
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 06 December 2019
Sec. Plant Breeding
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.01607
This article is part of the Research Topic Wild Plants as Source of New Crops View all 23 articles
 Yu Takahashi1
Yu Takahashi1 Hiroaki Sakai2Yuki Yoshitsu3Chiaki Muto1Toyoaki Anai4Muthaiyan Pandiyan5Natesan Senthil6Norihiko Tomooka1
Hiroaki Sakai2Yuki Yoshitsu3Chiaki Muto1Toyoaki Anai4Muthaiyan Pandiyan5Natesan Senthil6Norihiko Tomooka1 Ken Naito1*
Ken Naito1*Though crossing wild relatives to modern cultivars is a usual means to introduce alleles of stress tolerance, an alternative is de novo domesticating wild species that are already tolerant to various kinds of stresses. As a test case, we chose Vigna stipulacea Kuntze, which has fast growth, short vegetative stage, and broad resistance to pests and diseases. We developed an ethyl methanesulfonate–mutagenized population and obtained three mutants with reduced seed dormancy and one with reduced pod shattering. We crossed one of the mutants of less seed dormancy to the wild type and confirmed that the phenotype was inherited in a Mendelian manner. De novo assembly of V. stipulacea genome, and the following resequencing of the F2 progenies successfully identified a Single Nucleotide Polymorphism (SNP) associated with seed dormancy. By crossing and pyramiding the mutant phenotypes, we will be able to turn V. stipulacea into a crop which is yet primitive but can be cultivated without pesticides.
To feed the growing population in the world, we have to produce more food with less input. This is a challenging issue because of global climate change, limited water resource, and acquired resistance of pests and diseases against chemicals.
To achieve this, many scientists are now focusing on harnessing genetic diversity of genebank accessions including wild crop relatives and neglected crops (McCouch et al., 2013). One of the limitations to the above described issue is genetic vulnerability of modern cultivars. They have gone through strong bottleneck and often sacrificed resilience to biotic and abiotic stresses (Pingali, 2012). On the other hand, many wild species are well-adapted to ecological niche, which is often harsh to domesticated species (Zhang et al., 2018). Thus, utilizing the adaptability of such wild or semi-wild species will be a key to sustainable agriculture.
To fully exploit the genetic diversity of wild species, “de novo domestication” or “redomestication” are now proposed (Fernie and Yan, 2019). Until recently, the main idea of using wild genetic resources were to cross with cultivars to introduce resistant alleles. However, cross compatibility is often limited even between a cultivar and its close relatives. In addition, adaptation to a certain environment is often a complex trait with multiple genes involved. On the other hand, domestication-related traits often arose with loss-of-function mutations in single loci (Doebley et al., 2006). Thus, it might be easier to introduce domestication-related mutations into wild species than to introduce adaptation-related alleles into crops.
Although the technologies of sequencing and editing genomes are expected to facilitate de novo domestication (Shapter et al., 2013; Li et al., 2018), we still believe that simple mutagenesis + forward phenotype screening could be the easiest way to do it. To apply CRISPR/Cas9 system, one has to sequence the whole genome and develop transformation system of the plant to be domesticated. In addition, genes involved in domestication-related traits are not well-catalogued except Solanaceae, Brassicaceae, and Poaceae (Østerberg et al., 2017). As such, mutagenesis followed by forward screening is currently the only practical approach to de novo domesticate most of the potentially useful wild plants, such as wild legumes.
Thus, in this study, we tried to domesticate Vigna stipulacea Kuntze by ethyl methanesulfonate (EMS) mutagenesis followed by phenotype screening. V. stipulacea inhabits mainly in South Asia, and has fast growth, short vegetative stage, and resistance to pests and diseases (Tomooka et al., 2014). The seeds are edible, and some local people cultivate it mainly as pasture but sometimes as food. However, less and less farmers use it because of the high labor caused by strong behavior of pod shattering (Tomooka et al., 2011). In addition, it retains seed dormancy, which needs to be reduced for uniform germination. To improve these traits, we screened and obtained mutants with reduced pod-shattering and with reduced seed dormancy. We also identified a SNP associated with seed dormancy in one of the obtained mutants by whole genome analyses.
Table 1 summarizes the materials tested in this study. Besides V. stipulacea, we used two or three accessions of three domesticated species, soybean [Glycine max (L.) Merr.], common bean (Phaseolus vulgaris L.), and cowpea [Vigna unguiculata (L.) Walp.], to evaluate pod shattering and seed dormancy. All the materials were provided by the NARO gene bank (https://www.gene.affrc.go.jp/index_en.php). We cultivated V. stipulacea in a field or in a bucket with gardening soil in a greenhouse of our institute, Tsukuba, Japan (36.030577, 140.098021). We cultivated three plants of the domesticated species in a bucket with gardening soil in a greenhouse of our institute, Tsukuba, Japan.
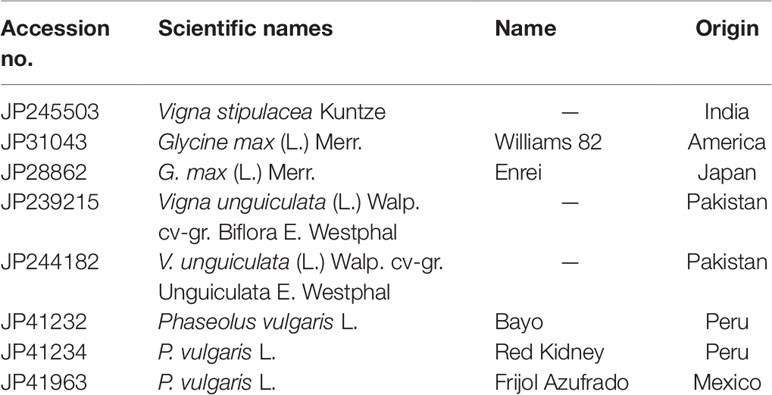
Table 1 Plant materials.
The process of mutant screening is shown in Figure 1. To increase mutation density, we adopted two cycles of successive chemical mutagenesis as described by Tsuda et al. (2015). We started with 12,000 seeds of V. stipulacea (JP245503). We first scratched the seed coat with a knife because it is hard and waterproof, treated the scratched seeds with 0.35% EMS solution for 8 h, and thoroughly washed them with distilled water (M1 seeds). The M1 seeds were incubated in a wet condition for 3 days and those with germination were sown. The M1 plants were cultivated in our greenhouse, where the temperature was kept above >20°C. Three pods per plant were harvested from 3,000 fertile M1 plants (M2 seeds). Of the harvested seeds, four per line (12,000 in total) were scratched and treated with 0.35% EMS solution as described above (M2M1 seeds). Germinating M2M1 seeds were sown and cultivated in a field, located in Tsukuba, Japan. Three thousand fertile plants were selected and three pods per plant were harvested (M2M2 seeds). Six seeds per M2M2 line were sown in a 1 L plastic pot filled with gardening soil and cultivated in the greenhouse until three pods per line were harvested (M2M3 seeds). Of the M2M3 lines, four lines with mutant phenotypes were selected and seven seeds per line were sown in a 7 L bucket filled with the gardening soil and cultivated in the greenhouse.

Figure 1 Schematic of domesticating V. stipulacea. After two rounds of EMS treatment, 3,000 M2M1 plants were selected to collect M2M2 seeds (six per line). The 18,000 M2M2 plants were grown in a field, where pod shattering mutants were screened for. Three pods (∼20 seeds) per plant was harvested, which were tested for seed-imbibition screening.
