 Sònia Garcia1,2
Sònia Garcia1,2 Jonathan F. Wendel3
Jonathan F. Wendel3 Natalia Borowska-Zuchowska4
Natalia Borowska-Zuchowska4 Malika Aïnouche5Alena Kuderova2
Malika Aïnouche5Alena Kuderova2 Ales Kovarik2*
Ales Kovarik2*- 1Institut Botànic de Barcelona (IBB, CSIC - Ajuntament de Barcelona), Barcelona, Spain
- 2Department of Molecular Epigenetics, Institute of Biophysics, Academy of Sciences of the Czech Republic, Brno, Czechia
- 3Department of Ecology, Evolution & Organismal Biology, Iowa State University, Ames, IA, United States
- 4Faculty of Natural Sciences, Institute of Biology, Biotechnology and Environmental Protection, University of Silesia in Katowice, Katowice, Poland
- 5UMR CNRS 6553 ECOBIO, Université de Rennes 1, Rennes, France
Introduction: Ribosomal DNA (rDNA) loci have been widely used for identification of allopolyploids and hybrids, although few of these studies employed high-throughput sequencing data. Here we use graph clustering implemented in the RepeatExplorer (RE) pipeline to analyze homoeologous 5S rDNA arrays at the genomic level searching for hybridogenic origin of species. Data were obtained from more than 80 plant species, including several well-defined allopolyploids and homoploid hybrids of different evolutionary ages and from widely dispersed taxonomic groups.
Results: (i) Diploids show simple circular-shaped graphs of their 5S rDNA clusters. In contrast, most allopolyploids and other interspecific hybrids exhibit more complex graphs composed of two or more interconnected loops representing intergenic spacers (IGS). (ii) There was a relationship between graph complexity and locus numbers. (iii) The sequences and lengths of the 5S rDNA units reconstituted in silico from k-mers were congruent with those experimentally determined. (iv) Three-genomic comparative cluster analysis of reads from allopolyploids and progenitor diploids allowed identification of homoeologous 5S rRNA gene families even in relatively ancient (c. 1 Myr) Gossypium and Brachypodium allopolyploids which already exhibit uniparental partial loss of rDNA repeats. (v) Finally, species harboring introgressed genomes exhibit exceptionally complex graph structures.
Conclusion: We found that the cluster graph shapes and graph parameters (k-mer coverage scores and connected component index) well-reflect the organization and intragenomic homogeneity of 5S rDNA repeats. We propose that the analysis of 5S rDNA cluster graphs computed by the RE pipeline together with the cytogenetic analysis might be a reliable approach for the determination of the hybrid or allopolyploid plant species parentage and may also be useful for detecting historical introgression events.
Introduction
It is well-established that all modern plant species have experienced at least one whole genome duplication and that many also have interspecific hybridization and recurrent introgression in their recent history (Wendel, 2015; Alix et al., 2017; Nieto Feliner et al., 2017; Van De Peer et al., 2017). Documenting recent allopolyploidy is relatively straightforward using cytogenetic analysis and genome size measurements, since allopolyploids have twice as many chromosomes (or more) as the parental species. Identification of homoploid hybrids is more difficult since the chromosome number and genome size are often similar to that of the parental species (Nieto Feliner et al., 2017). Evolutionary young allopolyploids and other hybrids tend to retain fixed polymorphisms at protein-coding and non-coding loci. These duplicated loci are called homoeologs (Glover et al., 2016) and are useful for documenting parentage as well as understanding the dynamics of polyploid genomes (Yoo et al., 2014; Wendel, 2015; Bourke et al., 2018). Older allopolyploids can have experienced episodes of intergenomic translocation, dysploidy, gene conversion, localized deletions, and other genetic events, leading eventually to diploidization of the genome (Wendel, 2015; Wendel et al., 2018).
Ribosomal RNA genes encoding 5S, 5.8S, 18S, and 26S ribosomal RNA are ubiquitous in plants and are organized into arrays containing hundreds to thousands of tandem repeats at one or more genomic loci (Hemleben and Zentgraf, 1994; Nieto Feliner and Rosselló, 2007; Roa and Guerra, 2012; Garcia et al., 2017). Due to their rapidly diverging intergenic (IGS) and internally transcribed spacers (ITS), rDNA loci have become popular taxonomic markers revealing allopolyploidy and other interspecific hybridization in many plant and animal systems (Alvarez and Wendel, 2003; Poczai and Hyvonen, 2010; Nieto Feliner and Rossello, 2012). The internet searches using *ITS* and *allopolyploidy* resulted in more than 650 hits in Web of Science for just 2019. Most studies have used classical single clone sequencing approaches whereas high-throughput data have only rarely been employed and are limited to the 35S (45S) rDNA (Matyasek et al., 2012; West et al., 2014; Boutte et al., 2016). The analysis of 5S rDNA is also informative and has been successfully used in many phylogenetic studies (Cronn et al., 1996; Fulnecek et al., 2002; Baum et al., 2004; Besendorfer et al., 2005; Volkov et al., 2007; Baum et al., 2012; Jang et al., 2016). Its analysis is complementary to that of 35S since both loci usually occur separately on chromosomes (Roa and Guerra, 2012; Garcia et al., 2017). The 5S rDNA is usually located in one chromosome pair in most angiosperms and can occupy variable chromosome positions. It is organized in tandemly arranged units comprising hundreds to tens of thousands of copies. Each unit is composed of a conserved c. 120 bp coding region separated by a variable intergenic spacer (Sastri et al., 1992). Similar to 35S loci, 5S rDNA loci undergo concerted evolution, a process maintaining high homogeneity within and often between arrays (Dover, 1982; Elder and Turner, 1995; Parks et al., 2019). Such a process may rapidly homogenize rDNA sequences and induce copy number variation (Bughio and Maggert, 2019) blurring their hybridogenic signatures in allopolyploids (Wendel et al., 1995a; Volkov et al., 1999; Muir et al., 2001; Matyasek et al., 2003). In contrast to 35S rDNA, the 5S rDNA loci appear to be less sensitive to homogenization in some allopolyploids (Fulnecek et al., 2002; Pedrosa-Harand et al., 2006; Weiss-Schneeweiss et al., 2008; Garcia et al., 2017), retaining diagnostic capacity with respect to their parental origin.
The clustering algorithm employed by RepeatExplorer (RE) (Novak et al., 2010; Novak et al., 2013) has become a tool of choice for the analysis of chromosome composition and genome evolution (Renny-Byfield et al., 2012; Weiss-Schneeweiss et al., 2015; Ribeiro et al., 2017; Mlinarec et al., 2019; Peska et al., 2019). The phylogenetic signal of the repeatome has also been exploited in phylogenetic studies (Dodsworth et al., 2015; Dodsworth et al., 2016; Grover et al., 2019; Vitales et al., 2019). The analysis of genomes by RE is based on an all-to-all comparison of sequence reads revealing their similarities. Subsequently, the data are used to build clusters of overlapping reads representing different repetitive elements. The TAREAN tool, recently introduced into the RepeatExplorer2 pipeline, allows repeat identification and reconstruction of tandem repeats solely from sequence reads (Novak et al., 2017). Graph theory and connected component methods lying in the heart of the computation algorithm produce graph structures reflecting genomic organization of repeats. Typically, tandem repeats exhibit circular (ring) shape topologies are characterized by high values of circularity parameters. Although the RepeatExplorer2/TAREAN tool was initially developed for identification of non-coding satellites, 5S rDNA can also be analyzed with the program. This is because 5S rDNA shows many features of satellite repeats: (I) its highly homogeneous units are tandemly arranged in a head to tail orientation, (II) it appears in high copy number, allowing analyses even at low coverage, and (III) the size of 5S rDNA monomers (c. 200–1,000 bp) (Sastri et al., 1992; Fulnecek et al., 2006) falls within the range defined for satellite DNA, allowing circularization of chains of overlapping reads.
In this study we investigated the 5S rDNA genomic organization and homogeneity in more than 80 plant diploids and polyploids, exploiting high-throughput reads available from read archives in public genomic databases and also de novo sequenced by us. Particular attention was paid on hybrid systems with well-defined evolutionary histories, both eudicots and monocots: (i) Brachypodium hybridum (Poaceae), Brassica carinata (Ethiopian mustard, Brassicaceae), Chenopodium quinoa (quinoa, Amaranthaceae), Gossypium hirsutum (cotton, Malvaceae), and Nicotiana rustica (Aztec tobacco, Solanaceae) allotetraploids. (ii) Spartina × townsendii (cordgrass, Poaceae) homoploid hybrid. (iii) Species with frequent introgression events included Gossypium gossypioides and Thinopyrum intermedium (intermediate wheatgrass, Poaceae). We used the RepeatExplorer2/TAREAN clustering pipeline and cluster graph computation methods to address the following questions: (1) What is the relationship between graph complexity and intragenomic heterogeneity of 5S rDNA repeats? (2) Can the full-length 5S rDNA units be assembled from short sequence reads? (3) Can allopolyploids and other interspecific hybrids be distinguished from their progenitors based on cluster graph topologies? We show that cluster graphs may represent a convenient and simple-use approach for identification of interspecific hybrids from high-throughput sequencing data.
Results
Relationship Between Cluster Graph Topology and Intragenomic Diversity of 5S rDNA
All of the 5S RNA gene families analyzed share a conserved c. 120 bp coding region while they differ in their intergenic spacers. We tested the hypothesis that the graph topologies of 5S rDNA clusters reflect the divergence and number of homoeologous gene families in allopolyploid genomes. Under this hypothesis, diploid species with a single gene family (and a locus) would generate a simple circular graph while allopolyploid and other hybrid genomes with multiple gene families (and loci) would display more complex graphs. To test this hypothesis we examined 5S rDNA cluster graph topologies in 84 plant species (Supplementary Table S1). Examples of cluster graph analyses in Gossypium, Brachypodium, and Spartina hybrid systems are shown in Figure 1. We have chosen these species because the parental genome donors, number of 5S rDNA loci, and approximate ages are known (Table 1). In the graphs, each vertex represents a sequence read and nodes connecting vertices depict sequence similarity between the reads. Simple circular 5S graphs with no or little deviation from regular circularity (referred as type 1 graphs) were observed in G. arboreum, G. raimondii, B. distachyon, and B. stacei diploid species. Except for the hexaploid S. alterniflora, the 5S graph topologies were more complex in polyploids (Figure 1). Specifically, two or more loops (rings) interconnected by a junction region (composed of 5S coding sequences) could be recognized. These complex structures are referred as type 2 graphs. Both loops were composed of vertices depicted in grey in Figure 1 representing variable IGS regions. The total k-mer coverage scores (mean of cluster homogeneity) were high in diploid species while they were lower in the allotetraploids (Table 1). The connected component index C (mean of graph circularity) was uniformly high across the species. The read richness varied between the loops. For example, in Brachypodium hybridum, the right loop contained far more reads than the left loop.
