 Mitsukazu Sakata1
,2
Noriko Takano-Kai1
Yuta Miyazaki1
†Hiroyuki Kanamori3
Mitsukazu Sakata1
,2
Noriko Takano-Kai1
Yuta Miyazaki1
†Hiroyuki Kanamori3
 Jianzhong Wu3
Takashi Matsumoto3
,4
Kazuyuki Doi1
,5
Jianzhong Wu3
Takashi Matsumoto3
,4
Kazuyuki Doi1
,5
 Hideshi Yasui1
Atsushi Yoshimura1
Hideshi Yasui1
Atsushi Yoshimura1
 Yoshiyuki Yamagata1
*
Yoshiyuki Yamagata1
*- 1Plant Breeding Laboratory, Faculty of Agriculture, Kyushu University, Fukuoka, Japan
- 2Faculty of Agriculture and Marine Science, Kochi University, Nankoku, Japan
- 3Institute of Crop Science, National Agriculture and Food Research Organization, Tsukuba, Japan
- 4Laboratory of Plant Molecular Breeding, Department of Bioscience, Tokyo University of Agriculture, Tokyo, Japan
- 5Graduate School of Bioagricultural Sciences, Nagoya University, Nagoya, Japan
Postzygotic reproductive isolation maintains species integrity and uniformity and contributes to speciation by restricting the free gene flow between divergent species. In this study we identify causal genes of two Mendelian factors S22A and S22B on rice chromosome 2 inducing F1 pollen sterility in hybrids between Oryza sativa japonica-type cultivar Taichung 65 (T65) and a wild relative of rice species Oryza glumaepatula. The causal gene of S22B in T65 encodes a protein containing DUF1668 and gametophytically expressed in the anthers, designated S22B_j. The O. glumaepatula allele S22B-g, allelic to S22B_j, possesses three non-synonymous substitutions and a 2-bp deletion, leading to a frameshifted translation at the S22B C-terminal region. Transcription level of S22B-j and/or S22B_g did not solely determine the fertility of pollen grains by genotypes at S22B. Western blotting of S22B found that one major band with approximately 46 kDa appeared only at the mature stage and was reduced on semi-sterile heterozygotes at S22B, implying that the 46 kDa band may associated in hybrid sterility. In addition, causal genes of S22A in T65 were found to be S22A_j1 and S22A_j3 encoding DUF1668-containing protein. The allele of a wild rice species Oryza meridionalis Ng at S22B, designated S22B_m, is a loss-of-function allele probably due to large deletion of the gene lacking DUF1668 domain and evolved from the different lineage of O. glumaepatula. Phylogenetic analysis of DUF1668 suggested that many gene duplications occurred before the divergence of current crops in Poaceae, and loss-of-function mutations of DUF1668-containing genes represent the candidate causal genetic events contributing to hybrid incompatibilities. The duplicated DUF1668-domain gene may provide genetic potential to induce hybrid incompatibility by consequent mutations after divergence.
Introduction
Hybrid incompatibilities (HIs), with respect to both intra‐ and interspecific hybridizations, are a widespread mechanism of postzygotic reproductive isolation, which restricts free gene flow between divergent species (Coyne and Orr, 2004). In plants, post-zygotic reproductive isolation occurs throughout the various life cycle stages of hybrids, from fertilization to sexual reproduction (inviability), gametogenesis and fertilization of gametes (sterility) in F1 hybrids, and in the subsequent generation (hybrid breakdown; Stebbins, 1950). In cross breeding for genetic improvement in crop species, HI frequently hinders unflagging efforts of hybridization between cultivated and wild species to exploit useful genes and quantitative trait loci (QTLs) from wild genetic resources. Therefore, understanding genetic and molecular basis of HI play an important role in broadening a gene pool for crop improvement and in understanding evolutionary pathway of postzygotic reproductive isolation in crop species. Chromosomal or genic changes that occur during species divergence from common ancestral species are considered to be the main causal events in the evolution of HI (Maheshwari and Barbash, 2011). With respect to genic incompatibility, incompatible combinations of genes, each of which is generally an ancestral and variant allele derived from two reproductively isolated species, combine in the sporophytes or gametophytes of hybrids, resulting in maladaptive phenotypes, leading to inviability, sterility, and/or hybrid breakdown. These incompatible zygotes or gametes with reduced fitness are subsequently eliminated in hybrid populations. However, nucleotide variants causing HI are heterozygous only at birth; thus, a simple genetic model assuming a single locus is insufficient to explain the evolution of HI (Coyne and Orr, 2004). Therefore, it is currently central question of evolutionary genetics of HI that how alleles causing HI can evolve and be maintained in a population without falling into fitness valleys.
Bateson-Dobzhansky-Muller (BDM) incompatibilities are a classical genetic model (Dobzhansky, 1937; Muller, 1942), which proposes that reciprocal genetic changes in divergent species at two or multiple loci allow for the maintenance of new alleles causing HI without negative selection in the intermediate step (Coyne and Orr, 2004; Noor and Feder, 2006). The BDM incompatibilities model emerged based on observations of genetic interactions among multiple loci using a forward genetics approach with hybrid populations derived from inter‐ or intraspecific crosses. The HI system is controlled by the interaction of multiple genes located on different chromosomes, and epistatic complementarity between two genetic loci is exhibited in many plant species such as lettuce (Jeuken et al., 2009), Arabidopsis (Bomblies et al., 2007), cotton (Deng et al., 2019), monkey flower (Zuellig and Sweigart, 2018), and wheat (Matsuda et al., 2012). Gene cloning studies have also supported the molecular basis of BDM incompatibilities in plants (Rieseberg and Blackman, 2010; Chen et al., 2016). Gene duplication via whole-genome duplication or segmental genome duplication is a primordial event for gene diversification by neofunctionalization and subfunctionalization, as well as for species diversification due to nonfunctionalization (Lynch and Conery, 2000). Reciprocal losses of duplicated genes in divergent plant species were shown to cause F1 pollen sterility at the S27/S28 (Yamagata et al., 2010), DPL1/DPL2 (Mizuta et al., 2010), and DGS1/DGS2 (Nguyen et al., 2017) loci, and could lead to hybrid breakdown (Bikard et al., 2009).
When incompatible genes from two divergent populations are closely linked and cause BDM incompatibilities in the hybrid, inheritance of HI appears as a genetic interaction of the gene complex (or haplotype) at a single Mendelian locus in heterozygotes in plants. For example, in rice, intra-subspecific hybrids of cultivated rice carry two adjacent genes encoding F-Box protein and SUMO E3 ligase-like protein at the Sa locus to induce pollen sterility (Long et al., 2008), and endoplasmic reticulum stress due to incompatible interactions among heat shock protein Hsp70, an unknown protein with a transmembrane region, and eukaryotic aspartic proteases resulted in embryo sac sterility governed by S5 (Yang et al., 2012). Recently, the S1 locus identified in hybrids between Oryza sativa and Oryza glaberrima that induces F1 pollen and embryo sac sterility was found to have originated from a gene complex consisting of S1A6 and S1A4 derived from an O. sativa allele (Xie et al., 2019) and S1TPR/SSP encoding a peptidase-like protein derived from an O. glaberrima allele at S1 (Xie et al., 2017; Koide et al., 2018). As another mechanism, multiple gene copies derived from tandem duplications can acquire new promoter sequences and suppress the expression of genes essential for pollen formation of alternative alleles (Shen et al., 2017).
It has been controversial whether specific protein families or domains are likely to induce HI, although dozens of HI genes have been isolated in plants to date (Rieseberg and Blackman, 2010). HI, including necrosis or weakness, is frequently caused by deleterious interactions of pathogen and insect resistance (R) genes in plants (Bomblies and Weigel, 2007), affecting autoimmune responses to ultimately reduce growth, deregulate cell death, and cause sterility (Bomblies et al., 2007; Alcázar et al., 2009; Jeuken et al., 2009; Yamamoto et al., 2010; Chen et al., 2014; Atanasov et al., 2018). In rice, the protease genes involved in hybrid pollen sterility are located at S1 (Xie et al., 2017; Koide et al., 2018) and involved in embryo sac sterility are located at S5 (Yang et al., 2012). Genes encoding domain unknown function DUF1618-containing protein at the Sc allele on chromosome 3 (Shen et al., 2017) and at HSA1A on chromosome 12 in an inter-subspecific hybrid of O. sativa (Kubo et al., 2016) were recognized as causal genes of male hybrid sterility. If a specific pattern of functional change or loss of function in a particular domain such as DUF1618 has the potential to induce HI, sporadic independent origins of HI might have occurred across species or genera, which would allow for prediction of the evolution of HI and speciation in geographically isolated species. However, these domains responsible for speciation have been little reported to date.
