 Weilin Cao
Weilin Cao Liming Gan
Liming Gan Chenchen Wang
Chenchen Wang Xuechen Zhao
Xuechen Zhao Changxiang Zhu
Changxiang Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 10 February 2021
Sec. Plant Pathogen Interactions
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.619062
This article is part of the Research TopicMachine Learning on Understanding the Epigenetic Mechanisms Underlying Plant Adaptation and DomesticationView all 7 articles
Long non-coding RNA (lncRNA) is a crucial regulatory mechanism in the plant response to biotic and abiotic stress. However, their roles in potato (Solanum tuberosum L.) resistance to Phytophthora infestans (P. infestans) largely remain unknown. In this study, we identify 2857 lncRNAs and 33,150 mRNAs of the potato from large-scale published RNA sequencing data. Characteristic analysis indicates a similar distribution pattern of lncRNAs and mRNAs on the potato chromosomes, and the mRNAs were longer and had more exons than lncRNAs. Identification of alternative splicing (AS) shows that there were a total of 2491 lncRNAs generated from AS and the highest frequency (46.49%) of alternative acceptors (AA). We performed R package TCseq to cluster 133 specific differentially expressed lncRNAs from resistance lines and found that the lncRNAs of cluster 2 were upregulated. The lncRNA targets were subject to KEGG pathway enrichment analysis, and the interactive network between lncRNAs and mRNAs was constructed by using GENIE3, a random forest machine learning algorithm. Transient overexpression of StLNC0004 in Nicotiana benthamiana significantly suppresses P. infestans growth compared with a control, and the expression of extensin (NbEXT), the ortholog of the StLNC0004 target gene, was significantly upregulated in the overexpression line. Together, these results suggest that lncRNAs play potential functional roles in the potato response to P. infestans infection.
The potato (Solanum tuberosum L.) is the fourth most important staple crop in global production after rice, maize, and wheat, and it is the most important non-grain crop for human consumption (Birch et al., 2012). However, multiple diseases can severely devastate the production and quality of potatoes, including late blight (Mizubuti and Fry, 2006), bacterial wilt (Wullings et al., 1998), and necrotic ringspot (Ohshima et al., 2000). The Irish famine of 1845–1852 caused by late blight led to around one million deaths and one million more people emigrated out of Ireland (Cantwell, 2017). Approximately billions of dollars are estimated as the cost of controlling the disease and damage caused by this disease per year (Haverkort et al., 2008).
Since the famine, potato late blight disease has attracted the attention of researchers, and more than 3,000 reports associated with the disease have been published (Fry et al., 2015). Control methods for the disease are divided into mainly two kinds: pesticide application and resistance breeding. The frequent use of systemic fungicides not only seriously pollutes the natural environment, but it also increases the probability of resistant populations by promoting the rapid evolution of numerous effector genes in the Phytophthora infestans genome (Raffaele et al., 2010). Therefore, breeding resistant varieties is the most effective control method. Investigating the mechanism of potato resistance to P. infestans plays an important role in the process of breeding resistant varieties.
Transcriptome sequencing is the main strategy for investigating the resistant mechanism at the genomic level. The differentially expressed genes associated with resistance pathways—salicylic acid-, jasmonic acid- (JA), and abscisic acid-mediated signaling pathways—were identified by sequencing the transcriptomes of potato infected by P. infestans at different stages (Duan et al., 2020). The hierarchies of resistance genes in the potato against late blight were investigated by integrating transcriptomic and metabolomic analysis (Yogendra and Kushalappa, 2016). The regulatory mechanism of miRNA in the tomato response to P. infestans was elucidated by high-throughput sequencing (Luan et al., 2015). Different resistance genes (R genes) and defense mechanisms against P. infestans infection between potato foliage and tuber were identified by the RNA-seq approach (Gao and Bradeen, 2016).
In addition to genes related to resistance pathways, R genes and miRNAs, long non-coding RNAs (lncRNAs) have also been proven to play an important role in gene expression and silencing pathways for several biological processes in response to pathogens (Zaynab et al., 2018). LncRNAs have no protein-coding ability, and their lengths are more than 200 nt (Sun et al., 2018). Based on their genomic location, lncRNAs are classified into three major categories, including long intergenic non-coding RNA (lincRNA), long non-coding natural antisense transcripts, and long intronic non-coding RNA (Cui et al., 2017). The functional roles of lncRNAs in plants are increasingly being unraveled, and a number of various reports associated with identification of lncRNAs in multiple plant species (Wang and Chekanova, 2017), including Zea mays (Li et al., 2014; Kim et al., 2019), Solanum lycopersicum (Cui et al., 2020), Arachis hypogaea (Zhao et al., 2019; Tian et al., 2020), Oryza sativa (Li et al., 2020), Populus (Ma et al., 2019), and Arabidopsis thaliana (Zhao et al., 2018), have been published. However, most experimental proofs focus on the functions of lncRNAs associated with plant development and abiotic stress responses, such as fruit ripening (Li et al., 2018; Yu et al., 2019), growth (Meng et al., 2018; Wang Y. et al., 2018), nutritional stimulation (Wang et al., 2017), and salt and drought stresses (Qin et al., 2017). In contrast, there are just beginning to emerge reports about lncRNAs associated with resistance regulatory mechanisms against pathogens (Xin et al., 2011; Zhu et al., 2014). To date, numerous pathogen-responsive lncRNAs have been identified from Triticum aestivum infected with powdery mildew and stripe rust (Zhang et al., 2016); Brassica napus with Sclerotinia sclerotiorum (Jain et al., 2017); Solanum lycopersicum with TYLCV (Joshi et al., 2016); Gossypium spp. with Verticillium dahliae (Wang J. et al., 2018); Oryza sativa with Magnaporthe (Zhang et al., 2018); and Oryzae, Arabidopsis thaliana with Fusarium oxysporum (Zhu et al., 2014). Kwenda et al. (2016) genome-wide identified potato lincRNAs and functionally analyzed the mechanism in response to Pectobacterium carotovorum subspecies brasiliense infection. A total of 1342 mRNAs and 22 lncRNAs of rice were identified to be differentially expressed after RBSDV infection (Zhang et al., 2020). An lncRNA of rice, ALEX1, whose expression is highly induced by Xoo infection, can active the JA pathway and promote resistance to bacterial blight (Yu et al., 2020). The differentially expressed lncRNAs (DELs) were identified, and lncRNA-mRNA networks were examined by performing comparative transcriptome analysis of P. infestans-resistant and -susceptible tomatoes. The tomato lncRNA16397 was confirmed to reduce reactive oxygen species accumulation and alleviate cell membrane injury by inducing SlGRX expression, leading to enhanced resistance to P. infestans (Cui et al., 2017). During P. infestans infection, the tomato Sl-lncRNA15492 inhibits the homeostasis of Sl-NBS-LRR1 through the infection of Sl-miR482a (Jiang et al., 2020). In the tomato, lncRNA33732 activated by transcription factor WRKY1 can induce H2O2 accumulation and participate in the mechanism of resistance to P. infestans (Cui et al., 2019). Cui et al. (2020) found that lncRNAs might regulate ceRNAs to decoy microRNAs (miRNAs) and function as their target genes in tomato plants, increasing resistance to P. infestans. In addition to mRNA and ceRNAs, other types of RNA were regulated by lncRNA to participate in the plant response to pathogen infection, such as circRNAs, and sRNAs (Zhou et al., 2020).
Although several studies have confirmed that lncRNAs play an important role in multiple plants’ resistance to pathogen infection, the regulatory mechanism of lncRNAs remains poorly understood. To date, compared with animal species, genome-wide identification and characterization of lncRNA in plants is still in its infancy (Kim and Sung, 2012). In this work, we genome-wide identified potato lncRNAs and investigated the regulatory mechanism of potato response to P. infestans infection for the first time. Our results provide insights into the valuable information for the basal plant defense mechanisms of lncRNAs and can benefit future molecular-based breeding approaches to acquire pathogen-resistant plants.
The Sequence Read Archive of the National Center for Biotechnology Information (NCBI) collection was used to obtain RNA data sets of potato (PRJNA203403), which were used to identify transcripts of the potato in response to P. infestans infection. WT (Russet Burbank) is the tuber late blight–susceptible non-transformed line, and RB tissue is the tuber late blight–resistant transgenic Russet Burbank line. Potato tubers of RB tissues infected by P. infestans or mock for 0, 24, and 48 h were named ht0/mht0, ht3/mht3, and ht4/mht4, respectively. Potato tubers of WT infected by P. infestans or mock for 0, 24, and 48 h were named rt0/mrt0, rt3/mrt3, and rt4/mrt4, respectively (Supplementary Table 1; Gao et al., 2013). There were three biological replicates for each time point.
We examined the quality of all RNA sequences by performing FASTQC software (v. 0.11.9)1. Trimmomatic software (v. 0.39) was used to remove the adaptors and low-quality bases (Bolger et al., 2014), and the command was trimmomatic-0.39.jar PE-threads 5 sample1.R1.fq sample2.R2.fq sample1.R1.clean.fq sample1.R1.unpaired.fq sample1.R2.clean.fq sample1.R2. unpaired.fq AVGQUAL:30 1> sample1.QC.log 2 > &1. The unpaired reads were discarded. We used the spliced read aligner HISAT (v2.1.0) (Pertea et al., 2016) to align clean reads from all samples to the potato reference genome (v4.03)2 released in 2011 (Xu et al., 2011) (hisat2 -p 10 –dta –x database –q -1 sample1.R1.clean.fq -2 sample1.R2.clean.fq -S sample1.sam –novel-splicesite-outfile sample1.splicesite 2> align_summary.txt). StringTie software (v2.1.3) was used to assemble the transcripts of each experiment separately (Pertea et al., 2016) (stringtie sample1.sorted.bam -p 10 -G potato_GM_V403.gtf -o sample1.transcript.gtf -l sample1). The assembled transcript isoforms detected in only one sample were removed to reduce transcriptional aberration. The assembled transcript isoforms were compared with the potato genome annotation by using Gffcompare (v. 0.11.2) (gffcompare -r potato_GM_V403.gtf -o./merged stringtie_merged.gtf). The assembled transcript sequences were uploaded to Transcriptome Shotgun Assembly (TSA) of NCBI (GIXO00000000).