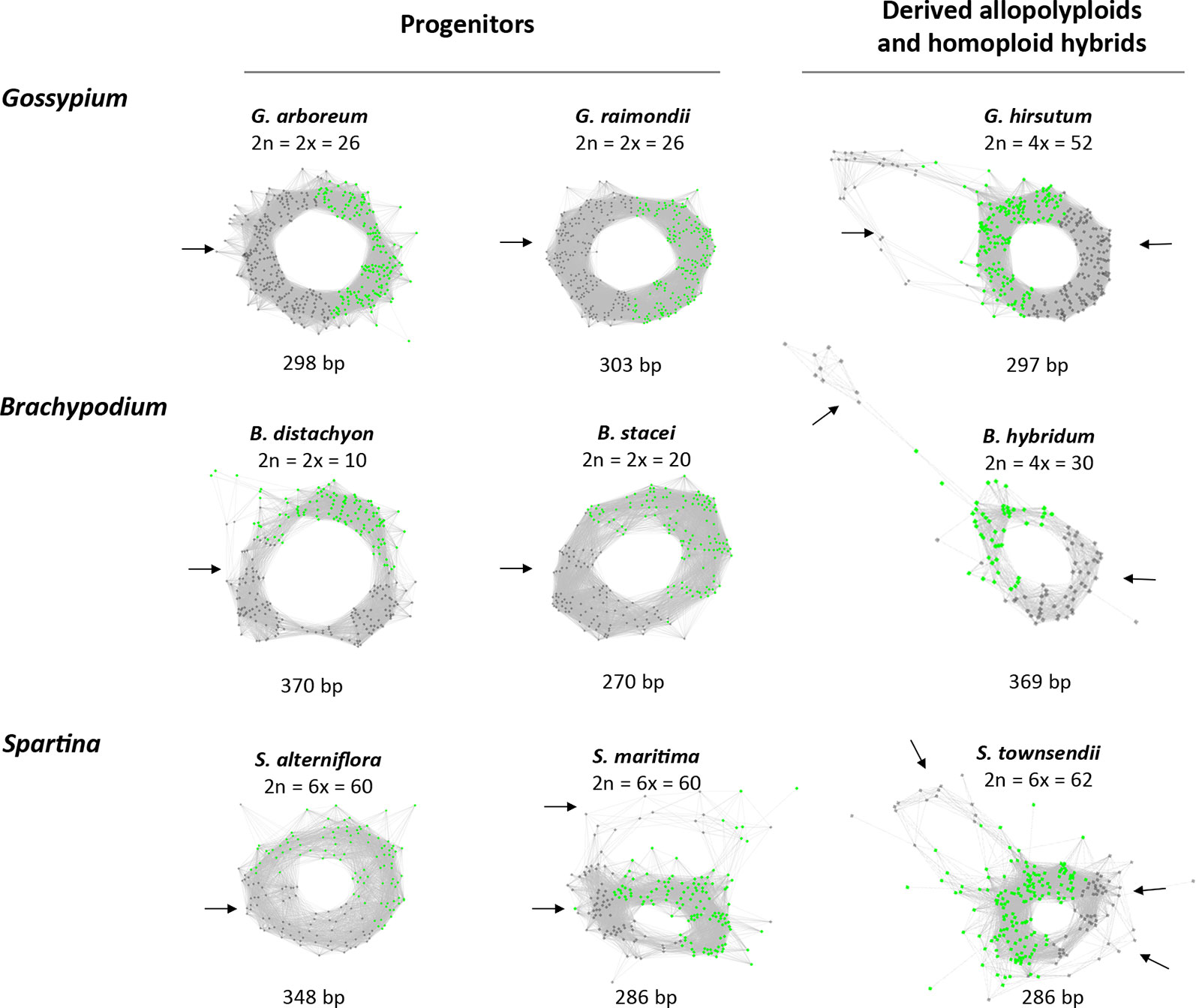
Figure 1 The 5S rDNA sequence reads organized in graph structures from the RepeatExplorer2 graphical output. Single reads are represented by vertices (nodes) and their sequence overlaps by edges. The 5S coding sequences and intergenic spacers are highlighted in green and grey vertices, respectively. Note, regular circular structures (referred to type 1) in most progenitors, and complex structures (referred to type 2) in derived allopolyploid and homoploid hybrids. Arrows indicate one or several intergenic spacers.
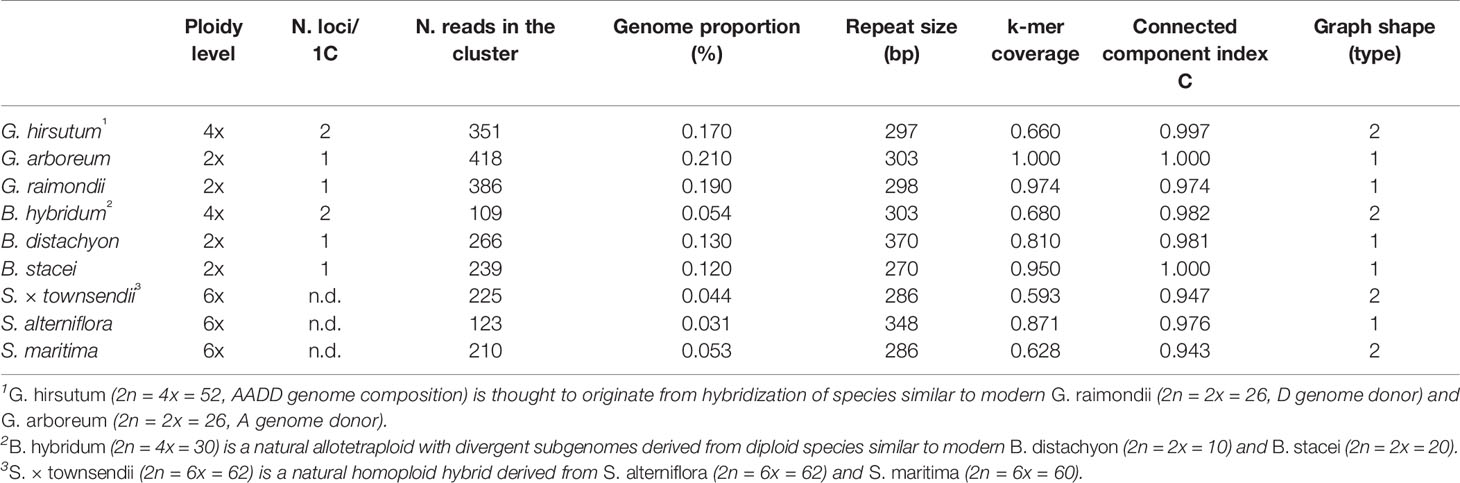
Table 1 Cytogenetic characteristics of 5S rDNA loci and cluster graph parameters in allopolyploid and homoploid species and their progenitors.
In the whole dataset (Supplementary Table S1) typical circular graph shapes of 5S rDNA clusters (type 1) were obtained in 81 (96%) species. The connected component index C parameter values (reported by a TAREAN) were high, ranging from 0.684 to 1.00 (average 0.959, s.d. 0.0599). In three species (4%), no circularization of 5S graphs clusters was obtained. Figure 2A shows the frequency of individual cluster types in allopolyploid and diploid species. The majority (87%) of diploid species showed type 1 structures while most (79%) allopolyploids displayed type 2 graphs (one-way ANOVA, F = 75.507, p < 0.001, Supplementary Table S2). About 95% single locus karyotypes displayed type 1 graphs while most (94%) karyotypes with two or more loci had type 2 structures (Figure 2B). There was a relationship between locus number and graph complexity (one-way ANOVA, F = 24.259, p < 0.001) with type 1 structures showing significantly lower locus numbers. The sequence homogeneity within each 5S rDNA cluster was estimated based on total k-mer coverage score reported by TAREAN and ranged 0.416–1.00 (average 0.760, s.d. 0.1392). The k-mer coverage values were significantly higher (one-way ANOVA, F = 200.363, p < 0.001, Supplementary Table S2) in the diploids compared to the allotetraploids and other hybrids (Supplementary Figure S1).
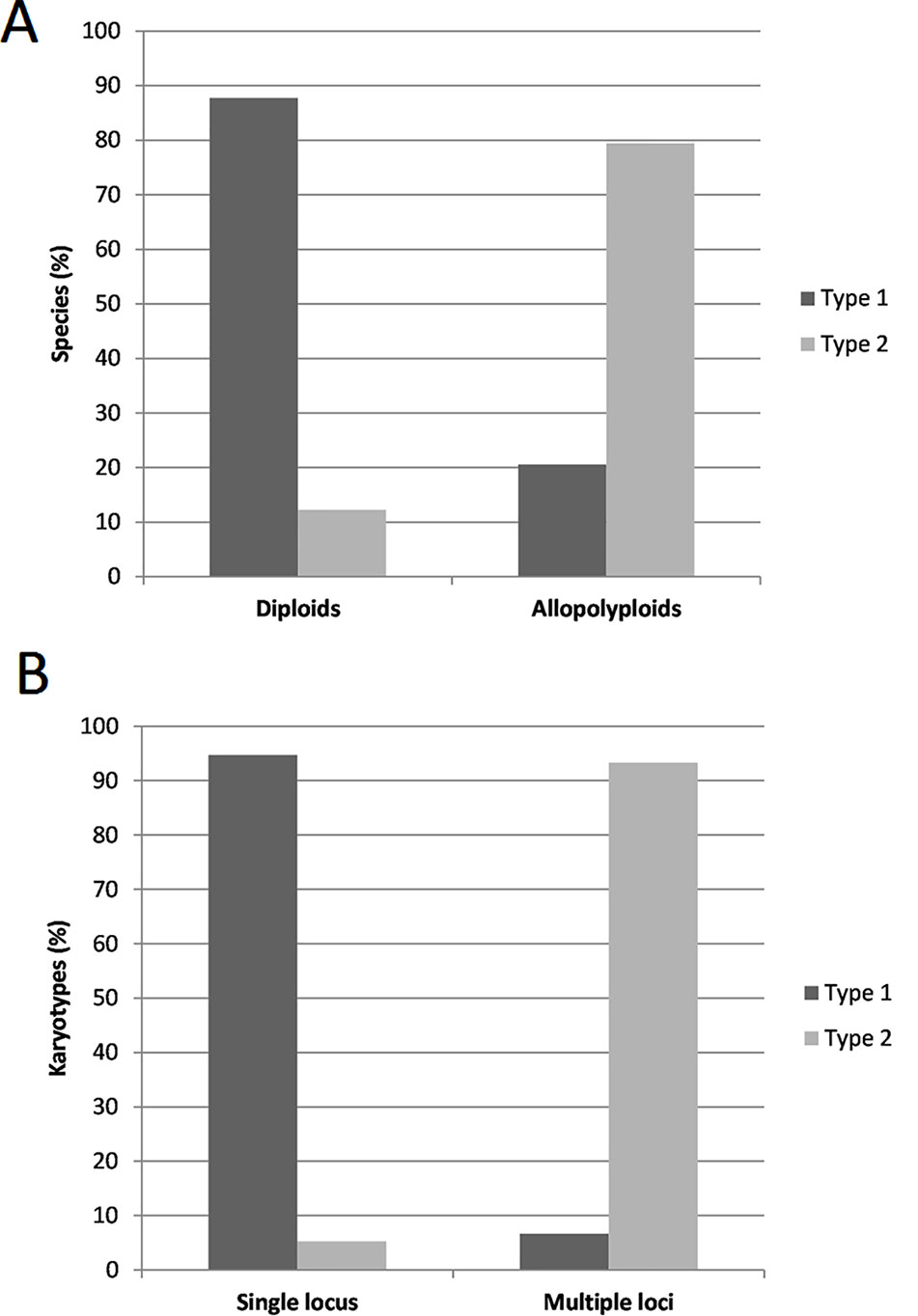
Figure 2 (A) Distribution of 5S rDNA type 1 and type 2 cluster graph structures between diploid (N = 50) and allopolyploid (including homoploid hybrids) (N = 32) species from the 84 species data set in this study. (B) Occurrence of type 1 and type 2 cluster graph structures in karyotypes with single (N = 38) and multiple (N = 30) 5S rDNA loci (data on 5S rDNA loci number only available for 68 species analyzed). The source data and the basic statistics are in Supplementary Table S2.
