 Veronika Cetlová1
Veronika Cetlová1 Judita Zozomová-Lihová
Judita Zozomová-Lihová Andrea Melichárková
Andrea Melichárková Lenka Mártonfiová
Lenka Mártonfiová Stanislav Španiel
Stanislav Španiel- 1Institute of Botany, Plant Science and Biodiversity Centre, Slovak Academy of Sciences, Bratislava, Slovakia
- 2Botanical Garden of P. J. Šafárik University in Košice, Košice, Slovakia
- 3Department of Botany, Faculty of Science, Charles University, Prague, Czechia
The Mediterranean Basin is a significant hotspot of species diversity and endemism, with various distribution patterns and speciation mechanisms observed in its flora. High species diversity in the Mediterranean is also manifested in the monophyletic lineage of Alyssum annuals (Brassicaceae), but little is known about its origin. These species include both diploids and polyploids that grow mainly in open and disturbed sites across a wide elevational span and show contrasting distribution patterns, ranging from broadly distributed Eurasian species to narrow island endemics. Here, we investigated the evolution of European representatives of this lineage, and aimed to reconstruct their phylogeny, polyploid and genome size evolution using flow cytometric analyses, chloroplast and nuclear high- and low-copy DNA markers. The origin and early diversification of the studied Alyssum lineage could be dated back to the Late Miocene/Pliocene and were likely promoted by the onset of the Mediterranean climate, whereas most of the extant species originated during the Pleistocene. The Aegean region represents a significant diversity center, as it hosts 12 out of 16 recognized European species and comprises several (sub)endemics placed in distinct phylogenetic clades. Because several species, including the closest relatives, occur here sympatrically without apparent niche differences, we can reject simple allopatric speciation via vicariance as well as ecological speciation for most cases. Instead, we suggest scenarios of more complex speciation processes that involved repeated range shifts in response to sea-level changes and recurrent land connections and disconnections since the Pliocene. In addition, multiple polyploidization events significantly contributed to species diversity across the entire distribution range. All seven polyploids, representing both widespread species and endemics to the western or eastern Mediterranean, were inferred to be allopolyploids. Finally, the current distribution patterns have likely been affected also by the human factor (farming and grazing). This study illustrates the complexity of evolutionary and speciation processes in the Mediterranean flora.
Introduction
The Mediterranean Basin represents one of the world’s major biodiversity hotspots with an exceptionally high species diversity and endemism rate (Myers et al., 2000; Thompson, 2020). This species richness can be attributed to a variety of different factors. Complex geological history, high topographical and ecological heterogeneity of this region have promoted allopatric and ecological diversification and speciation (Médail and Diadema, 2009; Hewitt, 2011; Nieto Feliner, 2014; Thompson, 2020). In addition, the impact of Quaternary climatic oscillations was less severe in the Mediterranean than in central and northern Europe, allowing preservation and accumulation of diversity through small-scale range shifts with minimized population and species extinctions (Nieto Feliner, 2011). Several major paleoclimatic and paleogeologic events have exerted crucial influences on Mediterranean flora, especially the Mediterranean Sea desiccation during the Messinian salinity crisis (ca. 5.96–5.33 mya; Krijgsman et al., 1999), the onset of the Mediterranean climate (ca. 3.2 mya, Suc, 1984), and sea-level oscillations that occurred during the glacial and interglacial periods of the Pleistocene (Lambeck et al., 2014). Sea-level drops resulted in the formation of temporary land connections between major peninsulas (e.g., between the Balkan and Apennine Peninsulas, Correggiari et al., 1996), between islands, and between the mainland and islands (e.g., Anatolia and East Aegean islands, Simaiakis et al., 2017). At the latest Pleistocene, the Aegean and Ionian sea level was ca. 120 m lower than today, and Aegean islands formed a kind of land bridges connecting mainland Greece and Turkey, separated locally by sea channels (Perissoratis and Conispoliatis, 2003), which enhanced plant migrations. Sea-level oscillations also fostered distribution range shifts, which led to secondary contacts and gene flow between otherwise isolated species and lineages (Nieto Feliner, 2011, 2014).
The Mediterranean Basin and the adjacent mountain ranges represent the center of species diversity for Alyssum L. (Dudley, 1965; Ball and Dudley, 1993; Jalas et al., 1996), which is the largest genus in the tribe Alysseae and comprises approximately 114 species, including 30 annuals and 84 perennials (Španiel et al., 2015, but still subject of taxonomic revisions). The genus Alyssum, in its traditional taxonomic treatment, was nearly twice as large (Al-Shehbaz, 2012); however, many species have recently been transferred into the genera Cuprella Salmerón-Sánchez, Mota & Fuertes, Meniocus Desv., and Odontarrhena C.A.Mey. ex Ledeb. (Španiel et al., 2015). In the current delimitation, the genus Alyssum includes three of its six original sections (Dudley, 1964b): Alyssum sect. Alyssum, A. sect. Gamosepalum (Hausskn.) T.R.Dudley, and A. sect. Psilonema (C.A.Mey.) Hook.f. Recent phylogenetic analyses, however, have revealed two major clades in Alyssum s.str., which do not agree with the traditional sectional classification (Rešetnik et al., 2013; Li et al., 2015; Salmerón-Sánchez et al., 2018). One clade includes annual species of A. sect. Psilonema and both annual and perennial species of A. sect. Alyssum, whereas the other clade includes perennial species from A. sect. Alyssum and A. sect. Gamosepalum and an annual species Alyssum dasycarpum Stephan ex. Willd. from A. sect. Psilonema. The present study focuses on the former clade, which comprises all annuals from sections Alyssum and Psilonema (except for the phylogenetically distant A. dasycarpum) and a nested Alyssum montanum–Alyssum repens perennial species complex. Whereas the taxonomy and evolutionary history of the A. montanum–A. repens species complex have been thoroughly explored in a series of recent studies (e.g., Španiel et al., 2011, 2017a,b, 2019; Magauer et al., 2014; Zozomová-Lihová et al., 2014, 2020; Arrigo et al., 2016; Melichárková et al., 2017), little is known about the phylogenetic relationships and speciation processes of the annual taxa. Here, we attempted to fill this gap by exploring the evolutionary history of all 16 annual taxa of Alyssum reported in Europe, some of which extend also to northern Africa or Asia (see Table 1). Altogether, the studied clade comprises tentatively 29 annual species, thus about 13 extra-European annuals distributed mainly in the Irano-Anatolian area, Caucasus and Central Asia (Dudley, 1964a,b, 1965; Rechinger, 1968) were not included in our study. Their circumscription and taxonomic treatment is namely uncertain in several cases, and detailed field and herbarium research is needed before including them in phylogenetic studies, which was beyond the scope of the present study.
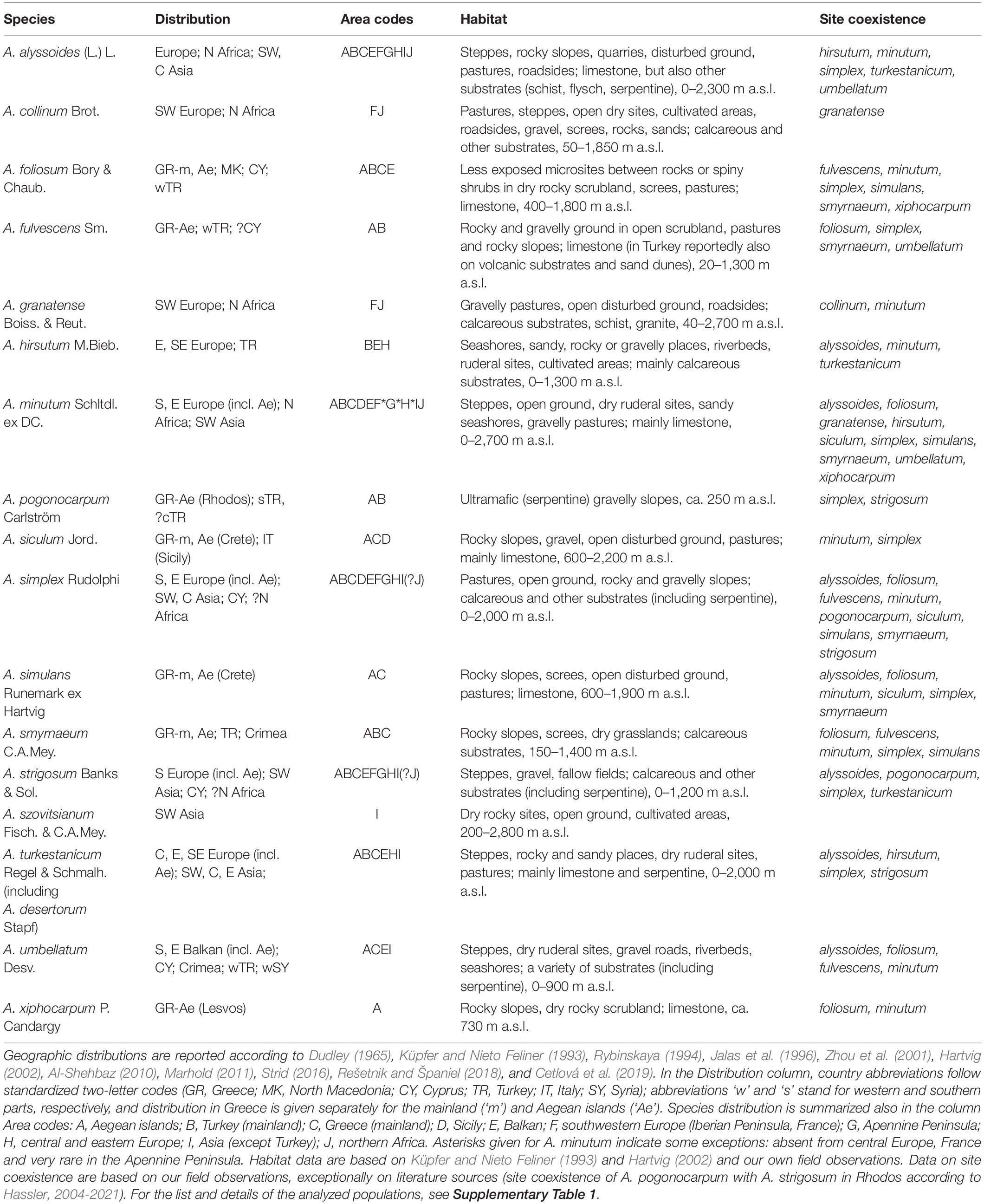
Table 1. Overview of the studied Alyssum annual species (listed alphabetically), including their distribution ranges, habitat characteristics and site coexistence with other species.
The here studied Alyssum annuals display contrasting distribution patterns in Europe, ranging from narrow endemics to widespread species (Table 1). Much species diversity is concentrated in the Aegean islands and adjacent mainland areas (Dudley, 1965; Hartvig, 2002; Strid, 2016). All these species grow preferentially on open, disturbed ground and, therefore, can be found on pastures, gravelly screes, rocky or stony slopes, phrygana, macchia, and typically (but not exclusively) on limestone at various elevations. Some species are more ubiquitous (e.g., Alyssum alyssoides and Alyssum minutum), whereas others are typically present in particular types of habitats (see Table 1 for details). Quite commonly three or even more annual Alyssum species can grow in the same locality within a close distance of few meters (field observations and the present sampling, see Table 1 and Supplementary Table 1). Small yellow flowers arranged in racemes are visited by a variety of insects, but nothing is known about the mating system of these Alyssum species. Phenological shifts observed between some co-occurring species suggest the existence of premating isolating barriers. Seed dispersal occurs primarily through gravity and zoochory, especially sheep and goats, as the seeds become mucilaginous under wet conditions and easily attach to the animal body (field observations). Three different ploidy levels were previously reported for these annuals, ranging from diploids to hexaploids (Table 2, Španiel et al., 2015).
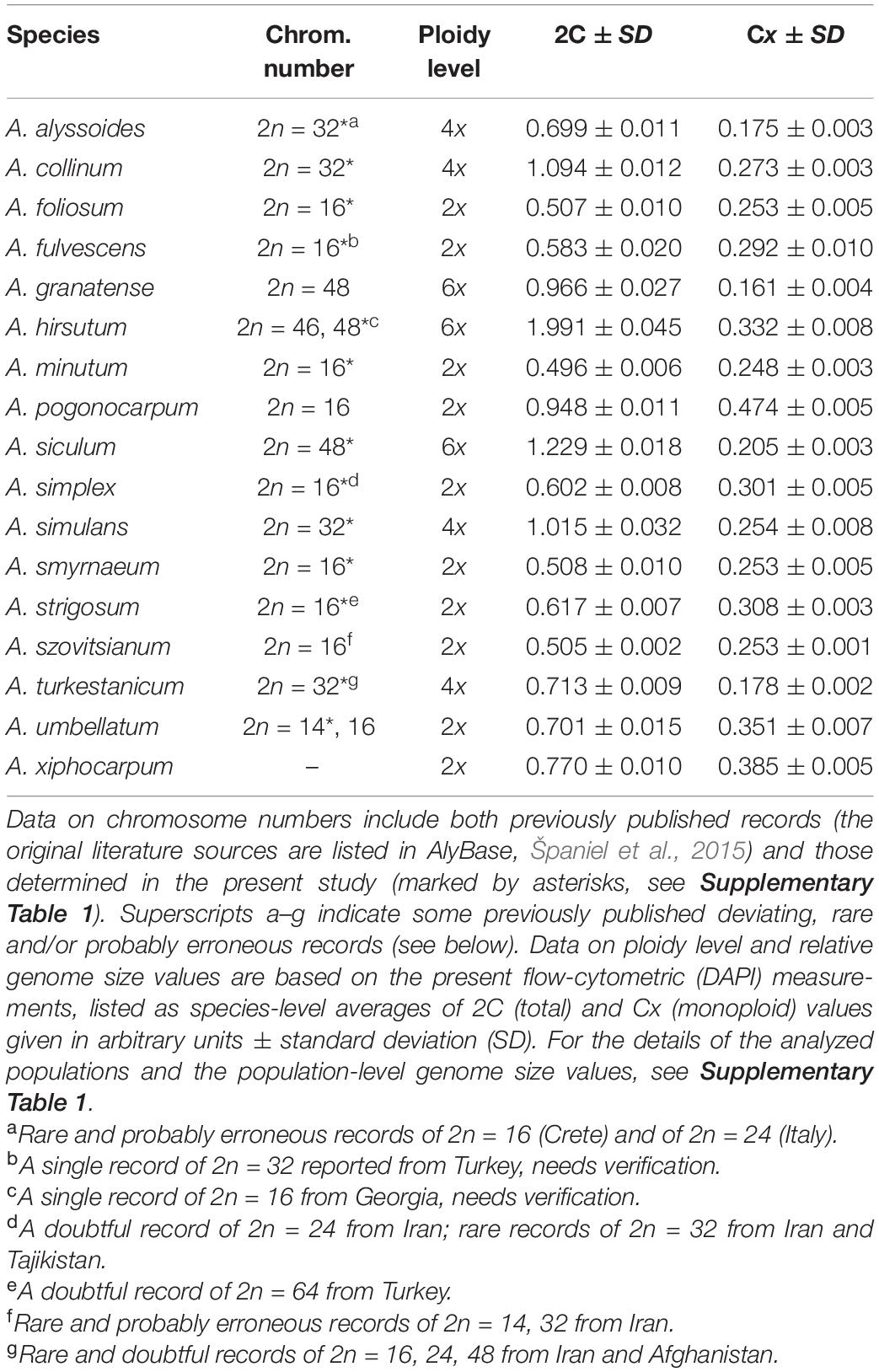
Table 2. Chromosome numbers, ploidy level data and relative genome size values of the studied Alyssum species.
Some hypotheses regarding species relationships can be drawn from morphological resemblance and the previously published phylogeny of Alysseae (Rešetnik et al., 2013). For example, available data suggest that the hexaploid species Alyssum siculum is either an allopolyploid derived from the tetraploid A. alyssoides and the diploid Alyssum simplex or an autopolyploid of A. alyssoides (Persson, 1971; Rešetnik et al., 2013). Another hexaploid taxon, A. granatense, morphologically resembles the tetraploid A. alyssoides (Küpfer and Nieto Feliner, 1993). Morphological similarity indicates close evolutionary relationships between the hexaploid Alyssum hirsutum and the diploid Alyssum pogonocarpum (Carlström, 1984), between the diploids A. simplex and Alyssum strigosum, and between the diploids Alyssum fulvescens and Alyssum smyrnaeum (Hartvig, 2002). The tetraploid Alyssum simulans is a suspected allopolyploid of the diploids Alyssum foliosum and A. minutum, due to its intermediate morphology (Hartvig, 2002). Alyssum xiphocarpum (the ploidy level of which has not yet been published) is morphologically very similar to the sympatric diploid Alyssum umbellatum and, therefore, has been treated by many authors as a synonym of the latter species (e.g., Dudley, 1965; Jalas et al., 1996); however, A. xiphocarpum exhibits much larger petals, anthers and styles than A. umbellatum (Hartvig, 2002). In contrast, the tetraploid Alyssum turkestanicum does not show obvious morphological affinity to any particular European species, and its phylogenetic relationships are unknown.