All transcripts from the transcriptome assemblies were used to identify lncRNAs using FEELnc software (v. 0.1.1). In the first step, the short (<200 bp) and single-exon transcripts were filtered using FEELncfilter (Harrow et al., 2012). In the second step, after the transcriptome reconstruction, we used FEELnccodpot predictors to compute a coding potential score of the assembled sequences. The UniProt database and CPC2 server were used to calculate the coding potential and remove transcripts with open reading frame (ORF) greater than 100 amino acids. We used blastn to filter the transcripts overlapping all mature miRNA sequences from miRBase, a high-confidence subset of miRBase obtained from miRBase (Kozomara et al., 2019)3 (E-value < 1 × 10–5). Finally, RNAs with length ≥200 bp, potential coding scores ≤0.5, coding potential <0, ORF <100 amino acids, transcripts per million (TPM) > 1 at least one sample, and ORF cover ≤50% are defined as lncRNAs. The target genes of lncRNAs were also predicted. The coding genes within 100 kb 5’ upstream or 3’ downstream of each lncRNA are identified as potential cis-targets (Hou et al., 2017). The Pearson correlation between the lncRNA and the gene were calculated based on expression patterns. A high Pearson correlation between lnRNA and gene represents that the expression patterns are similar in all samples, and they are more inclined to play the same role or have a regulatory relationship. Therefore, using highly correlated genes as targets of lncRNA is a good strategy for predicting the function of lncRNA. Genes with a Pearson correlation of r > 0.98, P ≤ 0.05 to lncRNA were considered as potential trans targets for the lncRNA. To further understand the biological process of target genes, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analysis by using KOBAS software 3.0 (prepareKEGGenrich.pl all.genes.list sig.genes.list geneName2KO KO2ko) (Mao et al., 2005; Xie et al., 2011) and topGO software (v 2.42.0) (topGO.enrichment.R -f sig.genes.go -b all.genes.go) (Frazee et al., 2015), respectively.
We used the ASTALAVISTA program4 to identify alternative splicing (AS) events (astalavista -t asta -i stringtie_merged.gtf -d 0 -o as.gtf.gz). The diverse categories of AS events, alternative acceptor (AA), alternative donor (AD), intron retention (IR), exon skipping (ES), and mutually exclusive exons (MX) (Feng et al., 2019) were identified using a Perl script generated in house.
We used Salmon (v 0.14.1) software to quantify all assembled transcripts, and the TPM was used to express the abundance (salmon quant -i transcripts_index -l A -1 reads_1.fastq -2 reads_2.fastq -o transcripts_quant) (Patro et al., 2017). We used tximport (v 1.18.0) to import transcript-level abundance and summarize it into expression matrices for downstream analysis. The read count of lncRNAs was used to calculate the difference by performing the R package DEseq2 (v1.30.0) with P-value ≤ 0.05 and log2 (fold change) > 1 (DEseq2.R rcound.txt group) (Love et al., 2014). R packages circlize(), ggplot(), and TCseq were used to plot distributions of lncRNAs and mRNAs in all potato chromosomes, bubble charts, heat maps, and expression pattern maps, respectively. A Venn gram was plotted by using TBtools software (v1.051) (Chen et al., 2020). We used Cytoscape software (v3.7.2) to construct an interactive network between lncRNAs and pathways associated with targets (Shannon et al., 2003). Gene regulatory networks (GRN) were generated by performing R package GENIE3 (v1.12.0) (Huynh-Thu and Geurts, 2018). Briefly, the transcripts with TPM > 1, at least one sample, and 16 lncRNAs from cluster 2 were selected to construct GRN based on expression patterns. We used GENIE3 to estimate the random forest regression for each transcript based on the lncRNAs as inputs and the default parameters (K = sqrt, nb.trees = 1000, input.idx = list of lncRNAs, importance. measure = IncNodePurity, seed = NULL) were applied5. Subsequently, the connectivity between transcript and lncRNA was calculated by GENIE3 software. For each lncRNA, all predicted targets (connectivity >0.3) were extracted (Ramírez-González et al., 2018).
The potatoes Désirée/Eshu 3 were maintained at the Shandong Agricultural University, Tai’an City, China, as the research material in this study. We propagated potato plantlets on Murashige-Skoog medium under growth chambers with 16 h of light at 20°C. Three-week-old plantlets were transplanted in a greenhouse and grown in individual pots at 20–26°C for 4–5 weeks for further assays. Nicotiana benthamiana plants were used to perform transient overexpression assays and grown in a long-day glass house with 16 h of light at 22°C; light intensity and humidity were 145 mE m–2 s–1 and 40%, respectively. The 5-week-old N. benthamiana was selected for P. infestans colonization and Agrobacterium tumefaciens infiltration.
The primers of StLNC0004 were designed (forward: acgggg gactctagaggatccAGAAATTGTGCAACTTCTATAGACATATCA, reverse: cgatcggggaaattcgagctcTCTCCTCCACCACACCATCA TC) and used to amplify the full length of StLNC0004 by performing PCR, and the products were recombined into the expressed vector pROK II to generate pROK II:StLNC0004 using the ClonExpress® Entry One Step Cloning Kit (Vazyme®, Vazyme Biotech Co., Ltd., China). We sequenced the constructed transient overexpression vectors for further confirmation. The electroporation was performed to transform pROK II:StLNC0004/pROK II into A. tumefaciens strain GV3101.
We carried out the A. tumefaciens transformation and P. infestans infection assays on N. benthamiana as described previously (Bos et al., 2010; Saunders et al., 2012; McLellan et al., 2013). Briefly, we used yeast extract and beef medium with appropriate antibiotics to culture the A. tumefaciens strains containing recombined vectors at 28°C (200 rpm, 24 h). We used sterile 10 mM MES and 10 mM MgCl2 buffer containing 200 μM acetosyringone to resuspend the bacteria pellet. Bacterial optical densities of OD600 = 0.3 were used for transient expression and P. infestans pathogenicity assays in N. benthamiana (Zhong et al., 2018).
The P. infestans isolate HLJ, a strongly pathogenic oomycete, was conserved at the State Key Laboratory of Shandong Agricultural University, Tai’an, Shandong, China (Wang et al., 2019). P. infestans HLJ was grown in Petri dishes with Rye A for 2 weeks at 18°C. Then, we used 5 mL sterile water to flood the dishes and used a glass rod to scrape sporangia for release. The clean Petri dish was poured into suspension, placed on ice, and stored at 4°C for 3 h to release the zoospores. We collected the resulting solution in a falcon tube and counted sporangia numbers by using a hemocytometer and adjusted to 15,000 sporangia/ml.
We placed the potato leaves in a Petri dish covered with moist filter paper and placed 3 leaves of the same cultivar in each dish. When inoculating, we dropped 10 μl of P. infestans HLJ spore suspension on both sides of the main vein of the leaves. After inoculation, we cultured at 18°C in a light incubator and inspected the disease condition of leaves after 5–7 days to determine whether it was pathogenic. Potatoes with diseased leaves and sporangia are susceptible materials, and leaves with only necrosis and no sporangia are resistant materials.
The potato leaves were inoculated with P. infestans at a concentration of 4 × 105 sporangia ml–1 for real-time quantitative PCR. Reverse transcription-polymerase chain reaction (RT-PCR) was used for the colonization of P. infestans in potatoes, and the primers of the conserved regions of the PI-O8 gene (Wang et al., 2019) of the P. infestans strains were designed (F: AAGATGATGTTGGATGATTG, R: TGCCTGATTTCTACCTTCT, 250 bp). Tobacco leaves were removed 24 h after infection and placed in sealed boxes. Each infiltration site was inoculated with 10 μl zoospores from P. infestans isolate 4 × 104 sporangia/ml (Zhong et al., 2018). As previously described, the degree of disease development was recorded in infected leaf assays (He et al., 2015; Tian et al., 2015). Fifty leaves from 15 individual plants were used for each of three replicates. ANOVA was used to analyze all data.
According to the manufacturer’s instructions on AG RNAex Pro Reagent AG21101 [Accurate Biotechnology (Hunan) Co., Ltd], we extracted the total RNA from all potato and N. benthamiana samples. We used the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States) to analyze the integrity, quality, and concentration of the RNAs.
The Evo M-MLV RT Kit with gDNA Clean for qPCR AG11705 [Accurate Biotechnology (Hunan) Co., Ltd] was used to reverse-transcribe the total RNA into a single-stranded RNA. We used Primer-BLAST in NCBI6 to design the gene-specific primers. Elongation factor 1-alpha-like protein (EF1α) was used as an internal reference gene. The reactions were conducted in a 20 μL volume containing 10 μL SYBR Green PCR Master Mix, 1 μL of each primer (10 μmol/ml), 3 μL dd H2O, and 5 μL of the template cDNA under the following conditions: 30 s at 72°C, 35 cycles of 5 s at 95°C, 30 s at 55°C, and 30 s at 72°C. Real-time RT-PCR was conducted using LightCycler96 (Switzerland). The real-time PCR analysis was performed using three biological replicates for each treated sample and at least three technical replicates of each biological replicate. Relative expression levels of the target genes were calculated using the 2–ΔΔCt method (Livak and Schmittgen, 2001).
The results are expressed as mean ± standard deviation (SD). The one-way ANOVA and Tukey’s test were used to analyze the differences between groups. P-value ≤ 0.05 is considered to be statistically significant.
To achieve a relatively comprehensive set of potato lncRNAs, we collected 36 public Illumina transcriptomes (Supplementary Table 1) and used the HISAT-StingTie-Salmon-FEELnc-CPC-Uniprot pipeline for further analysis (Figure 1A). After quality control, 0.85 billion clean reads (average 23.59 million) were obtained for lncRNA identification. The “Q30” value ranged from 97.72 to 98.32%, and their GC content ranged from 42.08 to 44% (Supplementary Table 2). All clean reads were mapped to the potato reference genome using HISAT2 software, and on average, 84.10% of the reads were successfully aligned with the potato reference genome sequence (Supplementary Table 3). Subsequently, we performed the assembly process using Stringtie software, and a total of 46,192 genes (71,446 transcripts) containing 33,150 mRNA were generated (Figure 1A and Supplementary Table 4). The transcript with TPM value >1 for at least one sample was considered to be expressed. The selected transcripts associated with each sample suggest that the distribution of transcript abundance in all samples was broadly similar (Supplementary Figure 1 and Supplementary Table 5). In total, there were 2857 lncRNAs (length ≥200 bp, ORF cover ≤0.5, potential coding scores ≤0.5, TPM > 1, coding potential <0) generated by using FEELnc, CPC2 software, and the UniProt database, and all lncRNAs were renamed (Supplementary Table 6). The lncRNAs were characterized according to the locations relative to the nearest protein-coding genes. The majority of lncRNAs (70.74%) were located in intergenic regions, and 29.26% of the lncRNAs (exonic: 17.01%, intronic: 12.25%) overlapped with protein-coding genes (Figure 1B). In all, 2021 intergenic lncRNAs, 28.25% and 23.95%, were at least 5 kb away upstream and downstream of genes, respectively. The remaining 47.80% of intergenic lncRNAs were located within 5 kb of genes (Figure 1C). The lengths of 76.79% lncRNAs ranged from 200 to 1000 bp; however, 57.43% mRNAs were longer ranging than 1000 bp (Figure 1D), suggesting that the sequence components of the mRNA exhibit differently when compared with the lncRNAs.