Tracking the Origin of 5S rDNA Families in Gossypium, Brachypodium, and Spartina Allopolyploids and Homoploid Hybrids by Comparative Cluster Analysis
Next, we investigated whether the homoeologous 5S genes can be visualized in cluster graph layouts and whether homoeologous gene families occur in assembled contigs. To address these questions we carried out a comparative three-genomic analysis (Figure 3) where inputs for clustering include reads from hybrids (allopolyploids) and their putative progenitor species. The overall cluster shapes were similar as in single genome analyses (Figure 1) indicating that the progenitor 5S rDNA sequences overlap with those of the derived hybrids and allopolyploids confirming, thus, their putative origin. Reads derived from 5S coding sequences (in green) were found in the junction region connecting both loops (Figures 3A, D, G). In Figures 3B, E, H reads originating from each progenitor (red and yellow) and hybrid (blue) genomes are labeled by different colors showing the 5S rDNA variants origin:
1. Figure 3B shows three-genomic cluster graph structures in Gossypium hirsutum and its progenitors. G. hirsutum is a 1–2 M years-old allotetraploid composed of subgenomes close to G. arboreum (A genome species) and G. raimondii (D genome species) (Wendel, 1989). The blue colored reads from the G. hirsutum allopolyploid were found in all parts of the graph—both in the junction region and loops; the red color reads from the G. arboreum diploid progenitor were located in the right loop and the junction region; the yellow colored reads originating from the other parental species, G. raimondii, were located in the left loop and the junction region. Similar cluster graph shapes were observed in remaining four Gossypium allotetraploids (G. barbandense, G. mustelinum, G. darwinii, and G. tomentosum, see Supplementary Figure S2), all having a similar AADD composition of the genome. In order to determine the identity of loop structures in the graphs we carried out a phylogenetic analysis of assembled 5S rDNA contigs (Figure 3C and Supplementary Figure S2). On the trees, sequences of both progenitors were well resolved forming separate branches, consistent with sequence divergence. The contigs from the G. hirsutum cluster grouped within the G. arboreum and G. raimondii branches, respectively.
2. Figure 3E shows three-genomic cluster graph structures in Brachypodium hybridum and its progenitors. B. hybridum is a c. 1 M years-old allotetraploid composed of subgenomes close to B. distachyon and B. stacei (Catalán et al., 2012). The comparative cluster graph displayed two loops composed of reads either from the B. distachyon (in red) or B. stacei (yellow). In contrast, reads from B. hybridum (blue) were shared between both loops. However, there were much less B. hybridum reads in the B. stacei loop compared to that of the B. distachyon. No B. stacei homoeologs were found among the contigs (Figure 3F).
3. Figure 3H shows three-genomic cluster graph structures in Spartina × townsendii and its progenitors. Spartina × townsendii is a less than 150 years-old homoploid hybrid composed of subgenomes inherited from S. alterniflora and S. martima hexaploids (Ainouche et al., 2004). The bottom read-rich circle contained sequences from the S. maritima (yellow) parent and S. × townsendii (blue); the upper read-poor circle was mostly formed by reads from S. alterniflora (red) parent and a few reads from S. × townsendii (blue). The junction region contained 5S genic sequences plus part of the IGS (grey) indicating short conserved sequences flanking the genic region. Another region of homoeologous genes similarity seems to exist in the middle of IGS indicated by interconnected reads from all three genomes (arrow). On the tree (Figure 3I), sequences from both progenitors were well resolved forming separate branches. However, all the assembled contigs grouped exclusively with the S. maritima branch.
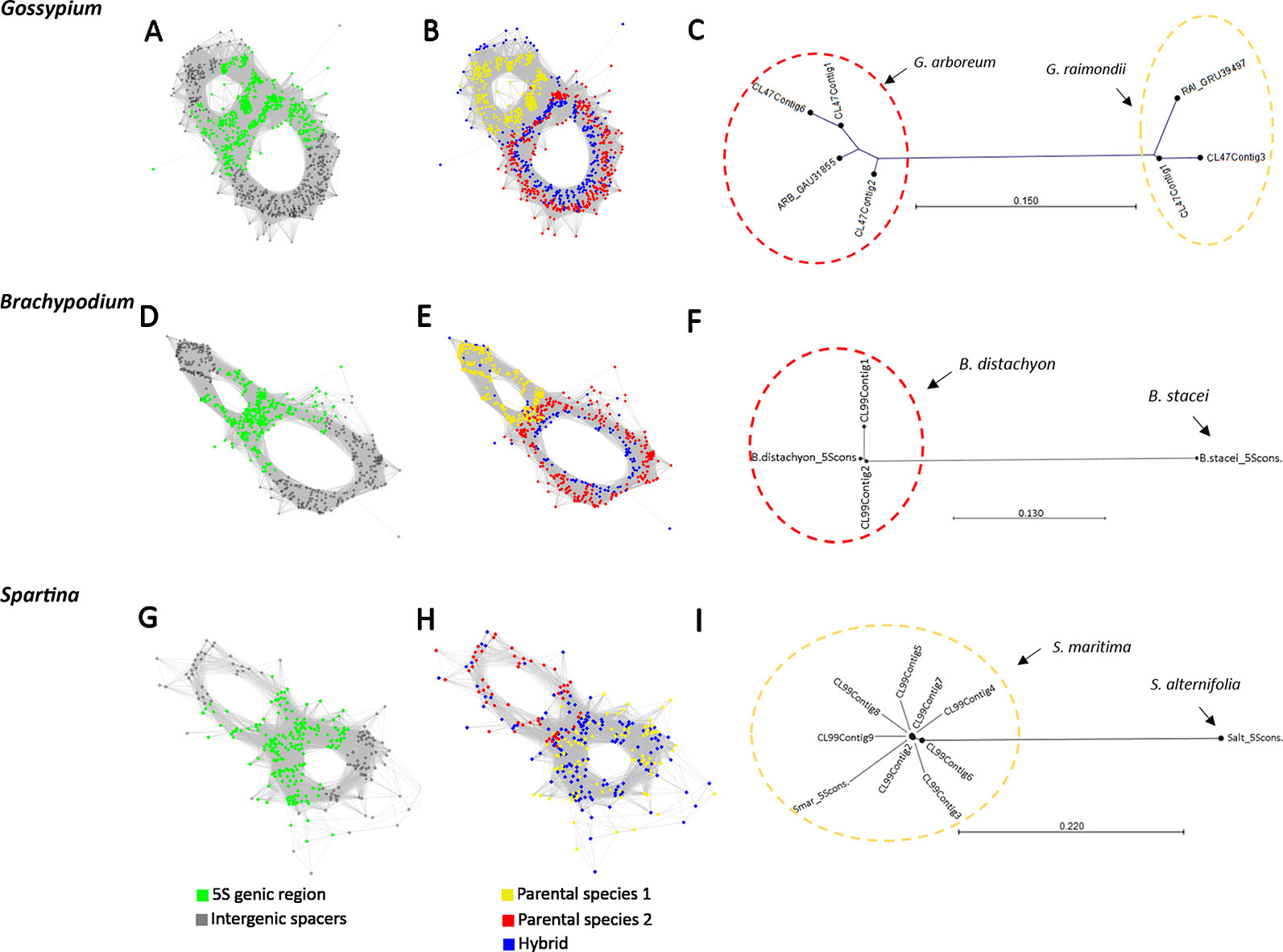
Figure 3 RepeatExplorer2 graphical output of the three-genomic comparative 5S rDNA analyses involving progenitor species and derived hybrids. (A, D, G) Graphs with highlighted 5S rDNA genic regions in green. (B, E, H) Cluster graphs with annotated reads origin: yellow vertices represent reads of one of the parental species (G. raimondii in B, B. stacei in E, and S. maritima in H); red vertices represent reads of the other putative parental species (G. arboreum in B, B. distachyon in E, and S. alterniflora in H); blue vertices represent reads from the hybrids and allotetraploids (G. hirsutum in B, B. hybridum in E, and S. × townsendii in H). (C, F, I) Neighbor joining phylogenetic trees constructed from aligned contigs from hybrids and progenitor 5S rDNA sequences.
Additional examples of 5S rDNA cluster analyses are shown in the Supplementary Figure S3 comprising the well-known allopolyploids, Brassica carinata (4x), Chenopodium quinoa (4x), and Nicotiana rustica (4x). All these species harbored complex type 2 graphs, in which at least one (Chenopodium) or both progenitors (Brassica and Nicotiana) could be identified.
Comparative Analysis Reveals Genetic Complexity in Species With Cryptic Introgression Histories
1. Gossypium gossypioides is a new world (D-genome) diploid species known to have experienced several rounds of introgressive hybridization from old world species (A genome) (Cronn et al., 2003). Its comparative cluster 5S rDNA graph of the three Gossypium species analyzed (Figures 4A, B) showed three loops where the G. gossipioides reads formed a unique loop (blue) that did not overlap with either A (red) or D (yellow) genome loops. Except for the genic junction region no significant interconnecting edges between the three genomes were visualized.
