
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 02 July 2020
Sec. Plant Pathogen Interactions
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.00973
 Alba Moreno-Pérez1,2†
Alba Moreno-Pérez1,2† Adrián Pintado1,2†
Adrián Pintado1,2† Jesús Murillo3*
Jesús Murillo3* Eloy Caballo-Ponce1,2
Eloy Caballo-Ponce1,2 Stefania Tegli4
Stefania Tegli4 Chiaraluce Moretti5
Chiaraluce Moretti5 Pablo Rodríguez-Palenzuela6,7
Pablo Rodríguez-Palenzuela6,7 Cayo Ramos1,2*
Cayo Ramos1,2*The study of host range determinants within the Pseudomonas syringae complex is gaining renewed attention due to its widespread distribution in non-agricultural environments, evidence of large variability in intra-pathovar host range, and the emergence of new epidemic diseases. This requires the establishment of appropriate model pathosystems facilitating integration of phenotypic, genomic and evolutionary data. Pseudomonas savastanoi pv. savastanoi is a model pathogen of the olive tree, and here we report a closed genome of strain NCPPB 3335, plus draft genome sequences of three strains isolated from oleander (pv. nerii), ash (pv. fraxini) and broom plants (pv. retacarpa). We then conducted a comparative genomic analysis of these four new genomes plus 16 publicly available genomes, representing 20 strains of these four P. savastanoi pathovars of woody hosts. Despite overlapping host ranges, cross-pathogenicity tests using four plant hosts clearly separated these pathovars and lead to pathovar reassignment of two strains. Critically, these functional assays were pivotal to reconcile phylogeny with host range and to define pathovar-specific genes repertoires. We report a pan-genome of 7,953 ortholog gene families and a total of 45 type III secretion system effector genes, including 24 core genes, four genes exclusive of pv. retacarpa and several genes encoding pathovar-specific truncations. Noticeably, the four pathovars corresponded with well-defined genetic lineages, with core genome phylogeny and hierarchical clustering of effector genes closely correlating with pathogenic specialization. Knot-inducing pathovars encode genes absent in the canker-inducing pv. fraxini, such as those related to indole acetic acid, cytokinins, rhizobitoxine, and a bacteriophytochrome. Other pathovar-exclusive genes encode type I, type II, type IV, and type VI secretion system proteins, the phytotoxine phevamine A, a siderophore, c-di-GMP-related proteins, methyl chemotaxis proteins, and a broad collection of transcriptional regulators and transporters of eight different superfamilies. Our combination of pathogenicity analyses and genomics tools allowed us to correctly assign strains to pathovars and to propose a repertoire of host range-related genes in the P. syringae complex.
The Pseudomonas syringae species complex is considered the most relevant phytopathogenic bacterium worldwide, as a prominent research model and as an economically important phytopathogen (Mansfield et al., 2012). It comprises at least 13 phylogroups (PGs) encompassing 15 different Pseudomonas species associated with plants and the water cycle (Berge et al., 2014; Gomila et al., 2017). The complex is also divided at an infrasubspecific level into ca. 65 pathovars defined by characteristic plant host ranges (Bull and Koike, 2015).
The very broad host range of the P. syringae complex contrasts with the host specialization of most individual isolates from agricultural crops. Conversely, environmental isolates show a wider host range (Morris et al., 2013; Berge et al., 2014; Monteil et al., 2016). Moreover, a recent study showed that P. syringae strains form an overlapping continuum of host range potential and that pathovar denominations do not correspond to the P. syringae biology (Morris et al., 2019). Understanding this complexity at the different levels requires the establishment of appropriate pathosystems, facilitating integration of phenotypic, genomic, and evolutionary data. Consequently, the P. syringae complex emerged as one of the most relevant models to study host specificity in bacterial phytopathogens (Vinatzer et al., 2014; Baltrus et al., 2017; Dillon et al., 2019b).
Pseudomonas savastanoi belongs to PG3 of the P. syringae complex. This is the only phylogroup including bacteria causing tumorous overgrowths (knots) in woody hosts, namely P. savastanoi pv. savastanoi (Psv), P. savastanoi pv. nerii (Psn) and P. savastanoi pv. retacarpa (Psr), P. meliae, P. tremae, and P. syringae pathovars cerasicola, daphniphylli, dendropanacis, myricae, and rhaphiolepidis (Lamichhane et al., 2014; Caballo-Ponce et al., 2017a). Additionally, isolates of P. savastanoi pv. fraxini (Psf) cause cankers accompanied by excrescences in ash (Fraxinus excelsior) (Janse, 1981; Gardan et al., 1992; Caballo-Ponce et al., 2017a). It was experimentally determined through pathogenicity tests that these four pathovars have different host ranges, namely: Psv induces knots in olive (Olea europaea) and ash but not in oleander (Nerium oleander); Psn in all three hosts; and Psr only in broom (Retama sphaerocarpa) (Álvarez et al., 1998). In turn, Psf induces symptoms both in ash and olive (Janse, 1982; Iacobellis et al., 1998; Ramos et al., 2012). Nevertheless, some P. savastanoi populations show heterogeneous host ranges, which can even drastically differ between strains of the same pathovar (Cinelli et al., 2014; Morris et al., 2019). At the same time, and likely because of the reported lack of genetic diversity and overlapping host ranges, various genetic and phenotypical analyses did not establish a clear-cut differentiation among these pathovars (Janse, 1991; Sisto et al., 2007; Cinelli et al., 2014; Moretti et al., 2017). Thus, this well-established delineation of the host range, converts these four P. savastanoi pathovars of woody hosts in an excellent system to study the basis of host range in knot- and canker-forming bacterial pathogens.
Here, we performed comparative genomics analyses of 20 P. savastanoi strains isolated from olive, oleander, ash, and broom, covering all four well-defined P. savastanoi pathovars of woody hosts. First, we obtained the complete chromosome sequence of Psv NCPPB 3335 and draft genome sequences of Psn Psn23, Psr CECT 4861 and Psf NCPPB 1006. Second, cross-pathogenicity tests allowed correlating the results of the genomics analyses with host specificity. We thus delineated the core and pan-genome of the four P. savastanoi pathovars of woody hosts and identified the set of pathovar- and strain-specific gene complements. Our results provide a collection of putative virulence genes that can now be experimentally tested for their contribution to the definition of host specificity in knot- and canker-forming bacterial pathogens and their evolution into pathovars.
P. savastanoi (Table 1) were routinely grown at 28°C in lysogeny broth (LB) medium (Bertani, 1951) without glucose and containing 0.5% NaCl. When required, media were supplemented with (final concentrations) nitrofurantoin (Nf, 20 µg/ml) and cycloheximide (Ch, 100 µg/ml).
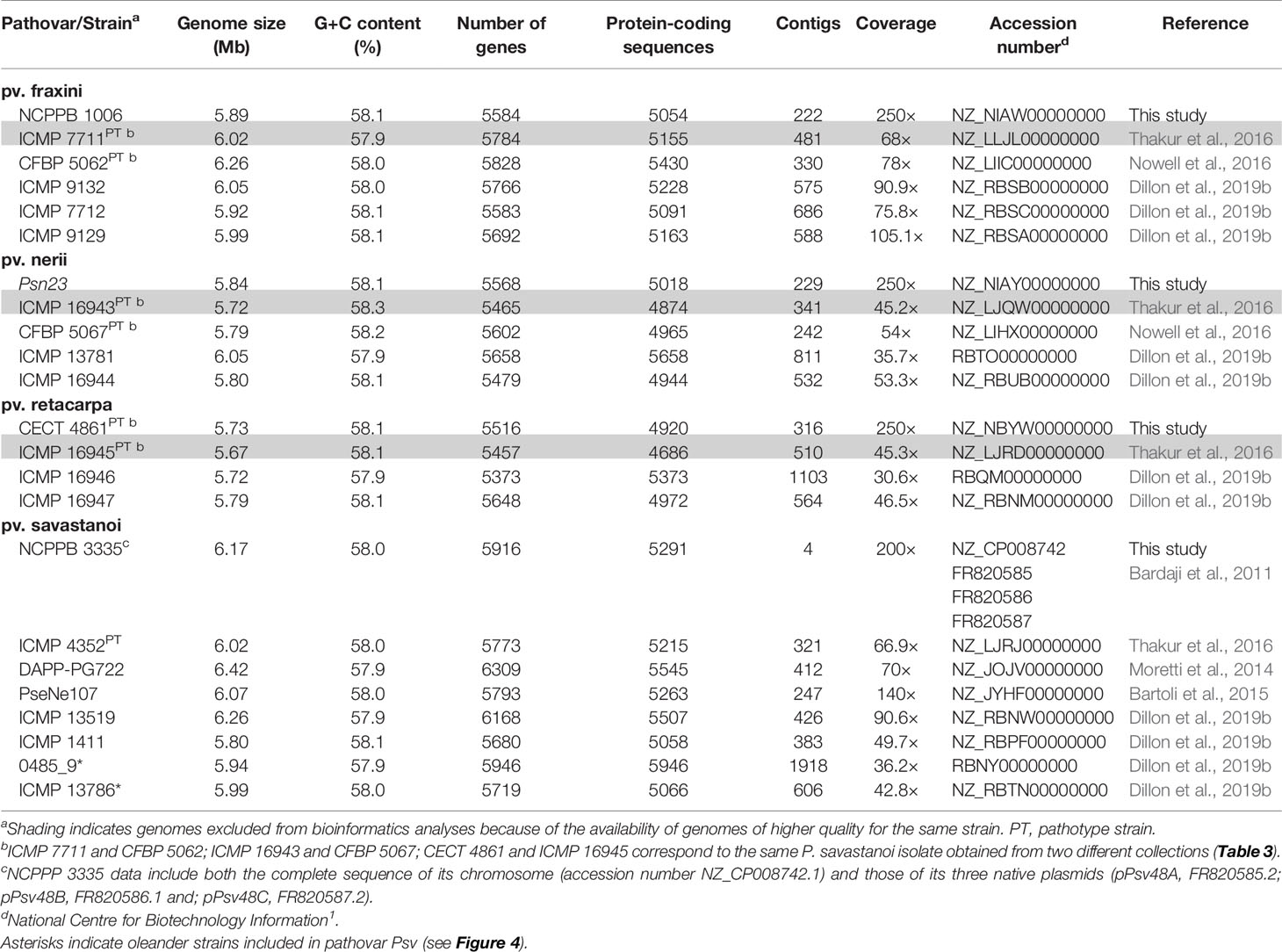
Table 1 General genome features of Pseudomonas savastanoi strains isolated from woody host.
Basic DNA and molecular techniques followed standard methods (Sambrook and Russell, 2001). DNA sequences were visualized and manipulated using Geneious 8.1.9 (Kearse et al., 2012) and Artemis 16.0.0 (Carver et al., 2012).
Genomic DNA from Psv NCPPB 3335, Psn Psn23, Psr CECT 4861, and Psf NCPPB 1006 was extracted from cultures grown overnight using a Genomic DNA Purification JETFLEX kit (Genomed GmbH, Löhne, Germany), and purified by extraction with phenol-chloroform.
The pathogenicity of P. savastanoi strains was analyzed on O. europaea plants derived from a seed germinated in vitro (originally collected from an “Arbequina” plant), N. oleander plants accession “pink” (single pink flowers) supplied by Viveros Guzmán (Málaga, Spain), and F. excelsior and R. sphaerocarpa plants native from Valladolid and supplied by Viveros Fuenteamarga (Valladolid, Spain). For inoculation, bacterial cells grown on LB plates for 48 h at 28°C were collected, washed twice with 10 mM MgCl2 and adjusted to a final concentration of 108 colony-forming unit (CFU)/ml in the same buffer. Olive, oleander, and ash plants were wounded with a scalpel along the stem and 106 CFU were placed per wound (Penyalver et al., 2006; Pérez-Martínez et al., 2007). For broom plants, 107 CFU were injected in each wound using a syringe with needle. For the cross-pathogenicity tests shown in Figure 3 and Supplementary Figure S3 we used two plants per strain. The number of wound sites infected per plant varied between 5 and 10, depending on the size of the plant. For the cross-pathogenicity tests of Psv and Psn strains shown in Figure 4 we used three olive and three oleander plants per strain, with between 10 and 12 wounds sites infected per strain and plant. Wounds were protected with parafilm (Bemis, Neenah, WI, USA) for seven days to maintain humidity. Plants were kept in a greenhouse under natural photoperiod (15 h light/9 h dark) at room temperature (26°C day/18°C night). Symptoms were scored at 90 dpi and captured with a high-resolution digital camera (Nikon DXM 1200; Nikon Corporation, Tokyo, Japan), for olive, oleander, and ash, or with a stereoscopic microscope (Leica MZ FLIII; Leica Microsystems, Wetzlar, Germany), for broom plants. P. savastanoi cells were recovered from olive infected plants as follows. Three inoculated sites excised from two (Figure 3, Supplementary Figure S3) or three (Figure 4) different plants were homogenized by mechanical disruption, and serial dilutions plated on LB containing Nf and Ch; bacterial populations were estimated by colony counts.
Genome sequencing and assembly of the Psv NCPPB 3335 chromosome was performed at BGI Tech Solutions Co., Ltd. (Hong Kong). A library of randomly sheared DNA fragments (0.5-2.0 kb) was subjected to Illumina GA II (Solexa) sequencing. Reads (coverage, 200×) were qualitatively assessed before assembling with SOAP de novo (Li et al., 2008). Assembly of the NCPPB 3335 chromosome was aided by subtraction of the complete sequences of its three native plasmids (Bardaji et al., 2011). Primer walking and PCR amplification were used to fill the remaining gaps and solve misassembled regions. Sequencing of the Psn Psn23, Psr CECT 4861 and Psf NCPPB 1006 draft genomes was performed at Centro de Investigación Biomédica (CIBIR), La Rioja, Spain. Paired-end (100 cycles) sequencing used the Illumina GA ll platform (coverage, 250×). The genomes were assembled using CLC Genomics Workbench. The four genomes were automatically annotated upon submission to GenBank at National Centre for Biotechnology Information1 (NCBI). The accession numbers of these four genomes are listed in Table 1.
Core− and pan-genome analyses were performed using BPGA v1.3 (Chaudhari et al., 2016) with assemblies downloaded from the NCBI1. In all comparative analyses, Psv NCPPB 3335 was used as reference with the sequence of the complete chromosome and of its three plasmids concatenated in a single file (5′-chromosome-pPsv48A-pPsv48B-pPsv48C-3′). Orthologous genes were identified using the USEARCH algorithm (Edgar, 2010) with a threshold of 0.9 (90% blastp identity). To determine whether pan-genomes were opened or closed, we used the medians of the total number of genes found and the curves were then fitted to Heap’s law model (Tettelin et al., 2008).