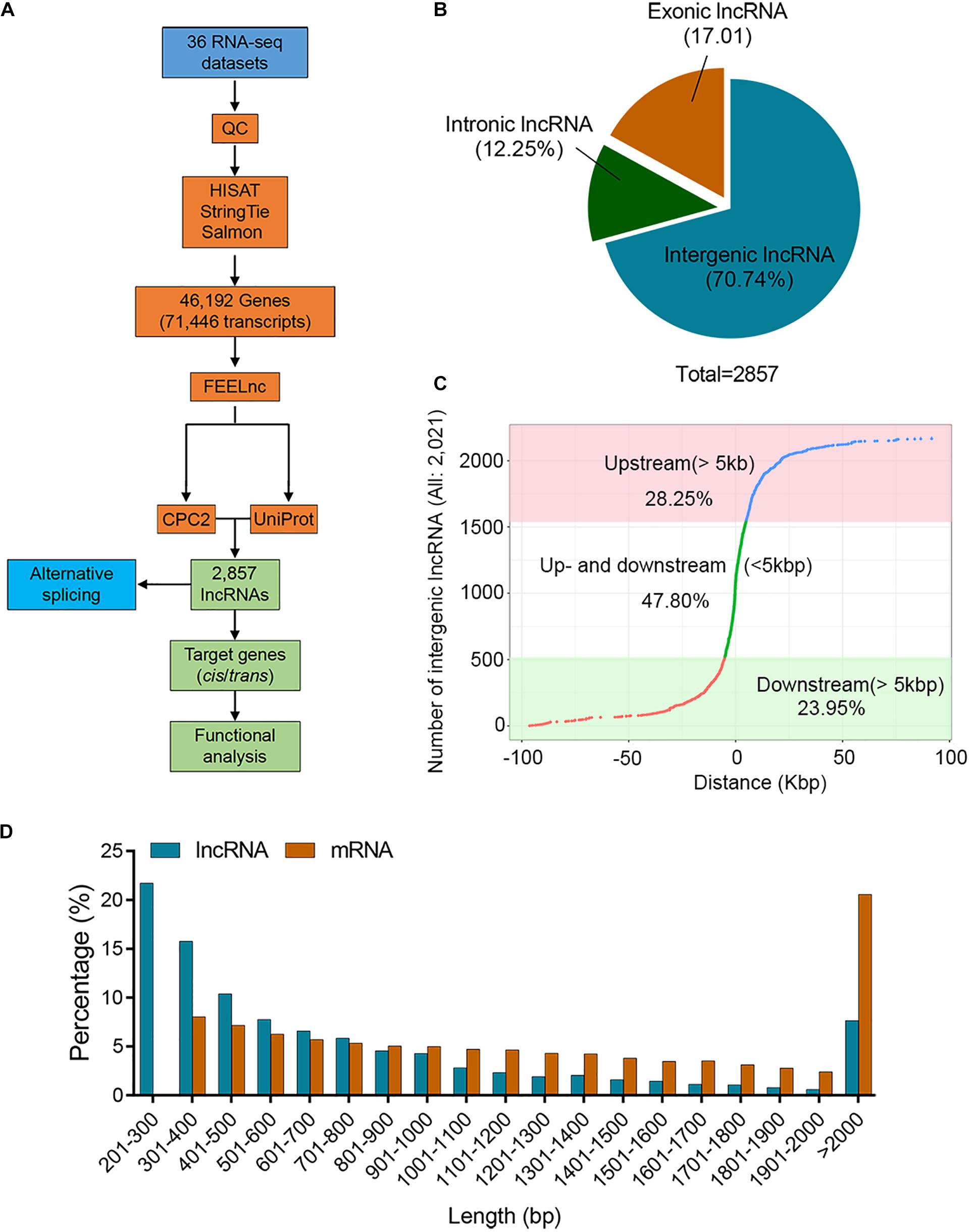
Figure 1. Identification of potato lncRNAs. (A) Pipeline for the identification of lncRNAs in the potato. (B) Proportion of lncRNAs located within 5 kb (upstream or downstream) or further than 5 kb from the nearest protein-coding genes. (C) Distribution of lincRNAs located upstream or downstream >5 kb and <5 kb. The X-axis represents the distance between the lncRNA and the nearest coding gene on the chromosome. The positive number represents that the lncRNA is located upstream of the gene, and a negative number represents downstream of the gene. The lncRNA with zero distance from the nearest gene belongs to intronic or exonic lncRNA. (D) Histogram showing the distribution of lncRNA and mRNA lengths.
We used R package Circlize (v. 0.4.10) to measure the distributions of lncRNA and mRNA on potato chromosome (Figure 2A). Higher densities of mRNA and lncRNAs were observed in the chromosome “arms” of most potato chromosomes than pericentromeric regions (Figure 2A). A total of 9323 (28.12%) mRNAs were composed of a single exon, and approximately 51.00% of lncRNAs were composed of two exons (Figure 2B). We detected 2491 lncRNAs derived from 563 AS events associated with 173 genes (Supplementary Table 7). Subsequently, the five major AS events, AA, AD, IR, ES, and MX, were identified by customizing a user-friendly program (Figure 2C). Figure 2C shows the statistics of AS events and corresponding gene models in the potato. We found that 49 (8.70%) IR events were identified with one retained intron for each transcript. There were 1158 (46.49%) lncRNAs undergoing AS associated with AA events (Figure 2C). The number of lncRNAs generated from ES was the second most common of AS events. The percentage of genes with two lncRNAs was significantly higher than other genes (47.98%) (Figure 2D).
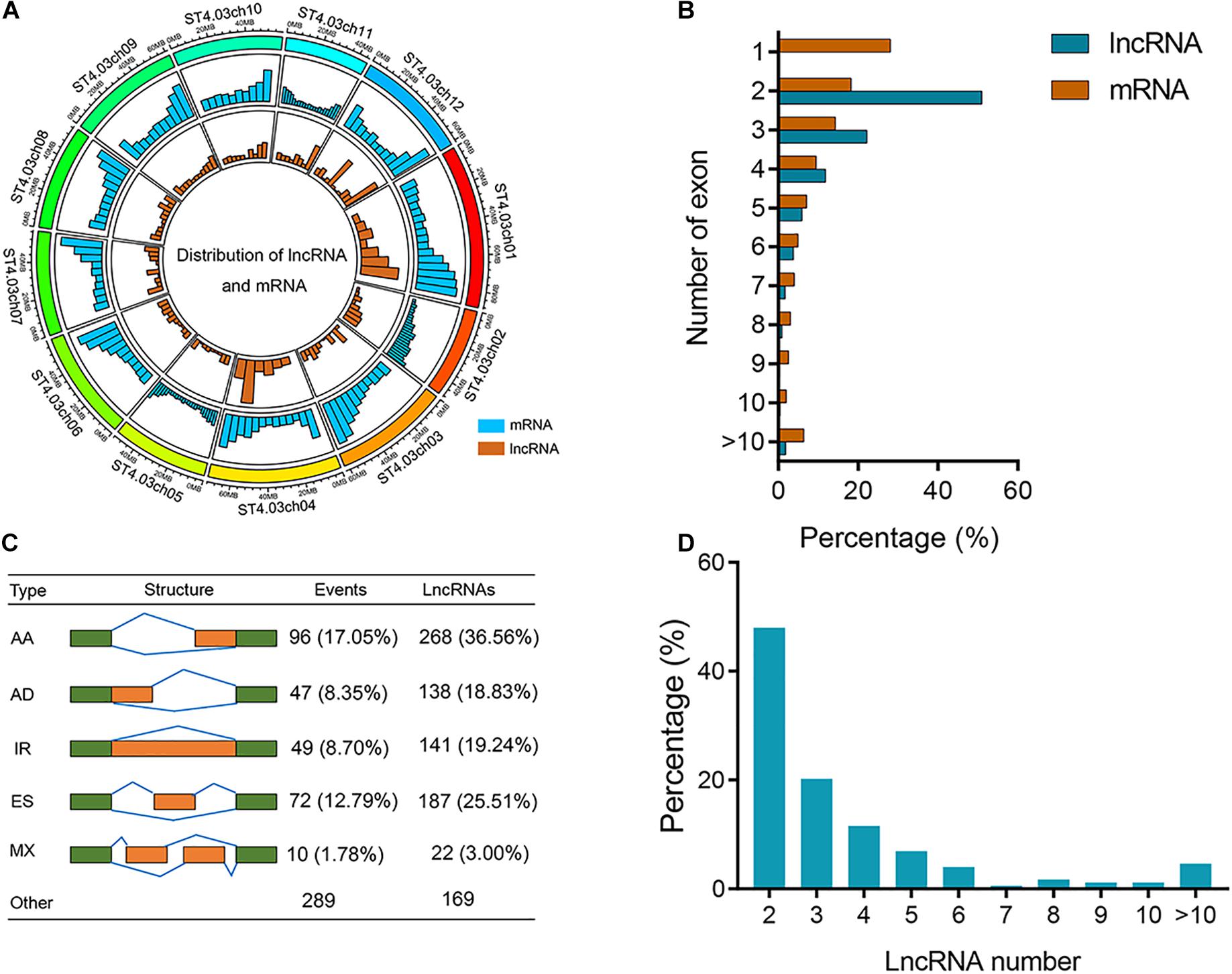
Figure 2. Characterization of potato lncRNAs. (A) Distribution of lncRNAs and mRNA along each potato chromosome. The abundance of lncRNA and mRNA in physical bins of 10 Mb for each chromosome (generated using Circlize). (B) Percentage of lncRNAs and mRNAs containing different numbers of exons. (C) Classification of AS events. Cartoons show five major types of AS events, including IR, AD, AA, ES, and MX. The numbers of AS events and associated lncRNAs are shown. Numbers in parentheses show the proportions of lncRNAs generated from different types of AS events occupying all AS lncRNAs. (D) Percentage of genes containing different numbers of lncRNAs.