2. The intermediate wheatgrass Thinopyrum intermedium (Poaceae) is a hexaploid species experiencing multiple introgression events, potentially including genome parts from several species. We therefore included candidate Aegilops tauschii and Hordeum vulgare progenitor species in our comparative analysis of 5S rDNA (Figures 4C, D) which had been suggested as potential genome contributors for T. intermedium (Tang et al., 2000; Mahelka et al., 2011). At least four loops could be recognized on the cluster graph. Two loops contained shared reads from T. intermedium (blue) and A. tauschii (yellow). In addition, there was a prominent read-rich Thinopyrum-specific loop (Figure 4D, all-blue loop) that did not contain reads from other genomes and may originate from Dasypirium (Mahelka et al., 2013) for which read archives were unavailable. No T. intermedium reads were present in the H. vulgare loop (red).

Figure 4 RepeatExplorer2 graphical output of the comparative analysis of the 5S rDNA clustering in species of hybrid origin and with complex evolutionary histories, in which introgressive hybridization may have been involved. Cluster graphs with highlighted 5S rDNA genic region in green (A, C) and annotated reads origin in (B, D) in Gossypium gossypioides and Thinopyrum intermedium, respectively. The comparative analysis in (B) mixing reads of the putative parental genome donors, G. arboreum as the A genome donor (red), G. raimondii as the D genome donor (yellow), and G. gossypioides as the hybrid (blue) shows that only few reads of the putative hybrid are placed in the A or D genome loops. The comparative analysis in (D) shows that the reads corresponding to the hexaploid Thinopyrum intermedium (blue) are partially shared with the reads of one of the putative parental genome donors, Aegilops tauschii (yellow) while there are no coincidences with the reads of the other putative parental genome donor, Hordeum vulgare (red). Arrows indicate two spacers of different sizes stemming from the Aegilops parental genome donor.
Quantification of 5S and 35S rDNA Homoeologs in Gossypium and Brachypodium Allotetraploids From High-Throughput Sequencing Data
In the cluster graphs of the hybrid species (Figures 1 and 3) we often observed differences in read-richness between both loops suggesting a skewed representation of homoeologous 5S rDNA variants. To validate this assumption we quantified homoeologous 5S rDNAs by mapping of Illumina reads to the reference sequences of 5S rDNA units (Supplementary Figure S4). In five Gossypium allopolyploids analyzed (Supplementary Figure S4A), the 5S rDNA homoeologs were slightly skewed toward the A genome units. This clearly contrasts with 35S rDNA (Supplementary Figure S4B), where all but one allotetraploids contained far fewer A-genome than D-genome ITS1 types, except of Gossypium mustelinum in which the homoelogous ratio was inversed. A similar analysis carried out in Brachypodium hybridum showed a higher representation of B. distachyon homoeologs (in this case, both 35S and 5S rDNA) than those of B. stacei (Supplementary Figure S4C).
Reconstitution of 5S rDNA Units and Gene Copy Number in Gossypium Allotetraploids
The unit length is an important characteristic of rDNA arrays. We compared the lengths of in silico assembled Gossypium units with those previously determined by conventional cloning and Sanger sequencing (Table 2). RepeatExplorer2/TAREAN generates consensus sequences of 5S rDNA units from the decomposition of read sequences into k-mers (Novak et al., 2017). The lengths of 5S rDNA units determined by cloning ranged from 295–303 bp while those calculated from sequence data by bioinformatics tools ranged from 265–303 bp. In general, there was congruence between both methods. In contrast, the copy number variation between species was extremely high (up to 10 fold), confirming previous findings (Cronn et al., 1996). In some cases, copy numbers determined by computation methods differed by more than five-fold from those of slot blot hybridization experiments (Cronn et al., 1996).
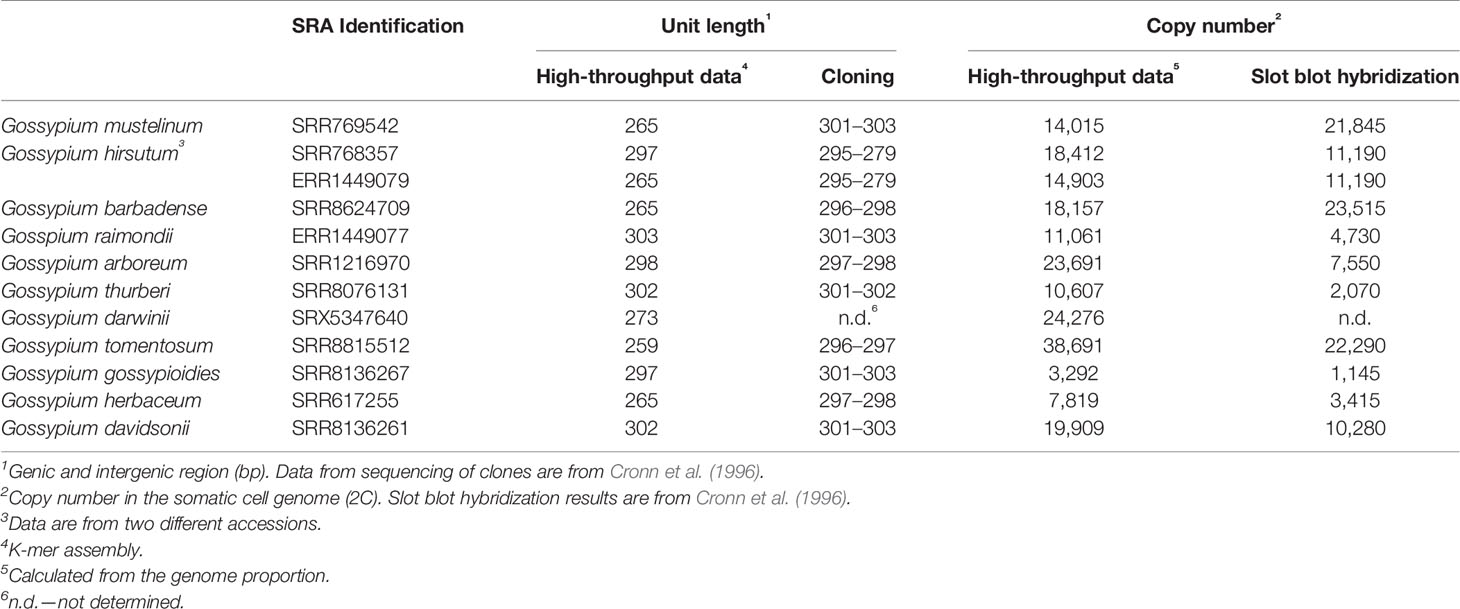
Table 2 5S rDNA unit lengths and copy number in Gossypium allotetraploids and diploids.
Southern Blot Hybridization Analysis of 5S rDNA in Spartina
Previous in silico analyses showed highly skewed 5S rDNA homoeologs in Spartina × townsendii hexaploid toward the S. maritima genome. In order to confirm this result, we carried out southern blot hybridization using genomic DNA from S. × townsendii (6x), the derived S. anglica allododecaploid (12x), and the progenitors of both species, S. maritima and S. alterniflora (Figure 5). Genomic DNA was digested with BamHI which has a conserved site in the angiosperm 5S rDNA units (Röser et al., 2001) (Figure 5A). The 5S rDNA probe generated ladders of bands, expected from a tandemly arranged sequence as the 5S rRNA genes. The probe hybridized strongly to the S. maritima DNA while the hybridization to S. alterniflora was relatively weak (Figure 5B). The S. maritima oligomers were slightly shorter than those of S. alterniflora consistent with shorter length of the S. maritima units (Figure 1). In both S. × townsendii and S. anglica oligomeric bands derived from both parents were visible indicating additivity.
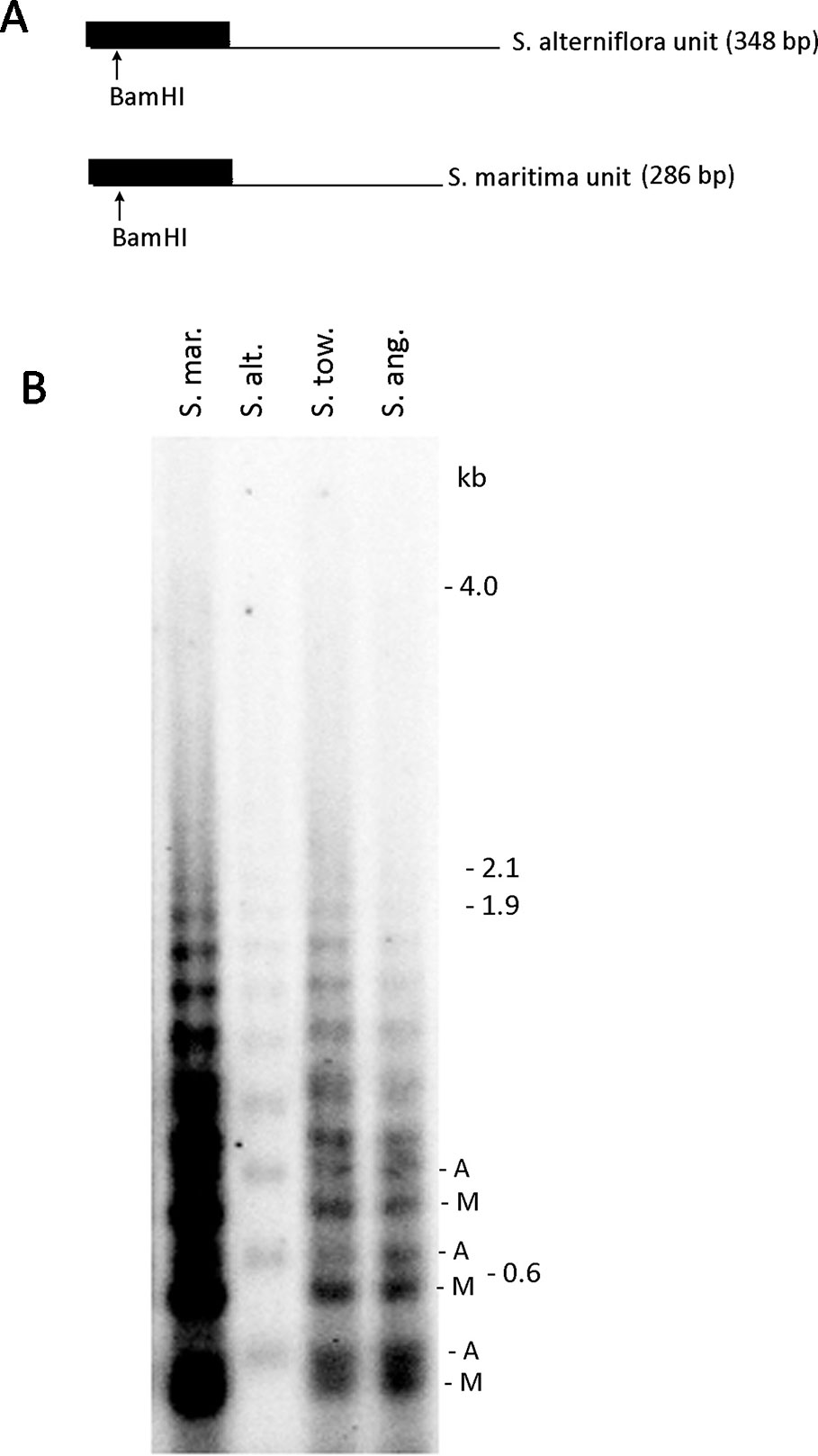
Figure 5 Southern blot analysis of 5S rDNA in Spartina hybrids and their progenitors. (A) Restriction maps of progenitor S. maritima and S. alterniflora of 5S rDNA. (B) The 5S rDNA probe hybridization to S. maritima (S. mar.), S. alterniflora (S. alt.), S. × townsendii homoploid hybrid (S. tow.), and S. anglica dodecaploid (S. ang.) DNAs. In S. × townsendii and S. anglica, hybridization fragments inherited from progenitors are marked as “A” (from S. alterniflora) and “M” (from S. maritima), respectively.