The core- and pan-genome were also estimated using Roary v3.12.0 (Page et al., 2015) with ≥ 90% blastp identity and using the same assemblies as with BPGA, but annotated with Prokka (Seemann, 2014) using Galaxy v1.12.02 (Afgan et al., 2016). The heat map depicting gene presence and absence was generated with Phandango (Hadfield et al., 2018).
Phylogenetic relationships were predicted by analysis of core genome single nucleotide polymorphisms (SNPs) using Parsnp v1.2 and Gingr v1.2 (Treangen et al., 2014), and 100 bootstrap replicates; trees were visualized and manipulated using MEGA 7 (Kumar et al., 2016). The P. savastanoi phylogeny was also analyzed by MLSA using 40 concatenated genes (Supplementary Table S1) whose sequences are complete in all 20 genomes (80,902-80,949 nt per strain). Sequence alignment using MUSCLE within MEGA 7 (Kumar et al., 2016) was hand curated to eliminate ambiguities. The tree was constructed using RaxML 7.2.8 within Geneious 8.1.9 (Kearse et al., 2012), with 200 bootstraps. All trees were rooted using P. syringae pv. ciccaronei ICMP 5710 (assembly LJPY01) as outgroup.
Codification of structural and regulatory components of the type III (T3SS), type IV (T4SS) and type VI (T6SS)secretion systems was investigated using T346 Hunter (Martínez-García et al., 2015a).
To search for known T3SS effectors (T3Es) we used PIFAR (Martínez-García et al., 2016). T3Es sequences were manually examined to search for truncations, disruptions, and frameshifts. To identify novel hop genes, we used and ad-hoc pipeline considering two different criteria: the N-terminal sequence features, using EffectiveDB3 (Eichinger et al., 2015), and presence of potential HrpL boxes 500 nucleotides upstream of the start codon, following previously described criteria (Fouts et al., 2002). Hierarchical clustering of P. savastanoi genomes based on their T3E content was performed using Morpheus4 with a presence-absence matrix with values of “1” (full-length CDS), “0.5” (truncated/disrupted/frameshifted CDS) and “0” (absence of T3E). Values of “2” and “1.5” were given for T3E paralogs encoded as two full-length CDSs or as one full-length and one truncated CDS, respectively. The matrix was used to measure the distance between strains using the Euclidean distance method. Then, the tree was constructed using the complete linkage hierarchical clustering method.
Variable regions (>3.9 kb) (Supplementary Figure S1) were identified with GView Pangenome Analysis tool5 (Petkau et al., 2010) using Psv NCPPB 3335 (5′-chromosome-pPsv48A-pPsv48B-pPsv48C-3′) as seed, and blastn with e-value ≤1 × 10−10 and ≥90% identity. IslandViewer4 (Bertelli et al., 2017) was used to predict genomic islands in the Psv NCPPB 3335 genome.
The gene content of P. savastanoi pathovars was compared using Roary v3.12.0 (Page et al., 2015) as described above. The resulting presence/absence matrix was used to obtain the lists of pathovar-exclusive genes and pathovar-absent genes. Genes were first classified by gene ontology (GO) enrichment analysis using Sma3s.v2 (Muñoz-Mérida et al., 2014) and then manually curated by blastx analysis (Supplementary Table S2).
Virulence gene repertoires were predicted using PIFAR (Martínez-García et al., 2016).
We reported the draft genome sequence of Psv NCPPB 3335 (Rodríguez-Palenzuela et al., 2010) and the complete sequence of its three plasmids (Bardaji et al., 2011). To use this genome as a reference in this and future comparative genomic analyses, we obtained the complete sequence of the Psv NCPPB 3335 chromosome. This yielded a single contig of 6.02 Mb, encoding 5,850 genes and 5,165 protein-coding sequences. Likewise, we generated draft genome sequences of three strains whose pathogenicity in their host of isolation was previously reported, i.e. Psn Psn23, Psf NCPPB 1006 and Psr CECT 4861 (Table 1). Additionally, we analyzed the previously published draft genomes of 19 other strains of the four P. savastanoi pathovars of woody hosts (Table 1). The relevant characteristics of all these genomes are shown in Table 1.
Six of the genomes correspond to three strains from different collections, independently sequenced by us and others (Table 1); we therefore selected those of the highest quality for subsequent genomic analyses. The resulting 20 genomes contained 114,601 genes, clustering into a hard pan-genome of 7,953 ortholog groups, of which 3,017 (38%) constitute the hard-core genome, 3,636 (46%) are accessory (encoded in 2-19 genomes), and the remaining 1,300 (16%) are strain-specific (Table 2, Figure 1). Nevertheless, there is a moderate strain-to-strain variation in the number of accessory genes (1,857 to 2,388) and strain-specific genes (14 to 236) (Figure 1B). We also estimated that only 19 additional ortholog groups are part of the P. savastanoi soft-core genome, i.e. 3,036 groups are present in at least 19 of the 20 genomes (95%).
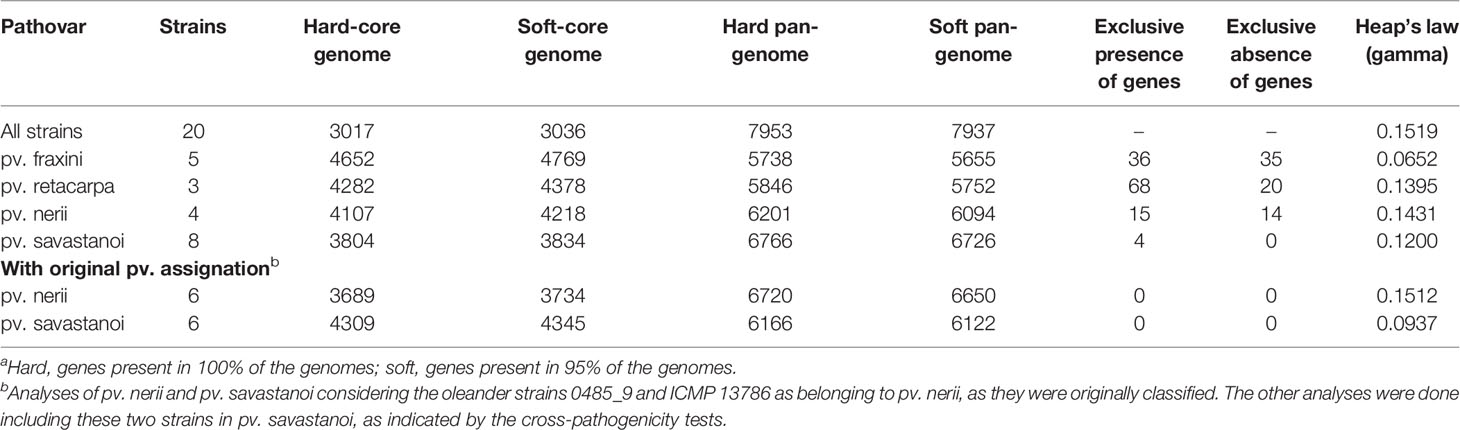
Table 2 Comparison of the core genomes and pan-genomes of Pseudomonas savastanoi pathovarsa.
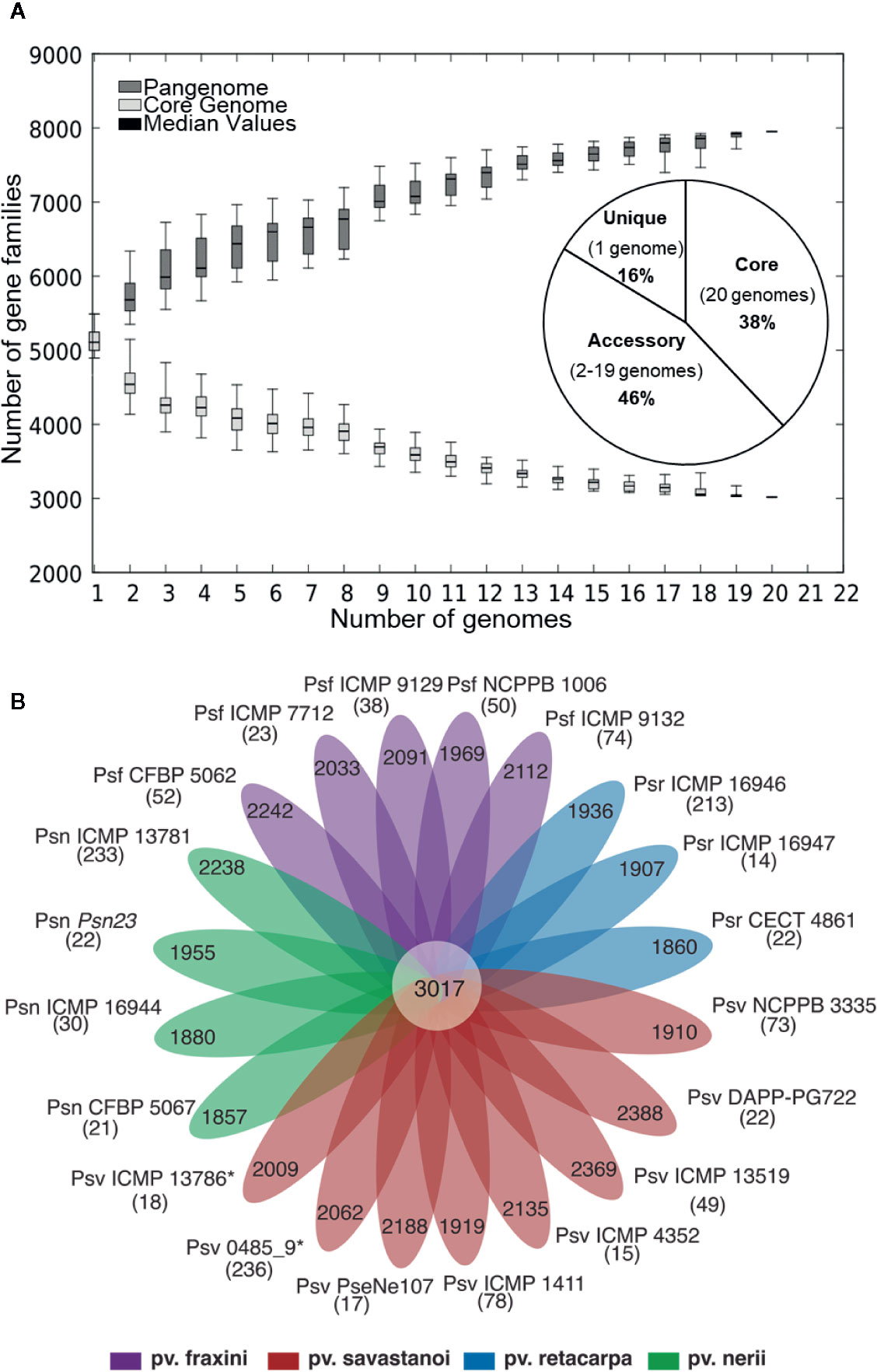
Figure 1 (A) Core- and pan-genome of P. savastanoi strains. Boxes indicate changes in number of gene families to the number of genes added sequentially, with median values denoted with a horizontal black line and standard deviation with vertical bars. The pie chart shows the frequency distribution of ortholog groups of genes. (B) Strain-specific P. savastanoi genes. Number of genes in the core-genome (center), accessory genes (petals) and strain-specific genes (in brackets), calculated using BPGA. Psv, Psn, Psf, and Psr, P. savastanoi pathovars savastanoi, nerii, fraxini, and retacarpa, respectively. Asterisks indicate oleander strains included in Psv (see Figure 4).
Using the Heap’s law model, we estimated a fitting parameter (γ) of 0.15 for all 20 genomes (Table 2), just above the threshold of γ=0 distinguishing open (γ > 0) from closed (γ < 0) genomes. Therefore, sampling more P. savastanoi genomes will likely not result in a significant increase of the pan-genome size nor in a substantial reduction of the core genome, because their accumulation curves tend towards an asymptote (Figure 1A). A heat map representing presence and absence of individual genes on each strain is shown in Figure 2.
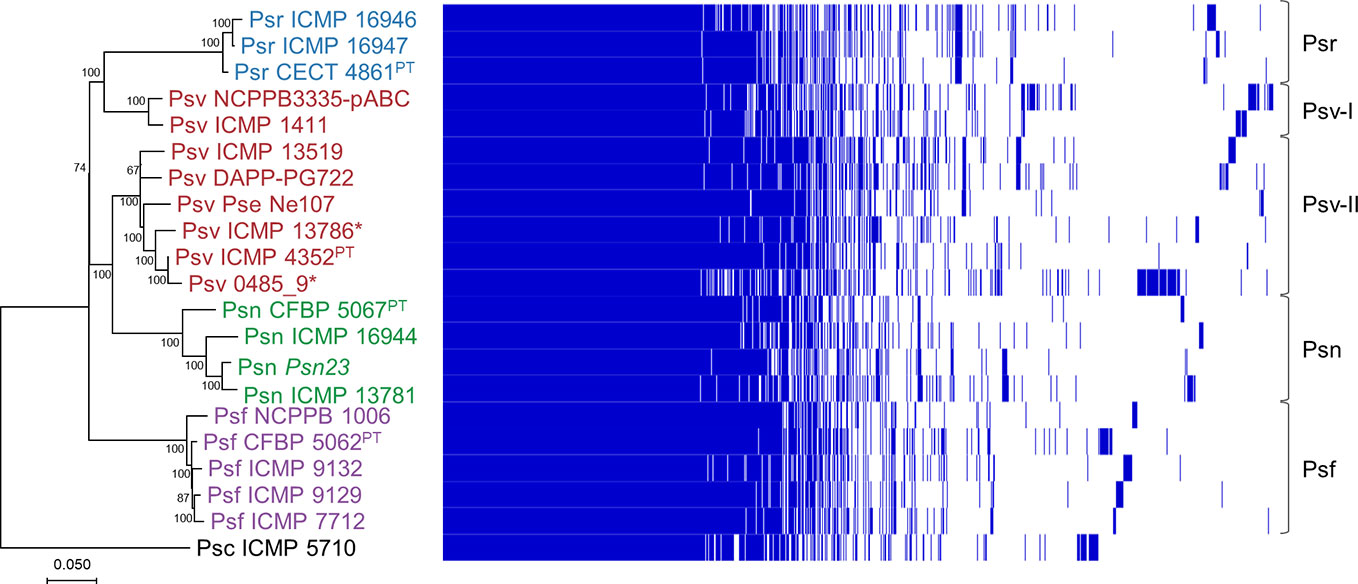
Figure 2 Phylogeny and gene content of P. savastanoi genomes. Maximum likelihood tree based on core genome SNPs, rooted with P. syringae pv. ciccaronei ICMP 5710. Values in nodes are bootstrap percentages from 100 replicates; the scale represents substitutions per site (left panel). Heat map representing presence (blue) or absence (blank) of individual genes (right panel). Strain abbreviations as in Figure 1; PT, pathotype strain.