To evaluate pod shattering, we harvested 20 pods of each domesticated accession and three pods per M2M2 plant and counted the number of shattered pods. Before evaluation, harvested pods were left at room temperature for a month and then completely dried at 40°C for 24 h in an incubator.
To evaluate seed dormancy, 20 seeds were soaked in distilled water and incubated in a dark incubator at 25°C, and imbibed seeds were counted daily until the 3rd day, twice a week until the 14th day, and weekly until the 28th day. We replicated the measurements by seven times and performed Dunnett test to compare averages of each time point between the wild type and each mutant line.
We observed the imbibing seeds to detect the imbibition start point. We also observed pod sclerenchyma (tissue with dead cells with thickened secondary wall) with a stereoscopic microscope (ECLIPSE Ci-L, Nikon, Tokyo, Japan). Three pods were collected from each individual and were sliced by a microtome (MTH-1, Nippon Medial & Chemical Instruments Co., Ltd. Osaka, Japan), and stained with Phloroglucinol-HCL solution (1 g phloroglucin in 50 mL ethanol + 25 mL concentrated hydrochloric acid).
We sequenced the whole genome of V. stipulacea (JP245503) with RSII sequencer (Pacific Biosciences, Menlo Park, CA), as we have done previously for azuki bean (Sakai et al., 2015). DNA was isolated from 1 g of unexpanded leaves with CTAB method and purified with Genomic Tip 20/G (Qiagen K. K. Tokyo). The extracted DNA was sheared into 20 kb fragments using g-TUBE (Covaris, MA, USA) and converted into 20 kb SMRTbell template libraries. The library was size-selected for a lower cutoff of 10 kb with BluePippin (Sage Science, MA, USA). Sequencing was performed on the PacBio RS II using P5 polymerase binding and C3 sequencing kits with 360 min acquisition. In total, 52 SMRT cells were used to obtain ∼19.6 Gb of subreads.
In total, ∼3.3 million PacBio reads were used for de novo assembly with Celera Assembler 8.3rc1 (asmOvlErrorRate = 0.1, asmUtgErrorRate = 0.06, asmCgwErrorRate = 0.1, asmCnsErrorRate = 0.1, asmObtErrorRate = 0.08, utgGraphErrorRate = 0.05, utgMergeErrorRate = 0.05) (Berlin et al., 2015). About 25.2x of the longest error-corrected and trimmed reads were assembled to contigs. Redundant contigs were discarded by conducting all-to-all BLASTN searches. The non-redundant contigs were polished by PacBio subreads by using Quiver in SMRT Analysis v2.2.0 (Pacific Biosciences of California, Inc.) and then further polished by Illumina short-reads using BWA 0.7.9a (Li and Durbin, 2009), Samtools 0.1.19 (Li et al., 2009), Picard 1.94 (http://picard.sourceforge.net/), and GATK 3.3 (McKenna et al., 2010). The polished contigs were scaffolded by Reference-Assisted Chromosome Assembly (RACA) program v.0.9.1.1 (Kim et al., 2013b) using the genome sequences of Vigna angularis and P. vulgaris as the reference and outgroup species, respectively.
Repetitive sequences in the genome assembly were predicted using Censor (Kohany et al., 2006) with a composite library consisting of de novo created library constructed by RepeatModeler 1.0.8 (http://www.repeatmasker.org) and the MIPS Repeat Element Database ver. 9.3 (Nussbaumer et al., 2013).
Ab initio gene prediction was done by BRAKER version 1.6 (Hoff et al., 2016) with RNA-Seq data. Besides, gene structures were predicted by genome-guided and de novo RNA-Seq data assembly approaches using TopHat 2.1.0 (Kim et al., 2013a), Cufflinks 2.2.1 (Trapnell et al., 2010), Trinity 2.1.1 (Grabherr et al., 2011), and PASA pipeline 2.0.2 (Haas et al., 2003; Rhind et al., 2011). Open reading frames were predicted by Transdecoder 2.0.1 and Trinotate 2.0.2 (Bryant et al., 2017). Protein sequences of the G. max (Wm82.a2.v1), P. vulgaris (v.1.0), Medicago truncatula Gaertn. (Mt4.0v1), and V. angularis (Willd.) Ohwi & H.Ohashi (VANGULARIS_V1.A1) were downloaded from Phytozome (G. max, P. vulgaris, and M. truncatula) (Goodstein et al., 2012) and VigGS (V. angularis) (Sakai et al., 2016) and mapped to the genome assembly by Exonerate 2.2.0 (Slater and Birney, 2005). The ab initio gene models and transcript and protein alignments were then combined by EvidenceModeler 1.1.1 (Haas et al., 2008) and the predicted gene models were updated by PASA. Gene models with extremely long introns and those merged by PASA were manually curated. BUSCO v3 (Waterhouse et al., 2017) was used to evaluate protein sequences of annotated genes.
We performed MutMap (Abe et al., 2012) to isolate the candidate gene for the mutant phenotype. We crossed one of the mutants of increased seed imbibition (isi1) to the wild type (JP245503) and obtained 280 F2 seeds. We cultivated all of them, extracted DNA from an unexpanded leaf of each F2 plants by CTAB method, and obtained F3 seeds. To evaluate seed dormancy, 20 seeds of each F3 line was soaked with distilled water for 24 h to test imbibition. DNA of 59 F2 plants with mutant phenotype (100% seed imbibition) and DNA of 221 F2 plants with wild type phenotype (0% imbibition) were pooled to make the MT pool and the WT pool, respectively, and were sequenced with Illumina HiSeq X (Illumina Co. Ltd, San Diego, USA).
The obtained sequences were mapped to the draft genome sequences with bwa-0.7.17 (Li and Durbin 2009) and formatted with samtools-1.9 (Li et al., 2009) as described in the distributed manuals. SNPs were called with bcftools-1.9, and the DP4 values were used to calculate the SNP-index. If a SNP site is responsible for the mutant phenotype, the locus must be fixed with the alternative allele [a] in the MT pool ([aa] shares 100%). On the other hand, in the WT pool, the expected ratio of the reference allele [A] and the alternative [a] is 2:1, because the WT pool [AA]:[Aa]:[aa] should be 1:2:0. Thus, at the responsible locus, the expected values of SNP-index are 1.0 in the MT pool and 0.33 in the WT pool. Thus, after calculating SNP-index, we screened all the SNP sites for those that met the criterion above.
We genotyped 280 F2 plants by Sanger sequencing for two SNPs whose SNP-index was 1.0 in the MutMap analysis. Two primer pairs were designed to amplify a 232 bp genomic region including 6,804,429 nt (5′-gagggaatacgaagagtttaaggtt-3′ and 5′-ttgaaaaccaggtcttttctctcta-3′) and to amplify a 243 bp genomic region including 7,009,873 nt (5′-acagagcaaaagattaaacgagaga-3′ and 5′-aaagccgcttcctagtccttac-3′) on scf0015. For Sanger sequencing, we amplified the template DNA with AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific K. K., Tokyo), performed sequencing reaction with BigDye Terminator v3.1 (Thermo Fisher Scientific K. K., Tokyo), and sequenced with ABI Genetic Analyzer 3130xl (Thermo Fisher Scientific K. K., Tokyo), according to the provider’s protocol.
Though many studies have investigated seed dormancy in legume crops, there are still some arguments on the sites of water entry (Smýkal et al., 2014). Thus, we evaluated seed imbibition and pod shattering in soybean, common bean, and cowpea.
