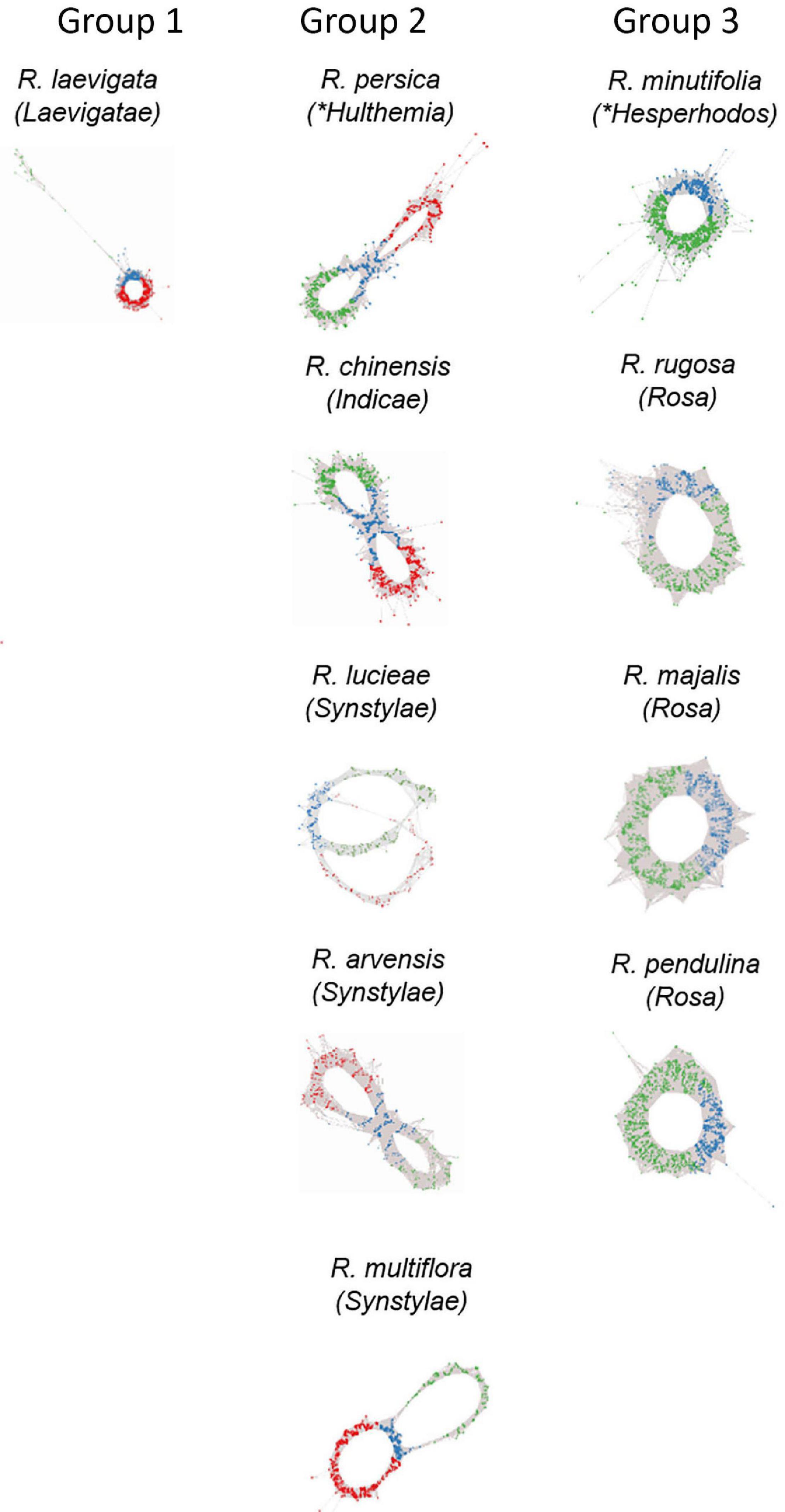
Figure 3. Genomic analysis of 5S rDNA variants in diploid (2n = 2x = 14) species of Rosa. Projections of 5S rDNA cluster graphs: Loops represent IGS reads colored according to the 5S_A (green) and 5S_B (red) IGS variants; genic regions are in blue. Group 1 represents species with dominance of 5S_B variant; Group 2 contains diploids with balanced ratio of both variants; and Group 3 shows species with only A variant in the genome. Name of the sections within subg. Rosa is given in brackets; other subgenera are marked by asterisks.
To determine the phylogenetic relationships between 5S rDNA families, we computed phylogenies based on aligned 5S rDNA consensus sequences (obtained from mapping experiments. Both the maximum-likelihood (Figure 4 and Supplementary Figure S2) and neighbor joining (Supplementary Figure S3) trees separate the A and B 5S rDNA families clearly into two well-supported clades (A and B). Both, clades A and B contained diploid and polyploid species. Except of sect. Rosa, whose members clustered exclusively within the A clade, members of other sections, including Synstylae, Indicae, Laevigatae, Pimpinellifoliae, and Caninae, partitioned their 5S rDNA between both clades. Sequences from R. persica (subg. Hulthemia) were consistently positioned on early diverging nodes at both subclades. The major 5S rDNA family of R. laevigata (sect. Laevigatae) branched off at a rather basal position in clade B. The 5S_B family of 4x R. spinosissima positioned as sister to R. persica. Five 5S rDNAs of 5x species from sect. Caninae clustered together in both clades with negligible resolution between species (Supplementary Figures S2, S4). Out of the diploids, 5S sequences of R. arvensis (sect. Synstylae, B clade) and R. pendulina in (sect. Rosa, A clade) were most closely related to those of the respective Caninae branches. To gauge the length of time these 5S rDNA variants have existed in the Rosa genomes, we used two calibration points (48.6 myrs for R. germerensis and 37.2 myrs for Acaena sp.). We estimated that a common ancestor of both A and B families lived about 32 myrs ago (Figure 4) relatively long before separation of modern clades.
