 Lian Wu†
Lian Wu† Quan Xu
Quan Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 April 2020
Sec. Plant Breeding
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.00303
Grain shape (GS) is an important agronomic trait that can improve rice breeding for optimal appearance quality, and it varies highly between indica and japonica subspecies. In this study, we conducted a genome sequencing of a series of recombination inbred lines (RILs) derived from a cross between japonica variety Shennong265 (SN265) and indica variety R99, and we successfully detected seven loci associated with GS. Subsequent analysis showed that a major quantitative trait locus (QTL) corresponded to the qSW5/GW5. To identify a main-effect locus, we conducted bulked segregant analysis (BSA) in two F2 populations. A 0.8-Mb region on chromosome 9 was identified as the candidate region of GS. There are 101 predicted genes in this region, and eight single nucleotide polymorphisms/insertions and deletions (SNPs/INDELs) caused frameshift. We found that a 637-bp stretch in exon 5 of the DENSE AND ERECT PANICLE 1 (DEP1) locus in SN265 was replaced by a 12-bp sequence. The two types of CRISPR/Cas9 gene-edited plants confirmed that DEP1 affected GS, and both Gγ and Cys-rich domains participated in this regulatory mechanism. These findings improve our understanding of the underlying mechanism of GS in rice and provide an effective and rapid strategy for the identification of main-effect loci of target traits.
Rice, a staple food crop cultivated worldwide, feeds over 50% of the world’s population. GS is one of the most important agronomic traits that influences yield and quality. The indica and japonica rice varieties differ in term of GS, and this trait has undergone extensive selection during rice domestication and breeding. Recent studies have shown that GS is controlled by multiple signaling pathways, and advances in the functional genomics have facilitated the cloning of a series of loci that control grain size. For example, qSW5/GW5 encodes a polyubiquitin-binding protein that is involved in the ubiquitin-proteasome pathway. This was shown to be a major loci of rice domestication and indica/japonica differentiation (Shomura et al., 2008; Weng et al., 2008). Phytohormones (i.e., cytokinins, brassinosteroids, and auxins) also participate in GS regulation. GS5, GS2/GL2, and GW5 have been reported to be involved in brassinosteroid signaling (Li et al., 2011; Che et al., 2015; Hu et al., 2015). GL3.1 regulates cyclin-T1-38 to influence GS, and qGL3 encodes OsPPKL1phosphatase, which has been suggested to be involved in brassinosteroid signaling (Qi et al., 2012). TGW6 encodes an IAA-glucose hydrolase activity protein, and BG1 encodes a membrane localized protein; both may be specifically induced by auxin (Ishimaru et al., 2013; Liu et al., 2015a). In G-protein signaling pathways, GS3 is known to be a G protein γ subunit, which has been identified as a GS regulator (Fan et al., 2006; Takano-Kai et al., 2009; Mao et al., 2010). These studies have demonstrated that GS3 is the most important determinant of grain length in breeding populations of cultivated rice, and the natural variants of GS3 have contributed to the global improvement of rice yield and quality. Furthermore, transcriptional regulatory factors have also been shown to play important roles in the regulation of GS, including GLW7, OsSPL16/GW8, and OsMKK4- OsMAPK5. GLW7 encodes the plant-specific transcription factor OsSPL13 that positively regulates cell size (Si et al., 2016). OsSPL16/GW8 directly binds to the GW7/GL7 promoter and represses its expression via a SBP-domain transcription factor (Wang et al., 2012, 2015). The OsMKK4- OsMAPK5 regulatory module also plays a key role in regulating GS by controlling cell proliferation of spikelet hulls (Duan et al., 2013; Liu et al., 2015b). Taken together, recent molecular studies have successfully identified numerous important genes that are associated with GS. However, their biological mechanisms and regulatory network remain largely unknown. Thus, identification and characterization of additional QTLs/genes that participate in the determination of GS is important not only to the elucidation of the molecular mechanisms underlying the regulation of grain traits but also to the generation of high yield and superior quality cultivars.
The present study conducted genome sequencing of a series of RILs that were derived from crossing the indica variety “R99” and the japonica variety “SN265” to identify the QTL/genes involved in the regulation of GS. Moreover, we conducted a BSA to identify the main-effect loci of GS. These findings improve our understanding of the underlying biological mechanisms of GS in rice and provide a rapid and cost-effective strategy for the identification of main-effect loci of target traits.
To identify the QTL/genes associated with GS, we generated an RIL population using the single-seed descendant method with at least 10 generations. The parent line was “R99” (indica) and “Shennong265” (japonica). A total of 151 RILs were constructed and used in the present study. The RILs were planted in three typical rice cultivated areas, namely, LN: Rice Research Institute of Shenyang Agricultural University (N41°, E123°), JS: the sub-base of China National Hybrid Rice R&D Center in Jiangsu Province (N32°, E120°), and SZ: the Agricultural Genomics Institute at Shenzhen (N22°, E114°) in the summer of 2016. The plots for each line were 5.4 m2 and included 120 plants with planting densities (hill per m2) of 22.2 (30 cm × 15 cm intervals). Plots were arranged in a randomized block design with three replications. The cultivation method and field management were as described in our previous report (Li et al., 2018). Twenty plants from the middle plot were harvested for each line at 45 days after heading. The plant materials for BSA were planted at the Rice Research Institute of Shenyang Agricultural University (SY; N41°, E123°) in 2018. To determine the GS of the experimental materials, we harvested the paddy rice and air-dried these at room temperature for at least 3 months before testing. Fully filled grains were used for measuring grain length, width, thickness, and weight. Ten randomly selected grains from each plant were lined up lengthwise along a Vernier caliper to measure grain length, and they were then arranged by breadth to measure grain width.
The young leaves of each line were sampled 15 days after transplanting. The cetyl trimethylammonium bromide (CTAB) method was used to obtain the high-quality genomic DNA. We constructed the sequencing libraries on the Illumina HiSeq2500 according the manufacturer’s instructions. The sequence data was aligned with the Nipponbare genome, as referenced1 by SOAP2. The HighMap software was used to combine the co-segregating SNP/InDel into bin for linkage map. The QTL analysis was conducted using R/qtl (version: 1.44-9) software via a composite interval mapping (CIM) model. The details of the linkage map construction and QTL analysis were described in our previous study (Li et al., 2018).