As a result, domesticated species showed variation in both imbibition start sites and time to imbibe (Figure 2). Among the domesticated accessions tested, soybean imbibed within a few minutes and was the quickest. In soybean, the whole testa seemed permeable. The second quickest was the cowpea “JP244182,” where the testa was partially permeable and imbibition was observed in an hour. The common bean “JP41232” showed imbibition in 2 to 3 h, which started at the site of micropyle. It took 4 to 5 h for the common bean “JP41234” and the cowpea “JP239215,” where imbibition started at the site of lens. All the seeds were fully imbibed within 48 h.
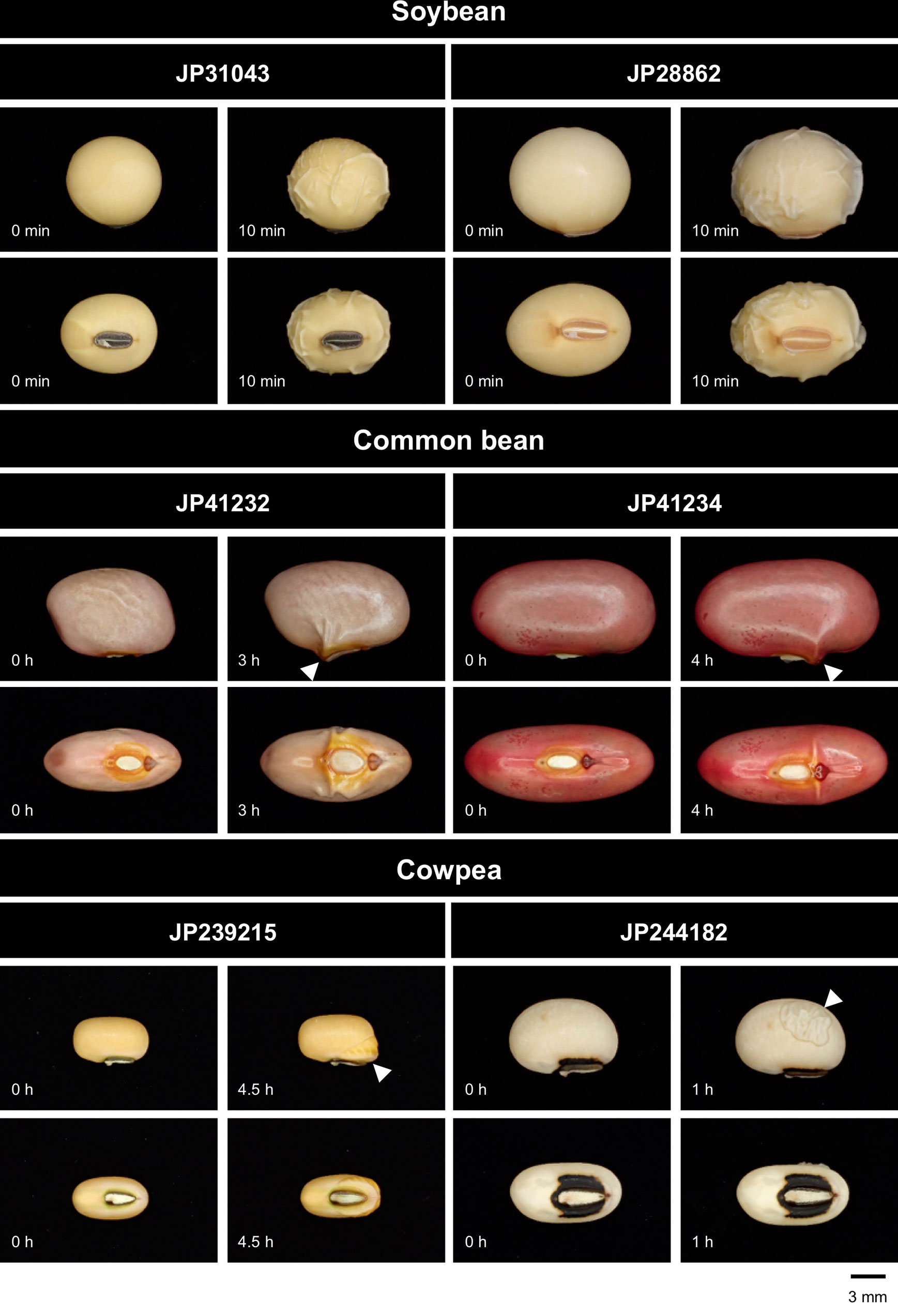
Figure 2 Water entry sites in domesticated legumes. The entry sites (indicated by arrowheads) varied across accessions, but did not vary within accessions.
We also observed abscission layers between the valves of seed pods and the sclerenchyma on the endocarp, because pod shattering is dependent on these tissues (Murgia et al., 2017). In soybean, as described by Dong et al. (2014), abscission layer was not completely formed at the fiber cap cells (Figure 3). Abscission layer was even less completely formed in the common bean accession JP41963 whereas it was fully formed in all other accessions (Figure 3). Pod sclerenchyma was thicker in the soybean accessions and the cowpea accession JP244182, whereas it was thinner in common bean accessions (Figure 3).
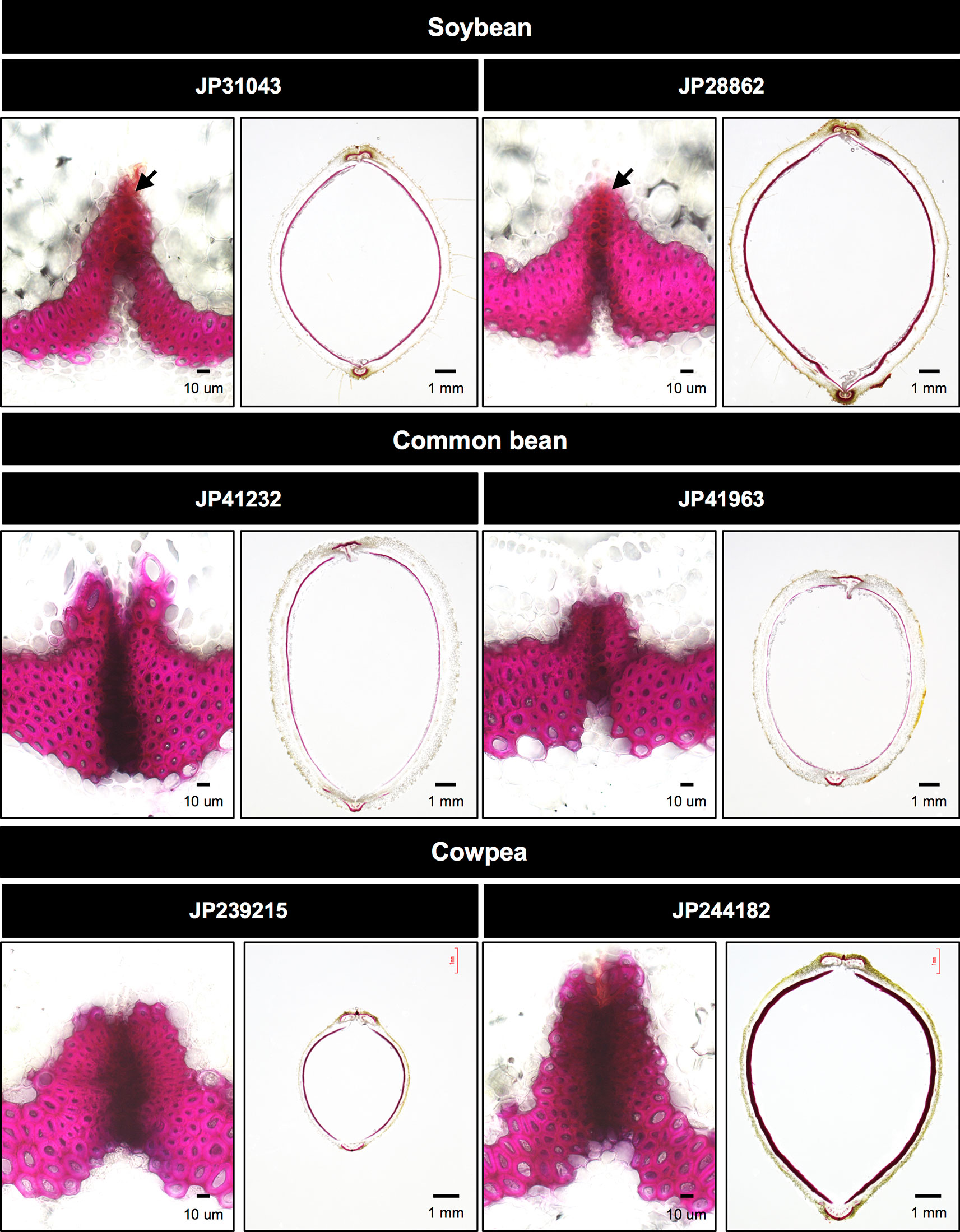
Figure 3 Cross-sections of seed pods in domesticated accessions. The left panel of each accession is a close-up of abscission zone. Phloroglucinol-HCl stains pod sclerenchyma with bright red while it stains abscission zone with dark red. Soybean accessions have brightly-stained sites at the tip of fiber cap cells (arrows), which indicates abscission layer is not completely formed.
