 Xiaohuan Mu
Xiaohuan Mu Jiankun Li
Jiankun Li Zhuangzhuang Dai
Zhuangzhuang Dai Liping Xu
Liping Xu Tianyuan Fan
Tianyuan Fan Teng Jing
Teng Jing Mengyao Chen
Mengyao Chen Mingyue Gou
Mingyue Gou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 17 May 2021
Sec. Plant Metabolism and Chemodiversity
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.638792
Disease lesion mimic (Les/les) mutants display disease-like spontaneous lesions in the absence of pathogen infection, implying the constitutive activation of defense responses. However, the genetic and biochemical bases underlying the activated defense responses in those mutants remain largely unknown. Here, we performed integrated transcriptomics and metabolomics analysis on three typical maize Les mutants Les4, Les10, and Les17 with large, medium, and small lesion size, respectively, thereby dissecting the activated defense responses at the transcriptional and metabolomic level. A total of 1,714, 4,887, and 1,625 differentially expressed genes (DEGs) were identified in Les4, Les10, and Les17, respectively. Among them, 570, 3,299, and 447 specific differentially expressed genes (SGs) were identified, implying a specific function of each LES gene. In addition, 480 common differentially expressed genes (CGs) and 42 common differentially accumulated metabolites (CMs) were identified in all Les mutants, suggesting the robust activation of shared signaling pathways. Intriguingly, substantial analysis of the CGs indicated that genes involved in the programmed cell death, defense responses, and phenylpropanoid and terpenoid biosynthesis were most commonly activated. Genes involved in photosynthetic biosynthesis, however, were generally repressed. Consistently, the dominant CMs identified were phenylpropanoids and flavonoids. In particular, lignin, the phenylpropanoid-based polymer, was significantly increased in all three mutants. These data collectively imply that transcriptional activation of defense-related gene expression; increase of phenylpropanoid, lignin, flavonoid, and terpenoid biosynthesis; and inhibition of photosynthesis are generalnatures associated with the lesion formation and constitutively activated defense responses in those mutants. Further studies on the identified SGs and CGs will shed new light on the function of each LES gene as well as the regulatory network of defense responses in maize.
Disease lesion mimics are a class of mutants that display disease-like spontaneous lesions in the absence of pathogen infection. A huge number of disease lesion mimic mutants have been found in higher plants including Arabidopsis, rice, barley, wheat, and maize (Johal et al., 1995; Lorrain et al., 2003; Rostoks et al., 2003; Gou et al., 2009; Matin et al., 2010; Tang et al., 2013). Apart from their close association with cell death in plants (Johal et al., 1995; Bruggeman et al., 2015), most of them confer enhanced resistance to diverse pathogens; disease lesion mimic mutants have therefore become keys for deciphering cell death and defense pathways in plants (Lorrain et al., 2003). In Arabidopsis, studies using over a series of lesion mimic mutants (also known as autoimmune mutants) have led to numerous breakthrough findings unraveling the complexity of plant defense responses (Lorrain et al., 2003; Van Wersch et al., 2016). For instance, bon1 (bonzai1) and cpr1/cpr30 (constitutive expressor of pathogenesis-related genes 1/30) mutants that show lesion mimic phenotypes all have elevated resistance to the bacterial pathogen Pseudomonas syringae. Further studies on related genes indicated that they acted as negative regulators of defense responses via modulating resistance (R) gene SNC1 (Suppressor Of NPR1-1 Constitutive 1) at transcriptional and/or protein level (Yang and Hua, 2004; Gou et al., 2009, 2012; Cheng et al., 2011; Gou and Hua, 2012). In rice, the lesion mimic mutant oscul3a (Oryza sativa cullin3a) is resistant to both Magnaporthe oryzae and Xanthomonas oryzae pv oryzae, and OsCUL3a (Cullin-3a) negatively regulates cell death and immunity by degrading OsNPR1 (Non-Expresser Of PR Genes1) (Liu et al., 2017). In barley, the mlo (mildew resistance locus O) mutant conferred race-non-specific resistance to powdery mildew pathogens (Erysiphe graminis f.sp. hordei) (Wolter et al., 1993). In wheat, the lesion mimic line Ning7840 and lm3 (lesion mimic 3) have enhanced resistance to leaf rust (Puccinia triticina) and powdery mildew (Blumeria graminis f. sp. tritici), respectively (Li and Bai, 2009; Wang et al., 2016).
As one of the most common classes of mutations in maize, a series of dominant disease lesion mimic (Les) and recessive disease lesion mimic (les) mutants have been found more than half a century ago (Neuffer and Calvert, 1975; Walbot et al., 1983). Although >200 Les/les loci likely exist in maize (Walbot, 1991), only several genes have been cloned so far. For instance, lls1 (lethal leaf spot 1) is a les mutant showing elevated resistance to both Cochliobolus heterostrophus and Puccinia sorghi infections at the leaf epidermis (Simmons et al., 1998). Rp1-D21 (Resistance to P sorghi-D21) mutant, a Les mutant resulting from an aberration in the maize Rp1 disease resistance gene, confers a non-specific resistance to all common rust biotypes tested (P. sorghi) (Hu et al., 1996; Smith et al., 2010). These studies indicate that maize Les mutants are invaluable tools to dissect the maize defense responses conferring elevated disease resistance especially broad-spectrum resistance. However, because limited genes have been cloned, Les/les mutants are far from well-explored. In particular, it remains to be determined the underlying bases of the commonly activated defense responses that lead to the constitutive lesion formation, stunted growth, and enhanced disease resistance.
Transcriptional and metabolic regulations are essential for priming plant defense responses. Previous transcriptomic analysis has identified thousands of genes being upregulated under pathogen invasion (Thilmony et al., 2006; Kawahara et al., 2012; Windram et al., 2012; Dobon et al., 2016; Kebede et al., 2018), with a series of transcriptional factors being involved in controlling plant defense responses (Buscaill and Rivas, 2014). Metabolically, enhanced plant immunity largely attributes to divergent antimicrobial secondary metabolites called phytoanticipin and phytoalexin (Grayer and Harborne, 1994; Vanetten et al., 1994), for instance, the phenylpropanoids, terpenoids, benzoxazinoids, saponins, glucosinolates, etc. (Piasecka et al., 2015).
The phenylalanine-derived phenylpropanoids including lignin, flavonoids, coumarins, lignans, etc. all get involved in plant defense (Naoumkina et al., 2010). When plants suffered from pathogen infection, phenylpropanoid-related gene expressions have a significant change. For instance, Phenylalanine Ammonia Lyase (PAL) and Cinnamyl Alcohol Dehydrogenase (CAD) gene expressions are upregulated after infection of Cercospora zeina or Rhizoctonia solani in maize (Meyer et al., 2017; Li et al., 2019). Consistently, phenylalanine-derived lignin was found to be often increased after pathogen infection (Southerton and Deverall, 1990; Tiburzy and Reisener, 1990; Mohr and Cahill, 2007; Zhang et al., 2007). After treatment of lignin synthesis inhibitor, the resistance to Puccinia graminis was decreased in wheat (Moerschbacher et al., 1990). Recently, there have been increasing evidence supporting the idea that lignin plays vital roles in disease resistance. For instance, in maize, the lignin biosynthetic enzymes Caffeoyl-Coenzyme A O-Methyltransferase (CCoAOMT) and Hydroxycinnamoyl Coenzyme A:Shikimate Hydroxycinnamoyl Transferase (HCT) regulated plant disease resistance by forming a complex with the Nucleotide Binding Leucine-Rich Repeat (NLR) protein Rpl (Wang et al., 2015; Wang and Balint-Kurti, 2016). A further study indicated that ZmCCoAOMT2 is involved in the resistance to multiple diseases including southern leaf blight, gray leaf spot, and northern leaf blight (Yang et al., 2017). Besides, a natural variation in the F-box gene (ZmFBL41) confers banded leaf and sheath blight resistance in maize, resulting from the accumulation of lignin and restriction of lesion expansion via ZmCAD activation (Li et al., 2019). With the discovery of new lignin biosynthetic components (Gou et al., 2018, 2019), the role of lignin in plant defense remains to be further elucidated. Other than lignin, flavonoids have been well-documented for their resistance against multiple pathogenic bacteria and fungi (Ferreyra et al., 2012; Mierziak et al., 2014). For instance, 3-deoxyanthocyanidins, one of flavonoid, accumulated in maize after Fusarium infection (Sekhon et al., 2006).
Other than phenylpropanoids, terpenoids and benzoxazinoids are the two most important antibiotic compound categories in maize. Terpenoids including the kauralexins class of diterpenoids and the zealexins class of sesquiterpenoids contribute largely to defense against pathogenic fungi like Rhizopus, Fusarium, Aspergillus, and Colletotrichum spp. in maize (Huffaker et al., 2011; Schmelz et al., 2011; Meyer et al., 2017). The biosynthesis of kauralexins and zealexins requires multiple genes, and their antiobiotic activities were strictly controlled (Ding et al., 2019, 2020). In addition, benzoxazinoids exhibits antibiotic activity against pathogenic fungi like Helminthosporium turcicum, Cephalosporium maydis, and P. graminis (Niemeyer, 2009). The biosynthesis of benzoxazinoids also consists of multiple steps via the actions of benzoxazinoneless 1 (BX1) to BX14 enzymes in maize (Handrick et al., 2016).
With the development of high-throughput transcriptomic and metabolomic techniques, the gene expression and metabolomic changes can be monitored simultaneously in order to dissect the underlying signaling and biochemical pathways in respect to specific traits (Li et al., 2010; Chen et al., 2014; Tamiru et al., 2016; Kaling et al., 2018; Mcloughlin et al., 2018; Wang et al., 2018; Castro-Moretti et al., 2020). Although Les/les mutants are considered invaluable models to dissect defense mechanisms, there is a lack of systemic characterization of maize Les mutants by multi-omics analysis. Therefore, in this work, we sought to determine the bases of the activated defense responses by transcriptomics and metabolomics analysis of three typical maize Les mutants, thereby providing new insights into the gene expression and metabolite changes associated with the lesion formation and defense-related traits in Les mutants.
The Les4 (227E Les4-N1375, maintained in W23/M14 background), Les10 (217I Les10-NA607, maintained in W23/M14 background), and Les17 (312B Les17-N2345, maintained in B73/A632 background) seeds were generously provided by Maize Genetics Cooperation Stock Center. All seeds were planted at the experimental station of Henan Agricultural University in Xinxiang, Henan Province, China. All Les mutant plants from the stock were self-pollinated for two generations. A typical line showing 1:3 segregations of wild type (WT) and mutant phenotypes in the third generation of each Les stock was considered to be heterozygous, and the plants with WT and mutant phenotypes were sampled separately in this generation. Specifically, the putative homozygous plants of Les4 and Les10 showed more intense lesions than the putative heterozygous plants, while for Les17, the lesion phenotype of putative homozygous and/or heterozygous plants were similar (Supplementary Figure 1). For all segregating populations, plants with uniquely intense lesion phenotype were mixed respectively to be the mutant pool, while plants with no lesion phenotype were mixed to be the WT pool. For RNA extraction and chemical analysis, the third and fourth above-ear leaves from four plants at 5 days after silking were pooled as one sample, and three replicates of sample were frozen in liquid nitrogen before use. For biomass quantification, three fully matured whole plants (without ear) per replicate were pooled as one sample and three replicates of samples were harvested and dried at 65°C to a constant weight and then weighed.
For diaminobenzidine (DAB) staining, the leaf sample harvested above were cut into 3-cm-wide pieces, soaked in 1 mg ml–1 DAB (pH = 3.8) solution for 8 h in the dark (Chintamanani et al., 2010). After removal of DAB solution, 90% ethanol was used to remove chlorophyll by incubating in a shaker. Images were taken using a stereomicroscope (Olympus SZX7). Cell wall was extracted from fresh leaves and lignin was quantified following Gou et al. (2019). Chlorophyll was measured according to Mu et al. (2016). For lignin and chlorophyll measurement, three replicates with four plants per replicate of mutant and WT were used.
We performed the pathogen test of C. lunata (Wakker) Boed. as previously described with some modifications (Huang et al., 2009). The C. lunata (Wakker) Boed. strain CX-3 was cultivated on Potato Dextrose Agar medium at 28°C for 1 week in a growth chamber. The spores were collected and suspended in distilled water with 0.02% Tween-20 and diluted into 1 × 106 spores ml–1. The above-ear leaves of maize plants at silking stage were sprayed with the suspension and pictures taken for each leaf 7 days later. The leaves of mock treatment were sprayed with distilled water with 0.02% Tween-20. The leaf spots of the fungal colony from three representative leaves were counted using Image J software and the colony forming unit (cfu) (cm2 leaf area)–1 is presented.
Total RNA was extracted from sample using the Trizol reagent as described by Gu et al. (2013). Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, United States) following the manufacturer’s manual and index codes were added to attribute sequences to each sample (Dong et al., 2019). The average insert size for the paired-end libraries was 150 bp. Paired-end sequencing was performed on an Illumina HiSeq platform (Illumina Hiseq X-ten).
Clean reads were derived after removal of low-quality regions and adapter sequences from raw reads. Then, clean reads were aligned to the maize reference genome (B73 RefGen_v4, available online: https://www.maizegdb.org/assembly/) using HISAT2 (Kim et al., 2015). Aligned reads from HISAT2 mapping were subjected to String Tie for DeNovo Transcript assembly (Pertea et al., 2015). The expression of each gene was normalized to fragments per kilobase of transcript per million reads (FPKM) to compare among different samples. The R package “DESeq2” was used to identify DEGs with fold changes (FC) above 2 and false discovery rate (FDR) lower than 0.05 (Love et al., 2014). The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were accomplished in R using the packages “clusterProfiler” and “pathview” (Yu et al., 2012). The significant GO terms in the biological process were further reduced with REVIGO1 (Supek et al., 2011) and visualized in Cytoscape (version 3.7.1) (Shannon et al., 2003). Venn diagram was generated from the web-based Venny2.12. The shared DEGs in Les4, Les10, and Les17 were defined as common genes (CGs). The subcellular location analysis of CGs was performed with the SUBA43 (Hooper et al., 2017). The heatmap was accomplished in R with the package “pheatmap.” The transcription factors (TFs) in CGs were identified based on the list obtained from PlantTFDB4 (Jin et al., 2017).