There was no information about functionally characterized lncRNAs in potato. Therefore, to investigate the potential functional roles or biological processes of the lncRNAs, their target genes were identified as cis and trans (Supplementary Table 8). LncRNAs with TPM ≥ 1 in at least one sample were considered to be expressed, and all samples were divided into two categories: infected samples, including ht3, ht4, rt3, and rt4, and non-infected samples, including ht0, mt0, mt3, mt4, rt0, mrt0, mrt3, and mrt4. The lncRNAs with TPM > 1 in at least one sample of infected samples and TPM ≤ 1 in all samples of non-infected samples were defined to be specifically expressed lncRNAs in infected samples. We analyzed the distribution of lncRNAs in two types of potato tissues and found that a total of 125 lncRNAs were specifically expressed in infected samples (Figure 3A). To understand the function of the specific lncRNA, we performed KEGG pathway and GO term enrichment analysis of the targets of these specific lncRNAs (Supplementary Table 9). The KEGG pathway analysis showed that a total of 15 pathways were enriched significantly (P-value ≤ 0.05), including “Sesquiterpenoid and triterpenoid biosynthesis,” “SNARE interactions in vesicular transport,” and “RNA polymerase” (Figure 3B). A total of 11, 1, and 32 GO terms for molecular function (MF), cellular component (CC), and biological process (BP) were obtained, respectively (Supplementary Table 9). For biological process, major categories were found for multicellular organism development (GO:0007275), anatomical structure development (GO:0048856), and developmental process (GO:0032502). For the cellular component, genes were only involved in the extracellular region (GO:0005576). For molecular function, S-adenosylmethionine–dependent methyltransferase activity (GO:0008757) was the most represented GO term, followed by ferredoxin-NADP+ reductase activity (GO:0004324) (Figure 3C).
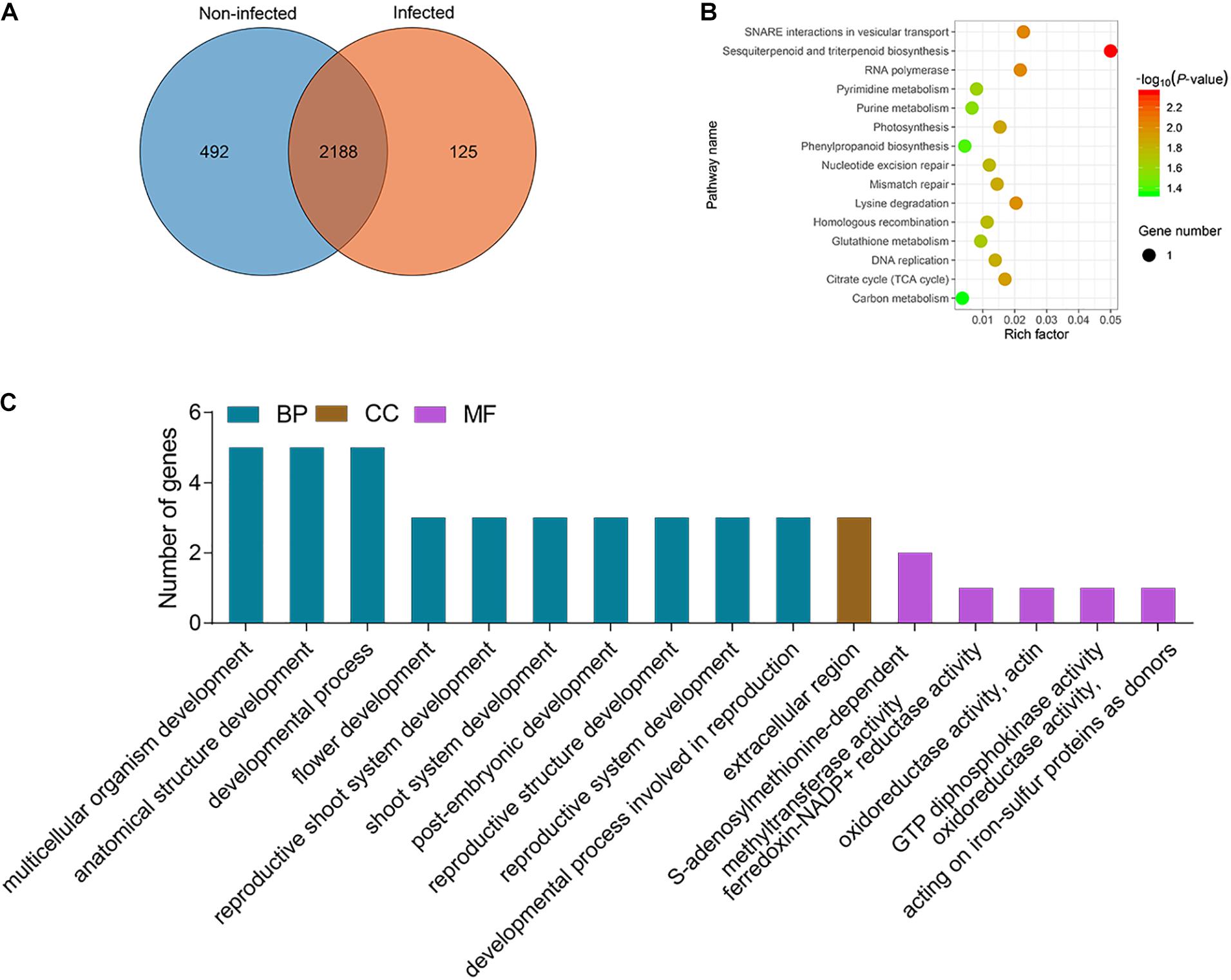
Figure 3. Analysis of specific lncRNAs from P. infestans infected samples. (A) Venn gram showing the distribution of lncRNAs in infected (ht3, ht4, rt3, and rt4) and non-infected (ht0, rt0, mht0, mht3, mht4, mrt0, mrt3, and mrt4) samples. (B) KEGG enrichment analysis of targets of 213 specific lncRNAs from infected samples. The circle size represents the gene number. Green to red represents the low to high P-value. (C) GO terms enrichment analysis of targets of 125 specific DELs and the top 10 of biological process (BP), and top 5 of molecular function (MF) and cellular component (CC) are shown.
To identify DELs between 24 h (ht3, rt3, mht3, and mrt3) and 48 h (ht4, rt4, mht4, and mrt4) samples and 0 h (ht0, rt0, mht0, and mrt0) samples, lncRNAs with at least a 2.0-fold change in expression and P-value ≤ 0.05 were considered to be differentially expressed (Supplementary Figure 2 and Supplementary Table 10). We analyzed the distribution of DELs in RB tissue and WT, respectively. In total, 308 specific DELs in RB tissue infected with P. infestans, including 86 specific DELs from ht3 vs. ht0, 118 specific DELs from ht4 vs. ht0, and 104 share DELs from the two comparison groups, were obtained (Figure 4A). These specific DELs may be related to the potato response to P. infestans infection. Furthermore, we investigated the functional roles of the specific DEL targets by KEGG and GO enrichment analysis (Supplementary Table 11). We found that multiple KEGG pathways and GO terms were significantly enriched, such as KEGG pathways “Photosynthesis-antenna proteins” and “Spliceosome” and GO terms photosynthesis and protein-chromophore linkage (Figure 4B). We observed that a total of 390 specific DELs (139 DELs in rt3 vs. rt0, 143 DELs in rt4 vs. rt0, both: 108 DELs in both comparisons) may be involved in the potato response to P. infestans infection in WT samples (Figure 4C). Functional enrichment analysis shows that KEGG pathways “Protein processing in endoplasmic reticulum” and “Spliceosome” and GO terms cellular process and response to stimulus were enriched significantly (Figure 4D and Supplementary Table 12).
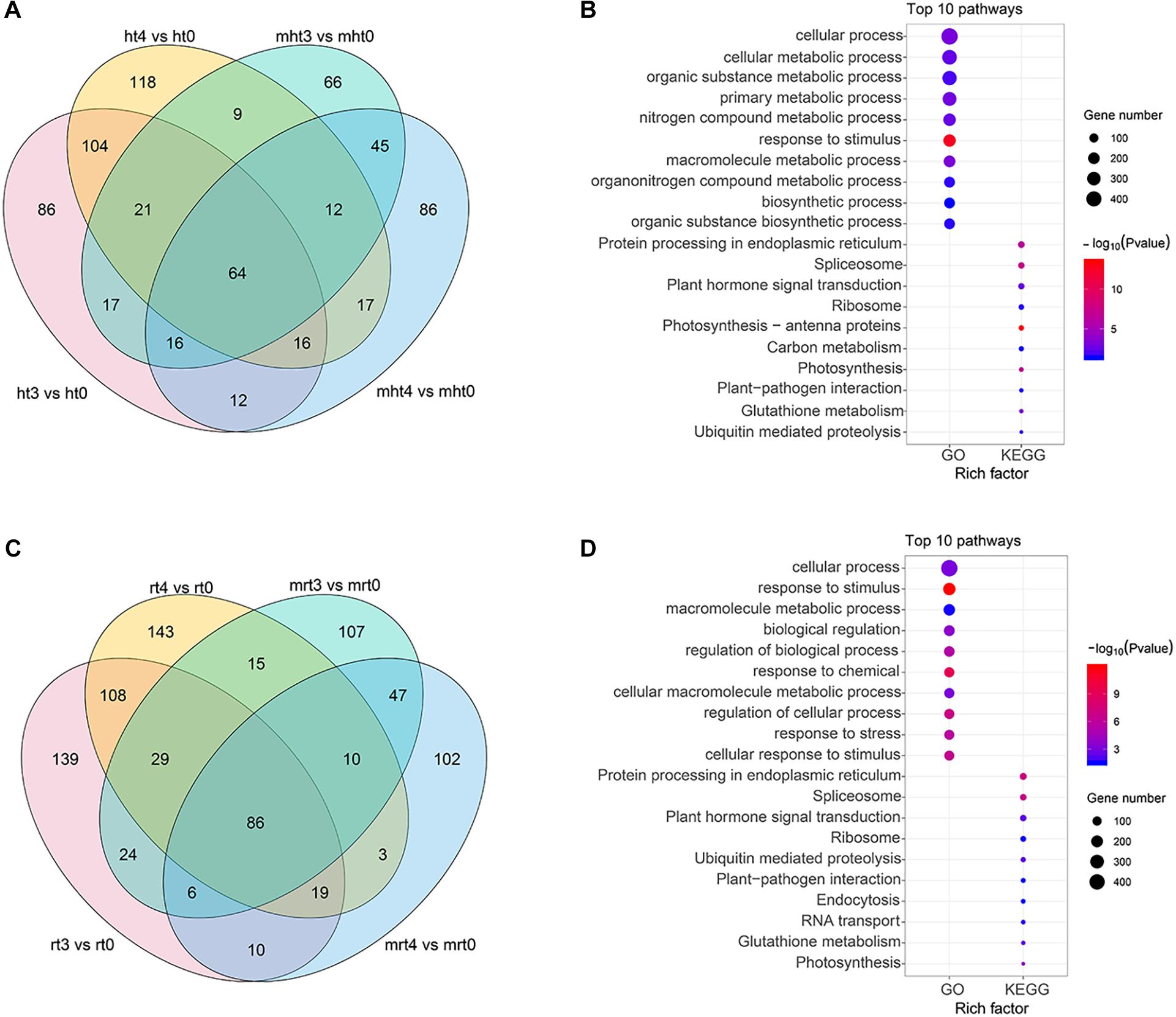
Figure 4. Identification and characterization of DELs. (A,C) Venn gram showing the distribution of DELs in WT and RB tissues, respectively. (B,D) GO and KEGG enrichment analysis were performed, and the top 10 GO terms and KEGG pathways are shown. The circle size represents the gene number. Blue to red represents the low to high P-value.