In the genus Oryza, six wild species with an AA genome show an allopatric or sympatric distribution in several continents. Oryza rufipogon Griff. and Oryza nivara Sharma et Shastry are wild species in South and Southeast Asia, Oryza longistaminata A. Chev. & Roehr. and Oryza barthii A. Chev. are wild species in Africa, Oryza glumaepatula Steud. is a wild species in South America, and Oryza meridionalis Ng is a wild species in Australia (Vaughan et al., 2005). Two cultivated species, O. sativa L. and O. glaberrima Steud., are considered to have been domesticated from O. rufipogon and O. barthii, respectively. More than 50 loci/QLTs for hybrid sterility have been reported in inter‐ and intraspecific hybrids of rice. Incompatible genotypes of the sporophyte or gametophyte determine sterility, and causal genes at 11 loci have been characterized based on molecular evidence (Chen et al., 2008; Long et al., 2008; Mizuta et al., 2010; Yamagata et al., 2010; Yang et al., 2012; Kubo et al., 2016; Yu et al., 2016; Nguyen et al., 2017; Shen et al., 2017; Koide et al., 2018; Xie et al., 2019).
In backcrossed hybrid progenies derived from a cross between O. sativa japonica-type cultivar Taichung 65 (T65) and O. glumaepatula accession IRGC105668 in the genetic background of T65, the F1 pollen sterility gene S22 was identified as a Mendelian genetic factor on the short-arm end of chromosome 2 (Sobrizal et al., 2000). The genomic regions of S22 responsible for pollen sterility were dissected into the two independent genetic loci: S22A and S22B (Sakata et al., 2014). In the T65 genetic background, plants with the S22A-T65+/S22A-glums|S22B-T65+/S22B-T65+ genotype (S22A_SS plants) showed approximately 50% pollen fertility (semi-sterility) due to sterility of pollen grains carrying the “sterile allele” S22A-glums. Similarly, plants carrying the S22A-T65+/S22A-T65+|S22B-T65+/S22B-glums genotype (S22B_SS plants) showed pollen semi-sterility because of sterility of pollen grains carrying the “sterile allele” S22B-glums. The coupling phase linkage of S22A-glums and S22B-glums on O. glumaepatula-derived chromosomal segments could explain the initial identification of S22 as a single Mendelian factor (Sakata et al., 2014).
In this study, to elucidate molecular players determining postzygotic reproductive isolation between O. sativa and O. glumaepatula, causal genes of S22A and S22B for F1 pollen sterility between these divergent species were identified by map-based cloning approach. The allelism of another allele in O. meridionalis possibly at S22B also investigated. Since causal genes of HI both at S22A and S22B were found to encode DUF1668-containing proteins, which were found to be diversified in Poaceae, phylogenetic analysis of DUF1668 domain in Poaceae was conducted to know evolutionary timing of their occurrence of duplicated copies during divergence of Poaceae.
Materials and Methods
Plant Materials and Phenotyping
The backcrossed progenies carrying chromosomal segments derived from O. glumaepatula accession IRGC105668 at S22A or S22B in the O. sativa L. cultivar T65 genetic background were developed in a previous study (Sakata et al., 2014). In this study, plants with the genotypes S22A-T65+/S22A-glums|S22B-T65+/S22B-T65+, S22A-T65+/S22A-T65+|S22B-T65+/S22B-glums, or S22A-T65+/S22A-glums|S22B-T65+/S22B-glums were designated S22A_SS, S22B_SS, or S22A+B_SS plants, respectively. These lines were maintained by marker-assisted selection using the simple sequence repeat (SSR) markers RM12317 and RM7451 for S22A_SS plants, RM7033 and RM279 for S22B_SS plants, and RM12317 and RM279 for S22A+B_SS plants. Phenotypes of S22A_SS, S22A+B_SS, and S22B_SS plants were discriminated according to the morphology of the sterile pollen grains stained with a 1% iodine-potassium iodide solution. Semi-sterile plants, in which almost all of the sterile pollen grains show no staining, stain slightly, and stain dark brown in color and are smaller than normal grains, were classified as S22A+B_SS, S22A_SS, and S22B_SS (Supplementary Figure S1). Transmission frequency of allele was estimated by maximum likelihood method (Supplementary Methods). The homozygous plants for S22B-glums and S22A-T65+ obtained in the previous study (Sakata et al., 2014) were used in this study.
Map-Based Cloning
Total genomic DNA was extracted according to the method described by Dellaporta et al. (1983), with minor modifications. Primer sequences of the polymerase chain reaction (PCR)-based DNA markers used in this study are listed in Supplementary Table S1. Each 15-μl reaction mixture consisted of 50 mM KCl, 10 mM Tris (pH 9.0), 1.5 mM MgCl2, 200 mM dNTPs, 0.2 mM primers, 0.75 units of Taq polymerase (Takara, Otsu, Japan), and 10 ng genomic DNA template. PCR was performed in a GeneAmp PCR System 9,700 (Applied Biosystems, Foster City, CA, United States). The cycling profile was an initial denaturation step at 95°C for 5 min; 35 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 40 s; and a final elongation step at 72°C for 7 min. Amplified products were electrophoresed on a 4% agarose gel in 0.5× TBE buffer. Agrobacterium-mediated transformation was conducted as described by Yamagata et al. (2010). In brief, genomic fragments digested by restriction enzymes were cloned into the Ti-plasmid binary vector pPZP2H-lac (Fuse et al., 2001), and were then transformed into S22A_SS and S22B_SS plants. The copy numbers of transgenes were analyzed by quantitative PCR (qPCR) in an MX3000P QPCR system (Agilent Technologies, Santa Clara, CA, United States) using QuantiTect SYBR Green PCR Kits (Qiagen, Venlo, The Netherlands) according to our previous analysis (Nguyen et al., 2017). The gene models in the mapping region were obtained from MSU Rice Genome Annotation Project Database Release 7 (MSU7; http://rice.plantbiology.msu.edu/) in O. sativa cv. Nipponbare Os-Nipponbare-Reference-IRGSP-1.0 and O. glumaepatula W1183 accession ALNU02000000 (Jacquemin et al., 2013). The homologous sequences were searched in bl2seq program with cut off score at 1e-50 (Altschul et al., 1990).
Expression Analysis
For temporal expression analysis of S22B, the developmental stages of male gametophytes in T65, S22B-glums heterozygotes, and S22B-glums homozygotes were observed under a light microscope without staining. Fifty anthers were sampled from the unicellular, bicellular, and mature stages, respectively, which were placed into 1.5-ml tubes and then frozen in liquid nitrogen. Total RNA was extracted using Trizol reagent (Life Technologies Japan, Tokyo, Japan) from the ground anther using a Multibeads shocker (Yasui Kikai, Osaka, Japan). Protein fractions of the Trizol extract were reserved for subsequent western blotting analysis. Extracted RNA was treated with DNase I (Takara, Otsu, Japan) to degrade contaminated genomic DNA. The first-strand cDNA was synthesized from approximately 100 ng of the extracted RNA and then reverse-transcribed using Revertra Ace (Toyobo, Otsu, Japan). Reverse transcription-quantitative PCR (RT-qPCR) was conducted in a MX3000P QPCR system (Agilent Technologies) using QuantiTect SYBR Green PCR Kits (Qiagen). For RT-qPCR of S22B, a pair of primers, 5'-CTC TGC CAA CTT CTG CAT CGC CAG G-3'/5'-GCT GAT AAG CTT GTA CAT CTC CGA C-3' and 5'-TGG AGG ATC CAT CTT GGC ATC AT-3'/5'-ACA GCT CCT CTT GGC TTA GCA-3' were used for amplification of S22B and the actin 1 gene (Os03g0718100) as an internal control, respectively.
For the promoter-β-glucuronidase (GUS) assay, genomic sequences of the 1,572-bp region upstream of the initiation codon (ATG) of S22B-T65+ were cloned into the Gateway-entry vector pENTR/TOPO using pENTR Directional TOPO Cloning Kits (Life Technologies Japan). The cloned insert was transferred into the destination vector pGWB3 (Nakagawa et al., 2007) using LR clonase (Life Technologies Japan) to fuse the promoter and GUS gene derived from pGWB3. The construct was transformed to T65 by Agrobacterium-mediated transformation.
Subcellular Localization
The coding sequence at S22B was amplified from the Nipponbare full-length cDNA clone J023058D10 (AK070727) provided by the National Agriculture and Food Research Organization (NARO), Japan, and was cloned into the Gateway-entry vector pENTR/TOPO (Life Technologies Japan). The GFP-fused gene at N-terminal under a control of 35S CaMV promoter was constructed using LR clonase (Life Technologies Japan) into the destination binary vector pGWB6 (Nakagawa et al., 2007). The construct was transformed into T65 by Agrobacterium-mediated transformation. The root of the obtained T0 plants was observed in a fluorescence microscope (Biozero, BZ-8000, Keyence, Osaka, Japan) after 500 nM of mitochondrial fluorescent dye MitoTracker Red CMXRos staining.
Western Blotting
For western blotting of the S22B product, we prepared a rabbit polyclonal antibody against two synthesized 14-amino acid peptide sequences: N-KLATPLDAGAHDG-C and N-ISGGRKPEQHSLLP-C (Eurofins Genomics, Tokyo, Japan). The antibody specificity was confirmed by western blotting of a 6×His + S22B-T65 fused recombinant protein expressed in Escherichia coli strain BL21. The protein fractions extracted by Trizol were mixed with 2× Laemmli sample buffer and 2-mercaptoethanol, and then run on a 12% TGX gel (Biorad, Hercules, CA, United States). The proteins were transferred onto a polyvinylidene difluoride membrane with a 0.2-μm pore size (ATTO, Tokyo, Japan). Goat anti-rabbit IgG (H + L) antibody and horseradish peroxidase-conjugated antibody (Bio-Rad) were used for the secondary antibody reactions, and detection was performed using chemiluminescence detection with Western BLoT Hyper HRP Substrate (Takara Bio, Shiga, Japan).