Three biological replicates of the total RNA used in the RNA-seq were treated with an RNase-free DNase Kit (Cat. # RR047A, TAKARA) to remove DNA contamination (He et al., 2019) and were verified by PCR amplification using the ZmACT1 intron primers. After being reverse transcribed into cDNA, the quantitative PCR was performed using a SYBR Green system. The primers used in the quantitative PCR analysis are listed in Supplementary Table 1. The maize ZmACT1 gene was used as internal controls for normalizing gene expression in maize.
Metabolomics was performed at Wuhan Metware Biotechnology Co., Ltd. (Wuhan, China). The metabolites were extracted using the method of Chen et al. (2013). Briefly, using an ultrahigh-performance liquid chromatography–electrospray ionization–tandem mass spectrometry (UPLC-ESI-MS/MS) system (UPLC, Shim-pack UFLC SHIMADZU CBM30A system5; MS, Applied Biosystems 4500 Q TRAP6, the 4-μl sample extracts were injected into a C18 column (1.8 μm, 2.1 mm × 100 mm), which was set to 40°C. The mobile phase was used as follows: A: pure water with 0.04% acetic acid, B: acetonitrile with 0.04% acetic acid. Sample measurements were performed with a gradient program as follows: 0–10 min, 5% B–95% B; 10–11 min, 95% B; 11–11.1 min, 95% B–5% B; 11.1–14 min, 5% B. The effluent was alternatively connected to an ESI-triple quadrupole-linear ion trap (QTRAP)-MS. The conditions and operation parameters were set as in previous studies (Zeng et al., 2020). Metabolite quantification was performed using multiple-reaction monitoring (MRM) mode (Shi et al., 2020). Partial least squares discriminant analysis (PLS-DA) was used to study the identified metabolites. Differentially accumulated metabolites (DAMs) were set with thresholds of variable importance in projection (VIP) ≥ 1 and log2(FC) ≥ 1 or ≤ -1. The shared DAMs in Les4, Les10, and Les17 were defined as common differentially accumulated metabolites (CMs). KEGG analysis of CMs was performed with the MBROLE 2.07 (Lopezibanez et al., 2016).
To explore the molecular bases of lesion formation and defense-related traits in Les mutants, we used three representative Les mutants Les4, Les10, and Les17 that have been previously mapped to distinct loci (Johal et al., 1995). Because the three mutants are maintained as heterozygotes in different background and the phenotype of Les mutants can be affected largely by genetic background according to previous report (Hoisington et al., 1982), all Les mutants from the original stock were self-pollinated for two generations, and the segregation population of the third generation was examined phenotypically and physiologically. Compared with their relative WT, Les4, Les10, and Les17 all showed spontaneous necrotic lesions on the leaves (Figure 1A and Supplementary Figure 1). In particular, Les4 mutant developed large necrotic lesion at later growth stage, Les10 displayed medium lesion at early stage, and Les17 showed small lesion at medium stage, consistent with a previous report (Johal et al., 1995). The shoot biomass was 86 and 48% lower than WT in Les10 and Les17, respectively (Figure 1B), while that of Les4 was only slightly but non-significantly lower than WT. Because all mutants showed yellowish phenotype, we measured their total chlorophyll content. Compared with their relative WT plants, all Les mutants have significantly reduced chlorophyll content (Figure 1C). Consistent with the necrotic lesions being observed, increased accumulation of H2O2 could be visualized in all three mutants by DAB staining following a previously described method (Chintamanani et al., 2010; Figure 1D).
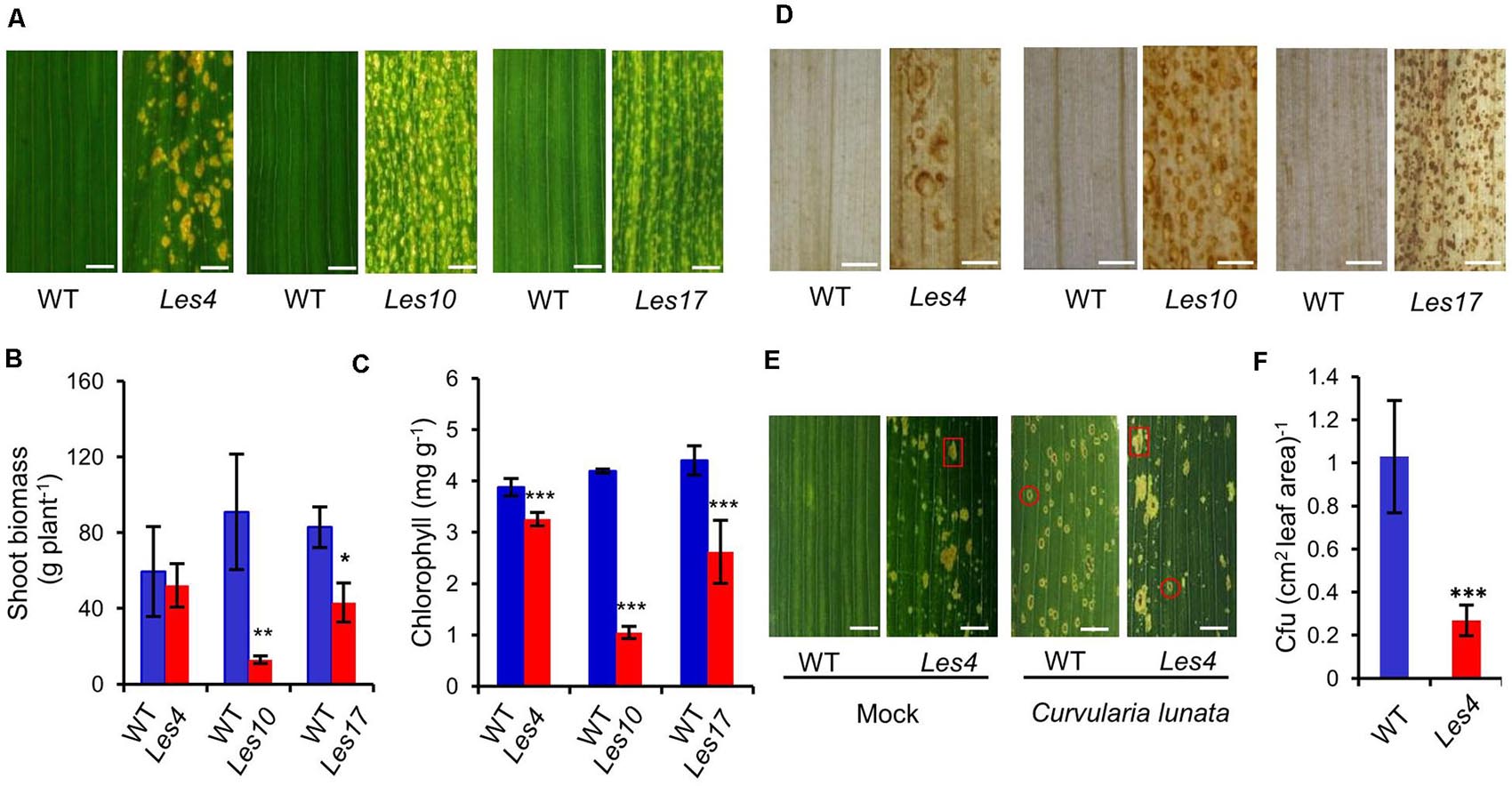
Figure 1. Phenotypic and physiological characterization of the Les4, Les10, and Les17. (A) Morphologies of Les4, Les10, and Les17 mutants and their respective wild type (WT). Scale bars = 5 mm. (B) The shoot biomass and content of chlorophyll in Les4, Les10, and Les17 mutants and their respective WT. (C) The total chlorophyll content in Les4, Les10, and Les17 mutants and their respective WT. (D) Images of DAB-stained leaves of in Les4, Les10, and Les17 mutants and their respective WT. Scale bars = 2 mm. (E) Morphologies of Mock and Curvularia lunata-infected WT and Les4 plant leaves 7 days after inoculation. A typical spontaneous lesion was indicated by a red square, and a typical curvularia-leaf spot-disease lesion was indicated by a red circle. Scale bars = 7.5 mm. (F) Quantification of Curvularia lunata colonies in Mock and Curvularia lunata-infected WT and Les4 plant leaves 7 days after inoculation. For (B,C,F), asterisks indicate significant differences compared with WT samples (Student’s t-test; *P < 0.05; **P < 0.01; ***P < 0.001). Error bars represent standard deviation.
We tested the disease resistance of Les4 using a C. lunata (Wakker) Boed. strain that causes curvularia leaf spot because the oval-shaped disease lesions are easily distinguishable from that of the big irregularly shaped spontaneous lesions in Les4. At 7 days after inoculation, the WT leaves displayed many disease lesions, while disease lesions observed in Les4 mutant were about 25% that of the WT, indicating significantly enhanced resistance of Les4 to curvularia leaf spot (Figures 1E,F).
We carried out transcriptomic analysis of Les mutants and their respective WT by RNA sequencing based on Illumina HiSeq platform. We used the third and fourth above-ear leaves at 5 days after silking because the lesion became easily visible at this stage, and the leaves were still in highly vigorous state. To eliminate the effect of background, the leaves of four plants were pooled as one sample, and three replicates of sample were used for RNA extraction and sequencing. After sequencing, 32,025, 33,031, and 32,035 expressed genes were detected in Les4, Les10, and Les17, respectively. The principal components analysis (PCA) plots clearly separated the WT samples from the mutant samples and the replicates of both WT and mutants were clustered into distinct patches (Supplementary Figure 2A), suggesting good reliability of our RNA-seq data.
A total of 1,714, 4,887, and 1,625 differentially expressed genes (DEGs) were identified in Les4, Les10, and Les17, compared to their respective WT, respectively (Figure 2A and Supplementary Tables 2, 3). Of these genes, 1,334, 2,861, and 1,134 were upregulated while 380, 2,026, and 491 were downregulated. More DEGs were identified in Les10 than in Les4 and Les17 (Supplementary Table 3). Furthermore, well-matched qRT-PCR results to the expression data of RNA-seq indicated reliability of our RNA-seq analysis (Supplementary Table 4). GO term enrichment analysis was performed to elucidate the functional enrichment of DEGs in each mutant. There were 187 biological processes (BPs), 17 cellular components (CCs), and 2 molecular functions (MFs) in GO analysis of DEGs of Les4. DEGs of Les4 were mainly related to “isoprenoid metabolic process,” “cellular aldehyde metabolic process,” “glyceraldehyde-3-phosphate metabolic process,” “isoprenoid biosynthetic process,” and “isopentenyl diphosphate biosynthetic process” (Figure 2C and Supplementary Table 5) in BP-GO terms. There were 270 BPs, 11 CCs, and 63 MFs in GO analysis of DEGs of Les10. DEGs of Les10 were mainly related to “response to wounding,” “response to drug,” “response to chitin,” “response to hormone levels,” and “hormone metabolic process” in BP-GO terms (Figure 2C and Supplementary Table 5). There were 129 BPs and 52 MFs in GO analysis of DEGs of Les17. DEGs of Les17 were mainly related to “defense response to bacterium,” “response to drug,” “response to wounding,” “defense response to oomycetes,” and “response to oomycetes” in BP-GO terms (Figure 2C and Supplementary Table 5). In general, the DEGs were mostly related to defense response and metabolic process.
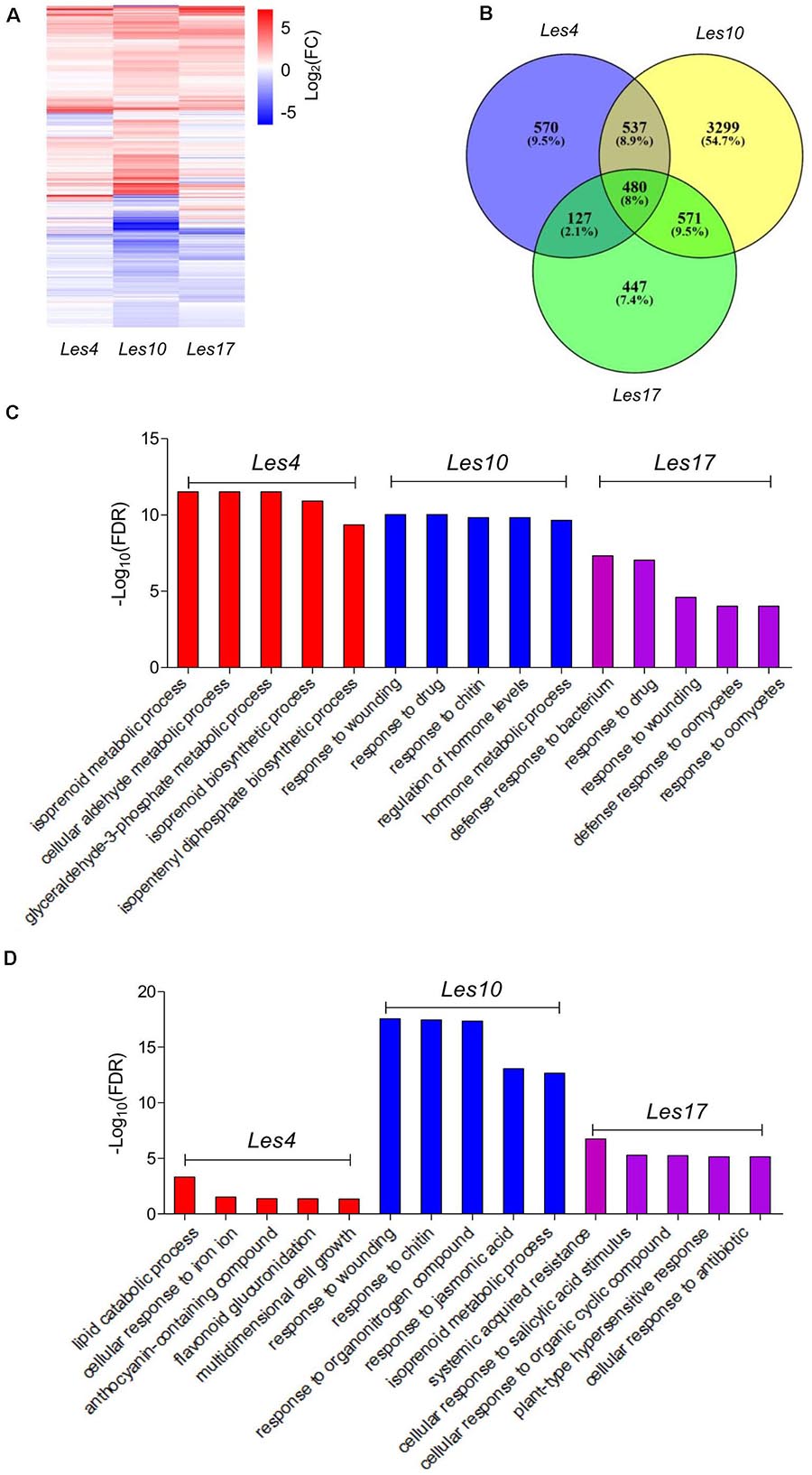
Figure 2. Transcriptomic analysis of Les4, Les10, and Le17. (A) Heatmap showing the differentially expressed genes (DEGs) in Les4, Les10, and Les17. (B) Venn diagram displaying DEGs unique and common in mutant vs. wild type in Les4, Les10, and Les17. (C) The top 5 GO terms of GO analysis of all DEGs in Les4, Les10, and Les17. (D) The top 5 GO terms of GO analysis of specific DEGs in Les4, Les10, and Les17.