We developed two DNA bulks by selecting extreme short-grain plants and extreme long-grain plants in the F2 populations. We then prepared the libraries for the DNA bulks following the Illumina TruSeq DNA sample Preparation v2 Guide. The MiSeq Reagent Kit v2 (500 cycles) of Illumina MiSeq platform (Illumina Inc., San Diego, CA, United States) was used to sequence the DNA libraries. We then aligned the short reads of the DNA bulks along with the parent lines to a Nipponbare reference genome (IRGSP 1.0) by the BWA software (Li and Durbin, 2009). To call SNPs, the reads of the two bulks were separately aligned to SN265 and R99 consensus sequence reads using SAM tools (Li and Durbin, 2009). We removed duplicate reads using the Picard tool according to the clean reads mapping results using the genome reference2. We performed the base recalibration and local realignment to guarantee the accuracy of SNP detection using GATK software (Mckenna et al., 2010). The SNPs between the reference genome and samples were detected by the GATK software3. We calculated the SNP-index and Euclidean distance (ED) to identify the candidate regions that are associated with GS (Hill et al., 2013). The ED algorithm’s calculation formula is as follows
where Amut, Cmut, Gmut, and Tmut are the bases A, C, G, and T’s frequency in the long-grain-type bulks, respectively. Awt, Cwt, Gwt, and Twt are the frequency of bases A, C, G, and T in the short-grain-type bulks, respectively. To eliminate the background noise, the ED value was powered, and the associated value was set as ED5. Then, we fit the data using a Loess curve with a polynomial exponent of 1 and a span parameter determined by minimizing the Aikaike Information Certerion (AICc). Peak regions are defined as regions where the Loess fitted values are greater than three standard deviations above the genome-wide median (Hill et al., 2013). SNP-index association analysis was used to calculate differences in genotype frequency between the two bulks (Rym et al., 2013; Takagi et al., 2013).
We performed vector construction to conduct CRISPR/Cas9 gene editing as described by Li et al. (2017). We selected the targeting sequence, including the PAM sequence (23 bp) in the first and fifth exons of the DEP1 gene. By performing a BLAST search against the rice genome, we confirmed the specificity of the targeting sequence4 (Hsu et al., 2013). We performed rice transformation as described elsewhere (Nishimura et al., 2006). The genomic DNA of the transformants was sequenced and analyzed using the Degenerate Sequence Decoding method (Ma et al., 2015).
In the present study, the indica variety “R99” and the japonica variety “SN265” were used to identify GS-related loci. A notable difference in GS was observed between SN265 and R99 (Figure 1A). The grain length of SN265 was 5.18 ± 0.03 mm, whereas that of R99 was 6.13 ± 0.03 mm. The grain widths of SN265 and R99 were 2.75 ± 0.01 mm and 2.33 ± 0.03 mm, respectively. The GS (grain length/grain width) of SN265 was 1.88 ± 0.02, whereas that of R99 was 2.6 ± 0.02. SN265 and R99 were then crossed and inbred for over 10 generations to develop the RIL population for QTL analysis of GS. The GS of RILs showed a typical normal distribution. Some lines exhibited transgressive segregation, which suggests that GS was controlled by multiple loci in this cross combination (Figure 1B).
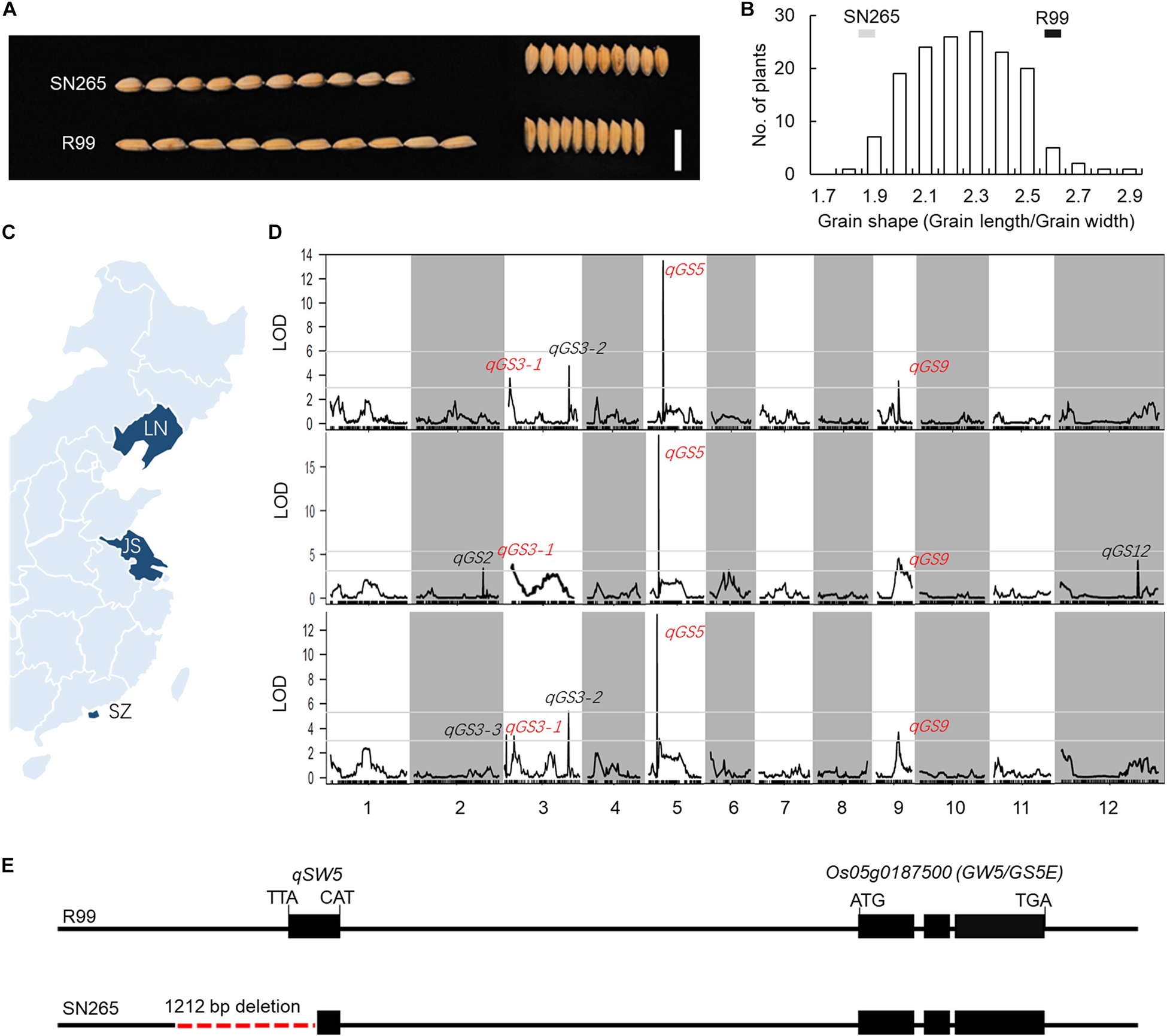
Figure 1. QTL analysis and candidate gene. (A) GS of SN265 and R99. Bar = 1 cm. (B) The distribution of GS in RILs. (C) The plant area of RILs (D) QTL analysis of GS. (E) The difference in the GW5/GS5E locus between SN265 and R99.