We analyzed the phylogenetic relationships among the 20 P. savastanoi strains (Table 1) using SNPs from a core genome alignment. This tree (Figure 2) shows a very similar topology to a MLSA-based tree constructed using 40 concatenated genes (Supplementary Table S1, Supplementary Figure S2).
We could identify five well-differentiated clades (Figure 2), two of which correspond to pathovars Psr and Psf. However, pathovar Psv appears not to be monophyletic being distributed into two different clades. Two strains of Psv (NCPPB 3335-pABC and ICMP 1411) clustered closely to the Psr strains (Psv-I cluster), well-separated from the remaining Psv strains (Psv-II cluster). This last group also includes two oleander strains (ICMP 13786 and 0485_9), whereas the remaining Psn strains clustered together in a separate clade.
The pathogenicity of the nine P. savastanoi strains whose genomes were available in 2017 (Figure 3, Table 3, Supplementary Figure S3) was compared with published host ranges (Janse, 1991; Álvarez et al., 1998; Iacobellis et al., 1998).
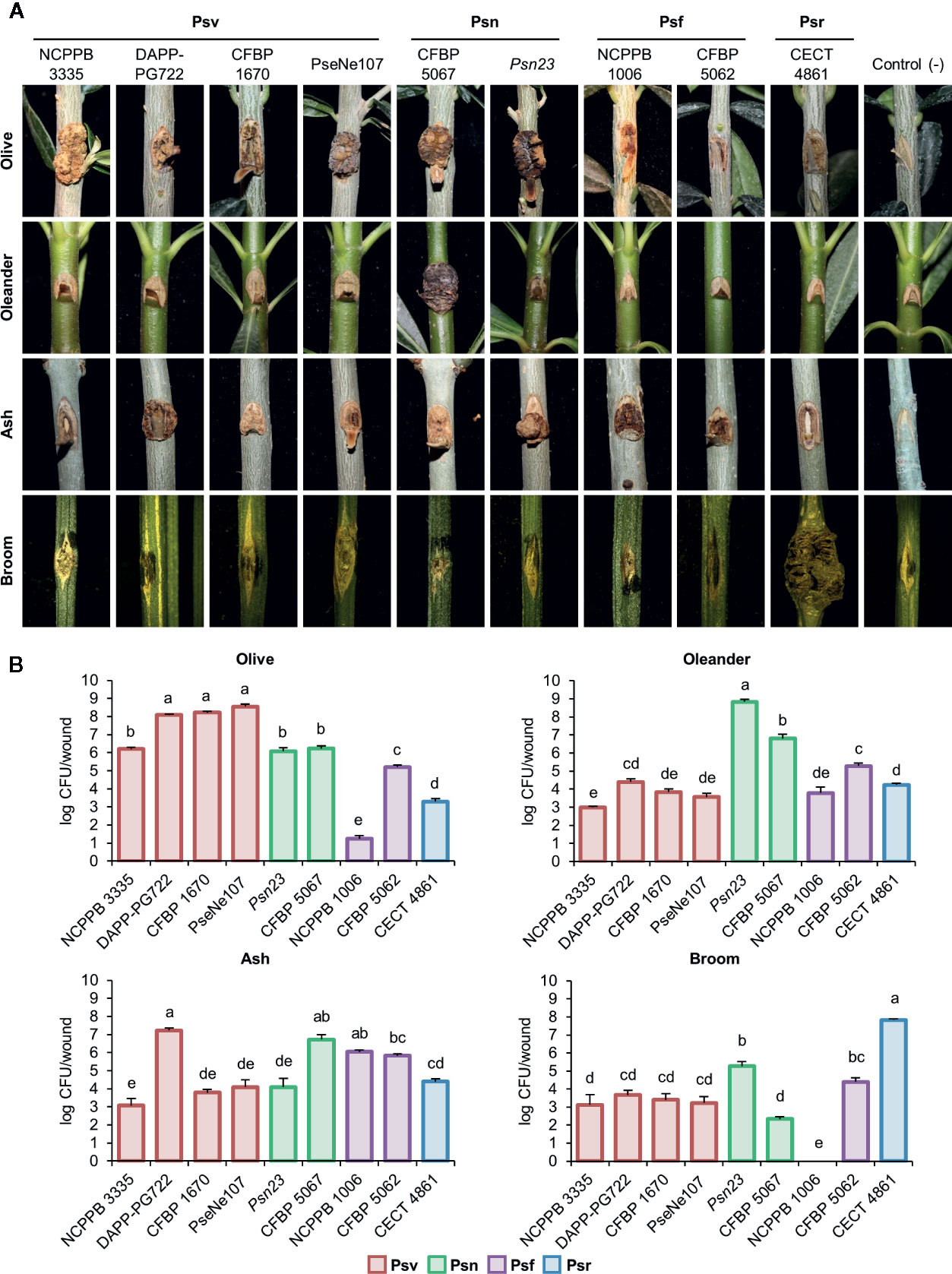
Figure 3 Cross pathogenicity tests of P. savastanoi strains. (A) Symptoms generated by P. savastanoi strains at 90 days post-inoculation. Control (-), negative control, plants inoculated with 10 mM MgCl2. (B) Populations of P. savastanoi strains in different hosts. Bars represent mean populations at 90 days post-inoculation from three biological replicates, with standard error; different letters indicate means that are significantly different using ANOVA test followed by the Bonferroni t-test (p < 0.05). Psv ICMP 4352 was obtained from the CFBP collection as strain CFBP 1670. Psv, Psn, Psf and Psr, P. savastanoi pathovars savastanoi, nerii, fraxini, and retacarpa, respectively.
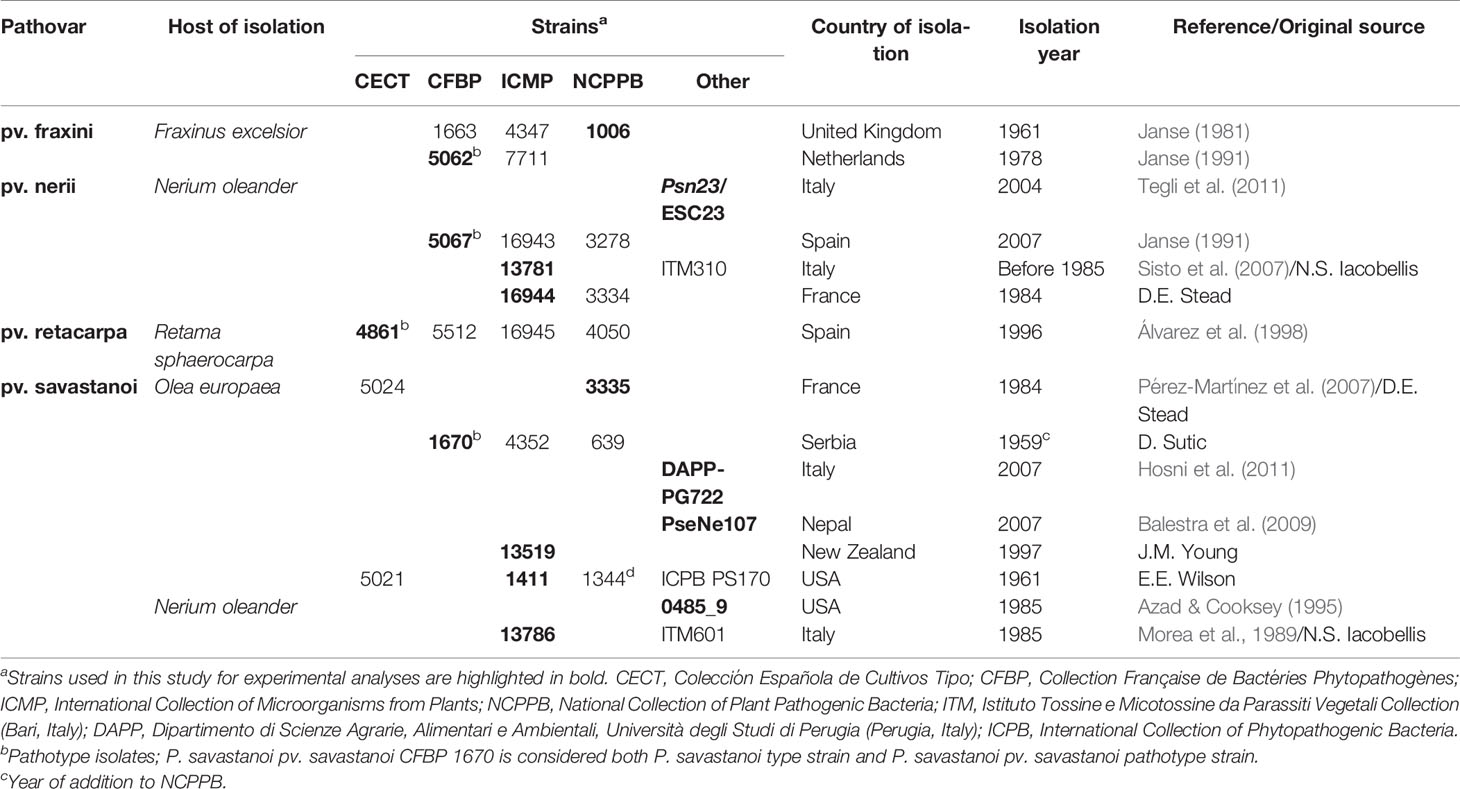
Table 3 Wild-type Pseudomonas savastanoi strains used in cross-pathogenicity tests.
Psr CECT 4861 induced well-defined knots at most inoculation sites only on broom plants (Figure 3A, Supplementary Figure S3). Ash and olive plants showed tissue swelling at most inoculation sites (80% and 60%, respectively), while infected oleander plants remained symptomless (Figure 3A, Supplementary Figure S3). These results agree with the narrow host range reported for P. savastanoi isolates from broom (Álvarez et al., 1998).
On ash plants, Psf strains NCPPB 1006 and CFBP 5062 induced excrescences (necrotic swellings) at most inoculation points (Figure 3A, Supplementary Figure S3). Excrescences also appeared in up to a third of infected sites on olive plants, supporting the reported ability of Psf to also induce symptoms in this host (Janse, 1981; Janse, 1982; Iacobellis et al., 1998). Inoculated oleander plants were symptomless.
All four Psv strains induced visible symptoms on olive and ash plants, although they were variable and strain dependent (Figure 3A, Supplementary Figure S3). For instance, Psv NCPPB 3335 and Psv PseNe107 generated knots on olive plants, which were more necrotic on wounds inoculated with the later, while Psv CFBP 1670 and Psv DAPP-PG722 primarily induced swelling of the tissues surrounding the wounds. This agrees with the reported strain- and cultivar-dependent severity of symptoms in inoculated olive plants (Penyalver et al., 2006; Pérez-Martínez et al., 2007). Ash plants primarily developed either knots (Psv DAPP-PG722) or swellings (remaining strains). Conversely, most Psv-infected oleander and broom plants remained symptomless and only a reduced number of wounds showed slightly swollen lesions (Figure 3A, Supplementary Figure S3).
Psn Psn23 and CFBP 5067 induced necrotic knots on olive plants and small overgrowths on ash. Most oleander plants inoculated with Psn Psn23 developed knots, however, few showed symptoms with Psn CFBP 5067 (Figure 3A, Supplementary Figure S3), perhaps reflecting the inability of some Psn isolates to induce knots in certain oleander cultivars (Janse, 1981). Nevertheless, both Psn CFBP 5067 and Psn23 appear to induce systemic infections because inoculated oleanders showed secondary symptoms, such as flower bud swellings, leaf curling, and tiny knots on the leaves (see Supplementary Figure S4 for CFBP 5067). Broom plants infected with Psn showed no symptoms (Figure 3A, Supplementary Figure S3).
All the strains reached the highest populations densities at 90 days post-inoculation (dpi) in their host of isolation, with medium to low populations in the other hosts. No viable counts were recovered from symptomless broom plants infected with Psf NCPPB 1006 (Figure 3B).
In summary, our results agree with the host range previously reported for the four well-established P. savastanoi pathovars of woody hosts. Additionally, we also found that some Psv strains can occasionally induce the formation of slightly swollen lesions on broom, and that strain Psr CECT 4861 also induces the formation of slight symptoms at some inoculation sites not only on broom, but also on olive and ash (Figure 3A, Supplementary Figure S3).
Psv strains did not cluster in a monophyletic lineage and two oleander strains clustered with four olive strains (Figure 2, Supplementary Figure S2). To confirm their pathovar assignation, we therefore examined the pathogenicity in olive and oleander of the six olive and oleander strains not included in the cross-pathogenicity tests shown in Figure 3.
Psn strains ICMP 16944 and ICMP 13781 induced the largest knots in olive plants, but their bacterial populations were the lowest (Figure 4). Additionally, they were the only strains inducing knots or swellings on oleander plants, were they reached very high populations. Conversely, oleander strains ICMP 13786 and 0485_9 induced symptoms in olive plants, were they reached the highest bacterial populations, but not in oleander. Thus, these two strains resemble the typical behavior of Psv strains and, although isolated from oleander, they belong to pathovar savastanoi.