A total of 570, 3,299, and 447 DEGs were specifically expressed in Les4, Les10, and Les17 mutants and were defined as specific genes (SGs) (Figure 2B). To look into the specificity of each Les mutant, GO enrichment analysis was also performed for SGs in each mutant. SGs of Les4 were mainly related to “lipid catabolic process,” “cellular response to iron ion,” “anthocyanin-containing compound,” and “flavonoid glucuronidation” (Figure 2D and Supplementary Table 5). SGs of Les10 were mainly related to “response to wounding,” “response to chitin,” “response to organonitrogen compound,” and “response to jasmonic acid” (Figure 2D and Supplementary Table 5). SGs of Les17, however, were mainly related to “systemic acquired resistance,” “cellular response to salicylic acid stimulus,” “cellular response to organic cyclic compound,” and “plant-type hypersensitive response” (Figure 2D and Supplementary Table 5). We searched for all SGs in the Pathogen Receptor Genes (PRGs) database8, a database of plant resistance genes, and found 31, 167, and 58 genes for Les4, Les10, and Les17, respectively (Supplementary Table 6), suggesting that those PRGs were specifically regulated in different Les mutants.
Since the three Les mutants are in different backgrounds, it is less likely that we could identify many common DEGs. However, 480 DEGs were commonly shared by the Les4, Les10, and Les17 mutants and were designated as CGs (Figure 2B). GO enrichment analysis was also performed for CGs. There were 114 BPs and 13 MFs in GO analysis of CGs (Supplementary Table 5). The 114 BP-GO terms were subjected to REVIGO software based on their relationship and then reassigned to 60 terms (Figure 3A). The top GO terms with high significance include “regulation of programmed cell death,” “regulation of immune system process,” “plant-type hypersensitive response,” “respiratory burst involved in defense response,” and “host programmed cell death induced by symbiont.” Interestingly, all these GO terms are related to plant defense responses. Many previously published defense-related genes in maize were upregulated in the Les mutants (Table 1) including the NLR genes, the receptor like kinase genes, the zealexins and kauralexins biosynthetic genes, and the pathogenesis-related (PR) genes. We specifically searched for the 480 CGs in the PRGs database and identified 58 putative PRGs upregulated in all three Les mutants (Supplementary Table 7). In addition, through a comparison of the 480 CG genes to the published transcriptomics data using Plant Regulomics online software9 (Ran et al., 2020), up to 384 out of 475 (∼81%) listed genes are shared with the published genes being induced by pathogen infection (Supplementary Table 8). Those data further indicate that the expression of defense-related genes was generally induced in all Les mutants, and substantial characterization of those genes may lead to interesting discovery dissecting the general nature of the lesion formation and constitutively activated defense responses in those mutants.
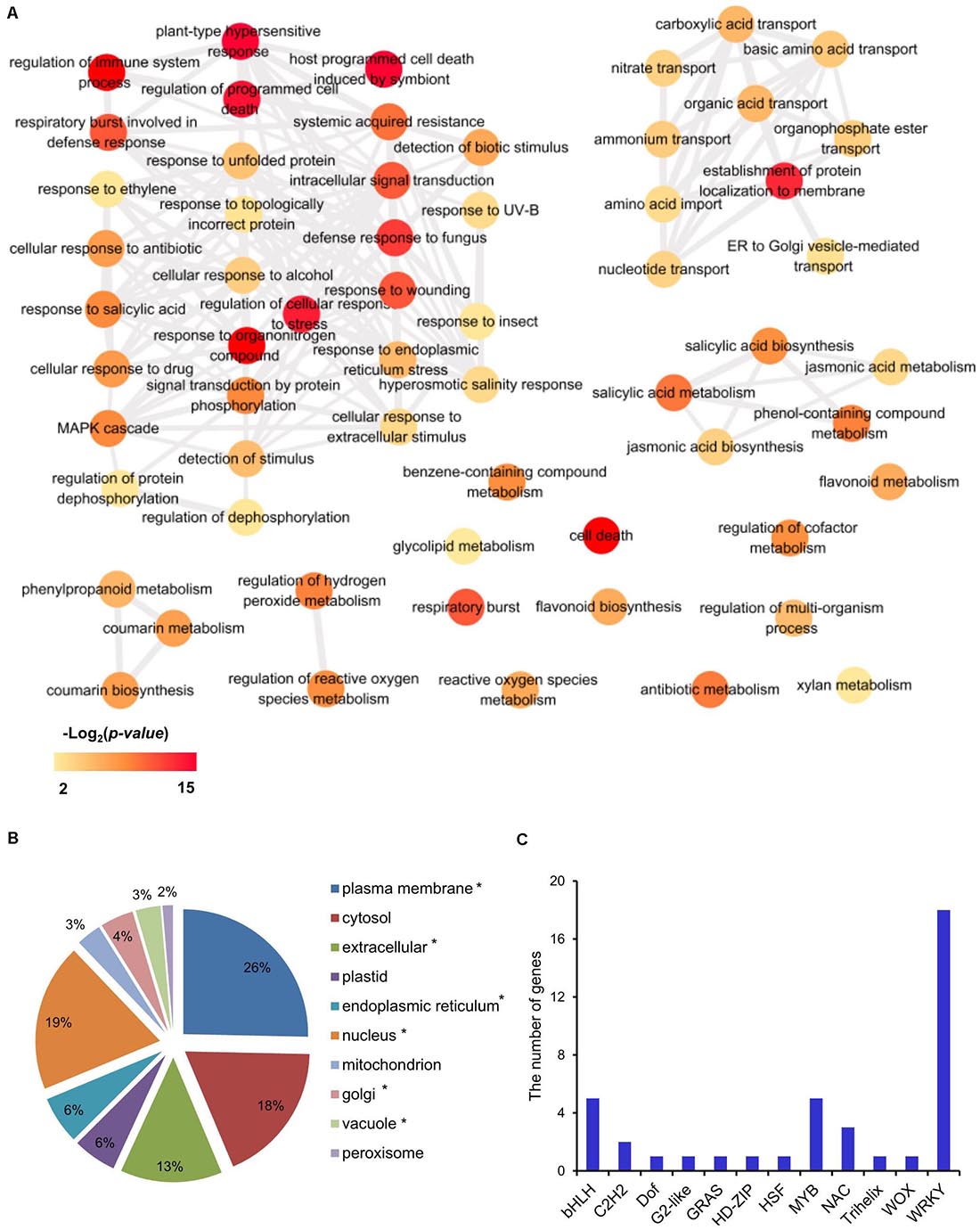
Figure 3. Transcriptomic analysis of common differentially expressed genes (CGs) in mutant vs. wild type in Les4, Les10, and Les17. (A) Biological process of gene ontology (GO) term analysis of CGs. The GO term analysis was conducted in R with packages “clusterProfiler.” The full list of significant GO terms (Supplementary Table 5) assigned into biological process were subjected into REViGO (http://revigo.irb.hr/) to redundant GO terms, and visualized in Cytoscape. The color indicated the significance [−Log2(p-value)] of GO terms. Only significant GO terms were shown. (B) The proportion of cellular location of proteins encoded by common genes (CGs) in Les4, Les10, and Les17. The asterisk behind each item indicates that the p-value of the hypergeometric distribution test was lower than 0.05. (C) The gene numbers of each transcription factor family in CGs.
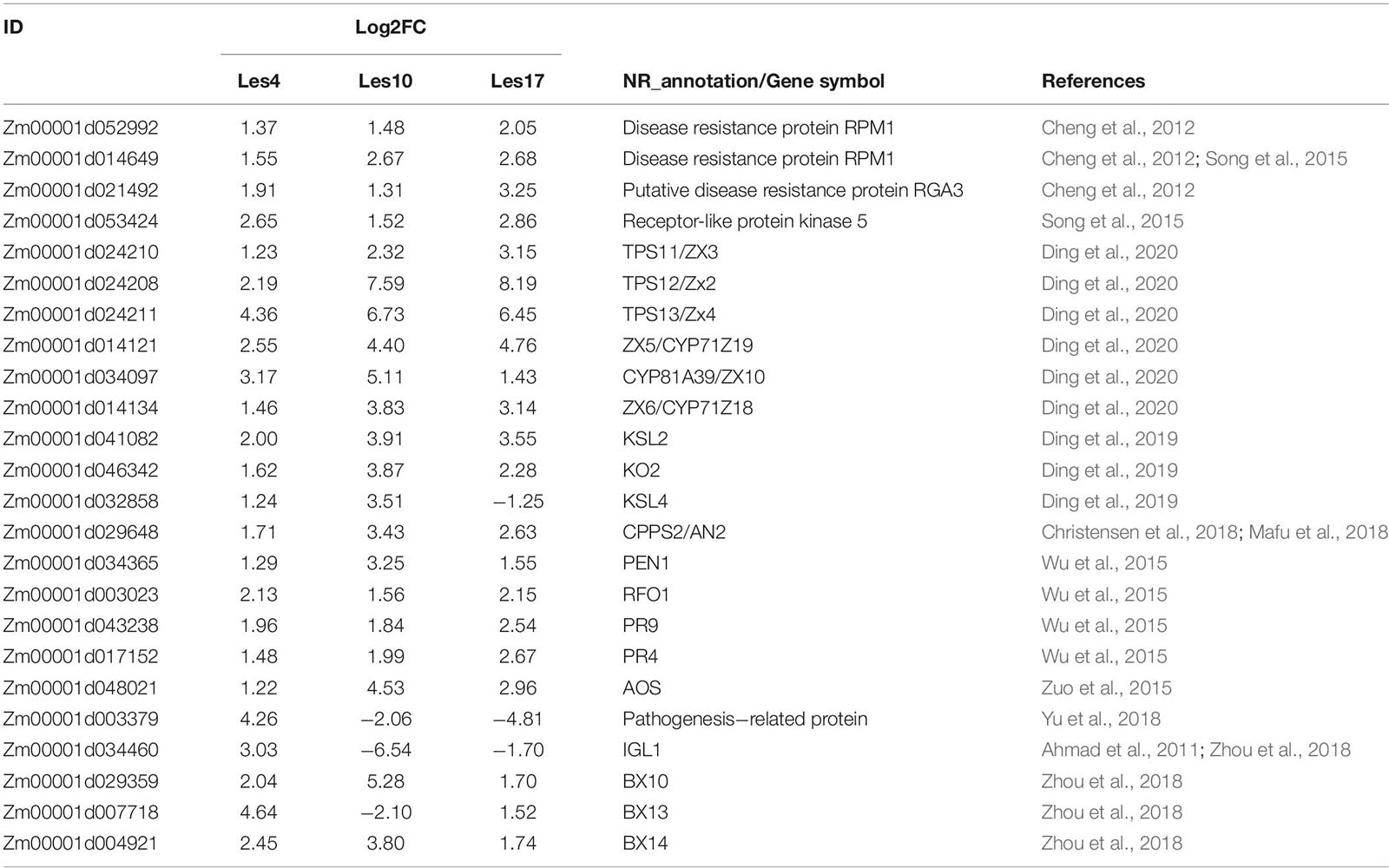
Table 1. Representative common differentially expressed genes involved in plant defense response.
In the GO category of metabolism of CGs, flavonoid, salicylic acid (SA), jasmonic acid (JA), and phenylpropanoid were significantly enriched (Figure 3A). Consistently, KEGG analysis also indicates that CGs were mainly enriched in pathways related to phenylpropanoid and flavonoid biosynthesis (Supplementary Table 9), and thiamine and diterpenoid biosynthesis were also enriched (Supplementary Table 9). In addition, almost all genes involved in regulating reactive oxygen species (ROS) production were upregulated in all Les mutants vs. their relative WT (Table 2), which is consistent with the increased H2O2 accumulation as shown by DAB staining in those Les mutants (Figure 1C).
