 Prasanna Kankanala
Prasanna Kankanala Raja Sekhar Nandety
Raja Sekhar Nandety Kirankumar S. Mysore
Kirankumar S. Mysore
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci., 30 October 2019
Sec. Plant Breeding
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.01345
This article is part of the Research TopicLegumes for Global Food SecurityView all 37 articles
The constant interactions between plants and pathogens in the environment and the resulting outcomes are of significant importance for agriculture and agricultural scientists. Disease resistance genes in plant cultivars can break down in the field due to the evolution of pathogens under high selection pressure. Thus, the protection of crop plants against pathogens is a continuous arms race. Like any other type of crop plant, legumes are susceptible to many pathogens. The dawn of the genomic era, in which high-throughput and cost-effective genomic tools have become available, has revolutionized our understanding of the complex interactions between legumes and pathogens. Genomic tools have enabled a global view of transcriptome changes during these interactions, from which several key players in both the resistant and susceptible interactions have been identified. This review summarizes some of the large-scale genomic studies that have clarified the host transcriptional changes during interactions between legumes and their plant pathogens while highlighting some of the molecular breeding tools that are available to introgress the traits into breeding programs. These studies provide valuable insights into the molecular basis of different levels of host defenses in resistant and susceptible interactions.
Legumes belong to the third-largest angiosperm family, Fabaceae or Leguminosae. This family comprises around 750 genera and 20,000 species, including grain, forage, and economically important legumes (Polhill et al., 1981). Legumes contribute 33% of human dietary protein (Vance et al., 2000). Although legumes are cultivated over 12 to 15% of the Earth’s arable land and account for 27% of the world’s primary crop production (Vance et al., 2000), their yield is limited due to environmental adaptability challenges and damage caused by pests and pathogens (Graham and Vance, 2003). Some of the major fungal diseases of legumes include rusts, mildews, root rots, wilts, blights, and anthracnoses. Bacterial diseases are mainly grouped into leaf blights, leaf spots, bacterial wilts, and a diverse group with symptoms such as dwarfing and rots (Rubiales et al., 2015; Wille et al., 2019). Viral diseases are caused by Bean pod mottle virus, Soybean mosaic virus, and Peanut stripe virus, among others. Cyst and root-knot nematodes are the devastating parasites of legumes (Rubiales et al., 2015).
Plants have evolved robust defense mechanisms against pathogen attack that are triggered by initial recognition of the pathogen. These mechanisms involve a cascade of signaling responses known as Pathogen-Associated Molecular Pattern (PAMP) Triggered Immune (PTI) response, which eventually leads to changes in the gene expression of the host. Depending on the type of interaction, this can result in either disease susceptibility or disease resistance. The pathogens, on the other end of the spectrum, have evolved several mechanisms involving effector delivery to evade the host defenses. The host defense response to effectors is called Effector-Triggered Immunity (ETI) (Young et al., 2005). The continuous arms race between the host and pathogen eventually determines the outcome of the interaction (Jones and Dangl, 2006). The host responses also vary based on pathogen infection strategies. The current understanding is that successful defense responses against biotrophic pathogens are predominantly mediated by the salicylic acid (SA)-dependent pathway and that those against hemibiotrophs and necrotrophs involve ethylene and jasmonic acid (JA) signaling (Glazebrook, 2005).
Leguminosae includes a diverse variety of plants. Medicago truncatula and Lotus japonicus have been chosen as model species to advance the study of legumes (Zhu et al., 2005). Several genetic and genomic resources have been developed in these two model legumes to assist breeding programs for enhanced tolerance/resistance to abiotic and biotic stresses in legume crop species. These include genome sequences (Sato et al., 2008; Young et al., 2011), expressed sequence tags (ESTs) (Asamizu et al., 2004; Gamas et al., 2006), physical and genetic maps (Choi et al., 2004; Yan et al., 2004; Young et al., 2005; Wang et al., 2008; Ohmido et al., 2010; Shah et al., 2016), and insertional mutagenesis lines (Tadege et al., 2008; Urbanski et al., 2013), among others. In addition to the model plants, the genome sequences of crop plants such as Glycine max (cultivated soybean), Glycine soja (wild soybean), Cajanus Cajun (pigeon pea), Cicer arietinum (chickpea), Vigna radiata (mung bean), V. angularis (adzuki bean), V. unguiculata (cowpea), Arachis hypogaea (cultivated peanut), A. duranensis (wild peanut A genome), A. ipaensis (wild peanut B genome), Medicago sativa (alfalfa), Phaseolus vulgaris (common bean), Trifolium pretense (red clover), Lupinus angustifolius (lupin), and Lens culinaris (lentil) are currently available at https://legumeinfo.org/genomes. The macrosynteny and microsynteny studies among some of these genomes have been useful for translating the knowledge from model to crop plants (Zhu et al., 2005). The availability of genome sequences coupled with recent advancements in affordable Next-Generation Sequencing (NGS) techniques and bioinformatics tools has enabled extensive study of genome-wide expression changes during plant–pathogen interactions to identify the pathways involved in plant defense. Macroarrays, microarrays, RNAseq, suppressive subtractive hybridization (Mehrtens et al., 2005), cDNA-amplified fragment length polymorphism (AFLP) techniques and gene-expression atlases have been used extensively to identify candidate genes for disease resistance. In this review, we focus on the interactions of legumes with plant pathogens such as fungi, oomycete, bacteria, nematodes, and viruses at the genomic level and the use of genomic technologies in breeding for resistance.
Fungi are among the most challenging plant pathogens to tackle owing to their genetic flexibility and plasticity, which allow them to adapt quickly to their changing environments (Perez-Nadales et al., 2014). Considerable effort has gone into understanding the plant–fungal interaction mechanisms in both model and crop legumes. Large-scale genomic studies have enabled understanding of the various plant disease-resistance mechanisms against hemibiotrophic, biotrophic, and necrotrophic fungal pathogens.