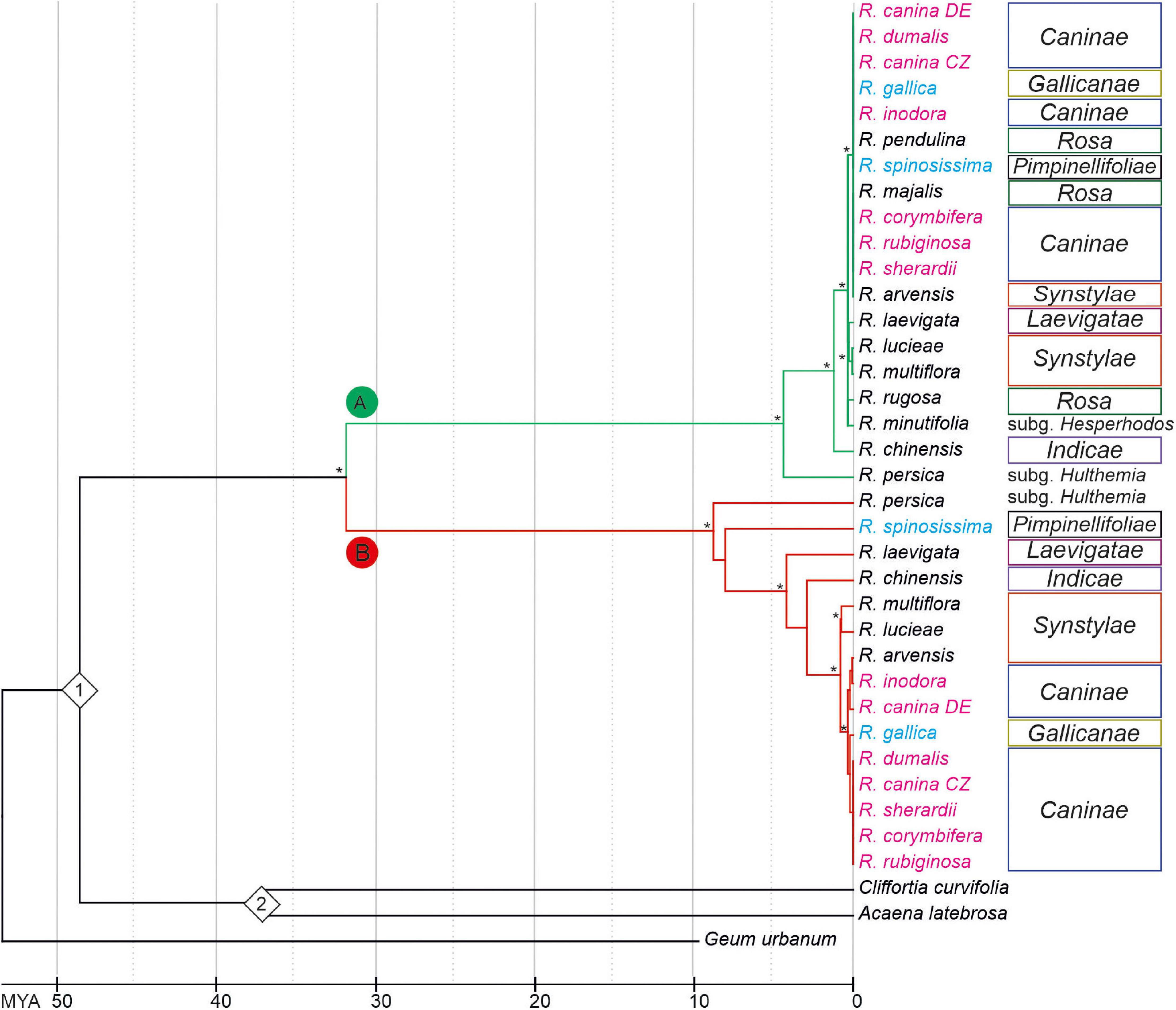
Figure 4. Chronogram based on a phylogeny among Rosa species inferred from Maximum-Likelihood analysis of 5S rDNA intergenic spacers. The tree was rooted with Geum urbanum. Bootstrap value of ≥ 70% is indicated by an asterisk. Species names are colored according to ploidy level (2x—black, 4x—light blue, 5x—pink). Color of branches represents 5S_A (green) and 5S_B (red) variants of IGS rDNA. Calibration points (1 = Rosa gemerensis; 48.6 myrs and 2 = Acaena sp.; 37.2 myrs) are indicated by diamonds.
Fluorescence in situ hybridization was conducted to visualize the position and number of 5S rDNA variants on chromosomes in several diploid and polyploid species. The diploids included representatives of subg. Rosa sect. Synstylae (R. arvensis and R. multiflora), sect. Rosa (R. rugosa, R. majalis, and R. nitida), and subg. Hulthemia (R. persica). Meiotic chromosomes from anthers (Figure 5A) were hybridized with rDNA probes derived from the 5S rDNA genic (Figure 5B, shown in white), 5S_B (red), and 5S_A (green) IGS subregions (Figure 5C). Additionally, the same chromosome spreads were re-hybridized with the 18S rDNA probe (shown in cyan, Figure 5D). In R. arvensis and R. multiflora, each 5S_A and 5S_B probe hybridized to one bivalent (one pair of chromosomes). The 5S_B probe was always co-localized on a chromosome bearing also the 18S rDNA signal. In R. rugosa and R. majalis, the 5S_A probe hybridized to a single bivalent, while we did not detect any hybridization signals with the 5S_B probe in accordance to genomic analyses (Figures 2, 3). In both species, the 18S and 5S rDNA loci were separate. However, in R. nitida, the 5S_A probe hybridized to one pair of chromosomes (mitotic metaphase from root tips, Supplementary Figure S5) which carried 18S rDNA signal (NOR). In R. persica, both variant-specific 5S rDNA probes hybridized to a single bivalent each, and these bivalents carried also 18S rDNA sites. The number and position of rDNA loci on chromosomes are summarized in Supplementary Table S4 and are diagrammatically depicted by ideograms (Figure 6).