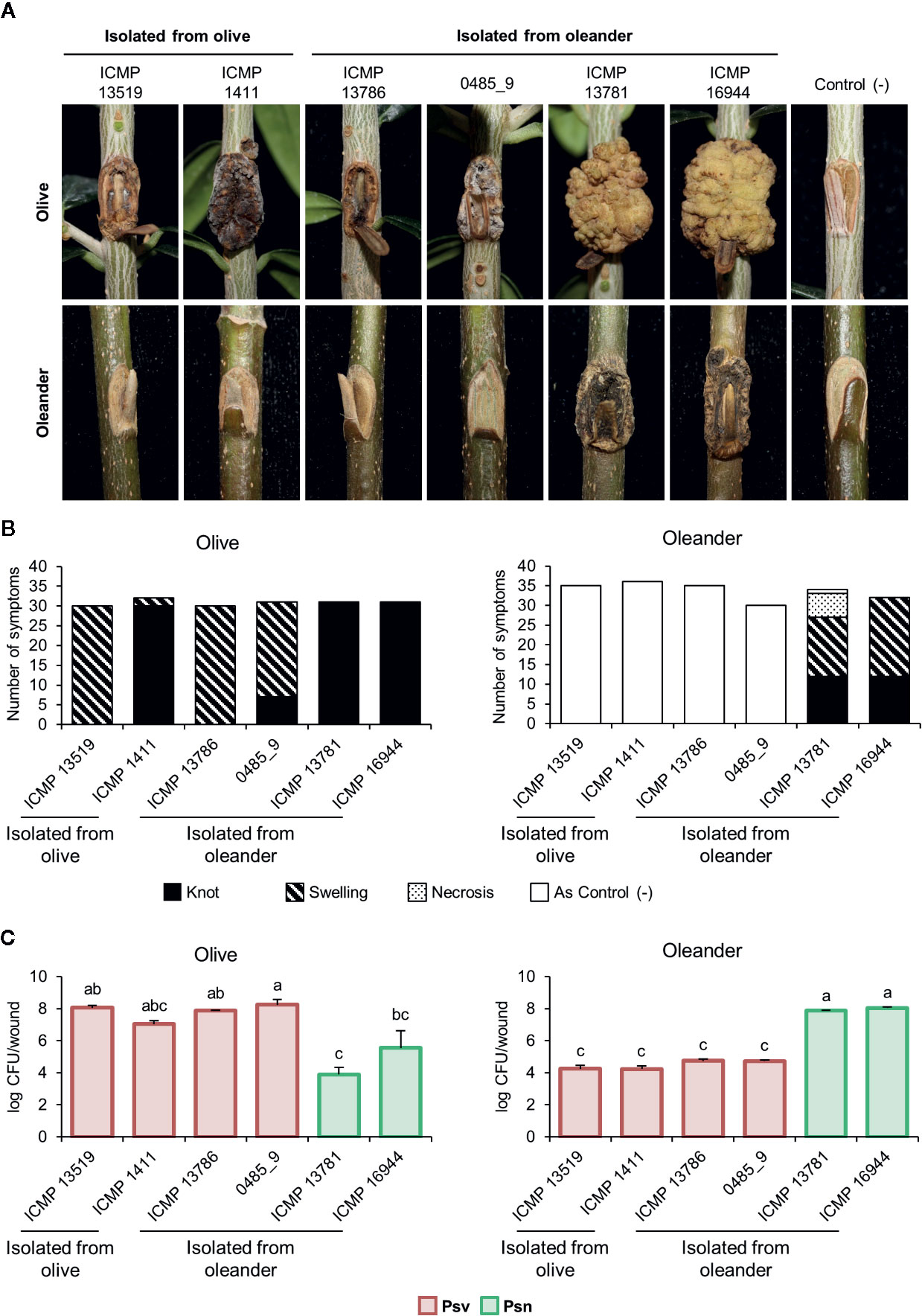
Figure 4 Cross pathogenicity tests of P. savastanoi pv. savastanoi and nerii strains. (A) Symptoms at 90 days post-inoculation. C(-), negative control, plants inoculated with 10 mM MgCl2. (B) Number of inoculation points showing differential symptoms. (C) Populations of P. savastanoi pv. savastanoi (Psv) and pv. nerii (Psn) strains at 90 days post-inoculation. Details as in Figure 3.
The distribution of the T3SS, T4SS, and T6SS determinants was investigated using the web-based tool T346Hunter (Martínez-García et al., 2015a).
As in Psv NCPPB 3335 (Rodríguez-Palenzuela et al., 2010), all the genomes encoded two different T3SS clusters: the tripartite pathogenicity island, the most prevalent T3SS within the P. syringae complex (Baltrus et al., 2017; Dillon et al., 2019b), and an additional T3SS resembling that found in Rhizobium species (Figure 5) (Joardar et al., 2005; Martínez-García et al., 2015b). Although most of the corresponding deduced products showed 100% identity among genomes, we also found amino acid variations characteristic of each of the five phylogenetic groups (Table 4).
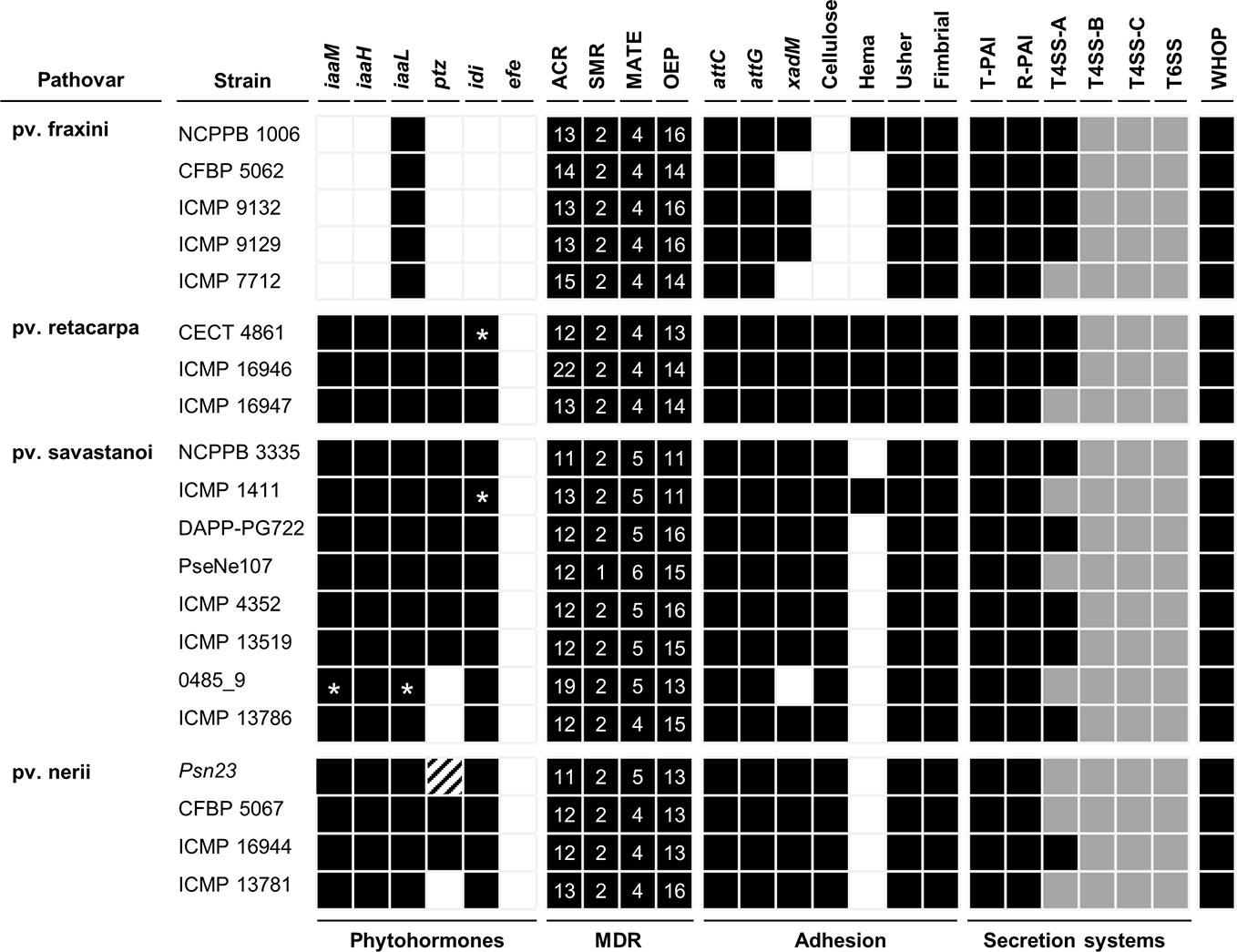
Figure 5 Bioinformatics prediction of the virulence gene repertoires of P. savastanoi strains using PIFAR. Black boxes and white boxes, presence or absence, respectively, of the indicated gene or gene set; asterisks, genes not found in the genome but identified by PCR; grey boxes, partial codification of core genes; striped boxes, genes not found in the assembly but found in the unassembled reads. MDR, multidrug resistance transporter; ACR, acridine-like transporter from the resistance/nodulation/cell division (RND) family; SMR, small multidrug transporter family; MATE, multidrug and toxic compound extrusion family; OEP, outer membrane efflux protein (RND family); attC and attG, attachment gene homologs (Agrobacterium tumefaciens); xadM, adhesion gene homolog (Xanthomonas oryzae); Cellulose, cellulose synthase; Hema, hemagglutinin-repeat protein; Usher, outer membrane usher protein; fimbrial, fimbrial protein. T-PAI, canonical tripartite T3SS; R-PAI, rhizobial T3SS; T4SS, type IV secretion system; T6SS, type VI secretion system. WHOP, genomic island carrying four operons and other genes involved in degradation of phenolics (Caballo-Ponce et al., 2017b).
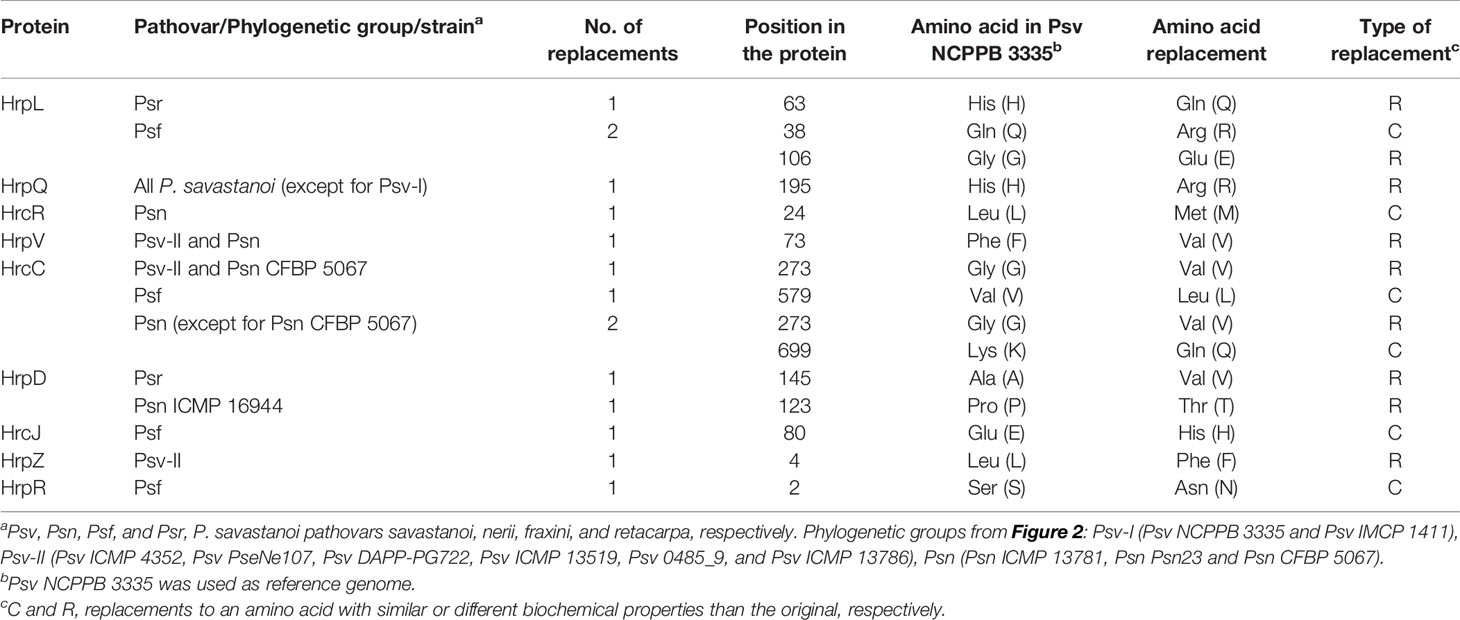
Table 4 Amino acids replacements in structural and regulatory proteins of the type III secretion system in P. savastanoi pathovars of woody hosts.
The complete set of T4SS-A genes (virB1-virB11 and virD4) carried by plasmid pPsv48B (Bardaji et al., 2011) was found in 12 genomes (Figure 5). Additionally, all 20 strains carried incomplete sets of T4SS-B genes (tra and/or trb genes) and T4SS-C genes (tfc genes) (Figure 5). The T4SS-C is involved in propagation of genomic islands (Juhas, 2015), and has not been previously described in the P. syringae complex. As described for Psv NCPPB 3335 (Rodríguez-Palenzuela et al., 2010), P. savastanoi strains encode two different T6SS clusters, one highly similar to that found in P. syringae pv. phaseolicola 1448A and a second homologous to the T6SS hcp1 cluster of P. syringae pv. tomato DC3000. Considering the composition of both T6SS clusters and additional chromosomal genes, all 20 genomes contains 12 of the 13 genes constituting the T6SS core (Boyer et al., 2009), only lacking that encoding the forkhead-associated (FHA) domain-containing protein VCA0112 (Figure 5).
The P. savastanoi pan-genome encodes 43 known T3Es (Figure 6), including truncated genes, distributed in 37 T3E families from the Hop database6 and 33 of the new merged families (Dillon et al., 2019a). T3E truncations were analyzed and assigned to groups according to their extent (Figure 7). All the strains contained the same truncations of hopAZ1, hopAA1, and hopM1. Conversely, T3E allelic variants encoded by specific pathovars, and therefore possibly involved in host specificity, include those only found in all Psn (avrPto1, hopAT1, and hopA2) or Psr strains (hopAF1-2 and hopAO1). Additionally, while all Psv and Psn strains encode a full-length coding sequence (CDS) for avrRpm2, Psr, and Psf strains either encode a truncated version of this gene or do not contain it. Specific truncations affecting single strains were also identified for hopA2, hopAF1-1, hopAO1, and hopW1 (Figure 7).