Phylogenetic Analysis of DUF1668-Containing Sequences
All protein sequence data deduced in Setaria viridis, Setaria italica, Panicum virgatum, Botryococcus distachyon, Sorghum bicolor, Zea mays, Botryococcus stacei, and Oryza sativa were downloaded from the Phytozome 12 database (Goodstein et al., 2012). The protein sequences harboring the DUF1668 domain were detected by hidden Markov model searches using hmmsearch software1 with a cutoff score of 1e−8. After the amino acid sequences of the DUF1668 domains were aligned using MUSCLE software with default parameters (Edgar, 2004), a phylogenetic tree based on maximum-likelihood inference was constructed in RAxML v. 8.2.8 software (Stamatakis, 2014) and drawn in FigTree v. 1.4.2 software.2
Results
Map-Based Cloning of S22B
To narrow down the candidate region of S22B, we conducted high-resolution mapping of S22B in the BC4F6 population in which both S22A and S22B segregated (n = 7,424; Figure 1). Since S22B was previously mapped between the SSR markers RM12329 and RM279 (Sakata et al., 2014), 308 recombinants obtained between the SSR markers RM12329 and RM279 were screened by genotyping at the seedling stage. Linkage analysis of the 308 recombinants demonstrated that five recombinants (9-7, 23-2, 28-1, 38-3, and 39-7) were the most informative plants to map S22B within 18.4 kb of the genomic region in the reference sequence Nipponbare between DNA markers M48 and M46 (Supplementary Figure S2; Figure 1B). For the complementation test of S22B, 13.1-kb Eco72I, 13.5-kb XmnI (XmnI_a), 13.7-kb SalI, and 10.7-kb XmnI (XmnI_b) fragments of the Nipponbare genomic fragment derived from BAC clone OSJNBa008C13 were introduced into S22B_SS plants by Agrobacterium-mediated transformation (Figure 1B). In the non-transgenic progeny of S22B_SS, a reduced transmission efficiency (k) of S22-glums alleles (k = 0.11) via pollen from the theoretical transmission (k = 0.5) was observed, which was attributed to the sterility of pollen grains carrying S22-glums (Table 1). Similarly, the T1 generation derived from the T0 plants transformed by the Eco72I, XmnI_a, and XmnI_b fragments showed segregation distortion of genotypes at RM7033 linking to S22B because of reduced transmission of S22-glums (Table 1). Meanwhile, transformation of the SalI fragment significantly altered the segregation of the genotype at RM7033 in the T1 generation (Table 1), suggesting that pollen grains harboring S22-glums recovered pollen fertility by the transformation of the SalI fragment. Pollen fertility of the T1 plants derived from the T0 plant with the SalI fragment or XmnI_a fragment was observed (Supplementary Figure S3). The two T1 lines, 6 and 7, were derived from the two independent T0 plants carrying a single copy and more than two copies of the SalI fragment, respectively. The heterozygotes at S22B harboring SalI fragment showed recovered pollen fertility as compared with null segregants of heterozygotes or the heterozygotes harboring XmnI_a fragment (Supplementary Figure S3). In the T1 line 6, transgene segregated at one locus and the heterozygous plants at S22B carrying two, one, and zero copies of the SalI fragment showed more than 90%, approximately 75, and 50% of pollen fertility, respectively, whereas the transformation of the XmnI_a fragment did not recover pollen fertility in T1 as a negative control (Supplementary Figure S4). LOC_Os02g04420 was predicted to be located on the SalI fragment but not on the other genomic fragments in the MSU7. These data demonstrated that the causal gene of S22B is LOC_Os02g04420, which encodes a protein containing DUF1668, designated S22B_j (Figures 1C,D). The genomic sequences of S22B including 2,500 bp of upstream region from the transcription initiation site are identical to those of Nipponbare. Sequencing of the O. glumaepatula (Acc. IRGC105668) BAC clone GL47D11 showed that the O. glumaepatula allele S22B-g, allelic to S22B_j, possesses three non-synonymous substitutions CCG(P) > GCG(A), TTC(F) > GTC (V), and CCC(P) > GCC(A) and a 2-bp deletion, leading to a frameshifted translation at the S22B C-terminal region (Supplementary Data 1). Many nucleotide substitutions at the promoter and first exon regions were also observed (Figures 1C,D; Supplementary Data 1). The S22B sequences of T65 and IRGC105668 were deposited to DNA databank of Japan (DDBJ; LC596092 and LC596094).
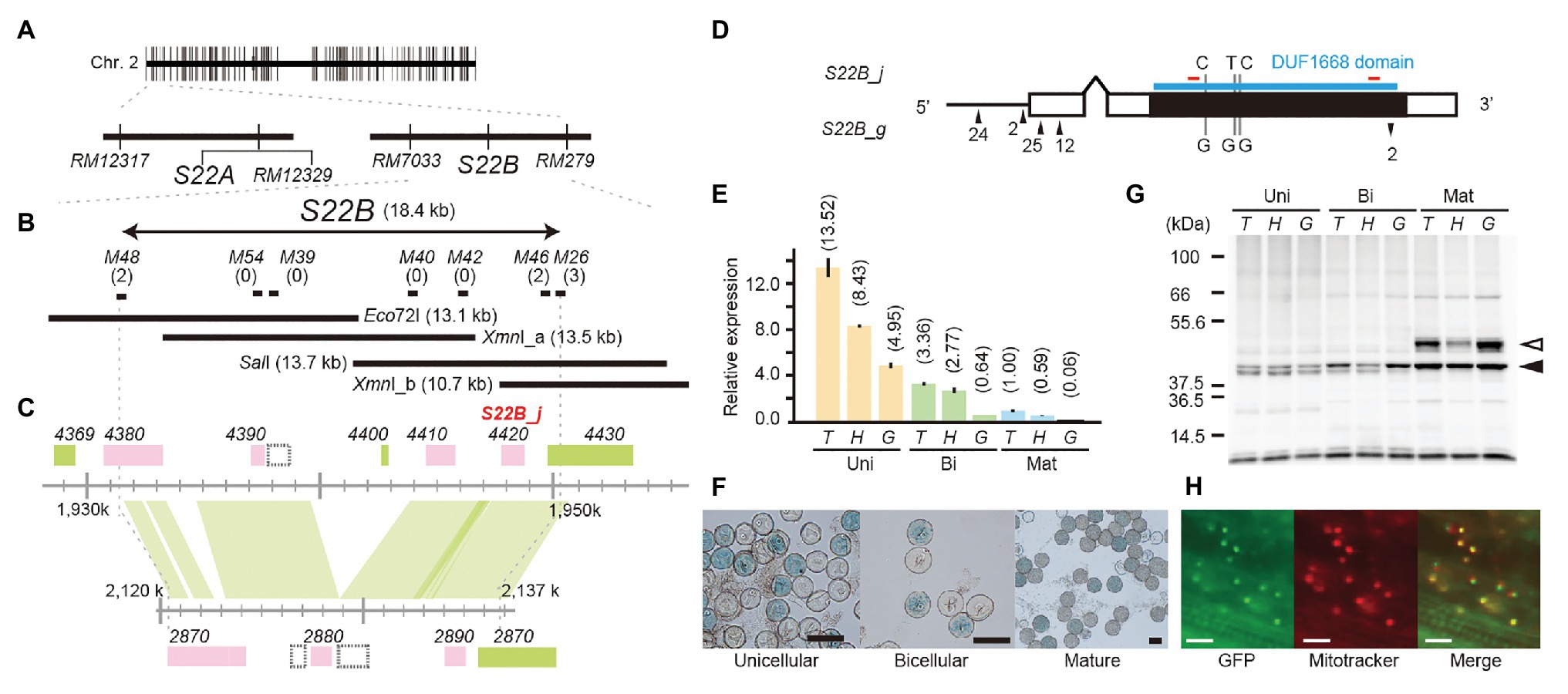
Figure 1. Map-based cloning of S22B. (A) Two tightly linked genes, S22A and S22B, independently induce F1 pollen sterility. (B) High-resolution linkage mapping of S22B. Numbers of recombinants between DNA markers and phenotypes are shown in parentheses. The restriction fragments Eco72I, XmnI_a, SalI, and XmnI_b were used for transformation experiments. (C) Genomic structures on the reference genome assembly of Oryza sativa cv. Nipponbare Os-Nipponbare-Reference-IRGSP-1.0 (Kawahara et al., 2013) and Oryza glumaepatula W1183 accession ALNU02000000 (Jacquemin et al., 2013). Green and pink rectangles represent annotated genes encoded in the plus and minus chain of the reference sequences, respectively. The hatched rectangles represent predicted transposable elements. (D) Nucleotide variations on the 1.5-kb promoter (black horizontal bar) and coding sequence (black rectangles) between S22B_j and S22B_g. Up and down arrowheads represent nucleotide insertions and deletions, respectively, in S22B_g against S22B_j. The DUF1668 domain is shown as a blue line. Red bars show the location of peptide sequences to develop S22B antibody. (E) Reverse transcription-quantitative PCR (RT-qPCR) of S22B in the anthers during male gametogenesis for each genotype at S22B. Relative expression compared to the transcript in homozygotes for T65 on mature stage is represented in parentheses. (F) Promoter-glucuronidase (GUS) assay using the S22B promoter. T, H, and G represent genotypes homozygous for the T65 allele, heterozygous, and homozygous for IRGC105668, respectively. Scale bar: 50 μm. (G) Western blotting of S22B protein in the anthers. Total protein levels among samples were adjusted according to the numbers of collected anthers from 50 spikelets. White and black arrowheads represent the two major bands mentioned in text with the estimated mass of 37 and 46 kDa, respectively. (H) Subcellular localization of N-GFP-S22B in root cells of the stable transformant (T0). Scale bar: 20 μm.