To detect the loci that are related to GS in the RILs, we planted the RILs in three areas and conducted QTL analysis via high-throughput sequencing (Figure 1C). Our previous study constructed a high-density bin map using the Highmap software (Li et al., 2018). The molecular linkage map, which consisted of 3,569 bins, was used to conduct the QTL analysis of GS. A total of seven loci were detected with a LOD >2.5 and were located on chromosomes 2, 3, 5, 6, 9, and 12, and some of these QTLs could be detected in multiple areas (Figure 1D; Supplementary Table S1). The LOD of one major QTL qGS5 on chromosome 5 ranged from 12.44 to 17.32; and this QTL explained 31.38–42.13% of the observed variations in three areas. Within this region, we found the key GS regulator qSW5/GW5, which has been shown to be the major GS loci for rice domestication and indica/japonica differentiation (Shomura et al., 2008; Weng et al., 2008). We then compared the qSW5/GW5 sequence in SN265 and R99. The results show that there was a 1,212-bp deletion in SN265 when compared with R99 (Figure 1E). These results indicate that qSW5/GW5 might be the target QTL/gene of the major QTL qGS5 detected in SN265/R99 RIL population in this study.
To detect the main-effect QTLs involved in GS regulation, we subsequently focused on the QTL located on chromosome 9 (qGS9) that could explain 11.32–12.63% of the observed variations (LOD = 3.64–4.40) and could be detected in all three areas (Figure 1D). We conducted the parental line selection based on genotypic data at qSW5/GW5 and qGS9. Line 155 (L155) shared the same type of qSW5/GW5 but a different allele of qGS9 compared to that of SN265. Moreover, the grains of L155 were significantly slimmer than SN265. Additionally, we found that L126 and L52 shared a R99-type allele at the qSW5/GW5 locus but differed in qGS9. L126 has shorter and wider grains than L152 (Figure 2A). Then, we crossed the L155 to SN265, and crossed L126 to L52 to develop two pairs of bulks for BSA analysis. We first sampled 30 plants that exhibited short grains as SN265 and long grains as L155 as bulks for high-throughput sequencing, respectively (Figure 2B). The Illumina high-throughput sequencing generated 70.91 Gb of data, covering 97.47% of whole genome. There was an average of 47.08-fold for each sample. There were 922,924 SNPs between SN265 and L155, and 22,119 of these were non-synonymous. A total of 116,993 SNPs were detected between the two bulks, and 1,860 of these were non-synonymous. We also detected 194,851 and 30,797 small INDELs between parental lines and between the two bulks, respectively. To improve the accuracy of BSA analysis, we used two approaches, the ED and SNP-index methods, to identify the candidate regions related to GS (Figure 3). The association threshold of the ED method was indicated by pink broken lines in Figure 3A, and only a region 3.26 Mb in length was significantly correlated with GS. Then, we calculated the SNP index of the two bulks for each SNP and computed an average SNP-index at a 1-Mb interval using a 10-kb sliding window; an 11.11-Mb region was identified that was significantly correlated with GS (Figure 3B). Thus, we obtained a 3.26-Mb candidate region by combining the result of ED and SNP-index methods using the F2 population from the cross between SN265 and L155. We subsequently sampled 30 plants having GS similar to L52 and 30 plants showing similar GS as L126 as two bulks for BSA. The sequencing generated 70.91-Gb of data, covering 97.18% of whole genome. There was an average of 47.08-fold genomic coverage for each sample. There were 784,722 SNPs between L126 and L52, and 19,902 of them were non-synonymous. Using the ED method, we detected a 1.81-Mb region from 15,764,348 to 17,574,044 on chromosome 9 that significantly correlated to GS (Figure 3C). By SNP-index methods, we identified a 1.80-Mb region from 14,811,581 to 16,614,006 on chromosome 9 related to GS (Figure 3D). Taken together, we obtained a 0.8-Mb candidate region using the ED and SNP-index method for the two pairs of the BSA population (Figure 4A). The candidate region consisted of 101 genes with 56 non-synonymous SNPs (Supplementary Table S2). Our previous study performed the de novo assembly of two high-quality genomes of SN265 and R99, which are the parental lines in the present study (Li et al., 2018). Thus, we can thoroughly compare the sequences of the 0.8-MB chromosome region between SN265 and R99. Among genes containing 56 non-synonymous SNPs, eight genes that exhibited frame shift mutations were chosen as candidate genes that might be more probably associated with GS (Figure 4B). Among the 56 non-synonymous SNPs, we subsequently found eight genes with frame shift mutations, and these eight genes were probably associated with GS (Figure 4B). Os09g0432033, Os09g0432601, Os09g0435700, and Os09g0438500 were non-protein coding transcript or hypothetically conserved genes. Os09g0431300 contained a Myb-like DNA-binding domain at the N-terminus, and the C/CTA difference between SN265 and R99 occurred at the C-terminus of the Os09g0431300. Similarly, the GTT/G difference in Os09g0433650 avoided the transcriptional activator of acetoin domain in Os09g0433650. A 9-bp deletion was detected in Os09g0438000 between the EF-hand domain and Ferric reductase-like component domain, but the deletion did not influence these two functional domains. Interestingly, there is a 637-bp stretch in the exon 5 of Os09g0441900 in the R99 genome that was replaced by a 12-bp sequence in SN265 (Figure 4C). This gene has been reported as a pleiotropic locus for panicle architecture and grain number, named as DENSE AND ERECT PANICLE 1 (DEP1) (Huang et al., 2009; Wang et al., 2009; Zhou et al., 2009). The substitution caused an elimination of Cys-rich domain at the C-terminus of DEP1. DEP1 might thus be the candidate gene for this main-effect QTL affecting GS.