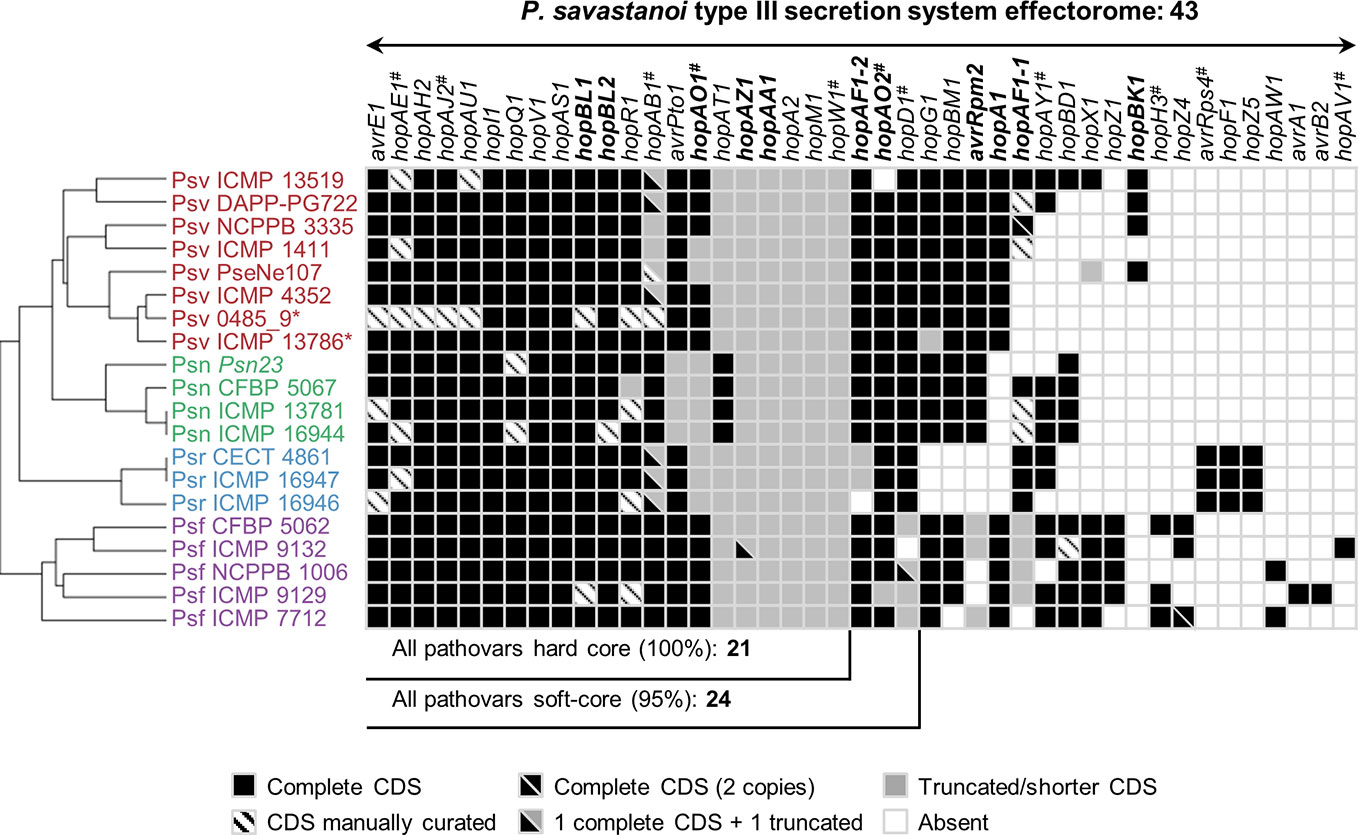
Figure 6 Distribution and hierarchical clustering of known type III secretion system effector (T3E) families. Bolded names, T3Es whose translocation has been previously demonstrated in P. savastanoi (Caballo-Ponce et al., 2017a). Hashes, T3E family names merged into new families (HopAE/HopW=HopW, HopAB/HopAY=HopAB, HopAO/HopD=HopD, HopK/AvrRps4=HopK) and T3E alleles established as novel families (HopAJ2=HopBV, HopH3=HopBQ, and HopAV1=HopBS) (Dillon et al., 2019a). Strain abbreviations as in Figure 1; CDS, coding sequence. The hierarchical clustering (left) was generated using Morpheus.
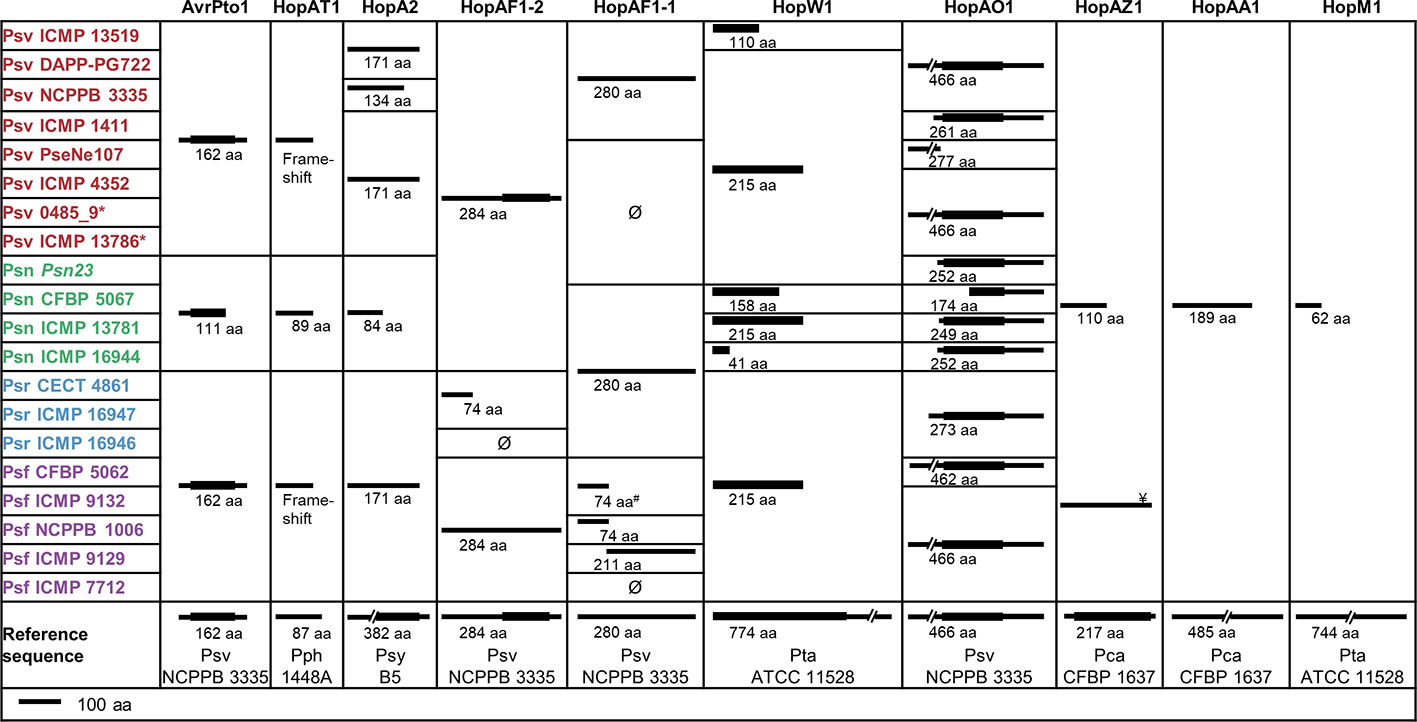
Figure 7 Truncated T3Es in P. savastanoi strains. Black lines represent the sequence length of the deduced T3E product and black boxes represent Pfam7 or HHPred8 protein domains. Ø and # indicate the absence and the truncation of hopAF1-1 by insertion of an IS5-like element, respectively. ¥Psf ICMP 9132 encodes a full-length hopAZ1; however, the encoded protein does not contain the functional domain found in the reference sequence. Strain abbreviations as in Figure 1. Pca, Pph, Psy, and Pta, P. cannabina pv. alisalensis, P. syringae pv. phaseolicola, P. syringae pv. syringae, and P. syringae pv. tabaci, respectively. 7https://pfam.xfam.org/ 8https://toolkit.tuebingen.mpg.de/tools/hhpred
We found two novel T3E candidates not homologous to known T3Es (Table 5) but preceded by a hrp box promoter, characteristic of genes regulated by HrpL (Fouts et al., 2002). Additionally, their deduced products meet at least two of the three features of N-terminal secretion signals found in Hop proteins (Lindeberg et al., 2005). The gene coding for WP_032074452 was found in all 20 P. savastanoi genomes, whereas that for WP_057446723 was exclusively found in Psr genomes.
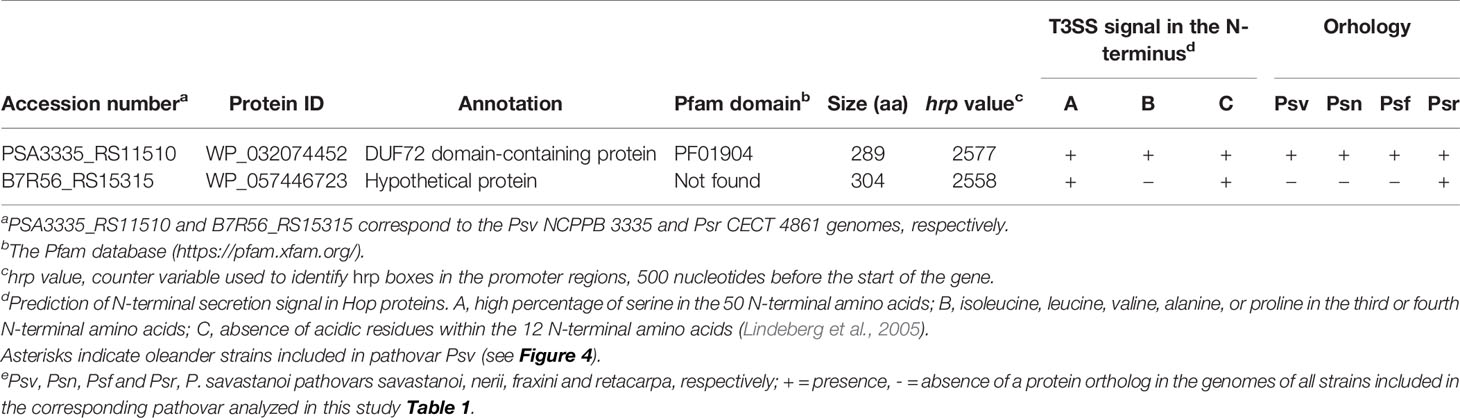
Table 5 Novel putative type III effectors identified in Pseudomonas savastanoi.
Hierarchical clustering based on T3E content identified three main clusters composed of i) all Psf genomes (Psf cluster), ii) all Psr genomes (Psr cluster) and iii) all Psv and Psn strains (Psv-Psn cluster) (Figure 6). The Psv-Psn cluster was divided into two sub-clusters, one comprising all eight Psv strains and the other the four Psn strains (Figure 6). Thus, T3E clustering of P. savastanoi genomes is similar to the core genome phylogeny (Figure 2) indicating that the four pathovars contain characteristic sets of T3Es, which are thus likely contributing to define their pathogenicity profile.
We found 21 core P. savastanoi T3E genes, encoded as full-length ORFs or truncated versions in all the genomes. Three additional T3Es (hopAF1-2, hopAO2, and hopD1) were found in 19 of the 20 genomes analyzed (95%) and were included in the soft-core effectorome. Four T3E genes are candidates to contribute to host range definition: the Psr-exclusive avrRps4, hopF1, and hopZ5 genes, and the hopG1 gene, only absent in all three Psr strains (Figure 6). Known T3E encoded in single pathovars but not in all strains, thus likely not having a relevant role in host specificity, are: hopBK1 (Psv strains), hopZ1, hopH3, hopZ4, hopAW1, avrA1, avrB2, and hopAV1 (Psf strains).
We identified several variable regions (VR) longer than 3.9 kb that were exclusively present or absent in distinct monophyletic groups (Figure 8). In general, the patterns of presence/absence of these VRs are congruent with the organismal phylogeny (Figure 2) and suggest their acquisition before pathovar differentiation followed by further vertical inheritance. Six VRs were of interest for being chromosomal and exclusively absent from all Psf strains (Figure 8, Supplementary Figure S1). VR3, VR4, VR5, and VR6 are enriched in hypothetical proteins (HPs) whereas VR1 and VR2 contain genes encoding putative virulence factors. VR1 encodes the iaaMH operon, responsible for the biosynthesis of indole-3-acetic acid (IAA) via indole-3-acetamide, and two genes involved in rhizobitoxine biosynthesis (rtxA and rtxC), a phytotoxin modulating plant-microbe interactions by inhibition of ethylene synthesis (Sugawara et al., 2006). In turn, VR2 encodes the type II secretion system proteins GspH and GspI and a pectate lyase (Figure 8, Supplementary Figure S1). With the exception of VR2, the other five VRs were predicted as genomic islands, being also often bordered by mobile genetic elements.
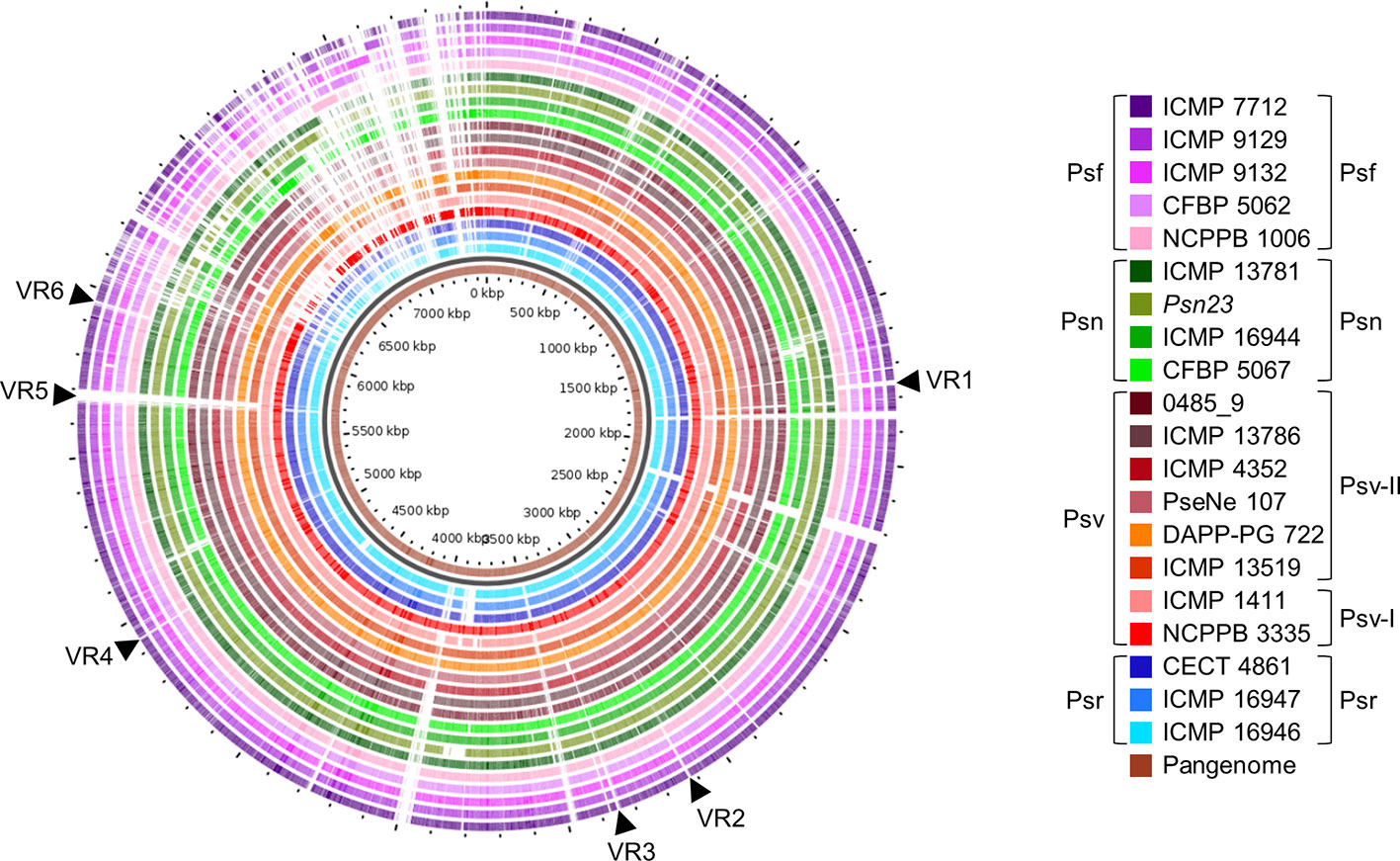
Figure 8 Genomic comparison of Pseudomonas savastanoi pathovars. Comparison made using the GView Pangenome analysis tool. Black arrows, selected variable regions (VR) that could be pathovar-specific (see Supplementary Figure S1). Strain abbreviations as in Figure 1, indicating the phylogenetic grouping from Figure 2 to the right of the legend.