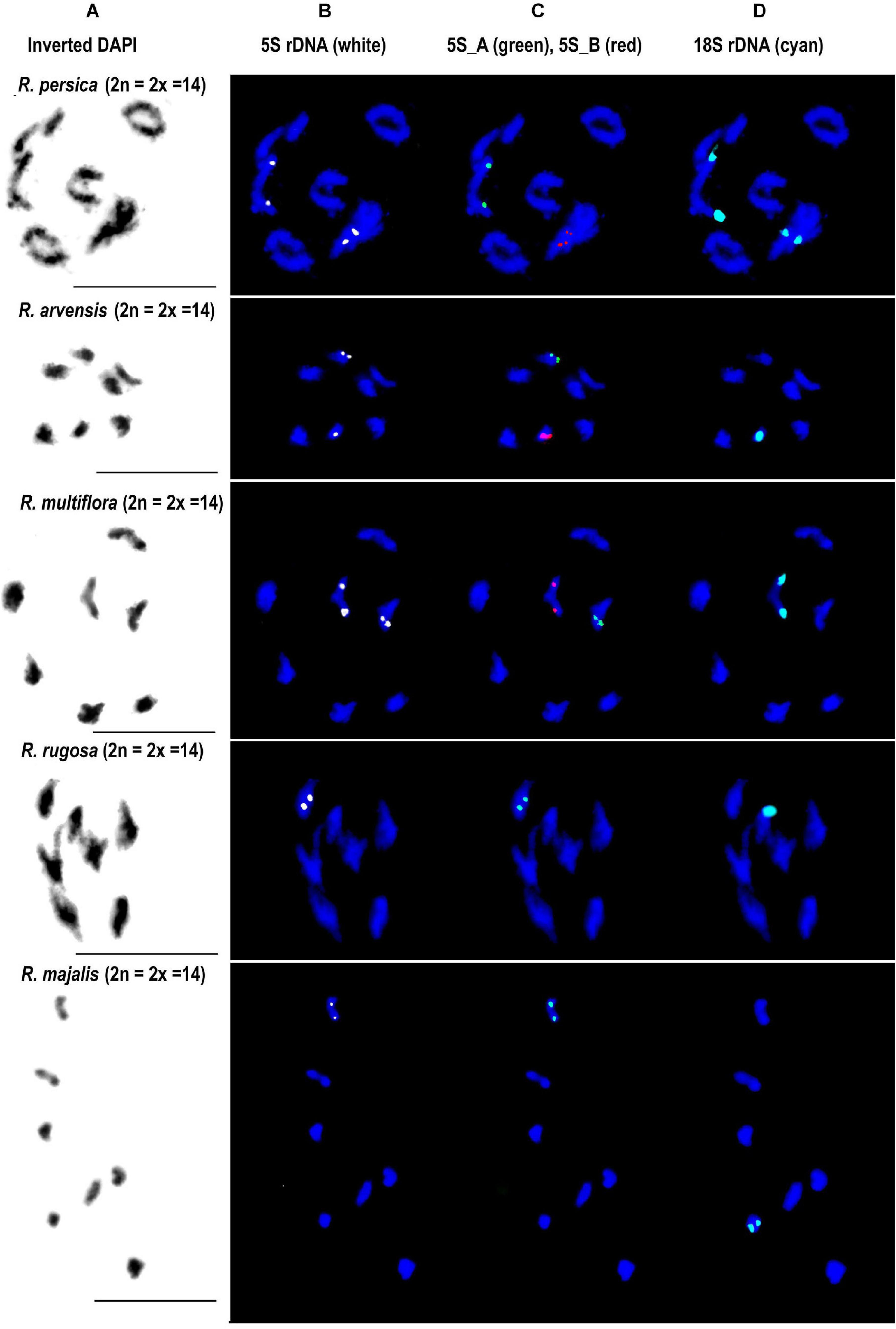
Figure 5. FISH analysis of 5S rDNA in diploid species. Diakinesis of meiotic phase I shown in R. persica (subgenera Hulthemia), R. arvensis and R. multiflora (section Synstylae), R. rugosa and R. majalis (section Rosa). In each species, the same metaphase was hybridized with the 5S genic (B), 5S intergenic (C), and 18S rDNA (D) probes. (A) DAPI staining (gray scale, inverted) of chromatin. Scale bar: 10 μm.
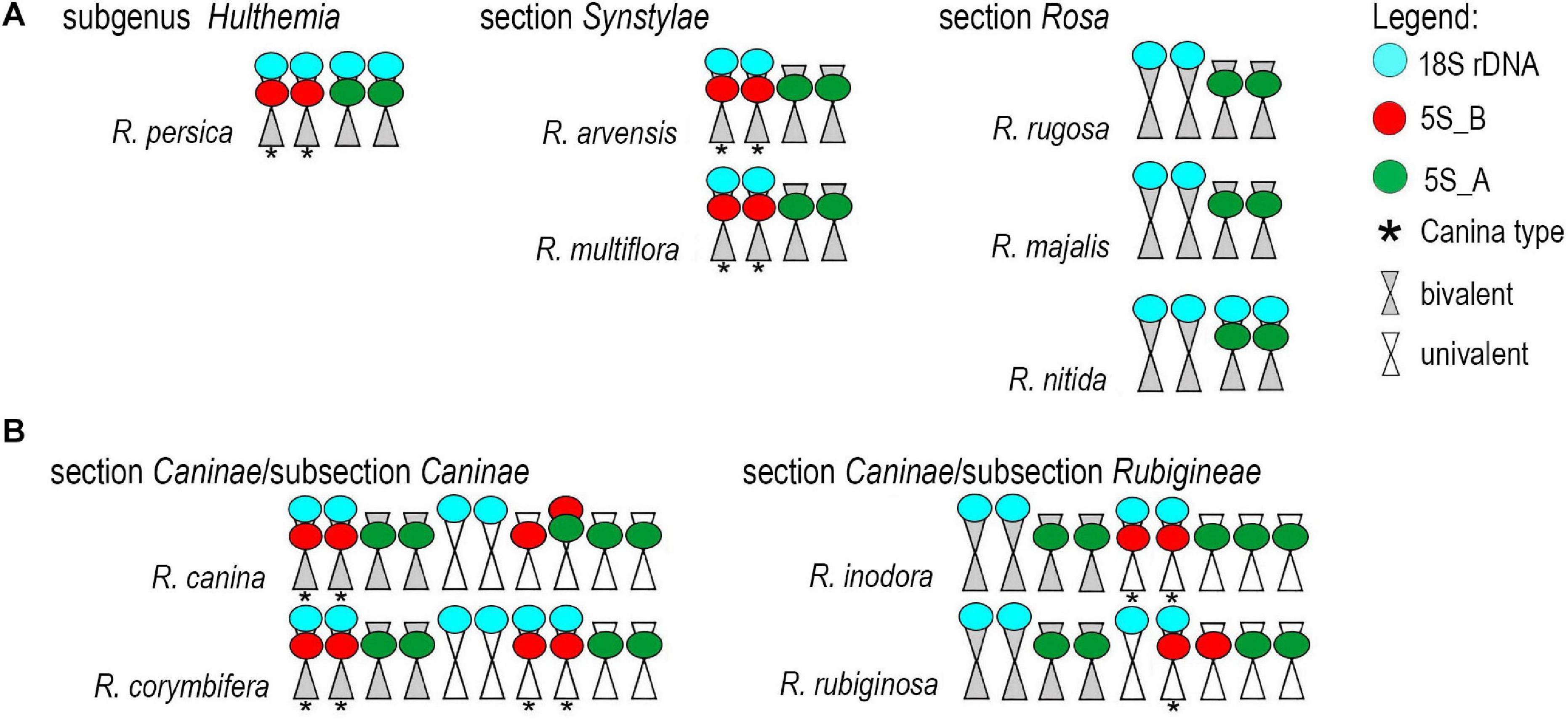
Figure 6. Ideograms of chromosomes carrying the rDNA loci. (A) Diploid species. (B) Pentaploid species from section Caninae.