Discussion
Here we studied the organization and evolution of 5S rDNA in 84 plant genomes. We found that the clustering analysis of high-throughput Illumina reads by RepeatExplorer2/TAREAN provides a comprehensive view about 5S rDNA origin and organization, corroborating classical cytogenetic and molecular studies. The 5S rDNA cluster graphs are typically circular, showing high values of circularity parameters underlining regular tandem arrangement of these genes. Graphs displayed no or little node irregularities (discussed further below) consistent with a high homogeneity of arrays and confirming the model of concerted evolution (Dover, 1982; Kellogg and Appels, 1995). The sequence and size of in silico reconstituted 5S rDNA units were congruent with those obtained from cloning and Sanger sequencing. Below we discuss a value-added information obtained from cluster analyses that is not obtainable (or with difficulty) by conventional molecular and cytogenetic analyses.
Dynamism of rDNA Loci in Hybrid Genomes
We investigated rDNAs in two relatively ancient (c.1 Myr) allopolyploid systems (Gossypium and Brachypodium) which already show substantial loss of 35S rDNA homoeologous units (Wendel et al., 1995a; Borowska-Zuchowska and Hasterok, 2017). Gossypium allotetraploids were represented by five species with a typical AADD genome composition originating from common diploid ancestors closely related to G. arboreum (A genome) and G. raimondii (D genome). In these allotetraploids, previous cloning analyses identified both homoeologous 5S rDNA sequences in G. hirsutum and G. mustelinum but not in G. barbadense and G. tomentosum, where only the A genome sequences were recovered (Cronn et al., 1996). However, comparative graph clustering of the 5S rDNA revealed both A and D genome homoeologs in these species, with a dominance of the A-genome units. Also, with respect to the 35S rDNA, the D-genome type of ITS in G. mustelinum sequence was barely detectable using Southern blot hybridization (Wendel et al., 1995a), but high-throughput sequencing recovers similar sequences at a frequency of about 15% (Supplementary Figure S4B). A similar example of skewed homoelog ratios is represented by B. hybridum and S. × townsendii, where one loop contained far more reads than the other in the 5S rDNA cluster graphs. Indeed, Southern blot analysis confirmed one strong and one weak 5S rDNA family in the S. × townsendii homoploid hybrid and S. anglica. Skewed gene ratios exist already in the progenitor genomes based on read abundance in the graph loops and differential intensity of Southern hybridization signals. A strong repeat-rich locus likely occurs in the S. maritima parent while a weaker locus may be present in the S. alterniflora parent. These examples demonstrate a higher sensitivity of a cluster graph-based approach over the de novo assembly or PCR-cloning approaches, where various technical biases may occur (Lunerova et al., 2017).
Evaluation of the Graph-Based Method for Identification of Allopolyploids and Hybrids
Diploid genomes show simple circular structures of 5S rDNA cluster graphs referred to type 1 (Figure 2A). In contrast, allopolyploids and homoploid hybrids display more complex graph structures (type 2) in which divergent gene families are visualized as distinct loops. In addition, there was a good correlation between cluster graph complexity and number of 5S rDNA loci (Figure 2B). These observations are consistent with a general view that most diploids carry a single 5S rDNA locus (and a single gene family) while allopolyploids tend to maintain multiple loci (and multiple 5S rDNA families) (Table 1) and (Roa and Guerra, 2015; Garcia et al., 2017). Thus, a simple visual inspection of the 5S rDNA cluster topology appears to be informative with respect to the putative hybridogenic origin of a species. Among the computation parameters, the k-mer coverage seems to reflect the intragenomic homogeneity—low k-mer scores associate with complex graph shapes and multiple gene families, while high k-mer scores associate with simple circular structures and single gene families. Thus, the k-mer coverage may be taken as a semi-quantitative parameter of 5S rDNA intragenomic homogeneity, although more studies are needed to validate the relationship.
One of the advantages of the clustering-based method is that it may provide initial information about the 5S rDNA homoeologs without prior knowledge of progenitor genomes based on the graph complexity. Certainly, the origin of 5S rDNA families in a hybrid is indicated by comparative clustering requiring sequences from candidate progenitor genomes. We observed similar graph complexities for the 35S rDNA encoding 18S-5.8S-26 rRNA genes (Supplementary Figure S5) suggesting that these clusters (particularly, 3' 26S region and the IGS) may be equally informative as that of 5S rDNA. These cases pose additional opportunities for studying the recombination dynamics of dispersed 5S and ITS arrays, which may be subject to complex and incomplete concerted evolutionary forces. Despite the apparent good correlation between cluster complexity and a hybrid character of the genome there were several notable exceptions from the rule:
1. About 21% of allopolyploids and homoploids showed simple type 1 graphs. These simple graphs can be explained by high similarity of progenitor units, preventing separation of reads. However, it can also be explained by locus loss and/or homogenization of 5S rDNA in allopolyploids over longer evolutionary times. Indeed, ancient (c.5 Myr) Nicotiana allotetraploids from section Repandae showed simple circular type 1 graphs (not shown) and a diploid character of 5S rDNA loci (Lim et al., 2007). Interestingly, Triticum turgidum (0.5 Myr) and Spartina alterniflora (3 Myr) (both Poaceae) polyploids also show simple cluster graphs despite their relatively young age, suggesting that the process of rDNA homogenization and diploidization may proceed at different rates in different systems. Frequent losses of 5S rDNA loci in Triticum (Baum et al., 2008), rice (Zhu et al., 2008) and Spartina (this work) polyploids may also suggest certain instability of 5S rDNA in Poaceae. Nevertheless, the Thinopyrum hybrid (Poaceae) displayed a highly complex cluster graph (Figures 4C, D) consistent with the retention of progenitor 5S rDNA families (Mahelka et al., 2013) and therefore arguing against generalization of these observations.
2. About 13% of diploids showed complex type 2 graphs, indicating intragenomic heterogeneity of 5S rDNA loci in these genomes. In at least some cases, the intragenomic heterogeneity of 5S rDNA in these diploids can be explained by homoploid hybridization and introgression events. This is probably the explanation for the complex graphs in Gossypium gossypioides (Figure 4A) which has a complex evolutionary history entailing at least two temporally widely separated divergence events (Wendel et al., 1995b; Cronn et al., 2003). Although introgression and hybridization is also relatively frequent in the banana genus (Němečková et al., 2018) a more likely explanation for complex graph structures in Musa acuminata (Figure 6C) is an exceptionally high number of 5S rDNA loci in this species (six per diploid genome) (Valarik et al., 2002; Garcia et al., 2012a) and probably inefficient interlocus recombination (Schlotterer and Tautz, 1994) leading to poor homogenization. Actually, the mechanisms of amplification of 5S rDNA loci across the chromosomes are still poorly understood (Schubert and Wobus, 1985; Symonova et al., 2017; Joachimiak et al., 2018; Souza et al., 2019).
3. The occurrence of non-rDNA sequences within the 5S rDNA clusters may potentially distort graph shapes. In Tragopogon porrifolius and Senecio campestris (both Asteraceae) the 5S rDNA clusters apparently contain traces of Cassandra transposable elements. These LTR elements are widespread in angiosperm genomes and carry a 5S rDNA related sequence (Kalendar et al., 2008). In cluster graphs the Cassandra element can be identified by divergent reads connected by only a few nodes to the 5S rDNA genic region (Figures 6A, B). The known high mobility of 5S rDNA in the Musa genus (Valarik et al., 2002) could be related to the activity of transposable elements whose remnants (TY1 copia/Tork family) are apparently found in some M. acuminata 5S rDNA units (Figure 6C). In general, the frequency of non-rDNA sequences was low (<4% analyses) in the major 5S rDNA clusters and likely does not represent significant source of artefacts.
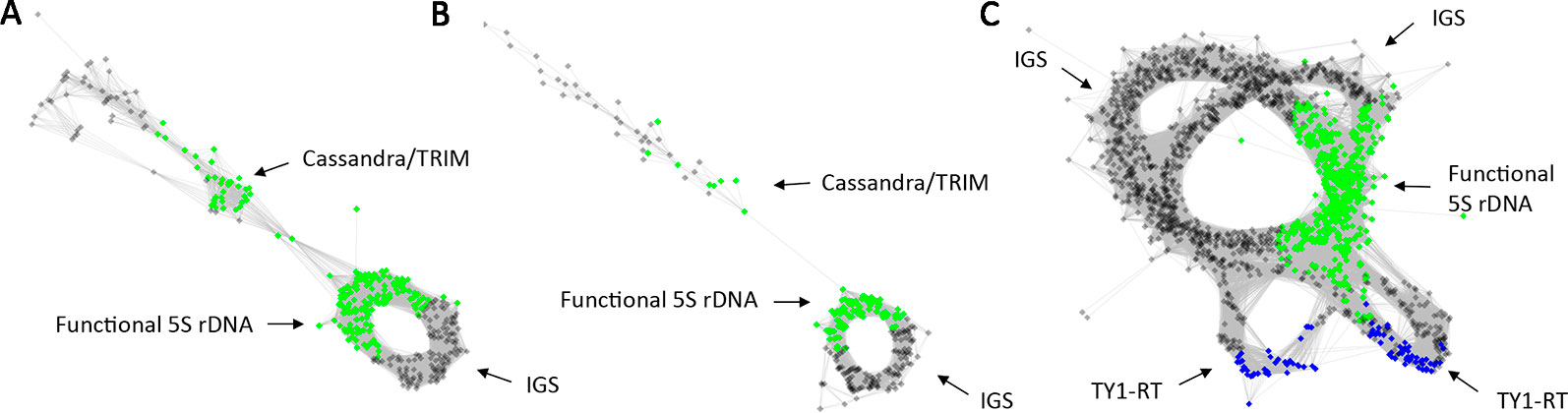
Figure 6 Graph structures of 5S rDNA clusters from the RepeatExplorer2 graphical output containing a significant number of retroelement sequences in Tragopogon porrifolius (A), Senecio vulgaris (B) and Musa acuminata (C). In (A, B) reads derived from a Cassandra/TRIM element. Note, the Cassandra/TRIM elements were relatively isolated and connected to 5S rDNA with 1–2 reads only. In (C) the highly complex structure of 5S rDNA in M. acuminata containing multiple IGS and regions of similarity to a reverse transcriptase domain of a TY-1 Copia retroelement.