The core and pan-genome profiles of the 20 strains were also analyzed at the pathovar level using BPGA (Table 2). Psf showed the largest pathovar soft-core genome size, with a nearly closed pan-genome (γ=0.065), suggesting that the gene content of Psf strains is more homogeneous than that of the other three pathovars. We also conducted this analysis considering the original assignation of strains 0485_9 and ICMP 13786 to pv. nerii (Table 2), which completely prevented the identification of exclusively present/absent genes in both Psn and Psv. This illustrates the confounding effects that might originate from an improper identification of the strains under analysis, and reinforces the need for appropriate phenotypic analyses to support conclusions derived exclusively from genomic data.
We explored the gene repertoire specific to pathovars using two complementary bioinformatics tools, Roary (Page et al., 2015) (Supplementary Table S2) and PIFAR (Figure 5) (Martínez-García et al., 2016). A total of 123 pathovar-exclusive genes and 69 pathovar-absent genes were identified and classified into eight categories, among which HPs and metabolism-related genes were the most common (Table 2, Supplementary Table S2). Many of the pathovar-exclusive and pathovar-absent genes are likely carried by plasmids and other mobile genetic elements (Figure 8, Supplementary Figure S1), because we found a fair number of genes characteristic of these elements, such as genes involved in DNA replication, recombination, mutation, and repair, and toxin/antitoxin systems (Bardaji et al., 2019). Although around 24% of the pathovar-exclusive/absent genes encoded HPs, other pathovar-exclusive genes encode proteins with a possible role in virulence and/or host specificity, such as 11 transcription factors, three c-di-GMP-related proteins, two chemotaxis-related proteins, 18 transporters belonging to eight superfamilies, the Psn-exclusive secretion systems chaperones PapD (type I secretion system; T1SS) and ShcF (T3SS), four Psr-exclusive T3Es (see also Figure 6 and Table 5) and the ferric-pseudobactin receptor PupB, also exclusive of Psr strains.
In addition to the absence in all Psf genomes of the iaaMH operon and the rhizobitoxin genes (Figure 5, Supplementary Table S2), other Psf-absent genes include those required for cytokinins (ptz) and cellulose biosynthesis, the type II secretion system (T2SS) genes hxcU, hxcP, and hxcV, the bacteriocin-related gene ALO43_200111, two genes involved in c-di-GMP turnover (gmr_3 and gmr_8), three genes encoding transporters of the ATP-binding cassette (ABC) and LysE superfamilies and cph2, encoding a bacteriophytochrome (Figure 5, Supplementary Table S2). All these genes are shared by all three tumorigenic P. savastanoi pathovars and could therefore be essential for knot induction on diverse plant hosts. Among others, Psn strains lack genes encoding an enzyme required for the biosynthesis of the phytotoxin phevamine A (O’Neill et al., 2018).
Genomics-based methods offer unprecedented resolution to analyze strain diversity and evolutionary relationships within the P. syringae complex, besides identifying candidate genes involved in pathogenicity and host range definition (Lindeberg et al., 2009; Baltrus et al., 2011; Bartoli et al., 2015; Monteil et al., 2016; Baltrus et al., 2017; Dillon et al., 2019a; Dillon et al., 2019b). However, understanding the biological meaning of the observed genomic differences depends on the availability of experimental data confirming specific phenotypes, such as virulence and host range (Baltrus and Orth, 2018). Specifically, testing more than one hundred P. syringae strains on different hosts showed that the host range is an overlapping continuum, with closely related strains exhibiting from extremely narrow to very broad host ranges (Morris et al., 2019). Our genomic analyses of 20 strains representing all four well-established P. savastanoi pathovars of woody hosts were combined with cross-pathogenicity tests, supporting the identification of candidate genes involved in host range definition. In fact, our combination of bioinformatics and functional analyses was critical to guarantee a correct pathovar assignation, ensuring the correct interpretation of the comparative data.
The P. savastanoi pan-genome (7,953 ortholog families for 20 genomes) (Table 2) is approximately 10 and 3.5 times smaller than those of the P. syringae complex (391 genomes) and PG3 (143 genomes), respectively (Dillon et al., 2019b). Additionally, the pan-genomes of the P. syringae complex and PG3 are still open (Dillon et al., 2019b), whereas the pan-genome for each P. savastanoi pathovar is nearly closed (Table 2). The large homogeneity among P. savastanoi genomes results from a low content of pathovar- and strain-specific genes (16% strain-specific genes vs. ca. 60% in the P. syringae complex; Figure 1), probably reflecting close phylogenetic relationships, a large repertoire of shared niche-specific genes and, perhaps, a natural inability to acquire foreign DNA (Pérez-Martínez et al., 2007). However, it is possible that the analyzed genomes may not represent the true diversity of the species and its four pathovars of woody hosts. Thus, the analysis of additional P. savastanoi genomes from strains isolated from woody host, as well as from the surface of herbaceous plant hosts or from non-agricultural environments, could contribute to the genetic diversity of this pathogen, resulting in more open pangenomes and reducing the number of host range determinants identified in this study. In fact, other authors have reported the existence of further genotypical and phenotypical variability among P. savastanoi strains, such as the ability to produce levan or an atypical host range (Marchi et al., 2005; Sisto et al., 2007; Cinelli et al., 2014; Moretti et al., 2017; Morris et al., 2019).
Seven of the strains analyzed here were repeatedly shown to cause distinct syndromes in their host of isolation (Psv NCPPB 3335, DAPP-PG722, and ICMP 1411; Psn Psn23; Psf NCPPB 1006 and CFBP 5062; Psr CECT 4861). However, pathogenicity data were not available for the remaining 13 strains, which were assigned to a P. savastanoi pathovar (Tables 1, 3) based on their host of isolation and, in some cases, on biochemical tests.
The cross-pathogenicity tests for nine P. savastanoi strains in olive, oleander, ash, and broom plants (Figure 3, Supplementary Figure S3) agree with the host range reported for the four well-established P. savastanoi pathovars of woody hosts. This supports using their genomes for comparative genomics analysis to identify candidate genes involved in host range definition. Psv, Psn, and Psf strains showed differences in virulence and in the ability to grow and survive in plant tissues, agreeing with previously reported cultivar-dependent virulence variability (Penyalver et al., 2000; Moretti et al., 2017). Additionally, we show that symptom severity is not always correlated with population levels in planta. In general, P. savastanoi strains showed the highest population level in their host of isolation (Figure 3B), regardless of their ability to also induce visible symptoms on other hosts. Nevertheless, maintenance of reduced bacterial density in non-host plants could favor the appearance of better-adapted variants and the development of emerging diseases in new hosts.
Agreeing with previous data (Gomila et al., 2017; Dillon et al., 2019b), Psf, Psn, Psr, and Psv cluster in a discrete genomic branch in the SNP ML tree (Figure 2) with non-tumorigenic pathovars as the phylogenetically closest relatives (Dillon et al., 2019b). This suggests that Psf, Psn, Psr, and Psv originate from a common ancestor, which likely acquired the ability to induce tumors in plants. These four pathovars also corresponded with well-defined genetic lineages indicating distinct evolutionary events leading to host specialization, although it is necessary to make two considerations. First, the Psv genomes are distributed in two separated clades, clustering with either Psr or Psn genomes (Figure 2). Although intriguing, the occurrence of different genetic lineages for a given pathovar is not new nor surprising (Gironde and Manceau, 2012; Martín-Sanz et al., 2012; Martín-Sanz et al., 2013; Hulin et al., 2018), because pathovars are defined for causing distinctive disease syndromes and not necessarily for their genetic relatedness (Young et al., 1992). Nevertheless, olive is a frequent host for the four pathovars and, since strains of P. savastanoi can cause knots in diverse species (Caballo-Ponce et al., 2017a), it is also possible that the two Psv genetic lineages show differing host ranges. In fact, pathogenicity assays on 15 herbaceous and woody hosts showed that the host range of Psv PseNe107 (Psv-II) is broader than that of Psv NCPPB 3335 (Psv-I) (Morris et al., 2019). A second relevant consideration is the clustering with Psv strains of two putative Psn strains (Figure 2). These two strains were more like Psv than Psn for several characteristics, such as their virulence gene repertoire (Figure 5) or their pattern of T3E genes (Figure 6). Nevertheless, the ability of strains 0485_9Ĕ and ICMP 13786 to cause knots in olive but not in oleander, and their better survival in olive than in oleander tissues (Figure 4) warrants their unequivocal classification as Psv strains (Janse, 1982).
All 20 genomes encode a canonical T-PAI T3SS cluster (Figure 5, Table 4). Since it is essential for knot formation and the induction of a visible hypersensitive response (HR) by Psv and Psn (Caballo-Ponce et al., 2017a), and given its high sequence and functional conservation (Table 4) (Büttner and He, 2009), we could expect a similar role for Psf and Psr. The number of T3Es in P. syringae complex strains varies from four to nearly 50 (Dillon et al., 2019a), and the average in woody hosts strains is 29 (Nowell et al., 2016). Similarly, P. savastanoi strains encode 28 to 35 T3E genes (Figure 6). As expected, the only three core T3E genes of the P. syringae complex primary PGs (avrE1, hopAA1, and hopAJ2) and the broadly distributed hopM1 (Dillon et al., 2019b), are also P. savastanoi core T3Es. P. savastanoi strains also encoded several T3E genes associated with pathogenicity on woody hosts, i.e. the core members hopAO1, hopBL1, and hopBL2 (Matas et al., 2014; Nowell et al., 2016; Castañeda-Ojeda et al., 2017), as well as hopAY1, hopH3, hopZ5, and hopAF1-1 (Figure 6). Additionally, all 20 genomes encode hopAN1 and hopJ1, present in all 11 P. syringae complex PGs, but removed as T3Es or reclassified as T3SS helpers (Dillon et al., 2019b).
Truncations of T3E genes are common in P. syringae (Baltrus et al., 2011; Dillon et al., 2019a), and we considered them here (Figure 7) because some of their products are translocated into plant cells and interfere with plant defenses (Zumaquero et al., 2010; Matas et al., 2014). T3Es are considered the primary determinants of host specificity in the P. syringae complex at both the pathovar-species and race-cultivar levels (Collmer et al., 2000; Fouts et al., 2003; Cunnac et al., 2009; Lindeberg et al., 2009; Mansfield, 2009). We therefore searched for exclusive T3E genes present/absent in specific pathovars. All three Psr genomes encode avrRps4, hopF1, hopZ5, and the putative novel T3E gene B7R56_RS15315. Additionally, they lack hopG1, a gene found in the remaining 17 strains. These four known T3E genes were shown to contribute to the virulence of P. syringae strains but also function as avirulence factors in soybean and Arabidopsis (avrRps4), bean (hopF1), Arabidopsis (hopZ5) and cherry trees (hopG1) (Hinsch and Staskawicz, 1996; Tsiamis et al., 2000; Jayaraman et al., 2017; Hulin et al., 2018). Thus, avrRps4, hopF1, and hopZ5 might contribute to Psr virulence in broom plants and/or restrict the infection of ash, oleander, and olive. Furthermore, Psr strains lack hopG1 and hopAF1-2, or encode a truncated HopAF1-2 protein lacking its catalytic domain (Figure 7). Therefore, the absence of these T3Es could be related to their recognition by broom resistance genes.
Although we did not find Psn-exclusive T3E genes, Psn strains encode a specific 3′ truncation of avrPto1 (Figure 7). Considering that point mutations affecting the C-terminal region of AvrPto abolish avirulence in tobacco (Shan et al., 2000), the specific Psn truncation might prevent avirulence in oleander. Other Psn-exclusive allelic variants possibly involved in host range definition are those of hopAT1 and hopA2 (Figure 7). Additionally, Psf and Psr strains do not encode a chimera of avrRpm2, with an N-terminal region homologous to that of the HopF4 family (Lo et al., 2017).
Using three complementary strategies, we identified a repertoire of pathovar-specific genes potentially associated with host specificity (Figures 5, 8, and Supplementary Table S2). GView identified several genomic regions exclusively present/absent in all strains from monophyletic branches of the P. savastanoi phylogeny, most of which correspond to single pathovars (Figure 8). Since this strategy is not suitable for the identification of individual genes encoded by specific pathovars, we used the additional bioinformatics tools Roary (Supplementary Table S2) and PIFAR (Figure 5). Nevertheless, many of the pathovar-exclusive and pathovar-absent genes identified by Roary were also detected using GView or PIFAR, increasing the reliability of the results obtained.