We further analyzed meiotic (Figure 7) and mitotic (Supplementary Figure S5) chromosomes in polyploid species. As expected meiotic chromosomes of four 5x dogrose species (sect. Caninae) were represented by seven bivalents (pairs of chromosomes) and 21 univalents (Figure 7). In R. canina and R. corymbifera (subsect. Caninae), the 5S_A probe hybridized to one bivalent and three sites on univalent chromosomes. The 5S_B probe hybridized to one bivalent carrying the 18S (NOR) signal and two sites on univalents. The 5S_A and 5S_B signals occurred on different chromosomes except of one univalent chromosome in R. canina where both probes were co-localized. In R. inodora (subsect. Rubigineae), the 5S_A probe hybridized to one bivalent and three univalent chromosomes. The 5S_B probe hybridized to two univalent chromosomes carrying the 18S rDNA signal. Rosa rubiginosa (subsect. Rubigineae) showed a similar distribution of signals like R. inodora except that only one out of two 5S_B univalent chromosomes co-localized with the 18S signal. In addition, there were only two 5S_A sites on univalents. Collectively, these observations indicate that the number of rDNA sites, their chromosome position, and their meiotic behavior differ between subsections Caninae and Rubigineae. FISH on mitotic chromosomes from 4x R. spinosissima is shown in Supplementary Figure S5. In this species, the 5S_B probe hybridized to a chromosome pair which also carried the 18S rDNA signal (NOR) (Supplementary Figure S5). Two other 5S_B signals were colocalized (but did not overlap) with that of 5S_A on non-NOR chromosomes. 18S rDNA and 5S_A signals were localized on two separate chromosomes. Results are summarized in Figure 6 and Supplementary Table S4.
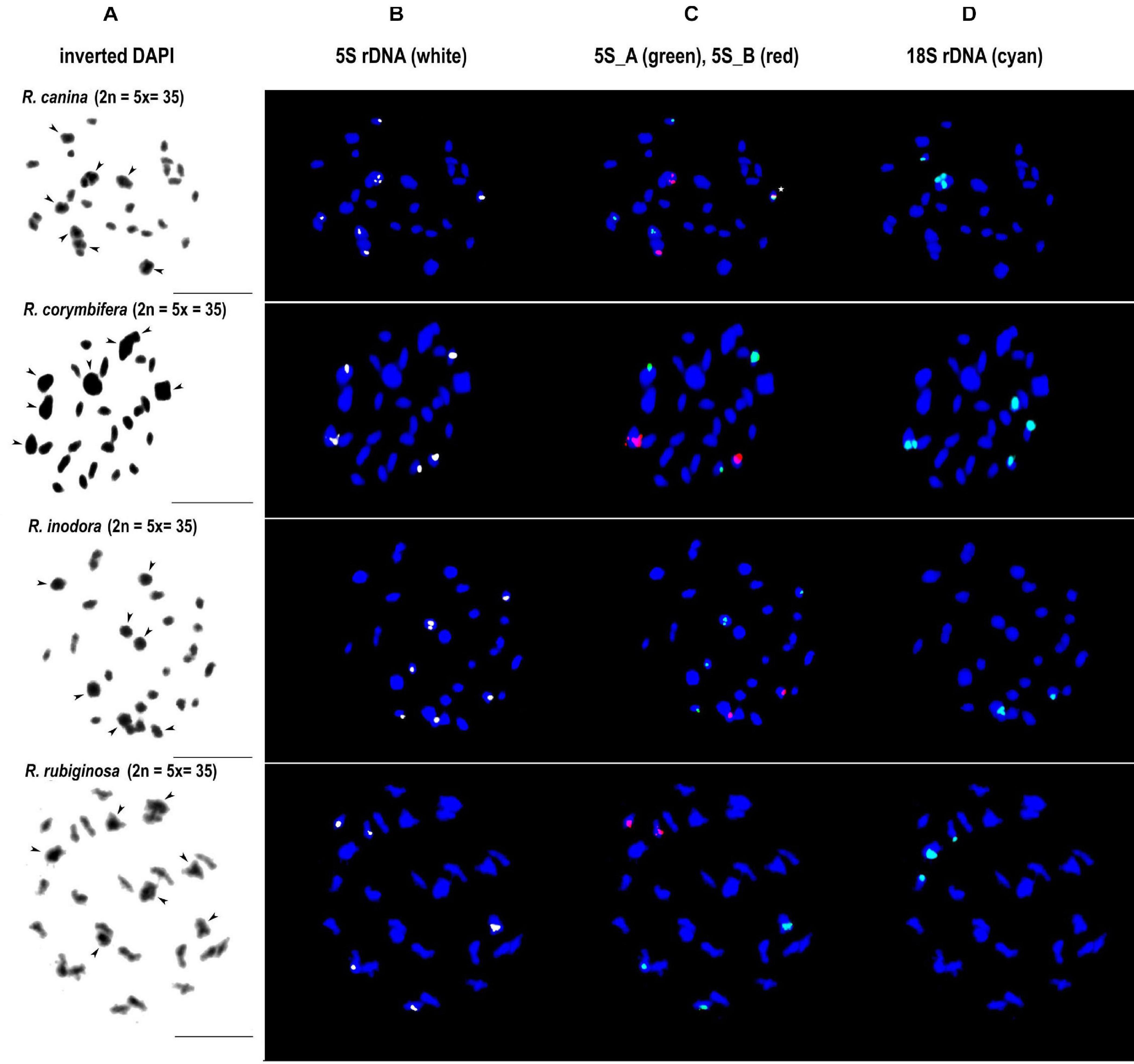
Figure 7. FISH analysis of meiotic chromosomes in pentaploid dogroses section Caninae: R. canina and R. corymbifera (subsection Caninae); R. inodora and R. rubiginosa (subsection Rubigineae). In each species, the same diakinesis was hybridized with the 5S genic (B), 5S intergenic (C), and 18S rDNA (D) probes. DAPI staining (gray scale, inverted) is to the left margin (A). Arrowheads indicate bivalent chromosomes. In R. canina, a chromosome with co-localized 5S_A and 5S_B signals is marked with an asterisk. Scale bar: 10 μm.
To study chromosome evolution and potential hybridization events in the genus Rosa, we analyzed the structure and organization of 5S rDNA in several diploid and polyploid species. We found that the genus is dominated by essentially two 5S rDNA families which markedly differ in IGSs and date back to the genus’ base.
The IGSs of rRNA genes are rapidly evolving sequences, and it is common to find variation even between closely related species. It was therefore striking to observe that the genus Rosa is dominated essentially by only two 5S rDNA families and that no other family was amplified in any of the species analyzed here. Both families occupy different chromosome loci: the 5S_B family was always co-localized with NOR (35S rDNA), while the 5S_A family was mostly but not exclusively (see below) separate (Figure 6). Moreover, R. persica (subg. Hulthemia) amplified both families at similar ratio in its genome (Figures 2, 3). In contrast to all other diploid species (Ma et al., 1997), R. persica is also exceptional in possessing two NORs instead of one per haploid chromosome set. Since R. persica was consistently identified as the earliest divergent lineage in most phylogenies (Fougere-Danezan et al., 2015; Debray et al., 2019), we presume that the configuration with NORs co-localizing with distinct 5S rDNA families (Figure 6) is an ancient condition, while the NOR chromosome without 5S rDNA locus is derived. This assumption is supported by the following observations: First, the Rosa and allies clade contained the 5S_A family which was co-localized with 18S rDNA locus in R. nitida but not in R. majalis and R. rugosa. Second, all members of Synstylae, Indicae, and Pimpinellifoliae contained two 5S rDNA families albeit at differing ratios. For example, R. multiflora (sect. Synstylae) had prevalent 5S_B family, while the 5S_A family dominated in R. chinensis, a member of the closely related sect. Indicae. Similarly, in R. lucieae ([R. wichurana]; sect. Synstylae), both families are likely to be represented by two loci out of which one co-localized with 18S rDNA on the same chromosome (Kirov et al., 2016). Third, R. laevigata (sect. Laevigatae) was dominated by the 5S_B family (Figures 2, 3). This species is sister to the remaining species of the Synstylae and allies clade (Fougere-Danezan et al., 2015; Debray et al., 2019).