In the present study, we aimed to resolve the evolutionary history of all 16 European Alyssum annuals described above. To accomplish this goal, we performed the following: (1) assessed the ploidy levels and genome size variation within and among the species; (2) inferred phylogenetic relationships among these species based on multilocus DNA data, including divergence time estimates; (3) proposed scenarios of speciation events based on distribution, phylogenetic, and polyploidy patterns; and (4) formulated consistent hypotheses for the origin of polyploids.
Materials and Methods
Plant Material
Material sampling comprised all 16 annual Alyssum species reported in Europe (Table 1 and Supplementary Table 1). In addition, two species from the well-explored perennial A. montanum-A. repens species complex, which is nested as a monophyletic clade within the lineage of the studied annuals, and one southwestern Asian annual species, Alyssum szovitsianum, were included. On the other hand, A. dasycarpum, which is an annual species that also occurs in Europe but is phylogenetically unrelated to the lineage being explored here (see section “Introduction” and Rešetnik et al., 2013) was not investigated. The sampling focused on the European Mediterranean, and particularly Greece and the Aegean area, which represent the regions with the highest species diversity of Alyssum annuals within Europe (Table 1). Two species of the related genus Odontarrhena were used as outgroups in the molecular analyses. In total, 191 populations of annual species were sampled and used for flow-cytometric (FCM) analyses. Of these, 76 populations, representing all sampled 17 annual species, were subjected to molecular analyses. A full list of the population samples, including detailed descriptions of their localities, can be found in Supplementary Table 1 and is shown on the map in Figure 1. Tissue samples were collected from five to ten individuals in each population. Leaves and sterile shoots were dried and preserved in silica gel for further use in FCM and DNA extraction procedures. All sampled individuals were analyzed by FCM, and typically, two individuals per population were subjected to molecular analyses. In several cases, when individuals at the sampled localities were in the fruiting stage, or the leaves were withered or damaged, we collected seeds. These seeds were later sown, and the plantlets that sprouted from the seeds were used to obtain vegetative tissues for FCM and molecular analyses. In addition, seeds were collected from most species (whenever available at localities) for chromosome counting. Voucher specimens of the plants analyzed during the present study have been deposited in the herbarium of the Institute of Botany, SAS, Bratislava, Slovakia (SAV).
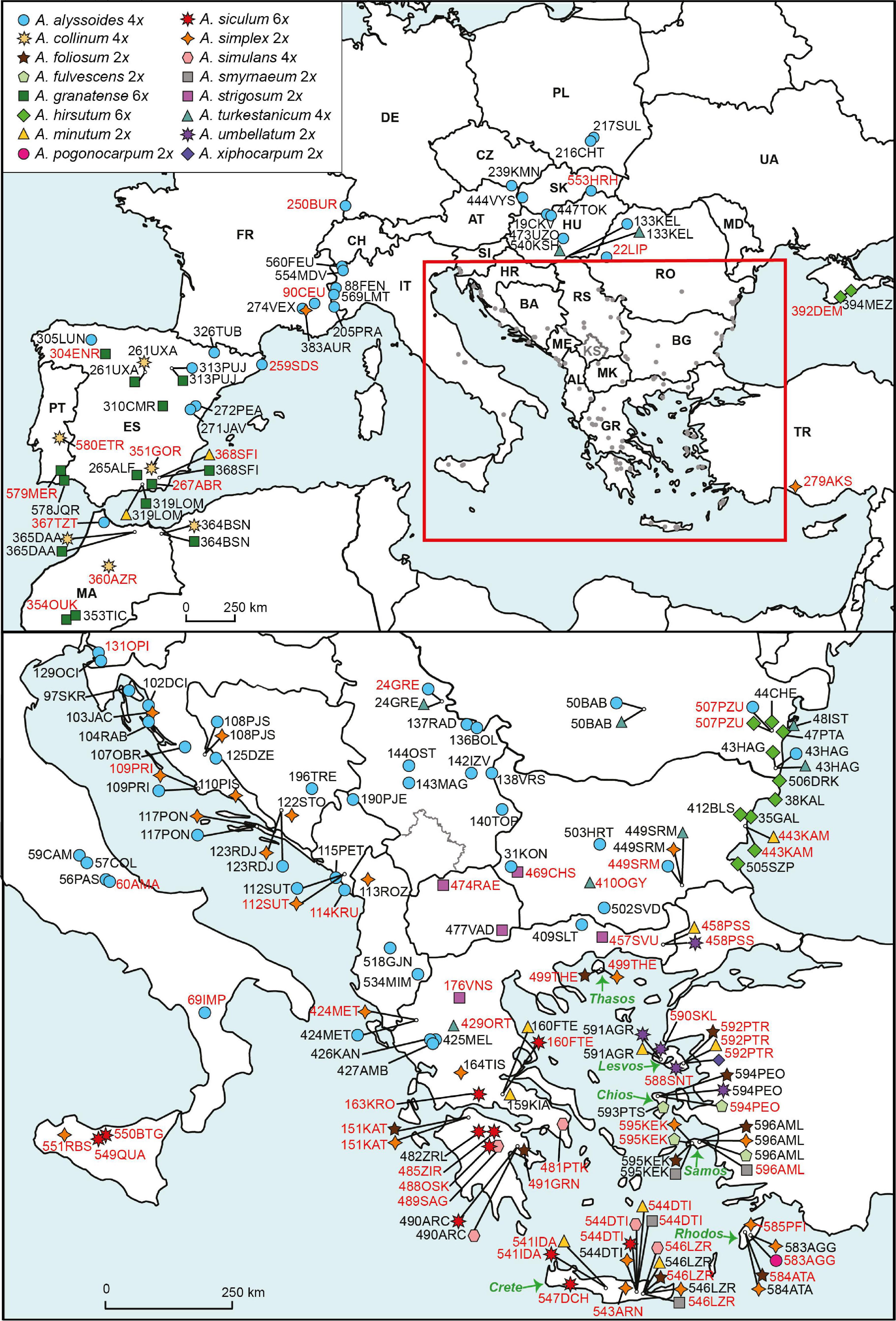
Figure 1. Map of the sample sites of the analyzed annual Alyssum species. Population assignments to the species are indicated by different symbol colors and shapes. Ploidy levels are indicated next to the species names. Alyssum szovitsianum, which was represented by one population sample (289TRJ-sz) from Iran, is not shown on the map. Population codes follow Supplementary Table 1 and are written in either black script (population samples used only for the FCM analyses) or red script (population samples used for both FCM and molecular analyses).
Chromosome Counting and Flow Cytometry
Chromosome numbers were determined in the mitotic metaphases of cells from root tips, following the protocol described in Španiel et al. (2018). FCM was used to determine relative genome sizes and to infer the ploidy levels of the investigated annual taxa and populations. Each sampled individual was analyzed separately, measured together with an internal standard (see below). Ploidy levels were inferred by comparing the relative genome size of each sample with the values measured in conspecific plants with known, counted chromosome numbers. In the case of four taxa for which we were unable to obtain chromosome numbers (A. granatense, A. pogonocarpum, A. szovitsianum, and A. xiphocarpum), their ploidy levels were deduced from the previously published chromosome counts for these species and/or through comparisons with the relative genome sizes of their closest relatives, for which the chromosome numbers were determined in this study. The relative genome size was measured based on the fluorescence intensity of nuclei stained with the AT-selective fluorochrome 4′,6-diamidino-2-phenylindole (DAPI). The relative genome size (relative 2C value) of a sample was computed as the ratio between the mean G0/G1 peak value of the sample and the mean G0/G1 peak value of the standard with a known genome size. A monoploid relative genome size (relative Cx value) was also inferred, representing the relative DNA content of a monoploid, unreplicated genome that comprises one set of chromosomes and corresponds to the base chromosome number (x) of the taxon (Greilhuber et al., 2005). One of three internal standards was selected for each individual measurement, depending on the genome size of the measured species. Lycopersicon esculentum ‘Stupické polní rané’ (2C = 1.96 pg; Doležel et al., 1992) was used as the primary standard for most taxa. In cases when Lycopersicon was not suitable (due to overlapping G0/G1 or G2 peaks between the sample and the standard), we used secondary standards, either Bellis perennis L. (2C = 3.38 pg; Schönswetter et al., 2007) or Solanum pseudocapsicum L. (2C = 2.59 pg; Temsch et al., 2010). The relative genome sizes of all samples that were measured using secondary standards were eventually recalculated relative to the primary standard to obtain a final value for the relative genome size (in arbitrary units), as reported in Section “Results.” The recalculation was performed based on the ratio between the relative genome sizes of the primary and secondary standards. This ratio was calculated based on repeated simultaneous measurements of the primary standard with each secondary standard. In addition to individual sample measurements, selected samples from two or three different species that showed divergent genome size values at the same ploidy level (see section “Results”) were analyzed simultaneously. These measurements were performed to prove and illustrate the differences in genome size between species. All analyses were performed using a Partec Cyflow ML instrument equipped with an HBO-100 mercury arc lamp (Partec, Münster, Germany) following the protocol described in Španiel et al. (2011). The relative genome sizes of the perennials A. repens and A. montanum were known and taken from previous studies (Španiel et al., 2018; Melichárková et al., 2019), used here in the analysis exploring phylogenetic patterns of relative genome size variation (see below).
PCR Amplification, Cloning, and Sequencing
Genomic DNA was isolated using the DNeasy 96 Plant Kit, according to the manufacturer’s protocol (Qiagen, Hilden, Germany). One chloroplast and two nuclear DNA regions were amplified and sequenced: the rpoB-trnCGCA intergenic spacer of chloroplast DNA (cpDNA), the multicopy internal transcribed spacer (ITS) region of rDNA (ITS1-5.8S-ITS2), and the single-copy DET1 (de-etiolated 1) nuclear gene. Universal primers were used to amplify the rpoB-trnC spacer (primers rpoB and trnCGCAR; Shaw et al., 2005) and the ITS region (primers ITS4 and ITS5; White et al., 1990). The partial sequence of the DET1 gene was amplified with the primer set published by Kuittinen et al. (2002), which binds to exons 5 and 8 in Arabidopsis thaliana and has previously been shown to perform well also in Alyssum (Melichárková et al., 2017). Polymerase chain reaction (PCR) amplifications were performed in a total volume of 20 μl containing 1 μl of gDNA, 0.58 U Pfu DNA polymerase (Thermo Fisher Scientific Inc., Waltham, MA, United States), 1× Pfu buffer with MgSO4, 0.2 mM dNTPs, and 0.2 μM each forward and reverse primers. The DET1 reaction mix also included 5% dimethyl sulfoxide (DMSO) to reduce possible recombination events during PCR. The following PCR conditions were used: 80°C for 5 min, 30 × (95°C for 1 min, 50°C for 1 min, followed by a 5% ramp to 65°C, 65°C for 4 min), 65°C for 5 min for the rpoB-trnC amplification; 94°C for 3 min, 35 × (94°C for 30 s, 50°C for 30 s, 72°C for 1 min), 72°C for 10 min for the ITS amplification; 94°C for 5 min, 30 × (94°C for 30 s, 51°C for 30 s, 72°C for 2 min), 72°C for 7 min for the DET1 amplification.
PCR products were purified either enzymatically (prior to direct sequencing) using a mixture of Exonuclease I and FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific Inc.) or with NucleoSpin Gel and PCR Clean-up column kit (Macherey-Nagel, Düren, Germany), when subjected to cloning before sequencing. ITS products from all individuals and DET1 products from all diploids were first sequenced directly, and if sequence polymorphisms (double peaks or unreadable parts) were detected in the electropherograms, the PCR products were cloned. DET1 products from the polyploid individuals were all cloned prior to sequencing. Purified PCR products were cloned with the CloneJET PCR Cloning kit using the pJET1.2/blunt vector, following the manufacturer’s protocol (Thermo Fisher Scientific), with transformation into JM109 competent cells (Promega, Madison, United States). Direct PCR from bacterial colonies was performed in a total volume of 13 μl, using 0.2 μM pJET1.2 sequencing primers, 0.65 U of DreamTaq DNA polymerase (Thermo Fisher Scientific), 1× DreamTaq Buffer with MgCl2, and 0.2 mM dNTPs. The cells were lysed by 95°C for 3 min, followed by 35 cycles (95°C for 30 s, 50°C for 30 s and 72°C for 2 min), and a 10-min final incubation step at 72°C. The PCR products were purified by enzymatic clean-up (as specified above) prior to sequencing. Multiple clones per sample were sequenced in an effort to recover all alleles and identify any potential PCR-mediated recombinations: six to eight clones per diploid, 16 clones per tetraploid, and 20 clones per hexaploid sample. Sequencing was performed using the ABI 3730xl DNA analyzer at Eurofins Genomics Company (Konstanz, Germany). The sequences were edited and aligned using Geneious software R7.1.9 (Biomatters Ltd., Auckland, New Zealand) and submitted to the GenBank nucleotide database (Supplementary Table 1). Indels present in the sequence alignments were coded as binary characters (except for those occurring in highly variable homopolymeric stretches that were ignored) according to the simple indel coding approach (Simmons and Ochoterena, 2000) using FastGap 1.2 software (Borchsenius, 2009). The indel coding datasets were appended to the nucleotide datasets and were included in the phylogenetic analyses. The final ITS and rpoB-trnC alignments contained sequence data from 152 individuals (76 populations); the DET1 alignment comprised data from 73 individuals (from a selection of 38 populations; Table 3 and Supplementary Table 1).
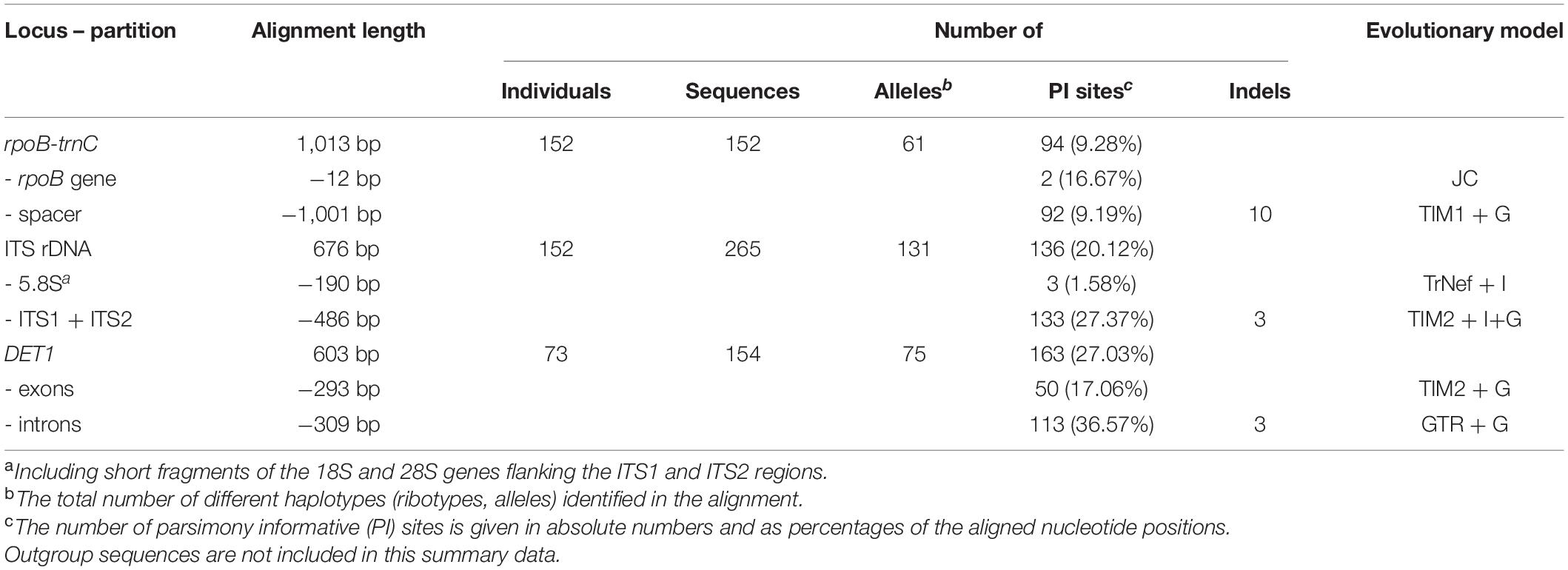
Table 3. Characteristics of the nucleotide alignments used in the present study.