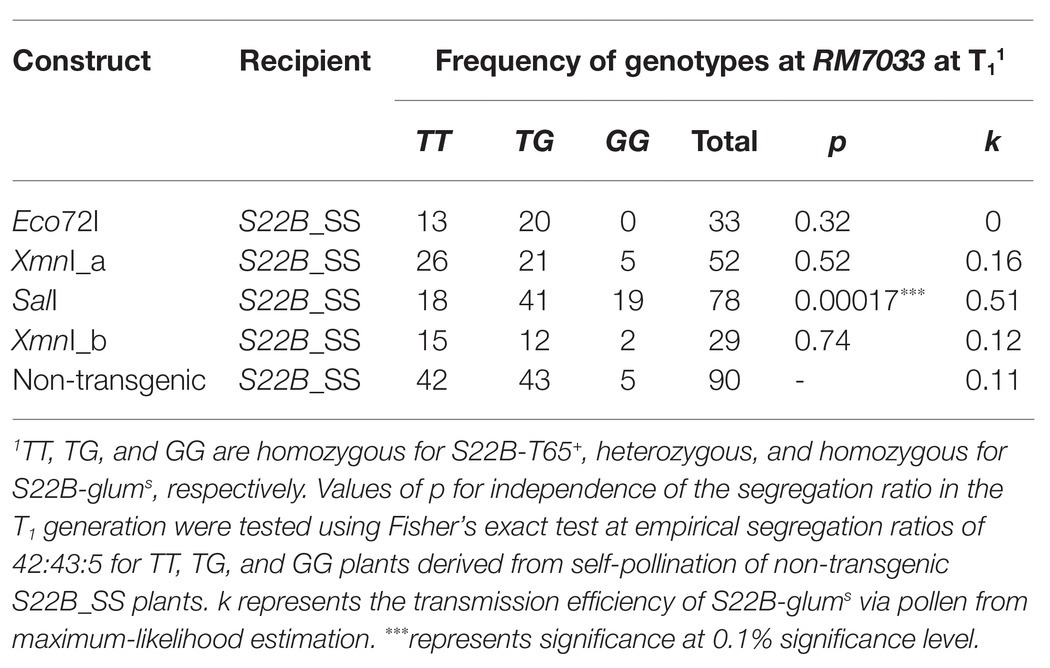
Table 1. Frequency of genotypes at RM7033 in the T1 generation for the complementation test of S22B.
Molecular Characterization of S22B
Expression of S22B from the anthers at the unicellular, bicellular, and mature stages was investigated by RT-qPCR among S22B-T65+ homozygotes, S22B-glums heterozygotes, and S22B-glums homozygotes. Transcript levels gradually decreased during the progression of post-meiotic male gametogenesis (Figure 1E). At the unicellular, bicellular, and mature stages, the S22B expression level was elevated in the order S22B-T65+ homozygotes, heterozygotes, and S22B-glums homozygotes, suggesting that transcription of S22B-glums was not as active as that of S22B-T65+. The promoter-GUS assay was conducted in T0 plants with 1.5 kb of endogenous genomic sequences upstream from the initial codon of S22B-T65 in the unicellular, bicellular, and mature pollen stages (Figure 1F). Half of the pollen grains displayed GUS signals in T0 transgenic plants carrying a single copy of the transgene, but no GUS signals were detected in the anther tissues, demonstrating that S22B is gametophytically expressed in the haploid generation. Together, the results from these expression analyses suggested that the level of gametophytic transcripts of S22B-T65+ or S22B-glums did not solely determine the fertility of pollen grains carrying S22B-T65+ or S22B-glums alleles.
Expression of S22B protein was investigated by western blotting using anti-S22B antibody in the anthers at the unicellular, bicellular, and mature stages (Figure 1G). The deduced molecular mass of S22B-T65+ and S22B-glums proteins was expected to beapproximately 39 kDa based on the coding nucleotide sequences. The density of approximately 37 kDa band estimated in western blotting (37 kDa) gradually increased during the pollen stage. Another major band at a molecular mass probably 46 kDa (46 kDa band) appeared only at the mature stage, and was reduced in the S22B-glums heterozygotes as compared with those of homozygotes for S22B-T65+ or S22B-glums alleles. Although the levels of S22B transcripts were reduced in homozygotes for the S22B-glums allele (Figure 1E), protein levels between homozygotes for the S22B-T65+ or S22B-glums allele were comparable (Figure 1G), implying that the transcript level was sufficient for expression of S22B protein and could be adjusted via feedback regulation.
The intracellular localization of S22B was investigated on root cells of the stable transformant (T0) transformed by the construct of 35SCaMV prom::N-GFP-S22B protein (Figure 1H). Green fluorescent protein (GFP) signals were colocalized with the mitochondrial fluorescent dye MitoTracker Red CMXRos on root cells. The program for subcellular localization prediction in plant cells, TargetP-2.0 (Almagro Armenteros et al., 2019), MitoFate (Fukusawa et al., 2015), and Localizer (Sperschneider et al., 2017) did not find apparent prediction of localization to the mitochondria. WolfPSORT programs (Horton et al., 2007) weakly suggested that S22B possesses mitochondrial-targeting peptide sequences.
Map-Based Cloning of Gametophytic Factors at S22A
High-resolution mapping of S22A was conducted using the BC4F7 population (n = 3,072) derived from the BC4F6 plants heterozygous at the S22A genomic region between the SSR markers RM12317 and RM12350, and homozygous for T65 at S22B in the genetic background of T65 (S22A_SS plants; Figure 2). Our previous study revealed that S22A_SS plants showed approximately 50% pollen sterility due to the sterility of pollen grains harboring S22A-glums; consequently, homozygotes for S22A-T65+, heterozygotes, and homozygotes for S22A-glums segregated at a 1:1:0 ratio (Sakata et al., 2014). The genomic region responsible for S22A was delimited within a 151.4-kb region between the DNA markers M24 and SSR33 (Figure 2A). Homozygous plants for S22A-glums were not obtained in the high-resolution mapping population, suggesting that male gametophytes with S22A-glums in heterozygotes are completely sterile. Within the candidate genomic region, three gene models, LOC_Os02g01790 designated S22A_j1, LOC_Os02g01870 designated S22A_j2, and LOC_Os02g01900 designated S22A_j3, were found to harbor DUF1668 in the reference genomic sequence of Nipponbare based on a Pfam search (Figure 2B). We speculated that the S22A-T65+ allele has a function to gametophytically provide fertility to pollen grains carrying the S22A-T65+ allele in spite of the haploid genotype, as in the case of S22B. Three Acc65I-digested genomic fragments Acc65I_a, Acc65I_b, and Acc65I_c, containing S22A_j1, S22A_j2, or S22A_j3, respectively, were subcloned from the Nipponbare BAC clone OSNBb0096M07 and transformed into S22A_SS plants. The T1 population for S22A_j1 showed segregation of homozygous genotypes for S22A-glums, demonstrating that pollen grains carrying the S22A-glums allele restored pollen fertility due to the effect of the S22A_j1 transgene (Table 2; Supplementary Table S2). The transgene in the T1 population for S22A_j3 resulted in segregation of two homozygotes for S22A-glums, the frequency of which was lower than that in the T1 population in S22A_j1. Moreover, the homozygous plants for S22A-glums were not observed in the T1 population derived from the T0 plants transformed by S22A_j2. If S22A_j2 has equivalent function to S22A_j1, non-segregation of the homozygous plants for S22A-glums means S22A_j2 does not function to restore pollen fertility with S22A-glums. Meanwhile, if S22A_j2 has a partial function to restore the sterility as shown in transformation of S22A_j3, the number of T1 individuals in this population might not have been sufficient to detect complementation of gene function. Therefore, further study is necessary to elucidate the function of this gene. Eventually, it was concluded that transgenes encoding protein with the DUF1668 domain derived from S22A-T65+, at least S22A_j1 and S22A_j3, gametophytically restored the fertility of pollen grains carrying the transgene. The amino acid sequences of S22A_j1, S22A_j2, and S22A_j3 deduced from O. glumaepatula genomic sequences lacked DUF1668, suggesting that these three alleles at S22A-glums lost their function as a DUF1668-harboring protein (Figure 2C).