To obtain mutants with reduced seed dormancy and reduced pod shattering, we treated 12,000 seeds of V. stipulacea with EMS and developed 3,000 M2M2 plants. Of the 3,000, we found one line which exhibited reduced pod shattering and designated it as reduced pod shattering1 (rps1) (Figure 4). We also screened M2M3 seeds for seed imbibition and obtained three lines which exhibited increased seed imbibition and designated them as increased seed imbibition1 (isi1), increased seed imbibition2 (isi2), and increased seed imbibition3 (isi3), respectively (Figure 5). Of them, the isi1 seeds had cracks in the hilum, and the isi3 seeds showed reduced pigmentation in the seed coat (Figure 5).
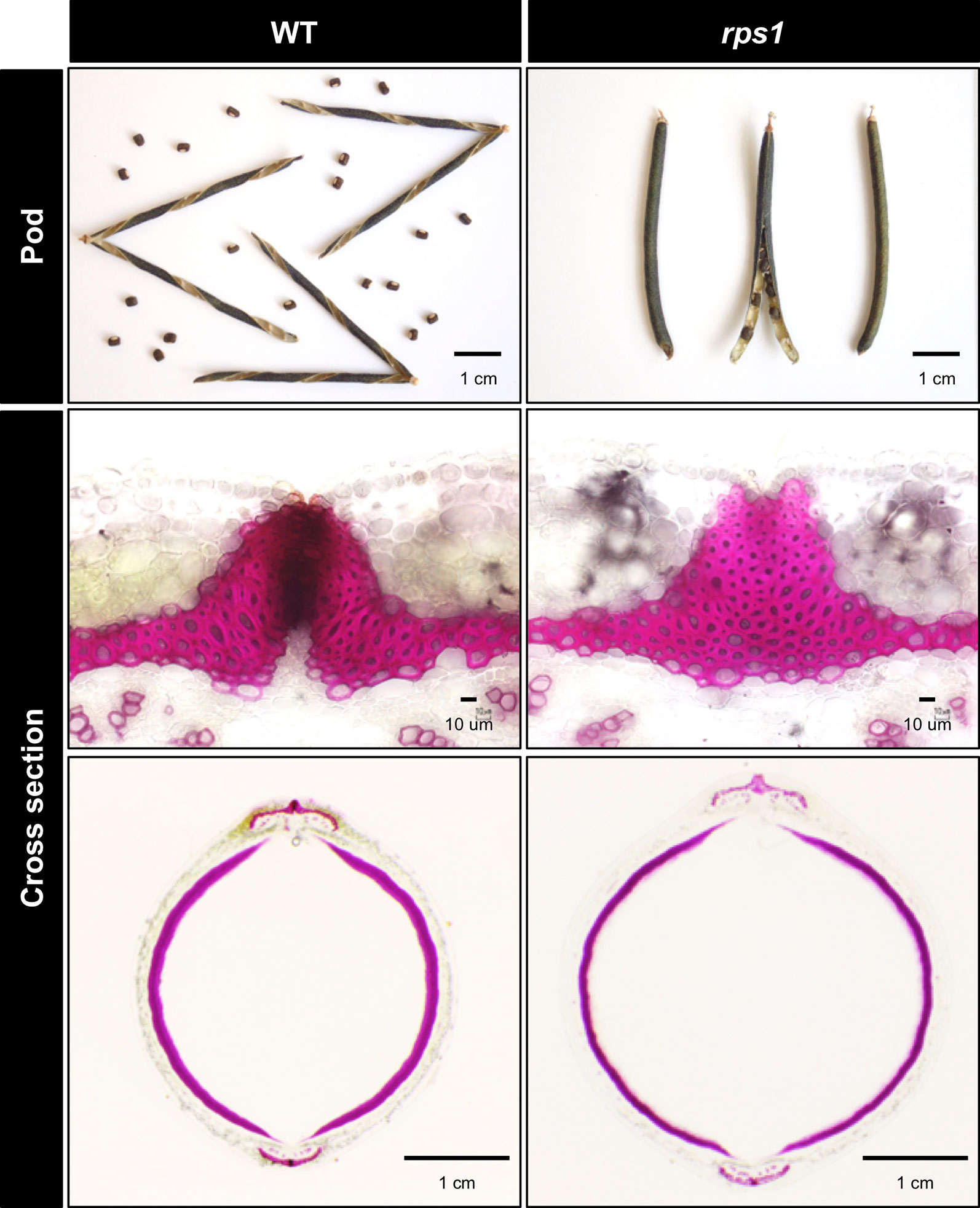
Figure 4 Non-shattering mutant of V. stipulacea. Pod shattering was strongly suppressed in the rps1 mutant, where the abscission layer between the valves were not formed at all, whereas the thickness of sclerenchyma did not greatly change.