Phylogenetic Analyses of the rpoB-trnC, ITS, and DET1 Sequences
The statistical parsimony-based TCS method (Clement et al., 2000) in PopART (Leigh and Bryant, 2015) was used for the identification of different alleles and haplotypes within the datasets, and to determine their frequencies and sharing patterns. A NeighborNet (NN) network (Huson and Bryant, 2006) was generated in SplitsTree4 v. 4.14.4 based on uncorrected P-distances. Phylogenetic trees were inferred using maximum-likelihood (ML, GARLI v.2.01; Zwickl, 2006) and Bayesian analyses (MrBayes v.3.2.6, Huelsenbeck and Ronquist, 2001), both of which were run at the CIPRES Science Gateway (Miller et al., 2010). The three studied DNA markers were analyzed separately, and three data partitions were defined within each dataset as follows: intergenic spacer of rpoB-trnC, a fragment of the rpoB gene, and indels within the rpoB-trnC dataset; non-coding ITS1 and ITS2, 5.8S of rDNA, and indels within the ITS dataset; introns, exons, and indels within the DET1 dataset. Best-fit models of nucleotide substitutions were assessed in jModelTest v.2.1.10 (Darriba et al., 2012) separately for each nucleotide data partition, using the Akaike information criterion (AIC; Akaike, 1974). Bayesian analyses were conducted with four MCMC (Markov chain Monte Carlo algorithm) chains for 10–15 million generations, with a sampling frequency of every 100th generation. The first 10% of trees were discarded as burn-in and a consensus tree was generated from the remaining trees, computing also the Bayesian posterior probabilities (BPP) for each node. For ML analyses, multiple searches were performed with random starting trees and by setting the program to stop after 20,000 generations if no improvement of the log-likelihood was detected (≤0.01), with a maximum of 215 million generations. Branch support was assessed by 2,000 bootstrap replicates (bootstrap support, BS).
Inference of Coalescent-Based Species Trees
A Bayesian coalescent-based approach was employed to estimate a species tree using STACEY package v. 1.2.2 (Jones, 2017) for BEAST v. 2.5.1 (Drummond and Rambaut, 2007). Since the multispecies coalescent accounts for incomplete lineage sorting as the primary source of gene tree discordance but not for hybridization events, we assembled two datasets avoiding reticulate evolutionary patterns in the markers and samples (especially in the case of DET1 data for polyploids, see section “Results”). The first dataset included the rpoB-trnC and ITS sequence data, obtained from both diploids and polyploids, whereas the second dataset included all three regions, rpoB-trnC, ITS, and DET1, from diploids only. The purpose of performing these analyses was to infer species relationships at the diploid level, as well as, to a limited extent, with polyploids involved and confront them with the multilabelled DET1 gene tree reconstruction. Two populations of diploid A. fulvescens sampled from different islands (Samos and Chios islands) were a priori defined as two ‘species’, because they were resolved in two divergent clades in each of the gene trees. The tetraploid A. simulans was excluded from the analyses because it displayed two divergent ITS copy variants, indicating an allopolyploid origin. All other species (both diploids and polyploids) appeared genetically coherent, without any indication of reticulation or polyphyletic origins when considering only the ITS and rpoB-trnC gene trees.
The tool BEAUti 2 (Bouckaert et al., 2014) was used to create a STACEY input file using the following settings and parameters: multiple unlinked data partitions (as defined above), the best-fit nucleotide substitution models, as determined with jModelTest (see above), lognormal relaxed clock, Yule species tree model, and lognormal priors. We ran three independent Markov chain Monte Carlo (MCMC) analyses, with 75 million generations each and a sampling frequency of 10,000 trees, using BEAST v. 2.5.1 through the CIPRES Science Gateway. The BEAST output was analyzed in Tracer v.1.7.1 (Rambaut et al., 2018) and verified for MCMC convergence and the ESS (effective sample sizes) values of parameters. Multiple runs were combined using LogCombiner v.2.5.2, after discarding the first 20% of the sampled trees as burn-in. A maximum clade credibility (MCC) tree was obtained in TreeAnnotator v.2.5.2 and visualized in FigTree v.1.4.4.
To explore the presence of a phylogenetic signal in the measured genome size values of the studied species (described above in section “Chromosome Counting and Flow Cytometry”), we calculated the tree transformation statistics, λ (Pagel, 1999), which estimates the statistical (in)dependence of trait (genome size) evolution and the reconstructed phylogeny. It ranges from 0 to 1, with values close to 0 indicating phylogenetic independence and values close to 1 indicating a phylogenetic signal in the examined trait. It was computed in BayesTraits v.3.0.2 (Pagel et al., 2004) on the combined post-burn-in set of 18,000 trees generated from the three BEAST runs used for MCC tree as described above (the dataset including both diploids and polyploids), with two independent MCMC runs each of 1 million generations and 100,000 burn-in. Since the two runs gave virtually the same results, the posterior distribution of λ values resulting from one MCMC run was depicted in a histogram.
Phylogenetic inference based on the concatenation of the three sequence alignments was discarded in the present study, as this approach can lead to biased topologies with false support, especially in recently diverged lineages (Kubatko and Degnan, 2007). The concatenation was also technically unfeasible, as it is generally not possible to know a priori which allele (DET1) should be concatenated with which ribotype (ITS) when multiple sequence variants are revealed even in diploid accessions (see Results).
Estimation of Divergence Times Based on ITS Sequence Data
To estimate the divergence times of the studied species, we assembled an additional ITS alignment with a broader, tribe-wide taxonomic coverage. This alignment followed the dataset compiled by Salmerón-Sánchez et al. (2018), which comprised representatives of almost all genera of the tribe Alysseae (Rešetnik et al., 2013; Španiel et al., 2015), complemented by the ITS sequences of annual Alyssum species generated in the present study and the ITS dataset of the perennial A. montanum–A. repens species complex (generated in Melichárková et al., 2019). For the tree calibration, we applied an approach described by Huang et al. (2020), who reconstructed a dated phylogeny of the Rosidae clade based on complete plastid sequences and multiple reliable non-Brassicales fossil records and, subsequently, inferred three secondary calibration points within Brassicaceae. These secondary calibrations were used as temporal anchor points for the molecular dating analyses of Brassicaceae tribes based on ITS sequences, representing major lineage splits within the family. Following this approach, the ITS sequences of two outgroup species (A. thaliana, tribe Camelineae, lineage I; and Clausia aprica, tribe Dontostemoneae, lineage III) were included in the assembled Alysseae dataset to define the ages of the respective nodes (node B representing the split between lineage I and Alysseae; node C representing the split between lineage III and Alysseae, following Huang et al., 2020). In total, the ITS alignment used in the present study comprised 702 sequences from 123 species and 25 genera (Supplementary Table 2). The tree topology and divergence times were estimated using the Bayesian MCMC approach, as implemented in BEAST v. 2.6.1 and run through the CIPRES Science Gateway (Miller et al., 2010). The input file was generated using the tool BEAUti 2. For the tree priors, we selected the general time-reversible (GTR + G) substitution model, the uncorrelated relaxed lognormal clock model, the birth-dead speciation model, and we set the ages of the two nodes with a normal distribution prior, as described by Huang et al. (2020). MCMC was run with 100 million generations and a sampling frequency of 10,000 trees. The BEAST output was analyzed in Tracer v.1.7.1 and checked for MCMC convergence and the ESS values of parameters. An MCC tree was obtained in TreeAnnotator v.2.6.0, discarding the first 25% of the sampled trees as burn-in, and visualized in FigTree v.1.4.4.
Results
Chromosome Numbers
Chromosome numbers were determined in 15 populations of 13 annual species (Supplementary Table 1, Table 2, and Figure 2). For the remaining four species (A. granatense, A. pogonocarpum, A. szovitsianum, and A. xiphocarpum), the seeds obtained from the studied populations did not germinate (were unripe or too old) and, therefore, were unusable for chromosome counting. Except for A. xiphocarpum, the chromosome numbers of these species were already known from the previously published records (see Table 2). Four different chromosome numbers were detected, 2n = 14, 16, 32, and 48, with the base chromosome number x = 8 or, exceptionally, x = 7 in A. umbellatum. They represented the diploid, tetraploid and hexaploid levels.
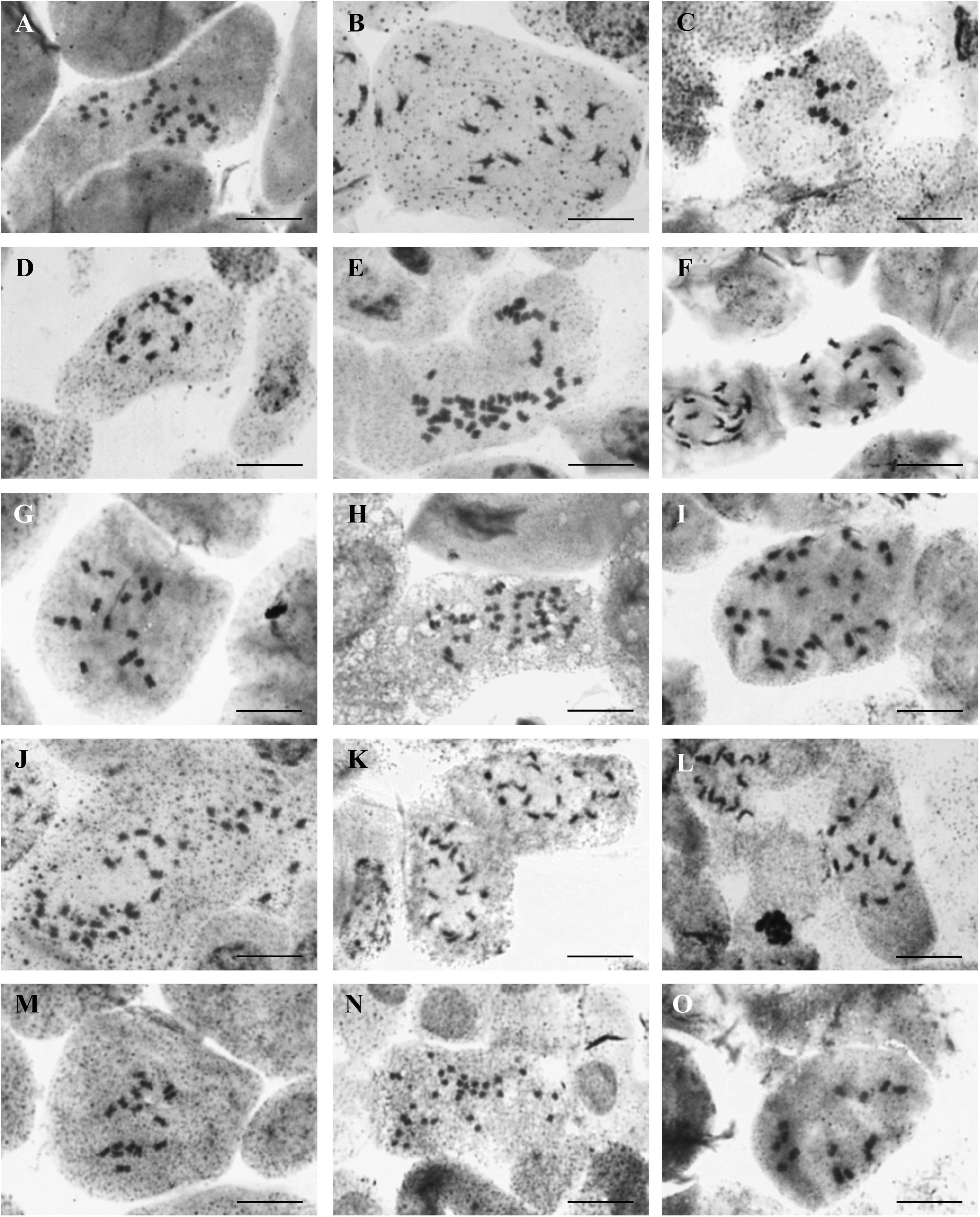
Figure 2. Photographs of chromosome metaphase plates from (A) Alyssum alyssoides (427AMB-al), 2n = 32; (B) Alyssum foliosum (151KAT-fo), 2n = 16; (C) Alyssum fulvescens (594PEO-fu), 2n = 16; (D) A. fulvescens (595KEK-fu), 2n = 16; (E) Alyssum hirsutum (506DRK-hi), 2n = 48; (F) Alyssum minutum (319LOM-mi), 2n = 16; (G) Alyssum simplex (551RBS-sx), 2n = 16; (H) Alyssum siculum (549QUA-sc), 2n = 48; (I) Alyssum collinum (261UXA-cl), 2n = 32; (J) Alyssum simulans (481PTK-ss), 2n = 32; (K) Alyssum smyrnaeum (544DTI-sy), 2n = 16; (L) Alyssum strigosum (457SVU-st), 2n = 16; (M) A. strigosum (477VAD-st), 2n = 16; (N) Alyssum turkestanicum (429ORT-t), 2n = 32; and (O) Alyssum umbellatum (458PSS-um), 2n = 14. Scale bar = 10 μm.
Ploidy Level and Genome Size Variation
The relative genome sizes were measured for all sampled 17 annual species (Table 2 and Supplementary Table 1). Both the chromosome number data and genome size values demonstrated uniform ploidy levels within individual species. The chromosome number and ploidy level of A. xiphocarpum were previously unknown; therefore, the diploid level inferred here represents the first ploidy record for this species. We also found that the studied annual species showed significant differences in their monoploid genome sizes, exhibiting approximately 2.9-fold variation (Table 2). Differences in genome sizes between several species (and groups within A. fulvescens) within the same ploidy level were large enough to yield separate peaks in the FCM histograms during simultaneous analyses, as shown in Figure 3. This variation among species also demonstrated some phylogenetic patterns (see below section “Coalescent-Based Species Trees” for details). The largest relative monoploid genome sizes were detected in A. hirsutum, A. umbellatum, A. xiphocarpum, and, especially, A. pogonocarpum (the chromosomes, unfortunately, could not be counted in this species; however, previous records reported the diploid level for this species). In contrast, the smallest relative Cx values were revealed in A. alyssoides, A. siculum, A. turkestanicum, and A. granatense (Table 2).
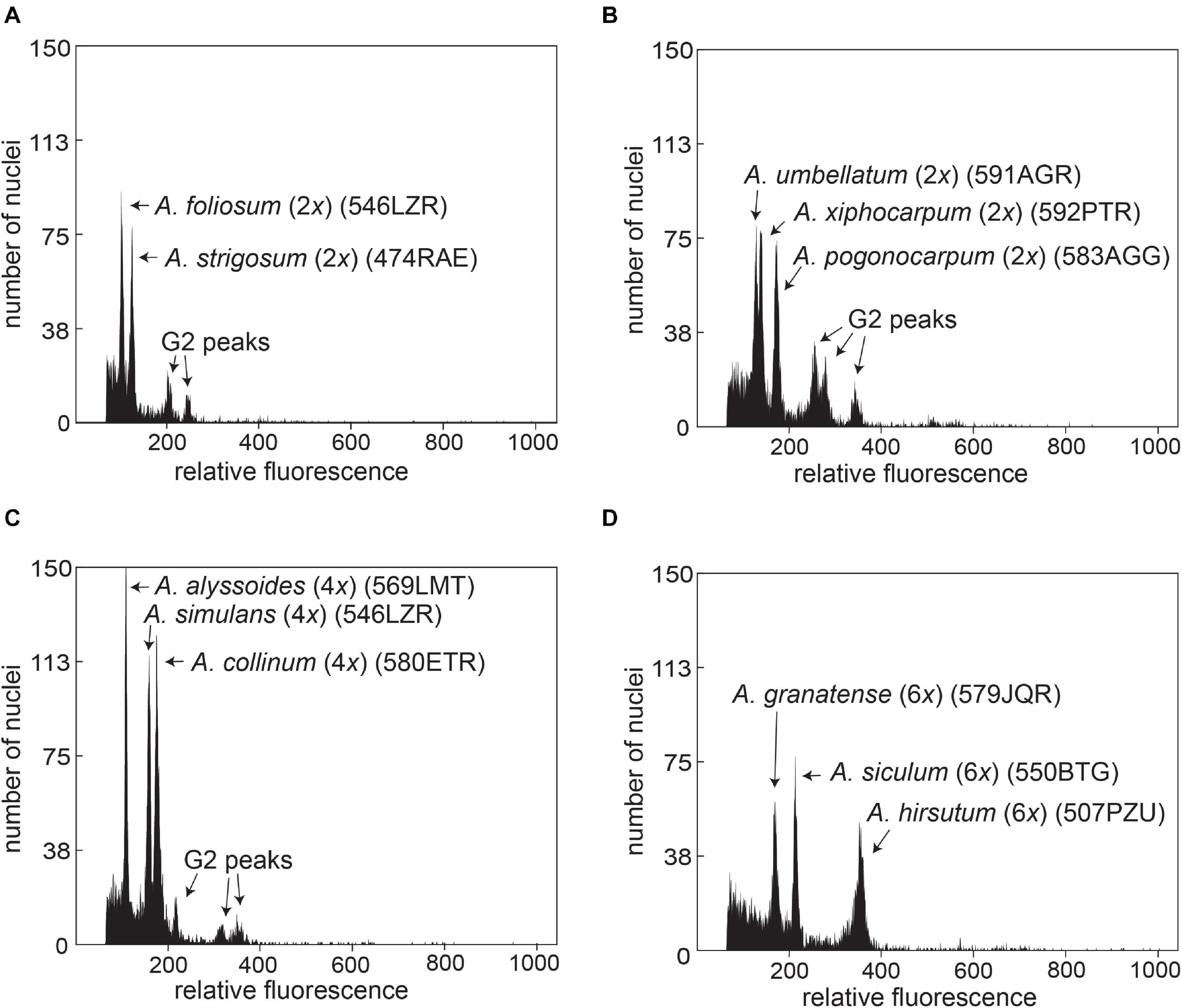
Figure 3. Simultaneous FCM analyses of Alyssum species, illustrating the differences in relative genome size values (relative fluorescence) between selected diploid (A,B), tetraploid (C), and hexaploid (D) species. For population codes, see Supplementary Table 1.