As expected, all non-tumorigenic Psf strains lack the iaaMH operon (Figure 5), required for the biosynthesis of IAA and knot formation by Psv and Psn strains (Comai and Kosuge, 1983; Iacobellis et al., 1998; Aragón et al., 2014). Tumorigenic P. savastanoi pathovars, encode a copy of this operon in VR1, a genomic island also encoding the rhizobitoxine operon and two glutamine biosynthesis-related genes that likely supply glutamine for rhizobitoxine biosynthesis (Sugawara et al., 2006). In Bradyrhizobium elkanii, this enol-ether amino acid toxin enhances nodulation by inhibiting ethylene biosynthesis in host roots. Remarkably, production of IAA in B. elkanii is restricted to strains also synthetizing rhizobitoxine. Therefore, rhizobitoxine might also be involved in knot formation.
All pathovars, except Psn, contained genes for the biosynthesis of the phytotoxin phevamine A. This molecule, composed of a modified spermidine, L-phenylalanine, and L-valine, contributes to the virulence of P. syringae DC3000 on Arabidopsis suppressing immune responses induced by flagellin (O’Neill et al., 2018). We did not find genes for the biosynthesis of other phytotoxins involved in virulence in woody hosts (Hulin et al., 2018).
Gene ptz, involved in cytokinins production, was neither found in Psf strains nor in the genomes of Psn ICMP 13781 and the oleander strains here reassigned into Psv (0485_9 and ICMP 13786) (Figure 5). Gene ptz is plasmid encoded in most Psv and Psn strains, and is absent in some Psn strains (Pérez-Martínez et al., 2008). In addition, failure to produce cytokinins is correlated to symptoms attenuation, both on olive and oleander (Surico et al., 1985). However, Psn ICMP 13781 consistently induced tumors in olive as large as those produced by other Psn strains carrying gene ptz (Figure 4), suggesting that induction of full symptoms by Psn on olive is also dependent on other virulence determinants, as reported for Psv strains (Caballo-Ponce et al., 2017a).
In conclusion, besides T3SS genes whose role in host specificity has already been demonstrated in the P. syringae complex, our results suggest the existence of other putative virulence factors that could also contribute to host range definition, such as proteins secreted by the T2SS and T6SS, genes involved in the biosynthesis of the phytotoxins rhizobitoxine and phevamine A, a large number of transcriptional regulators, proteins involved in the metabolism of c-di-GMP, signal-transduction proteins and transporters of eight different superfamilies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JM, ST, CM, PR-P, and CR planned and designed the research and analyzed and interpreted the data. AM-P, AP and EC-P performed experiments. AM-P, AP, JM, PR-P, and CR carried out bioinformatics analyses. AM-P, AP, JM, and CR designed and prepared Figures and Tables. AM-P, AP, EC-P, JM, and CR wrote the manuscript.
AM-P, AP, CR, and JM were supported by grants FPI/BES-2015-074847, FPU14/05551, AGL2017-82492-C2-1-R and AGL2017-82492-C2-2-R, respectively, from Ministerio de Ciencia, Innovación y Universidades (Spain), cofinanced by the Fondo Europeo de Desarrollo Regional (FEDER); CM was supported by DSA3 research funds “Fondo di base” (Italy).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank I. Matas for the isolation of Psv NCPPB 3335 genomic DNA; C. Cortese, for PCR amplification of diverse T3Es genes from Psv DAPP-PG722; L. Díaz and J. Sánchez-Colmenero for help with bioinformatics pipelines; P. García-Vallejo, for technical assistance; and T.H. Osinga for advice on English usage.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00973/full#supplementary-material
Álvarez, F., García de los Ríos, J. E., Jimenez, P., Rojas, A., Reche, P., Troya, M. T. (1998). Phenotypic variability in different strains of Pseudomonas syringae subsp. savastanoi isolated from different hosts. Eur. J. Plant Pathol. 104 (6), 603–609. doi: 10.1023/A:1008604011154
Afgan, E., Baker, D., van den Beek, M., Blankenberg, D., Bouvier, D., Čech, M., et al. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3–W10. doi: 10.1093/nar/gkw343
Aragón, I. M., Pérez-Martínez, I., Moreno-Pérez, A., Cerezo, M., Ramos, C. (2014). New insights into the role of indole-3-acetic acid in the virulence of Pseudomonas savastanoi pv. savastanoi. FEMS Microbiol. Lett. 356 (2), 184–192. doi: 10.1111/1574-6968.12413
Azad, H. R., Cooksey, D. A. (1995). A semiselective medium for detecting epiphytic and systemic population of Pseudomonas savastanoi from oleander. Phytopathology 85, 740–745. doi: 10.1094/Phyto-85-740
Büttner, D., He, S. Y. (2009). Type III protein secretion in plant pathogenic bacteria. Plant Physiol. 150 (4), 1656–1664. doi: 10.1104/pp.109.139089
Balestra, G. M., Lamichhane, J. R., Kshetri, M. B., Mazzaglia, A., Varvaro, L. (2009). First report of olive knot caused by Pseudomonas savastanoi pv. savastanoi in Nepal. Plant Pathol. 58 (2), 393. doi: 10.1111/j.1365-3059.2008.02007.x
Baltrus, D. A., Orth, K. N. (2018). Understanding genomic diversity in Pseudomonas syringae throughout the forest and on the trees. New Phytol. 219 (2), 482–484. doi: 10.1111/nph.15269
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukhtar, M. S., Cherkis, K. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PloS Pathog. 7 (7), e1002132–e1002132. doi: 10.1371/journal.ppat.1002132
Baltrus, D. A., McCann, H. C., Guttman, D. S. (2017). Evolution, genomics and epidemiology of Pseudomonas syringae. Mol. Plant Pathol. 18 (1), 152–168. doi: 10.1111/mpp.12506
Bardaji, L., Pérez-Martínez, I., Rodríguez-Moreno, L., Rodríguez-Palenzuela, P., Sundin, G. W., Ramos, C., et al. (2011). Sequence and role in virulence of the three plasmid complement of the model tumor-inducing bacterium Pseudomonas savastanoi pv. savastanoi NCPPB 3335. PloS One 6 (10), e25705. doi: 10.1371/journal.pone.0025705
Bardaji, L., Añorga, M., Echeverría, M., Ramos, C., Murillo, J. (2019). The toxic guardians - multiple toxin-antitoxin systems provide stability, avoid deletions and maintain virulence genes of Pseudomonas syringae virulence plasmids. Mobile DNA 10, 7. doi: 10.1186/s13100-019-0149-4
Bartoli, C., Carrere, S., Lamichhane, J. R., Varvaro, L., Morris, C. E. (2015). Whole-genome sequencing of 10 Pseudomonas syringae strains representing different host range spectra. Genome Announcements 3 (2), e00379–e00315. doi: 10.1128/genomeA.00379-15
Berge, O., Monteil, C. L., Bartoli, C., Chandeysson, C., Guilbaud, C., Sands, D. C. (2014). A user’s guide to a data base of the diversity of Pseudomonas syringae and its application to classifying strains in this phylogenetic complex. PloS One 9, e105547. doi: 10.1371/journal.pone.0105547
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62 (3), 293–300. doi: 10.1128/JB.62.3.293-300.1951
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45 (W1), W30–W35. doi: 10.1093/nar/gkx343
Boyer, F., Fichant, G., Berthod, J., Vandenbrouck, Y., Attree, I. (2009). Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 10, 104. doi: 10.1186/1471-2164-10-104
Bull, C. T., Koike, S. T. (2015). Practical benefits of knowing the enemy: modern molecular tools for diagnosing the etiology of bacterial diseases and understanding the taxonomy and diversity of plant-pathogenic bacteria. Annu. Rev. Phytopathol. 53 (1), 157–180. doi: 10.1146/annurev-phyto-080614-120122
Caballo-Ponce, E., Murillo, J., Martínez-Gil, M., Moreno-Pérez, A., Pintado, A., Ramos, C. (2017a). Knots untie: molecular determinants involved in knot formation induced by Pseudomonas savastanoi in woody hosts. Front. Plant Sci. 8, 1089. doi: 10.3389/fpls.2017.01089
Caballo-Ponce, E., van Dillewijn, P., Wittich, R. M., Ramos, C. (2017b). WHOP, a genomic region associated with woody hosts in the Pseudomonas syringae complex contributes to the virulence and fitness of Pseudomonas savastanoi pv. savastanoi in olive plants. Mol. Plant-Microbe Interact. 30 (2), 113–126. doi: 10.1094/mpmi-11-16-0233-r
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28 (4), 464–469. doi: 10.1093/bioinformatics/btr703
Castañeda-Ojeda, M. P., Moreno-Pérez, A., Ramos, C., López-Solanilla, E. (2017). Suppression of plant immune responses by the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 type III effector tyrosine phosphatases HopAO1 and HopAO2. Front. Plant Sci. 8, 680. doi: 10.3389/fpls.2017.00680
Chaudhari, N., Gupta, V., Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6, 24373. doi: 10.1038/srep24373
Cinelli, T., Marchi, G., Cimmino, A., Marongiu, R., Evidente, A., Fiori, M. (2014). Heterogeneity of Pseudomonas savastanoi populations infecting Myrtus communis in Sardinia (Italy). Plant Pathol. 63 (2), 277–289. doi: 10.1111/ppa.12096
Collmer, A., Badel, J. L., Charkowski, A. O., Deng, W. L., Fouts, D. E., Ramos, A. R., et al. (2000). Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc. Natl. Acad. Sci. 97 (16), 8770–8777. doi: 10.1073/pnas.97.16.8770
Comai, L., Kosuge, T. (1983). Transposable element that causes mutations in a plant pathogenic Pseudomonas sp. J. Bacteriol. 154 (3), 1162–1167. doi: 10.1128/JB.154.3.1162-1167.1983
Cunnac, S., Lindeberg, M., Collmer, A. (2009). Pseudomonas syringae type III secretion system effectors: repertoires in search of functions. Curr. Opin. Microbiol. 12 (1), 53–60. doi: 10.1016/j.mib.2008.12.003
Dillon, M. M., Almeida, R. N. D., Laflamme, B., Martel, A., Weir, B. S., Desveaux, D., et al. (2019a). Molecular evolution of Pseudomonas syringae type III secreted effector proteins. Front. Plant Sci. 10, 418. doi: 10.3389/fpls.2019.00418
Dillon, M. M., Thakur, S., Almeida, R. N. D., Wang, P. W., Weir, B. S., Guttman, D. S. (2019b). Recombination of ecologically and evolutionarily significant loci maintains genetic cohesion in the Pseudomonas syringae species complex. Genome Biol. 20 (1), 3. doi: 10.1186/s13059-018-1606-y
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 (19), 2460–2461. doi: 10.1093/bioinformatics/btq461
Eichinger, V., Nussbaumer, T., Platzer, A., Jehl, M.-A., Arnold, R., Rattei, T. (2015). EffectiveDB—updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 44 (D1), D669–D674. doi: 10.1093/nar/gkv1269
Fouts, D. E., Abramovitch, R. B., Alfano, J. R., Baldo, A. M., Buell, C. R., Cartinhour, S., et al. (2002). Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc. Natl. Acad. Sci. 99 (4), 2275–2280. doi: 10.1073/pnas.032514099
Fouts, D. E., Badel, J. L., Ramos, A. R., Rapp, R. A., Collmer, A. (2003). A Pseudomonas syringae pv. tomato DC3000 Hrp (type III secretion) deletion mutant expressing the Hrp system of bean pathogen P. syringae pv. syringae 61 retains normal host specificity for tomato. Mol. Plant-Microbe Interact. 16 (1), 43–52. doi: 10.1094/mpmi.2003.16.1.43
Gardan, L., Bollet, C., Abu Ghorrah, M., Grimont, F., Grimont, P. A. D. (1992). DNA relatedness among the pathovar strains of Pseudomonas syringae subsp. savastanoi Janse, (1982) and proposal of Pseudomonas savastanoi sp. nov. Int. J. Systemat. Bacteriol. 42, 606–612. doi: 10.1099/00207713-42-4-606
Gironde, S., Manceau, C. (2012). Housekeeping gene sequencing and multilocus variable-number tandem-repeat analysis to identify subpopulations within Pseudomonas syringae pv. maculicola and Pseudomonas syringae pv. tomato that correlate with host specificity. Appl. Environ. Microbiol. 78 (9), 3266–3279. doi: 10.1128/aem.06655-11
Gomila, M., Busquets, A., Mulet, M., García-Valdés, E., Lalucat, J. (2017). Clarification of taxonomic status within the Pseudomonas syringae species group based on a phylogenomic analysis. Front. Microbiol. 8, 2422. doi: 10.3389/fmicb.2017.02422
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34 (2), 292–293. doi: 10.1093/bioinformatics/btx610
Hinsch, M., Staskawicz, B. J. (1996). Identification of a new Arabidopsis disease resistance locus, RPS4, and cloning of the corresponding avirulence gene, avrRps4, from Pseudomonas syringae pv. pisi. Mol. Plant-Microbe Interact. 9 (1), 55–61. doi: 10.1094/mpmi-9-0055
Hosni, T., Moretti, C., Devescovi, G., Suarez-Moreno, Z. R., Fatmi, M. B., Guarnaccia, C., et al. (2011). Sharing of quorum-sensing signals and role of interspecies communities in a bacterial plant disease. ISME J. 5 (12), 1857–1870. doi: 10.1038/ismej.2011.65
Hulin, M. T., Armitage, A. D., Vicente, J. G., Holub, E. B., Baxter, L., Bates, H. J., et al. (2018). Comparative genomics of Pseudomonas syringae reveals convergent gene gain and loss associated with specialization onto cherry (Prunus avium). New Phytol. 219 (2), 672–696. doi: 10.1111/nph.15182
Iacobellis, N. S., Caponero, A., Evidente, A. (1998). Characterization of Pseudomonas syringae ssp. savastanoi strains isolated from ash. Plant Pathol. 47 (1), 73–83. doi: 10.1046/j.1365-3059.1998.00202.x
Janse, J. D. (1981). The bacterial disease of ash (Fraxinus excelsior), caused by Pseudomonas syringae subsp. savastanoi pv. fraxini II. Etiology and taxonomic considerations. Eur. J. For. Pathol. 11 (7), 425–438. doi: 10.1111/j.1439-0329.1981.tb00115.x
Janse, J. D. (1982). Pseudomonas syringae subsp. savastanoi (ex Smith) subsp. nov., nom. rev., the bacterium causing excrescences on Oleaceae and Nerium oleander L. Int. J. Systemat. Evolution. Microbiol. 32 (2), 166–169. doi: 10.1099/00207713-32-2-166
Janse, J. D. (1991). Pathovar discrimination within Pseudomonas syringae subsp. savastanoi using whole cell fatty acids and pathogenicity as criteria. Systemat. Appl. Microbiol. 14 (1), 79–84. doi: 10.1016/S0723-2020(11)80365-6
Jayaraman, J., Choi, S., Prokchorchik, M., Choi, D. S., Spiandore, A., Rikkerink, E. H., et al. (2017). A bacterial acetyltransferase triggers immunity in Arabidopsis thaliana independent of hypersensitive response. Sci. Rep. 7 (1), 3557. doi: 10.1038/s41598-017-03704-x
Joardar, V., Lindeberg, M., Jackson, R. W., Selengut, J., Dodson, R., Brinkac, L. M., et al. (2005). Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J. Bacteriol. 187 (18), 6488–6498. doi: 10.1128/JB.187.18.6488-6498.2005
Juhas, M. (2015). Type IV secretion systems and genomic islands-mediated horizontal gene transfer in Pseudomonas and Haemophilus. Microbiol. Res. 170, 10–17. doi: 10.1016/j.micres.2014.06.007
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lamichhane, J. R., Varvaro, L., Parisi, L., Audergon, J.-M., Morris, C. E. (2014). “Disease and frost damage of woody plants caused by Pseudomonas syringae: seeing the forest for the trees,” in Advances in Agronomy. Ed. Sparks, D. L. (San Diego, CA: Academic Press), 235–295.