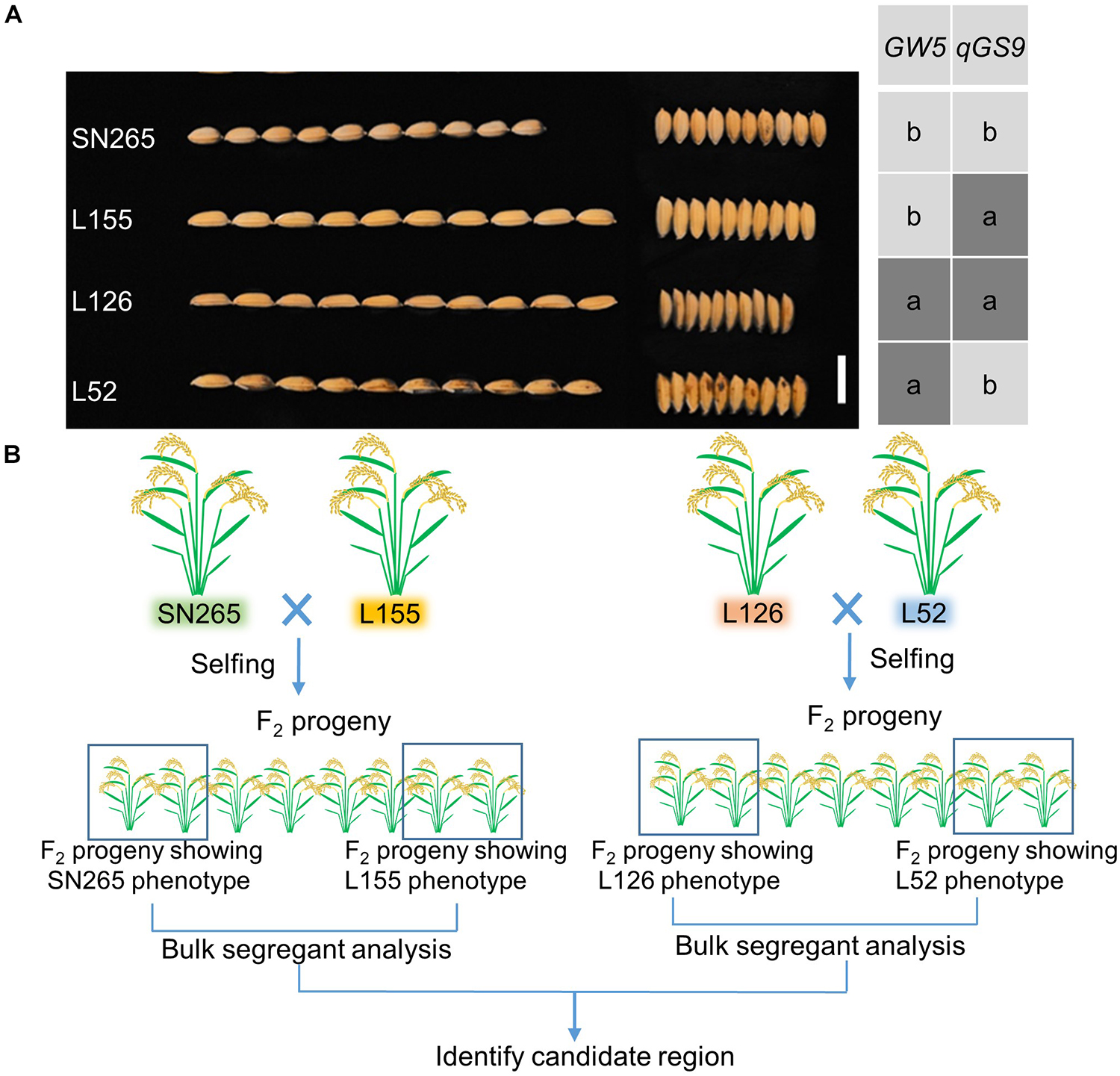
Figure 2. The population construction for BSA. (A) The GS of SN265 and L155. Bar = 1 cm. (B) Simplified scheme for the application of BSA.
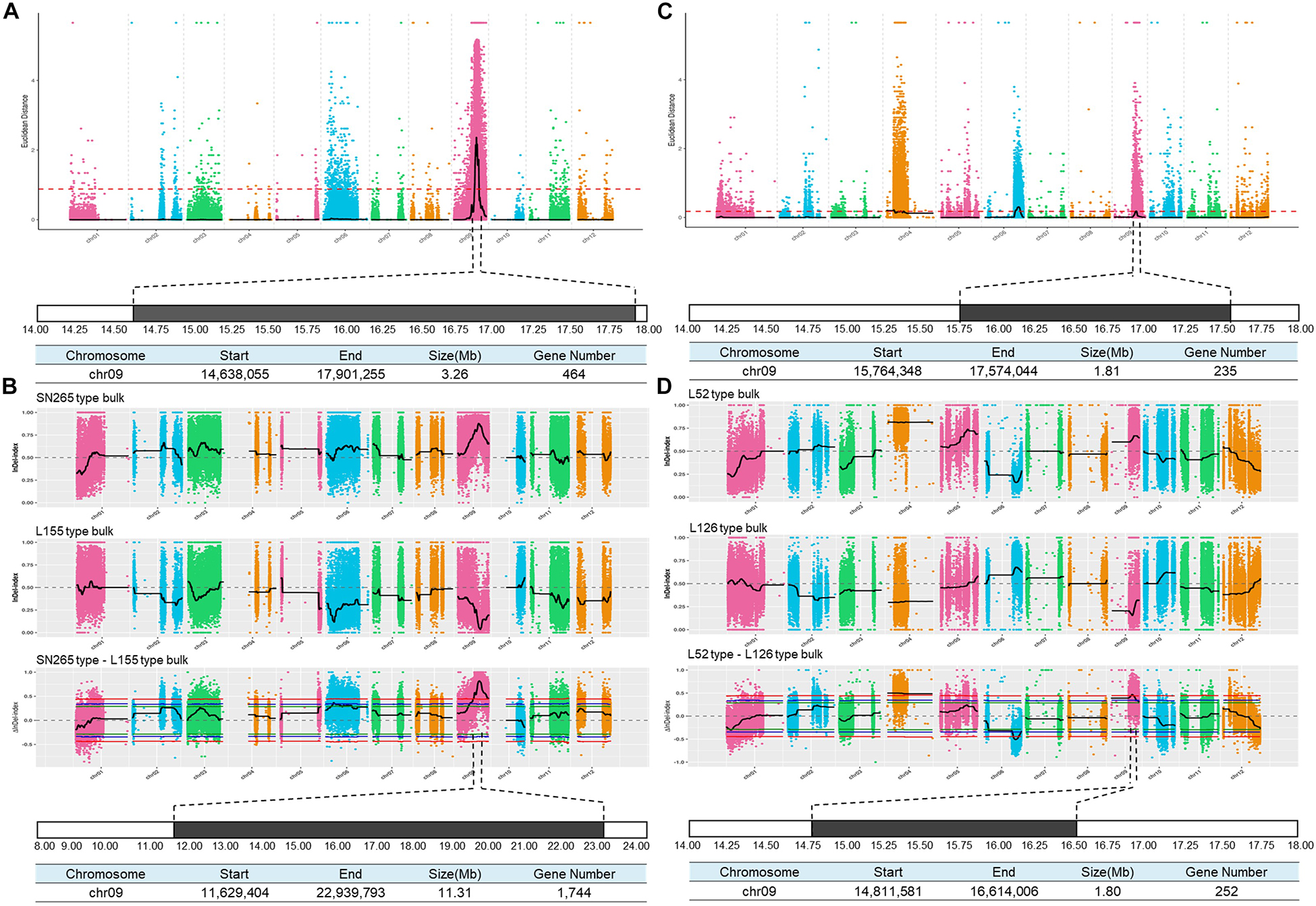
Figure 3. BSA analysis of GS. (A) Identification of the hot-region for GS via ED association analysis of the cross between SN265 and L155. (B) Identification of the candidate gene for GS via the SNP-index method of the cross between SN265 and L155. (C) Identification of the hot-region for GS via