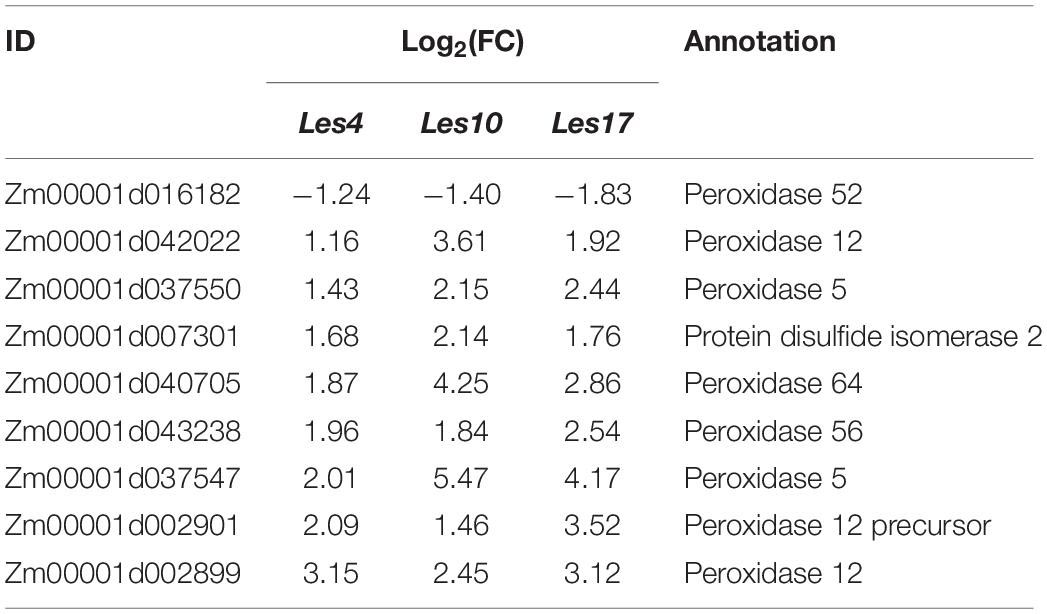
Table 2. Common differentially expressed genes involved in generation of reactive oxygen species.
Based on SUBA4 analysis, most proteins encoded by those CGs were estimated to localize in plasma membrane and nucleus (Figure 3B). Consistent with their dominant nucleus localization, 40 TFs were found, as predicted by PlantTFDB software (Jin et al., 2017). These TFs belong to 12 gene families, among which, WRKY, MYB, and bHLH rank the top three most enriched TF families (Figure 3C and Supplementary Table 10).
We carried out widely targeted metabolomics assay of Les mutants and their relative WT by UPLC-ESI-MS/MS. In this assay, 455 metabolites were collectively identified, including ∼30% flavonoids, 13% phenolic acids, 12% lipid, 11% alkaloids, 11% amino acids and derivatives, 6% organic acids, 5% nucleotides and derivatives, 2% lignans and coumarins, and 10% others (Supplementary Figure 3 and Supplementary Table 11). PCA showed that all biological replicates for each group were clustered closely (Supplementary Figure 2B), suggesting high reproducibility and reliability. We found 97, 184, and 91 differentially accumulated metabolites (DAMs) in Les4, Les10, and Les17, respectively. Of these metabolites, 94, 134, and 66 were upregulated while 3, 50, and 25 were downregulated in Les4, Les10, and Les17, respectively (Supplementary Tables 2, 11). Venn diagram indicates that 42 DAMs were commonly shared by all three Les mutants and were defined as common metabolites (CMs) (Figure 4A and Supplementary Table 12). In those CMs, only 3 DAMs were downregulated while 39 were upregulated (Figure 4B and Supplementary Table 12). Interestingly, the fold change of metabolites in Les10 was larger than in Les4 and Les17. This is consistent with the larger amount of DEGs in Les10 (Figure 2A). Among all CMs, flavonoids, phenolic acids, and organic acids ranked the top 3 most represented metabolites (Figure 4C). While for KEEG pathway enrichment analysis, pathways including phenylpropanoid biosynthesis and flavonoid biosynthesis were significantly enriched (Figure 4D).
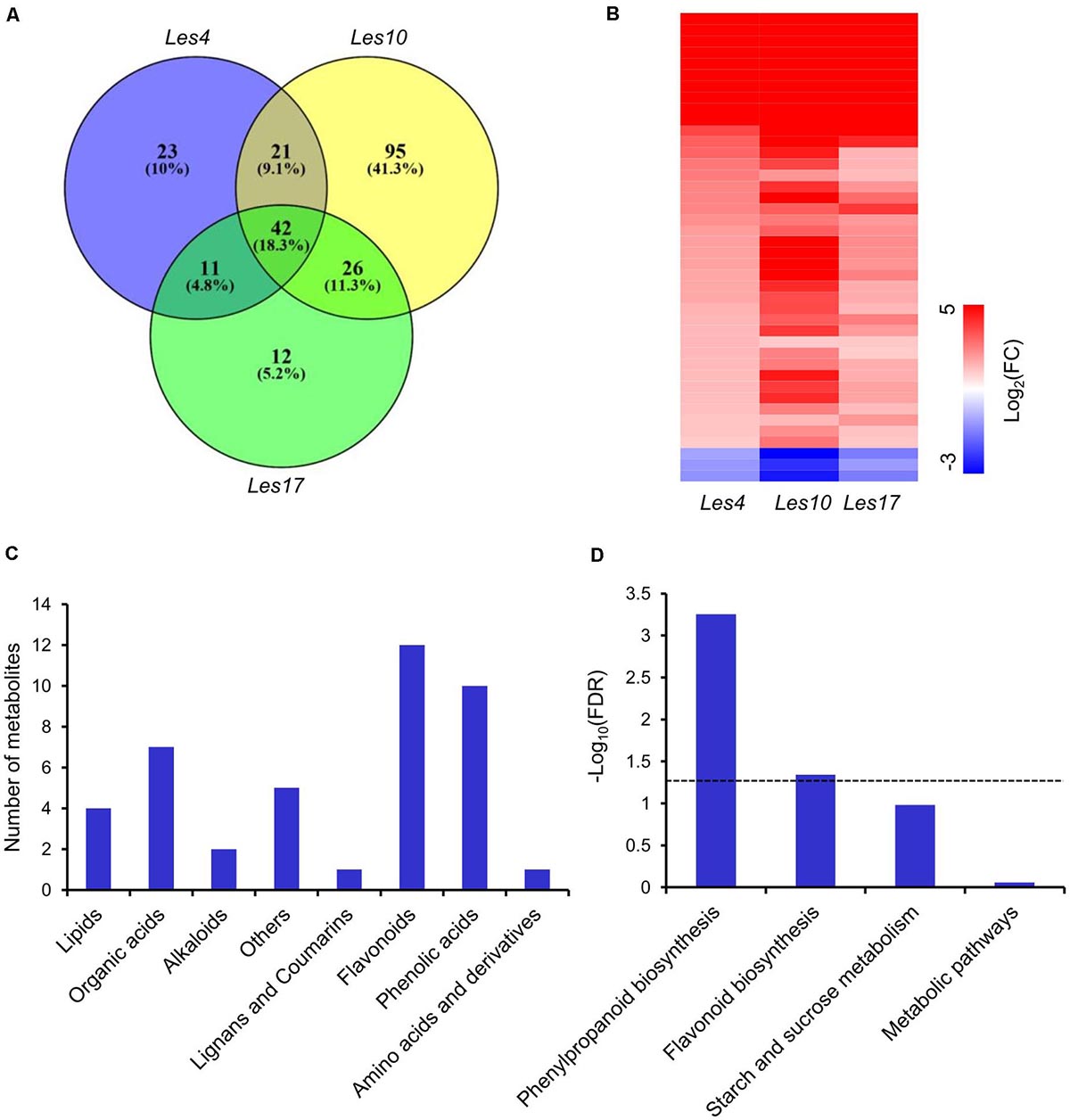
Figure 4. Metabolomics analysis of Les4, Les10, and le17. (A) Venn diagram displaying differentially accumulated metabolites unique and common in mutant vs. wild type in Les4, Les10, and Les17. (B) Heatmap showing the differentially accumulated common metabolites in Les4, Les10, and Les17. (C) The number of common differentially accumulated metabolites (CMs) in different metabolite categories. (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of CMs. The dash line represents a cutoff of p = 0.05.
Based on KEGG and GO enrichment analysis of CGs and CMs, we found that terms of phenylpropanoids and flavonoids were most highly enriched (Figure 4D and Supplementary Tables 5, 9). Therefore, an integrated transcriptomics and metabolomics analysis specifically on phenylpropanoid, lignin, and flavonoid biosynthesis was applied in this study.
Most of the genes encoding the key enzymes in phenylpropanoid and lignin biosynthesis, including PAL, Cinnamate 4-Hydroxylase (C4H), 4-4-Coumarate: Coenzyme A Ligase (4CL), HCT, CAD, Laccase (LAC), etc., displayed increased expression in Les4, Les10, and Les17 mutant compared to their respective WT (Figure 5A and Supplementary Table 13). Most of the phenylpropanoid compounds, e.g., p-coumaric acid, caffeic acid, and coniferyl alcohol, were increased in all Les mutants compared with WT (Figure 5A and Supplementary Table 14). p-coumaric acid, for instance, was increased 2.72, 9.09, and 5.89 times in Les4, Les10, and Les17 mutants, respectively. Besides, we performed acetyl bromide lignin measurement of all Les mutants and their respective WT. The lignin content was 33, 85, and 31% higher in mutant vs. WT in Les4, Les10, and Les17, respectively (Figure 6). Taken together, these data indicate that all three Les mutants have altered phenylpropanoid biosynthetic gene expression as well as higher phenylpropanoid and lignin accumulation.
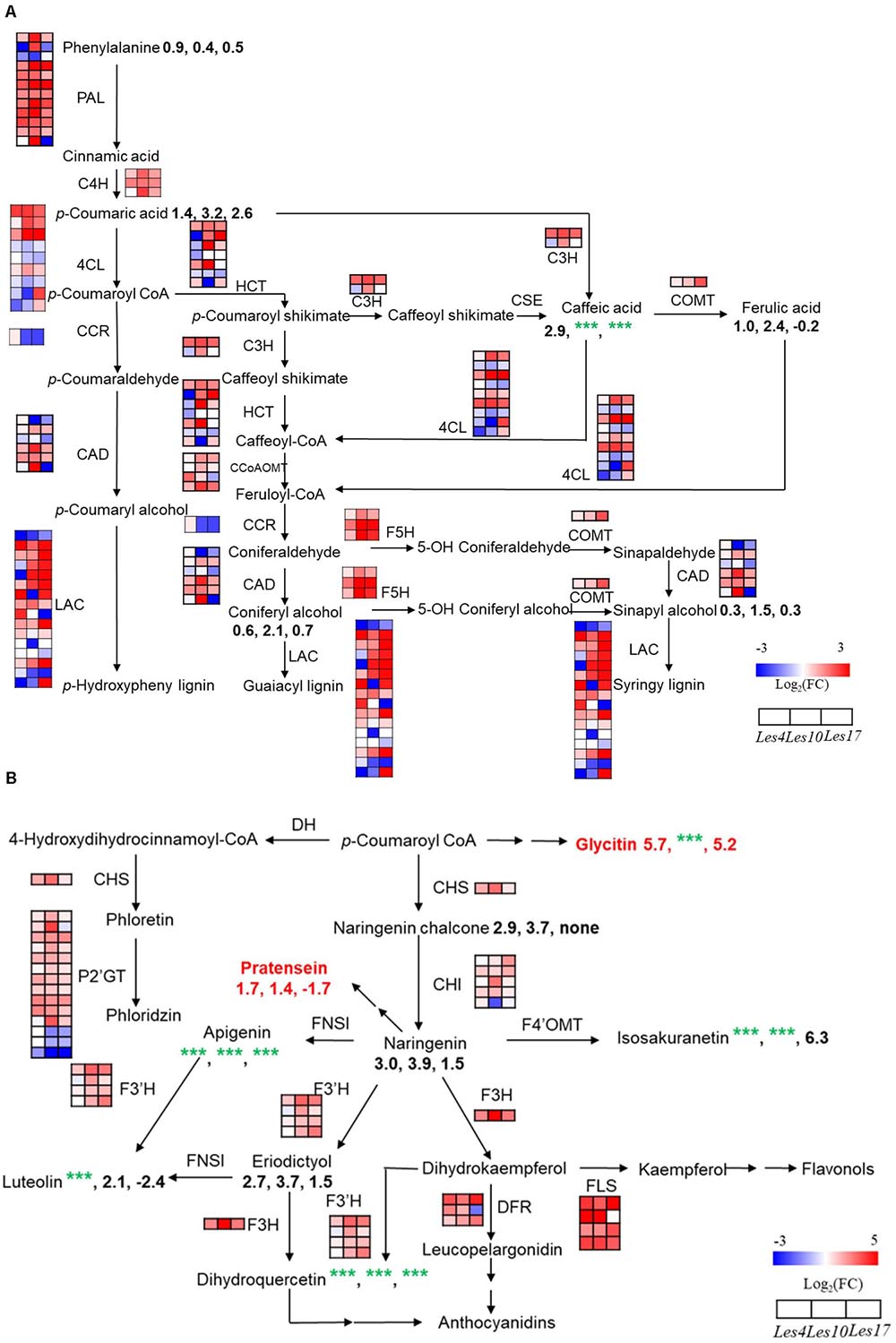
Figure 5. Integrated transcriptomics and metabolomics analysis of lignin and flavonoid biosynthesis. (A) Genes and metabolites involved in lignin biosynthetic pathway in the comparisons of Le4, Les10, and Les17 mutants and their respective wild type (WT). (B) Genes and metabolites involved in flavonoid biosynthetic pathway in the comparisons of Le4, Les10, and Les17 mutants and their respective WT. For (A,B), the different rows of each heatmap represent the different homolog genes for each enzyme, with each column representing different genotypes and different colors indicating different gene expression levels. The number below each metabolite is the log2 (fold change) value of it. Red text highlights the compounds belonging to isoflavone. The detailed information of genes and metabolites involved in lignin and flavonoid biosynthesis are available in Supplementary Tables 13–16, respectively. Double arrows in sequence represent omitted metabolites and genes. nd, not detected. CAD, Cinnamyl Alcohol Dehydrogenase; CCR, Cinnamoyl-Coenzyme A Reductase; C3H, Cinnamate 3-Hydroxylase; C4H, Cinnamate 4-Hydroxylase; CHS, Chalcone Synthase; CHI, Chalcone Isomerase; 4CL, 4-Coumarate:Coenzyme A Ligase; CCoAOMT, Caffeoyl-Coenzyme A O-Methyltransferase; COMT, Caffeic Acid O-Methyltransferase; CSE, Caffeoyl Shikimate Esterase; DFR, Dihydroflavonol 4-Reductase; DH, Dehydrogenase; FLS, Flavonol Synthase; FNS, Flavone Synthase; F3H, Flavanone 3-Hydroxylase; F3’H, Flavanoid 3’-Hydroxylase; F4’OMT, Flavonoid 4’-O-Methyltransferase; F5H, Ferulate 5-Hydroxylase; HCT, Hydroxycinnamoyl Coenzyme A:Shikimate Hydroxycinnamoyl Transferase; LAC, Laccase; PAL, Phenylalanine Ammonia Lyase; P2’GT, UDP-Glucose:Phloretin 2’-O-Glucosyltransferase.