Li, R., Li, Y., Kristiansen, K., Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24 (5), 713–714. doi: 10.1093/bioinformatics/btn025
Lindeberg, M., Stavrinides, J., Chang, J. H., Alfano, J. R., Collmer, A., Dangl, J. L., et al. (2005). Proposed guidelines for a unified nomenclature and phylogenetic analysis of type III Hop effector proteins in the plant pathogen Pseudomonas syringae. Mol. Plant-Microbe Interact. 18 (4), 275–282. doi: 10.1094/mpmi-18-0275
Lindeberg, M., Cunnac, S., Collmer, A. (2009). The evolution of Pseudomonas syringae host specificity and type III effector repertoires. Mol. Plant Pathol. 10 (6), 767–775. doi: 10.1111/j.1364-3703.2009.00587.x
Lo, T., Koulena, N., Seto, D., Guttman, D. S., Desveaux, D. (2017). The HopF family of Pseudomonas syringae type III secreted effectors. Mol. Plant Pathol. 18 (3), 457–468. doi: 10.1111/mpp.12412
Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, P., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13 (6), 614–629. doi: 10.1111/j.1364-3703.2012.00804.x
Mansfield, J. W. (2009). From bacterial avirulence genes to effector functions via the hrp delivery system: an overview of 25 years of progress in our understanding of plant innate immunity. Mol. Plant Pathol. 10 (6), 721–734. doi: 10.1111/j.1364-3703.2009.00576.x
Marchi, G., Viti, C., Giovannetti, L., Surico, G. (2005). Spread of levan-positive populations of Pseudomonas savastanoi pv. savastanoi, the causal agent of olive knot, in central Italy. Eur. J. Plant Pathol. 112 (2), 101–112. doi: 10.1007/s10658-005-0804-0
Martínez-García, P. M., Ramos, C., Rodríguez-Palenzuela, P. (2015a). T346Hunter: a novel web-based tool for the prediction of type III, type IV and type VI secretion systems in bacterial genomes. PloS One 10 (4), e0119317. doi: 10.1371/journal.pone.0119317
Martínez-García, P. M., Rodríguez-Palenzuela, P., Arrebola, E., Carrión, V. J., Gutiérrez-Barranquero, J. A., Pérez-García, A., et al. (2015b). Bioinformatics analysis of the complete genome sequence of the mango tree pathogen Pseudomonas syringae pv. syringae UMAF0158 reveals traits relevant to virulence and epiphytic lifestyle. PloS One 10 (8), e0136101. doi: 10.1371/journal.pone.0136101
Martínez-García, P. M., López-Solanilla, E., Ramos, C., Rodríguez-Palenzuela, P. (2016). Prediction of bacterial associations with plants using a supervised machine-learning approach. Environ. Microbiol. 18 (12), 4847–4861. doi: 10.1111/1462-2920.13389
Martín-Sanz, A., Pérez de la Vega, M., Murillo, J., Caminero, C. (2012). Genetic, biochemical and pathogenic diversity of Pseudomonas syringae pv. pisi strains. Plant Pathol. 61 (6), 1063–1072. doi: 10.1111/j.1365-3059.2012.02604.x
Martín-Sanz, A., de la Vega, M. P., Murillo, J., Caminero, C. (2013). Strains of Pseudomonas syringae pv. syringae from pea are phylogenetically and pathogenically diverse. Phytopathology 103 (7), 673–681. doi: 10.1094/phyto-08-12-0196-r
Matas, I. M., Castañeda-Ojeda, M. P., Aragón, I. M., Antúnez-Lamas, M., Murillo, J., Rodríguez-Palenzuela, P., et al. (2014). Translocation and functional analysis of Pseudomonas savastanoi pv. savastanoi NCPPB 3335 type III secretion system effectors reveals two novel effector families of the Pseudomonas syringae complex. Mol. Plant-Microbe Interact. 27 (5), 424–436. doi: 10.1094/MPMI-07-13-0206-R
Monteil, C. L., Yahara, K., Studholme, D. J., Mageiros, L., Méric, G., Swingle, B., et al. (2016). Population-genomic insights into emergence, crop adaptation and dissemination of Pseudomonas syringae pathogens. Microbial. Genomics 2 (10), e000089. doi: 10.1099/mgen.0.000089
Morea, M., Sisto, A., Iacobellis, N. S., Surico, G., Morea, A. (1989). Introduction of cosmid pVK102 into Pseudomonas syringae pv. savastanoi. Phytopathol. Mediterr. 28 (2), 140–146.
Moretti, C., Cortese, C., Passos da Silva, D., Venturi, V., Ramos, C., Firrao, G., et al. (2014). Draft genome sequence of Pseudomonas savastanoi pv. savastanoi strain DAPP-PG 722, isolated in Italy from an olive plant affected by knot disease. Genome Announcements 2 (5), e00864–e00814. doi: 10.1128/genomeA.00864-14
Moretti, C., Vinatzer, B. A., Onofri, A., Valentini, F., Buonaurio, R. (2017). Genetic and phenotypic diversity of Mediterranean populations of the olive knot pathogen, Pseudomonas savastanoi pv. savastanoi. Plant Pathol. 66 (4), 595–605. doi: 10.1111/ppa.12614
Morris, C. E., Monteil, C. L., Berge, O. (2013). The life history of Pseudomonas syringae: linking agriculture to earth system processes. Annu. Rev. Phytopathol. 51 (1), 85–104. doi: 10.1146/annurev-phyto-082712-102402
Morris, C. E., Lamichhane, J. R., Nikolić, I., Stanković, S., Moury, B. (2019). The overlapping continuum of host range among strains in the Pseudomonas syringae complex. Phytopathol. Res. 1 (1), 4. doi: 10.1186/s42483-018-0010-6
Muñoz-Mérida, A., Viguera, E., Claros, M. G., Trelles, O., Pérez-Pulido, A. J. (2014). Sma3s: a three-step modular annotator for large sequence datasets. DNA Res. 21 (4), 341–353. doi: 10.1093/dnares/dsu001
Nowell, R. W., Laue, B. E., Sharp, P. M., Green, S. (2016). Comparative genomics reveals genes significantly associated with woody hosts in the plant pathogen Pseudomonas syringae. Mol. Plant Pathol. 17 (9), 1409–1424. doi: 10.1111/mpp.12423
O’Neill, E. M., Mucyn, T. S., Patteson, J. B., Finkel, O. M., Chung, E.-H., Baccile, J. A., et al. (2018). Phevamine A, a small molecule that suppresses plant immune responses. Proc. Natl. Acad. Sci. 115 (41), E9514–E9522. doi: 10.1073/pnas.1803779115
Pérez-Martínez, I., Rodríguez-Moreno, L., Matas, I. M., Ramos, C. (2007). Strain selection and improvement of gene transfer for genetic manipulation of Pseudomonas savastanoi isolated from olive knots. Res. Microbiol. 158 (1), 60–69. doi: 10.1016/j.resmic.2006.09.008
Pérez-Martínez, I., Zhao, Y., Murillo, J., Sundin, G. W., Ramos, C. (2008). Global genomic analysis of Pseudomonas savastanoi pv. savastanoi plasmids. J. Bacteriol. 190 (2), 625–635. doi: 10.1128/JB.01067-07
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31 (22), 3691–3693. doi: 10.1093/bioinformatics/btv421
Penyalver, R., García, A., Ferrer, A., Bertolini, E., López, M. M. (2000). Detection of Pseudomonas savastanoi pv. savastanoi in olive plants by enrichment and PCR. Appl. Environ. Microbiol. 66 (6), 2673–2677. doi: 10.1128/aem.66.6.2673-2677.2000
Penyalver, R., García, A., Ferrer, A., Bertolini, E., Quesada, J. M., Salcedo, C. I., et al. (2006). Factors affecting Pseudomonas savastanoi pv. savastanoi plant inoculations and their use for evaluation of olive cultivar susceptibility. Phytopathology 96 (3), 313–319. doi: 10.1094/PHYTO-96-0313
Petkau, A., Stuart-Edwards, M., Stothard, P., Van Domselaar, G. (2010). Interactive microbial genome visualization with GView. Bioinformatics 26 (24), 3125–3126. doi: 10.1093/bioinformatics/btq588
Ramos, C., Matas, I. M., Bardaji, L., Aragón, I. M., Murillo, J. (2012). Pseudomonas savastanoi pv. savastanoi: some like it knot. Mol. Plant Pathol. 13 (9), 998–1009. doi: 10.1111/j.1364-3703.2012.00816.x
Rodríguez-Palenzuela, P., Matas, I. M., Murillo, J., López-Solanilla, E., Bardaji, L., Pérez-Martínez, I., et al. (2010). Annotation and overview of the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 draft genome reveals the virulence gene complement of a tumour-inducing pathogen of woody hosts. Environ. Microbiol. 12 (6), 1604–1620. doi: 10.1111/j.1462-2920.2010.02207.x
Sambrook, J., Russell, D. (2001). Molecular cloning: a laboratory manual (New York, USA: Cold Spring Harbor Laboratory Press).
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
Shan, L., Thara, V. K., Martin, G. B., Zhou, J.-M., Tang, X. (2000). The pseudomonas AvrPto protein is differentially recognized by tomato and tobacco and is localized to the plant plasma membrane. Plant Cell 12 (12), 2323–2337. doi: 10.1105/tpc.12.12.2323
Sisto, A., Cipriani, M. G., Tegli, S., Cerboneschi, M., Stea, G., Santilli, E. (2007). Genetic characterization by fluorescent AFLP of Pseudomonas savastanoi pv. savastanoi strains isolated from different host species. Plant Pathol. 56 (3), 366–372. doi: 10.1111/j.1365-3059.2007.01567.x
Sugawara, M., Okazaki, S., Nukui, N., Ezura, H., Mitsui, H., Minamisawa, K. (2006). Rhizobitoxine modulates plant–microbe interactions by ethylene inhibition. Biotechnol. Adv. 24 (4), 382–388. doi: 10.1016/j.biotechadv.2006.01.004
Surico, G., Iacobellis, N. S., Sisto, A. (1985). Studies on the role of indole-3-acetic acid and cytokinins in the formation of knots on olive and oleander plants by Pseudomonas syringae pv. savastanoi. Physiol. Plant Pathol. 26 (3), 309–320. doi: 10.1016/0048-4059(85)90006-2
Tegli, S., Gori, A., Cerboneschi, M., Cipriani, M. G., Sisto, A. (2011). Type three secretion system in Pseudomonas savastanoi pathovars: does timing matter? Genes 2 (4), 957. doi: 10.3390/genes2040957
Tettelin, H., Riley, D., Cattuto, C., Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11 (5), 472–477. doi: 10.1016/j.mib.2008.09.006
Thakur, S., Weir, B. S., Guttman, D. S. (2016). Phytopathogen genome announcement: draft genome sequences of 62 Pseudomonas syringae type and pathotype strains. Mol. Plant-Microbe Interact. 29 (4), 243–246. doi: 10.1094/MPMI-01-16-0013-TA
Treangen, T. J., Ondov, B. D., Koren, S., Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15 (11), 524. doi: 10.1186/s13059-014-0524-x
Tsiamis, G., Mansfield, J. W., Hockenhull, R., Jackson, R. W., Sesma, A., Athanassopoulos, E., et al. (2000). Cultivar-specific avirulence and virulence functions assigned to avrPphF in Pseudomonas syringae pv. phaseolicola, the cause of bean halo-blight disease. EMBO J. 19 (13), 3204–3214. doi: 10.1093/emboj/19.13.3204
Vinatzer, B. A., Monteil, C. L., Clarke, C. R. (2014). Harnessing population genomics to understand how bacterial pathogens emerge, adapt to crop hosts, and disseminate. Annu. Rev. Phytopathol. 52 (1), 19–43. doi: 10.1146/annurev-phyto-102313-045907
Young, J. M., Takikawa, Y., Gardan, L., Stead, D. E. (1992). Changing concepts in the taxonomy of plant pathogenic bacteria. Annu. Rev. Phytopathol. 30, 67–105. doi: 10.1146/annurev.py.30.090192.000435
Keywords: Pseudomonas syringae, Pseudomonas savastanoi pathovars, woody hosts, host range, virulence factors, comparative genomics, type III effectors, phytotoxins
Citation: Moreno-Pérez A, Pintado A, Murillo J, Caballo-Ponce E, Tegli S, Moretti C, Rodríguez-Palenzuela P and Ramos C (2020) Host Range Determinants of Pseudomonas savastanoi Pathovars of Woody Hosts Revealed by Comparative Genomics and Cross-Pathogenicity Tests. Front. Plant Sci. 11:973. doi: 10.3389/fpls.2020.00973
Received: 24 April 2020; Accepted: 15 June 2020;
Published: 02 July 2020.
Edited by:
Andrea Chini, National Center for Biotechnology (CNB), SpainReviewed by:
Boris Alexander Vinatzer, Virginia Tech, United StatesCopyright © 2020 Moreno-Pérez, Pintado, Murillo, Caballo-Ponce, Tegli, Moretti, Rodríguez-Palenzuela and Ramos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Murillo, amVzdXMubXVyaWxsb0B1bmF2YXJyYS5lcw==; Cayo Ramos, Y3JyQHVtYS5lcw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.