Mycosphaerella pinodes is a broad-host range fungal pathogen that causes ascochyta blight disease. It is known to have a transient biotrophic phase in some hosts and to behave like a necrotrophic pathogen in other hosts (Fondevilla et al., 2011; Almeida et al., 2015). M. truncatula-based microarrays were used to study resistant interactions of pea with M. pinodes. The functional gene categories involved in the resistance mechanism included phytohormones, Pathogenesis Related (PR) genes, the phenylpropanoid pathway, cell-wall fortification, and genes involved in ethylene- and jasmonic-acid(JA)-related defense pathways (Fondevilla et al., 2011). This work was later augmented by investigating the transcriptome in the host pea plants using deep SuperSAGE analysis to enrich for transcripts in the pea–M. pinodes interactions, followed by next-generation sequencing (NGS) of the transcripts (Fondevilla et al., 2014). Several factors that play key roles in resistance were identified, such as the WRKY protein in pathogen perception, proteases as an active defense against fungal toxins, and the roles of ethylene, abscisic acid, and indole-3-acetic acid as phytohormones in defense. Flavonoids, terpenoids, reactive oxygen species (ROS) and phytoalexins were identified as antifungal components that inhibit hyphal growth and destroy toxins (Fondevilla et al., 2014).
In Vigna unguiculata (cowpea) and orphan legumes such as Lathyrus sativus (grass pea) and Vicia faba (fava bean) where whole-genome sequence information is not available, SuperSAGE and Deep SuperSAGE coupled with NGS sequencing has been used successfully to study transcriptomes during Ascochyta infections (Madrid et al., 2013; Almeida et al., 2015). Early gene expression profiling in a resistant variety of grass pea during Ascochyta infection identified that classical defense-response genes involved in cell-wall fortification and the phenylpopanoid pathway were differentially expressed in resistant interactions. In addition, homologs of several candidate resistance genes such as receptor kinases containing thaumatin-like protein (TLP) domains, leucine-rich repeat (LRR) domains, and a gene homologous to Resistance to Pseudomonas syringae pv maculicola 1 (RPM1) involved in conferring resistance to P. syringae in Arabidopsis were identified (Almeida et al., 2015). During resistant interactions in the fava bean-Ascochyta fabae infection process, genes involved in JA signaling and pectin esterase-encoding genes were identified with the SuperSAGE technique (Madrid et al., 2013). In a later study, de novo transcriptome assembly was used to identify transcripts in susceptible and resistant interactions of fava bean and A. fabae. Genes encoding LRR proteins, Rho2 GTPase-activating protein 2 (RGA2), several plant growth regulators, heat shock proteins, chitin elicitor-binding protein, and those genes that produce chlorogenic acid, scopoletin, and flavonoids were among the significant genes involved in fava bean defenses (Ocaña et al., 2015). Functional characterization of these candidate genes will be the next essential step toward including them in breeding programs.
Anthracnose disease is caused by a hemibiotrophic fungal pathogen, Colletotrichum spp. The genomics of Colletotrichum–host and –nonhost interactions were investigated using microarray analysis in M. truncatula (Jaulneau et al., 2010). In this study, resistant and susceptible M. truncatula varieties were infected with pathogenic strain Colletotrichum trifolii and non-adapted strains C. higginsianum and C. lindemuthianum. Resistance responses to non-adapted Colletotrichum spp. were similar to the incompatible responses induced by the adapted strain on the resistant line. The nonhost responses included localized oxidative burst and fluorescent compound release. The host resistance response was characterized through defense gene signaling and SA accumulation (Jaulneau et al., 2010). To identify the genetic components of bean immunity against C. lindemuthianum, EST analysis was carried out in common bean with putative A. thaliana orthologs (Oblessuc et al., 2012). This study suggested that ETI-triggered hypersensitive response is mediated by downregulation of FLS2-like and MKK-5 like putative orthologs of A. thaliana genes involved in pathogen perception (Oblessuc et al., 2012). The resistant and susceptible interactions of P. vulgaris with C. lindemuthianum were investigated using NGS methods (Padder et al., 2016). Most of the DEGs were expressed in the biotrophic phase in the susceptible interaction, while most of the DEGs were expressed in the necrotrophic phase in the resistant interaction. DEGs in the resistant interaction were over-represented by genes expressing PR proteins and peroxidases, while the susceptible interaction was over-represented by genes encoding sugar transporters (Padder et al., 2016). The genomics of partially resistant and susceptible interactions of Lens culinaris and C. lentis were studied using EST analysis (Bhadauria et al., 2017). Twenty-six resistance genes were identified during the symptomatic phase of infection in the compatible interaction. Further, a complex interplay of plant hormone pathways was also observed in this study (Bhadauria et al., 2017).
Fusarium wilt is a destructive disease in several legumes that is caused by host-specific Fusarium oxysporum strains. Extensive studies have been done to understand the molecular basis of this disease interaction in various legumes. In Phaseolus vulgaris (common bean), the cDNA-AFLP technique was used to determine transcriptionally regulated genes in response to F. oxysporum f. sp. phaseoli (Fop) infection in resistant and susceptible interactions (Xue et al., 2015). This study identified 122 defense-related gene fragments that are distributed across the genome. This distribution could serve to tag defense-related molecular markers in breeding programs (Xue et al., 2015). RNAseq analysis of Glycine max infected with both pathogenic and non-pathogenic strains of F. oxysporum identified over-representation of defense-related genes corresponding to necrosis in resistant interactions (Lanubile et al., 2015). RNAseq was carried out to understand the molecular differences in defense responses between cultivated and wild species of Glycine max against the pathogenic F. oxysporum Schltdl (Chang et al., 2019). That study identified the role of secondary metabolites and plant hormones in wild-type germplasm that could be adapted into cultivated species for enhanced resistance.