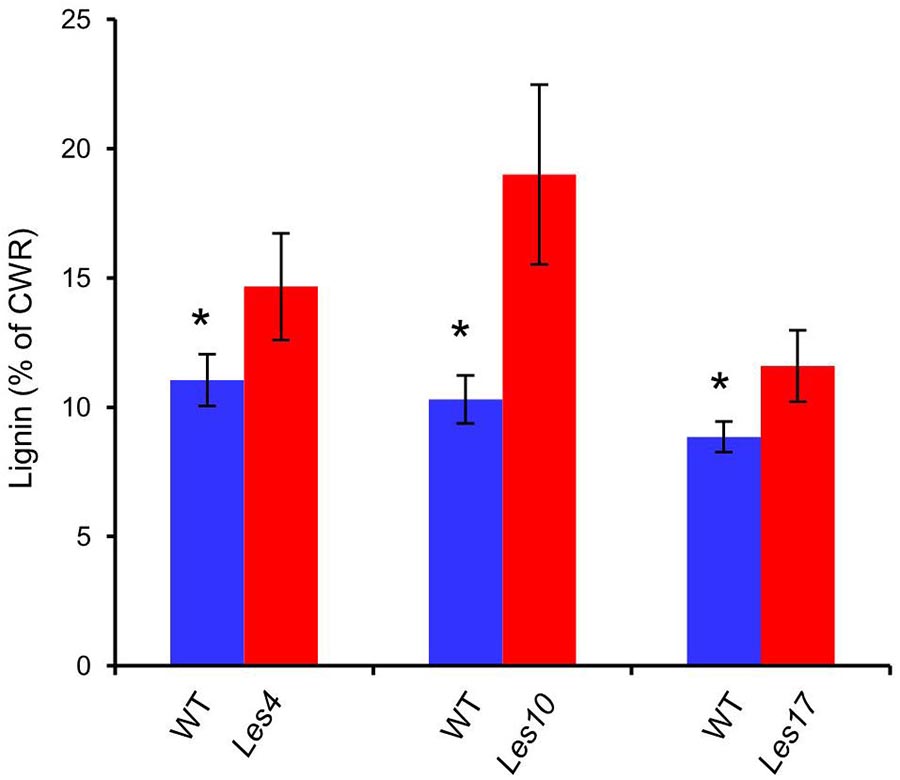
Figure 6. Total lignin content in Les4, Les10, and Les17 mutants and their respective wild type. CWR, cell wall residues. Asterisks indicate significant differences compared with wild type (WT) samples (Student’s t-test; *P < 0.05). Error bars represent standard deviation.
Most flavonoid biosynthetic genes, including Chalcone Synthase (CHS), Chalcone Isomerase (CHI), Flavonol Synthase (FLS), UDP-Glucose:Phloretin 2’-O-Glucosyltransferase (P2’GT), Flavanone 3-Hydroxylase (F3H), Flavonoid 3’-Hydroxylase (F3’H), and Dihydroflavonol 4-Reductase (DFR), were greatly increased in Les mutants (Figure 5B and Supplementary Table 15). The levels of associated metabolites including dihydroquercetin, naringenin, apigenin, eriodictyol, luteolin, etc. were increased dramatically in the Les mutants (Figure 5B and Supplementary Table 16). For example, eriodictyol was increased 6. 30-, 13. 05-, and 2.84-fold in Les4, Les10, and Les17 mutants compared with their respective WT (Supplementary Table 16), and this is consistent with the substantial rises in the F3H expression level. Besides, the isoflavone compounds, glycitin, and pratensein, also accumulated significantly in Les4 and Les10 mutants (Figure 5B and Supplementary Table 11).
We specifically looked into the biosynthesis of terpenoids (zealexins and kauralexins) and benzoxazinoid since both are very important antibiotic compounds in maize. Because zealexins and kauralexins were not detected due to technical issue in this study, we specifically checked the transcriptional changes of zealexins and kauralexins biosynthetic genes. Surprisingly, all of the zealexins biosynthetic genes, Zealexin (ZX) 1-10 (ZX1-ZX10), were upregulated in all Les mutants (Figure 7 and Supplementary Table 17). Similarly, all kauralexins biosynthetic genes including Anther Ear 2 (AN2), Kaurene Synthase-Like (KSL2), Cytochrome P450 (CYP) Z16/18 (CYP71Z16/18), Kauralexin Reductase 2 (KR2), and Kauralexin Oxidase 2 (KO2) were also upregulated in Les mutants (Figure 7 and Supplementary Table 17). These data collectively indicate that biosynthesis of zealexins and kauralexins are generally activated in Les mutants. In addition, according to the metabolomics data, there was a common decrease of DIMBOA-Glucoside (Supplementary Figure 4 and Supplementary Table 11), the dominant benzoxazinoid compound with strong antifungal activity, in all three Les mutants. Interestingly, the upstream benzoxazinoid biosynthetic genes Benzoxazinless (BX) 1–6 (BX1–BX6) were mostly downregulated in almost all three Les mutants (Supplementary Figure 4 and Supplementary Table 18). In contrast, the downstream benzoxazinoid biosynthetic genes BX10, BX11, BX12, and BX14 genes were mostly upregulated in those Les mutants (Supplementary Figure 4 and Supplementary Table 18). These data indicate that while the biosynthesis of DIMBOA-Glucoside was repressed, the processing of DIMBOA-Glucoside into HDMBOA-Glucoside was generally activated in Les mutants.
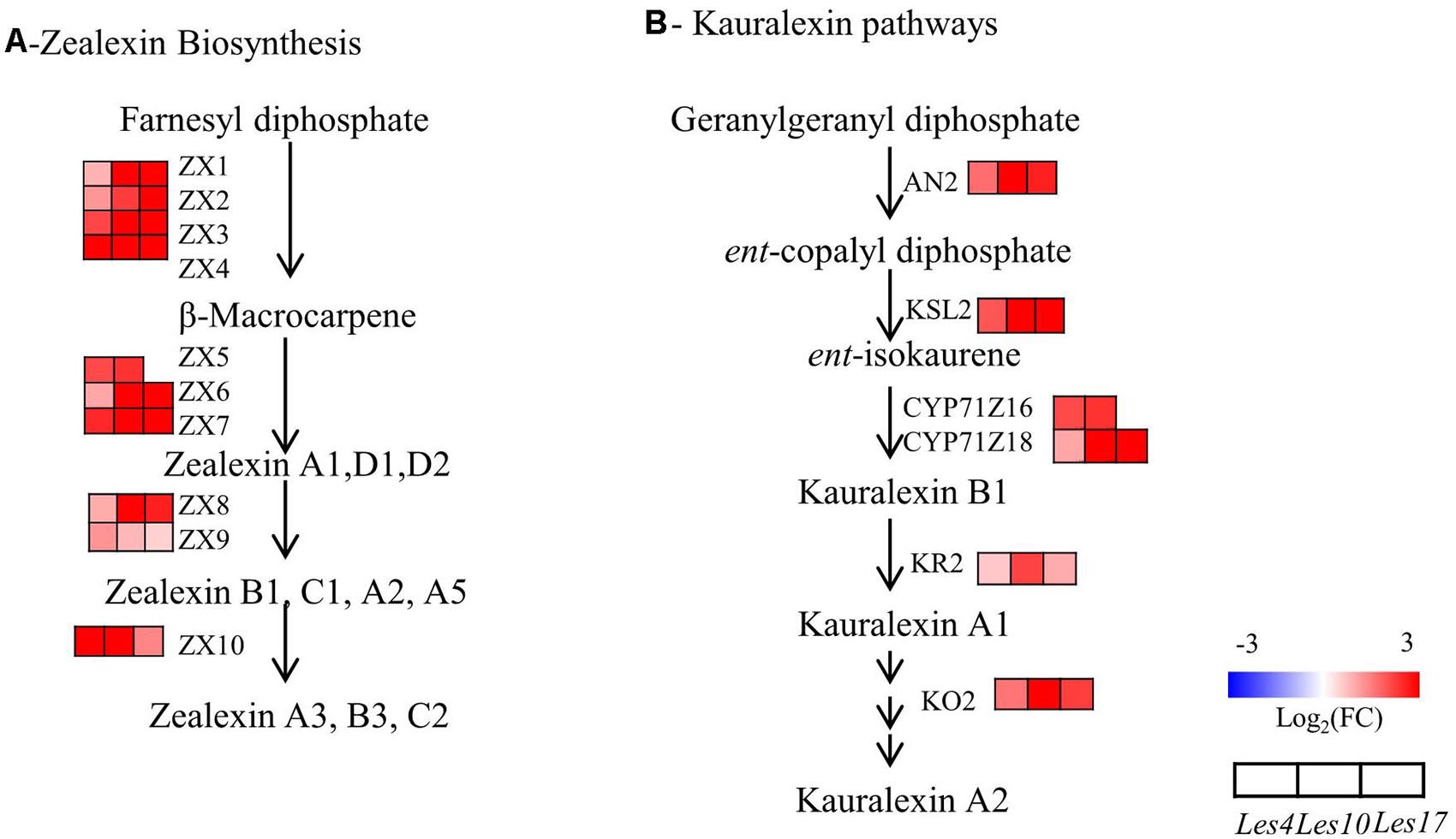
Figure 7. The transcriptomics analysis of zealexins and kauralexins biosynthesis. (A) Transcriptional changes of zealexins biosynthetic genes in Le4, Les10, and Les17 mutants compared to their respective wild type (WT). (B) Transcriptional changes of kauralexins biosynthetic genes in Le4, Les10, and Les17 mutants compared to their respective WT. For (A,B), different rows represent the different homolog genes encoding the related enzyme, with each column representing different genotypes and different colors indicating different gene expression levels. The number below each metabolite is the log2 (fold change) value. The detailed information of genes involved in zealexins and kauralexins biosynthesis is available in Supplementary Table 17. Double arrows in sequence represent omitted metabolites and genes. ZX, Zealexin; TPS, Terpene Synthase; AN2, Anther Ear 2; KSL, Kaurene Synthase-Like, CYP, Cytochrome P450, KO2, Kaurene Oxidase 2; KR2, Kauralexin Reductase 2.
Plant Les/les mutants show spontaneous lesion and enhanced disease resistance that are often accompanied by stunted plant growth, implying the constitutive activation of defense responses in those mutants. To make better use of the maize Les/les mutants, it is crucial to systematically understand the molecular bases of commonly and specifically activated defenses leading to those traits. In this study, high-throughput transcriptomics and metabolomics analysis were utilized to explore the specific and common transcriptional and metabolic changes associated with the Les-related traits including necrotic lesions, stunted plant growth, and enhanced disease resistance.
In this study, we identified 1,714, 4,887, and 1,625 DEGs in Les4, Les10, and Les17, respectively. According to GO enrichment analysis, most of those DEGs were related to defense response and metabolic process (Figure 2C and Supplementary Table 5). To dissect the specificity of each Les mutant and the potential function of each LES gene, we looked into those SGs in each mutant (570, 3,299, and 447 are SGs for Les4, Les10, and Les17, respectively). GO enrichment analysis of SGs indicate that LES4’s function may be more directly related to secondary metabolism regulation since lipid, anthocyanin, and flavonoid metabolic processes were most significantly changed in Les4. LES10 is probably involved in the regulation of defense responses to necrotrophic pathogen since wounding and JA-related processes were most significantly changed. LES17, however, is likely involved in the regulation of SA-mediated systemic acquired resistance and hypersensitive responses since representative genes were captured. Using PRG database, we identified 31, 167, and 58 PRGs for Les4, Les10, and Les17, respectively (Supplementary Table 6), which also support the specific regulation of defense responses in different Les mutants. Further studies on those SGs will provide more clues to the biological function of each LES gene.
We found that the total DEGs and DAMs in Les10 are generally more abundant than Les4 and Les17, likely due to more intense lesion observed in Les10 since its lesion formed at earlier stage. While designing the experiments, two aspects may affect the results in general. Firstly, different timing of sampling will surely affect the intensity of lesion and thus the gene expression change and metabolite accumulation. We sampled the third and fourth above-ear leaves at 5 days after silking, although the leaves of all mutants are still in good vigor, the relatively “matured” state may lead to substantial changes of gene expression and metabolite levels. Secondly, because of the non-uniform nature of the genetic backgrounds of the three mutants, there is lack of common reference for changes in gene expression levels and metabolite levels. Both aspects argue against the joint analysis of the three mutants; however, we did identify a significantly large amount of CGs (480) for all three Les mutants. More interestingly, ∼81% of CGs are shared with the published genes being induced by pathogen infection (Supplementary Table 8), representing robust changes of general defense-related gene expression. Although it is possible that those robust changes of general defense-related gene expression are largely due to general responses accompanying the lesion formation, those changes allow us to digest the shared regulatory network controlling the generally activated defense responses in Les mutants. Based on further in-depth analysis, we found that the transcriptional and metabolic changes are closely associated with each common trait of the Les mutants, which are organized and discussed below.