ED association analysis of the cross between L126 and L52. (D) Identification of the candidate gene for GS via the SNP-index method of the cross between L126 and L52.
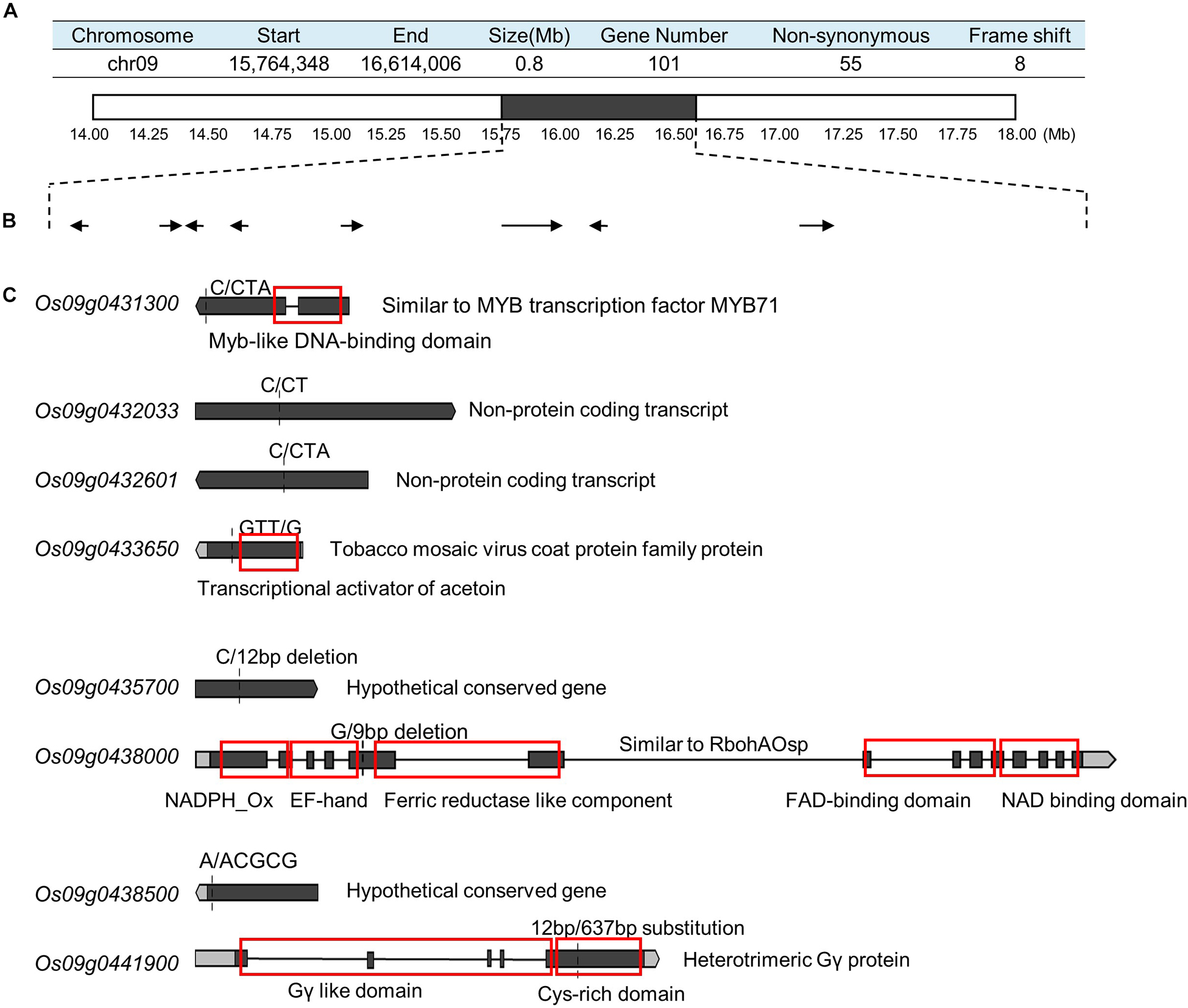
Figure 4. Identification of candidate gene. (A) The candidate region identified by ED and SNP-index methods using two pairs of BSA populations. (B) The genes in which the SNP/InDel result in a frame shift. (C) The detail of sequence analysis of the eight genes. The red boxes indicate the functional domains.