Concluding Remarks
To summarize, we infer that the visual inspection of rDNA cluster graph topologies coupled with calculation of graph parameters is highly informative for the assessment of rDNA genomic organization, number of rRNA gene families, and loci. The method may provide clues for testable hypotheses about evolutionary histories of interspecific hybrids and allopolyploids, especially in biological systems with unknown or not well defined genome donors (Mahelka et al., 2011; Kaplan et al., 2013; Fredotovic et al., 2014; Belyayev et al., 2018). It is necessary to stress that a robust evaluation of hybridization and polyploidy cannot be solely based on read clustering, but should involve a combination of various cytogenetic, molecular and genomic methods.
Materials and Methods
DNA Isolation, High-Throughput Sequencing, and Read Archive Accessions
Most sequences used in this study were downloaded from sequence read archives at the EBI server (Supplementary Table S1). Six genomes were sequenced de novo as follows: genomic DNA from leaf tissue was isolated by a modified CTAB method and sequenced by Illumina technology at BGI. The Spartina DNAs originated from natural samples collected in Southampton area, UK: S. maritima (Isle of Wight), S.× townsendii (Hythe), S. alterniflora, and S. anglica (both from Eling Marchwood) (Huska et al., 2016); Cardamine × insueta and C. amara were from natural populations in Urnerboden, Switzerland (Zozomova-Lihova et al., 2014); C. flexuosa was from Zelezne, Slovakia, and C. hirsuta from Gehausen, Germany (Mandakova et al., 2014). Details of sequencing are provided in Supplementary Table S3.
In Silico Identification of 5S rDNA Repeats
The fastq reads were initially filtered for quality and trimmed to a uniform length by pre-processing and QC tools by RepeatExplorer2 (Novak et al., 2013). The pipeline is implemented in the Galaxy environment (https://galaxy-elixir.cerit-sc.cz/). For computation, resources at the international ELIXIR infrastructure (European research infrastructure for biological information) were used. Read length ranged 100–150 bp depending on sequencing library and platform. After the fastq > fasta conversion reads were analyzed with RepeatExplorer2 using default parameters. The RepeatExplorer2 pipeline runs a graph-based clustering algorithm (Novak et al., 2013) to assemble the groups of frequently overlapping reads into clusters of reads, representing a repetitive element, or part of a repetitive element with a higher order genome structure. It uses a BLAST threshold of 90% similarity across 55% of the read to identify reads to each cluster by default (minimum overlap = 55, cluster threshold = 0.01%, minimum overlap for assembly = 40), and the clusters are identified based on a principle of maximum modularity. Typically, 200,000 of pair-end reads were used as input for clustering. This number typically yields a cluster comprising several hundreds of 5S rDNA specific reads. Analysis of larger (>2 Gb/1C) genomes requires an increase of the number of input reads (up to 2 million), as the 5S rDNA coverage decreases. Although this prolongs computation times (typically 5–6 hours on the MetaCentrum ELIXIR computer clusters) we were able to reconstruct 5S rDNA units in the large (50 GB) Fritillaria imperialis genome (Zonneveld, 2010) although, in this case, the number of reads in the cluster was too low, preventing graph analysis. High coverages may also help to reveal rare 5S rDNA variants and pseudogenes that are frequent in gymnosperms (Wang et al., 2016; Wang et al., 2019) while they rarely occur in angiosperms. In interspecific comparisons, the usage of a standard fraction of genome (0.1–1.0%) is recommended to prevent biases in interspecific comparisons.
The 5S rDNA clusters were searched among the cluster annotation files using “rDNA” search keyword. Alternatively, 5S rDNA clusters were found in TAREAN tandem reports (a specific tool for the analysis of tandem repeats implemented in RepeatExplorer2). The shapes of cluster graphs were characterized by a connected component index parameter (C) which is calculated as the proportion of the largest strongly connected component in graph composed of oriented reads (Novak et al., 2017). Cluster graph topologies were visually inspected and categorized into two groups (simple, type 1, and complex, type 2, graphs). The k-mer score was calculated by the RepeatExplorer2/TAREAN program as the sum of frequencies of all k-mers used for consensus sequence reconstruction.
Identification and Quantification of Homoeologous 5S rRNA Gene Families
5S rDNA homoeologous families were quantified by mapping analysis using CLC Genomics Workbench (QIAGEN), CLC onwards. Trimmed reads (typically >7 million) were mapped to the corresponding reference with following parameters: insertion and deletion costs = 3, lengths fraction = 0.5, similarity fraction = 0.9, deletion cost = 2. As reference sequences we used Gossypium arboreum (GenBank no. GAU31855) and G. raimondii (GRU39497) clones. Since no GenBank 5S rDNA clones were available for Brachypodium we used consensus sequences of B. distachyon (370 bp) and B. stacei (270 bp) generated by RepeatExplorer2 as a reference.
Phylogenetic analysis was carried out using assembled contigs computed by RepeatExplorer2. Briefly, BLAST libraries of contigs from hybrids and allopolyploids were BLASTed against the 5S rDNA sequences: For Gossypium, these were GenBank clones (#GAU31855 and #GRU39497); for Brachypodium and Spartina contigs generated by RepeatExplorer were used. The IGS subregions were extracted from BLAST outputs by a selection command in CLC, grouped and aligned. Alignments were manually edited and neighbor joining phylogeny trees constructed (CLC).
Because homoeologous ITS1 (internally transcribed spacer 1 of 35S rDNA) cannot be quantified by mapping procedures due to their overall similarity, we calculated the ITS1 homoeologous ratios from the number of nodes in genome-specific clades of phylogeny trees: (i) ITS sequences from Gossypium and Brachypodium allotetraploids were extracted from mapped reads, yielding typically hundreds to thousands of sequences. (ii) Stand-alone BLAST databases were generated from the ITS sequences. The databases were queried with reference sequences derived from variable 50–70 bp central subregions of ITS1. The ITS1 consensus sequences were obtained from the alignment of GenBank clones: B. stacei (JX665827-JX665832), B. hybridum (JX665718-JX665731), G. arboreum (GAU12712), and G. raimondii (GTU12711). (iii) Reads extracted from BLAST outputs were trimmed to uniform length, sampled (100–500 reads), and NJ trees constructed using a phylogeny tool of CLC. Homoeologous sequences in distinct clades were extracted, counted, and expressed as a ratio.
Statistical Methods
We analyzed the data by one-way ANOVA statistical test implemented within the MS Office package (XL-Toolbox NG). Box-plots were constructed using an online BoxPlotR tool (www.shiny.chmgid.org/boxplotr/).
Southern Blot Hybridization
Total genomic DNA was extracted from fresh young leaves using a modified CTAB method following procedures described previously (Kovarik et al., 1997). Genomic DNA was digested with the BamHI restriction enzyme and hybridized on blots. The DNAs were digested with BamHI and hybridized with the radioactively labeled ([32P] dCTP, Dekaprimer labeling kit (Thermo Fischer, USA) 5S rDNA probe. The probe was a trimer of the 5S rRNA gene from Artemisia tridentata [S4 clone, GenBank # JX101915.1, (Garcia et al., 2012b)]. Hybridization signals were visualized using a PhosphorImager (Fuji, FLA 9000).
Data Availability Statement
The datasets generated for this study can be found in the GenBank Sequence Read Archives (Bioprojects “Chromosome evolution in invasive Spartina plants”, ID: PRJNA575642 and “Chromosome evolution in Cardamine hybrids and polyploids”, ID: PRJNA575831).
Author Contributions
Conceived and designed the study: AKo, SG. Performed the experiments and collected material: SG, AKo, and MA. Analyzed the data: SG, MA, JW, NB-Z, AKu, and AKo. Wrote the manuscript: AKo, SG.