Necrotic lesion is caused by programmed cell death, a process including cytoplasmic shrinkage, chromatin condensation, mitochondrial swelling, vacuolization, and chloroplast disruption (Coll et al., 2011). Indeed, our transcriptomic analysis indicates that many DEGs in Les mutants are cell death related. Specially, genes involved in ROS accumulation including several peroxidase coding genes are mostly upregulated in all tested Les mutants (Table 2). Consistently, all tested Les mutants have higher H2O2 accumulation and lower chlorophyll content (Figures 1C,D). Previous studies indicate that chlorophyll perturbation can also lead to ROS production and consequently plant cell death. For instance, LES22 encodes a key enzyme in the biosynthetic pathway of chlorophyll, and accumulation of photo-excitable uroporphyrin leads to minute necrotic spots in Les22 (Hu et al., 1998). Similarly, the light-dependent cell death of the les mutant lls1 is caused by failure of chlorophyll breakdown (Gray et al., 1997, 2004). The reduced shoot biomass may be partly due to the lower photosynthesis accompanied by lower chlorophyll content since chlorophyll plays a vital role in photosynthesis (Mu et al., 2016). We found 57 differentially regulated photosynthesis-related genes, including light reaction, Calvin cycle, and carbon concentration, while 3, 42, and 7 of them were downregulated in Les4, Les10, and Les17, respectively (Supplementary Table 19). These data support the tight correlation between lower photosynthesis and compromised plant growth since Les4, Les10, and Les17 show mild, intense, and medium reduction of biomass, respectively. In summary, the upregulation of cell death-related genes and the accumulation of ROS is associated with the necrotic lesion formation while the lower photosynthesis rate likely contributes to the reduced plant growth in Les mutants.
In this study, we found that Les4, Les10, and Les 17 mutants showed constitutively activated defense responses. For instance, Les4 was highly resistant to curvularia leaf spot (Figures 1E,F). Consistently, GO enrichment analysis of CGs indicates that most terms were related to plant defense (Supplementary Table 5). Besides, most CGs are shared with genes differentially expressed after pathogen infection according to the Plant Regulomics database (Supplementary Table 8), and PRGs were enriched in both CGs and SGs (Supplementary Tables 6, 7). Although relatively less defense-related genes have been verified in maize, we did find a few previous reported maize defense genes in CGs, including NLR genes, the receptor-like kinase genes, the zealextins and kauralexins biosynthetic genes, and PR genes (Table 1).
Of course, we should always be cautious about the identified DEGs considering that many genes could be consequences of general metabolic changes due to the lesion formation of Les mutants. We therefore tried to look at the whole regulatory network by checking TFs that likely control downstream defense gene expression and metabolic changes. Among the 480 CGs, there are 40 TFs mainly belonging to WRKY, bHLH, and MYB families (Figure 3C and Supplementary Table 10). WRKY and MYB TFs have previously been found to play broad and pivotal roles in regulating plant disease resistance (Eulgem and Somssich, 2007; Pandey and Somssich, 2009; Liu et al., 2013). In our research, 18 WRKY and 5 MYB TFs were upregulated in all three Les mutants (Supplementary Table 10), implying their contributions to the enhanced resistance. Interestingly, WRKY79 (Zm00001d020137), which was previously shown to positively regulate zealexins and kauralexins biosynthesis (Fu et al., 2018), was upregulated in all Les mutants (Supplementary Table 10), consistent with the upregulation of the terpenoid biosynthetic genes (Figure 7 and Supplementary Table 17). Previous studies also indicated that a series of MYB TFs were involved in the transcriptional regulation of lignin and flavonoid biosynthesis (Zhao and Dixon, 2011; Geng et al., 2020; Wang et al., 2020); some of the MYB TFs identified in this study may be potentially involved in lignin and flavonoid biosynthesis in maize. Taken together, we propose that the upregulation of defense-related genes and secondary metabolite biosynthetic genes potentially mediated by WRKY and MYB TFs is closely associated with the enhanced disease resistance in Les mutants.
Genes involved in secondary metabolite biosynthesis are also highly enriched in CGs by KEGG pathway analysis (Supplementary Table 9). For instance, the large families of phenylpropanoid biosynthetic genes like PAL, HCT, CAD, and Caffeic Acid O-Methyltransferase (COMT) and the flavonoid biosynthetic genes like CHS, DFR, F3’H, and FLS were upregulated in all three Les mutants (Figure 5 and Supplementary Tables 13, 15). Besides, genes encoding laccases, which promote the oxidative coupling of monolignols to form lignin (Zhao et al., 2013), are mostly co-upregulated (Figure 5A and Supplementary Table 13). In addition, almost all genes involved in the biosynthesis of kauralexins and zealexins were upregulated in all three Les mutants (Figure 7 and Supplementary Table 17). Interestingly, for those benzoxazinoid biosynthetic genes, BX1–BX6 are generally downregulated, while BX10, BX11, BX12, and BX14 were generally upregulated (Supplementary Figure 4 and Supplementary Table 18), suggesting the differential regulation of DIMBOA-Glucoside and HDMBOA-Glucoside biosynthesis in Les mutants. It is possible that the upregulation of BX11–BX14 may lead to the accumulation of HDMBOA-Glucoside, which was previously known to dominantly accumulate upon insect herbivory or fungal infestation (Handrick et al., 2016). Although the absence of early BX genes in the CGs was not expected, it represents a truly negative control supporting the relative importance of phenopropanoids and terpenoids in Les-conferred disease resistance. It will be also interesting to determine the mechanism of differential regulation of upstream and downstream BX genes in Les mutants in the future.
Consistent with the transcriptomic data, we detected a large amount of CMs potentially involved in plant defense response (Figure 5). For instance, significantly increased accumulation of phenylpropanoid compounds, e.g., p-coumaric acid, caffeic acid, and coniferyl alcohol, were detected in all Les mutants compared with WT (Figure 5A and Supplementary Table 14), most of those compounds have antifungal activities according to previous reports (Konig et al., 2014; Riaz et al., 2018; Yuan et al., 2019). Additionally, we also detected dramatic increases of the phenylpropanoid-based lignin accumulation in all three les mutants (Figure 6). It has long been proposed that lignin is involved in plant disease resistance because of its enhancement of cell wall rigidity, thus providing barriers against pathogen infection and restricting the infiltration of fungal enzymes and toxins into plant cells (Vance et al., 1980); this concept has been manifested by a recent finding that a lignin-deposited structure functions as a physical barrier similar to the Casparian strip, trapping pathogens and thereby terminating their growth (Lee et al., 2019), while MYB15 is a key TF that controls the defense-induced lignification in Arabidopsis (Chezem et al., 2017; Kim et al., 2020).
As branch products of the phenylpropanoid biosynthesis pathway, many flavonoids act as defensive compounds in response to pathogen (Middleton, 1996; Treutter, 2005, 2006). In this study, accumulation of flavonoids including dihydroquercetin, naringenin, apigenin, eriodictyol, luteolin, etc. has been dramatically increased in Les4, Les10, and Les17 mutants (Figure 5B). Two isoflavone compounds, glycitin and pratensein, which were not reported in maize previously, were also found significantly accumulated in Les4 and Les10 mutant (Figure 5B and Supplementary Table 11). We were unable to directly detect kauralexins and zealexins due to technical limit, although all the biosynthetic genes appeared to be upregulated. Interestingly, the amount of DIMBOA-Glucoside was reduced in all Les mutants (Supplementary Figure 4 and Supplementary Table 11), consistent with the commonly downregulation of BX1–BX6 genes in Les mutants (Supplementary Figure 4 and Supplementary Table 18). Besides, although less represented, some other potential defense-related secondary metabolites like lignans and coumarins were also detected in Les mutants (Supplementary Table 11). Taken together, our metabolomics study suggests that the secondary metabolites, including the phenylpropanoids, lignin, flavonoids, kauralexins, zealexins, etc., are closely associated with the enhanced disease resistance of Les mutants.
In this study, while the identification of SGs in each Les mutant gave clues to the function of each Les gene, the detailed analysis of the overlapped robust changes of defense-related gene expression and metabolite levels in all three Les mutants led to the interesting discovery of a regulatory and metabolic network associated with Les-related traits including spontaneous lesion formation, stunted growth, and enhanced disease resistance. Since both genetic background and timing of sampling those Les mutants could affect the general gene expression and metabolite changes, the regulation is not likely direct and straightforward, instead, the feedback regulation likely plays a pivotal role in amplifying the signaling, resulting in the constitutive activation of defense responses and cell death, and finally exhausting the plants. The above hypothesis is worthy of testing using some better-characterized Les mutant in pure background and sampling of Les mutants at different stages of lesion formation while introducing “days after lesion formation” as a parameter.
Based on our study, a working model is proposed (Supplementary Figure 5). Firstly, as consequences of LES gene mutations, a series of WRKY and MYB TFs were activated in yet unknown mechanisms. Those TFs could then regulate the expression of cell death and defense-related genes including immune receptors, defense signaling components, PR genes, ROS production genes, and secondary metabolite biosynthetic genes, leading to lesion formation and enhanced disease resistance. Secondly, the activation of secondary metabolite biosynthetic gene expression would result in high accumulations of metabolites including phenylpropanoids, lignin, flavonoids, kauralexins, zealexins, etc., and the metabolite change accompanied by lesion formation likely also causes amplified gene expression changes in a feedback manner. Thirdly, the stunted plant growth of Les mutants is probably caused by the reduced expression of photosynthetic genes followed by lower photosynthesis rate in Les mutants, which is also likely related to the cell death and metabolite changes. However, the proposed regulatory network is preliminary and remains to be validated and fixed in the future when the more LES genes are cloned and more direct evidence including the protein–protein (or DNA) interaction and genetic analysis is provided. In addition, further studies on the identified SGs and CGs will shed new light on the function of each LES gene as well as the general regulatory network of defense responses in maize.
Data of RNA-Seq in this study have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (accession no. GSE148220).
MG and XM designed the experiments, interpreted the data, and wrote the article. XM performed most of the experiments and analyzed the data. JL prepared the materials. LX and TF performed the pathogen test. ZD, TJ, and MC worked on the lignin measurement and RNA extraction. All authors contributed to the article and approved the submitted version.
This work was supported by the Startup Grant of Henan Agricultural University (Grant No. 30601732 to MG), the Zhongyuan Thousand Talents Program (Grant No. ZYQR201912168 to MG), the National Natural Science Foundation of China (Grant No. U2004207 to MG), the Fund for Distinguished Young Scholars in Henan (Grant No. 212300410007), and the Henan Province Tackling Key Problems in Science and Technology (Grant No. 192102110018 to LX and Grant No. 212102110039, to JL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Maize Genetics Cooperation Stock Center for kindly providing Les mutant seeds for this study. We thank Dr. Chunsheng Xue for providing the Curvularia lunata (Wakker) Boed strain CX-3.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.638792/full#supplementary-material
Supplementary Figure 1 | The phenotype of typical wild type, putative homozygous, and putative heterozygous plants of Les4, Les10, and Les17.
Supplementary Figure 2 | Principal components analysis of the gene expression profiles (A) and metabolites (B) in mutant vs. wild type of Les4, Les10, and Les17.
Supplementary Figure 3 | Distribution of metabolites measured by ultra-high performance liquid chromatography-electrospray ionization-tandem mass spectrometry (UPLC-ESI-MS/MS).
Supplementary Figure 4 | Integrated transcriptomics and metabolomics analysis of benzoxazinoids biosynthesis. The changes of gene expression and metabolite accumulation in benzoxazinoids biosynthetic pathway in Le4, Les10, and Les17 mutants compared to their respective wild type (WT). The different rows represent the different genes for each enzyme, with each column representing different genotypes and different colors indicating different gene expression levels. The number below each metabolite is the log2 (fold change) value. The detailed information of genes involved in benzoxazinoids biosynthesis is available in Supplementary Tables 18. BX1-14, Benzoxazinless 1-14; IGPS, Indole-3-Glycerolphosphate Synthases; IGL1, Indole-3-Glycerolphosphate Lyase 1; TSA, Tryptophan Synthase Alpha-Subunit, TSB, Tryptophan Synthase Beta-Subunit.
Supplementary Figure 5 | The proposed model for the regulatory network associated with Les-conferred defense response. NLRs, Nucleotide Binding Leucine-Rich Repeat Proteins; PRs, Pathogenesis-Related Proteins; PRRs, Pattern Recognition Receptors; ROS, reactive oxygen species.
Supplementary Table 1 | Primers used in the PCR and qRT-PCR.
Supplementary Table 2 | Number of differentially expressed genes and metabolites in leaves of different maize mutant vs. wild type of Les4, Les10, and Les17.
Supplementary Table 3 | Significantly differentially expressed genes in the comparisons of mutant vs. wild type of Les4, Les10, and Les17.
Supplementary Table 4 | Fold change of gene expression profiles detected by qRT-PCR.
Supplementary Table 5 | Gene ontology term analysis of differentially expressed genes in Les4, Les 10, and Les17.
Supplementary Table 6 | Predicted Pathogen Receptor Genes (PRGs) in specific differentially expressed genes of Les4, Les10, and Les17.
Supplementary Table 7 | Predicted Pathogen Receptor Genes (PRGs) in common differentially expressed genes of Les4, Les10, and Les17.
Supplementary Table 8 | Transcriptomic comparison enrichment analysis of common differentially expressed genes.
Supplementary Table 9 | Kyoto Encyclopedia of Genes and Genomes analysis of common differentially expressed genes.
Supplementary Table 10 | Transcription factors in the common differentially expressed genes.
Supplementary Table 11 | The detected metabolites in Les4, Les10, and Les17 compared to their respective wild type by LC-MS.
Supplementary Table 12 | Common differentially accumulated metabolites in the comparisons of mutant vs. WT in Les4, Les10, and Les17.
Supplementary Table 13 | Fold change of gene expressions involved in phenylpropanoid and lignin synthesis.
Supplementary Table 14 | Fold change of accumulated metabolites involved in phenylpropanoid and lignin synthesis.
Supplementary Table 15 | Fold change of gene expressions involved in flavonoid biosynthetic genes.
Supplementary Table 16 | Fold change of accumulated metabolites involved in flavonoid synthesis.
Supplementary Table 17 | Fold change of expressions level for zealexins and kauralexins biosynthetic genes in Les mutants compared to their respective wild type.