To identify the potential functional roles of specific DELs in the resistant sample of RB tissue, we analyzed the distribution of specific DELs in RB tissue infected by P. infestans. Figure 5A shows that a total of 133 DELs were observed only in RB tissue, indicating these DELs may participate in the resistance mechanism of the potato to P. infestans infection. Many KEGG pathways and BP of GO terms were involved in the targets of these specific DELs, such as “Plant hormone signal transduction,” “Ubiquitin mediated proteolysis,” and “Photosynthesis” from KEGG pathways and response to stimulus and biological regulation from GO terms (Figure 5B). The expression patterns of the specific DELs were clustered into six types by performing R package TCseq (Supplementary Figure 3). The expressed levels of 16 DELs in cluster 2 were increased at 48 h compared with 0 h (Figure 5C). To further explore the regulatory patterns of lncRNAs from clusters 2 (regulatory factors) and mRNA (target genes) during P. infestans infection in the potato, we used GENIE3 to generate GRNs, which are directed networks of lncRNAs and their regulatory genes (Supplementary Table 13). As the top-performing method in the DREAM4 and -5 GRN reconstruction challenges, GENIE3 takes advantage of the random forest machine learning algorithm. All targets were subjected to KEGG enrichment analysis and the interactive network between lncRNAs, and a total of 25 significantly enriched pathways were constructed by Cytoscape 3.7.2 software (Figure 5D). The lncRNAs may participate in the potato resistance mechanism by regulating these pathways.
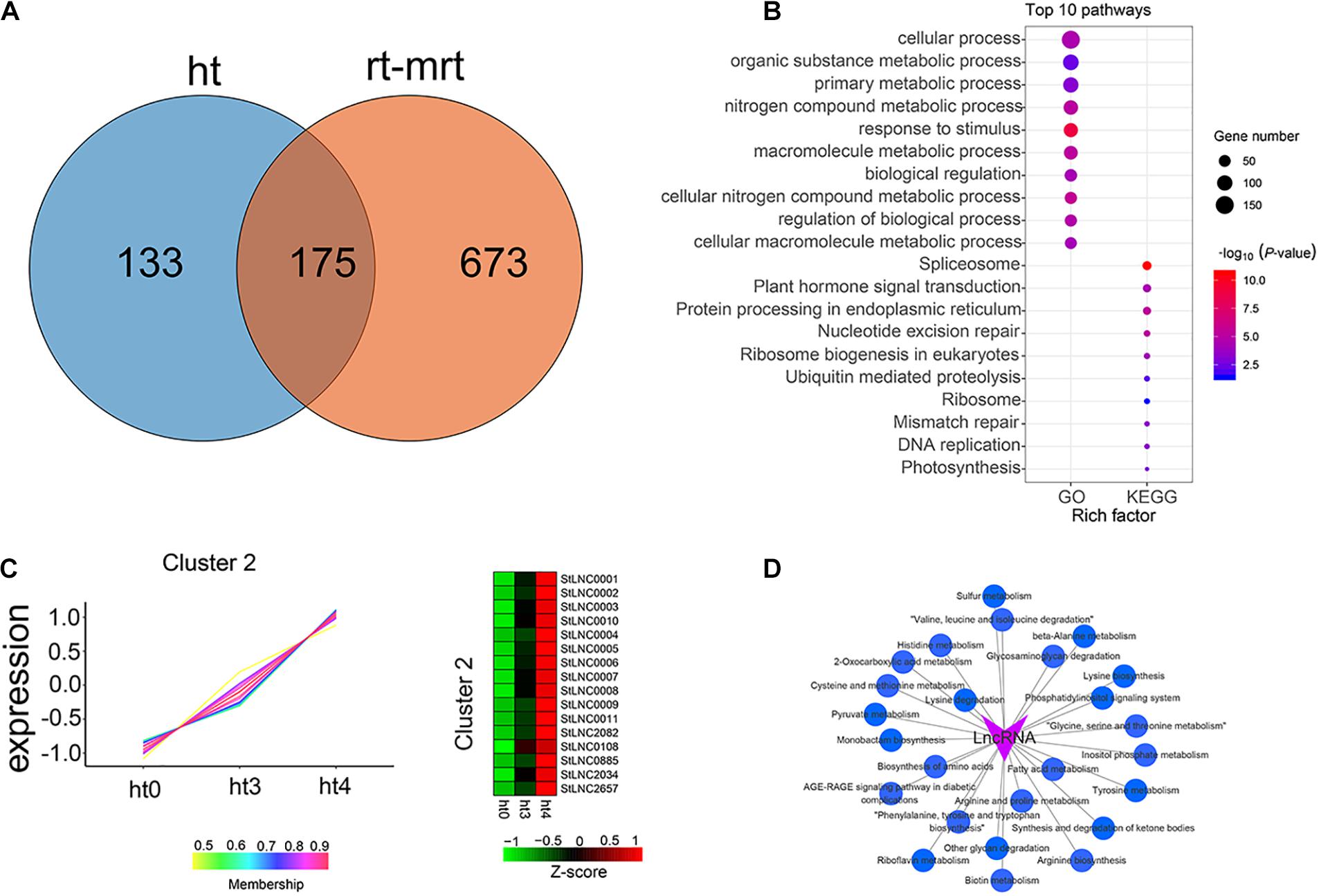
Figure 5. Identification and characterization of specific lncRNAs from P. infestans–infected RB tissue. (A) Venn gram showing the distribution of DELs in ht (P. infestans-infected RB tissues) and rt-mrt (P. infestans-infected WT and mock-treated RB tissues and WT). (B) GO and KEGG enrichment analysis of targets of 133 specific DELs in RB tissue infected by P. infestans and the top 10 pathways are shown. The circle size represented the gene number. Blue to red represents the low to high P-value. (C) TCseq was performed to cluster 133 specific DELs, and heat maps show the expression patterns of lncRNAs from cluster 2. Green color represents the low expression level and red is the high expression level. (D) The interactive network between lncRNAs from cluster 2 and the significantly enriched pathways.
The expression patterns of 11 specific DELs from cluster 2 were randomly selected to identify by performing qRT-PCR, and the lncRNAs were renamed (Supplementary Table 14). EF1α was used as the reference gene. Because the potato materials (WT/RB) used in this work were not obtained, we used leaves of susceptible/resistant potatoes Désirée/Eshu 3 identified by detecting separated leaves (Supplementary Figure 4) to detect the expressed levels of selected lncRNAs. Sequence comparison showed that the full length of the 11 lncRNAs in the RNA-seq data and PCR results from Désirée and Eshu 3 share high nucleic acid similarity (99%) (Supplementary Table 13). The RT-PCR data indicates the colonization of P. infestans in potato leaves was successful (Supplementary Figure 5). The selected lncRNAs were upregulated in resistant potatoes infected by P. infestans (Figure 6), and there was no significant difference in the susceptible potato, indicating that the DELs obtained in this study can be used for research on the Désirée/Eshu 3 potatoes.
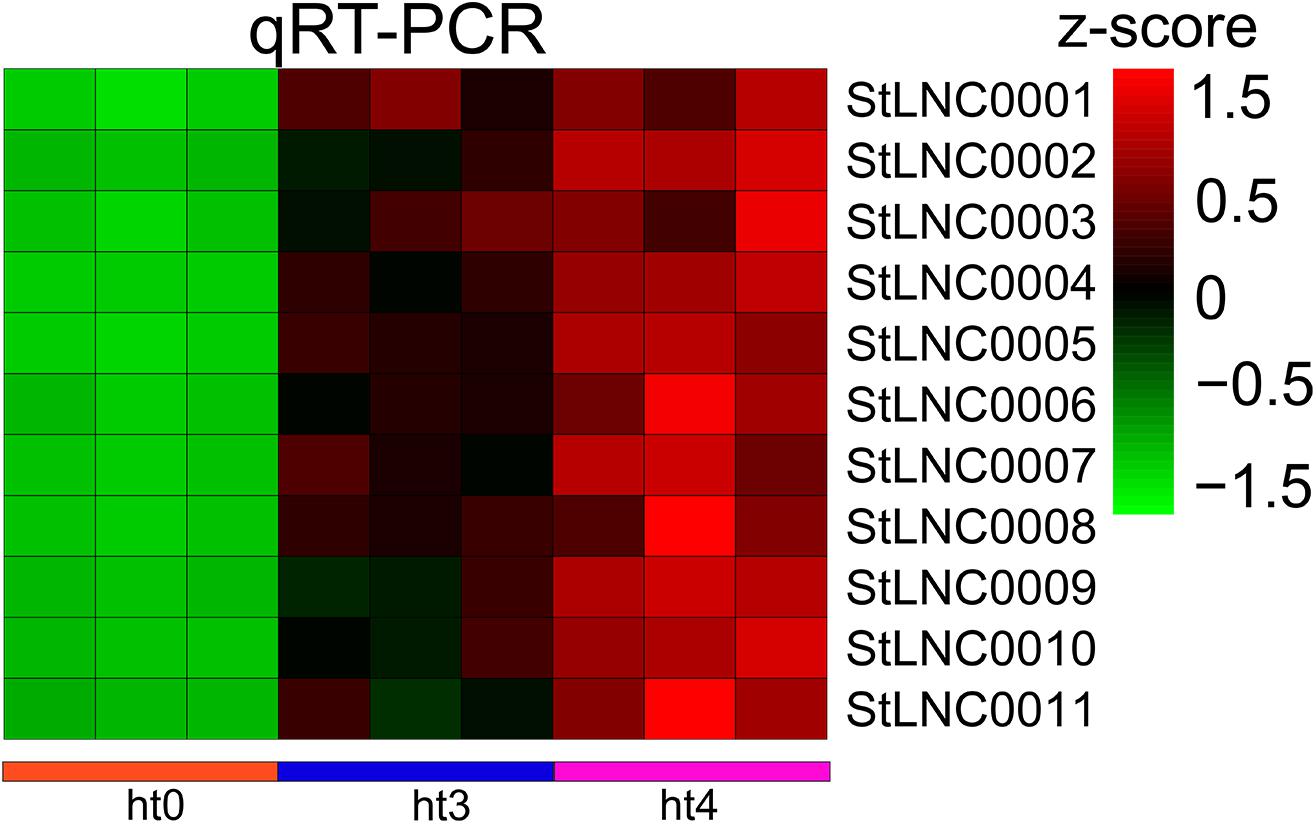
Figure 6. Comparison of 11 specific DELs from cluster 2 expressions determined by RNA-seq and qRT-PCR. Relative transcript levels of the DELs were determined by qRT-PCR. Data is shown by a heat map with z-score.