Several races of F. oxysporum f. sp. Ciceri (Foc) have been identified across the chickpea-growing regions of the world. While most F. oxysporum strains are considered as necrotrophic or hemibiotrophic pathogens, Foc race 1 (Foc1) is reported to be an obligate biotrophic pathogen of chickpea (Gupta et al., 2009). cDNA-AFLP-based analyses, cDNA-based microarrays, and cDNA RAPD methods have been used to study chickpea interactions with Foc1 (Nimbalkar et al., 2006; Ashraf et al., 2009; Gupta et al., 2009; Gupta et al., 2010; Gurjar et al., 2012). Although host responses during biotrophic infections are mediated by SA-dependent pathways, gene expression analyses in the above-referenced studies indicate non-traditional responses. The cDNA-AFLP method identified that genes encoding sucrose synthases, invertases, and β-amylase were induced in resistant interactions. The 14-3-3 gene expression was overrepresented in the susceptible interaction, indicating potential nutrient starvation, and the resistant interaction potentially copes with this sugar starvation by over-inducing sugar-metabolism genes. This study was highly suggestive that sugar also acts as a signaling molecule in response to pathogen perception (Gupta et al., 2010). A comparative study of resistant and susceptible interactions with Foc races 1, 2, and 7 was conducted in chickpea using the cDNA-RAPD method (Gurjar et al., 2012). This study identified a role for plant glucosyltransferase genes in resistance response. Further, race-dependent defense responses were observed (Gurjar et al., 2012). A similar study with resistant and susceptible interactions with Foc race 1, 2, and 4 was conducted recently with the LongSAGE method (Upasani et al., 2017). Clustering analysis and interaction networks of differentially expressed genes (DEGs) identified that the resistant interaction is characterized by ROS production, SA production, lignification, R-gene expression, and hormone homeostasis. The susceptible interaction was enriched for actin depolymerization genes, aquaporin genes, and tetrapyrrole synthesis genes (Upasani et al., 2017). Another study detailing the transcriptional changes during resistant and susceptible chickpea interactions with Foc1 was done using RNAseq analysis (Gupta et al., 2017). Plant pathogen interaction networks constructed with this transcriptional data identified several nodal hub genes that modulate defense responses and could be further characterized for resistance (Gupta et al., 2017). A microarray-based study of resistant and susceptible chickpea interaction transcriptomes with Foc1 was used to create regulatory gene networks (Ashraf et al., 2018). This study identified 76 disease- and immunity-related genes. The gene regulatory networks identified transcriptional plasticity in immune pathways and disease pathways during wilt interactions. This work also highlighted that the primary metabolic components are shared between defense and disease (Ashraf et al., 2018).
Asian soybean rust (ASR), caused by Phakospora pachyrhizi, is a devastating disease that is listed among the top five biotic threats to agriculture (Pennisi, 2010). Although six resistance genes, Resistance to Phakospora pachyrhizi (Rpp1-6), have been identified in soybean that confer resistance to ASR in a race-specific manner, no single soybean genotype can confer resistance to all races of the rust fungus (Langenbach et al., 2016a). Several studies have been conducted to identify key players in resistance to ASR. Initial studies to identify key players in the R-gene-mediated response of ASR employed the SSH complementary DNA (cDNA) method (Choi et al., 2008; Soria-Guerra et al., 2010b). These studies indicated a time-dependent coordinated gene expression pattern in Rpp-mediated resistant and susceptible interactions and identified the role of peroxidases and lipoxygenases in resistance. Later work using whole-genome microarrays confirmed these two findings and provided further detail (Van De Mortel et al., 2007; Panthee et al., 2009; Soria-Guerra et al., 2010a; Soria-Guerra et al., 2010b; Morales et al., 2013). Several of these studies have reported an overrepresentation of transcription factors (TF) and the roles of flavonoids and cell-wall lignification in the active resistance mechanisms. Metabolite analysis of the ASR interactions has confirmed some of these findings (Lygin et al., 2009). Several genomic studies involving TFs identified roles for WRKY, the Basic Leucine Zipper (bZIP) domain, and predicted TF families in resistant interactions (Pandey et al., 2011; Aoyagi et al., 2014; Bencke-Malato et al., 2014; Alves et al., 2015). The SuperSAGE technique was used to identify several antimicrobial peptides such as defensins, thionin, and lipid transfer protein (LTP) family genes in cowpea and soybean infected with the rust pathogen P. pachyrhizi (Kido et al., 2010).
Nonhost resistance (NHR) is a type of resistance that is displayed by plants against most potential pathogens. This type of resistance can be multi-layered, and plants can exhibit this either prior to infection (pre-invasive NHR) or post-infection (post-invasive NHR) (Senthil-Kumar and Mysore, 2013; Gill et al., 2015; Lee et al., 2017; Fonseca and Mysore, 2019). The NHR responses of Arabidopsis and M. truncatula against P. pachyrhizi were explored to identify sources of durable disease resistance. Studies with Arabidopsis have indicated that the ASR fungus exploits the necrotrophic pathway by inducing JA-mediated responses to evade host defense responses (Loehrer et al., 2008; Campe et al., 2014). The roles of PENETRATION 1-4 (PEN1-4), SENESCENCE-ASSOCIATED GENE 101, BRIGHT TRICHOMES 1, and POSTINVASION-INDUCED NONHOST RESISTANCE GENES4/5/9 (PING4/5/9) in pre-invasive and post-invasive NHR mechanisms have been explored (Loehrer et al., 2008; Langenbach et al., 2013; Langenbach et al., 2016b). Furthermore, the potential of transferring NHR PING genes to soybeans and conferring enhanced ASR resistance has been demonstrated (Langenbach et al., 2016b). Transcriptional changes during the interaction of P. pachyrhizi with M. truncatula were used to identify genes involved in NHR (Ishiga et al., 2015). A combination of transcriptome and metabolite analysis indicated the role of the secondary metabolite, medicarpin, in inhibiting the germination and differentiation of rust urediniospores. Transcriptome analysis also indicated the role of chlorophyll catabolism genes in disease resistance. Further characterization of the STAY GREEN gene indicated its role in the hypersensitive-like response during the resistance interaction (Ishiga et al., 2015). Further, a forward genetics-based screening of M. truncatula Tnt1 insertion lines (Tadege et al., 2008; Sun et al., 2019) for alterations in response against P. pachyrhizi identified the inhibitior of rust germ tube differentiation1 (irg1) mutant (Uppalapati et al., 2012). IRG1 encodes a Cys(2)His(2) zinc finger transcription factor, PALM1, which plays a role in regulating epicuticular wax metabolism and transport, and epicuticular wax is important for ASR spore differentiation (Uppalapati et al., 2012; Ishiga et al., 2013).
An interesting study involving resistant interaction with two foliar pathogens, Colletotrichum trifolii (hemibiotrophic pathogen) and Erysiphe pisi (biotrophic pathogen) and a partially resistant root pathogen, Phytophthora medicaginis (necrotrophic pathogen), with M. truncatula identified three Pathogenesis Related (PR) 10 genes, a TLP, and a gene encoding hevein-like protein to be upregulated (Samac et al., 2011). The phenylpropanoid pathway involving isoflavonoid synthesis was also upregulated. Further characterization of these genes using RNAi lines identified the role of the Chalcone Synthase gene in the phenylpropanoid pathway in conferring resistance to necrotrophic pathogens. (Samac et al., 2011).