Supplementary Table 18 | Fold change of expression level for benzoxazinoid biosynthetic genes in Les mutants compared to their respective wild type.
Supplementary Table 19 | Differentially expressed genes involved in photosynthesis in mutant vs. wildtype of Les4, Les10, and Les17.
Ahmad, S., Veyrat, N., Gordon-Weeks, R., Zhang, Y. H., Martin, J., Smart, L., et al. (2011). Benzoxazinoid metabolites regulate innate immunity against aphids and fungi in maize. Plant Physiol. 157, 317–327. doi: 10.1104/pp.111.180224
Bruggeman, Q., Raynaud, C. C., Benhamed, M., and Delarue, M. (2015). To die or not to die? Lessons from lesion mimic mutants. Front. Plant Sci. 6:24.
Buscaill, P., and Rivas, S. (2014). Transcriptional control of plant defence responses. Curr. Opin. Plant Biol. 20, 35–46. doi: 10.1016/j.pbi.2014.04.004
Castro-Moretti, F. R., Gentzel, I. N., Mackey, D., and Alonso, A. P. (2020). Metabolomics as an emerging tool for the study of plant–pathogen interactions. Metabolites 10:52. doi: 10.3390/metabo10020052
Chen, W., Gao, Y., Xie, W., Gong, L., Lu, K., Wang, W., et al. (2014). Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46, 714–721. doi: 10.1038/ng.3007
Chen, W., Gong, L., Guo, Z., Wang, W., Zhang, H., Liu, X., et al. (2013). A novel integrated method for Large-Scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 6, 1769–1780. doi: 10.1093/mp/sst080
Cheng, Y. T., Li, Y., Huang, S., Huang, Y., Dong, X., Zhang, Y., et al. (2011). Stability of plant immune-receptor resistance proteins is controlled by SKP1-CULLIN1-F-box (SCF)-mediated protein degradation. Proc. Natl. Acad. Sci. U.S.A. 108, 14694–14699. doi: 10.1073/pnas.1105685108
Cheng, Y., Li, X., Jiang, H., Ma, W., Miao, W., Yamada, T., et al. (2012). Systematic analysis and comparison of nucleotide-binding site disease resistance genes in maize. Febs. J. 279, 2431–2443. doi: 10.1111/j.1742-4658.2012.08621.x
Chezem, W. R., Memon, A., Li, F. S., Weng, J. K., and Clay, N. K. (2017). SG2-Type R2R3-MYB transcription factor MYB15 controls defense-induced lignification and basal immunity in Arabidopsis. Plant cell 29, 1907–1926. doi: 10.1105/tpc.16.00954
Chintamanani, S., Hulbert, S. H., Johal, G. S., and Balintkurti, P. J. (2010). Identification of a maize locus that modulates the hypersensitive defense response, using mutant-assisted gene identification and characterization. Genetics 184, 813–825. doi: 10.1534/genetics.109.111880
Christensen, S. A., Sims, J., Vaughan, M. M., Hunter, C., Block, A., Willett, D., et al. (2018). Commercial hybrids and mutant genotypes reveal complex protective roles for inducible terpenoid defenses in maize. J. Exp. Bot. 69, 1693–1705. doi: 10.1093/jxb/erx495
Coll, N. S., Epple, P., and Dangl, J. L. (2011). Programmed cell death in the plant immune system. Cell Death Differ. 18, 1247–1256. doi: 10.1038/cdd.2011.37
Ding, Y., Murphy, K. M., Poretsky, E., Mafu, S., Yang, B., Char, S. N., et al. (2019). Multiple genes recruited from hormone pathways partition maize diterpenoid defences. Nat. Plants 5, 1043–1056. doi: 10.1038/s41477-019-0509-6
Ding, Y., Weckwerth, P. R., Poretsky, E., Murphy, K. M., Sims, J., Saldivar, E., et al. (2020). Genetic elucidation of interconnected antibiotic pathways mediating maize innate immunity. Nat. Plants 6, 1375–1388. doi: 10.1038/s41477-020-00787-9
Dobon, A., Bunting, D. C. E., Cabreraquio, L. E., Uauy, C., and Saunders, D. G. O. (2016). The host-pathogen interaction between wheat and yellow rust induces temporally coordinated waves of gene expression. BMC Genomics 17:380.
Dong, J., Ma, G., Sui, L., Wei, M., Satheesh, V., Zhang, R., et al. (2019). Inositol pyrophosphate InsP8 acts as an intracellular phosphate signal in arabidopsis. Mol. Plant 12, 1463–1473. doi: 10.1016/j.molp.2019.08.002
Eulgem, T., and Somssich, I. E. (2007). Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 10, 366–371. doi: 10.1016/j.pbi.2007.04.020
Ferreyra, M. L. F., Rius, S. P., and Casati, P. (2012). Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3:222.
Fu, J., Liu, Q., Wang, C., Liang, J., Liu, L., and Wang, Q. (2018). ZmWRKY79 positively regulates maize phytoalexin biosynthetic gene expression and is involved in stress response. J. Exp. Bot. 69, 497–510. doi: 10.1093/jxb/erx436
Geng, P., Zhang, S., Liu, J., Zhao, C., Wu, J., Cao, Y., et al. (2020). MYB20, MYB42, MYB43, and MYB85 regulate phenylalanine and lignin biosynthesis during secondary cell wall formation. Plant Physiol. 182, 1272–1283. doi: 10.1104/pp.19.01070
Gou, M., and Hua, J. (2012). Complex regulation of an R gene SNC1 revealed by autoimmune mutants. Plant Signal Behav. 7, 213–216.
Gou, M., Ran, X., Martin, D. W., and Liu, C. J. (2018). The scaffold proteins of lignin biosynthetic cytochrome P450 enzymes. Nat. Plants 4, 299–310. doi: 10.1038/s41477-018-0142-9
Gou, M., Shi, Z., Zhu, Y., Bao, Z., Wang, G., and Hua, J. (2012). The F−box protein CPR1/CPR30 negatively regulates R protein SNC1 accumulation. Plant J. 69, 411–420. doi: 10.1111/j.1365-313x.2011.04799.x
Gou, M., Su, N., Zheng, J., Huai, J., Wu, G., and Zhao, J. (2009). An F-box gene, CPR30, functions as a negative regulator of the defense response in Arabidopsis. Plant J. 60, 757–770. doi: 10.1111/j.1365-313x.2009.03995.x
Gou, M., Yang, X., Zhao, Y., Ran, X., Song, Y., and Liu, C. J. (2019). Cytochrome b5 is an obligate electron shuttle protein for syringyl lignin biosynthesis in Arabidopsis. Plant Cell 31, 1344–1366. doi: 10.1105/tpc.18.00778
Gray, J., Close, P. S., Briggs, S. P., and Johal, G. S. (1997). A novel suppressor of cell death in plants encoded by the lls1 gene of maize. Cell 89, 25–31. doi: 10.1016/s0092-8674(00)80179-8
Gray, J., Wardzala, E., Yang, M., Reinbothe, S., Haller, S., and Pauli, F. (2004). A small family of LLS1-related non-heme oxygenases in plants with an origin amongst oxygenic photosynthesizers. Plant Mol. Biol 54, 39–54. doi: 10.1023/b:plan.0000028766.61559.4c
Grayer, R. J., and Harborne, J. B. (1994). A survey of antifungal compounds from higher plants, 1982–1993. Phytochemistry 37, 19–42. doi: 10.1016/0031-9422(94)85005-4
Gu, R., Duan, F., An, X., Zhang, F., Von Wiren, N., and Yuan, L. (2013). Characterization of AMT-mediated high-affinity ammonium uptake in roots of maize (Zea mays L.). Plant Cell Physiol. 54, 1515–1524. doi: 10.1093/pcp/pct099
Handrick, V., Robert, C. A., Ahern, K. R., Zhou, S., Machado, R. A., Maag, D., et al. (2016). Biosynthesis of 8-O-methylated benzoxazinoid defense compounds in maize. Plant Cell 28, 1682–1700.
He, J., Zhang, C., Dai, H., Liu, H., Zhang, X., Yang, J., et al. (2019). A LysM receptor heteromer mediates perception of arbuscular mycorrhizal symbiotic signal in rice. Mol. Plant 12, 1561–1576. doi: 10.1016/j.molp.2019.10.015
Hoisington, D. A., Neuffer, M. G., and Walbot, V. (1982). Disease lesion mimics in maize: I. Effect of genetic background, temperature, developmental age, and wounding on necrotic spot formation with Les1. Dev. Biol. 93, 381–388. doi: 10.1016/0012-1606(82)90125-7
Hooper, C. M., Castleden, I., Tanz, S. K., Aryamanesh, N., and Millar, A. H. (2017). SUBA4: The interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res. 45, D1064–D1074.
Hu, G., Richter, T., Hulbert, S. H., and Pryor, T. (1996). Disease lesion mimicry caused by mutations in the rust resistance gene rp1. Plant Cell 8, 1367–1376. doi: 10.2307/3870307
Hu, G., Yalpani, N., Briggs, S. P., and Johal, G. S. (1998). A porphyrin pathway impairment is responsible for the phenotype of a dominant disease lesion mimic mutant of maize. Plant Cell 10, 1095–1105. doi: 10.2307/3870714
Huang, X., Liu, L., Zhai, Y., Liu, T., and Chen, J. (2009). Proteomic comparison of four maize inbred lines with different levels of resistance to Curvularia lunata (Wakker) Boed infection. Prog. Nat. Sci. 19, 353–358. doi: 10.1016/j.pnsc.2008.04.020
Huffaker, A., Kaplan, F., Vaughan, M. M., Dafoe, N. J., Ni, X., Rocca, J. R., et al. (2011). Novel acidic sesquiterpenoids constitute a dominant class of pathogen-induced phytoalexins in maize. Plant Physiol. 156, 2082–2097. doi: 10.1104/pp.111.179457
Jin, J., Tian, F., Yang, D., Meng, Y., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045.
Johal, G. S., Hulbert, S. H., and Briggs, S. P. (1995). Disease lesion mimics of maize: A model for cell death in plants. BioEssays 17, 685–692. doi: 10.1002/bies.950170805
Kaling, M., Schmidt, A., Moritz, F., Rosenkranz, M., Witting, M., Kasper, K., et al. (2018). Mycorrhiza-triggered transcriptomic and metabolomic networks impinge on herbivore fitness. Plant Physiol. 176, 2639–2656. doi: 10.1104/pp.17.01810
Kawahara, Y., Oono, Y., Kanamori, H., Matsumoto, T., Itoh, T., Minami, E., et al. (2012). Simultaneous RNA-Seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One 7:e49423. doi: 10.1371/journal.pone.0049423
Kebede, A. Z., Johnston, A., Schneiderman, D., Bosnich, W., and Harris, L. J. (2018). Transcriptome profiling of two maize inbreds with distinct responses to Gibberella ear rot disease to identify candidate resistance genes. BMC Genomics 19:131.
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, S. H., Lam, P. Y., Lee, M. H., Jeon, H. S., Tobimatsu, Y., and Park, O. K. (2020). The Arabidopsis R2R3 MYB transcription factor MYB15 is a key regulator of lignin Biosynthesis in effector-triggered immunity. Front. Plant Sci. 11:583153.
Konig, S., Feussner, K., Kaever, A., Landesfeind, M., Thurow, C., Karlovsky, P., et al. (2014). Soluble phenylpropanoids are involved in the defense response of Arabidopsis against Verticillium longisporum. New Phytol. 202, 823–837. doi: 10.1111/nph.12709
Lee, M., Jeon, H. S., Kim, S. H., Chung, J. H., Roppolo, D., Lee, H. J., et al. (2019). Lignin-based barrier restricts pathogens to the infection site and confers resistance in plants. EMBO J. 38:e101948.
Li, N., Lin, B., Wang, H., Li, X., Yang, F., Ding, X., et al. (2019). Natural variation in ZmFBL41 confers banded leaf and sheath blight resistance in maize. Nat. Genet. 51, 1540–1548. doi: 10.1038/s41588-019-0503-y
Li, P., Ponnala, L., Gandotra, N., Wang, L., Si, Y., Tausta, S. L., et al. (2010). The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 42, 1060–1067. doi: 10.1038/ng.703
Li, T., and Bai, G. (2009). Lesion mimic associates with adult plant resistance to leaf rust infection in wheat. Theor. Appl. Genet. 119, 13–21. doi: 10.1007/s00122-009-1012-7
Liu, Q., Ning, Y., Zhang, Y., Yu, N., Zhao, C., Zhan, X., et al. (2017). OsCUL3a negatively regulates cell death and immunity by degrading OsNPR1 in rice. Plant Cell 29, 345–359. doi: 10.1105/tpc.16.00650
Liu, X., Yang, L., Zhou, X., Zhou, M., Lu, Y., Ma, L., et al. (2013). Transgenic wheat expressing Thinopyrum intermedium MYB transcription factor TiMYB2R-1 shows enhanced resistance to the take-all disease. J. Exp. Bot. 64, 2243–2253. doi: 10.1093/jxb/ert084
Lopezibanez, J., Pazos, F., and Chagoyen, M. (2016). MBROLE 2.0-functional enrichment of chemical compounds. Nucleic Acids Res. 44, W201–W204.
Lorrain, S., Vailleau, F., Balague, C., and Roby, D. (2003). Lesion mimic mutants: Keys for deciphering cell death and defense pathways in plants? Trends Plant Sci. 8, 263–271. doi: 10.1016/s1360-1385(03)00108-0
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Mafu, S., Ding, Y. Z., Murphy, K. M., Yaacoobi, O., Addison, J. B., Wang, Q., et al. (2018). Discovery, biosynthesis and stress-related accumulation of dolabradiene-derived defenses in maize. Plant Physiol. 176, 2677–2690. doi: 10.1104/pp.17.01351
Matin, M. N., Pandeya, D., Baek, K., Lee, D., Lee, J., Kang, H., et al. (2010). Phenotypic and genotypic analysis of rice lesion mimic mutants. Plant Pathol. J. 26, 159–169. doi: 10.5423/ppj.2010.26.2.159
Mcloughlin, F., Augustine, R. C., Marshall, R. S., Li, F., Kirkpatrick, L. D., Otegui, M. S., et al. (2018). Maize multi-omics reveal roles for autophagic recycling in proteome remodelling and lipid turnover. Nat. Plants 4, 1056–1070. doi: 10.1038/s41477-018-0299-2
Meyer, J., Berger, D. K., Christensen, S. A., and Murray, S. L. (2017). RNA-Seq analysis of resistant and susceptible sub-tropical maize lines reveals a role for kauralexins in resistance to grey leaf spot disease, caused by Cercospora zeina. BMC Plant Biol. 17:197.