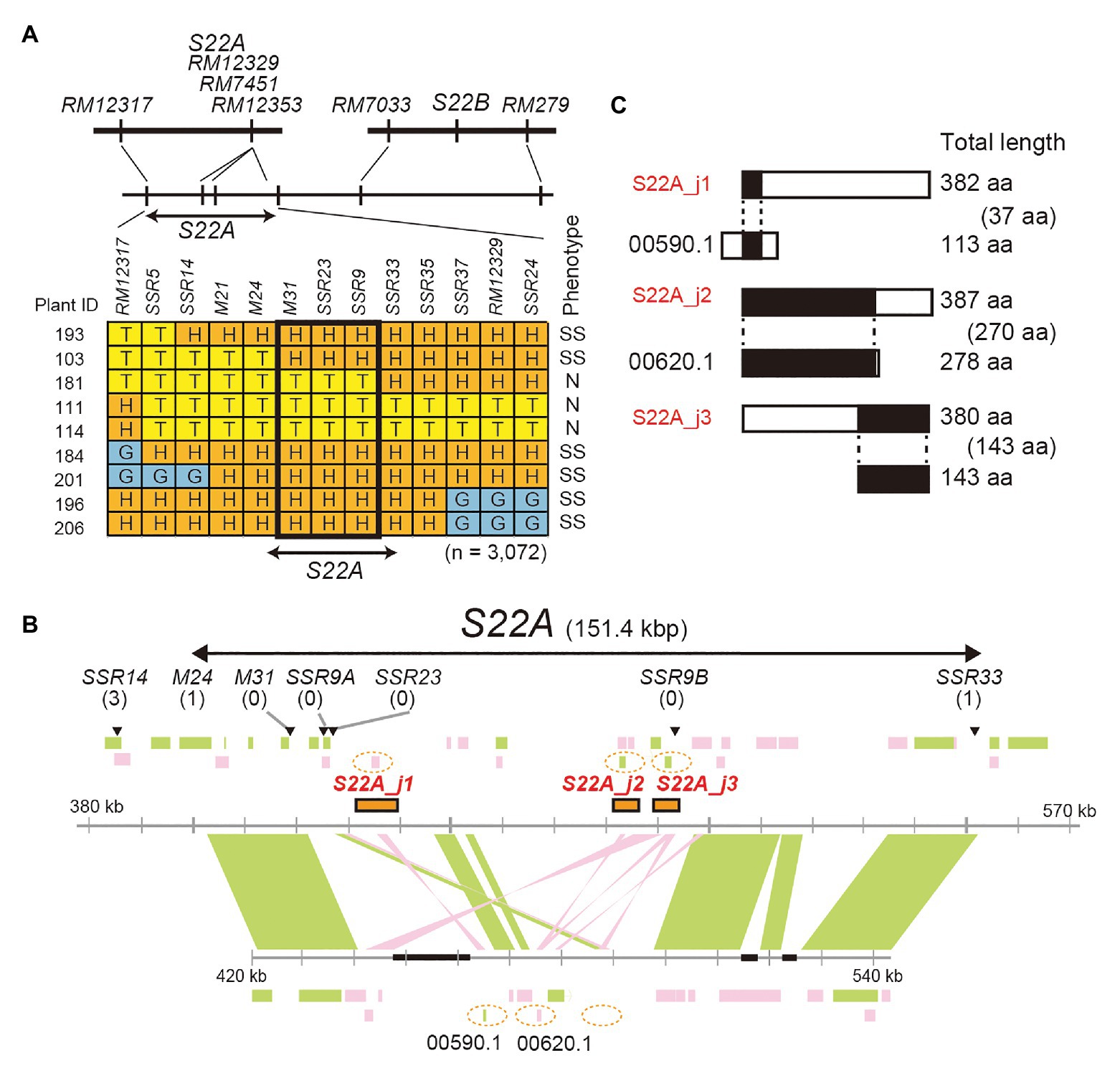
Figure 2. Map-based cloning of S22A. (A) High-resolution linkage mapping of S22A. T, H, and G represent genotypes homozygous for the T65 allele, heterozygous, and homozygous for IRGC105668, respectively. N and SS represent a pollen phenotype of normal fertility and semi-sterility, respectively. (B) Genomic structures on the reference genome assembly of O. sativa cv. Nipponbare Os-Nipponbare-Reference-IRGSP-1.0 and O. glumaepatula W1183 accession ALNU02000000. Three restriction fragments, Acc65I_a, Acc65I_b, and Acc65I_c, including S22A_j1, S22A_j2, and S22A_j3, respectively, were used for transformation experiments. These fragments are shown as orange rectangles. Black boxes represent a sequence gap in the reference sequence. Numbers of recombinants between DNA markers and phenotypes are shown in parentheses. Green and pink rectangles represent annotated genes encoded in the plus and minus chain of the reference sequences, respectively. (C) Putative pseudogenes for DUF1668-containing protein in the O. glumaepatula genome at S22A. A similarity search using the BLASTn program revealed that only parts of DUF1668-containing protein S22A_j1, S22A_j2, and S22A_j3 showed sequence similarity in the O. glumaepatula genome. S22A_j1 and S22A_j2 appear to be allelic to OGLUM02G00590.1 and OGLUM02G00620.1, respectively. However, DUF1668 domains were lost in the O. glumaepatula genome. Alignment lengths between proteins are shown in parentheses.
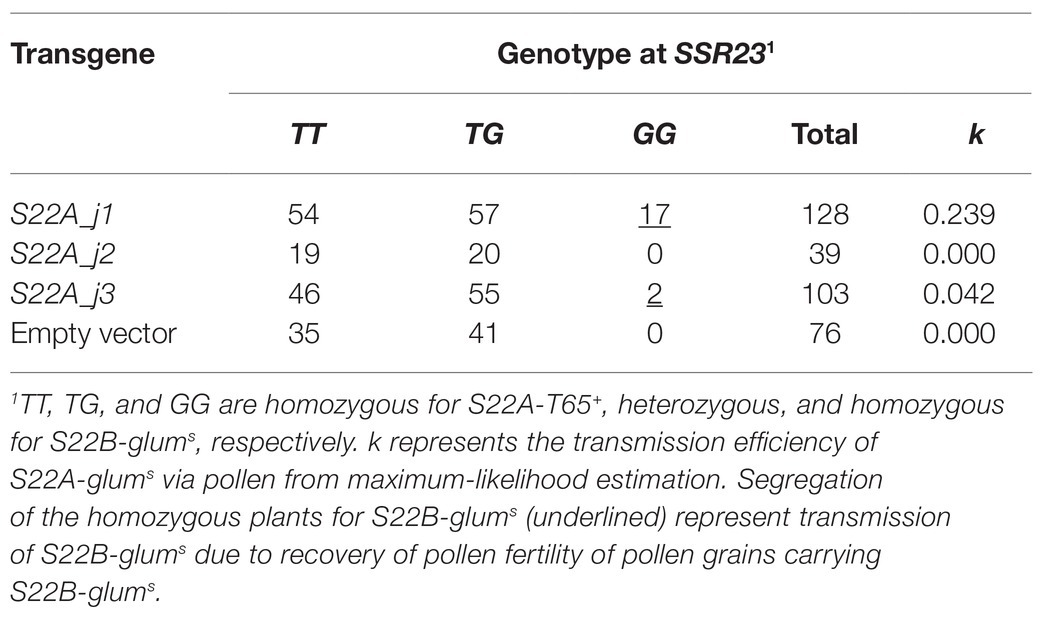
Table 2. Complementation test of S22A by transmission analysis at SSR23.
Independent Origin of Sterile Alleles in O. meridionalis at S22B
During the process of development of introgression lines of O. meridionalis accession W1625 (MER-ILs) in the genetic background of T65 (Yoshimura et al., 2010), pollen semi-sterile plants carrying W1625 chromosomal segments on the short arm of chromosome 2 with T65 cytoplasm were observed in the BC4F2 population (Supplementary Figure S5). Linkage mapping using the BC4F3 progeny demonstrated that semi-sterile and normal fertile pollen completely co-segregated to RM7033, and one single Mendelian factor close to RM7033 located in the genomic region between SSR markers RM7451 and RM5984 controls pollen sterility in this population. This genetic factor was designated as S22-mer. Further genetic dissection of the Mendelian factor S22-mer using 753 plants of the mapping population indicated that S22-mer is located between SSR24 and RM5984 containing the S22B locus. The O. meridionalis allele at LOC_Os02g04420 corresponding to S22B_j in O. sativa was designated S22B_m. Sequencing of the O. meridionalis BAC clone OMERIa-82O19 containing S22B_j allelic region showed that O. meridionalis has lost the upstream gene region, including the promoter, transcription initiation site, and N-terminal region of the DUF1668-containing protein (Supplementary Data 1). However, S22B_m does not possess the non-synonymous substitutions and a 2-bp deletion found in O. glumaepatula (Supplementary Data 1) except the CCG (P) > GCG (A) mutation. These results suggest that S22B_m is a loss-of-function allele and evolved from the different lineage of O. glumaepatula. The S22B sequence of W1625 was deposited to DDBJ (LC596093).