DNA Sequence Data Variation
The detailed characteristics of the rpoB-trnC, ITS, and DET1 alignments, such as the alignment length, number of sequences, alleles, polymorphic sites, and indels, are summarized in Table 3.
The rpoB-trnC dataset comprised 152 sequences (individuals) with 61 different haplotypes. Haplotype sharing most commonly occurred within populations, as well as among different populations of the same species. In two cases, haplotype sharing was observed between different species: between the tetraploid A. alyssoides and the hexaploid A. siculum, and between the diploid A. foliosum and the tetraploid A. simulans. The analyzed species displayed one to six different haplotypes, and the most diverse species were the diploid A. simplex and the hexaploids A. granatense and A. siculum.
Molecular cloning revealed multiple ITS copy variants (ribotypes) within several accessions, in both diploids and polyploids (Supplementary Table 1). Up to eight different ribotypes were, for instance, found in one individual of the hexaploid A. granatense; and up to five ribotypes in one individual of the diploid A. fulvescens. Altogether, the final ITS alignment comprised 265 sequences retrieved from 152
individuals, which represented 131 different ribotypes. The ribotype sharing patterns were similar to those observed in the rpoB-trnC dataset. The same ribotypes were frequently revealed within populations and among different populations of the same species. Ribotype sharing between different species was observed only in two cases: between the tetraploid A. alyssoides and the hexaploid A. siculum, and between the diploid A. minutum and the tetraploid A. simulans. The analyzed species displayed one to 15 different ribotypes; the most diverse species were the diploid A. fulvescens (12 ribotypes) and the polyploids A. granatense (15 ribotypes), and A. alyssoides (11 ribotypes).
The DET1 alignment included four exons and introns among them (although the first intron was missing in several accessions, which was also observed for some perennials by Melichárková et al., 2017). All diploid accessions, except for A. fulvescens, appeared homozygous. Multiple alleles were revealed by cloning in the polyploids, with up to six alleles in the hexaploids (see Supplementary Table 1). Altogether, the alignment comprised 154 sequences from 73 individuals, which represented 75 different alleles. No alleles were shared between different diploid species, but allele sharing occurred in several cases between diploids and polyploids (reflecting progenitor-polyploid derivative relationships, see below), and between different polyploid species [those with presumably shared progenitor(s)].
Phylogenetic Analyses of the rpoB-trnC, ITS, and DET1 Sequences
The NN networks, the ML, and Bayesian phylogenetic trees exhibited largely congruent topologies for each of the three markers used. Here we present the ML trees (Figures 4–6) with both the bootstrap support (BS) and the Bayesian posterior probability (BPP) values plotted on them. Only the clades that were supported by BS (≥50%) and BPP (≥0.85) are described and interpreted. The NN networks are shown in the Supplementary Figures 1–3.
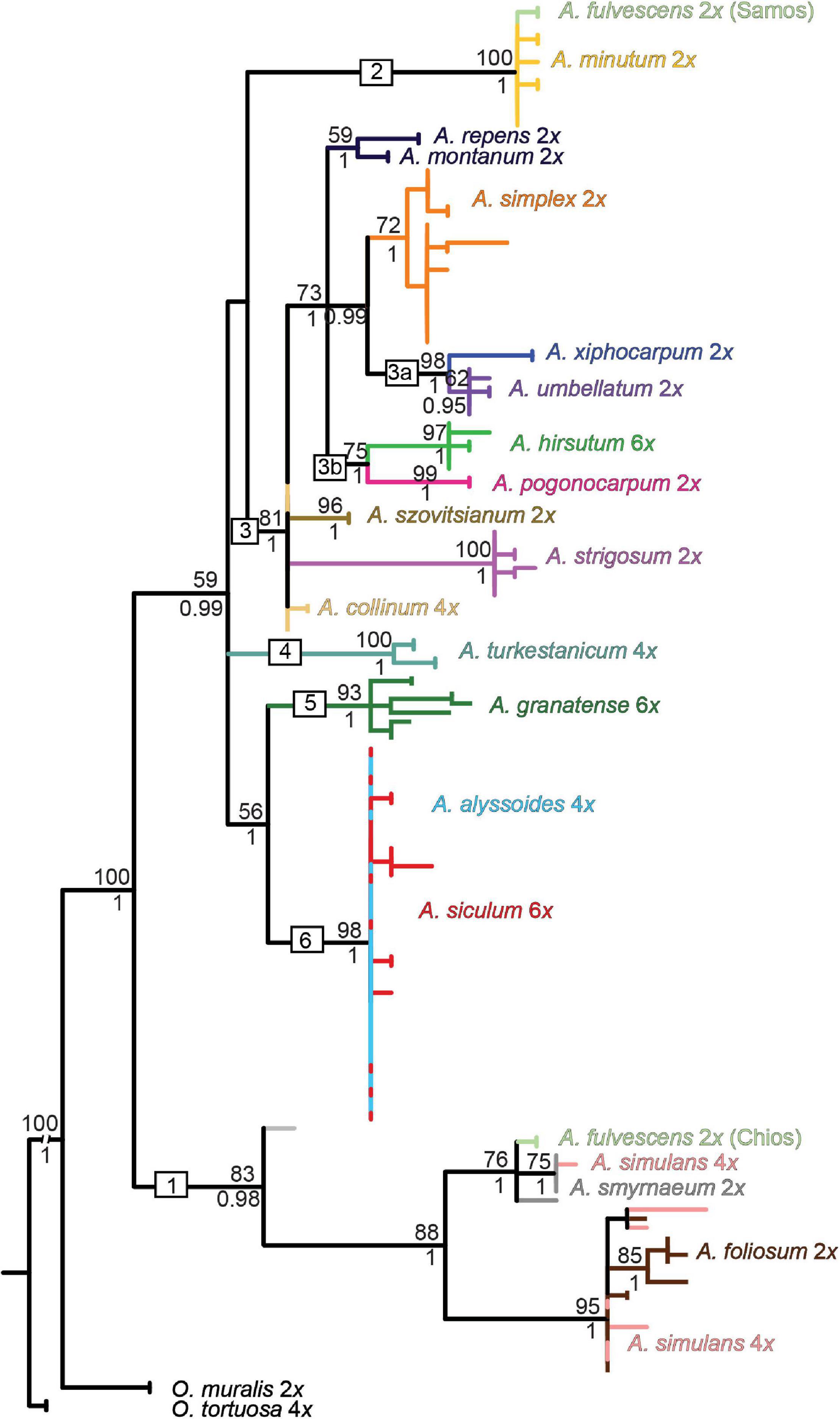
Figure 4. Maximum-likelihood tree based on the rpoB-trnC sequences of cpDNA for the studied Alyssum species. The values above the branches are bootstrap support (BS) ≥ 50%; those below the branches are Bayesian posterior probability (BPP) values ≥ 0.75 taken from the Bayesian majority-rule consensus tree (not shown). BS and BPP are indicated only for major and species-specific clades, whereas they are skipped for the most terminal clades. Branches and clades are colored according to the species assignment, omitting the terminal labels of individual sequences for the sake of readability (see Supplementary Figure 1A for the fully labeled version of the tree). Each species name is followed by its ploidy level. The geographic origins of the two populations of diploid A. fulvescens that exhibited genetic divergence in each of the three DNA regions are indicated (Samos and Chios islands). The numbers 1-6 in boxes denote the clades, as described in the text.
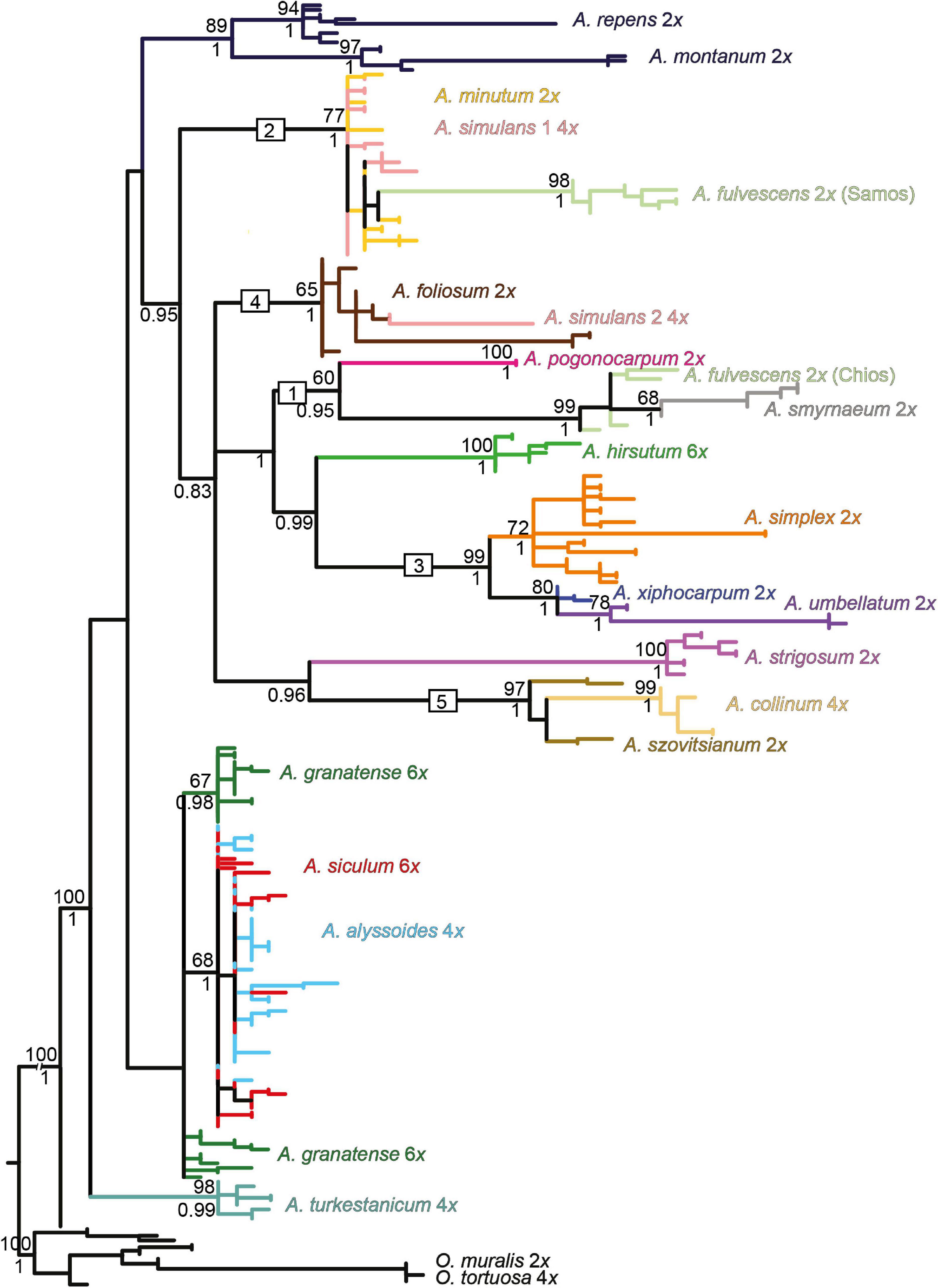
Figure 5. Maximum-likelihood tree based on the ITS sequences of nrDNA for the studied Alyssum species. For the tree description, see the legend of Figure 4. Divergent ITS sequences were observed in the tetraploid A. simulans, which are placed in distinct clades (nr. 2 and 4), marked here as A. simulans 1 and A. simulans 2. The fully labeled version of the tree is shown in Supplementary Figure 2A.
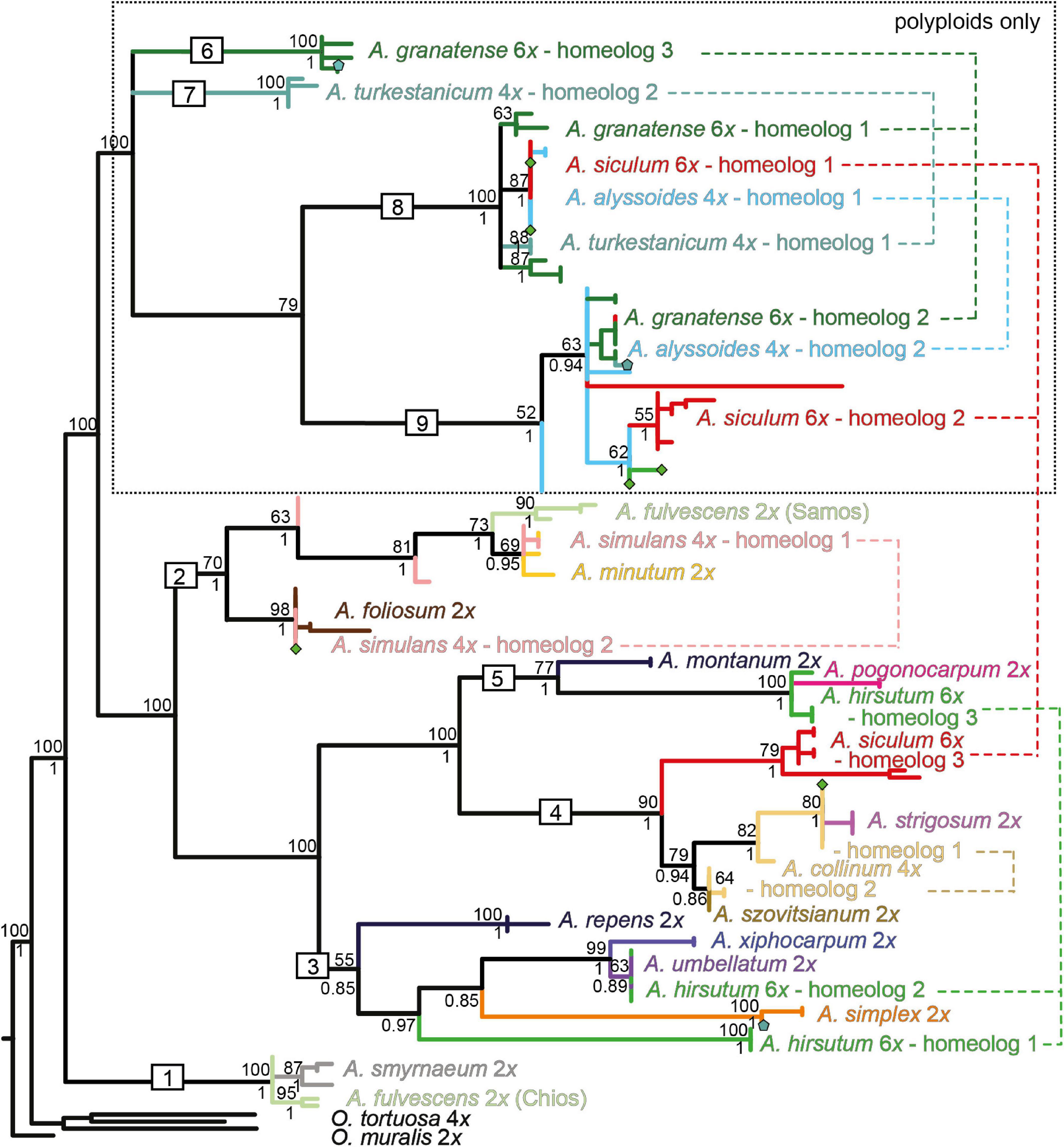
Figure 6. Maximum-likelihood tree based on the DET1 sequence data for the studied Alyssum species. For the tree description, see the legend of Figure 4. The different homeologs identified in the polyploid species are numbered and connected by dashed lines. Symbols (pentagons and diamonds) show the alleles that deviated from the observed homeolog variation in A. turkestanicum and A. hirsutum, respectively. The fully labeled version of the tree is shown in Supplementary Figure 3A.