A quantitative PCR-based TF platform in M. truncatula was used to conduct TF expression profiling during interactions with Uromyces striatus (Madrid et al., 2010; Villegas-Fernández et al., 2014). The TF profiling in resistant interactions of M. truncatula with U. striatus identified genes encoding pathogenesis-related ethylene response factor (PR-ERF), WRKY, and the Myb class of TFs to be differentially expressed (Madrid et al., 2010). Comparing the TF expression profiling in the two pathosystems, Botrytis spp. and U. striatus, there were higher constitutively expressed TFs in M. truncatula-Botrytis spp. interactions, indicating an NHR-like response, although this resistance was compromised in the lab-experimental system, allowing infection. This may also be indicative of the differences in the host response to biotrophic versus necrotrophic pathogens (Madrid et al., 2010).
The availability of whole-genome microarrays of the model legume M. truncatula has advanced the understanding of various other fungal pathogen interactions in legumes (Uppalapati et al., 2009; Samac et al., 2011). M. truncatula is a susceptible host for Phymatotrichopsis root rot caused by the fungus Phymatotrichopsis omnivora (Uppalapati et al., 2010). Microarray analysis of this interaction identified JA- and ethylene-responsive genes, indicating a necrotrophic infection strategy (Uppalapati et al., 2009). Secondary metabolite genes involved in isoflavonoid synthesis were upregulated at the early infection stage but were eventually reduced to basal level during later disease progression stages, indicating the role of fungal manipulation of host defenses (Uppalapati et al., 2009).
Although M. truncatula is a nonhost for Botrytis spp., by screening several M. truncatula genotypes under lab conditions, a partially resistant genotype and a susceptible genotype were identified (Villegas-Fernández et al., 2014). This study identified Botrytis fabae as a more aggressive pathogen compared to B. cinerea on M. truncatula. However, microscopic studies indicate there is higher spore germination of the later pathogen species. Transcription factor (TF) profiling indicated that the host perceives B. fabae to be a more virulent pathogen by upregulating diverse TFs involved in stress responses even before the visible symptoms appear (Villegas-Fernández et al., 2014).
An integrated omics approach using RNAseq and metabolomics (1H NMR) data was used to understand the primary metabolism regulation of soybean in response to Rhizoctonia solani infection (Copley et al., 2017). A significant flux of responses in redox reactions and ROS signaling along with changes in peroxidases, post-infection, were observed in soybean leaves (Copley et al., 2017).
The oomycete pathogens of legumes that have been most studied using genomic tools are Phytophthora spp and Aphanomyces spp. Some of the early studies of gene expression changes in soybean with Phytophthora sojae reflected the hemibiotrophic infection strategy of the pathogen at the molecular level (Moy et al., 2004). A cDNA microarray with genes from both host plant and pathogen was custom-built, and a time course of susceptible interaction studies revealed the expression of active defenses in the host mediated by SA-triggered pathways at early infection stages. This included expression of PR1a gene and genes involved in the phenylpropanoid pathway. The host and pathogen responses peaked at around 24 hpi, followed by a reduction in the host responses, indicating the shift from biotrophy to necrotrophy (Moy et al., 2004). With the availability of the Affymetrix® gene chip for soybean, a detailed mapping of the soybean transcriptome change was carried out using three genotypes – resistant, partially resistant, and susceptible soybean varieties. This experiment was conducted with 72 biological replicates to understand the effect of genotypic variation on transcriptome changes (Zhou et al., 2009). The large number of replicates coupled with detailed statistical analysis demonstrated that almost the entire genome underwent low-level transcriptional changes in response to disease and genetic variation, yet most of the differences were less than two-fold in magnitude. This work hypothesized that these pervasive and statistically significant low-level changes may reflect the genotype-specific host adaptive changes in response to the pathogen and that studying these might be valuable. A macroarray study of resistant and susceptible interactions in the same disease system identified a role for putative regulators of chromosome condensation 1 protein family in the resistant interaction, suggesting the suppression of nucleocytoplasmic trafficking as one of the host strategies for combating disease (Narayanan et al., 2009). A more recent attempt to enrich the transcripts differentially expressed during the disease process employed the SSH cDNA library coupled with NGS (Xu et al., 2012). This study identified genes encoding several traditional proteins involved in disease-resistance strategies, including various PR-like proteins, the WRKY class of transcription factors, and proteins involved in the phenylpropanoid pathway. A novel discovery of this work involved identifying the allergen gene Pru ar 1 (Prunus armeniaca) in soybean, which could be involved in resistance. Functional characterization of the Pru ar 1 gene identified it as a novel gene encoding PR10 protein (Fan et al., 2015).
MicroRNAs (miRNAs) are 20- to 24-nucleotide long, single-stranded non-coding RNAs that play critical roles in various biological functions, including plant innate immunity. miRNAs-mRNA complexes regulate these responses (Navarro et al., 2008). Microarray analysis was conducted with susceptible, qualitative-resistant, and quantitative-resistant cultivars of soybean infected with Phytophthora sojae (Guo et al., 2011). This study identified different microRNAs in the three different interactions. The bioinformatics search indicated that some of the targets involved diverse categories such as defense response genes, kinases, transcriptional factors, etc. Several microRNAs have inverse expression patterns to their putative target genes. These data indicated a role for microRNAs in regulating plant defense responses in resistant interactions. To understand the single dominant gene in Resistance to Phytophthora sojae (Rps)-mediated resistance mechanisms in soybean-P. sojae interactions, researchers conducted transcriptome analysis of 10 near-isogenic lines, each with a unique Rps gene/allele (Lin et al., 2014). This study identified that Rps recognition was characterized by induction of SA-, ethylene-, and brassinosteroid phytohormone-signaling pathways, repression of JA pathways, ROS, WRKY transcription factors, MAP kinase-signaling pathways, and phytoalexin production. The compatible reaction was characterized by the induction of the JA pathway, repression of the ethylene pathway, and no changes to the SA and brassinosteroid pathways (Lin et al., 2014).