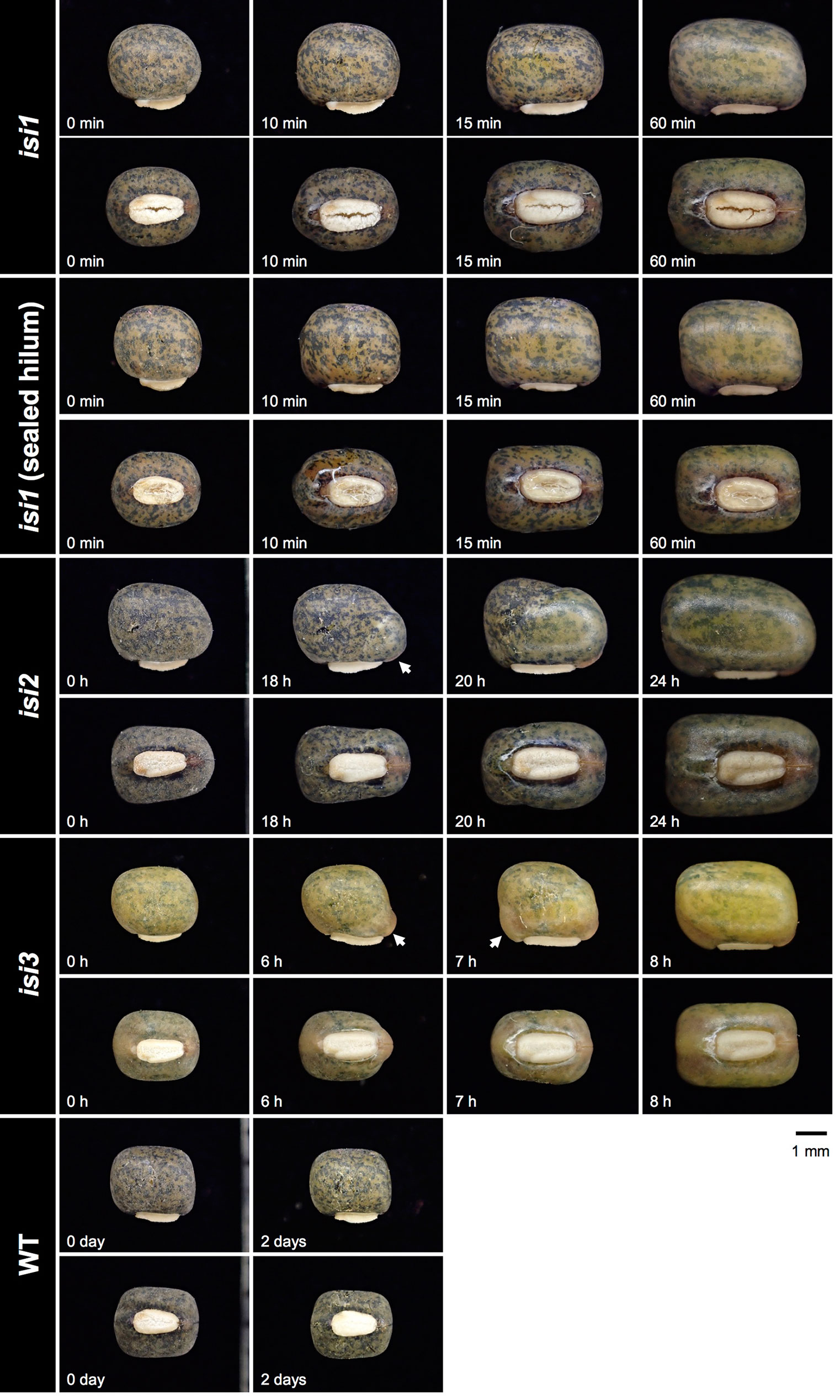
Figure 5 Increased seed imbibition mutants of V. stipulacea. Time after watering is indicated at the bottom-left of each photo. Arrows indicate where water entry was initiated.
The mode of imbibition also varied across the mutants (Figure 5). In the isi1, the water entry was not necessarily through the cracks in the hilum but through the whole seed coat. Even though we sealed the hilum with glue, imbibition was quickly initiated [see the “isi1 (sealed hilum)” in Figure 5]. In isi2, the imbibition was initiated at the lens. In isi3, the imbibition started near the hilum, but not necessarily through the micropyle or the lens.
To elucidate the extent of seed dormancy in the mutant lines, we put the seeds on wet filter paper and evaluated the rate of imbibed seeds (Figure 6). We also tested the seeds of 1 and 6 months old to elucidate the effect of duration time after harvesting. When we did the experiment with the wild type seeds, the imbibition rate was zero for at least 4 weeks in 1-month-old seeds and was 11% in the 6-month-old seeds (Figure 6).
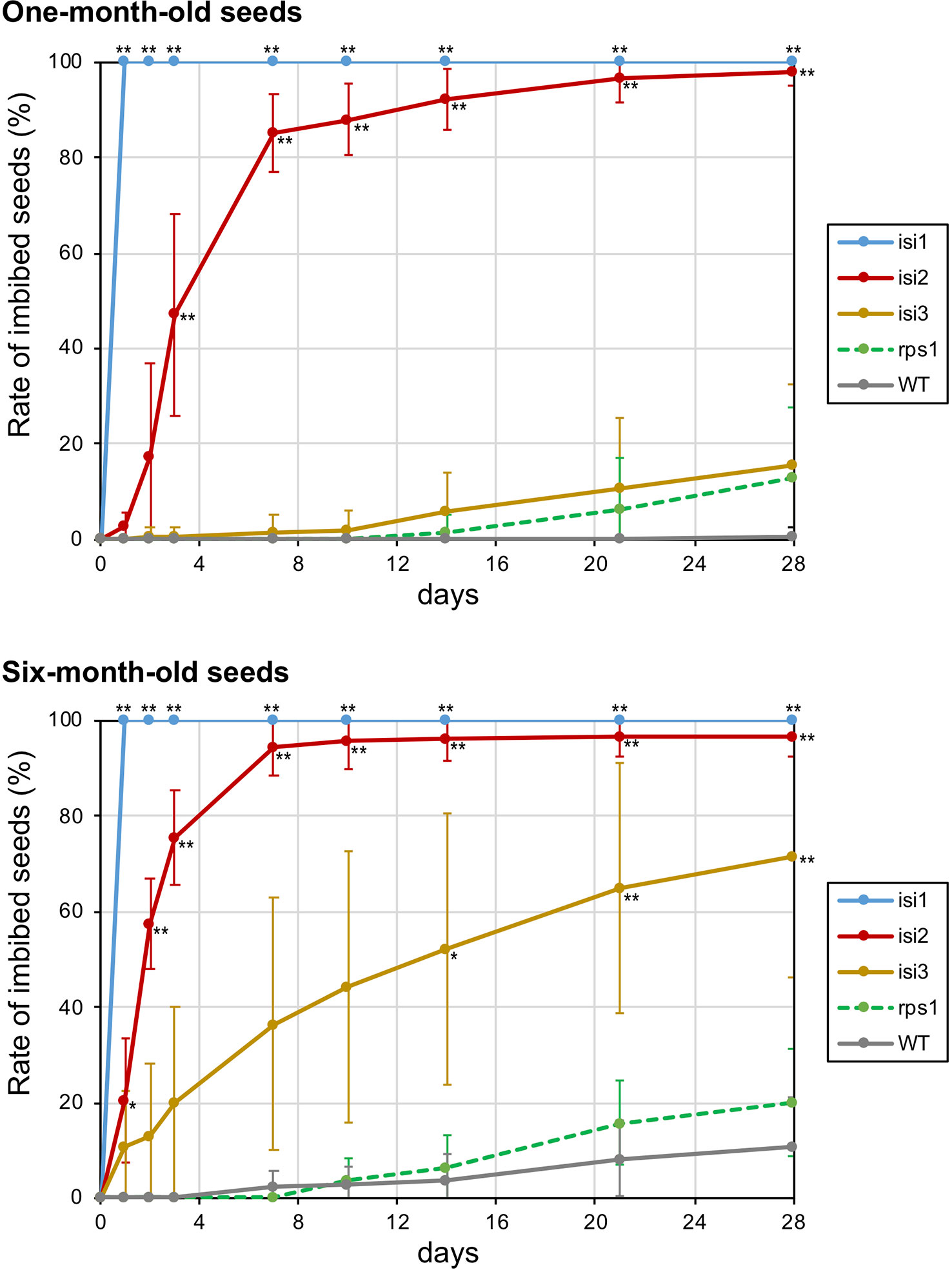
Figure 6 Rate of imbibed seeds in the mutant plants over time. Twenty seeds of 1 and 6 months old were soaked in distilled water and number of imbibed seeds were manually counted twice a week for 4 weeks. The error bars indicate standard deviation of replicated evaluations (n = 7). Asterisks indicate that the mean values are significantly different from the wild type (** for p < 0.01 and * for p < 0.05).