The ML tree based on the chloroplast rpoB-trnC data (Figure 4) showed a relatively well-resolved structure, in which most of the species harbored monophyletic sequences (but see A. fulvescens), and several clades could be highlighted. One clade (denoted as clade 1) was composed of four Aegean (sub-)endemics, the diploids A. foliosum, A. fulvescens from Chios island (594PEO population) and A. smyrnaeum, and the tetraploid A. simulans. In contrast, A. fulvescens from Samos island (595KEK) was placed in another clade together with the more widespread A. minutum (clade 2). A. xiphocarpum endemic to Lesvos formed a clade with the more widespread Balkan to western Asian A. umbellatum (clade 3a). As for the polyploids, the widespread hexaploid A. hirsutum was sister to the Aegean diploid subendemic A. pogonocarpum (clade 3b). The other four polyploids, A. turkestanicum, A. granatense, A. alyssoides, and A. siculum (clades 4–6) were clearly differentiated from all here analyzed diploids. While the first two polyploids formed two distinct clades (clades 4 and 5), the last two polyploids appeared completely intermingled (clade 6).
The ML tree based on the ITS data displayed poorly supported backbone relationships (Figure 5), therefore, here we focus only on some well supported patterns. Multiple ITS copy variants obtained from several individuals by cloning were phylogenetically close even in polyploids, placed within the same clades (with a single exception of tetraploid A. simulans, see below). Two Aegean diploid subendemics, A. fulvescens from Chios island and A. smyrnaeum, formed a well-supported clade in a sister position to another Aegean subendemic, A. pogonocarpum (clade 1). A. fulvescens from Samos island, in congruence with cpDNA data, was placed in a different clade together with A. minutum (clade 2). A. xiphocarpum endemic to Lesvos together with A. umbellatum were resolved in a clade with the more widespread A. simplex (clade 3). As for the polyploids, clearly polyphyletic ITS sequences were observed only in tetraploid A. simulans, placed intermingled either in the clade with A. minutum (clade 2) or in the clade with A. foliosum (clade 4), the latter position being in congruence with the rpoB-trnC data. The tetraploid Alyssum collinum, so far reported from southwestern Europe and northern Africa, was placed with much support within the clade of Asian A. szovitsianum (clade 5). Four polyploids, A. turkestanicum, A. granatense, A. alyssoides, and A. siculum, similarly as in the rpoB-trnC data, appeared differentiated from all here analyzed diploids, but only A. turkestanicum formed a separate well-supported clade. The NN network (Supplementary Figure 2B) displayed a structure that supported the topology of the ML tree, but, in addition, indicated conflicting, reticulated patterns for two polyploids: the tetraploid A. simulans against the clades 2 and 4; and the hexaploid A. hirsutum against the clades 1 and 3.
The ML tree based on the DET1 data (Figure 6) showed low resolution at the backbone (in congruence with the NN network structure, see Supplementary Figure 3B), but more terminal clades and the relationships among them generally received high support. The diploid species were placed in five distinct clades (clades 1–5), in agreement with the splits resolved in the NN network. In congruence with the ITS data, two Aegean subendemics, A. fulvescens from Chios island and A. smyrnaeum, formed one clade (clade 1), whereas A. fulvescens from Samos island was sister to A. minutum (within clade 2). A. xiphocarpum endemic to Lesvos together with A. umbellatum were resolved in a clade with the more widespread A. simplex (clade 3). Finally, the remaining three annual diploids, A. pogonocarpum, A. strigosum, and A. szovitsianum, were placed within the clades 4 and 5. The sequences obtained from the polyploids were scattered across the tree, placed within four of the five above-mentioned clades of the diploids, plus in four additional clades (clades 6–9) comprising only polyploids. Within each polyploid, we revealed divergent alleles, which were placed in separate clades of the trees, indicating the presence of two or three different homeologs, i.e., gene copies derived from the parental species (Figure 6). In four polyploids, A. alyssoides, A. granatense, A. siculum, and A. turkestanicum, we observed two to three divergent homeologs, which were placed in the clades comprising exclusively polyploids, i.e., not matching any of the analyzed diploids.
Coalescent-Based Species Trees
The species tree inferred from all three DNA markers of diploids is depicted in Figure 7A. It includes one larger clade (BPP = 0.95) comprising two Aegean (sub)endemics (A. pogonocarpum and A. xiphocarpum) and two species with wider ranges (A. simplex and A. umbellatum), together with the nested A. montanum and A. repens perennials. The rest of the tree displayed low resolution at higher-level relationships, and only smaller clades can be highlighted as follows. The widespread southern Europe-southwestern Asian A. strigosum formed a clade with southwestern Asian A. szovitsianum (BPP = 1). In agreement with the individual gene trees, A. fulvescens from Samos was resolved in the sister position to widespread A. minutum (BPP = 1), whereas A. fulvescens from Chios was sister to the Aegean subendemic A. smyrnaeum (BPP = 1). Another Aegean subendemic, A. foliosum, clustered with the latter two species, albeit with somewhat lower support (BPP = 0.88). The species tree based on cpDNA and ITS alignments and including both diploids and polyploids (Figure 7B) showed virtually the same topology as the former species tree (in terms of the relationships among the diploids), but with lower BPP for some clades. Two polyploids, A. hirsutum and A. collinum, were placed within the clades of diploids. In contrast, the polyploids A. granatense, A. siculum, and A. alyssoides formed a distinct clade (BPP = 0.93), and together with another polyploid A. turkestanicum, were separated from the diploids.
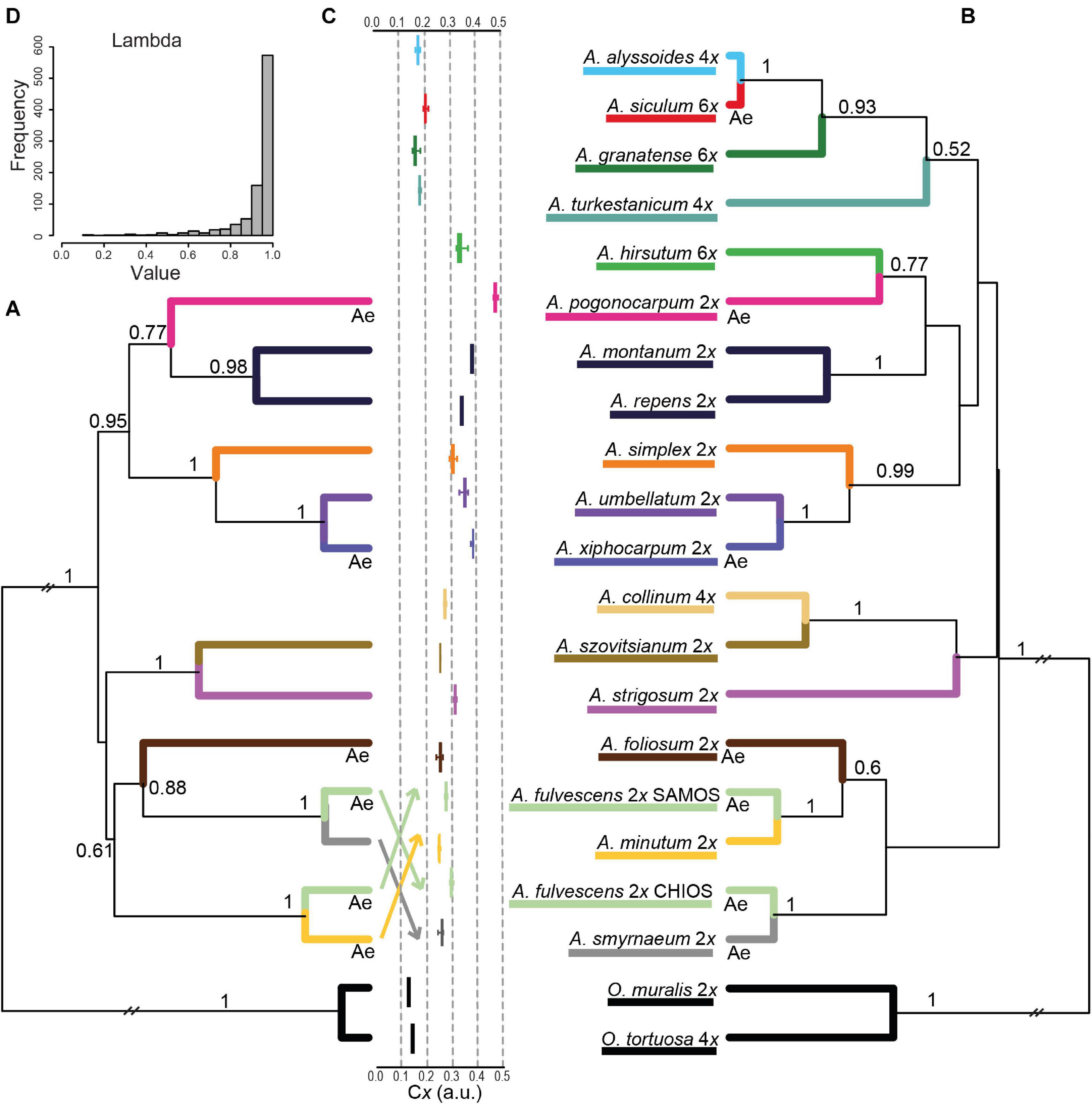
Figure 7. Results of the species tree inference in the studied Alyssum species. The maximum clade credibility species trees, which were obtained from the coalescent analysis in BEAST are shown, based on (A) cpDNA, ITS, and DET1 datasets for diploids, and (B) cpDNA and ITS datasets for both diploids and polyploids (excluding the tetraploid A. simulans, see section “Materials and Methods”). Posterior probability values ≥ 0.5 are indicated above the branches. Ploidy levels are shown next to the species names. ‘Ae’ below branches indicates the placements of Aegean subendemics referred to in the text. (C) Boxplots depicting the relative monoploid genome size (Cx) values measured for the studied species. See Table 2 and Supplementary Table 1 for the precise population- and species-level Cx values. (D) Posterior distribution of the tree transformation statistic λ (Pagel, 1999), computed by MCMC analyses in BayesTraits, quantifying the phylogenetic signal in genome size variation.
The boxplots showing the relative monoploid genome size values (Cx) measured in the studied species (Figure 7C) and plotted next to the species tree indicate that the observed genome size variation is phylogenetically structured. The presence of a strong phylogenetic signal in the genome size data was, indeed, supported by the posterior distribution of λ values, which were skewed toward 1 (Figure 7D).
Estimation of Divergence Times Based on ITS Sequence Data
The dated phylogeny based on the ITS sequence data (Supplementary Figure 4) suggested that the most recent common ancestor (MRCA) of the genus Alyssum evolved during the Tortonian Age of the Late Miocene epoch [median age of 9.58 mya, 95% high posterior density (HPD): 7.11–14.09]. The origin and early diversification of the annual lineage examined in this study, including the nested subclade of the perennial A. montanum-A. repens species complex, could be dated back to the Late Miocene (Messinian Age) up to the Early Pliocene (median age of the MRCA: 5.38 mya, 95% HPD: 4.15–7.85). The annuals were split into three clades. The first clade, comprising most of the species, originated and began to diversify as early as in the Early Pliocene (median age of 4.66 mya, 95% HPD: 3.36–6.44), followed by further speciation during the Pliocene and Pleistocene. The MRCA of the two other clades, comprising the remaining four and three species, were inferred to appear later (median ages of 3.51 mya, 95% HPD: 2.59–5.39; and 1.71 mya, 95% HPD: 0.91–3.47, respectively). In summary, when confronted with the clades resolved in the species tree (Figure 7A, see also Table 4), we can derive that the annual species examined in this study have likely originated in the Late Pliocene and the Pleistocene epochs.
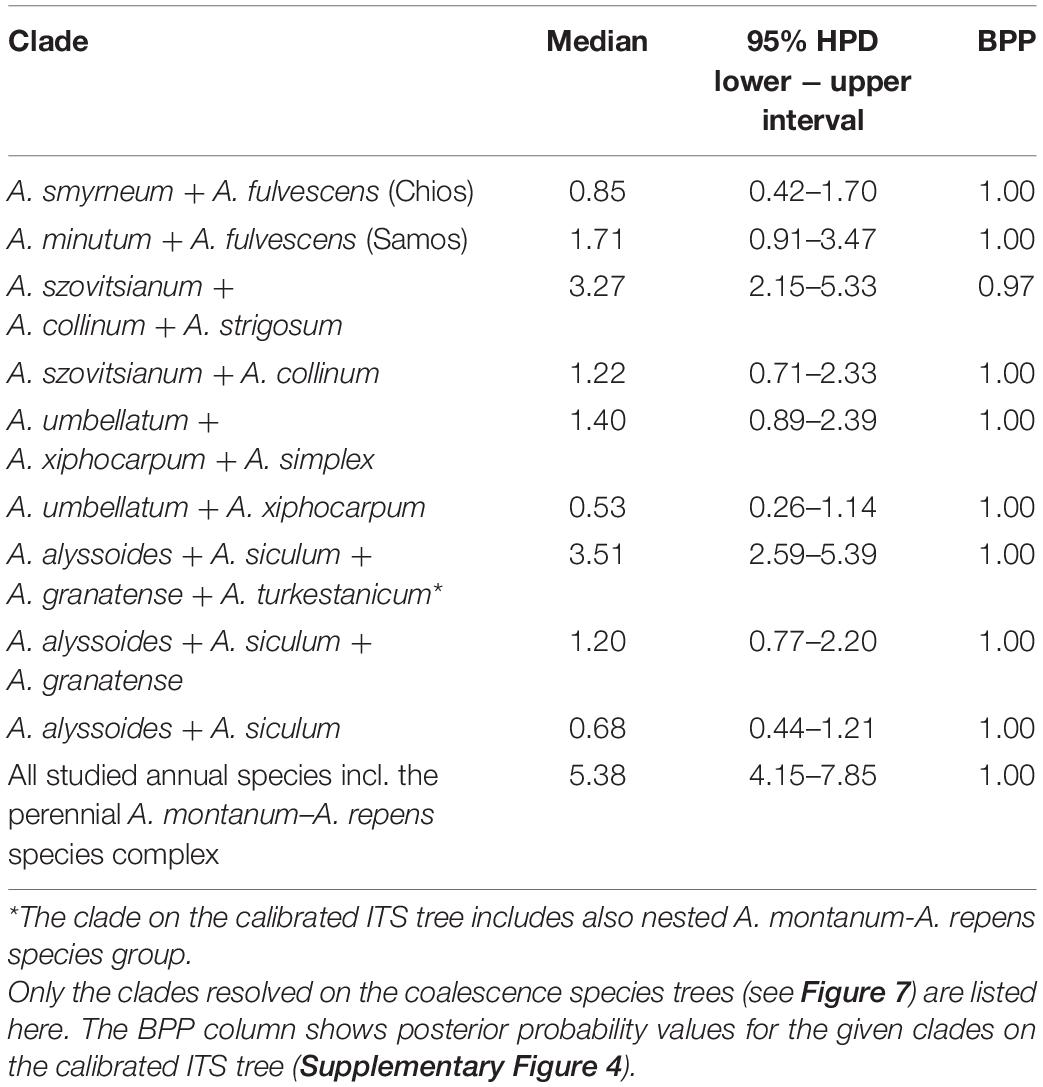
Table 4. Age estimates (median and 95% of the highest posterior density, HPD) in millions of years for the nodes of the given clades of the studied Alyssum species, as inferred from the relaxed molecular-clock analysis, performed in BEAST and based on ITS sequence data (see Supplementary Figure 4 and Supplementary Table 2).
Discussion
Distribution and Diversity Patterns of the Studied Alyssum Annuals: An Evolutionary Hotspot in the Aegean Region
The phylogenetic lineage of the Alyssum species examined in the present study includes 16 species growing in Europe. Their distribution is centered in southern Europe, especially in the eastern Mediterranean and the Aegean archipelago (Jalas et al., 1996; Hartvig, 2002; Strid, 2016). As many as 12 species occur in the Aegean region, seven of which are subendemics that are either restricted to this region or extend only to the adjacent mainland regions of Greece, North Macedonia, western Turkey or Sicily (Dudley, 1965; Hartvig, 2002; Strid, 2016). Our phylogenetic reconstructions revealed that these Aegean subendemics do not form a single monophyletic group but are placed in several distinct clades across the inferred species trees (Figure 7), and include both diploids and polyploids. Along with the divergence time estimates, these results suggest that the Aegean subendemics likely originated at different times and through different modes. Thus, it seems that the Aegean region has repeatedly acted as a significant evolutionary center favoring multiple speciation events in this phylogenetic lineage. Indeed, this area is recognized to be one of the major biodiversity hotspots of the Mediterranean Basin, featuring high species richness and a significant proportion of narrow endemics (Médail and Quézel, 1997; Panitsa et al., 2018). Several factors have been highlighted that are likely to drive plant diversification and distribution patterns in the Aegean area, especially its complex climatic and geological history, high topographical and geological heterogeneity, which contributes to the formation of large habitat diversity, as well as the long-term presence and influence of humans (Sfenthourakis and Triantis, 2017; Panitsa et al., 2018).