The expression level of StLNC0004 at 24 and 48 h from RNA-seq and qRT-PCR data was highest among the 11 specific DELs from cluster 2; therefore, we selected StLNC0004 to further investigate the potential function in response to P. infestans in N. benthamiana. The transcription level of StLNC0004 was significantly increased after A. tumefaciens strain GV3101 containing pROK II:StLNC0004 transformed into N. benthamiana for 36 h (Figure 7A). Then, we carried out the P. infestans pathogenicity assays and found that transient overexpression StLNC0004 in N. benthamiana significantly suppresses P. infestans growth compared with empty vector (pROK II) control (Figures 7B,C). The result suggests that StLNC0004 act as positive regulators in N. benthamiana resistance to P. infestans. The predicted target of StLNC0004 was gene PGSC0003DMG400000776 encoding extensin precursor (StEXT). To obtain the ortholog of StEXT from N. benthamiana, we blasted the coding region of StEXT in the N. benthamiana Genome and Transcriptome7 and found that the nucleic acid of StEXT and NbEXT (Nbv5.1tr6234951) were highly similar (80%), indicating that NbEXT was the ortholog of StEEXT. After infection with P. infestans for 48 and 72 h, the expression level of NbEXT in N. benthamiana containing pROK II:StLNC0004 was significantly higher than that of the control (Figure 7D). This suggests that StLNC0004 may act as a positive regulator in potato resistance to P. infestans by regulating the transcription level of StEXT.
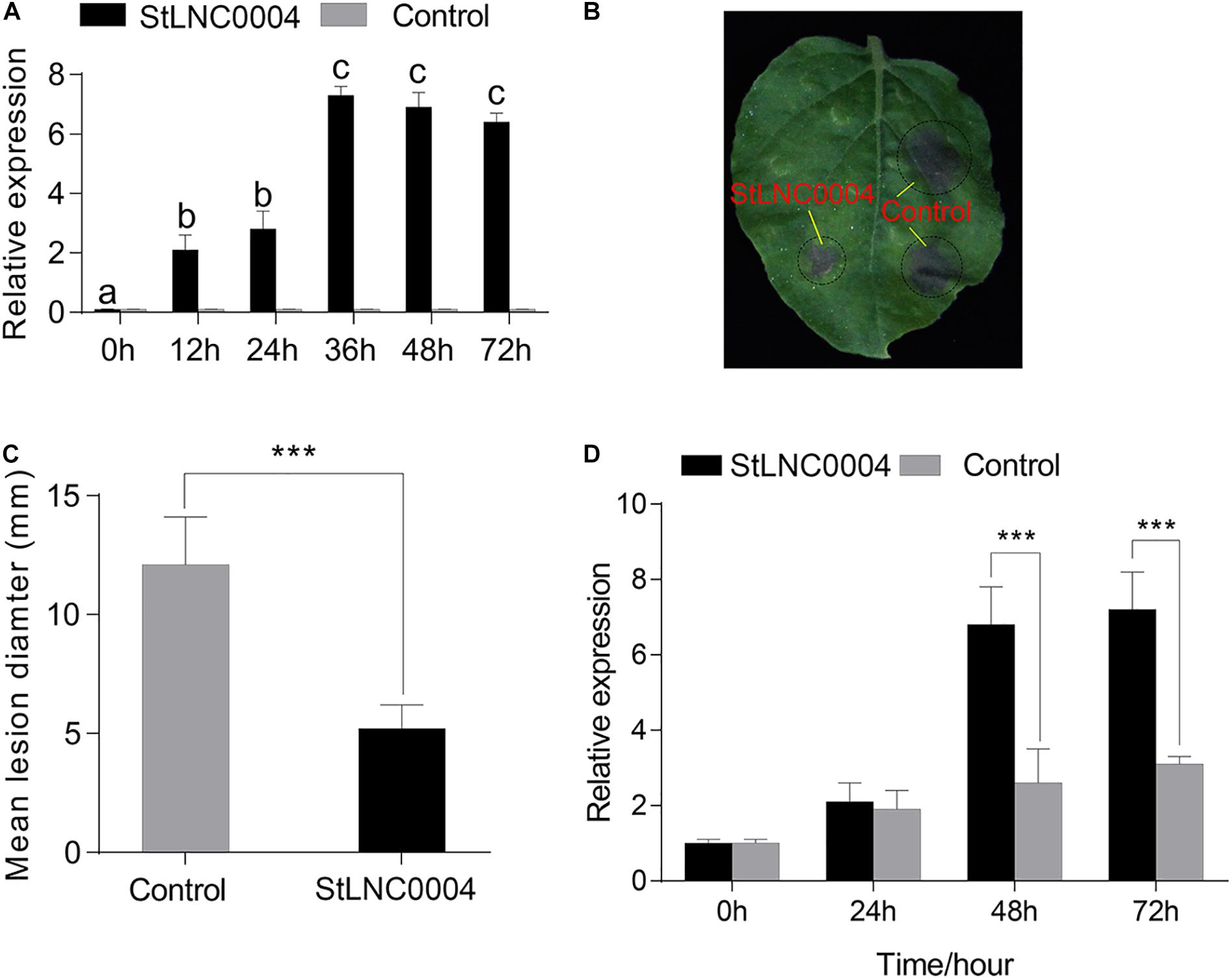
Figure 7. Transient expression of StLNC0004 inhibits P. infestans colonization in Nicotiana benthamiana. (A) Quantitative real-time PCR (qRT-PCR) was performed to identify the expression level of StLNC0004 in N. benthamiana. StLNC0004 and Control represent the N. benthamiana containing pROK II: StLNC0004 and pROK II, respectively. (B) Representative leaf images of P. infestans lesions following expression of StLNC0004/Control in N. benthamiana. (C) Mean P. infestans lesion diameter (mm) on sites of transient expression of the StLNC0004 and Control. (D) QRT-PCR identified the expression level of NtEXT, the ortholog of StEXT, the target gene of StLNC0004. Different letters in the columns represent a significant difference in the qRT-PCR results between StLNC0004 and Control (P ≤ 0.05). *** represents a significant difference between StLNC0004 and Control (P ≤ 0.001).
In general, the functions of most of the lncRNAs that have been identified are still unstudied. The lncRNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection in potato and P. infestans infection in tomato were identified, but no information on lncRNAs associated with the potato response to P. infestans infection is available. In this work, we obtained 36 published RNA-seq data sets from different cultivars of potato treated with P. infestans or mock from NCBI. We identified novel candidate lncRNAs that might be important components in potato defense response mechanisms during P. infestans infection.
A total of 2857 candidate lncRNAs from all samples were identified in this study. The average length of mRNAs in potato plants was longer than that of lncRNAs, and the percentage of lincRNA was 70.74%. Total lncRNAs distributed throughout 11 potato chromosomes suggest that lncRNA was the functional component of the potato genome. AS is an important functional role of post-transcriptional gene regulation (Reddy et al., 2013). Previous reports prove that AS events can regulate multiple aspects of plants, such as development, growth, flowering, environmental cue responses, circadian clock function, signal transduction, and plant immunity (Remy et al., 2014; Yang et al., 2014; Zhang et al., 2014; Tang et al., 2016). In plants and animals, the transcribed sequences of lncRNAs can be generated by AS events, which are similar to protein-encoding genes (Yang et al., 2017; Ma et al., 2018). LncRNA-PXN-AS1 without exon 4 can inhibit PXN mRNA translation by binding to the coding sequences of the mRNA, and lncRNAPXN-AS1 with exon 4 can protect PXN mRNA degradation, leading to increased expression levels of the mRNA by binding to the 3’ untranslated region of PXN mRNA, preferentially (Yuan et al., 2017). Here, a total of 2491 lncRNAs were generated by 563 AS events from potato RNA-seq data. We found that AA were the most frequently occurring AS events in producing lncRNA. The results suggest that AS events may be an important regulation of lncRNAs.
The functional characterization of potato lncRNAs was improved by predicting target genes in cis and trans. In total, 308 and 390 specific DELs from RB tissues and WT infected by P. infestans at different stages were obtained, respectively, indicating that the specific DELs may respond to P. infestans infection in the potato. To further investigate resistance-related lncRNAs, we identified a total of 133 DELs specifically expressed in RB tissue. We used R package TCseq to cluster the DELs and found that the expression of 16 lncRNAs from cluster 2 were higher in ht4 than that in ht0 and ht3, respectively, implying that the 16 lncRNAs may participate in the resistance mechanism in the potato to P. infestans. We constructed a directed network of the 16 lncRNAs and their regulated genes by performing GENIE3. A total of 92 KEGG pathways were significantly enriched by the regulated genes of the 16 lncRNAs. The pathway “Plant hormone signal transduction” was enriched with 5 targets, including PGSC0003DMT400004165 encoding SAUR family protein, PGSC0003DMT400064343 encoding the auxin responsive GH3 gene family, PGSC0003DMT400021210 encoding protein phosphatase 2C (PP2C), PGSC0003DMT400049445 encoding DELLA protein, and PGSC0003DMT400058305 encoding the two-component response regulator ARR-A family. The SAUR family may integrate various hormonal and environmental signals to regulate leaf senescence in Arabidopsis (Wen et al., 2020). The PP2C-SnRK2-ABF signaling module plays an important role in the ABA signaling pathway (Sun et al., 2020), which can promote plant defense to biotic and drought stresses by regulating stomatal opening and closure (Lim et al., 2015). To investigate the potential regulatory mechanism of lncRNAs, we performed the transient expression assay in N. benthamiana using the lncRNA StLNC0004 with the highest expression level in the 11 lncRNAs from cluster 2 and found that StLNC0004 inhibited the colonization of P. infestans on N. benthamiana. The target gene of StLNC0004 was extensin precursor (StEXT), which participates in the plant response to multiple stresses, such as drought (Wu et al., 1996), disease (Ding et al., 2008), and high temperature (Xu et al., 2007). The expression level of NbEXT, the ortholog of StEXT, was significantly upregulated by P. infestans infection in transient expression lines compared with the control, suggesting that StLNC0004 may enhance the potato defense to P. infestans by regulating the transcription level of StEXT. These results show that the specifically upregulated lncRNAs may participate in potato resistance mechanisms by regulating many of the targets involved in multiple pathways. The potential lncRNAs of the potato identified from our analysis were relatively robust. These lncRNAs may be used for further functional genomics studies or analysis of potential functional differences between different potato varieties.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA203403.