When we tested the mutant seeds of 1 month old, the imbibition was quick in isi1 and isi2, whereas it was slow in isi3. In isi1, all the seeds fully imbibed within 1 h (Figure 6). In isi2, 85% of the seeds imbibed within a week, and almost 100% did so within 4 weeks. In contrast, the imbibition rate of the isi3 seeds was only 16% in 4 weeks, which was not significantly different from the wild type. The imbibition rate of the reduced shattering mutant rps1 was also higher but not significantly different compared to the wild type (Figure 6).
When we tested the mutant seeds of 6 months old, the overall imbibition rate was increased compared to the experiment with 1-month-old seeds (Figure 6). In isi1, the seeds also fully imbibed within 1 h. In isi2, the imbibition rate was already significantly different from the wild type in 1 day, more than 90% of the seeds imbibed within a week, and reached plateau (∼95%) in two weeks. In isi3, 20% of the seeds imbibed in 3 days, 36% in a week, and 71% in 4 weeks. However, this line exhibited a large standard deviation between the replicates and was not significantly different before 2 weeks (Figure 6). In rps1, ∼20% of the seeds imbibed in 4 weeks, which was twice as high as the wild type but not significantly different from the wild type was not significant.
To evaluate pod shattering in the mutant lines, we calculated the rate of shattering of the harvested pods which were completely dried in the incubator. Whereas the shattering rate was 100% in the wild type, it was 0% in the rps1 mutant (Table 2). The rps1 mutant also showed a reduced twisting of the seed pod. The number of twists/cm in the pods was 0.371 ± 0.018 in the rps1 mutant, which was less than half of the wild type (0.866 ± 0.022) (Table 2).
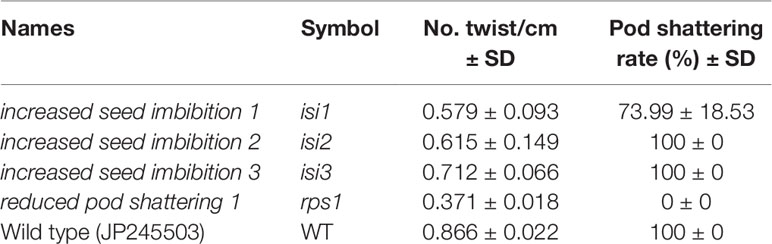
Table 2 Phenotypic data of mutant lines.
Interestingly, the isi1, one of the mutants of seed imbibition, also exhibited slightly reduced shattering rate (73.99 ± 18.53%) and number of twists/cm (0.579 ± 0.093) (Table 2). Other mutants were also slightly reduced in number of twists/cm, but their shattering rate was 100% (Table 2).
We also observed cross-sections of seed pods and found the rps1 mutant did not form abscission layer between the valves at all (Figure 4).
To build a reference sequence of V. stipulacea, we sequenced the genomic DNA with a PacBio sequencer, assembled, and annotated. We obtained 19.6 Gbp of subreads using 52 SMRT cells. Genome size of the V. stipulacea was estimated to be ∼445.1 Mbp based on the k-mer frequency distribution obtained from 10.3 Gbp of the Illumina short-reads. The assembled contigs showed N50 length of ∼1.9 Mbp and mean length of ∼169.8 kbp and covered ∼387.7 Mbp (87.9%) of the estimated genome size (Table 3). Out of the 2,102 scaffolds, 52 scaffolds were comprised of 233 contigs scaffolded by RACA.
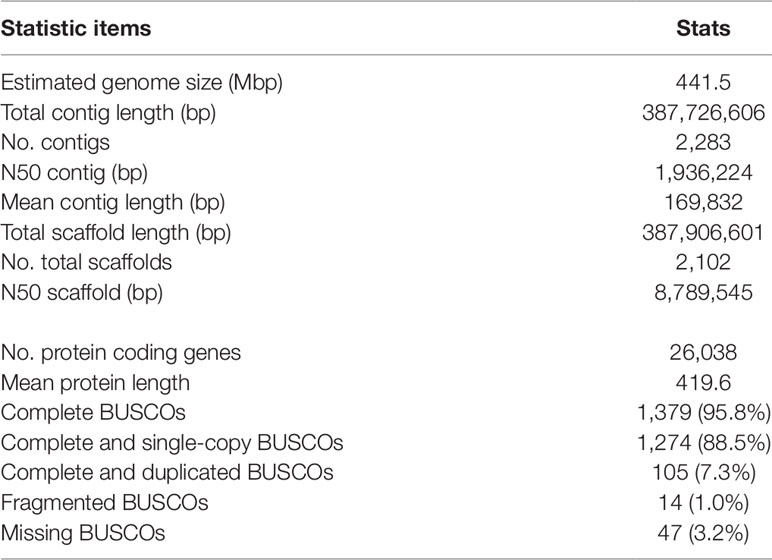
Table 3 Stats of the assembled genome sequence of V. stipulacea.
By combining the ab initio gene models and transcript and protein alignments, 26,038 protein coding genes were predicted on the assembled genome (Table 3). We assessed the completeness of the gene set by BUSCO and found that 95.8% of the BUSCO gene models were completely detected (Table 3). The BUSCO metrics was almost the same as that assessed for the recently published high quality genome sequence of cowpea (Lonardi et al., 2019), suggesting that our genome assembly covered nearly complete gene space of the V. stipulacea genome.
To identify the responsible genes for the mutant phenotype, we performed MutMap analysis. Of the 280 F2 plants derived from isi1 x WT, 221 showed the wild type phenotype and 59 showed the mutant phenotype, which was close to the expected 3:1 ratio (p = 0.13). We sequenced the genomic DNA of the wild type plants (WT pool) and the mutants (MT pool) and identified 33,936 SNPs across the whole genome. We then calculated SNP-index of each SNP and screened for those where it was 1.0 in MT pool and 0.3 ± 0.1 in WT pool.
As a result, we found only two on scf0015, a C to T substitution at 6,804,429 nt and a G to A at 7,009,873 nt. The C to T SNP was present in the 8th exon of Vigst.0015s042600.01, which encoded CELLULOSE SYNTHASES A 7 (CesA7) protein, and turned the Glu codon into a STOP codon. The G to A SNP was present in Vigst.0015s044500.01, which encoded a nucleoporin-like protein, but was synonymous.
For further mapping, we looked for recombinants between the two candidate SNPs by direct sequencing of the SNP-containing regions in all the F2 plants. As a result, one F2 plant, which showed the wild type phenotype, was heterozygous at Vigst.0015s042600.01 but was fixed with the mutant A allele at Vigst.0015s044500.01 (see #130 in Supplementary Table 1). Another F2 plant, which showed the mutant phenotype, was fixed with the alternative T allele at Vigst.0015s042600.01 but was heterozygous at Vigst.0015s044500.01 (see #251 in Supplementary Table 1). The result was further confirmed by genotyping and phenotyping F3 plants derived from the F2 plants with recombination between the two candidate SNPs (#130, #251, and #275 in Supplementary Table 1). As shown in Supplementary Table 2, the phenotype was completely linked with the SNP at scf0015_6804429 on CesA7 gene but not with the other.