To confirm the function of DEP1 in GS regulation, we generated a series of dep1 mutations using the CRISPR/Cas9 gene editing technology under the genetic background of Sasanishiki (a famous japonica variety) as wild-type control (WT). The DEP1 has a Gγ at its N-terminus and a Cys-rich domain at its C-terminus (Figure 5a). As a previous study showed that the truncated dep1 allele that lost the cys-rich domain was a gain-of-function mutant, we generated two types of dep1 mutants. As shown in Figure 4A, the dep1△cys allele loses the Cys-rich region but maintains the Gγ domain, whereas the dep1△full allele loses both the Cys-rich and Gγ domains. At least three independent transgenic lines were obtained (Figure 5b). The results show that the dep1△cys plants exhibited a semi-dwarf and erect panicle architecture, and panicle length and grain length decreased relative to the WT (Figures 5c–e). The dep1△full plants showed a slight decrease in plant height, panicle length, and grain length. No significant differences in grain width were observed among WT, dep1△cys, and dep1△full. These results indicate that the DEP1 locus indeed participates in the regulation of GS, and both the Gγ and the Cys-rich domains played essential roles in GS regulation.
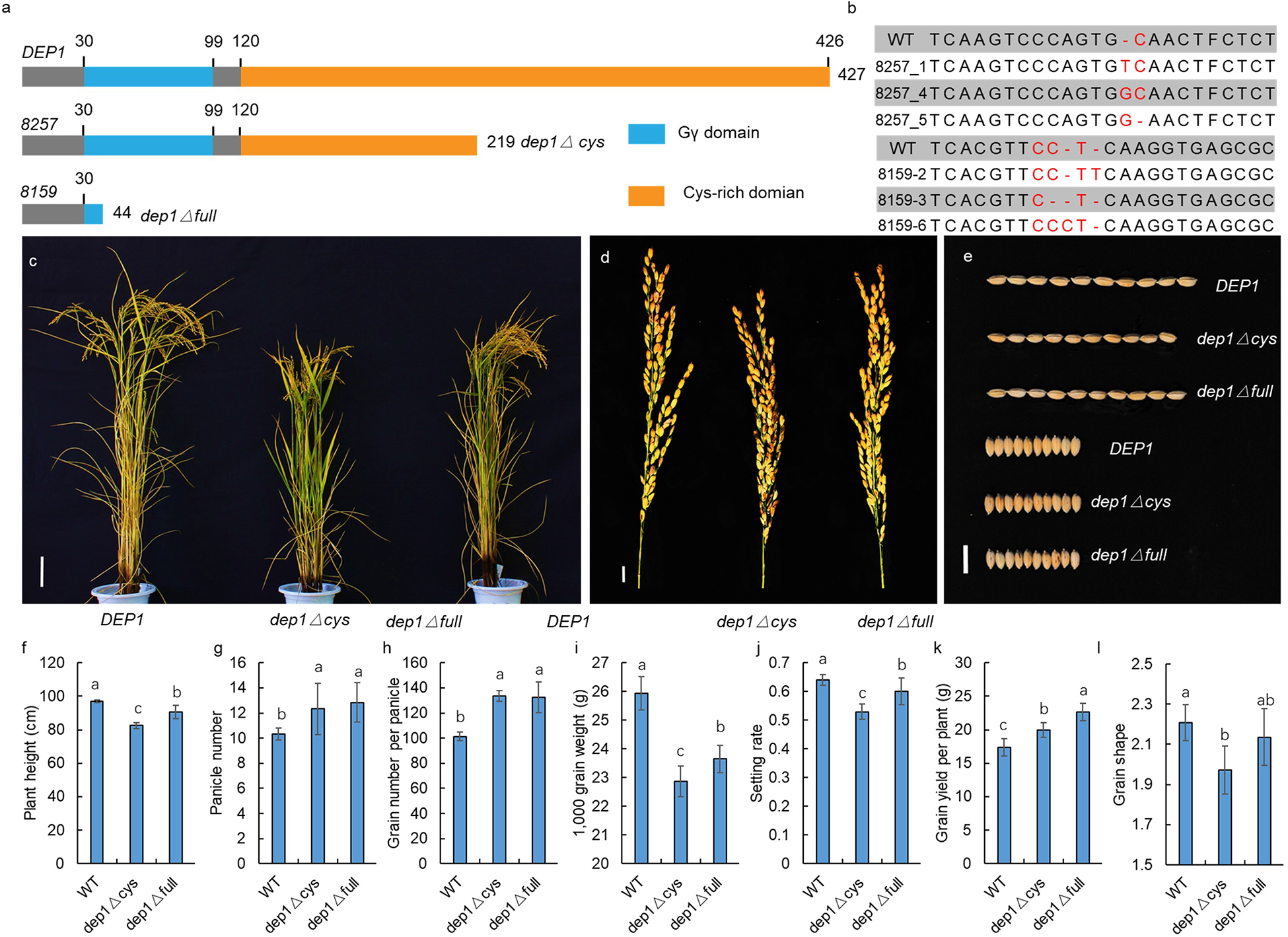
Figure 5. Confirmation by CRISPR/Cas9 gene editing technology. (a) The protein structure of DEP1, dep1△ cys, and dep1△ full. (b) Sequence mutation of the CRISPR/Cas9 gene edited plant. (c) The plant architecture of DEP1, dep1△ cys, and dep1△ full. (d) The panicle of DEP1, dep1△ cys, and dep1△ full. (e) The GS of DEP1, dep1△ cys, and dep1△ full. (f–l) The plant height, panicle number, grain number per panicle, 1,000 grain weight, setting rate, grain yield per plant and GS of DEP1, dep1△ cys, and dep1△ full plants. Data are means ± SE (n = 20), different letters denote significant differences (P < 0.05, Duncan multiple-range test).