Diversity Analysis of DUF1668
A BLASTP search using the amino acid sequences of S22B_j as a query in the proteome of angiosperm species in the Phytozome 12 database (Goodstein et al., 2012) found homologous sequences of proteins only in Poaceae species, including the PACMAD clade species S. viridis, S. italica, P. virgatum, S. bicolor, and Z. mays, and the BOP clade species B. distachyon, B. stacei, and O. sativa (Supplementary Figure S6). Phylogenetic relationships based on DUF1668-containing sequences in Poaceae species revealed that each genome showed more than 40 copies of DUF1668-containing genes except for Z. mays (Supplementary Figure S6). The phylogenetic tree of DUF1668 domain sequences did not exhibit apparent monophyletic branches by species (Figure 3A), suggesting that active gene duplication and diversification of multiple copies of DUF1668-containing genes likely started before the diversification of Poaceae. However, S22B_j, S22A_j1, S22A_j2, and S22A_j3 were included in a single clade (Clade 1) harboring other members from Setaria, Panicum, Botryococcus, and Oryza species (Figure 3, yellow-shaded area). Reconstruction of the phylogenetic tree of DUF1668 members in Clade 1 revealed that DUF1668 sequences deduced from S22A_j1, S22A_j2, and S22A_j3 formed a monophyletic clade, but S22B_j was found to have an independent origin from the clade containing S22A_j1, S22A_j2, and S22A_j3 (Figure 3B).
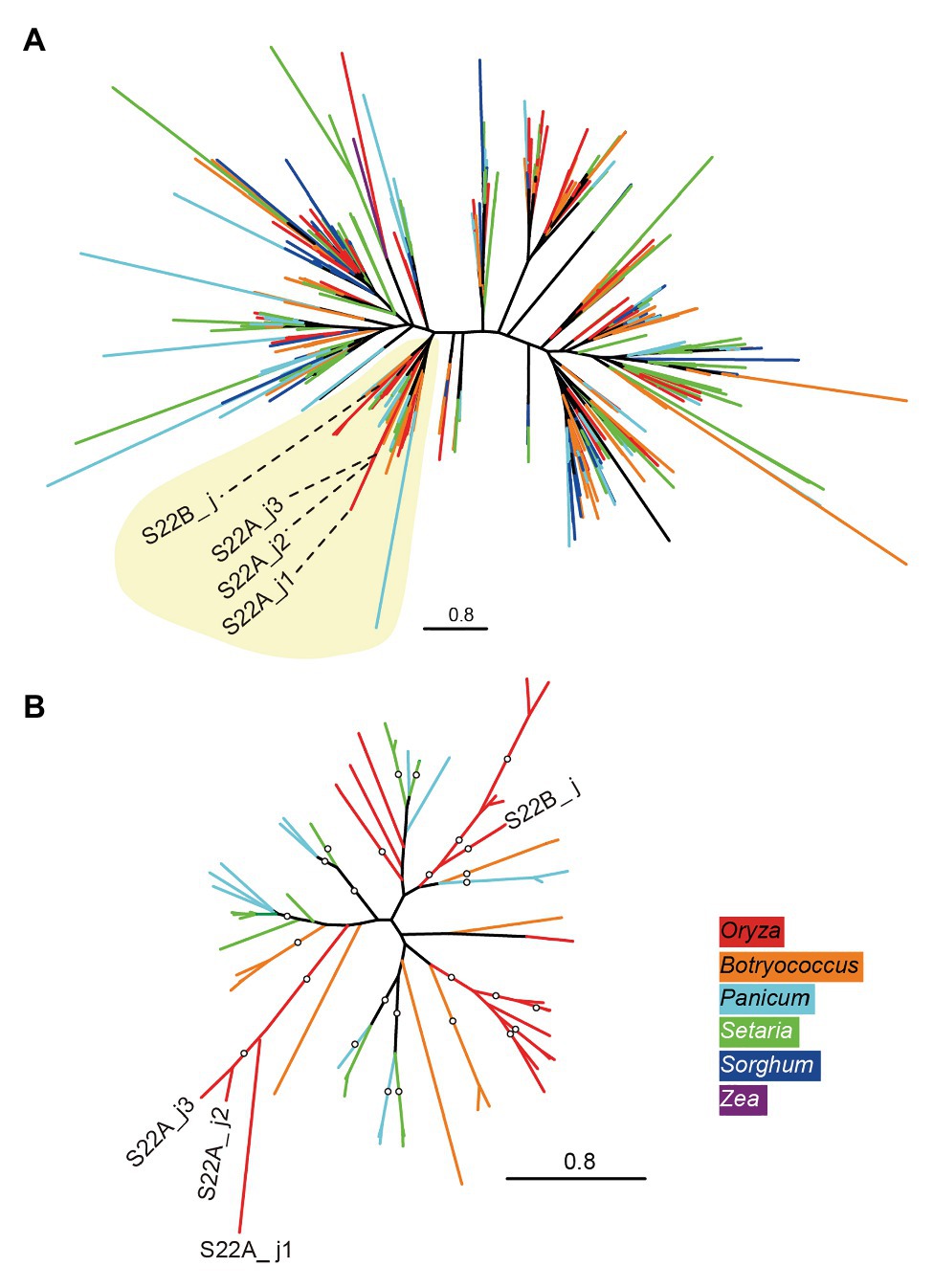
Figure 3. Phylogenetic tree of DUF1668 domain in Poaceae species. (A) Maximum-likelihood trees showing the phylogenetic relationships of DUF1668 domain found in Poaceae species were constructed. The colors of external nodes (leaves) indicate the genus of DUF1668 domain members: red (Oryza), orange (Botryococcus), light blue (Panicum), green (Setaria), dark blue (Sorghum), and purple (Zea). Clade 1 (shaded in yellow) includes the DUF1668 members from the genera Oryza, Botryococcus, Panicum, and Setaria. (B) Reconstructed tree of DUF1668 members belonging to Clade 1. The branches supported by more than 75% bootstrap values are indicated by circles.
Discussion
Here, we revealed that the genes S22B_j, S22A_j1, and S22A_j3 encoding DUF1668-containing protein are causal genes of F1 pollen sterility controlled by S22A or S22B in hybrids derived from a cross between O. sativa cultivar T65 and O. glumaepatula accession IRGC105668. Our previous genetic analysis demonstrated that S22A and S22B independently induced pollen semi-sterility on heterozygotes, and that pollen grains carrying the sterility allele S22A-glums and S22B-glums in S22A_SS and S22B_SS plants are, respectively, sterile (Sakata et al., 2014). The transformation of S22B_j to S22B_SS plants recovered the fertility of pollen grains harboring sterile alleles (Supplementary Table S3) and transmission of the sterile allele via pollen increased (Table 1). Although the sterile allele S22A-glums had never transmitted via pollen in the high-resolution linkage mapping, the transformation of S22A_j1 or S22A_j3 to S22A_SS plants also archived transmission of the sterile allele via pollen (Table 2), demonstrating recovery of the fertility of pollen grains harboring sterile alleles by the transgene. Since S22B_j, S22A_j1, and S22A_j3 are linked in the coupling phase, S22A and S22B were previously considered to represent the single Mendelian locus S22 based on genetic mapping (Sobrizal et al., 2000). Although S22A and S22B harbor the same domain, DUF1668, the DUF1668-containing proteins at S22A and S22B are not functionally redundant. Phylogenetic analysis of DUF1668-containing sequences in Poaceae revealed that gene duplication and diversification of DUF1668-containing genes occurred in ancestral species of Poaceae, and the sterility-causing genes S22A and S22B belong to a specific clade, Clade 1 (Figure 3A). Since Clade 1 contains both species from the BOP and PACMAD clades (including the genera Setaria, Panicum, Botryococcus, and Oryza), ancestral DUF1668-containing genes likely originated before divergence of the BOP and PACMAD clades and are thus shared among these species. Since S22A_j1, S22A_j2, and S22A_j3 also formed a monophyletic clade within Clade 1 (Figure 3B), they likely originated from gene duplications at S22A after divergence of the genus Oryza. Therefore, the genetic functions of these DUF1668-containing gene copies at S22A are likely to be redundant, and transformation of these copies restored the fertility of sterile pollen grains carrying S22A-glums. By contrast, gene duplication between S22A (S22A_j1, S22A_j2, and S22A_j3) and S22B may have at least an ancestral population of the divergence of the BOP and PACMAD clades. Although this study demonstrated that genetic variants at both S22A and S22B induce HI, it will be necessary to further examine whether the DUF1668 domain has evolved as an HI factor in other Poaceae species.
Molecular Behavior of S22B
S22B transcript levels were reduced in the order of homozygotes for S22B-T65+, heterozygotes, and homozygotes for S22B-glums in the anthers at each of the unicellular, bicellular, and mature stages using forward and reverse primers targeting the same sequences at S22B_j and S22B_g (Figure 1E). These data suggest that the expression level of S22B_g is lower than that of S22B_j. The expression level of S22B also decreased as male gametogenesis progressed. The S22B transcripts are likely mainly contributed from male gametophytes rather than from sporophytic tissues (anthers) based on the results of the promoter-GUS assay (Figure 1F). On the other hand, S22B broadly expressed in vegetative tissues in RiceXpro expression database (Supplementary Figure S6; Sato et al., 2013). Comparison of the genomic regions of S22B_j and S22B_g revealed many nucleotide substitutions in the promoter region and the 5' untranslated region near the transcriptional start site. These substitutions may be involved in regulating interspecific differences in transcription between the two alleles.