CZ and SZ designed the work. LG performed the data analysis. WC and CW wrote the manuscript and performed the experiments. XZ, MZ, and JD edited the manuscript and provided valuable suggestions during the experiment. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (31720103912), Shandong “Double Tops” Program (SYL2017XTTD11), National Natural Science Foundation of China (No. 31801270), and Natural Science Foundation of Shandong Province, China (No. ZR2019MC006).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.619062/full#supplementary-material
Supplementary Figure 1 | The expression level of transcripts in each sample based on log10 (TPM).
Supplementary Figure 2 | Expression patterns of DELs in each comparison.
Supplementary Figure 3 | TCseq wasperformed to cluster specific DELs in RB tissue infected by P. infestans.
Supplementary Figure 4 | Identifying susceptible/resistant potatoes Désirée/Eshu 3 by detecting separated leaves.
Supplementary Figure 5 | Identifying the colonization of P. infestans in potato leaves by using RT-PCR.
Supplementary Table 1 | Details of each sample.
Supplementary Table 2 | Statistics of RNA-seq data.
Supplementary Table 3 | Information on Illumina transcriptome-mapped potato reference genome sequence.
Supplementary Table 4 | The annotated information on all transcripts.
Supplementary Table 5 | The expression level of all transcripts based on TPM.
Supplementary Table 6 | Identification of lncRNAs.
Supplementary Table 7 | Alternative splicing (AS) event analysis of all lncRNAs.
Supplementary Table 8 | Predicting the target genes of lncRNAs.
Supplementary Table 9 | KEGG and GO enrichment analysis of specific lncRNAs from P. infestans-infected samples.
Supplementary Table 10 | Identification of DELs at 24 and 48 h compared with 0 h under the same treatment in WT or RB tissue.
Supplementary Table 11 | KEGG and GO enrichment analysis of specific DELs from P. infestans–infected samples in RB tissue.
Supplementary Table 12 | KEGG and GO enrichment analysis of specific DELs from P. infestans–infected samples in WT tissue.
Supplementary Table 13 | Constructing GRN using lncRNAs and other mRNAs by performing GENIE3.
Supplementary Table 14 | The primers of this work are listed.
Birch, P. R. J., Bryan, G., Fenton, B., Gilroy, E. M., Hein, I., Jones, J. T., et al. (2012). Crops that feed the world 8: potato: are the trends of increased global production sustainable? Food Sec. 4, 477–508. doi: 10.1007/s12571-012-0220-1
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bos, J. I., Armstrong, M. R., Gilroy, E. M., Boevink, P. C., Hein, I., Taylor, R. M., et al. (2010). Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc. Natl. Acad. Sci. U.S.A. 107, 9909–9914. doi: 10.1073/pnas.0914408107
Cantwell, J. D. (2017). A great-grandfather’s account of the Irish potato famine (1845-1850). Proc. (Bayl. Univ. Med. Cent.) 30, 382–383. doi: 10.1080/08998280.2017.11929657
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cui, J., Jiang, N., Hou, X., Wu, S., Zhang, Q., Meng, J., et al. (2020). Genome-Wide identification of lncRNAs and analysis of ceRNA networks during tomato resistance to Phytophthora infestans. Phytopathology 110, 456–464. doi: 10.1094/phyto-04-19-0137-r
Cui, J., Jiang, N., Meng, J., Yang, G., Liu, W., Zhou, X., et al. (2019). LncRNA33732-respiratory burst oxidase module associated with WRKY1 in tomato- Phytophthora infestans interactions. Plant J. 97, 933–946. doi: 10.1111/tpj.14173
Cui, J., Luan, Y., Jiang, N., Bao, H., and Meng, J. (2017). Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 89, 577–589. doi: 10.1111/tpj.13408
Ding, X., Cao, Y., Huang, L., Zhao, J., Xu, C., Li, X., et al. (2008). Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell 20, 228–240. doi: 10.1105/tpc.107.055657
Duan, Y., Duan, S., Armstrong, M. R., Xu, J., Zheng, J., Hu, J., et al. (2020). Comparative transcriptome profiling reveals compatible and incompatible patterns of Potato toward Phytophthora infestans. G3 (Bethesda) 10, 623–634. doi: 10.1534/g3.119.400818
Feng, S., Xu, M., Liu, F., Cui, C., and Zhou, B. (2019). Reconstruction of the full-length transcriptome atlas using PacBio Iso-Seq provides insight into the alternative splicing in Gossypium australe. BMC Plant Biol. 19:365. doi: 10.1186/s12870-019-1968-7
Frazee, A. C., Pertea, G., Jaffe, A. E., Langmead, B., Salzberg, S. L., and Leek, J. T. (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 33, 243–246. doi: 10.1038/nbt.3172
Fry, W. E., Birch, P. R., Judelson, H. S., Grunwald, N. J., Danies, G., Everts, K. L., et al. (2015). Five reasons to consider Phytophthora infestans a Reemerging Pathogen. Phytopathology 105, 966–981. doi: 10.1094/phyto-01-15-0005-fi
Gao, L., and Bradeen, J. M. (2016). Contrasting potato foliage and tuber defense mechanisms against the late blight Pathogen Phytophthora infestans. PLoS One 11:e0159969. doi: 10.1371/journal.pone.0159969
Gao, L., Tu, Z. J., Millett, B. P., and Bradeen, J. M. (2013). Insights into organ-specific pathogen defense responses in plants: RNA-seq analysis of potato tuber-Phytophthora infestans interactions. BMC Genomics 14:340. doi: 10.1186/1471-2164-14-340
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
Haverkort, A. J., Boonekamp, P. M., Hutten, R., Jacobsen, E., Lotz, L. A. P., Kessel, G. J. T., et al. (2008). Societal costs of late blight in potato and prospects of durable resistance through cisgenic modification. Potato Res. 51, 47–57. doi: 10.1007/s11540-008-9089-y
He, Q., Mclellan, H., Boevink, P. C., Sadanandom, A., Xie, C., Birch, P. R., et al. (2015). U-box E3 ubiquitin ligase PUB17 acts in the nucleus to promote specific immune pathways triggered by Phytophthora infestans. J. Exp. Bot. 66, 3189–3199. doi: 10.1093/jxb/erv128
Hou, X., Du, Y., Liu, X., Zhang, H., Liu, Y., Yan, N., et al. (2017). Genome-Wide analysis of long non-coding RNAs in potato and their potential role in tuber sprouting process. Int. J. Mol. Sci. 19:101 doi: 10.3390/ijms19010101
Huynh-Thu, V. A., and Geurts, P. (2018). dynGENIE3: dynamical GENIE3 for the inference of gene networks from time series expression data. Sci. Rep. 8:3384. doi: 10.1038/s41598-018-21715-0
Jain, P., Sharma, V., Dubey, H., Singh, P. K., Kapoor, R., Kumari, M., et al. (2017). Identification of long non-coding RNA in rice lines resistant to Rice blast pathogen Maganaporthe oryzae. Bioinformation 13, 249–255. doi: 10.6026/97320630013249
Jiang, N., Cui, J., Hou, X., Yang, G., Xiao, Y., Han, L., et al. (2020). Sl-lncRNA15492 interacts with Sl-miR482a and affects Solanum lycopersicum immunity against Phytophthora infestans. Plant J. 103, 1561–1574. doi: 10.1111/tpj.14847
Joshi, R. K., Megha, S., Basu, U., Rahman, M. H., and Kav, N. N. (2016). Genome wide identification and functional prediction of long non-coding RNAs responsive to Sclerotinia sclerotiorum infection in Brassica napus. PLoS One 11:e0158784. doi: 10.1371/journal.pone.0158784
Kim, E., Xiong, Y., Kang, B. H., and Sung, S. (2019). Identification of long noncoding RNAs in the developing endosperm of Maize. Methods Mol. Biol. 1933, 49–65. doi: 10.1007/978-1-4939-9045-0_3
Kim, E. D., and Sung, S. (2012). Long noncoding RNA: unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 17, 16–21. doi: 10.1016/j.tplants.2011.10.008
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Kwenda, S., Birch, P. R., and Moleleki, L. N. (2016). Genome-wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genomics 17:614. doi: 10.1186/s12864-016-2967-9
Li, L., Eichten, S. R., Shimizu, R., Petsch, K., Yeh, C.T., Wu, W., et al. (2014). Genome-wide discovery and characterization of maize long non-coding RNAs. Genome. Biol. 15:R40. doi: 10.1186/gb-2014-15-2-r40
Li, M., Cao, A., Wang, R., Li, Z., Li, S., and Wang, J. (2020). Genome-wide identification and integrated analysis of lncRNAs in rice backcross introgression lines (BC(2)F(12)). BMC Plant Biol. 20:300. doi: 10.1186/s12870-020-02508-y
Li, R., Fu, D., Zhu, B., Luo, Y., and Zhu, H. (2018). CRISPR/Cas9-mediated mutagenesis of lncRNA1459 alters tomato fruit ripening. Plant J. 94, 513–524. doi: 10.1111/tpj.13872
Lim, C. W., Baek, W., Jung, J., Kim, J. H., and Lee, S. C. (2015). Function of ABA in stomatal defense against biotic and drought stresses. Int. J. Mol. Sci. 16, 15251–15270. doi: 10.3390/ijms160715251
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Luan, Y., Cui, J., Zhai, J., Li, J., Han, L., and Meng, J. (2015). High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta 241, 1405–1416. doi: 10.1007/s00425-015-2267-7
Ma, J., Bai, X., Luo, W., Feng, Y., Shao, X., Bai, Q., et al. (2019). Genome-Wide identification of long noncoding RNAs and their responses to salt stress in two closely related poplars. Front. Genet. 10:777. doi: 10.3389/fgene.2019.00777
Ma, W., Chen, C., Liu, Y., Zeng, M., Meyers, B. C., Li, J., et al. (2018). Coupling of microRNA-directed phased small interfering RNA generation from long noncoding genes with alternative splicing and alternative polyadenylation in small RNA-mediated gene silencing. New Phytol. 217, 1535–1550. doi: 10.1111/nph.14934
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
McLellan, H., Boevink, P. C., Armstrong, M. R., Pritchard, L., Gomez, S., Morales, J., et al. (2013). An RxLR effector from Phytophthora infestans prevents re-localisation of two plant NAC transcription factors from the endoplasmic reticulum to the nucleus. PLoS Pathog. 9:e1003670. doi: 10.1371/journal.ppat.1003670