Recent molecular studies have identified a number of major QTL/genes that control GS. However, further efforts to identify main-effect QTL/genes are necessary. Our study used genome sequencing and a BSA strategy to successfully identify seven QTLs that were related to GS. Among the QTLs, one major locus qGS5 was co-located with GW5/GS5E, which has been reported to be related to rice grain width and grain weight. Weng et al. (2008) and Shomura et al. (2008) fine mapped GW5 to a 2,263-bp and 21-kb genomic region, respectively. Subsequent studies demonstrated that a gene encoding a calmodulin-binding protein, which was located approximately 5 kb downstream of the 1,212-bp deletion, corresponded to qSW5/GW5 (Duan et al., 2017; Liu et al., 2017). Our study confirmed qSW5/GW5 as a major locus that explained 42.13% of the observed variation in the JS experimental site as the highest. The G protein has been shown to possess the ability to regulate GS and rice yield. The GS3, a Gγ subunit protein-encoding gene, is a major QTL for GS, and it functions as a negative regulator of GS and organ size (Fan et al., 2006; Mao et al., 2010). Another Gγ subunit protein-encoding gene, RGG2, has been reported to participate in the regulation of GS and rice yield (Miao et al., 2019). Sun et al. (2018) used multiple mutation combinations of the G protein encoding gene to propose a model to explain how these Gγ proteins work in GS regulation. Taken together, the G-protein pathway plays an important role in GS regulation. Our study also detected QTLs in chromosome 9 that were associated with GS and explained 11.35% of the observed variations, and the additive effects ranged from 0.06–0.07 among different experimental sites. The candidate gene was identified as DEP1 using the BSA strategy. The DEP1 locus was first reported as a panicle architecture regulator by three research teams (Huang et al., 2009; Wang et al., 2009; Zhou et al., 2009). A subsequent study showed that DEP1 is also involved in nitrogen utilization (Sun et al., 2014). A “self-inhibition” model was constructed at the C-terminal domain that inhibited the N-terminal domain in non-canonical Gγ proteins in DEP1 (Mao et al., 2010; Botella, 2012). Recent molecular studies employing CRISPR/Cas9 gene editing have been conducted to verify the function of truncated DEP1 (Li et al., 2016, 2018, 2019; Shen et al., 2017; Wang et al., 2017; Sun et al., 2018). The plants with a Gγ domain but no cys-rich domains exhibited an erect panicle architecture and an increase in the grain number per panicle. The plants that had lost both the Gγ and cys-rich domains, however, showed reductions in grain number per panicle and setting rate. Additionally, the truncated dep1 allele also exhibited a negative effect in grain number per panicle under the genetic background of Wuyunjing 8 and Non-gken 57 (Zhou et al., 2009; Yi et al., 2011). These results suggested that the truncated dep1 alleles generated an opposite phenotypic change on grain number per panicle under a different genetic background. The present study showed that both the dep1△cys and dep1△full plants exhibited an increase in grain number per panicle compared to the WT (Figure 5). Li et al. (2019) showed that three lines harboring different lengths of truncation at the C-terminal of DEP1 by CRISPR/Cas9 gene editing technology exhibited similar phenotypic changes of the GS. Taken together, we hypothesize that both the dep1△cys and dep1△full could increase grain number per panicle, generate a semi-dwarf phenotype, and reduce panicle length and grain length. Since the 1980s, a number of high-yielding japonica rice strains with dense and erect panicles have been released as commercial varieties. In China, these japonica ideotypes, such as SN265 and Jiahua, 1 have dominated the japonica rice acreage, and almost all of these varieties harbored the truncated allele of DEP1, which is similar to dep1△cys (Zhao et al., 2016). Interestingly, we found that the dep1△full transgenic plants that lost both Gγ and Cys-rich domains exhibited an enhanced phenotype compared to the WT and dep1△cys plants (Figures 5f–l). These findings suggest a potential method to develop important germplasm with novel allele of DEP1 locus and suggest a novel strategy for the flexible utilization of DEP1 in rice breeding.
The datasets supporting the conclusions of this study are included within the report.
QX and ZX designed this study and contributed to the original concept of the project. LW and YC performed most of the experiments. QX wrote the manuscript. All of the authors have read and approved the final manuscript.
The Excellent Youth Science Foundation of the Natural Science Foundation of Liaoning Province (2019-YQ-06) supported this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00303/full#supplementary-material
Botella, J. R. (2012). Can heterotrimeric G proteins help to feed the world? Trends Plant Sci. 17, 563–568. doi: 10.1016/j.tplants.2012.06.002
Che, R., Tong, H., Shi, B., Liu, Y., Fang, S., Liu, D., et al. (2015). Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2:15195. doi: 10.1038/nplants.2015.195
Duan, P., Rao, Y., Zeng, D., Yang, Y., and Li, Y. (2013). SMALL GRAIN 1, which encodes a mitogen-activated protein kinase kinase 4 (MKK4), influences grain size in rice. Plant J. 77, 547–557. doi: 10.1111/tpj.12405
Duan, P., Xu, J., Zeng, D., Zhang, B., and Li, Y. (2017). Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol. Plant 10, 685–694. doi: 10.1016/j.molp.2017.03.009
Fan, C., Xing, Y., Mao, H., Lu, T., Han, B., Xu, C., et al. (2006). GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theoret. Appl. Genet. 112, 1164–1171. doi: 10.1007/s00122-006-0218-1
Hill, J. T., Demarest, B. L., Bisgrove, B. W., Gorsi, B., Su, Y.-C., and Yost, H. J. (2013). MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697. doi: 10.1101/gr.146936.112
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi: 10.1038/nbt.2647
Hu, J., Wang, Y., Fang, Y., Zeng, L., Xu, J., Yu, H., et al. (2015). A rare allele of GS2 enhances grain size and grain yield in rice. Mol. Plant 8, 1455–1465. doi: 10.1016/j.molp.2015.07.002
Huang, X., Qian, Q., Liu, Z., Sun, H., He, S., Luo, D., et al. (2009). Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 41, 494–497. doi: 10.1038/ng.352
Ishimaru, K., Hirotsu, N., Madoka, Y., Murakami, N., Hara, N., Onodera, H., et al. (2013). Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 45, 707–711. doi: 10.1038/ng.2612
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, M., Li, X., Zhou, Z., Wu, P., Fang, M., Pan, X., et al. (2016). Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 7:377. doi: 10.3389/fpls.2016.00377
Li, W., Zhu, Z., Chern, M., Yin, J., Yang, C., Ran, L., et al. (2017). A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 170, 114–126. doi: 10.1016/j.cell.2017.06.008
Li, X., Tao, Q., Miao, J., Yang, Z., Gu, M., Liang, G., et al. (2019). Evaluation of differential qPE9-1/DEP1 protein domains in rice grain length and weight variation. Rice 12:5. doi: 10.1186/s12284-019-0263-4
Li, X., Wu, L., Wang, J., Sun, J., Xia, X., Geng, X., et al. (2018). Genome sequencing of rice subspecies and genetic analysis of recombinant lines reveals regional yield- and quality-associated loci. BMC Biol. 16:102. doi: 10.1186/s12915-018-0572-x
Li, Y., Fan, C., Xing, Y., Jiang, Y., Luo, L., Sun, L., et al. (2011). Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 43, 1266–1269. doi: 10.1038/ng.977
Liu, J., Chen, J., Zheng, X., Wu, F., Lin, Q., Heng, Y., et al. (2017). GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 3:17043. doi: 10.1038/nplants.2017.43
Liu, L., Tong, H., Xiao, Y., and Che, R. (2015a). Activation of Big Grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. U.S.A. 112, 11102–11107.