Middleton, E. (1996). Biological properties of plant flavonoids: an overview. Pharm Biol. 34, 344–348. doi: 10.1076/phbi.34.5.344.13245
Mierziak, J., Wojtasik, W., Kostyn, K., Czuj, T., Szopa, J., and Kulma, A. (2014). Crossbreeding of transgenic flax plants overproducing flavonoids and glucosyltransferase results in progeny with improved antifungal and antioxidative properties. Mol. Breeding 34, 1917–1932. doi: 10.1007/s11032-014-0149-5
Moerschbacher, B. M., Noll, U., Gorrichon, L., and Reisener, H. (1990). Specific inhibition of lignification breaks hypersensitive resistance of wheat to stem rust. Plant Physiol. 93, 465–470. doi: 10.1104/pp.93.2.465
Mohr, P. G., and Cahill, D. M. (2007). Suppression by ABA of salicylic acid and lignin accumulation and the expression of multiple genes, in Arabidopsis infected with Pseudomonas syringae pv. Tomato. Funct. Integr. Genomic 7, 181–191. doi: 10.1007/s10142-006-0041-4
Mu, X., Chen, Q., Chen, F., Yuan, L., and Mi, G. (2016). Within-Leaf nitrogen allocation in adaptation to low nitrogen supply in maize during Grain-Filling stage. Front. Plant Sci. 7:699.
Naoumkina, M., Zhao, Q., Gallegogiraldo, L., Dai, X., Zhao, P. X., and Dixon, R. A. (2010). Genome-wide analysis of phenylpropanoid defence pathways. Mol. Plant Pathol. 11, 829–846.
Neuffer, M. G., and Calvert, O. H. (1975). Dominant disease lesion mimics in maize. J. Hered 66, 265–270. doi: 10.1093/oxfordjournals.jhered.a108627
Niemeyer, H. M. (2009). Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: key defense chemicals of cereals. J. Agric. Food Chem. 57, 1677–1696. doi: 10.1021/jf8034034
Pandey, S. P., and Somssich, I. E. (2009). The role of WRKY transcription factors in plant immunity. Plant Physiol. 150, 1648–1655. doi: 10.1104/pp.109.138990
Pertea, M., Pertea, G., Antonescu, C., Chang, T. C., Mendell, J. T., Salzberg, S. L., et al. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Piasecka, A., Jedrzejczakrey, N., and Bednarek, P. (2015). Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 206, 948–964. doi: 10.1111/nph.13325
Ran, X., Zhao, F., Wang, Y., Liu, J., Zhuang, Y., Ye, L., et al. (2020). Plant regulomics: a data-driven interface for retrieving upstream regulators from plant multi-omics data. Plant J. 101, 237–248. doi: 10.1111/tpj.14526
Riaz, U., Kharal, M. A., Murtaza, G., Zaman, Q. U., Javaid, S., Malik, H. A., et al. (2018). Prospective roles and mechanisms of caffeic acid in counter plant stress: a mini review. Pak. J. Agr. Sci. 32, 8–19. doi: 10.17582/journal.pjar/2019/32.1.8.19
Rostoks, N., Schmierer, D., Kudrna, D. A., and Kleinhofs, A. (2003). Barley putative hypersensitive induced reaction genes: genetic mapping, sequence analyses and differential expression in disease lesion mimic mutants. Theor. Appl. Genet. 107, 1094–1101. doi: 10.1007/s00122-003-1351-8
Schmelz, E. A., Kaplan, F., Huffaker, A., Dafoe, N. J., Vaughan, M. M., Ni, X., et al. (2011). Identity, regulation, and activity of inducible diterpenoid phytoalexins in maize. Proc. Natl. Acad. Sci. U.S.A. 108, 5455–5460. doi: 10.1073/pnas.1014714108
Sekhon, R. S., Kuldau, G. A., Mansfield, M. A., and Chopra, S. (2006). Characterization of Fusarium-induced expression of flavonoids and PR genes in maize. Physiol. Mol. Plant P. 69, 109–117. doi: 10.1016/j.pmpp.2007.02.004
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shi, T., Zhu, A., Jia, J., Hu, X., Chen, J., Liu, W., et al. (2020). Metabolomic analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 103, 279–292. doi: 10.1111/tpj.14727
Simmons, C. R., Hantke, S., Grant, S., Johal, G. S., and Briggs, S. P. (1998). The maize lethal leaf spot 1 mutant has elevated resistance to fungal infection at the leaf epidermis. Mol. Plant Microbe In. 11, 1110–1118. doi: 10.1094/mpmi.1998.11.11.1110
Smith, S. M., Steinau, M., Trick, H. N., and Hulbert, S. H. (2010). Recombinant Rp1 genes confer necrotic or nonspecific resistance phenotypes. Mol. Genet. Genomics 283, 591–602. doi: 10.1007/s00438-010-0536-5
Song, W., Wang, B., Li, X., Wei, J., Chen, L., Zhang, D., et al. (2015). Identification of immune related LRR-containing genes in maize (Zea mays L.) by genome-wide sequence analysis. Int. J. Genomics 2015, 231358.
Southerton, S. G., and Deverall, B. J. (1990). Histochemical and chemical evidence for lignin accumulation during the expression of resistance to leaf rust fungi in wheat. Physiol. Mol. Plant P. 36, 483–494. doi: 10.1016/0885-5765(90)90021-o
Supek, F., Bosnjak, M., Skunca, N., and Smuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Tamiru, M., Takagi, H., Abe, A., Yokota, T., Kanzaki, H., Okamoto, H., et al. (2016). A chloroplast-localized protein LESION and LAMINA BENDING affects defence and growth responses in rice. New Phytol. 210, 1282–1297. doi: 10.1111/nph.13864
Tang, C., Wang, X., Duan, X., Wang, X., Huang, L., Huang, L., et al. (2013). Functions of the lethal leaf-spot 1 gene in wheat cell death and disease tolerance to Puccinia striiformis. J. Exp. Bot. 64, 2955–2969. doi: 10.1093/jxb/ert135
Thilmony, R., Underwood, W., and Sheng, Y. H. (2006). Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonas syringae pv. Tomato DC3000 and the human pathogen Escherichia coli O157:H7. Plant J. 46, 34–53.
Tiburzy, R., and Reisener, H. J. (1990). Resistance of wheat to Puccinia graminis f.sp. Tritici: association of the hypersensitive reaction with the cellular accumulation of lignin-like material and callose. Physiol. Mol. Plant P. 36, 109–120. doi: 10.1016/0885-5765(90)90100-c
Treutter, D. (2005). Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Biol. 7, 581–591. doi: 10.1055/s-2005-873009
Treutter, D. (2006). Significance of flavonoids in plant resistance: a review. Environ. Chem. Lett. 6, 147–157. doi: 10.1007/s10311-006-0068-8
Van Wersch, R., Li, X., and Zhang, Y. (2016). Mighty dwarfs: Arabidopsis autoimmune mutants and their usages in genetic dissection of plant immunity. Front. Plant Sci. 7:1717.
Vance, C. P., Kirk, T. K., and Sherwood, R. T. (1980). Lignification as a mechanism of disease resistance. Annu. Rev. Phytopathol. 18, 259–288. doi: 10.1146/annurev.py.18.090180.001355
Vanetten, H. D., Mansfield, J. W., Bailey, J. A., and Farmer, E. E. (1994). Two Classes of Plant antibiotics: Phytoalexins versus “Phytoanticipins”. Plant Cell 6, 1191–1192. doi: 10.1105/tpc.6.9.1191
Walbot, V., Hoisington, D. A., and Neuffer, M. G. (1983). Disease Lesion Mimic Mutations. Berlin: Springer, 431–442.
Wang, F., Wu, W., Wang, D., Yang, W., Sun, J., Liu, D., et al. (2016). Characterization and genetic analysis of a novel Light-Dependent lesion mimic mutant, lm3, showing Adult-Plant resistance to powdery mildew in common wheat. PLos One 11:e155358.
Wang, G. F., and Balint-Kurti, P. J. (2016). Maize homologs of CCoAOMT and HCT, two key enzymes in lignin biosynthesis, form complexes with the NLR rp1 protein to modulate the defense response. Plant Physiol. 171, 2166–2177. doi: 10.1104/pp.16.00224
Wang, G. F., He, Y., Strauch, R., Olukolu, B. A., Nielsen, D., Li, X., et al. (2015). Maize homologs of hydroxycinnamoyltransferase, a key enzyme in lignin biosynthesis, bind the nucleotide binding Leucine-Rich repeat rp1 proteins to modulate the defense response. Plant Physiol. 169:2230.
Wang, X. C., Wu, J., Guan, M. L., Zhao, C. H., Geng, P., and Zhao, Q. (2020). Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 101, 637–652. doi: 10.1111/tpj.14570
Wang, Y., Lysoe, E., Armaregomarriott, T., Erban, A., Paruch, L., van Eerde, A., et al. (2018). Transcriptome and metabolome analyses provide insights into root and root-released organic anion responses to phosphorus deficiency in oat. J. Exp. Bot. 69, 3759–3771.
Windram, O. P., Madhou, P., Mchattie, S., Hill, C., Hickman, R., Cooke, E., et al. (2012). Arabidopsis defense against Botrytis cinerea: chronology and regulation deciphered by high-resolution temporal transcriptomic analysis. Plant Cell 24, 3530–3557. doi: 10.1105/tpc.112.102046
Wolter, M., Hollricher, K., Salamini, F., and Schulzelefert, P. (1993). The mlo resistance alleles to powdery mildew infection in barley trigger a developmentally controlled defence mimic phenotype. Mol. Genet. Genomics 239, 122–128. doi: 10.1007/bf00281610
Wu, X., Xu, L., Zhao, P., Li, N., Wu, L., He, Y., et al. (2015). Comparative transcriptome profiling of two maize near-isogenic lines differing in the allelic state for bacterial brown spot disease resistance. J. Integr. Agr. 14, 610–621. doi: 10.1016/s2095-3119(14)60873-x
Yang, Q., He, Y., Kabahuma, M. K., Chaya, T., Kelly, A., Borrego, E., et al. (2017). A gene encoding maize caffeoyl-CoA O -methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 49, 1364–1372. doi: 10.1038/ng.3919
Yang, S., and Hua, J. (2004). A haplotype-specific resistance gene regulated by BONZAI1 mediates temperature-dependent growth control in arabidopsis. Plant Cell 16, 1060–1071. doi: 10.1105/tpc.020479
Yu, G., Wang, L., Han, Y., and He, Q. (2012). ClusterProfiler: an r package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Yu, Y., Shi, J., Li, X., Liu, J., Geng, Q., Shi, H., et al. (2018). Transcriptome analysis reveals the molecular mechanisms of the defense response to gray leaf spot disease in maize. BMC Genomics 19:742.
Yuan, S., Li, W., Li, Q., Wang, L., Cao, J., Jiang, W., et al. (2019). Defense responses, induced by p-coumaric acid and Methyl p-Coumarate, of Jujube (Ziziphus jujuba Mill.) fruit against black spot rot caused by Alternaria alternata. J. Agr. Food Chem. 67, 2801–2810. doi: 10.1021/acs.jafc.9b00087
Zeng, X., Yuan, H., Dong, X., Peng, M., Jing, X., Xu, Q., et al. (2020). Genome-wide dissection of co-selected UV-B responsive pathways in the UV-B adaptation of qingke. Mol. Plant 13, 112–127. doi: 10.1016/j.molp.2019.10.009
Zhang, S. H., Yang, Q., and Ma, R. C. (2007). Erwinia carotovora ssp. Carotovora infection induced “defense lignin” accumulation and lignin biosynthetic gene expression in Chinese cabbage (Brassica rapa L. Ssp. Pekinensis). J. Integr. Plant Biol. 49, 993–1002. doi: 10.1111/j.1672-9072.2007.00478.x
Zhao, Q., and Dixon, R. A. (2011). Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends plant sci. 16, 227–233. doi: 10.1016/j.tplants.2010.12.005
Zhao, Q., Nakashima, J., Chen, F., Yin, Y., Fu, C., Yun, J., et al. (2013). LACCASE is necessary and nonredundant with PEROXIDASE for lignin polymerization during vascular development in arabidopsis. Plant Cell 25, 3976–3987. doi: 10.1105/tpc.113.117770
Zhou, S., Richter, A., and Jander, G. (2018). Beyond defense: multiple functions of benzoxazinoids in maize metabolism. Plant Cell Physiol. 59, 1528–1537. doi: 10.1093/pcp/pcy064
Keywords: maize, disease lesion mimics, defense responses, transcriptomics, metabolomics, phenylpropanoid, lignin, terpenoid
Citation: Mu X, Li J, Dai Z, Xu L, Fan T, Jing T, Chen M and Gou M (2021) Commonly and Specifically Activated Defense Responses in Maize Disease Lesion Mimic Mutants Revealed by Integrated Transcriptomics and Metabolomics Analysis. Front. Plant Sci. 12:638792. doi: 10.3389/fpls.2021.638792
Received: 07 December 2020; Accepted: 29 March 2021;
Published: 17 May 2021.
Edited by:
Anton R. Schäffner, Helmholtz-Gemeinschaft Deutscher Forschungszentren (HZ), GermanyReviewed by:
Dereje Worku Mekonnen, Independent Researcher, Munich, GermanyCopyright © 2021 Mu, Li, Dai, Xu, Fan, Jing, Chen and Gou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingyue Gou, bWluZ3l1ZWdvdUBoZW5hdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.