Of note, R. nitida differed from R. majalis and R. rugosa in having the 5S_A family co-localized with NOR. Traditionally, R. nitida has been classified into a separate section called Carolinae (Crépin, 1889). However, more recent taxonomies based on molecular markers failed to support this distinction and all three species are now placed within the Rosa and allies clade (Wissemann and Ritz, 2005; Joly and Bruneau, 2006). Interestingly, members of sect. Rosa tend to have much smaller loci of the centromeric satellite repeat CANR4 compared to other species of the genus (Lunerova et al., 2020). However, R. nitida is exceptional in having large abundance of the CANR4 satellite (10 out 14 chromosomes carried strong FISH signals, not shown). These features suggest chromosomal rearrangements accompanying speciation events in sect. Rosa although the basic chromosome number (x = 7) remained unchanged.
Neither A nor B type sequences were found in 5S rDNA of the genera Prunus, Rubus, Fragaria, Cliffortia, Acaena, and Sanguisorba (all Rosaceae). These observations suggest that both 5S rDNA families have their origin in the early evolution of the genus Rosa because the common ancestor of both families was dated at app. 32 myrs ago. Furthermore, molecular dating of diversification within both 5S rDNA clades was dated to app. 5–9 myrs each (Figure 4) matching at least roughly the origin of Synstylae and allies and Rosa and allies (Fougere-Danezan et al., 2015).
Despite considerable interest, the composition of pentaploid (2n = 5x = 35) dogrose genome remains enigmatic (Wissemann, 1999; Nybom et al., 2004; Ritz et al., 2005; Herklotz et al., 2018; Lunerova et al., 2020). Previous studies based on microsatellite markers revealed genetic distinction between bivalent- and univalent-forming chromosomes (Nybom et al., 2006). The analysis of 35S rDNA markers confirmed these assumptions revealing two highly divergent ITS types (named Canina type and Rubiginosa type) present at variable ratios in subsections Caninae and Rubigineae, respectively (Ritz et al., 2005; Kovařik et al., 2008). Here we show that the Canina type ITS is co-localized with 5S_B rDNA locus (NOR), while the Rubiginosa ITS type is not. This Canina type of configuration [equivalent to marker chromosome 1 (Lim et al., 2005) or chromosome 7 (Kirov et al., 2016)] is typical for the bivalent-forming chromosomes in R. canina and R. corymbifera, both from subsection Caninae (Figure 6). In contrast, the Canina-type configuration of both rDNAs is found on univalent chromosomes in R. rubiginosa and R. inodora (subsection Rubigineae). The bivalent-forming chromosomes in this subsection carry the 5S_A family on a non-NOR chromosome. These data support the hypothesis that dogroses from both subsections arose by reciprocal hybridization of closely related species (Bruneau et al., 2007; Herklotz and Ritz, 2017). A very similar composition of dogrose genomes is also supported by the shallow nodes among dogroses in the 5S rDNA phylogeny (Figure 4). However, within the subsections, we detected some variation in number and distribution of loci especially between the univalent sets. For example, R. inodora carried two univalent-forming chromosomes with Canina type configuration, while R. rubiginosa contained only one. Additionally, one of the R. canina univalent chromosomes carried both 5S_A and 5S_B chromosomes co-localized, while this chromosome was not found in the closely related R. corymbifera. The variation between univalent chromosomes is consistent with increased diversity of microsatellite markers on univalent genomes (Nybom et al., 2004) and could be either related to divergence of genome donors and/or to partial degeneration of univalent chromosomes due to their exclusion from recombination in meiosis. Although 5S rDNA pseuodogenes seem to be present in R. rugosa (Tynkevich and Volkov, 2014b), there is no indication for extensive 5S rDNA pseuodogenization in R. canina (Lim et al., 2005) and in other dogroses (this work).
Although it might be preliminary to trace potential genome donors of allopolyploid dogroses, it is notable that the tetraploid species with a regular meiosis, R. gallica (sect. Gallicanae), tends to cluster with dogroses. It is therefore possible that pentaploid dogroses actually arose by pollination of an unreduced non-dogrose tetraploid egg cell by a reduced male gamete from a diploid donor. In support, R. gallica ITS types were occasionally found in dogroses (Herklotz et al., 2018) and R. gallica belonged to a clade together with dogroses in plastid phylogenies (Fougere-Danezan et al., 2015). Moreover, R. gallica shares the distinct morphological feature of partially pinnate sepals with dogroses, which is absent in the remaining species of the genus. However, there are also tetraploid cytotypes within dogroses (e.g., R. villosa L., R. canina) forming triploid egg cells and haploid sperm cells, which would not fit in the proposed scenario so far but might have arisen by another combination of partially reduced gametes. The also occurring higher ploidy (6x) levels which are less frequently found in dogroses rather originated from hybridizations within dogrose species involving unreduced egg cells (Herklotz and Ritz, 2017).
The maintenance of two 5S rDNA families in the Rosa genomes is consistent with the increased stability of 5S loci as compared to 35S loci in allopolyploid genomes documented in several allopolyploid systems (Pedrosa-Harand et al., 2006; Weiss-Schneeweiss et al., 2008; Garcia et al., 2016; Amosova et al., 2019). The reasons for relative stasis of 5S rDNA loci are not well understood, while their position on chromosomes (Garcia et al., 2016) and epigenetic modifications (5S rDNA loci carry mostly heterochromatic landmarks; Simon et al., 2018) have been discussed. One also has to consider the relative scarcity of meiosis driving genetic recombination (and homogenization) in these long-living perennial shrubs. For example, a R. canina individual known as “Rose of Hildesheim” (North Germany) is estimated to be more than 700 years old (Peters and Peters, 2013). Interestingly, Gossypium allopolyploids which represent also perennial shrubs tend to maintain 5S rDNA loci relatively intact over millions of years, while they homogenized their 35S rDNA loci (Cronn et al., 1996).
We identified two 5S rDNA families which are widespread across the Rosa genus. The molecular and cytogenetic observations lead us to propose that both families have their origin deep in the genus history probably close to its base. A remarkably slow tempo of 5S rDNA evolution differs from other systems where these loci show considerable dynamics. The retention of a large number of ancient rDNA sequences in Rosa genomes resonates with drastic allelic heterozygosity encountered in previous studies of microsatellites (Nybom et al., 2006), protein coding genes sequences (Joly and Bruneau, 2006), and more recent whole genome sequencing projects (Raymond et al., 2018). In future, it will be interesting to analyze expression of alleles inherited from deep evolutionary times.