Funding
The work was supported by the Czech Science Foundation (grant 19-03442S), the Polish OPUS project of the National Science Centre (2018/31/B/NZ3/01761) and by the Dirección General de Investigación Científica y Técnica (CGL2016-75694-P AEI/FEDER, UE) from the government of Spain. SG benefited from a Ramón y Cajal contract (RYC-2014-16608) from the government of Spain.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Acknowledgments
We thank members of the Malika Ainouche, Andrew R. Leitch and Martin Lysak laboratories for their kind help with Spartina and Cardamine sampling. We further thank the editor and reviewers for their helpful comments and the organizers of annual RepeatExplorer meetings in Ceske Budejovice (Czech Republic). Access to computing and storage facilities owned by parties and projects contributing to the National Grid Infrastructure MetaCentrum provided under the program Projects of Large Research, Development, and Innovations Infrastructures (CESNET LM2015042) is greatly appreciated.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00041/full#supplementary-material
References
Ainouche, M. L., Baumel, A., Salmon, A. (2004). Spartina anglica Schreb.: a natural model system for analysing early evolutionary changes that affect allopolyploid genomes. Biol. J. Linn. Soc 82, 474–475. doi: 10.1111/j.1095-8312.2004.00334.x
Alix, K., Gerard, P. R., Schwarzacher, T., Heslop-Harrison, J. S. (2017). Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann. Bot. 120, 619–619. doi: 10.1093/aob/mcx096
Alvarez, I., Wendel, J. W. (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434. doi: 10.1016/S1055-7903(03)00208-2
Baum, B. R., Bailey, L. G., Belyayev, A., Raskina, O., Nevo, E. (2004). The utility of the nontranscribed spacer of 5S rDNA units grouped into unit classes assigned to haplomes - a test on cultivated wheat and wheat progenitors. Genome 47, 590–599. doi: 10.1139/g03-146
Baum, B. R., Edwards, T., Johnson, D. A. (2008). Loss of 5S rDNA units in the evolution of agropyron, pseudoroegneria, and douglasdeweya. Genome 51, 589–598. doi: 10.1139/G08-045
Baum, B. R., Edwards, T., Mamuti, M., Johnson, D. A. (2012). Phylogenetic relationships among the polyploid and diploid Aegilops species inferred from the nuclear 5S rDNA sequences (Poaceae: Triticeae). Genome 55, 177–193. doi: 10.1139/g2012-006
Belyayev, A., Pastova, L., Fehrer, J., Josefiova, J., Chrtek, J., Mraz, P. (2018). Mapping of Hieracium (Asteraceae) chromosomes with genus-specific satDNA elements derived from next-generation sequencing data. Plant Syst. Evol. 304, 387–396. doi: 10.1007/s00606-017-1483-y
Besendorfer, V., Krajacić-Sokol, I., Jelenić, S., Puizina, J., Mlinarec, J., Sviben, T., et al. (2005). Two classes of 5S rDNA unit arrays of the silver fir, Abies alba Mill.: structure, localization and evolution. Theor. Appl. Genet. 110, 730–741. doi: 10.1007/s00122-004-1899-y
Borowska-Zuchowska, N., Hasterok, R. (2017). Epigenetics of the preferential silencing of Brachypodium stacei-originated 35S rDNA loci in the allotetraploid grass Brachypodium hybridum. Sci. Rep. 7, 5260. doi: 10.1038/s41598-017-05413-x
Bourke, P. M., Voorrips, R. E., Visser, R. G. F., Maliepaard, C. (2018). Tools for genetic studies in experimental populations of polyploids. Front. Plant Sci. 9, 513. doi: 10.3389/fpls.2018.00513
Boutte, J., Aliaga, B., Lima, O., de Carvalho, J. F., Ainouche, A., Macas, J., et al. (2016). Haplotype detection from Next-generation sequencing in high-ploidy-level species: 45S rDNA gene copies in the hexaploid Spartina maritima. Genes Genomes Genet. 6, 29–40. G3-. doi: 10.1534/g3.115.023242
Bughio, F., Maggert, K. A. (2019). The peculiar genetics of the ribosomal DNA blurs the boundaries of transgenerational epigenetic inheritance. Chromosome Res. 27, 19–30. doi: 10.1007/s10577-018-9591-2
Catalán, P., Müller, J., Hasterok, R., Jenkins, G., Mur, L. A., Langdon, T., et al. (2012). Evolution and taxonomic split of the model grass Brachypodium distachyon. Ann. Bot. 109, 385–405. doi: 10.1093/aob/mcr294
Cronn, R. C., Zhao, X., Paterson, A. H., Wendel, J. F. (1996). Polymorphism and concerted evolution in a tandemly repeated gene family: 5S ribosomal DNA in diploid and allopolyploid cottons. J. Mol. Evol. 42, 685–705. doi: 10.1007/BF02338802
Cronn, R., Small, R. L., Haselkorn, T., Wendel, J. F. (2003). Cryptic repeated genomic recombination during speciation in Gossypium gossypioides. Evolution 57 (11), 2475–2489. doi: 10.1111/j.0014-3820.2003.tb01493.x
Dodsworth, S., Chase, M. W., Kelly, L. J., Leitch, I. J., Macas, J., Novak, P., et al. (2015). Genomic repeat abundances contain phylogenetic signal. Syst. Biol. 64, 112–126. doi: 10.1093/sysbio/syu080
Dodsworth, S., Chase, M. W., Sarkinen, T., Knapp, S., Leitch, A. (2016). Using genomic repeats for phylogenomics: a case study in wild tomatoes (Solanum section Lycopersicon: Solanaceae). Biol. J. Linn. Soc 117, 96–105. doi: 10.1111/bij.12612
Dover, G. A. (1982). Molecular drive: a cohesive mode of species evolution. Nature 299, 111–117. doi: 10.1038/299111a0
Elder, J. F., Turner, B. J. (1995). Concerted evolution of repetitive DNA-sequences in eukaryotes. Q. Rev. Biol. 70, 297–320. doi: 10.1086/419073
Fredotovic, Z., Samanic, I., Weiss-Schneeweiss, H., Kamenjarin, J., Jang, T. S., Puizina, J. (2014). Triparental origin of triploid onion, Allium x cornutum (Clementi ex Visiani, 1842), as evidenced by molecular, phylogenetic and cytogenetic analyses. BMC Plant Biol. 14, 14. doi: 10.1186/1471-2229-14-24
Fulnecek, J., Lim, K. Y., Leitch, A. R., Kovarik, A., Matyasek, R. (2002). Evolution and structure of 5S rDNA loci in allotetraploid Nicotiana tabacum and its putative parental species. Heredity 88, 19–25. doi: 10.1038/sj.hdy.6800001
Fulnecek, J., Matyasek, R., Kovarik, A. (2006). Plant 5S rDNA has multiple alternative nucleosome positions. Genome 49, 840–850. doi: 10.1139/g06-039
Garcia, S., Garnatje, T., Kovarik, A. (2012a). Plant rDNA database: ribosomal DNA loci data including other karyological and cytogenetic information in plants. Chromosoma 121, 389–394. doi: 10.1007/s00412-012-0368-7
Garcia, S., Crhak Khaitova, L., Kovarik, A. (2012b). Expression of 5S rRNA genes linked to 35S rDNA in plants, their epigenetic modification and regulatory element divergence. BMC Plant Biol. 12, 95. doi: 10.1186/1471-2229-12-95
Garcia, S., Kovarik, A., Leitch, A. R., Garnatje, T. (2017). Cytogenetic features of rRNA genes across land plants: analysis of the Plant rDNA database. Plant J. 89, 1020–1030. doi: 10.1111/tpj.13442
Glover, N. M., Redestig, H., Dessimoz, C. (2016). Homoeologs: what are they and how do we infer them? Trends Plant Sci. 21, 609–621. doi: 10.1016/j.tplants.2016.02.005
Grover, C. E., Arick, M. A., Thrash, A., Conover, J. L., Sanders, W. S., Peterson, D. G., et al. (2019). Insights into the evolution of the New world diploid cottons (Gossypium, subgenus Houzingenia) based on genome sequencing. Genome Biol. Evol. 11, 53–71. doi: 10.1093/gbe/evy256
Hemleben, V., Zentgraf, U. (1994). “Structural organisation and regulation of transcription by RNA polymerase I of plant nuclear ribosomal genes,” in Results and problems in cell differentiation 20: plant promoters and transcription factors, vol. 3-24 . Ed. Lapitan, N. (Berlin/Heidelberg: Springer-Verlag). doi: 10.1007/978-3-540-48037-2_1
Huska, D., Leitch, I. J., de Carvalho, J. F., Leitch, A. R., Salmon, A., Ainouche, M., et al. (2016). Persistence, dispersal and genetic evolution of recently formed Spartina homoploid hybrids and allopolyploids in Southern England. Biol. Invasions 18, 2137–2151. doi: 10.1007/s10530-015-0956-6
Jang, T. S., McCann, J., Parker, J. S., Takayama, K., Hong, S. P., Schneeweiss, G. M., et al. (2016). rDNA loci evolution in the genus Glechoma (Lamiaceae). PloS One 11, e0167177. doi: 10.1371/journal.pone.0167177
Joachimiak, A. J., Hasterok, R., Sliwinska, E., Musial, K., Grabowska-Joachimiak, A. (2018). FISH-aimed karyotype analysis in Aconitum subgen. Aconitum reveals excessive rDNA sites in tetraploid taxa. Protoplasma 255, 1363–1372. doi: 10.1007/s00709-018-1238-9
Kalendar, R., Tanskanen, J., Chang, W., Antonius, K., Sela, H., Peleg, O., et al. (2008). Cassandra retrotransposons carry independently transcribed 5S RNA. Proc. Natl. Acad. Sci. U.S.A. 105, 5833–5838. doi: 10.1073/pnas.0709698105
Kaplan, Z., Jarolimova, V., Fehrer, J. (2013). Revision of chromosome numbers of Potamogetonaceae: a new basis for taxonomic and evolutionary implications. Preslia 85, 421–482. doi: 10.1007/s12228-012-9299-0
Kellogg, E. A., Appels, R. (1995). Intraspecific and interspecific variation in 5S RNA genes are decoupled in diploid wheat relatives. Genetics 140 (1), 325–343.