Aphanomyces euteiches, the causal organism of Aphanomyces root rot, is a major soilborne oomycete pathogen that infects various legume species, including pea, lentil, and alfalfa (Pilet-Nayel et al., 2009). In an attempt to understand the strategies employed during A. euteuches interactions with M. truncatula in a compatible interaction, a cDNA-AFLP approach was first employed to understand the optimal infection time for evaluation, followed by cDNA enrichment with SSH (Nyamsuren et al., 2003). This study identified classical PR- and defense genes. The molecular analysis indicated abscisic acid-mediated signaling that could induce PR-10 protein. The PR-4 protein-containing hevein domain, which could bind chitin, was also identified. A more recent study was conducted to compare the transcriptional responses in compatible interactions of pea plants with both the oomycete pathogens discussed here—Phytophthora pisi and A. euteiches—using a M. truncatula microarray (Hosseini et al., 2015). The study revealed different recognition and signaling components in the host against the two pathogens. PTI and ETI responses were detected in the early stages of infection with both pathogens. JA- and ET-hormone signaling were involved in both interactions, while the auxin-induced SAUR family proteins were specific to A. euteiches. The interactions of downy mildew pathogen, Peranospora viciae f. sp. Pisi, with pea leaves were investigated with SSH cDNA libraries (Feng et al., 2012). The study identified downy mildew resistance genes RPP6/6/27 involved in this interaction.
The application of genomic tools in understanding bacterial pathogenesis in legumes is relatively limited compared to in fungal pathogenesis studies. NGS technologies have been employed to understand the interactions of Xanthomonas axonopodis pv. glycines (Xag), which causes bacterial leaf pustule (BLP) disease in soybean (Kim et al., 2011; Chatnaparat et al., 2016). Kim et al. (2011) studied the transcriptome profiling in near-isogenic lines (NILs) of resistant and susceptible cultivars of BLP while Chatnaparat et al. (2016) studied the gene expression of the pathogen in susceptible host leaves. In the former study, several genes involved in PTI response such as EF-TU RECEPTOR (EFR)- and FLAGELLIN SENSING 2 (FLS2)-, ATPASE 4 (ACA4)-, ACA11-, MAP KINASE 4 (MPK4)-, MPK6-, and RESPIRATORY BURST OXIDASE HOMOLOGUE (RBOH)-like genes, and Damage-Associated Molecular Pattern (DAMP) receptors such as PLASMA MEMBRANE LRR RECEPTOR KINASE 1(PEPR1) and PEPR2, were induced at 0 hours post inoculation (hpi) in BLP-resistant NILs and not in BLP-susceptible NILs. Defense response genes such as RPP-, RPM1-, and Mildew Locus O (MLO)-like genes also were induced at this time point in BLP-resistant NILs. The authors speculate that this early up-regulation of PTI-related genes potentiates immune response during pathogen attack. Although the Xanthomonas species is known to be a biotroph, Xag behaves like a necrotrophic pathogen in soybean, as demonstrated by the molecular mechanisms in this study. Several genes encoding jasmonate-zim domain (JAZ)-like and MYC2 TF proteins were also induced at 0 hpi (Kim et al., 2011). This work demonstrates that the activity of PTI components at the early stage of infection is an important defense mechanism in the resistant soybean NIL tested.
Bacterial wilt caused by Ralstonia solanacearum is an important pathogen of peanuts, and genomic tools have been employed to understand the host–pathogen interactions. Early studies in this system were done using cDNA libraries where both the roots and leaves of peanuts were challenged with the pathogen, while in nature Ralstonia is a root pathogen (Huang et al., 2012). Ethylene and JA pathway genes were induced in both the roots and leaves of a highly resistant peanut cultivar. Several secondary metabolite genes were induced in roots and not in leaves, indicating the natural adaptation of the host to a root pathogen (Huang et al., 2012). In a more recent study of this pathosystem, NGS technology was used to study the gene expression differences between susceptible and resistant cultivars (Chen et al., 2014). In this study, the suppression of primary metabolism, especially carbohydrate metabolism, was an important feature in the resistant interaction, indicating the shift of energy investment from the primary metabolism to defense mechanisms. The PTI defense pathway was triggered in both resistant and susceptible interactions, and its partial suppression by the pathogen was observed. The expression patterns of secondary metabolites and defense response genes and hormone analysis indicated that resistance was primarily conferred by defense response genes in the ETI response cascade. Bacterial blight disease of soybean is caused by Pseudomonas syringae pv. glycinea (Psg). In a cDNA microarray study of both resistant and susceptible interactions of Psg with soybean using a virulent and avirulent strain of Psg, a three-phase response was studied. In phase I, which corresponded to the induction stage at 2 hpi, no significant differences were seen between susceptible and resistant interactions. Phase II, which lasted from 3 to 10 hpi, and phase III, up to 24 hpi, corresponded to the effector stage and programmed cell death (PCD) stages in resistant interaction, respectively. Several gene expression changes were observed in phases II and III. An important reported observation was a 92% reduction in the expression of chloroplast-related genes in the resistant interaction event at 8 hpi with no visible symptoms. Physiological measurements supported these data. There was a lack of ROS in the susceptible interaction at phase II. This study suggested the role of photosystem centers as a potential source of the secondary ROS or the oxidative burst response that eventually leads to PCD in phase III (Zou et al., 2005). Pseudomonas syringae pv. syringae causes bacterial stem blight in alfalfa. RNAseq was conducted to study the host–pathogen interactions in resistant and susceptible alfalfa cultivars at two different time points (Nemchinov et al., 2017). The timing of resistance response differed in both cultivars. The ZG9830 cultivar triggered ETI responses much earlier than the Maverick cultivar. The resistance response in cultivar ZG9830 may involve NBS-LRR, TIR-unknown (TX), and nematode-resistance proteins named based on their homology to Hs1pro-1 (HSPRO2)-like R genes, while the cultivar Maverick may involve the CNL class of R genes (Nemchinov et al., 2017).