Western blotting showed that the level of S22B protein increased with the progression of male gametogenesis, in contrast to the decrease observed at the transcription level (Figure 1G). No obvious difference in S22B protein accumulation was observed between the two homozygotes for S22B-T65+ and S22B-glums. These data suggest that the level of S22B protein is under control by a post-translational regulation mechanism or that S22B-glums is sufficient for male gametogenesis. By contrast, the level of S22B protein was reduced in heterozygotes (Figure 1G). As one example of reduced expression only in heterozygotes in HI systems, genetic variants of a single gene between two diverged alleles at the Sc locus were reported to constitute the HI system (Shen et al., 2017). The japonica allele Sc-j contains a pollen-essential gene, and the indica allele Sc-i contains two or three tandem duplicates of an Sc-j homolog with a distinct promoter. In Sc-j/Sc-i hybrids, the high expression level of Sc-i in sporophytic cells causes suppression of Sc-j expression in pollen and selective abortion of Sc-j-pollen (Shen et al., 2017). Their study further revealed that feedback-mediated regulation of genes or proteins may result in the misregulation of gene expression between differentiated alleles in heterozygotes. Similar to the Sc system, the reduced S22B protein level in the anthers of heterozygotes observed in the present study could be due to allelic suppression of S22B protein via incompatible feedback modulation between the two alleles.
Our western blotting analysis of S22B also revealed that the 46 kDa band appeared specifically at the mature stage. In S22B_SS, development of pollen grains carrying the S22B-glums allele starts to delay from the late bicellular stages, and this delay is particularly apparent at the mature stage as compared with normal genotypes (Sakata et al., 2014). Almost all of the pollen grains carrying the S22B-glums allele could reach the tricellular stage but did not complete the formation of the male germ unit and failed to produce the pollen tube. We speculate that the reduction of the 46 kDa band in heterozygotes is involved in pollen semi-sterility. It is possible that the 46 kDa band resulted from alternative splicing of S22B_j and/or S22B_g, or from post-translational modification of the 37 kDa band, such as phosphorylation, lipidation, or glycosylation. The genetic effects of nucleotide substitutions and the 2-bp deletion on the S22B coding sequence for protein modification, and the mitochondrial localization of S22B have not yet been elucidated. Thus, these biochemical properties require further study.
In the linkage analysis of S22A (n = 3,072), homozygotes for S22A-glums were not obtained (Supplementary Figure S2). The total seed set of self-pollinated S22A_SS plants was fertile, and self-pollinated seeds of S22A_SS showed normal seed germination. These results demonstrate that the S22A-glums allele is insufficient for male gametogenesis in heterozygotes. Alternatively, the S22B-glums allele is transmitted via male gametophytes in heterozygotes and homozygotes, which segregates at low frequency. The S22A-glums allele encodes a truncated protein with loss of the complete DUF1668 domain (Figure 2C). In contrast, the S22B-glums allele has a few single nucleotide substitutions and a 2-bp frameshift mutation at the C-terminal, but the DUF1668 domain was predicted to exist in the Pfam search (Finn et al., 2014). S22B_m is likely a loss-of-function allele, and a homozygous plant for the S22-mer allele segregated and showed normal pollen fertility (Supplementary Figure S4). Therefore, an unidentified causative mutation in O. glumaepatula may inactivate gene function, although the DUF1668 domain was predicted in silico.
Hybrid Incompatibility at S22A and S22B
Diverged haplotypes, including multiple tightly linked genes, are known to induce HI in intra-specific and interspecific hybrids of rice, such as Sa, S5, and S1. The incompatible gene complex includes sporophytic genetic factors and gametophytic genetic factors that determine pollen fertility of its own gametophytes in heterozygotes. The BLAST similarity search using the cloned genes at the known HI as a query did not find homologous sequences within the S22A and S22B mapping regions. If HI systems conferred by S22A and S22B also evolve incompatible haplotypes to induce HI in hybrids, the genes cloned in this study only represent a portion of gametophytic members acting as a protector and were not sufficient to induce HI. To further identify other possible genes including a killer factor involved in HI at S22A or S22B, it is necessary to conduct defective mutant experiments using genome-editing methods such as CRISPR/Cas9 in heterozygotes showing HI. As suggested in the BDM model, nucleotide variants causing HI are heterozygous only at birth, which is necessary to escape from negative selection due to their own maladaptive phenotypes owing to this incompatibility. When S22A_j1, S22A_j2, and S22A_j3 are considered as the ancestral types and the truncated genes at S22A-glums are considered as the variant types, nucleotide variants of S22A-glums occurring only at birth need to escape from natural selection via BDM partners such as duplicated genes or interacting genes in other genomic regions of O. glumaepatula or ancestral species. Since near-isogenic lines of S22A and S22B in the T65 genetic background were used in this study, the genetic phenomena may appear as monogenic events, and other BDM partners have not yet been identified.
In contrast, a single gene can also cause an HI system. Allelic suppression of the japonica allele at the Sc locus results from the feedback-mediated regulation of gene expression between differentiated alleles in inter-subspecific hybrids between japonica and indica rice (Shen et al., 2017). If the allelic suppression occurred on the semi-sterile S22B_SS, transcription level on heterozygote would show half of the homozygotes for S22B-T65+ because transcription of S22B-glums is suppressed on heterozygote. However, expression level on the S22B_SS was closed to average of homozygotes of S22B-T65+ and S22B-glums, suggesting that allelic suppression has not occurred on transcription level. Instead, we suggest that alternative splicing of S22B_j and/or S22B_g or production of the 46 kDa band resulted from post-translational modification is suppressed in pollen grains harboring S22B-glums in allele-specific manner. The further studies may reveal this opinion. Another possibility may be that copy number variation of functional copies of DUF1668-containing genes in pollen grains results in competitive transmission efficiencies between fertile and sterile alleles. The allelic differences in development and starch absorption capacity may also result in the biased allocation of nutrient resources between gametophytes, leading to the distorted transmission efficiency of alleles.
In summary, HI at S22A and S22B could be caused by gene complexes within the candidate region, or by structural changes in a single gene. Further genetic analysis to identify other genetic loci interacting with S22A or S22B, and biochemical and genetic characterization of DUF1668-containing genes may reveal the HI mechanism via DUF1668.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
MS: investigation, development of genetic materials, review, writing draft manuscript, and editing of the manuscript. NS and YM: investigation and development of plant materials. HK, JW, and TM: investigation and sequencing. HY and AY: project administration, funding acquisition, and supervision. YY: conceptualization, methodology, investigation, data curation, and writing draft manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation, QTL-5002 to AY), Ministry of Education, Culture, Sports, Science and Technology of Japan [Grant-in-Aid for Scientific Research (A) grant number 24248002] and Japan Society for the Promotion of Science KAKENHI (grant number JP18K05576 to YY). This work was partially supported by a Grant-in-Aid from the Japan Agency for Medical Research and Development [National Bioresource Project (Rice); grant number JP19km0210105j0003 to HY].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The wild rice species accessions used in this study were distributed by the National Institute of Genetics supported by the National Bioresource Project (NBRP), AMED, Japan. Part of the genetic materials used in this study was grown in the Biotron Application Center, Kyushu University, Japan.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.632420/full#supplementary-material
Footnotes
References
Alcázar, R., García, A. V., Parker, J. E., and Reymond, M. (2009). Incremental steps toward incompatibility revealed by Arabidopsis epistatic interactions modulating salicylic acid pathway activation. Proc. Natl. Acad. Sci. U. S. A. 106, 334–339. doi: 10.1073/pnas.0811734106