In this study, we have inferred that the origin and early diversification of the studied lineage of annual Alyssum species could be dated back to the Late Miocene and Pliocene, between the Messinian salinity crisis (Krijgsman et al., 1999) and the establishment of the Mediterranean climate (Suc, 1984). The onset of the Mediterranean climate was a major climatic change, which likely promoted the further diversification of this lineage, in a similar manner as identified for some other Mediterranean-adapted plant groups (Fiz-Palacios and Valcárcel, 2013; see also Carnicero et al., 2017). Most extant Alyssum species, including those that are distributed throughout the Aegean, originated at the end of the Pliocene or during the Pleistocene. This was the era when the climatic oscillations caused sea-level changes and recurrent land connections and disconnections (Perissoratis and Conispoliatis, 2003). They affected the speciation processes by facilitating species dispersal during sea-level drops in glacial periods, and increasing the degree of range fragmentation and population isolation during interglacial and postglacial periods (Nieto Feliner, 2014; Simaiakis et al., 2017; Panitsa et al., 2018). The Alyssum species do not bear specific adaptations to facilitate the long-distance dispersal of seeds; instead, field observations suggest that seed dispersal occurs primarily through gravity and zoochory (sheep and goats), and thus also through human activities, mainly cattle farming and grazing. Therefore, geographic distance and barriers may efficiently reduce gene flow, which is manifested by the existence of a few narrow endemics. Interestingly, most species that occur in the Aegean region are largely sympatric (Table 1), with several species recorded within one island (e.g., four species in Lesvos and Samos, and six in Crete), with as many as five species recorded from a single locality in Crete (Supplementary Table 1). Thus, a simple vicariance scenario associated with allopatric differentiation that has generally been favored in the fragmented Mediterranean landscape (Thompson, 2020) and has been observed for a number of Mediterranean plant groups (e.g., Bittkau and Comes, 2009; Crowl et al., 2015), cannot explain the Alyssum species diversity observed in the Aegean region. Considering the common co-occurrence of multiple species within a single site, ecological speciation (Coyne and Orr, 2004) does not appear to play a determinant role either, despite the wide range of occupied habitats, the substrate-specificity observed in some cases (Hartvig, 2002; Strid, 2016, see also Table 1), and the large elevational gradient. However, detailed ecological niche characterization and modeling to compare the potential and realized niche spaces of the species (see, e.g., López-Jurado et al., 2019; Castro et al., 2020) will be needed in future to address this issue in detail. We assume that the restricted distribution of some narrow endemics is more likely due to low-dispersal ability rather than environmental constraints [as inferred e.g., by Crowl et al. (2015)].
The distribution ranges can change over time, either naturally or due to human factor, which both can be expected in the biogeographically and geologically complex Aegean area with the long history of human presence (Panitsa et al., 2018; Thompson, 2020). When inspecting the phylogenetic relationships in comparison with the distributional patterns, a specific pattern emerged in some cases. Several diploid species and their closest relatives (also diploids) occur parapatrically or sympatrically, with one species characterized by a restricted distribution (being a narrow endemic), whereas the other occupying a much broader range. This finding was observed for the species pair A. umbellatum and A. xiphocarpum, as well as for species from the clade comprising A. fulvescens, A. smyrnaeum, A. minutum, and A. foliosum (Figure 7). This pattern resembles a specific type of allopatric speciation, which is referred to as founder-event speciation (Templeton, 2008), and was recently inferred in the Mediterranean genus Cymbalaria (Carnicero et al., 2017). This mode of speciation involves the establishment of a new (peripheral) population from a larger ancestral population, and its genetic divergence due to genetic drift, as well as divergent selective response (Naciri and Linder, 2020). We propose that such events may have been facilitated by recurrent range shifts, expansion or fragmentation, in response to sea-level changes in the Pleistocene (Nieto Feliner, 2014). In addition, since the studied Alyssum annuals commonly grow on disturbed sites and in pastures, human-driven colonization due to cattle transport may also have shaped and changed the distribution patterns. Thus, even though these sister species now grow sympatrically, they may have evolved through allopatric pathways at a finer spatial scale. The development of some premating isolating barriers (Coyne and Orr, 2004) have also likely played an important role in the speciation process, as phenological shifts have been observed among some of these co-occurring, closely related species (personal observations, see also below).
A few recent biogeographic studies that have focused on the Aegean area have investigated the diversification, speciation, and colonization processes of some plant groups in detail (Crowl et al., 2015; Jaros et al., 2018; Carnicero et al., 2021). These studies have suggested primarily allopatric, non-adaptive lineage diversification and speciation, driven by geographic isolation and random drift. These processes have been associated with the formation of the mid-Aegean Trench in the Middle Miocene (12–9 mya), when the ancient Aegean landmass became fragmented (Sfenthourakis and Triantis, 2017), and the Pleistocene-era sea-level oscillations, depending on the age of the studied species (Bittkau and Comes, 2005, 2009; Crowl et al., 2015; Jaros et al., 2018). In other cases, sympatric ecological speciation and founder-event speciation have also been inferred (Carnicero et al., 2017, 2021). Thus, the few detailed studies that have been performed so far, along with the here presented scenarios, highlight the complexity of speciation processes in the Aegean region.
Multiple Allopolyploid Speciation Events Increased the Species Diversity Across the Whole Distribution Range
We revealed the presence of diploid, tetraploid, and hexaploid levels among the studied Alyssum annuals, in accordance with earlier reports (summarized in AlyBase, see Španiel et al., 2015). We did not observe ploidy level variation within populations or within species. The diploids and tetraploids that were traditionally classified within A. simplex were recently split into two separate species, A. simplex (diploids) and A. collinum (tetraploids), which was substantiated by preliminary molecular data, by differences in the monoploid genome size, and some morphological traits (Cetlová et al., 2019). Indeed, the here presented phylogenetic analyses placed these two species in two distinct clades. In addition, the parentage of the tetraploid A. collinum did not appear to involve the diploid A. simplex (see also below). Some ploidy level variation was previously reported within a few species (see Table 2); however, these observations remain to be confirmed by future studies. In this study, all examined annual polyploids (seven species in total) appeared to evolve through allopolyploidy. Interestingly, only three of these polyploids occur in the Aegean region, thus, it seems that although the polyploidization played a certain evolutionary role in this area as well, it was not the prevalent speciation mechanism there.
The allopolyploid origins of the studied annuals were derived from the cpDNA–ITS incongruence, from ITS intragenomic polymorphisms, and from single-copy (DET1) homeolog variation. The combination of multiple markers demonstrating different patterns of inheritance and molecular evolution has been shown to be the most efficient approach for reconstructing reticulate evolution (Naciri and Linder, 2015; e.g., Záveská et al., 2016; Díaz-Pérez et al., 2018; Mandák et al., 2018). In this study, we combined the nrDNA region that is prone to sequence homogenization by concerted evolution and may show complex patterns in polyploids (Álvarez and Wendel, 2003; Nieto Feliner and Rosselló, 2012), cpDNA with uniparental inheritance, and a biparentally inherited single-copy nuclear gene with the potential to retain parental subgenomes (Small et al., 2004; Nieto Feliner and Rosselló, 2012). In some cases, we were able to identify the most likely parental species or at least the phylogenetic lineages involved in the polyploid origins, which are discussed below and summarized in Figure 8.
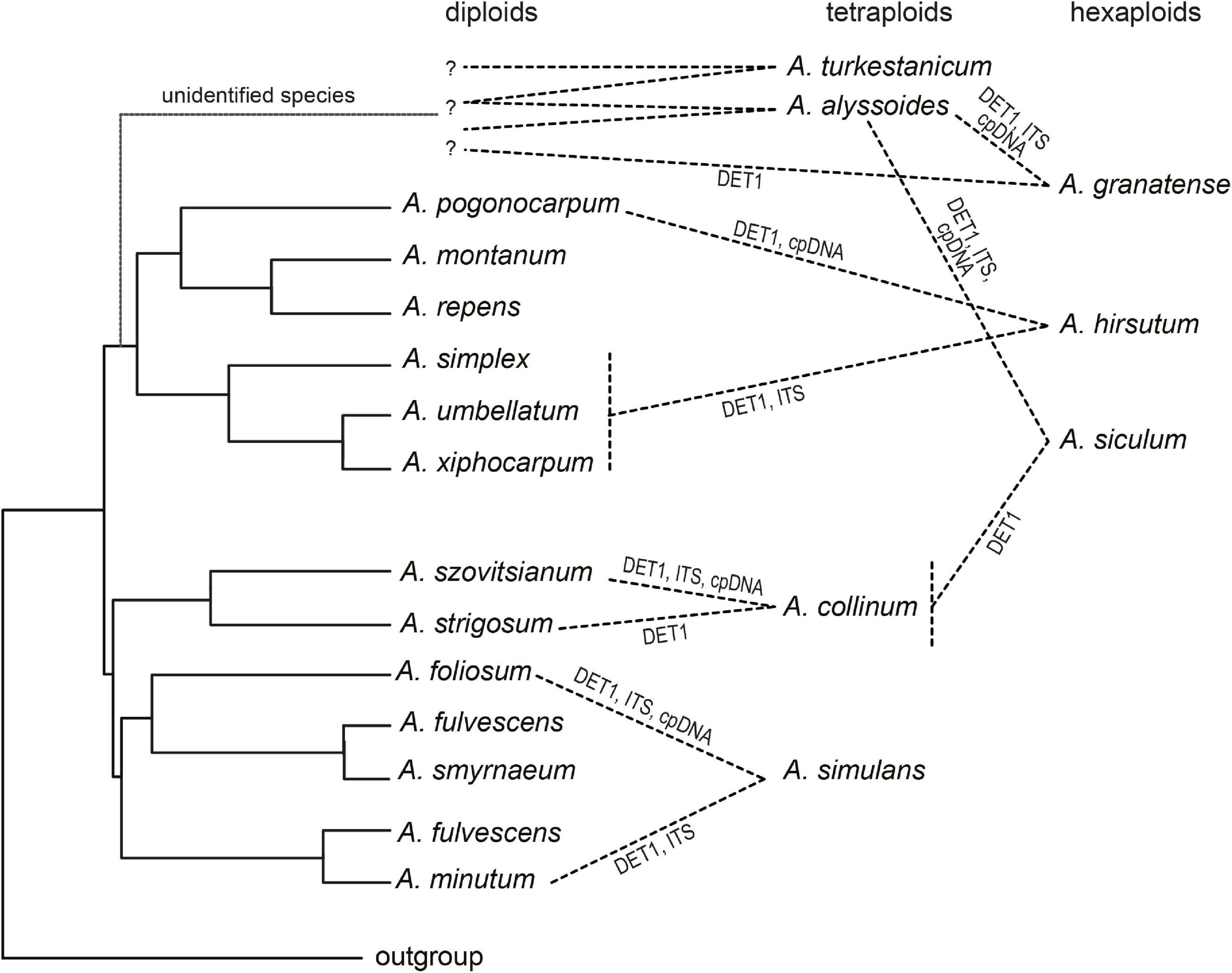
Figure 8. Scheme summarizing the allopolyploidization events in the studied Alyssum species, as inferred from DNA sequence data. Diploid species are depicted in the coalescent species tree, as shown in Figure 7A, based on all three markers used, whereas polyploids are connected to their inferred diploid or lower-ploidy progenitors. Question marks indicate unidentified diploid progenitors (species currently not occurring in Europe, possibly of Asian origin or extinct). See the text for further details.
The tetraploid A. simulans, which is distributed in southeastern mainland Greece, Peloponnese, Evvia, and Crete, was the only species in which divergent, non-homogenized ITS variants were revealed, clustering with two distinct diploids, Aegean subendemic A. foliosum and widespread Mediterranean A. minutum, in concert with the identified DET1 homeologs. The former diploid was revealed as the probable maternal parent, based on the cpDNA patterns. All three species occur in sympatry, and these distribution patterns, non-homogenized nrDNA, and almost identical genome size (monoploid Cx values) suggested a recent, likely Late Pleistocene or even postglacial allopolyploid origin. Alyssum simulans was described only recently, and its hybrid origin was already hypothesized based on morphological intermediacy (Hartvig, 2002). It remains uncertain, however, whether A. simulans originated multiple times, i.e., independently in Crete and in mainland Greece, or just at a single site and expanded to its current range after its origin.
The hexaploid A. hirsutum is widely distributed across eastern Europe, especially in the Black Sea region, extending to the Caucasus region (Grossheim, 1949; Jalas et al., 1996), but does not grow in the Aegean area. One of its parental species was suggested to originate from the clade comprising A. xiphocarpum (which is endemic to Lesvos), A. umbellatum (Aegean subendemic), and A. simplex (throughout the entire Mediterranean, reaching the Caucasus). Thus, either A. simplex (being more similar to A. hirsutum morphologically than the other two species, which have short umbellate fruiting racemes) or a common ancestor of that clade appear to be the most likely parental species. All three species have similar monoploid genome size values, which are also close to A. hirsutum. The identity of the other parental species remains puzzling because both cpDNA and DET1 point to A. pogonocarpum, a species restricted to Rhodos and southwestern Turkey (Kleinsteuber et al., 2016), which might be supported by the morphological resemblance between these species (very long hairs on fruits, Carlström, 1984), but occurs allopatrically and also has a markedly larger genome size (Figure 7C and Table 2). Nevertheless, also an unsampled, related species from the Irano-Anatolian or Caucasus areas could have been involved in the origin of hexaploid A. hirsutum (see also below).
The tetraploid A. collinum contained ITS and cpDNA sequences that both clustered together with the diploid A. szovitsianum. In the DET1 locus, two slightly divergent groups of alleles were identified, clustering with the sister species A. szovitsianum (southwestern Asia) and A. strigosum (southern Europe-southwestern Asia). This pattern favors an allopolyploid origin involving progenitors close to these two relatives. Also the monoploid genome size of A. collinum is clearly intermediate between these two species (Figure 7C and Table 2). The distribution of A. collinum remains poorly understood because it has only recently been taxonomically segregated from the widespread Mediterranean diploid A. simplex (Cetlová et al., 2019). The occurrence of A. collinum is so far confirmed only from the western Mediterranean (Cetlová et al., 2019, and the present samples), and no records exist from eastern Europe. Some tetraploid individuals (under the name A. simplex) were reported from Iran and Tajikistan (see Table 2), but it is unclear if they could be conspecific with A. collinum or represent autotetraploids of A. simplex, or even another species. Since the present sequence data suggest that the parental species of A. collinum came from the eastern regions, it is evident that the taxonomic identity of the Asian tetraploids should be resolved to delimit the distribution range of A. collinum completely, and to understand its origin and evolution.
The remaining four polyploids (the tetraploids A. alyssoides and A. turkestanicum; the hexaploids A. granatense and A. siculum) did not show closer affinities to any of the European diploids; however, their divergent DET1 sequences (Figure 6), which were placed in clearly distinct clades, strongly favored independent allopolyploid origins. The most widespread tetraploid A. alyssoides displayed two divergent DET1 copies, which were both present in the hexaploids A. siculum and A. granatense, and one in the tetraploid A. turkestanicum. Thus, A. alyssoides was most likely involved in the origin of both hexaploids, i.e., the western Mediterranean A. granatense (which is in congruence with their similar morphology, Küpfer and Nieto Feliner, 1993) and the eastern Mediterranean A. siculum. The third DET1 homeolog of A. granatense, in agreement with the distinct clade formed by its cpDNA sequences, pointed to another progenitor, which, however, remained unidentified. The hexaploid A. siculum, based on previous morphological and molecular data, was suggested to be either an allopolyploid, derived from A. alyssoides and A. simplex, or an autopolyploid of A. alyssoides (Persson, 1971; Rešetnik et al., 2013). In this study, we found that A. siculum, which is distributed in Crete, Peloponnese, mainland Greece, and Sicily, indeed, displayed lot of shared variation in all three markers with A. alyssoides, which may suggest a very recent polyploid origin. In contrast, we did not find any evidence that A. simplex could be the second parent. Instead, one of the DET1 homeologs revealed in A. siculum was sister to the clade containing A. collinum, A. szovitsianum, and A. strigosum, which suggests that one of these species may have been the paternal parent of the hexaploid A. siculum. Based on the present records, only A. strigosum occurs sympatrically (in mainland Greece as well as Crete; Hartvig, 2002; Strid, 2016) with the hexaploid; however, the highest morphological resemblance points to the southwestern Asian A. szovitsianum (personal observation). We did not observe any genetic differentiation between the accessions of A. siculum from mainland Greece, Crete, and Sicily that could suggest the independent, polytopic origins of this hexaploid. However, the present analyses were based on only a few loci and may not be sufficient to detect multiple polyploid origins. Where this polyploid originated and how it reached its current disjunct distribution also remains an open question. The sea barriers between mainland Greece, Crete, and Sicily have persisted since the late Miocene; therefore, either a natural long-distance dispersal event occurred or introduction by man was possible, as it commonly grows on disturbed sites and pastures. Finally, the tetraploid A. turkestanicum, which is distributed from the Balkan Peninsula to eastern Europe, Black Sea Region, Caucasus, Near East, central Asia, and Siberia (Rechinger, 1968 as Alyssum desertorum; Rybinskaya, 1994; Jalas et al., 1996; Zhou et al., 2001; German, 2003; Marhold, 2011), formed a distinct clade according to both the ITS and cpDNA-based trees, with no obvious affinities to any European diploids. One DET1 homeolog was in a clade with A. alyssoides, which suggests several scenarios: A. alyssoides may have been involved in its origin, both species may share a common diploid ancestor of extra-European origin, or homeolog sharing may also reflect local hybridization events. Interestingly, these four polyploids (A. alyssoides, A. turkestanicum, A. granatense, and A. siculum) have the smallest monoploid genome sizes from all here analyzed species (Figure 7C), which may suggest that their unidentified diploid progenitors also have small genomes, and/or that genome-downsizing occurred as a result of diploidization, a process commonly observed in polyploids (Dodsworth et al., 2016). Genome-downsizing, however, was not observed in the other three polyploids (A. collinum, A. simulans, and A. hirsutum); their genome sizes were rather correlated with the phylogenetic positions and, thus, reflected the genome size variation among their diploid ancestors (Table 2 and Figure 7C).