Kovarik, A., Matyasek, R., Leitch, A., Gazdova, B., Fulnecek, J., Bezdek, M. (1997). Variability in CpNpG methylation in higher plant genomes. Gene 204, 25–33. doi: 10.1016/S0378-1119(97)00503-9
Lim, K. Y., Kovarik, A., Matyasek, R., Chase, M. W., Clarkson, J. J., Grandbastien, M. A., et al. (2007). Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytol. 175, 756–763. doi: 10.1111/j.1469-8137.2007.02121.x
Lunerova, J., Renny-Byfield, S., Matyasek, R., Leitch, A., Kovarik, A. (2017). Concerted evolution rapidly eliminates sequence variation in rDNA coding regions but not in intergenic spacers in Nicotiana tabacum allotetraploid. Plant Syst. Evol. 303, 1043–1060. doi: 10.1007/s00606-017-1442-7
Mahelka, V., Kopecky, D., Pastova, L. (2011). On the genome constitution and evolution of intermediate wheatgrass (Thinopyrum intermedium: Poaceae, Triticeae). BMC Evol. Biol. 11, 127. doi: 10.1186/1471-2148-11-127
Mahelka, V., Kopecky, D., Baum, B. R. (2013). Contrasting Patterns of Evolution of 45S and 5S rDNA families uncover new aspects in the genome constitution of the agronomically important grass Thinopyrum intermedium (Triticeae). Mol. Biol. Evol. 30, 2065–2086. doi: 10.1093/molbev/mst106
Mandakova, T., Marhold, K., Lysak, M. A. (2014). The widespread crucifer species Cardamine flexuosa is an allotetraploid with a conserved subgenomic structure. New Phytol. 201, 982–992. doi: 10.1111/nph.12567
Matyasek, R., Lim, K. Y., Kovarik, A., Leitch, A. R. (2003). Ribosomal DNA evolution and gene conversion in Nicotiana rustica. Heredity 91 (3), 268–275. doi: 10.1038/sj.hdy.6800333
Matyasek, R., Renny-Byfield, S., Fulnecek, J., Macas, J., Grandbastien, M. A., Nichols, R., et al. (2012). Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids. BMC Genomics 13, 722. doi: 10.1186/1471-2164-13-722
Mlinarec, J., Skuhala, A., Jurković, A., Malenica, N., McCann, J., Weiss-Schneeweiss, H., et al. (2019). The repetitive DNA composition in the natural pesticide producer Tanacetum cinerariifolium: interindividual variation of subtelomeric tandem repeats. Front. Plant Sci. 10, 613. doi: 10.3389/fpls.2019.00613
Muir, G., Fleming, C. C., Schlotterer, C. (2001). Three divergent rDNA clusters predate the species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol. Biol. Evol. 18, 112–119. doi: 10.1093/oxfordjournals.molbev.a003785
Němečková, A., Christelová, P., Čížková, J., Nyine, M., Van den Houwe, I., Svačina, R., et al. (2018). Molecular and cytogenetic study of east African highland banana. Front. Plant Sci. 9, 1371. doi: 10.3389/fpls.2018.01371
Nieto Feliner, G., Rosselló, J. A. (2007). Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol. Phylogenet. Evol. 44, 911–919. doi: 10.1016/j.ympev.2007.01.013
Nieto Feliner, G., Rossello, J. A. (2012). “Concerted evolution of multigene families and homeologous recombination,” in Plant Genome Diversity. Ed. Wendel, J. F. (Wien: Springer-Verlag), 171–194. doi: 10.1007/978-3-7091-1130-7_12
Nieto Feliner, N., Alvarez, I., Fuertes-Aguilar, J., Heuertz, M., Marques, I., Moharrek, F., et al. (2017). Is homoploid hybrid speciation that rare? An empiricist's view. Heredity 118, 513–516. doi: 10.1038/hdy.2017.7
Novak, P., Neumann, P., Macas, J. (2010). Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinf. 11, 378. doi: 10.1186/1471-2105-11-378
Novak, P., Neumann, P., Pech, J., Steinhaisl, J., Macas, J. (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793. doi: 10.1093/bioinformatics/btt054
Novak, P., Robledillo, L. A., Koblizkova, A., Vrbova, I., Neumann, P., Macas, J. (2017). TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucl. Acids Res. 45, E111. doi: 10.1093/nar/gkx257
Parks, M. M., Kurylo, C. M., Batchelder, J. E., Vincent, C. T., Blanchard, S. C. (2019). Implications of sequence variation on the evolution of rRNA. Chromosome Res. 27, 89–93. doi: 10.1007/s10577-018-09602-w
Pedrosa-Harand, A., de Almeida, C. C. S., Mosiolek, M., Blair, M., Schweizer, D., Guerra, M. (2006). Extensive ribosomal DNA amplification during Andean common bean (Phaseolus vulgaris L.) evolution. Theor. Appl. Genet. 112, 924–933. doi: 10.1007/s00122-005-0196-8
Peska, V., Mandakova, T., Ihradska, V., Fajkus, J. (2019). Comparative dissection of three giant genomes: Allium cepa, Allium sativum, and Allium ursinum. Int. J. Mol. Sci. 20, 25. doi: 10.3390/ijms20030733
Poczai, P., Hyvonen, J. (2010). Nuclear ribosomal spacer regions in plant phylogenetics: problems and prospects. Mol. Biol. Rep. 37, 1897–1912. doi: 10.1007/s11033-009-9630-3
Renny-Byfield, S., Kovarik, A., Chester, M., Nichols, R. A., Macas, J., Novak, P., et al. (2012). Independent, rapid and targeted loss of highly repetitive DNA in natural and synthetic allopolyploids of Nicotiana tabacum. PloS One 7, 722. doi: 10.1371/journal.pone.0036963
Ribeiro, T., Marques, A., Novak, P., Schubert, V., Vanzela, A. L., Macas, J., et al. (2017). Centromeric and non-centromeric satellite DNA organisation differs in holocentric Rhynchospora species. Chromosoma 126, 325–335. doi: 10.1007/s00412-016-0616-3
Röser, M., Winterfeld, G., Grebenstein, B., Hemleben, V. (2001). Molecular diversity and physical mapping of 5S rDNA in wild and cultivated oat grasses (Poaceae: Aveneae). Mol. Phylogenet. Evol. 2, 198–217. doi: 10.1006/mpev.2001.1003
Roa, F., Guerra, M. (2012). Distribution of 45S rDNA sites in chromosomes of plants: structural and evolutionary implications. BMC Evol. Biol. 12, 225. doi: 10.1186/1471-2148-12-225
Roa, F., Guerra, M. (2015). Non-random distribution of 5S rDNA sites and its association with 45S rDNA in plant chromosomes. Cytogenet. Genome Res. 146, 243–249. doi: 10.1159/000440930
Sastri, D. C., Hilu, K., Appels, R., Lagudah, E. S., Playford, J., Baum, B. R. (1992). An overview of evolution in plant 5S-DNA. Plant Syst. Evol. 183, 169–181. doi: 10.1007/BF00940801
Schlotterer, C., Tautz, D. (1994). Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Curr. Biol. 4, 777–783. doi: 10.1016/S0960-9822(00)00175-5
Schubert, I., Wobus, U. (1985). In situ hybridization confirms jumping nucleolus organizing regions in Allium. Chromosoma 92, 143–148. doi: 10.1007/BF00328466
Souza, G., Marques, A., Ribeiro, T., Dantas, L. G., Speranza, P., Guerra, M., et al. (2019). Allopolyploidy and extensive rDNA site variation underlie rapid karyotype evolution in Nothoscordum section Nothoscordum (Amaryllidaceae). Bot. J. Linn. Soc 190, 215–228. doi: 10.1093/botlinnean/boz008
Symonova, R., Ocalewicz, K., Kirtiklis, L., Delmastro, G. B., Pelikanova, S., Garcia, S., et al. (2017). Higher-order organisation of extremely amplified, potentially functional and massively methylated 5S rDNA in European pikes (Esox sp.). BMC Genomics 18, 391. doi: 10.1186/s12864-017-3774-7
Tang, S., Li, Z., Jia, X., Larkin, P. J. (2000). Genomic in situ hybridization (GISH) analyses of Thinopyrum intermedium, its partial amphiploid Zhong 5, and disease-resistant derivatives in wheat. Theor. Appl. Genet. 100, 344–352. doi: 10.1007/s001220050045
Valarik, M., Simkova, H., Hribova, E., Safar, J., Dolezelova, M., Dolezel, J. (2002). Isolation, characterization and chromosome localization of repetitive DNA sequences in bananas (Musa spp.). Chromosome Res. 10, 89–100. doi: 10.1023/A:1014945730035
Van De Peer, Y., Mizrachi, E., Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Vitales, D., Garcia, S., Dodsworth, S. (2019). “Recostructing phylogenetic relatioships based on repeat sequence similarities,” in BioRxiv (USA: Cold Spring Harbour Laboratory publishers). doi: 10.1101/624064
Volkov, R. A., Borisjuk, N. V., Panchuk, I. I., Schweizer, D., Hemleben, V. (1999). Elimination and rearrangement of parental rDNA in the allotetraploid Nicotiana tabacum. Mol. Biol. Evol. 16, 311–320. doi: 10.1093/oxfordjournals.molbev.a026112
Volkov, R. A., Komarova, N. Y., Hemleben, V. (2007). Ribosomal DNA in plant hybrids: inheritance, rearrangement, expression. Syst. Biodivers. (NHM London) 5, 261–276. doi: 10.1017/S1477200007002447
Wang, W. C., Ma, L., Becher, H., Garcia, S., Kovarikova, A., Leitch, I. J., et al. (2016). Astonishing 35S rDNA diversity in the gymnosperm species Cycas revoluta Thunb. Chromosoma 125, 683–699. doi: 10.1007/s00412-015-0556-3
Wang, W., Wan, T., Becher, H., Kuderova, A., Leitch, I. J., Garcia, S., et al. (2019). Remarkable variation of ribosomal DNA organization and copy number in gnetophytes, a distinct lineage of gymnosperms. Ann. Bot. 123, 767–781. doi: 10.1093/aob/mcy172
Weiss-Schneeweiss, H., Tremetsberger, K., Schneeweiss, G. M., Parker, J. S., Stuessy, T. F. (2008). Karyotype diversification and evolution in diploid and polyploid South American Hypochaeris (Asteraceae) inferred from rDNA localization and genetic fingerprint data. Ann. Bot. 101, 909–918. doi: 10.1093/aob/mcn023
Weiss-Schneeweiss, H., Leitch, A. R., McCann, J., Jang, T. S., Macas, J. (2015). “Employing next-generation sequencing to explore the repeat landscape of the plant genome,” in Next generation sequencing in plant systematics. Eds. Hörandl, E., Appelhans, M. S. (Germany: Königstein), 155–179. doi: 10.14630/000006
Wendel, J. F., Schnabel, A., Seelanan, T. (1995a). Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc. Natl. Acad. Sci. U.S.A. 92, 280–284. doi: 10.1073/pnas.92.1.280
Wendel, J. F., Schnabel, A., Seelanan, T. (1995b). An unusual ribosomal DNA-sequence from Gossypium gossypioides reveals ancient, cryptic, intergenomic introgression. Mol. Phylogenet. Evol. 4, 298–313. doi: 10.1006/mpev.1995.1027
Wendel, J. F., Lisch, D., Hu, G. J., Mason, A. S. (2018). The long and short of doubling down: polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 49, 1–7. doi: 10.1016/j.gde.2018.01.004
Wendel, J. F. (1989). New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 86, 4132–4136. doi: 10.1073/pnas.86.11.4132
Wendel, J. F. (2015). The wondrous cycles of polyploidy in plants. Am. J. Bot. 102, 1753–1756. doi: 10.3732/ajb.1500320
West, C., James, S. A., Davey, R. P., Dicks, J., Roberts, I. N. (2014). Ribosomal DNA sequence heterogeneity reflects intraspecies phylogenies and predicts genome structure in two contrasting yeast species. Syst. Biol. 63, 543–554. doi: 10.1093/sysbio/syu019
Yoo, M. J., Liu, X. X., Pires, J. C., Soltis, P. S., Soltis, D. E. (2014). Nonadditive gene expression in polyploids. Annu. Rev. Genet. 48, 485–517. doi: 10.1146/annurev-genet-120213-092159
Zhu, X. Y., Cai, D. T., Ding, Y. (2008). Molecular and cytological characterization of 5S rDNA in Oryza species: genomic organization and phylogenetic implications. Genome 51, 332–340. doi: 10.1139/G08-016
Zonneveld, B. J. (2010). New record holders for maximum genome size in eudicots and monocots. J. Bot. article ID 527357. (Hindawi publishing corporation). doi: 10.1155/2010/527357
Keywords: 5S rRNA genes, allopolyploidy, hybridization, evolution, graph structure clustering, high-throughput sequencing, repeatome
Citation: Garcia S, Wendel JF, Borowska-Zuchowska N, Aïnouche M, Kuderova A and Kovarik A (2020) The Utility of Graph Clustering of 5S Ribosomal DNA Homoeologs in Plant Allopolyploids, Homoploid Hybrids, and Cryptic Introgressants. Front. Plant Sci. 11:41. doi: 10.3389/fpls.2020.00041
Received: 17 October 2019; Accepted: 13 January 2020;
Published: 10 February 2020.
Edited by:
Hanna Weiss-Schneeweiss, University of Vienna, AustriaReviewed by:
Tony Heitkam, Dresden University of Technology, GermanyJelena Mlinarec, University of Zagreb, Croatia
Copyright © 2020 Garcia, Wendel, Borowska-Zuchowska, Aïnouche, Kuderova and Kovarik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ales Kovarik, a292YXJpa0BpYnAuY3o=