In this study we demonstrated a practical approach to de novo domesticate a wild legume species V. stipulacea by chemically-induced mutations. We successfully obtained three independent mutations in seed dormancy and one in pod shattering. In addition, we identified a SNP in the candidate gene, which encoded CELLULOSE SYNTHASE A 7, in one of the three mutants of reduced seed dormancy.
We demonstrated that forward screening following mutagenesis was practical enough to screen for mutants in domestication-related traits. This could be easily predicted from the fact that legume crops have reduced seed dormancy and pod shattering in various manners (Figures 2 and 3). This indicates that many genetic loci are involved in these traits, and loss-of-function mutation in any of them could cause such phenotypes. This is why we identified three mutants for seed dormancy and one for pod shattering from only 3,000 M2M2 lines (18,000 plants) (Figure 1). We also note that we obtained the mutants within 3 years since we initiated the first mutagenesis.
Of the three mutants for seed dormancy, isi1 showed the severest phenotype and imbibition was always completed within hours (Figures 5 and 6). This was because of water-permeability in the seed coat, which were probably caused by the loss-of-function mutation in CesA7. CesA7 is involved in cellulose synthesis and its malfunction might have disrupted development of hilum and seed coat. The phenotype of cracked hilum had not been observed in other legume crops and makes the seed almost completely non-dormant. However, the seeds might not be suitable for long-term storage, because the cotyledons are exposed to the air and could be easily oxidized. In addition, isi1 showed slightly lower rate of pod shattering. This could be a pleiotropic effect of the mutation, because cellulose fibers are also important for pod shattering (Suanum et al., 2016). However, we cannot exclude the possibility that isi1 contains other mutations involved in pod shattering.
Compared to isi1, isi2, and isi3 showed milder phenotypes (Figure 6). Because isi2 seeds imbibe through lens, the mutation disturbed the same pathway as in the common bean “JP41234” or the cowpea “JP239215” (Figure 2). However, it took much longer time for isi2 to complete imbibition than for those cultivars. This might be because the domesticated accessions have accumulated multiple mutations involved in seed coat permeability (Jang et al., 2015; Sun et al., 2015). As for isi3, the seed coat color was retarded but imbibition did not always initiate in the seed coat. It sometimes did through the lens and sometimes through the micropyle. Given the water entry sites are basically stable in the cultivated accessions, the isi3 mutant might be a novel phenotype of seed dormancy. Or, it is also possible that isi3 have multiple mutations involved in seed imbibition, given we are currently aware of only the seed color in this mutant. If so, such mutant alleles (except the one involved in seed pigmentation) could be segregating and lead to variation of water entry sites and imbibition rate within the mutant line (Figure 6). Though isi2 and isi3 have only partial effect on seed imbibition, double-mutant of both might be able to complete imbibition within a week with smaller effect on long-term storability.
The rps1 mutant almost completely lost the pod shattering behavior because of suppressed formation of the abscission layer between the valves (Figure 4). This phenotype was similar to the effect of the domestication-type allele of SHAT1-5 in soybean (Figure 2) (Dong et al., 2014). Thus, the mutation in rps1 might be in a gene involved in the SHAT1-5 pathway. The responsible gene for rps1 phenotype might be useful for improving shattering problem in other legumes because rps1 phenotype was severer than soybean SHAT1-5. On the other hand, however, severe disruption in development of abscission layer could increase labor to thresh. In addition, though not significant, we repeatedly observed that the rps1 mutant exhibited slightly increased seed imbibition compared to the wild type (Figure 6). Such pleiotropy, unless it has other mutations involved in seed dormancy, might be because secondary wall thickening plays important roles in shattering behavior in seed pod (Suanum et al., 2016; Parker et al., 2019; Rau et al., 2019; Takahashi et al., 2019) and water permeability in seed coat (Smýkal et al., 2014; Hradilová et al., 2019).
Last but not least, the technology of long-read sequencing (Eid et al., 2009) enabled us to obtain high-quality genome assembly of V. stipulacea. Although we did not use further scaffolding techniques such as optical mapping (Lam et al., 2012) or Hi-C (Burton et al., 2013), it achieved the N50 of nearly 2 Mbp and the BUSCO score of 96.8 (Table 3). The high-quality reference genome should be the reason for that we successfully identified the candidate SNP by a single resequencing analysis.
As described above, here we report that we have achieved the first step of domesticating the wild legume V. stipulacea. As reported in Tomooka et al. (2011), this legume showed resistance to broad range of pests and diseases in Tamil Nadu, India, and does not need pesticides to spray. Broad range of pests and diseases resistance of V. stipulacea was confirmed during the mutant screening procedure conducted in our experimental field in Tsukuba, Japan. Thus, if we cross and pyramid the mutant phenotypes obtained in this study, we will be able to make V. stipulacea easier to cultivate and easier to harvest, and we believe it will be a step forward to accomplish low input agriculture. By screening for more useful traits from more mutagenized population, we hope this kind of “domesticated wild plants” will be popular and prevalent.
The raw and assembled sequence data (DRA009127) are all available from DNA Data Bank of Japan (https://www.ddbj.nig.ac.jp/dra/index.html) or VigGS (https://viggs.dna.affrc.go.jp/).
NT and KN provided the idea of the study. YT, MP, NS, NT and KN planned the study. YT, CM, TA, NT and KN cultivated plants and collected data. YT, YY, HS and KN analyzed data. YT, HS, MP, NS, NT and KN wrote the paper.