The datasets presented in this study can be found in online repositories. The names of the repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW349696; https://www.ncbi.nlm.nih.gov/genbank/, MW349697.
AK, CR, and RAV conceived and designed the study. CR, JL, RAV, VH, and YT performed the experiments and collected material. JL, RV, RAV, VH, and YT analyzed the data. AK, CR, JL, and RAV wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the German Science Foundation (DFG Ri 2090/3-1), the Czech Science Foundation (GAČR 20-14133J), the Ministry of Education and Science of Ukraine (Grant No. 0106U003620), and DAAD (German Academic Exchange Service) Research Fellowship for Yuri Tynkevich.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are thankful to M. Schwager, M. Laufer, and J. Lorenz (Senckenberg Museum of Natural History Görlitz) for their excellent help in the laboratory. We thank V. Wissemann (Justus Liebig University Gießen, Germany), G. Vogg (Botanical Garden of University Würzburg, Germany), S. Arndt (Botanical Garden of Friedrich Schiller University Jena, Germany), U. Pietzarka (Forstpark Tharandt, TU Dresden, Germany), and M. I. Vykliuk (Botanical Garden of the Yuriy Fedkovych Chernivtsi National University, Ukraine) for their kind help in obtaining access to plant material. We also thank P. Hotka and P. Ferus (Botanical Garden Mlynany of the Slovak Academy of Sciences) for their kind help and for enabling the collecting of the samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.643548/full#supplementary-material
Supplementary Figure 1 | Genomic analysis of 5S rDNA variants. Projections of 5S rDNA cluster graphs in polyploid Rosa species. Loops representing the IGS reads are colored according to the 5S_A (green) and 5S_B (red) IGS sequences. Blue nodes represent the 5S genic regions.
Supplementary Figure 2 | Phylogenic relationships among Rosa species inferred from Maximum-Likelihood analyses of 5S rDNA intergenic spacers. The tree was rooted with Geum urbanum. Species names are colored according to ploidy level (2x—black, 4x—light blue, 5x—pink). Branch colors represent 5S_A (green) and 5S_B (red) lineages. Bootstrap support >70% is indicated above branches.
Supplementary Figure 3 | A Neighbor-Joining phylogeny tree constructed from the 5S rDNA consensus sequences. The same data set as in Figure 4 was analyzed using the Juke Cantor model. One thousand repetitions were allowed for the statistical support. Bootstrap support levels >80% are indicated.
Supplementary Figure 4 | Phylogenetic NJ trees constructed from type A and B 5S rDNA sequences, respectively. Note a similar position of R. persica on both trees (early separating). Note clustering of Caninae species (red asterisks after the names) in an unresolved branch. Note incongruent placement of the R. spinosissima variants.
Supplementary Figure 5 | FISH analysis of root tip mitotic chromosomes from R. spinosissima and R. nitida. Arrows indicate NOR chromosomes with co-localized 5S rDNA loci. Note odd (5) number of 5S rDNA sites in the assumingly R. spinosissima allotetraploid.
Supplementary Table 1 | Information about the taxonomy and origin of plants used in this study.
Supplementary Table 2 | Details of bioinformatic procedures leading to 5S rDNA genome proportion and copy number estimation. Sheet 1—5S_A variant. Sheet 2—5S_B variant.
Supplementary Table 3 | Summary of genome proportions and copy number of 5S rDNA families in Rosa genomes.
Supplementary Table 4 | Summary of cytogenetic FISH analyses.
Amosova, A. V., Zoshchuk, S. A., Rodionov, A. V., Ghukasyan, L., Samatadze, T. E., Punina, E. O., et al. (2019). Molecular cytogenetics of valuable Arctic and sub-Arctic pasture grass species from the Aveneae/Poeae tribe complex (Poaceae). BMC Genet. 20:92. doi: 10.1186/s12863-019-0792-2
Benson, C. W., Mao, Q., and Huff, D. R. (2020). Global DNA methylation predicts epigenetic reprogramming and transgenerational plasticity in Poa annua L. Crop Sci. 2020, 1–12. doi: 10.1002/csc2.20337
Blackburn, K. B., and Harrison, J. W. H. (1921). The status of the British rose forms as determined by their cytological behaviour. Ann. Bot. 35, 159–188. doi: 10.1093/oxfordjournals.aob.a089753
Bruneau, A., Starr, J. R., and Joly, S. (2007). Phylogenetic relationships in the genus Rosa: New evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst. Bot. 32, 366–378. doi: 10.1600/036364407781179653
Cronn, R. C., Zhao, X., Paterson, A. H., and Wendel, J. F. (1996). Polymorphism and concerted evolution in a tandemly repeated gene family: 5S ribosomal DNA in diploid and allopolyploid cottons. J. Mol. Evol. 42, 685–705. doi: 10.1007/BF02338802
Debray, K., Marie-Magdelaine, J., Ruttink, T., Clotault, J., Foucher, F., and Malecot, V. (2019). Identification and assessment of variable single-copy orthologous (SCO) nuclear loci for low-level phylogenomics: a case study in the genus Rosa (Rosaceae). BMC Evol. Biol. 19:152. doi: 10.1186/s12862-019-1479-z
Dover, G. (1982). Molecular drive: a cohesive mode of species evolution. Nature 299, 111–117. doi: 10.1038/299111a0
Edelman, D. W. (1975). The Eocene Germer Basin Flora of South-Central Idaho. Ph. D. thesis, Moscow: University of Idaho.
Eickbush, T. H., and Eickbush, D. G. (2007). Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175, 477–485. doi: 10.1534/genetics.107.071399
Fernandez-Romero, M. D., Torres, A. M., Millan, T., Cubero, J. I., and Cabrera, A. (2001). Physical mapping of ribosomal DNA on several species of the subgenus Rosa. Theor. Appl. Genet. 103, 835–838. doi: 10.1007/s001220100709
Fougere-Danezan, M., Joly, S., Bruneau, A., Gao, X. F., and Zhang, L. B. (2015). Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann. Bot. 115, 275–291. doi: 10.1093/aob/mcu245
Fulnecek, J., Lim, K. Y., Leitch, A. R., Kovařik, A., and Matyasek, R. (2002). Evolution and structure of 5S rDNA loci in allotetraploid Nicotiana tabacum and its putative parental species. Heredity 88, 19–25. doi: 10.1038/sj.hdy.6800001
Garcia, S., Borowska-Zuchowska, N., Wendel, J. F., Ainouche, M., Kuderova, A., and Kovařik, A. (2020). The utility of graph clustering of 5S ribosomal DNA homoeologs in plant allopolyploids, homoploid hybrids and cryptic introgressants. Front. Plant Sci. 11:41. doi: 10.3389/fpls.2020.00041
Garcia, S., Kovařik, A., Leitch, A. R., and Garnatje, T. (2016). Cytogenetic features of rRNA genes across land plants: analysis of the Plant rDNA database. Plant J. 89, 1020–1030. doi: 10.1111/tpj.13442
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Hemleben, V., Ganal, M., Gersnter, J., Schiebel, K., and Torres, R. A. (1988). “Organization and length heterogeneity of plant ribosomal RNA genes,” in The architecture of Eukaryotic Gene, ed. G. Kahl (Weinheim: VHC), 371–384.