Meng, X., Zhang, P., Chen, Q., Wang, J., and Chen, M. (2018). Identification and characterization of ncRNA-associated ceRNA networks in Arabidopsis leaf development. BMC Genomics 19:607. doi: 10.1186/s12864-018-4993-2
Ohshima, K., Sako, K., Hiraishi, C., Nakagawa, A., Matsuo, K., Ogawa, T., et al. (2000). Potato tuber necrotic ringspot disease occurring in Japan: its association with Potato virus Y Necrotic Strain. Plant Dis. 84, 1109–1115. doi: 10.1094/pdis.2000.84.10.1109
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Qin, T., Zhao, H., Cui, P., Albesher, N., and Xiong, L. (2017). A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance. Plant Physiol. 175, 1321–1336. doi: 10.1104/pp.17.00574
Raffaele, S., Farrer, R. A., Cano, L. M., Studholme, D. J., Maclean, D., Thines, M., et al. (2010). Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330, 1540–1543. doi: 10.1126/science.1193070
Ramírez-González, R. H., Borrill, P., Lang, D., Harrington, S. A., Brinton, J., Venturini, L., et al. (2018). The transcriptional landscape of polyploid wheat. Science 361:eaar6089. doi: 10.1126/science.aar6089
Reddy, A. S., Marquez, Y., Kalyna, M., and Barta, A. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. doi: 10.1105/tpc.113.117523
Remy, E., Cabrito, T. R., Batista, R. A., Hussein, M. A., Teixeira, M. C., Athanasiadis, A., et al. (2014). Intron retention in the 5’UTR of the novel ZIF2 transporter enhances translation to promote zinc tolerance in arabidopsis. PLoS Genet. 10:e1004375. doi: 10.1371/journal.pgen.1004375
Saunders, D. G., Breen, S., Win, J., Schornack, S., Hein, I., Bozkurt, T. O., et al. (2012). Host protein BSL1 associates with Phytophthora infestans RXLR effector AVR2 and the Solanum demissum immune receptor R2 to mediate disease resistance. Plant Cell 24, 3420–3434. doi: 10.1105/tpc.112.099861
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sun, X., Zheng, H., and Sui, N. (2018). Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 503, 402–407. doi: 10.1016/j.bbrc.2018.07.072
Sun, Y., Pri-Tal, O., Michaeli, D., and Mosquna, A. (2020). Evolution of abscisic acid signaling module and its perception. Front. Plant Sci. 11:934. doi: 10.3389/fpls.2020.00934
Tang, W., Zheng, Y., Dong, J., Yu, J., Yue, J., Liu, F., et al. (2016). Comprehensive transcriptome profiling reveals long noncoding RNA expression and alternative splicing regulation during fruit development and ripening in kiwifruit (Actinidia chinensis). Front. Plant Sci. 7:335. doi: 10.3389/fpls.2016.00335
Tian, H., Guo, F., Zhang, Z., Ding, H., Meng, J., Li, X., et al. (2020). Discovery, identification, and functional characterization of long noncoding RNAs in Arachis hypogaea L. BMC Plant Biol. 20:308. doi: 10.1186/s12870-020-02510-4
Tian, Z., He, Q., Wang, H., Liu, Y., Zhang, Y., Shao, F., et al. (2015). The potato ERF transcription factor StERF3 negatively regulates resistance to Phytophthora infestans and salt tolerance in Potato. Plant Cell Physiol. 56, 992–1005. doi: 10.1093/pcp/pcv025
Wang, H. V., and Chekanova, J. A. (2017). Long Noncoding RNAs in Plants. Adv. Exp. Med. Biol. 1008, 133–154. doi: 10.1007/978-981-10-5203-3_5
Wang, J., Gao, C., Li, L., Cao, W., and Chu, Z. (2019). Transgenic RXLR effector PITG_15718.2 suppresses immunity and reduces Vegetative growth in Potato. Int. J. Mol. Ences 20:3031. doi: 10.3390/ijms20123031
Wang, J., Yang, Y., Jin, L., Ling, X., Liu, T., Chen, T., et al. (2018). Re-analysis of long non-coding RNAs and prediction of circRNAs reveal their novel roles in susceptible tomato following TYLCV infection. BMC Plant Biol. 18:104. doi: 10.1186/s12870-018-1332-3
Wang, Y., Luo, X., Sun, F., Hu, J., Zha, X., Su, W., et al. (2018). Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 9:3516. doi: 10.1038/s41467-018-05829-7
Wang, T., Zhao, M., Zhang, X., Liu, M., Yang, C., Chen, Y., et al. (2017). Novel phosphate deficiency-responsive long non-coding RNAs in the legume model plant Medicago truncatula. J. Exp. Bot. 68, 5937–5948. doi: 10.1093/jxb/erx384
Wen, Z., Mei, Y., Zhou, J., Cui, Y., Wang, D., and Wang, N. N. (2020). SAUR49 can positively regulate leaf senescence by suppressing SSPP in Arabidopsis. Plant Cell Physiol. 61, 644–658. doi: 10.1093/pcp/pcz231
Wullings, B. A., Van Beuningen, A. R., Janse, J. D., and Akkermans, A. D. (1998). Detection of Ralstonia solanacearum, which causes brown rot of potato, by fluorescent in situ hybridization with 23S rRNA-targeted probes. Appl. Environ. Microbiol. 64, 4546–4554. doi: 10.1128/aem.64.11.4546-4554.1998
Wu, Y., Sharp, R. E., Durachko, D. M., and Cosgrove, D. J. (1996). Growth maintenance of the maize primary root at low water potentials involves increases in cell-wall extension properties, expansin activity, and wall susceptibility to expansins. Plant Physiol. 111, 765–772. doi: 10.1104/pp.111.3.765
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xin, M., Wang, Y., Yao, Y., Song, N., Hu, Z., Qin, D., et al. (2011). Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 11:61. doi: 10.1186/1471-2229-11-61
Xu, J., Tian, J., Belanger, F. C., and Huang, B. (2007). Identification and characterization of an expansin gene AsEXP1 associated with heat tolerance in C3 Agrostis grass species. J. Exp. Bot. 58, 3789–3796. doi: 10.1093/jxb/erm229
Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., Ni, P., et al. (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. doi: 10.1038/nature10158
Yang, S., Tang, F., and Zhu, H. (2014). Alternative splicing in plant immunity. Int. J. Mol. Sci. 15, 10424–10445. doi: 10.3390/ijms150610424
Yang, T., Zhou, H., Liu, P., Yan, L., Yao, W., Chen, K., et al. (2017). lncRNA PVT1 and its splicing variant function as competing endogenous RNA to regulate clear cell renal cell carcinoma progression. Oncotarget 8, 85353–85367. doi: 10.18632/oncotarget.19743
Yogendra, K. N., and Kushalappa, A. C. (2016). Integrated transcriptomics and metabolomics reveal induction of hierarchies of resistance genes in potato against late blight. Funct. Plant Biol. 43, 766–782. doi: 10.1071/fp16028
Yu, T., Tzeng, D. T. W., Li, R., Chen, J., Zhong, S., Fu, D., et al. (2019). Genome-wide identification of long non-coding RNA targets of the tomato MADS box transcription factor RIN and function analysis. Ann. Bot. 123, 469–482. doi: 10.1093/aob/mcy178
Yu, Y., Zhou, Y. F., Feng, Y. Z., He, H., Lian, J. P., Yang, Y. W., et al. (2020). Transcriptional landscape of pathogen-responsive lncRNAs in rice unveils the role of ALEX1 in jasmonate pathway and disease resistance. Plant Biotechnol. J. 18, 679–690. doi: 10.1111/pbi.13234
Yuan, J. H., Liu, X. N., Wang, T. T., Pan, W., Tao, Q. F., Zhou, W. P., et al. (2017). The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat. Cell Biol. 19, 820–832. doi: 10.1038/ncb3538
Zaynab, M., Fatima, M., Abbas, S., Umair, M., Sharif, Y., and Raza, M. A. (2018). Long non-coding RNAs as molecular players in plant defense against pathogens. Microb. Pathog. 121, 277–282. doi: 10.1016/j.micpath.2018.05.050
Zhang, H., Hu, W., Hao, J., Lv, S., Wang, C., Tong, W., et al. (2016). Genome-wide identification and functional prediction of novel and fungi-responsive lincRNAs in Triticum aestivum. BMC Genomics 17:238. doi: 10.1186/s12864-016-2570-0
Zhang, L., Wang, M., Li, N., Wang, H., Qiu, P., Pei, L., et al. (2018). Long noncoding RNAs involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 16, 1172–1185. doi: 10.1111/pbi.12861
Zhang, T., Liang, Q., Li, C., Fu, S., Kundu, J. K., Zhou, X., et al. (2020). Transcriptome analysis of rice reveals the lncRNA-mRNA regulatory network in response to rice black-streaked Dwarf virus infection. Viruses 12:951. doi: 10.3390/v12090951
Zhang, Z., Liu, Y., Ding, P., Li, Y., Kong, Q., and Zhang, Y. (2014). Splicing of receptor-like kinase-encoding SNC4 and CERK1 is regulated by two conserved splicing factors that are required for plant immunity. Mol. Plant. 7, 1766–1775. doi: 10.1093/mp/ssu103
Zhao, X., Gan, L., Yan, C., Li, C., Sun, Q., Wang, J., et al. (2019). Genome-Wide identification and characterization of long non-coding RNAs in peanut. Genes (Basel) 10:536. doi: 10.3390/genes10070536
Zhao, X., Li, J., Lian, B., Gu, H., Li, Y., and Qi, Y. (2018). Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 9:5056. doi: 10.1038/s41467-018-07500-7
Zhong, C., Ren, Y., Qi, Y., Yu, X., Wu, X., and Tian, Z. (2018). PAMP-responsive ATL gene StRFP1 and its orthologue NbATL60 positively regulate Phytophthora infestans resistance in potato and Nicotiana benthamiana. Plant Sci. 270, 47–57. doi: 10.1016/j.plantsci.2018.01.016
Zhou, X., Cui, J., Meng, J., and Luan, Y. (2020). Interactions and links among the noncoding RNAs in plants under stresses. Theor. Appl. Genet. 133, 3235–3248. doi: 10.1007/s00122-020-03690-1
Keywords: long non-coding RNA, Solanum tuberosum L., Phytophthora infestans, alternative splicing, TCseq, transiently overexpression
Citation: Cao W, Gan L, Wang C, Zhao X, Zhang M, Du J, Zhou S and Zhu C (2021) Genome-Wide Identification and Characterization of Potato Long Non-coding RNAs Associated With Phytophthora infestans Resistance. Front. Plant Sci. 12:619062. doi: 10.3389/fpls.2021.619062
Received: 20 October 2020; Accepted: 06 January 2021;
Published: 10 February 2021.
Edited by:
Giorgio Gambino, National Research Council (CNR), ItalyReviewed by:
Yushi Luan, Dalian University of Technology, ChinaCopyright © 2021 Cao, Gan, Wang, Zhao, Zhang, Du, Zhou and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changxiang Zhu, emhjaHhAc2RhdS5lZHUuY24=; Shumei Zhou, emhvdXNtQHNkYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.