Liu, S., Hua, L., Dong, S., Chen, H., Zhu, X., Jiang, J., et al. (2015b). OsMAPK6, a mitogen-activated protein kinase, influences rice grain size and biomass production. Plant J. 84, 672–681. doi: 10.1111/tpj.13025
Ma, X., Zhang, Q., Zhu, Q., Liu, W., Chen, Y., Qiu, R., et al. (2015). A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 8, 1274–1284. doi: 10.1016/j.molp.2015.04.007
Mao, H., Sun, S., Yao, J., Wang, C., Yu, S., Xu, C., et al. (2010). Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. U.S.A. 107, 19579–19584. doi: 10.1073/pnas.1014419107
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Miao, J., Yang, Z., Zhang, D., Wang, Y., Xu, M., Zhou, L., et al. (2019). Mutation of RGG2, which encodes a type B heterotrimeric G protein gamma subunit, increases grain size and yield production in rice. Plant Biotechnol. J. 17, 650–664. doi: 10.1111/pbi.13005
Nishimura, A., Aichi, I., and Matsuoka, M. (2006). A protocol for Agrobacterium-mediated transformation in rice. Nat. Protoc. 1:2796. doi: 10.1038/nprot.2006.469
Qi, P., Lin, Y.-S., Song, X.-J., Shen, J.-B., Huang, W., Shan, J.-X., et al. (2012). The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res. 22, 1666–1680. doi: 10.1038/cr.2012.151
Rym, F., Hiroki, T., Muluneh, T., Akira, A., Satoshi, N., Hiroki, Y., et al. (2013). MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One 8:e68529. doi: 10.1371/journal.pone.0068529
Shen, L., Hua, Y., Fu, Y., Li, J., Liu, Q., Jiao, X., et al. (2017). Rapid generation of genetic diversity by multiplex CRISPR/Cas9 genome editing in rice. Sci. China Life Sci. 60, 506–515. doi: 10.1007/s11427-017-9008-8
Shomura, A., Izawa, T., Ebana, K., Ebitani, T., Kanegae, H., Konishi, S., et al. (2008). Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1028. doi: 10.1038/ng.169
Si, L., Chen, J., Huang, X., Gong, H., Luo, J., Hou, Q., et al. (2016). OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456. doi: 10.1038/ng.3518
Sun, H., Qian, Q., Wu, K., Luo, J., Wang, S., Zhang, C., et al. (2014). Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nat. Genet. 46, 652–656. doi: 10.1038/ng.2958
Sun, S., Wang, L., Mao, H., Shao, L., Li, X., Xiao, J., et al. (2018). A G-protein pathway determines grain size in rice. Nat. Commun. 9:851. doi: 10.1038/s41467-018-03141-y
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL−seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Takano-Kai, N., Jiang, H., Kubo, T., Sweeney, M., Matsumoto, T., Kanamori, H., et al. (2009). Evolutionary history of GS3, a gene conferring grain length in rice. Genetics 182, 1323–1334. doi: 10.1534/genetics.109.103002
Wang, J., Nakazaki, T., Chen, S., Chen, W., Saito, H., Tsukiyama, T., et al. (2009). Identification and characterization of the erect-pose panicle gene EP conferring high grain yield in rice (Oryza sativa L.). Theor. Appl. Genet. 119, 85–91. doi: 10.1007/s00122-009-1019-0
Wang, S., Wu, K., Yuan, Q., Liu, X., Liu, Z., Lin, X., et al. (2012). Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 44, 950–954. doi: 10.1038/ng.2327
Wang, Y., Geng, L., Yuan, M., Wei, J., Jin, C., Li, M., et al. (2017). Deletion of a target gene in Indica rice via CRISPR/Cas9. Plant Cell Rep. 36, 1–11. doi: 10.1007/s00299-017-2158-4
Wang, Y., Xiong, G., Hu, J., Jiang, L., Yu, H., Xu, J., et al. (2015). Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 47, 944–948. doi: 10.1038/ng.3346
Weng, J., Gu, S., Wan, X., Gao, H., Guo, T., Su, N., et al. (2008). Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 18, 1199–1209. doi: 10.1038/cr.2008.307
Yi, X., Zhang, Z., Zeng, S., Tian, C., Peng, J., Li, M., et al. (2011). Introgression of qPE9-1 allele, conferring the panicle erectness, leads to the decrease of grain yield per plant in japonica rice (Oryza sativa L.). J. Genet. Genom. 38, 217–223. doi: 10.1016/j.jgg.2011.03.011
Zhao, M., Sun, J., Xiao, Z., Cheng, F., Xu, H., Tang, L., et al. (2016). Variations in dense and erect panicle 1 (DEP1) contribute to the diversity of the panicle trait in high-yielding japonica rice varieties in northern China. Breed. Sci. 66, 599–605. doi: 10.1270/jsbbs.16058
Keywords: rice, grain shape, high-throughput sequence, BSA, DEP1
Citation: Wu L, Cui Y, Xu Z and Xu Q (2020) Identification of Multiple Grain Shape-Related Loci in Rice Using Bulked Segregant Analysis With High-Throughput Sequencing. Front. Plant Sci. 11:303. doi: 10.3389/fpls.2020.00303
Received: 15 December 2019; Accepted: 03 March 2020;
Published: 03 April 2020.
Edited by:
Hanwei Mei, Shanghai Agrobiological Gene Center, ChinaReviewed by:
Lijun Meng, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2020 Wu, Cui, Xu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Xu, a29iZXh1MzRAc3lhdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.