Herklotz, V., and Ritz, C. M. (2017). Multiple and asymmetrical origin of polyploid dog rose hybrids (Rosa L. sect. Caninae (DC.) Ser.) involving unreduced gametes. Ann. Bot. 120, 209–220. doi: 10.1093/aob/mcw217
Herklotz, V., Kovařik, A., Lunerova, J., Lippitsch, S., Groth, M., and Ritz, C. M. (2018). The fate of ribosomal RNA genes in spontaneous polyploid dogrose hybrids [Rosa L. sect. Caninae (DC.) Ser.] exhibiting non-symmetrical meiosis. Plant J. 94, 77–90. doi: 10.1111/tpj.13843
Joly, S., and Bruneau, A. (2006). Incorporating allelic variation for reconstructing the evolutionary history of organisms from multiple genes: An example from Rosa in north America. Syst. Biol. 55, 623–636. doi: 10.1080/10635150600863109
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kirov, I. V., Van Laere, K., Van Roy, N., and Khrustaleva, L. I. (2016). Towards a FISH-based karyotype of Rosa L. (Rosaceae). Comp. Cytogenet. 10, 543–554. doi: 10.3897/CompCytogen.v10i4.9536
Koopman, W. J. M., Wissemann, V., De Cock, K., Van Huylenbroeck, J., De Riek, J., Sabatlno, G. J. H., et al. (2008). AFLP markers as a tool to reconstruct complex relationships: A case study in Rosa (Rosaceae). Am. J. Bot. 95, 353–366. doi: 10.3732/ajb.95.3.353
Kovařik, A., Werlemark, G., Leitch, A. R., Souckova-Skalicka, K., Lim, Y. K., Khaitova, L., et al. (2008). The asymmetric meiosis in pentaploid dogroses (Rosa sect. Caninae) is associated with a skewed distribution of rRNA gene families in the gametes. Heredity 101, 359–367. doi: 10.1038/Hdy.2008.63
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/Nmeth.1923
Lim, K. Y., Werlemark, G., Matyasek, R., Bringloe, J. B., Sieber, V., El Mokadem, H., et al. (2005). Evolutionary implications of permanent odd polyploidy in the stable sexual, pentaploid of Rosa canina L. Heredity 94, 501–506. doi: 10.1038/sj.hdy.6800648
Liu, C. Y., Wang, G. L., Wang, H., Xia, T., Zhang, S. Z., Wang, Q. G., et al. (2015). Phylogenetic relationships in the genus Rosa revisited based on rpl16, trnL-F, and atpB-rbcL sequences. HortScience 50, 1618–1624. doi: 10.21273/Hortsci.50.11.1618
Long, E. O., and Dawid, I. B. (1980). Repeated genes in eukaryotes. Annu. Rev. Biochem. 49, 727–764. doi: 10.1146/annurev.bi.49.070180.003455
Lunerova, J., Herklotz, V., Laudien, M., Vozarova, R., Groth, M., Kovařik, A., et al. (2020). Asymmetrical canina meiosis is accompanied by the expansion of a pericentromeric satellite in non-recombining univalent chromosomes in the genus Rosa. Ann. Bot. 125, 1025–1038. doi: 10.1093/aob/mcaa028
Ma, Y., Crane, C. F., and Byrne, D. H. (1997). Karyotypic relationships among some Rosa species. Caryologia 50, 317–326. doi: 10.1080/00087114.1997.10797405
Matsumoto, S., Kouchi, M., Yabuki, J., Kusunoki, M., Ueda, Y., and Fukui, H. (1998). Phylogenetic analyses of the genus Rosa using the matK sequence: molecular evidence for the narrow genetic background of modern roses. Sci. Hortic. 77, 73–82. doi: 10.1016/S0304-4238(98)00169-1
Millan, T., Osuna, F., Cobos, S., Torres, A. M., and Cubero, J. I. (1996). Using RAPDs to study phylogenetic relationships in Rosa. Theor. Appl. Genet. 92, 273–277. doi: 10.1007/Bf00223385
Nagylaki, T. (1990). Gene conversion, linkage, and the evolution of repeated genes dispersed among multiple chromosomes. Genetics 126, 261–276.
Nakamura, N., Hirakawa, H., Sato, S., Otagaki, S., Matsumoto, S., Tabata, S., et al. (2018). Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 25, 113–121. doi: 10.1093/dnares/dsx042
Nieto Feliner, G., and Rossello, J. A. (2012). “Concerted evolution of multigene families and homeologous recombination,” in Plant Genome Diversity, ed. J. F. Wendel (Wien: Springer-Verlag), 171–194.
Novak, P., Neumann, P., and Macas, J. (2010). Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 11:378. doi: 10.1186/1471-2105-11-378
Novak, P., Neumann, P., Pech, J., Steinhaisl, J., and Macas, J. (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793. doi: 10.1093/bioinformatics/btt054
Nybom, H., Esselink, G. D., Werlemark, G., and Vosman, B. (2004). Microsatellite DNA marker inheritance indicates preferential pairing between two highly homologous genomes in polyploid and hemisexual dog-roses, Rosa L. sect. Caninae DC. Heredity 92, 139–150. doi: 10.1038/sj.hdy.6800332
Nybom, H., Esselink, G. D., Werlemark, G., Leus, L., and Vosman, B. (2006). Unique genomic configuration revealed by microsatellite DNA in polyploid dogroses, Rosa sect. Caninae. J. Evol. Biol. 19, 635–648. doi: 10.1111/j.1420-9101.2005.01010.x
Ohta, T. (1984). Some models of gene conversion for treating the evolution of multigene families. Genetics 106, 517–528.
Pachl, Š (2011). Variablita botanických druhů rodu Rosa L., a možnosti jejich využití v krajinářské tvorbê. Ph. D.thesis, Nitra, SK: Slovak University of Agriculture.