This study was supported by JSPS KAKENHI Grant Number 13J09808, 19KT0016, and 26850006. It was also partially supported by Research Supporting Program of the Advanced Analysis Center, National Agriculture and Food Research Organization (NARO) and the Genebank Project, NARO.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful for Ms. Motoyoshi, Ms. Asano and Ms. Yamamoto for taking good care of plant materials. We also appreciate for the support provided by Research Supporting Program of the Advanced Analysis Center, National Agriculture and Food Research Organization (NARO) and the Genebank Project, NARO.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01607/full#supplementary-material
Østerberg, J. T., Xiang, W., Olsen, L. I., Edenbrandt, A. K., Vedel, S. E., Chistiansen, A., et al. (2017). Accelerating the domestication of new crops. Trends Plant Sci. 22, 373–384. doi: 10.1016/j.tplants.2017.01.004
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt2095
Berlin, K., Koren, S., Chin, C. S., Drake, J. P., Landolin, J. M., Phillippy, A. M. (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630. doi: 10.1038/nbt3238
Bryant, D. M., Johnson, K., Ditommaso, T., Tickle, T., Couger, M. B., Payzin-Dogru, D., et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776. doi: 10.1016/j.celrep.2016.12.063
Burton, J. N., Ady, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt2727
Doebley, J. F., Gaut, B. S., Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Dong, Y., Yang, X., Liu, J., Wang, B. H., Liu, B. L., Wang, Y. Z. (2014). Pod shattering resistance associated with domestication is mediated by NAC gene in soybean. Nat. Commun. 5, 3352. doi: 10.1038/ncomms4352
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Fernie, A. R., Yan, J. (2019). De novo domestication: an alternative route toward new crops for the future. Mol. Plant 12, 615–631. doi: 10.1016/j.molp.2019.03.016
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt1883
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M., Stanke, M. (2016). BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769. doi: 10.1093/bioinformatics/btv661
Hradilová, I., Duchoslav, M., Brus, J., Pechanec, V., Hýbl, M., Kopecký, P., et al. (2019). Variation in wild pea (Pisum sativum subsp. elatius) seed dormancy and its relationship to the environment and seed coat traits. PeerJ 7, e6263. doi: 10.7717/peerj6263
Jang, S. J., Sato, M., Sato, K., Jitsuyama, Y., Fujino, K., Mori, H., et al. (2015). A Single-Nucleotide Polymorphism in an Endo-1,4-β-Glucanase Gene Controls Seed Coat Permeability in Soybean. PloS One 10, e0128527. doi: 10.1371/journal.pone.0128527
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., Salzberg, S. L. (2013a). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1186/gb-2013-14-4-r36
Kim, J., Larkin, D. M., Cai, Q., Asan, Zhang, Y., Ge, R. L., et al. (2013b). Reference-assisted chromosome assembly. Proc. Natl. Acad. Sci. U.S.A. 110, 1785–1790. doi: 10.1073/pnas.1220349110
Kohany, O., Gentles, A. J., Hankus, L., Jurka, J. (2006). Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinf. 7, 474. doi: 10.1186/1471-2105-7-474
Lam, E. T., Hastie, A., Lin, C., Ehrlich, D., Das, S. K., Austin, M. D., et al. (2012). Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30, 771–776. doi: dio: 10.1038/nbt.2303
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, T., Yang, X., Yu, Y., Si, X., Zhai, X., Zhang, H., et al. (2018). Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 36, 1160–1163. doi: 10.1038/nbt4273
Lonardi, S., Munoz-Amatriain, M., Liang, Q., Shu, S., Wanamaker, S. I., Lo, S., et al. (2019). The genome of cowpea (Vigna unguiculata [L.] Walp.). Plant J. 98, 767–782. doi: 10.1111/tpj.14349
McCouch, S., Baute, G. J., Bradeen, J., Bramel, P., Bretting, P. K., Buckler, E., et al. (2013). Agriculture: Feeding the future. Nature 499, 23–24. doi: 10.1038/499023a
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Murgia, M. L., Attene, G., Rodriguez, M., Bitocchi, E., Bellucci, E., et al. (2017). A comprehensive phenotypic investigation of the "pod-shattering syndrome" in common bean. Front. Plant Sci. 8, 251. doi: 10.3389/fpls.2017.00251
Nussbaumer, T., Martis, M. M., Roessner, S. K., Pfeifer, M., Bader, K. C., Sharma, S., et al. (2013). MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res. 41, D1144–D1151. doi: 10.1093/nar/gks1153
Parker, T. A., Mier y Teran, J. C. B., Palkovic, A., Jernstedt, J., Gepts, P. (2019). Genetic control of pod dehiscence in domesticated common bean: associations with range expansion and local aridity conditions. biorxiv, 517516. doi: 10.1101/517516
Pingali, P. L. (2012). Green revolution: impacts, limits and the path ahead. Proc. Natl. Acad. Sci. U.S.A. 109, 12302–12308. doi: 10.1073/pnas.0912953109
Rau, D., Murgia, M. L., Rodriguez, M., Bitocchi, E., Bellucci, E., Fois, D., et al. (2019). Genomic dissection of pod shattering in common bean: mutations at non-orthologous loci at the basis of convergent phenotypic evolution under domestication of leguminous species. Plant J. 97, 693–714. doi: 10.1111/tpj.14155
Rhind, N., Chen, Z., Yassour, M., Thompson, D. A., Haas, B. J., Habib, N., et al. (2011). Comparative functional genomics of the fission yeasts. Science 332, 930–936. doi: 10.1126/science.1203357
Sakai, H., Naito, K., Ogiso-Tanaka, E., Takahashi, Y., Iseki, K., Muto, C., et al. (2015). The power of single molecule real-time sequencing technology in the de novo assembly of a eukaryotic genome. Sci. Rep. 5, 16780. doi: 10.1038/srep16780
Sakai, H., Naito, K., Takahashi, Y., Sato, T., Yamamoto, T., Muto, et al. (2016). The Vigna Genome Server, ‘VigGS’: a genomic knowledge base of the genus vigna based on high-quality, annotated genome sequence of the azuki bean, vigna angularis (Willd) Ohwi & Ohashi. Plant Cell Physiol. 57, e2. doi: 10.1093/pcp/pcv189
Shapter, F. M., Cross, M., Ablett, G., Malory, S., Chivers, I. H., King, G. J., et al. (2013). High-throughput sequencing and mutagenesis to accelerate the domestication of Microlaena stipoides as a new food crop. PloS One 8, e82641. doi: 10.1371/journal.pone.0082641
Slater, G. S., Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinf. 6, 31. doi: 10.1186/1471-2105-6-31
Smýkal, P., Vernoud, V., Blair, M. W., Soukup, A., Thompson, R. D. (2014). The role of the testa during development and in establishment of dormancy of the legume seed. Front. Plant Sci. 5, 351. doi: 10.3389/fpls.2014.00351
Suanum, W., Somta, P., Kongjaimun, A., Yimram, T., Kaga, A., Tomooka, N., et al. (2016). Co-localization of QTLs for pod fiber content and pod shattering in F2 and backcross populations between yardlong bean and wild cowpea. Mol. Breed. 36, 80. doi: 10.1007/s11032-016-0505-8
Sun, L., Miao, Z., Cai, C., Zhang, D., Zhao, M., Wu, Y., et al. (2015). GmHs1-1, encoding a calcineurin-like protein, controls hard-seededness in soybean. Nat. Genet. 47, 939–943. doi: 10.1038/ng3339
Takahashi, Y., Kongjaimun, A., Muto, C., Kobayashi, Y., Kumagai, M., Sakai, H., et al. (2019) Genetic factor for twisting legume pods identified by fine-mapping of shattering-related traits in azuki bean and yard-long bean. bioRxiv doi: 10.1101/774844
Tomooka, N., Pandiyan, M., Senthil, N. (2011). Conservation of leguminous crops and their wild relatives in Tamil Nadu, India. Annu. Rep. Explor. Introduction Plant Genet. Resour. 27, 111–127.
Tomooka, N., Naito, K., Kaga, A., Sakai, H., Isemura, T., Ogiso-Tanaka, E., et al. (2014). Evolution, domestication and neo-domestication of the genus Vigna. Plant Genet. Resour. 12, S168–S171. doi: 10.1017/S1479262114000483
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt1621
Tsuda, M., Kaga, A., Anai, T., Shimizu, T., Sayama, T., Takagi, K., et al. (2015). Construction of a high-density mutant library in soybean and development of a mutant retrieval method using amplicon sequencing. BMC Genomics 16, 1014. doi: 10.1186/s12864-015-2079-y
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2017). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Keywords: plant domestication, wild species, legume, Vigna, mutant screening, seed dormancy, pod shattering, bulked segregant analysis
Citation: Takahashi Y, Sakai H, Yoshitsu Y, Muto C, Anai T, Pandiyan M, Senthil N, Tomooka N and Naito K (2019) Domesticating Vigna Stipulacea: A Potential Legume Crop With Broad Resistance to Biotic Stresses. Front. Plant Sci. 10:1607. doi: 10.3389/fpls.2019.01607
Received: 30 August 2019; Accepted: 15 November 2019;
Published: 06 December 2019.
Edited by:
Eric Von Wettberg, University of Vermont, United StatesReviewed by:
R. Varma Penmetsa, University of California, Davis, United StatesCopyright © 2019 Takahashi, Sakai, Yoshitsu, Muto, Anai, Pandiyan, Senthil, Tomooka and Naito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken Naito, a25haXRvQGFmZnJjLmdvLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.