The significance of recent polyploidization events (neopolyploidy) for the diversification and speciation processes has been inferred for a number of Mediterranean genera, which have been especially linked to climate-induced range shifts and secondary contacts during the Pleistocene (e.g., Cecchi et al., 2013; Frajman et al., 2016; Wagner et al., 2019). Except for two polyploids, A. siculum and A. simulans, which are both narrowly distributed and restricted to certain parts of the Mediterranean regions (SE Greece, Crete, plus Sicily, in the case of A. siculum) and supposedly of very recent origins, the other studied polyploids have broader distributions, some also occurring in extra-Mediterranean areas (e.g., the Caucasus, central Asia). This finding may support the hypothesis that allopolyploids, harboring greater genetic variation, complex and dynamic genomes, could become successful colonizers that exhibit larger distribution areas and different, often wider, ecological niches than diploids (Soltis et al., 2014; see e.g., Arrigo et al., 2016; Cornille et al., 2016). Still, it may be premature to favor or reject this hypothesis with the present data, as in the case of these widespread Alyssum polyploids, either at least one of its diploid progenitors also shows widespread occurrence (cases of A. hirsutum and A. granatense), or the diploid progenitors (and their ecological and chorological characteristics) remain unknown (cases of A. turkestanicum and A. alyssoides).
Phylogenetic Patterns, Cryptic Diversity, and Further Perspectives
The phylogenetic trees that were generated based on the three genetic markers employed in this study differed in their resolution. The best resolved hierarchical structure was obtained from the cpDNA tree, in which most species were monophyletic, and the higher-level relationships received at least moderate support. Few species appeared paraphyletic, which most likely reflects lack of resolution or shared ancestral variation, as this pattern was only observed in some closely related diploids (e.g., A. minutum and A. fulvescens from Samos; A. smyrnaeum and A. fulvescens from Chios) or indicated the maternal origins of some polyploids (e.g., A. siculum and A. alyssoides; A. simulans and A. foliosum). In contrast, both the ITS- and DET1-based trees displayed a low resolution in the tree backbone, whereas the more terminal clades of pairs or groups of closely related species were well-supported and largely congruent among all three markers. The low resolution of the nuclear trees, despite the sufficient number of variable sites, most likely mirrors the more complicated evolution of biparentally inherited markers, in which past interspecific gene flow, during and after speciation, polyploidization, recombination, and, in the case of ITS also processes of sequence homogenization, may have obscured the divergence history, and resulted in conflicting, reticulate patterns (Nieto Feliner and Rosselló, 2012; Naciri and Linder, 2015). In agreement with the individual gene trees, the initial diversification patterns were poorly resolved in the species tree, although the terminal clades of the closest relatives received high support. The close relatedness of some species, as was revealed in this study, was already suggested based on their morphological similarity (e.g., A. fulvescens and A. smyrnaeum; A. xiphocarpum and A. umbellatum; Hartvig, 2002). An intriguing case remains the eastern Aegean endemic A. fulvescens (distributed in Samos, Chios, and two smaller islands of Patmos and Kalymnos, Strid, 2016). The populations from the Chios and Samos islands that were analyzed in this study were genetically differentiated, despite no obvious morphological distinction. Consistently for each genetic marker, the samples from Chios were genetically closest to A. smyrnaeum (the more widespread Aegean subendemic, which grows in both Samos and Chios islands, Strid, 2016 and present records), whereas those from Samos clustered with A. minutum (a widespread Mediterranean species, including the Aegean region, but not recorded from Samos and Chios). This pattern cannot be explained by a recent hybridization or introgression event with either of these species, considering also the fact that A. fulvescens from Samos grows at the same site, together with A. smyrnaeum (pop. 595KEK), but they apparently do not hybridize nowadays and they display some phenological shifts (our field observations). Thus, we can only speculate whether the present patterns are due to past hybridization events that caused genetic divergence within A. fulvescens or whether they indicate the existence of two independent and separately evolving entities with low levels of morphological differentiation, also known as cryptic species (Fišer et al., 2018; see, e.g., Crowl et al., 2015, 2017; Padilla-García et al., 2018). Considerable among-population variation was observed in genome size values in this species, although it does not correlate precisely with genetic patterns. Still, these patterns may indicate some hidden evolutionary processes in this species. In the future, more detailed investigations of morphological and genetic variation in A. fulvescens should be undertaken to solve this issue.
Genome size variation observed among the analyzed species was congruent with the phylogenetic patterns (Figure 7). The monoploid relative genome size of the annual species studied here ranged from 0.161 in A. granatense to 0.474 in A. pogonocarpum (Table 2). Perennial species of the A. montanum–A. repens complex, according to previously published studies, showed a much narrower range of values, between 0.296 ± 0.004 (in some populations of A. repens, Melichárková et al., 2019) and 0.389 ± 0.006 (in Alyssum cacuminum, Španiel et al., 2018, 2019). These values are within the range of those of the here studied annuals, in agreement with the phylogenetic position of the perennial A. montanum–A. repens group, nested within the lineage of annuals, suggesting that these perennials could have evolved from the annuals. It has been proposed and also documented in several cases that annuals tend to have smaller genomes compared to perennials (e.g., Frajman et al., 2015; Zahradníček et al., 2018), albeit this correlation may be also due to the common association of annual life history with higher rates of selfing (Albach and Greilhuber, 2004). Here, we did not support this tendency, in congruence with the findings by Cacho et al. (2021), as genome size variation displayed significant phylogenetic correlation, irrespective of the annual or perennial life history. It has been shown that most of genome size variation in plants is attributable to the differential evolution of repetitive DNA components, which may represent a highly dynamic process that is correlated with species relatedness and phylogeny (McCann et al., 2020).
One limitation of the present study, however, is that we did not include species and populations from the extra-European range (except of A. szovitsianum). About 12 species from northern Africa and southwestern to central Asia not sampled here (Dudley, 1964a, 1965), will be needed in future to resolve the evolutionary and biogeographic history of these Alyssum annuals completely. So far, up to five extra-European species were included in previous tribus- or genus-wide phylogenetic studies (Li et al., 2015; Huang et al., 2020), which supported their placement within the here studied clade of annuals, and appeared close to some more widespread Eurasian species (A. minutum, A. simplex, and A. strigosum), similarly as resolved here for A. szovitsianum from Iran. Still, the existence of an independent Asian lineage cannot be ruled out either. Taxonomic identification of some Asian species, however, may be tentative, and thorough taxonomic revision of Asian species is necessarily needed as first. The complete Eurasian sampling in future studies may help to identify the missing parental species for some polyploids (see above), reinforce or weaken the phylogenetic signal in genome size, and will allow to gain deeper insights into the colonization and speciation processes in this group.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW070213–MW070364; https://www.ncbi.nlm.nih.gov/genbank/, MW022541–MW022804; and https://www.ncbi.nlm.nih.gov/genbank/, MW070365–MW070517.
Author Contributions
SŠ and JZ-L conceived and designed the study and wrote the manuscript. SŠ and VC sampled plant material. All authors generated data and performed data analyses. All authors have read, revised, and approved the final manuscript.
Funding
This study was financially supported by the Slovak Research and Development Agency (APVV; Grant No. APVV-17-0616), Czech Science Foundation (GAČR; Grant No. 19-06632S), and Grant Agency VEGA, Bratislava, Slovakia (Grant No. 2/0133/17).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Marek Šlenker, Pavol Mered’a Jr., Dominik Roman Letz (all three from Bratislava), and Gianniantonio Domina (Palermo) for their assistance during field trips. We also thank to Arne Strid (Ørbæk), Ioannis Bazos (Athens), and Michael Hassler (Bruchsal) for advice on localities.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.627909/full#supplementary-material
References
Akaike, H. (1974). A new look at the statistical model identification. IEEE Trans. Automat. Contr. 19, 716–723. doi: 10.1109/TAC.1974.1100705
Albach, D. C., and Greilhuber, J. (2004). Genome size variation and evolution in Veronica. Ann. Bot. 94, 897–911. doi: 10.1093/aob/mch219
Al-Shehbaz, I. A. (2010). “Alyssum linnaeus,” in Flora of North America, North of Mexico, Magnoliophyta: Salicaceae to Brassicaceae, Vol. 7, ed. Flora of North America Editorial Committee (New York, NY: Oxford University Press, Inc.), 247–251.
Al-Shehbaz, I. A. (2012). A generic and tribal synopsis of the Brassicaceae (Cruciferae). Taxon 61, 931–954. doi: 10.1002/tax.615002
Álvarez, I., and Wendel, J. (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogen. Evol. 29, 417–434. doi: 10.1016/S1055-7903(03)00208-2
Arrigo, N., de La Harpe, M., Litsios, G., Zozomová-Lihová, J., Španiel, S., Marhold, K., et al. (2016). Is hybridization driving the evolution of climatic niche in Alyssum montanum? Am. J. Bot. 103, 1348–1357. doi: 10.3732/ajb.1500368
Ball, P. W., and Dudley, T. R. (1993). “Alyssum L,” in Flora Europaea, 2nd Edn. Vol. 1, eds T. G. Tutin, N. A. Burges, A. O. Chater, J. R. Edmondson, V. H. Heywood, D. M. Moore, et al. (Cambridge: Cambridge University Press), 359–369.
Bittkau, C., and Comes, H. P. (2005). Evolutionary processes in a continental island system: molecular phylogeography of the Aegean Nigella arvensis alliance (Ranunculaceae) inferred from chloroplast DNA. Mol. Ecol. 14, 4065–4083. doi: 10.1111/j.1365-294X.2005.02725.x
Bittkau, C., and Comes, H. P. (2009). Molecular inference of a late Pleistocene diversification shift in Nigella s. lat. (Ranunculaceae) resulting from increased speciation in the Aegean archipelago. J. Biogeogr. 36, 1346–1360. doi: 10.1111/j.1365-2699.2008.02003.x
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Cacho, N. I., McIntyre, P. J., Kliebenstein, D. J., and Strauss, S. Y. (2021). Genome size evolution is associated with climate seasonality and glucosinolates, but not life history, soil nutrients or range size, across a clade of mustards. Ann. Bot. (in press). doi: 10.1093/aob/mcab028
Carlström, A. (1984). New species of Alyssum, Consolida, Origanum and Umbilicus from the SE Aegean sea. Willdenowia 14, 15–26.
Carnicero, P., Garcia-Jacas, N., Sáez, L., Constantinidis, T., and Galbany-Casals, M. (2021). Disentangling relationships among eastern Mediterranean Cymbalaria including description of a novel species from the southern Peloponnese (Greece). Plant Syst. Evol. 307:13. doi: 10.1007/s00606-020-01730-3
Carnicero, P., Sáez, L., Garcia-Jacas, N., and Galbany-Casals, M. (2017). Different speciation types meet in a Mediterranean genus: the biogeographic history of Cymbalaria (Plantaginaceae). Taxon 66, 393–407. doi: 10.12705/662.7
Castro, M., Loureiro, J., Figueiredo, A., Serrano, M., Husband, B. C., and Castro, S. (2020). Different patterns of ecological divergence between two tetraploids and their diploid counterpart in a parapatric linear coastal distribution polyploid complex. Front. Plant Sci. 11:315. doi: 10.3389/fpls.2020.00315
Cecchi, L., Colzi, I., Coppi, A., Gonnelli, C., and Selvi, F. (2013). Diversity and biogeography of Ni-hyperaccumulators of Alyssum section Odontarrhena (Brassicaceae) in the central western Mediterranean: evidence from karyology, morphology and DNA sequence data. Bot. J. Linn. Soc. 173, 269–289. doi: 10.1111/boj.12084
Cetlová, V., Fuertes-Aguilar, J., Iudova, D., and Španiel, S. (2019). Overlooked morphological variation and a proposal for a new taxonomic circumscription of Alyssum simplex (Brassicaceae). Phytotaxa 416, 149–166. doi: 10.11646/phytotaxa.416.2.3
Clement, M., Posada, D., and Crandall, K. (2000). TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x
Cornille, A., Salcedo, A., Kryvokhyzha, D., Glémin, S., Holm, K., Wright, S. I., et al. (2016). Genomic signature of successful colonization of Eurasia by the allopolyploid shepherd’s purse (Capsella bursa-pastoris). Mol. Ecol. 25, 616–629. doi: 10.1111/mec.13491
Correggiari, A., Field, M. E., and Trincardi, F. (1996). Late Quaternary transgressive large dunes on the sediment-starved Adriatic shelf. Geol. Soc. Spec. Publ. 117, 155–169. doi: 10.1144/GSL.SP.1996.117.01.09
Crowl, A. A., Myers, C., and Cellinese, N. (2017). Embracing discordance: phylogenomic analyses provide evidence for allopolyploidy leading to cryptic diversity in a Mediterranean Campanula (Campanulaceae) clade. Evolution 71, 913–922. doi: 10.1111/evo.13203
Crowl, A. A., Visger, C. J., Mansion, G., Hand, R., Wu, H. H., Kamari, G., et al. (2015). Evolution and biogeography of the endemic Roucela complex (Campanulaceae: Campanula) in the Eastern Mediterranean. Ecol. Evol. 5, 5329–5343. doi: 10.1002/ece3.1791
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Díaz-Pérez, A., Lopez-Alvarez, D., Sancho, R., and Catalan, P. (2018). Reconstructing the origins and the biogeography of species’ genomes in the highly reticulate allopolyploid-rich model grass genus Brachypodium using minimum evolution, coalescence and maximum likelihood approaches. Mol. Phylogen. Evol. 127, 256–271. doi: 10.1016/j.ympev.2018.06.003
Dodsworth, S., Chase, M. W., and Leitch, A. R. (2016). Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 180, 1–5. doi: 10.1111/boj.12357
Doležel, J., Sgorbati, S., and Lucretti, S. (1992). Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 85, 625–631. doi: 10.1111/j.1399-3054.1992.tb04764.x
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Dudley, T. R. (1964a). Studies in Alyssum: Near Eastern representatives and their allies, I. J. Arnold Arbor. 45, 57–100.
Dudley, T. R. (1965). “Alyssum L,” in Flora of Turkey, Vol. I, ed. P. H. Davis (Edinburgh: Edinburgh University press), 362–409.
Fišer, C., Robinson, C. T., and Malard, F. (2018). Cryptic species as a window into the paradigm shift of the species concept. Mol. Ecol. 27, 613–635. doi: 10.1111/mec.14486
Fiz-Palacios, O., and Valcárcel, V. (2013). From Messinian crisis to Mediterranean climate: a temporal gap of diversification recovered from multiple plant phylogenies. Perspect. Plant Ecol. 15, 130–137. doi: 10.1016/j.ppees.2013.02.002
Frajman, B., Rešetnik, I., Niketić, M., Ehrendorfer, F., and Schönswetter, P. (2016). Patterns of rapid diversification in heteroploid Knautia sect. Trichera (Caprifoliaceae, Dipsacoideae), one of the most intricate taxa of the European flora. BMC Evol. Biol. 16:204. doi: 10.1186/s12862-016-0773-2
Frajman, B., Rešetnik, I., Weiss-Schneeweiss, H., Ehrendorfer, F., and Schönswetter, P. (2015). Cytotype diversity and genome size variation in Knautia (Caprifoliaceae, Dipsacoideae). BMC Evol. Biol. 15:140. doi: 10.1186/s12862-015-0425-y
German, D. A. (2003). Zametki po rodu Alyssum L. (Cruciferae) Kazakhstana / Notes on the genus Alyssum L. (Cruciferae) in Kazakhstan. Turczaninowia 6, 45–57. [in Russian],Google Scholar
Greilhuber, J., Doležel, J., Lysak, M. A., and Bennett, M. D. (2005). The origin, evolution and proposed stabilization of the terms ‘genome size’and ‘C-value’to describe nuclear DNA contents. Ann. Bot. 95, 255–260. doi: 10.1093/aob/mci019
Grossheim, A. A. (1949). Opredelitel’ Rastenii Kavkaza / Key to the Plants of the Caucasus. Moscow: Sovietskaia nauka. [in Russian]
Hartvig, P. (2002). “Alyssum L,” in Flora Hellenica, Vol. 2, eds A. Strid and K. Tan (Ruggell: A.R.G. Gantner Verlag K. G.), 199–227.