Heterodera glycines, commonly known as soybean cyst nematode (SCN), is among the most devastating soybean pathogens. In an attempt to engineer resistance against SCN, detailed characterization of the molecular changes during the infection process in both soybean and the pathogen have been performed. cDNA-based microarrays were initially used to profile the transcriptome changes in the soybean roots during different infection stages in both compatible and incompatible interactions with SCN (Alkharouf et al., 2004; Khan et al., 2004; Alkharouf et al., 2006). These studies identified compatible interaction-specific and incompatible interaction-specific genes as well as time-based induction of genes. Stress-induced gene PR-10 was identified in both compatible and incompatible interactions. Several genes belonging to carbohydrate metabolism, plant defense response, and signaling were indicated in compatible interaction. In a time-course study by Alkharouf et al. (2006), plant responses were documented prior to feeding cell selection (pre-FCS) as well as after feeding cell selection (post-FCS) during compatible interaction. The pre-FCS stages induced PR-10 genes, stress-related genes, carbohydrate-metabolism genes, and secondary metabolism genes. The differentially expressed genes during post-FCS were involved in transcription and protein synthesis. Later studies employing the Affymetrix® gene chip identified differentially expressed genes like PR-5, PR1a, Expansins, cell wall-fortification genes, and phenylpropanoid pathway genes during the post-FCS stage (Ithal et al., 2007). Differential gene expression changes were observed in different genotypes even during the pre-FCS based on the interaction type (compatible/incompatible) in whole-root analysis. Genes belonging to No Apical Meristem (NAM) domain-containing TFs, the WRKY class of TFs, Nucleotide Binding Site-Leucine Rich Repeat (NBS-LRR) kinases, signal transduction, cell wall-fortification, and GC-enriched elements in promoters of genes were identified in the incompatible interaction (Klink et al., 2007b; Mazarei et al., 2011; Wan et al., 2015). In an attempt to enrich for genes differentially expressed during pathogenesis, RNA isolated from laser-capture microdissection samples of syncytial cells was used for microarray analysis with the soybean Affymetrix® gene chip to understand the defense responses in both compatible and incompatible interactions (Klink et al., 2007a; Klink et al., 2009b; Klink et al., 2010; Kandoth et al., 2011; Matsye et al., 2011). The developmental stages of syncytium are divided into a parasitism phase where the syncytium develops and a second phase when the resistance develops. Microarray studies of gene expression during these specific stages indicated that the whole-root analysis masked several key players that are involved in the specific interactions. There were no significant changes in gene expression in the parasitism stage in the compatible and incompatible interactions (Klink et al., 2010). Lipoxygenases, 14-3-3, and genes involved in JA and ethylene biosynthesis, the S-adenosyl methionine pathway, the flavonoid pathway, and coumarin and cellulose biosynthesis were highly induced in the resistant interaction at different stages of resistance response (Klink et al., 2007a; Klink et al., 2009b; Klink et al., 2010). The gene expression profiling in Resistance to H. glycines (Rhg1)-mediated soybean resistance utilizing laser capture microdissections identified apoptosis-related, hypersensitive, and SA-induced defense response genes in the resistant interaction. Several of these genes were either partially or completely suppressed during susceptible interactions with SCN (Kandoth et al., 2011). Genotype-specific defense response studies in soybean indicated two different types of resistant responses involving the varieties Peking and PI88788. Resistance in the Peking variety involved rapid and potent cell wall appositions, while resistance in the PI88788 variety involved potent but slow response without cell wall appositions (Matsye et al., 2011). Microarray studies in these two varieties indicated the role of amino acid transporter and alpha soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein in plant defense (Matsye et al., 2011). Another study with two different SCN populations that invoke a resistant and susceptible response in the same soybean genotype, Peking, revealed that SCN might have evolved different mechanisms to overcome host resistance (Klink et al., 2009a). Approximately 71 genes were induced and 44 genes were suppressed in the SCN strain that triggered a resistant reaction in the host during pre-infection stage. As the infection progressed, many SCN genes were suppressed in the resistant interaction. These data indicate that the feeding and nutritional uptake mechanisms of SCN might be the targets of the host defense. A recent study conducted by Tian et al. identified the microRNAs that are differentially expressed between two soybean cultivars, KS4313N and KS4607, which have differential resistance response to SCN. They identified a total of 60 differentially expressed miRNAs belonging to 25 families correlating to the response of the cultivars (Tian et al., 2017). Black soybean, Huipizhi Heidou, has different grades of resistance to SCN. RNAseq analyses at three different infection time points were conducted in two cultivars representing resistant and susceptible interactions (Li et al., 2018b). The study suggested roles for five plant hormones in the resistance. While SCN is a pathogen of soybean, it can also infect and reproduce in the roots of common bean, causing yield reductions. Gene expression profiling of common bean roots upon infection with SCN resulted in the differential expression of genes encoding nucleotide-binding site leucine-rich repeat resistance (NLR) proteins, WRKY TFs, PR proteins, and heat shock proteins (Jain et al., 2016).
Meloidogyne spp., commonly known as root-knot nematodes (RKN), are biotrophic parasites of soybean and cause major crop losses. The resistance mechanisms of incompatible soybean interactions with Meloidogyne incognita indicated auxin-mediated defense responses (Beneventi et al., 2013). Based on transcript profiling, a potential defense model was proposed in which ROS-mediated calcium signaling and nucleoside sugar formation play a critical role in plant hormone signaling. ROS homeostasis was proposed through a balance of auxin-mediated gibberellic acid (GA) and ROS-mediated JA and GA pathway signaling, which maintains low oxidative stress in plants and allows for plant growth. DELLA-like protein was proposed to be a key element in the plant hormone signaling pathway. On the other hand, the gene expression studies on the compatible interactions with RKN indicated an induction of genes involved in the cell cycle, sugar metabolism, and cell wall metabolism. These processes are involved in the successful establishment of giant cells during the infection process. Host defense response genes involving JA-mediated pathways were induced in the early infection stages, while most of them were downregulated by the time the infection had progressed, indicating that RKN actively manipulates host defenses (Ibrahim et al., 2011). The studies on the Rk locus-mediated resistant interaction of RKN indicated that most of the host defenses were suppressed during the infection and feedings stages. Based on this work, it was proposed that the host defenses are triggered against the nematode infection, likely due to the high accumulation of toxins involving unique resistance mechanisms (Das et al., 2010). NGS studies during the early and late stages of the compatible interaction of M. incognita in common bean identified biotic and abiotic stress responses (Santini et al., 2016). Enhanced expression of wound responsive genes at early stages and the TMV resistance protein encoding N gene indicated an active host response to block pathogen infection. This basal response was broken by suppression of ET/JA pathways and at later infection stage (Santini et al., 2016). M. incognita can also infect alfalfa and cause disease in some varieties or accessions. Resistant and susceptible interactions of M. incognita with alfalfa were profiled using both cDNA libraries and through NGS using Illumina Hiseq 2000 (Potenza et al., 2001; Postnikova et al., 2015). There was a high induction of defense-related and stress-response genes in susceptible interaction, indicating basal defense responses. Analysis with the bioinformatics platform for plant resistance (R) gene analysis, PRGdb, identified two potential resistance (R) genes specific to the resistant interaction. Recently, NGS was used to study the genomics of resistance in wild diploid peanut Arachis stenosperma that harbors resistance to M. arenaria (Guimaraes et al., 2015). This study identified components of genetic resistance and induced resistance that could be integrated into breeding programs for durable resistance to RKN.