Pastova, L., Belyayev, A., and Mahelka, V. (2019). Molecular cytogenetic characterisation of Elytrigia xmucronata, a natural hybrid of E. intermedia and E. repens (Triticeae, Poaceae). BMC Plant. Biol. 19:230. doi: 10.1186/s12870-019-1806-y
Pedrosa-Harand, A., de Almeida, C. C. S., Mosiolek, M., Blair, M., Schweizer, D., and Guerra, M. (2006). Extensive ribosomal DNA amplification during Andean common bean (Phaseolus vulgaris L.) evolution. Theor. Appl. Genet. 112, 924–933. doi: 10.1007/s00122-005-0196-8
Peters, H., and Peters, H. (2013). Tausendjähriger Rosenstock from Hildesheim (the thousand-year-oldrosebush). Available online at: www.webcitation.org/6PIIQmfN8?url=http://www.worldrose.org/heritage/HeritageNumber10.pdf
Raymond, O., Gouzy, J., Just, J., Badouin, H., Verdenaud, M., Lemainque, A., et al. (2018). The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50:772. doi: 10.1038/s41588-018-0110-3
Ritz, C. M., Schmuths, H., and Wissemann, V. (2005). Evolution by reticulation: European dogroses originated by multiple hybridization across the genus Rosa. J. Hered. 96, 4–14. doi: 10.1093/jhered/esi011
Roa, F., and Guerra, M. (2012). Distribution of 45S rDNA sites in chromosomes of plants: structural and evolutionary implications. BMC Evol. Biol. 12:225. doi: 10.1186/1471-2148-12-225
Roberts, A. V., Gladis, T., and Brumme, H. (2009). DNA amounts of roses (Rosa L.) and their use in attributing ploidy levels. Plant Cell Rep. 28, 61–71. doi: 10.1007/s00299-008-0615-9
Rogers, S. O., and Bendich, A. J. (1985). Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant-tissues. Plant Mol. Biol. 5, 69–76. doi: 10.1007/Bf00020088
Saint-Oyant, L. H., Ruttink, T., Hamama, L., Kirov, I., Lakhwani, D., Zhou, N. N., et al. (2018). A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 4, 473–484. doi: 10.1038/s41477-018-0166-1
Schwarzacher, T., and Heslop-Harrison, P. (2000). Practical in situ hybridization. Oxford: BIOS Scientific Publishers.
Simon, L., Rabanal, F. A., Dubos, T., Oliver, C., Lauber, D., Poulet, A., et al. (2018). Genetic and epigenetic variation in 5S ribosomal RNA genes reveals genome dynamics in Arabidopsis thaliana. Nucl. Acids Res. 46, 3019–3033. doi: 10.1093/nar/gky163
Tamura, K. (1992). Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 9, 678–687.
Tamura, K., Tao, Q., and Kumar, S. (2018). Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates. Mol. Biol. Evol. 35, 1770–1782. doi: 10.1093/molbev/msy044
Tynkevich, Y. O., and Volkov, R. A. (2014a). Novel structural class of 5S rDNA of Rosa wichurana Crep. Rep. Natl. Acad. Sci. Ukraine 5, 143–148. doi: 10.15407/dopovidi2014.05.143
Tynkevich, Y. O., and Volkov, R. A. (2014b). Structural organization of 5Sribosomal DNA in Rosa rugosa. Cytol. Genet. 48, 1–6. doi: 10.3103/S0095452714010095
Volkov, R. A., Panchuk, I. I., Borisjuk, N. V., Hosiawa-Baranska, M., Maluszynska, J., and Hemleben, V. (2017). Evolutional dynamics of 45S and 5S ribosomal DNA in ancient allohexaploid Atropa belladonna. BMC Plant Biol. 17:6. doi: 10.1186/s12870-017-0978-6
Volkov, R. A., Zanke, C., Panchuk, I. I., and Hemleben, V. (2001). Molecular evolution of 5S rDNA of Solanum species (sect. Petota): application for molecular phylogeny and breeding. Theor. Appl. Genet. 103, 1273–1282. doi: 10.1007/s001220100670
Wang, G. L. (2007). A study on the history of Chinese roses from ancient works and images. Acta Hortic. 751, 347–356. doi: 10.17660/ActaHortic.2007.751.44
Weiss-Schneeweiss, H., Tremetsberger, K., Schneeweiss, G. M., Parker, J. S., and Stuessy, T. F. (2008). Karyotype diversification and evolution in diploid and polyploid South American Hypochaeris (Asteraceae) inferred from rDNA localization and genetic fingerprint data. Ann. Bot. 101, 909–918. doi: 10.1093/aob/mcn023
Wissemann, V. (1999). Genetic constitution of Rosa sect. Caninae (R. canina, R. jundzillii) and sect. Gallicanae (R. gallica). Angew. Bot. 73, 191–196.
Wissemann, V. (2003). “Conventional taxonomy (wild roses),” in Encyclopedia of Rose Science, eds A. V. Roberts, T. Debener, and S. Gudin (Oxford: Elsevier Academic Press).
Wissemann, V., and Ritz, C. M. (2005). The genus Rosa (Rosoideae, Rosaceae) revisited: molecular analysis of nrITS-1 and atpB-rbcL intergenic spacer (IGS) versus conventional taxonomy. Bot. J. Linn. Soc. 147, 275–290. doi: 10.1111/j.1095-8339.2005.00368.x
Xiang, Y. Z., Huang, C. H., Hu, Y., Wen, J., Li, S. H., Yi, T. S., et al. (2017). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 1026–1026. doi: 10.1093/molbev/msx093
Yokoya, K., Roberts, A. V., Mottley, J., Lewis, R., and Brandham, P. E. (2000). Nuclear DNA amounts in roses. Ann. Bot. 85, 557–561. doi: 10.1006/ambo.1999
Zetter, R., Hofmann, C., Draxler, I., Durango, de Cabrera, J., Vergel, M., et al. (1999). A rich Middle Eocene Microflora at Arroyo de los Mineros, near Cañadón Beta, NE Tierra del Fuego Province, Argentina. Abh. Geol. Bundesanst. 56, 439–460.
Zhang, J., Esselink, G. D., Che, D., Fougere-Danezan, M., Arens, P., and Smulders, M. J. M. (2013). The diploid origins of allopolyploid rose species studied using single nucleotide polymorphism haplotypes flanking a microsatellite repeat. J. Hortic. Sci. Biotechnol. 88, 85–92. doi: 10.1080/14620316.2013.11512940
Keywords: 5S rDNA, evolution, Rosa, genomics, cytogenetics, repeatome, Rosaceae
Citation: Vozárová R, Herklotz V, Kovařík A, Tynkevich YO, Volkov RA, Ritz CM and Lunerová J (2021) Ancient Origin of Two 5S rDNA Families Dominating in the Genus Rosa and Their Behavior in the Canina-Type Meiosis. Front. Plant Sci. 12:643548. doi: 10.3389/fpls.2021.643548
Received: 18 December 2020; Accepted: 15 February 2021;
Published: 08 March 2021.
Edited by:
Susann Wicke, Humboldt University of Berlin, GermanyReviewed by:
Hai-Qin Zhang, Sichuan Agricultural University, ChinaCopyright © 2021 Vozárová, Herklotz, Kovařík, Tynkevich, Volkov, Ritz and Lunerová. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jana Lunerová, amFuYS5sdW5lckBpYnAuY3o=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.