Almagro Armenteros, J. J., Salvatore, M., Emanuelsson, O., Winther, O., von Heijne, G., Elofsson, A., et al. (2019). Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2:e201900429. doi: 10.26508/lsa.201900429
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Atanasov, K. E., Liu, C., Erban, A., Kopka, J., Parker, J. E., and Alcázar, R. (2018). NLR mutations suppressing immune hybrid incompatibility and their effects on disease resistance. Plant Physiol. 177, 1152–1169. doi: 10.1104/pp.18.00462
Bikard, D., Patel, D., Le Metté, C., Giorgi, V., Camilleri, C., Bennett, M. J., et al. (2009). Divergent evolution of duplicate genes leads to genetic incompatibilities within A. thaliana. Science 323, 623–626. doi: 10.1126/science.1165917
Bomblies, K., Lempe, J., Epple, P., Warthmann, N., Lanz, C., Dangl, J. L., et al. (2007). Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 5:e236. doi: 10.1371/journal.pbio.0050236
Bomblies, K., and Weigel, D. (2007). Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 8, 382–393. doi: 10.1038/nrg2082
Chen, C., Chen, H., Lin, Y. S., Shen, J. B., Shan, J. X., Qi, P., et al. (2014). A two-locus interaction causes interspecific hybrid weakness in rice. Nat. Commun. 5:3357. doi: 10.1038/ncomms4357
Chen, J., Ding, J., Ouyang, Y., Du, H., Yang, J., and Cheng, K. (2008). A triallelic system of S5 is a major regulator of the reproductive barrier and compatibility of indica-japonica hybrids in rice. Proc. Natl. Acad. Sci. U. S. A. 105, 11436–11441. doi: 10.1073/pnas.0804761105
Chen, C., Zhiguo, E., and Lin, H. X. (2016). Evolution and molecular control of hybrid incompatibility in plants. Front. Plant Sci. 7:1208. doi: 10.3389/fpls.2016.01208
Dellaporta, S., Wood, J., and Hicks, J. (1983). A plant DNA minipreparation: version II. Plant Mol. Biol. Report. 1, 19–21. doi: 10.1007/BF02712670
Deng, J., Fang, L., Zhu, X., Zhou, B., and Zhang, T. (2019). A CC-NBS-LRR gene induces hybrid lethality in cotton. J. Exp. Bot. 70, 5145–5156. doi: 10.1093/jxb/erz312
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fukasawa, Y., Tsuji, J., Fu, S. C., Tomii, K., Horton, P., and Imai, K. (2015). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell. Proteomics 14, 1113–1126. doi: 10.1074/mcp.M114.043083
Fuse, T., Sasaki, T., and Yano, M. (2001). Ti-plasmid vectors useful for functional analysis of rice genes. Plant Biotechnol. 18, 219–222. doi: 10.5511/plantbiotechnology.18.219
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Jacquemin, J., Bhatia, D., Singh, K., and Wing, R. A. (2013). The international Oryza map alignment project: development of a genus-wide comparative genomics platform to help solve the 9 billion-people question. Curr. Opin. Plant Biol. 16, 147–156. doi: 10.1016/j.pbi.2013.02.014
Jeuken, M. J., Zhang, N. W., McHale, L. K., Pelgrom, K., den Boer, E., Lindhout, P., et al. (2009). Rin4 causes hybrid necrosis and race-specific resistance in an interspecific lettuce hybrid. Plant Cell 21, 3368–3378. doi: 10.1105/tpc.109.070334
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:4. doi: 10.1186/1939-8433-6-4
Koide, Y., Ogino, A., Yoshikawa, T., Kitashima, Y., Saito, N., Kanaoka, Y., et al. (2018). Lineage-specific gene acquisition or loss is involved in interspecific hybrid sterility in rice. Proc. Natl. Acad. Sci. U. S. A. 115, E1955–E1962. doi: 10.1073/pnas.1711656115
Kubo, T., Takashi, T., Ashikari, M., Yoshimura, A., and Kurata, N. (2016). Two tightly linked genes at the hsa1 locus cause both F1 and F2 hybrid sterility in rice. Mol. Plant 9, 221–232. doi: 10.1016/j.molp.2015.09.014
Long, Y., Zhao, L., Niu, B., Su, J., Wu, H., Chen, Y., et al. (2008). Hybrid male sterility in rice controlled by interaction between divergent alleles of two adjacent genes. Proc. Natl. Acad. Sci. U. S. A. 105, 18871–18876. doi: 10.1073/pnas.0810108105
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Maheshwari, S., and Barbash, D. A. (2011). The genetics of hybrid incompatibilities. Annu. Rev. Genet. 45, 331–355. doi: 10.1146/annurev-genet-110410-132514
Matsuda, R., Iehisa, J. C., and Takumi, S. (2012). Application of real-time PCR-based SNP detection for mapping of Net2, a causal D-genome gene for hybrid necrosis in interspecific crosses between tetraploid wheat and Aegilops tauschii. Genes Genet. Syst. 87, 137–143. doi: 10.1266/ggs.87.137
Mizuta, Y., Harushima, Y., and Kurata, N. (2010). Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proc. Natl. Acad. Sci. U. S. A. 107, 20417–20422. doi: 10.1073/pnas.1003124107
Nakagawa, T., Kurose, T., Hino, T., Tanaka, K., Kawamukai, M., Niwa, Y., et al. (2007). Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 104, 34–41. doi: 10.1263/jbb.104.34
Nguyen, G. N., Yamagata, Y., Shigematsu, Y., Watanabe, M., Miyazaki, Y., Doi, K., et al. (2017). Duplication and loss of function of genes encoding RNA polymerase III subunit C4 causes hybrid incompatibility in rice. G3 (Bethesda) 7, 2565–2575. doi: 10.1534/g3.117.043943
Noor, M. A., and Feder, J. L. (2006). Speciation genetics: evolving approaches. Nat. Rev. Genet. 7, 851–861. doi: 10.1038/nrg1968
Rieseberg, L. H., and Blackman, B. K. (2010). Speciation genes in plants. Ann. Bot. 106, 439–455. doi: 10.1093/aob/mcq126
Sakata, M., Yamagata, Y., Doi, K., and Yoshimura, A. (2014). Two linked genes on rice chromosome 2 for F1 pollen sterility in a hybrid between Oryza sativa and O. glumaepatula. Breed. Sci. 64, 309–320. doi: 10.1270/jsbbs.64.309
Sato, Y., Takehisa, H., Kamatsuki, K., Minami, H., Namiki, N., Ikawa, H., et al. (2013). RiceXPro version 3.0: expanding the informatics resource for rice transcriptome. Nucleic Acids Res. 41, D1206–D1213. doi: 10.1093/nar/gks1125
Shen, R., Wang, L., Liu, X., Wu, J., Jin, W., Zhao, X., et al. (2017). Genomic structural variation-mediated allelic suppression causes hybrid male sterility in rice. Nat. Commun. 8:1310. doi: 10.1038/s41467-017-01400-y
Sobrizal,, Matsuzaki, Y., Sanchez, P. L., Ikeda, K., and Yoshimura, A. (2000). Mapping of F1 pollen semi-sterility gene found in backcross progeny of Oryza sativa L. and Oryza glumaepatula Steud. Rice Genet. Newsl. 17, 61–63.
Sperschneider, J., Catanzariti, A. M., DeBoer, K., Petre, B., Gardiner, D. M., Singh, K. B., et al. (2017). LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Sci. Rep. 7:44598. doi: 10.1038/srep44598
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Vaughan, D. A., Kadowaki, K., Kaga, A., and Tomooka, N. (2005). On the phylogeny and biogeography of the genus Oryza. Breed. Sci. 55, 113–122. doi: 10.1270/jsbbs.55.113
Xie, Y., Tang, J., Xie, X., Li, X., Huang, J., Fei, Y., et al. (2019). An asymmetric allelic interaction drives allele transmission bias in interspecific rice hybrids. Nat. Commun. 10:2501. doi: 10.1038/s41467-019-10488-3
Xie, Y., Xu, P., Huang, J., Ma, S., Xie, X., Tao, D., et al. (2017). Interspecific hybrid sterility in rice is mediated by OgTPR1 at the S1 locus encoding a peptidase-like protein. Mol. Plant 10, 1137–1140. doi: 10.1016/j.molp.2017.05.005
Yamagata, Y., Yamamoto, E., Aya, K., Win, K. T., Doi, K., Sobrizal,, et al. (2010). Mitochondrial gene in the nuclear genome induces reproductive barrier in rice. Proc. Natl. Acad. Sci. U. S. A. 107, 1494–1499. doi: 10.1073/pnas.0908283107
Yamamoto, E., Takashi, T., Morinaka, Y., Lin, S., Wu, J., Matsumoto, T., et al. (2010). Gain of deleterious function causes an autoimmune response and Bateson-Dobzhansky-Muller incompatibility in rice. Mol. Gen. Genomics. 283, 305–315. doi: 10.1007/s00438-010-0514-y
Yang, J., Zhao, X., Cheng, K., Du, H., Ouyang, Y., Chen, J., et al. (2012). A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science 337, 1336–1340. doi: 10.1126/science.1223702
Yoshimura, A., Nagayama, H., Sobrizal,, Kurakazu, T., Sanchez, P. L., Doi, K., et al. (2010). Introgression lines of rice (Oryza sativa L.) carrying a donor genome from the wild species, O. glumaepatula Steud. And O. meridionalis Ng. Breed. Sci. 60, 597–603. doi: 10.1270/jsbbs.60.597
Yu, Y., Zhao, Z., Shi, Y., Tian, H., Liu, L., Bian, X., et al. (2016). Hybrid sterility in rice (Oryza sativa L.) involves the tetratricopeptide repeat domain containing protein. Genetics 203, 1439–1451. doi: 10.1534/genetics.115.183848
Keywords: hybrid incompatibility, reproductive isolation, rice, Oryza, pollen sterility, DUF1668, domain unknown function
Citation: Sakata M, Takano-Kai N, Miyazaki Y, Kanamori H, Wu J, Matsumoto T, Doi K, Yasui H, Yoshimura A and Yamagata Y (2021) Domain Unknown Function DUF1668-Containing Genes in Multiple Lineages Are Responsible for F1 Pollen Sterility in Rice. Front. Plant Sci. 11:632420. doi: 10.3389/fpls.2020.632420
Edited by:
Dayun Tao, Yunnan Academy of Agricultural Sciences, ChinaReviewed by:
Gang Zhi Zhao, Nanjing Agricultural University, ChinaZhang Yu, Yunnan Academy of Agricultural Sciences, China
Kinya Toriyama, Tohoku University, Japan
Copyright © 2021 Sakata, Takano-Kai, Miyazaki, Kanamori, Wu, Matsumoto, Doi, Yasui, Yoshimura and Yamagata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiyuki Yamagata, eW9zaGl5dWtAYWdyLmt5dXNodS11LmFjLmpw
†Present address: Yuta Miyazaki, Saga Prefectural Agriculture Research Center, Kawagoe, Saga, Japan