Soybean mosaic virus (SMV) is an RNA virus and is one of the most prevalent viral pathogens of soybean. A handful of genomics studies have been conducted to understand the molecular changes involved in this disease interaction, including transcriptomics, degradome-seq, and smallRNA-seq (sRNA-seq). One of the earliest genomic studies of SMV-soybean interaction was conducted using cDNA microarrays to investigate the transcriptional changes from early to late infection stages. This study revealed that the plant immune responses are activated at late infection stages in the compatible interaction and that this delayed defense response may be critical to establishing systemic infection (Babu et al., 2008). To study the impact of elevated ozone on the SMV–soybean compatible interaction, gene expression profiling was conducted using soybean microarrays. Increasing ozone concentrations delayed the onset of disease, and this delay corresponded to the expression of basal defense response genes (Bilgin et al., 2008). Comprehensive RNA-seq, sRNAseq, and degradome-seq were performed in soybean during compatible and incompatible interactions with SMV in two different studies (Chen et al., 2016; Chen et al., 2017). An miRNA-mRNA regulatory network was developed based on these data to elucidate the role of miRNAs in the SMV infection process. This study further identified 71 genes that potentially play a role in defense during SMV infection (Chen et al., 2016). One of the differentially expressed genes, Eukaryotic Elongation Initiation Factor 5A (ElF5A), was further characterized, and the knockout mutant of this gene was hyper-susceptible to SMV (Chen et al., 2017). A time-course RNA-seq study during the soybean–SMV compatible interaction identified roles for SA and NLR family genes that were downregulated during compatible interaction and upregulated during incompatible interactions (Zhao et al., 2018).
Transcriptional responses during Bean common mosaic virus (BCMV) interaction with common bean were investigated with two known and one unknown strains of BCMV. The known strains that caused moderate disease symptoms induced more transcriptional changes than the unknown strain that caused severe symptoms (Martin et al., 2016). More recently, a study was conducted to identify miRNAs during the infection of Mungbean yellow mosaic India virus (MYMIV) in common bean employing high-throughput sequencing and identified 107 differentially expressed miRNAs during infection and 3,367 potential target genes for these miRNAs (Patwa et al., 2018).
Engineering or breeding for resistance against plant diseases and nematodes is a more economical and eco-friendly approach than is the use of pesticides. The selective breeding process depends on the type of trait and whether the information for such resistance can be inherited in a qualitative or quantitative manner (Poland and Rutkoski, 2016). Qualitative disease-resistance breeding involves large screening assays that are often laborious and require extensive knowledge of plant–pathogen interactions. Lately, introgression of resistance genes into selective breeding material have relied on the use of molecular markers to assist breeders in the breeding process, which is often called Marker Assisted Selection (MAS) (Cobb et al., 2019). Molecular markers, including AFLPs, simple sequence repeats (SSRs), and more commonly single nucleotide polymorphisms (SNPs), have been developed in a variety of crops and used for different breeding programs. Nucleotide binding site (NBS) profiling, a new marker technology that improves the detection of molecular markers for disease resistance, was developed to identify markers by using NBS regions in the genomes (Van Der Linden et al., 2004). Due to its gene-targeting nature, NBS-profiling directs a PCR reaction to NBS domains through which a large number of R genes can be identified as molecular markers (Van Der Linden et al., 2004). More recently, the availability of genome sequence information for a number of plant species, including legumes, has helped the identification of molecular markers such as SNP markers that can be integrated into breeding programs for resistance screening. Some of the recent genomic resources available in legumes such as M. truncatula, L. japonicus, soybean, chickpea, and pigeon pea are described here (Sato et al., 2007; Sato et al., 2008; Schmutz et al., 2010; Young et al., 2011; Varshney et al., 2012; Varshney et al., 2013; Wang et al., 2013; Pecrix et al., 2018). Markers developed in a variety of ways are integrated into legume breeding programs for resistance against plant pathogenic fungi, oomycetes, bacteria, and nematodes. Though a common approach for engineering resistance into plants is through the integration of race-specific resistance against a known pathogen, this may not impart long or durable resistance, as the single R gene-mediated resistance can be overcome in an arms race by the rapidly evolving pathogens (Fonseca and Mysore, 2019). Hence, a better approach to create a durable resistance is through the deployment of quantitative trait loci (QTLs) through breeding strategies (Kou and Wang, 2010; Kou and Wang, 2012; Zhou et al., 2018) or through a transgenic approach using genes involved in NHR from different plant species (Fonseca and Mysore, 2019). Previous studies identified several genes involved in NHR against important legume pathogens (Fonseca and Mysore, 2019). Phytophthora sojae is a fungal pathogen that causes root rot in soybean and is non-pathogenic on M. truncatula, alfalfa, and Arabidopsis. Penetration mutants (pen1-1) in Arabidopsis were found to be compromised to Phytophthora sojae, thus establishing the case for pre-invasive non-host resistance (Sumit et al., 2012). This gene, when transferred to soybean, resulted in enhanced resistance to Fusarium virguliforme (Wang et al., 2018). Similarly, the Asian soybean rust pathogen, P. pachyrhizi, was not able to infect M. truncatula, alfalfa, or Arabidopsis (Loehrer et al., 2008; Langenbach et al., 2013; Ishiga et al., 2015). The strategy of transferring genes involved in NHR was used to increase the resistance of soybean to P. pachyrhizi. In this case study, 10 PING genes were overexpressed in soybean, resulting in enhanced resistance to P. pachyrhizi infections (Langenbach et al., 2016b).
In this section, we will explore the use of markers and QTLs in legume resistance breeding. Bulk segregant analysis (BSA) was used to map resistance to A. euteiches (AER1) in M. truncatula (Pilet-Nayel et al., 2009). Meta-QTL analysis in pea resulted in the identification of 27 meta-QTLs for resistance to A. euteiches. Six of them were found to co-localize with six of the meta-QTL regions identified for plant height and earliness (Hamon et al., 2013). Two major QTLs Ae-Ps7.6 and Ae-Ps4.5 were identified in pea near-isogenic lines (NILs) that were able to delay the symptoms of oomycete pathogen A. euteiches in pea (Lavaud et al., 2016).