Hassler, M. (2004–2021). Flora of Rhodos and Chalki. Picture Atlas and Database. Version 3.05; Last Update 9.1.2021. Available online at: www.flora-germanica.de/rhodos/ (accessed January 19, 2021).
Hewitt, G. M. (2011). Quaternary phylogeography: the roots of hybrid zones. Genetica 139, 617–638. doi: 10.1007/s10709-011-9547-3
Huang, X. C., German, D. A., and Koch, M. A. (2020). Temporal patterns of diversification in Brassicaceae demonstrate decoupling of rate shifts and mesopolyploidization events. Ann. Bot. 125, 29–47. doi: 10.1093/aob/mcz123
Huelsenbeck, J. P., and Ronquist, F. (2001). MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jalas, J., Suominen, J., and Lampinen, R. (1996). Atlas Florae Europaeae, Vol. 11. Helsinki: The Committee for Mapping the Flora of Europe and Societas Biologica Fennica Vanamo.
Jaros, U., Tribsch, A., and Comes, H. P. (2018). Diversification in continental island archipelagos: new evidence on the roles of fragmentation, colonization and gene flow on the genetic divergence of Aegean Nigella (Ranunculaceae). Ann. Bot. 121, 241–254. doi: 10.1093/aob/mcx150
Jones, G. (2017). Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. J. Math. Biol. 74, 447–467. doi: 10.1007/s00285-016-1034-0
Kleinsteuber, A., Ristow, M., and Hassler, M. (2016). Flora von Rhodos und Chalki. Band 1. Karlsruhe: Verlag Kleinsteuber Books.
Krijgsman, W., Hilgen, F. J., Raffi, I., Sierro, F. J., and Wilson, D. S. (1999). Chronology, causes and progression of the Messinian salinity crisis. Nature 400, 652–655. doi: 10.1038/23231
Kubatko, L., and Degnan, J. H. (2007). Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56, 17–24. doi: 10.1080/10635150601146041
Kuittinen, H., Aguadé, M., Charlesworth, D., Haan, A. D. E., Lauga, B., Mitchell-Olds, T., et al. (2002). Primers for 22 candidate genes for ecological adaptations in Brassicaceae. Mol. Ecol. Notes 2, 258–262. doi: 10.1046/j.1471-8286.2002.00210.x
Küpfer, P., and Nieto Feliner, G. (1993). “Alyssum L,” in Flora Iberica, Vol. 4, eds S. Castroviejo, C. Aedo, C. Gómez Campo, M. Laínz, P. Montserrat, R. Morales, et al. (Madrid: Real Jardín Botánico, C.S.I.C.), 167–184.
Lambeck, K., Rouby, H., Purcell, A., Sun, Y., and Sambridge, M. (2014). Sea level and global ice volumes from the last glacial maximum to the Holocene. Proc. Natl. Acad. Sci. U.S.A. 111, 15296–15303. doi: 10.1073/pnas.1411762111
Leigh, J. W., and Bryant, D. (2015). PopART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Li, Y., Feng, Y., Lv, G., Liu, B., and Qi, A. (2015). The phylogeny of Alyssum (Brassicaceae) inferred from molecular data. Nord. J. Bot. 33, 715–721. doi: 10.1111/njb.00588
López-Jurado, J., Mateos-Naranjo, E., and Balao, F. (2019). Niche divergence and limits to expansion in the high polyploid Dianthus broteri complex. New Phytol. 222, 1076–1087. doi: 10.1111/nph.15663
Magauer, M., Schönswetter, P., Jang, T.-S., and Frajman, B. (2014). Disentangling relationships within disjunctly distributed Alyssum ovirense/A. wulfenianum group (Brassicaceae), including description of a novel species from the north-eastern Alps. Bot. J. Linn. Soc. 176, 486–505. doi: 10.1111/boj.12214
Mandák, B., Krak, K., Vít, P., Lomonosova, M. N., Belyayev, A., Habibi, F., et al. (2018). Hybridization and polyploidization within the Chenopodium album aggregate analysed by means of cytological and molecular markers. Mol. Phylogen. Evol. 129, 189–201. doi: 10.1016/j.ympev.2018.08.016
Marhold, K. (2011). “Brassicaceae,” in Euro+Med Plantbase – The Information Resource for Euro-Mediterranean Plant Diversity. Available online at: http://www.emplantbase.org/home.html (accessed June 1, 2020).
McCann, J., Macas, J., Novák, P., Stuessy, T. F., Villaseñor, J. L., and Weiss-Schneeweiss, H. (2020). Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 11:362. doi: 10.3389/fpls.2020.00362
Médail, F., and Diadema, K. (2009). Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J. Biogeogr. 36, 1333–1345. doi: 10.1111/j.1365-2699.2008.02051.x
Médail, F., and Quézel, P. (1997). Hot-Spots analysis for conservation of plant biodiversity in the Mediterranean Basin. Ann. Miss. Bot. Gard. 84, 112–127. doi: 10.2307/2399957
Melichárková, A., Španiel, S., Brišková, D., Marhold, K., and Zozomová-Lihová, J. (2017). Unravelling allopolyploid origins in the Alyssum montanum–A. repens species complex (Brassicaceae): low-copy nuclear gene data complement plastid DNA sequences and AFLPs. Bot. J. Linn. Soc. 184, 485–502. doi: 10.1093/botlinnean/box039
Melichárková, A., Španiel, S., Marhold, K., Hurdu, B. I., Drescher, A., and Zozomová-Lihová, J. (2019). Diversification and independent polyploid origins in the disjunct species Alyssum repens from the Southeastern Alps and the Carpathians. Am. J. Bot. 106, 1499–1518. doi: 10.1002/ajb2.1370
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), (New Orleans, LA: Institute of Electrical and Electronics Engineers (IEEE)), 1–8.
Myers, N., Mittermeier, R. A., Mittermeier, C. G., da Fonseca, G. A., and Kent, J. (2000). Biodiversity hotspots for conservation priorities. Nature 403, 853–858. doi: 10.1038/35002501
Naciri, Y., and Linder, H. P. (2015). Species delimitation and relationships: the dance of the seven veils. Taxon 64, 3–16. doi: 10.12705/641.24
Naciri, Y., and Linder, H. P. (2020). The genetics of evolutionary radiations. Biol. Rev. 4, 1055–1072. doi: 10.1111/brv.12598
Nieto Feliner, G. (2011). Southern European glacial refugia: a tale of tales. Taxon 60, 365–372. doi: 10.1002/tax.602007
Nieto Feliner, G. (2014). Patterns and processes in plant phylogeography in the Mediterranean basin. A review. Perspect. Plant Ecol. 16, 265–278. doi: 10.1016/j.ppees.2014.07.002
Nieto Feliner, G., and Rosselló, J. A. (2012). “Concerted evolution of multigene families and homoeologous recombination,” in Plant Genome Diversity, Vol. 1, eds J. F. Wendel, J. Greilhuber, J. Doležel, and I. J. Leitch (Vienna: Springer), 171–193. doi: 10.1007/978-3-7091-1130-7_12
Padilla-García, N., Rojas-Andrés, B. M., López-González, N., Castro, M., Castro, S., Loureiro, J., et al. (2018). The challenge of species delimitation in the diploid-polyploid complex Veronica subsection Pentasepalae. Mol. Phylogen. Evol. 119, 196–209. doi: 10.1016/j.ympev.2017.11.007
Pagel, M., Meade, A., and Barker, D. (2004). Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 53, 673–684.
Panitsa, M., Kagiampaki, A., and Kougioumoutzis, K. (2018). “Plant diversity and biogeography of the Aegean Archipelago: a new synthesis,” in Biogeography and Biodiversity of the Aegean. In honour of Prof. Moysis Mylonas, eds M. Moysis, P. Pafilis, A. Parmakelis, N. Poulakakis, S. Sfenthourakis, and K. Triantis (Nicosia: Broken Hill Publishers, Ltd.), 269–278.
Perissoratis, C., and Conispoliatis, N. (2003). The impacts of sea-level changes during latest Pleistocene and Holocene times on the morphology of the Ionian and Aegean seas (SE Alpine Europe). Mar. Geol. 196, 145–156. doi: 10.1016/S0025-3227(03)00047-1
Persson, J. (1971). Studies in the Aegean flora XIX. Notes on Alyssum and some other genera of Cruciferae. Bot. Not. 124, 399–418.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rešetnik, I., Satovic, Z., Schneeweiss, G. M., and Liber, Z. (2013). Phylogenetic relationships in Brassicaceae tribe Alysseae inferred from nuclear ribosomal and chloroplast DNA sequence data. Mol. Phylogenet. Evol. 69, 772–786. doi: 10.1016/j.ympev.2013.06.026
Rešetnik, I., and Španiel, S. (2018). The new circumscription of the genus Alyssum L. (Brassicaceae) in the flora of Croatia. Glas. Hrvatskog Bot. Društva 6, 4–16.
Rybinskaya, E. V. (1994). “Alyssum L,” in Flora Sibiri: Berberidaceae–Grossulariaceae, Vol. 7, eds L. I. Malyshev and G. A. Peshkova (Novosibirsk: VO Nauka), 103–106.
Salmerón-Sánchez, E., Fuertes-Aguilar, J., Španiel, S., Pérez-García, F. J., Merlo, E., Garrido-Becerra, J. A., et al. (2018). Plant evolution in alkaline magnesium-rich soils: a phylogenetic study of the Mediterranean genus Hormathophylla (Cruciferae: Alysseae) based on nuclear and plastid sequences. PLoS One 13:e0208307. doi: 10.1371/journal.pone.0208307
Schönswetter, P., Suda, J., Popp, M., Weiss-Schneeweiss, H., and Brochmann, C. (2007). Circumpolar phylogeography of Juncus biglumis (Juncaceae) inferred from AFLP fingerprints, cpDNA sequences, nuclear DNA content and chromosome numbers. Mol. Phylogenet. Evol. 42, 92–103. doi: 10.1016/j.ympev.2006.06.016
Sfenthourakis, S., and Triantis, K. A. (2017). The Aegean archipelago: a natural laboratory of evolution, ecology and civilisations. J. Biol. Res. (Thessalon) 24:4. doi: 10.1186/s40709-017-0061-3
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Simaiakis, S. M., Rijsdijk, K. F., Koene, E. F., Norder, S. J., Van Boxel, J. H., Stocchi, P., et al. (2017). Geographic changes in the Aegean Sea since the Last Glacial Maximum: postulating biogeographic effects of sea-level rise on islands. Palaeogeogr. Palaeocl. 471, 108–119. doi: 10.1016/j.palaeo.2017.02.002
Simmons, M. P., and Ochoterena, H. (2000). Gaps as characters in sequence-based phylogenetic analyses. Syst. Biol. 49, 369–381. doi: 10.1093/sysbio/49.2.369
Small, R. L., Cronn, R. C., and Wendel, J. F. (2004). Use of nuclear genes for phylogeny reconstruction in plants. Aust. Syst. Bot. 17, 145–170. doi: 10.1071/SB03015
Soltis, P. S., Liu, X., Marchant, D. B., Visger, C. J., and Soltis, D. E. (2014). Polyploidy and novelty: Gottlieb’s legacy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130351. doi: 10.1098/rstb.2013.0351
Španiel, S., Kaplan, K., Bovio, M., Mártonfiová, L., and Cetlová, V. (2018). Alyssum rossetii (Brassicaceae), a new species from the Aosta Valley in Italy based on morphological and genome size data. Phytotaxa 360, 269–281. doi: 10.11646/phytotaxa.360.3.7
Španiel, S., Kempa, M., Salmerón-Sánchez, E., Fuertes-Aguilar, J., Mota, J. F., Al-Shehbaz, I. A., et al. (2015). AlyBase: database of names, chromosome numbers, and ploidy levels of Alysseae (Brassicaceae), with a new generic concept of the tribe. Plant Syst. Evol. 301, 2463–2491. doi: 10.1007/s00606-015-1257-3
Španiel, S., Marhold, K., Passalacqua, N. G., and Zozomová-Lihová, J. (2011). Intricate variation patterns in the diploid-polyploid complex of Alyssum montanum-A. repens (Brassicaceae) in the Apennine Peninsula: evidence for long-term persistence and diversification. Am. J. Bot. 98, 1887–1904. doi: 10.3732/ajb.1100147
Španiel, S., Marhold, K., and Zozomová-Lihová, J. (2017a). The polyploid Alyssum montanum-A. repens complex in the Balkans: a hotspot of species and genetic diversity. Plant Syst. Evol. 303, 1443–1465. doi: 10.1007/s00606-017-1470-3
Španiel, S., Marhold, K., and Zozomová-Lihová, J. (2019). Polyphyletic Alyssum cuneifolium (Brassicaceae) revisited: morphological and genome size differentiation of recently recognized allopatric taxa. J. Syst. Evol. 57, 287–301. doi: 10.1111/jse.12464
Španiel, S., Zozomová-Lihová, J., and Marhold, K. (2017b). Revised taxonomic treatment of the Alyssum montanum-A. repens complex in the Balkans: a multivariate morphometric analysis. Plant Syst. Evol. 303, 1413–1442. doi: 10.1007/s00606-017-1468-x
Suc, J. P. (1984). Origin and evolution of the Mediterranean vegetation and climate in Europe. Nature 307, 429–432. doi: 10.1038/307429a0
Templeton, A. R. (2008). The reality and importance of founder speciation in evolution. Bioessays 30, 470–479. doi: 10.1002/bies.20745
Thompson, J. D. (2020). Plant Evolution in the Mediterranean: Insights for Conservation, 2nd Edn. Oxford: Oxford University Press.
Wagner, F., Ott, T., Zimmer, C., Reichhart, V., Vogt, R., and Oberprieler, C. (2019). ‘At the crossroads towards polyploidy’: genomic divergence and extent of homoploid hybridization are drivers for the formation of the ox-eye daisy polyploid complex (Leucanthemum, Compositae-Anthemideae). New Phytol. 223, 2039–2053. doi: 10.1111/nph.15784
White, T., Bruns, T., Lee, S., and Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. 18, 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1
Zahradníček, J., Chrtek, J., Ferreira, M. Z., Krahulcová, A., and Fehrer, J. (2018). Genome size variation in the genus Andryala (Hieraciinae, Asteraceae). Folia Geobot. 53, 429–447. doi: 10.1007/s12224-018-9330-7
Záveská, E., Fér, T., Šída, O., Marhold, K., and Leong-Škorničková, J. (2016). Hybridization among distantly related species: examples from the polyploid genus Curcuma (Zingiberaceae). Mol. Phylogen. Evol. 100, 303–321. doi: 10.1016/j.ympev.2016.04.017
Zhou, T., Lu, L., Yang, G., Al-Shehbaz, I. A., and Dorofeyev, V. I. (2001). “Brassicaceae (Cruciferae),” in Flora of China (Brassicaceae Through Saxifragaceae), Vol. 8, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press), 1–193.
Zozomová-Lihová, J., Marhold, K., and Španiel, S. (2014). Taxonomy and evolutionary history of Alyssum montanum (Brassicaceae) and related taxa in southwestern Europe and Morocco: diversification driven by polyploidy, geographic and ecological isolation. Taxon 63, 562–591. doi: 10.12705/633.18
Zozomová-Lihová, J., Melichárková, A., Svitok, M., and Španiel, S. (2020). Pleistocene range disruption and postglacial expansion with secondary contacts explain the genetic and cytotype structure in the western Balkan endemic Alyssum austrodalmaticum (Brassicaceae). Plant Syst. Evol. 306, 47.
Keywords: Aegean area, allopolyploidy, Alyssum, annual species, endemics, Mediterranean, phylogeny, sympatry
Citation: Cetlová V, Zozomová-Lihová J, Melichárková A, Mártonfiová L and Španiel S (2021) Multiple Drivers of High Species Diversity and Endemism Among Alyssum Annuals in the Mediterranean: The Evolutionary Significance of the Aegean Hotspot. Front. Plant Sci. 12:627909. doi: 10.3389/fpls.2021.627909
Received: 10 November 2020; Accepted: 22 March 2021;
Published: 27 April 2021.
Edited by:
Božo Frajman, University of Innsbruck, AustriaReviewed by:
Pau Carnicero, University of Innsbruck, AustriaVirginia Valcárcel, Autonomous University of Madrid, Spain
Andreas Tribsch, University of Salzburg, Austria
Copyright © 2021 Cetlová, Zozomová-Lihová, Melichárková, Mártonfiová and Španiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stanislav Španiel, c3RhbmlzbGF2LnNwYW5pZWxAc2F2YmEuc2s=