Ascochyta blight (AB) of pea is caused by complex of fungal pathogens including Didymella pinodes and Phoma medicaginis var pinodella. QTLs of resistance to the blight complex pathogens were identified as QTLs based on two QTL mapping populations, A26 × Rovar and A88 × Rovar. QTL peaks, for the Asc2.1, Asc4.2, Asc4.3, and Asc7.1 QTLs, were defined by four of the pea defense candidate genes (Timmerman-Vaughan et al., 2016). These regions were identified on linkage group I in the vicinity of markers c206 and sB17-655, on linkage group III in the vicinity of markers M2P5-169 and PI39, and on linkage group VII in the vicinity of markers Z12-2400, HSP18.1, and MAPKinase (Timmerman-Vaughan et al., 2016). Similarly, AB of dry pea is predominantly caused by Didymella pisi. In an effort to identify QTLs that show consistency across locations and years, Jha et al. (2016) identified two QTLs, abIII-1 and abI-IV-2, for AB resistance. AB is also an important disease in faba bean, resulting in yield losses of 35-40% (Atienza et al., 2016). Two QTLs governing resistance to Ascochyta fabae were identified on chromosome II (Af2) and chromosome II (Af3) of faba bean (Atienza et al., 2016).
Rusts in pea are caused by pea rust pathogen Uromyces pisi. Using DArT-Seq and 8,514 SNP markers, two QTLs, UpDSII and UpDSIV, were identified in the Linkage Groups (LGs) II and IV that controlled resistance to Uromyces pisi (Barilli et al., 2018). In cowpea, rust is caused by the Uromyces vignae pathogen. A single dominant R gene (Ruv2) that confers resistance against U. vignae was found to be inherited in RILs against the U. vignae isolate, Auv-LS (Wu et al., 2018).
Powdery mildew in pea is caused by Erysiphe pisi. The infection results in the formation of small diffused spots on the upper surface of the leaves and at advanced stages covers the entire plant surfaces as a white powdery growth (Ek et al., 2005). Powdery mildew resistance in pea is governed by a pair of recessive alleles “er1er1” (W.H, 1948; Tiwari et al., 1997). Humphry et al. (2011) identified the Er1 locus as PsMLO1 and established through complementation that the loss of PsMLO1 function conditions durable broad-spectrum powdery mildew resistance in pea. Besides the recessive allele er1, another recessive allele er2 and a dominant gene Er3 was recently identified and reviewed by Fondevilla and Rubiales (Fondevilla and Rubiales, 2012). Er3, the dominant gene conferring resistance to powdery mildew in pea, was mapped to pea linkage group IV (Cobos et al., 2018). A variety of molecular markers closer to the Er locus were developed to screen the genotypes of pea for powdery mildew resistance (Ek et al., 2005; Sun et al., 2016a; Ganopoulos et al., 2018). A novel er1-7 allele conferring pea powdery mildew resistance was identified through a 10-bp deletion in PsMLO1 cDNA (Sun et al., 2016b). Other natural variations of er1 alleles have been identified, and markers have been designed to screen for powdery mildew resistance in pea (Sudheesh et al., 2015; Sun et al., 2016a; Sun et al., 2016b).
Pea root rot is caused by a variety of fungal plant pathogens, and the causal agent has been identified as Fusarium solani fsp. pisi (Fsp). A strong QTL, Fsp-Ps 2.1, governing resistance to Fsp has been detected in the recombinant inbred line (RIL) populations of pea (Baccara × PI 180693). The QTL Fsp-Ps 2.1 has been identified along with two other minor variance QTLs using three criteria: root disease severity, ratios of diseased vs. healthy shoot heights, and dry plant weights under controlled conditions (Coyne et al., 2019).
Soybean cultivation is significantly affected by SCN and by the sudden death syndrome (SDS) caused by the soilborne fungus F. virguliforme. SDS of soybean results in necrosis/rot of roots, while SCN infection results in yellow dwarf symptoms in soybean. Using soybean plant populations resistant to SCN and SDS, QTL mapping populations have been developed to identify QTLs for both SDS and SCN (Swaminathan et al., 2018).
Verticillium wilt, caused by the soil borne fungus Verticillium alfalfae, is one of the most serious diseases of alfalfa. Through the use of BSA on the alfalfa genotypes and by using marker-trait associations with the help of SSRs and SNPs, 17 SNP markers linked to Verticillium wilt resistance were identified (Zhang et al., 2014). Similarly, using M. truncatula as a model to develop QTLs for resistance against Verticillium, a population of recombinant inbred lines (RILs) from a cross between resistant line F83005.5 and susceptible line A17 were inoculated with a potato isolate of V. albo-atrum, LPP0323. Following the inoculation and screening, a set of four QTLs were identified for the area under the disease progress curve and for maximum symptom score (Negahi et al., 2014). A similar study design was used to identify three distinct QTLs (MtVa1, MtVa2 and MtVa3) that confer resistance to V. albo-atrum in a population of A17 and DZA45.5 (Ben et al., 2013). A recent transcriptomic study conducted on the early root responses of M. truncatula lines A17 (resistant) and a susceptible line (F83005.5) identified core transcriptional responses against root pathogens and showed that the resistance line A17 displayed higher defense-related genes upon inoculation with V. alfalfae V31-2 (Toueni et al., 2016). Phytophthora root rot is caused by an oomycete pathogen Phytophthora sojae, resulting in damping-off, yellowing and wilting diseases in soybean (Li et al., 2017). A QTL, Resistance to Phytophthora sojae (RpsQ), which confers resistance against P. sojae in soybean cultivar Qichadou 1, was mapped using SSR markers to a 118-kb region on the soybean chromosome 3. This 118-kb mapped region consists of 11 candidate genes, and one of them, Glyma.03g027200, was found to encode a serine threonine receptor-like kinase (RLK), which was later confirmed as a likely candidate gene of RpsQ. In a study of M. truncatula roots colonized by pathogenic oomycete Phytophthora palmivora, SNP markers associated with plant colonization response were identified upstream of a Required for Arbuscule Development 1 (RAD1) locus, a positive regulator of arbuscular mycorrhizal (AM) fungus (Rey et al., 2017). The rad1 mutant was impaired in colonization by AM fungi as well as by P. palmivora (Rey et al., 2017). This is one example showing how the use of association mapping in legumes can help identify the genes responsible for genetic resistance against an oomycete pathogen. Readers are encouraged to read the more recent review on fungal root diseases in grain legumes and the implications of plant genetic variation in plant breeding (Wille et al., 2019).
Anthracnose of lentils is caused by Colletotrichum lentis and accounts for 70% of the crop loss in lentils (Bhadauria et al., 2019). Recent genomic sequencing studies on one of the pathogenic races of C. lentis (virulent race 0) combined with QTL mapping led to the identification of a single QTL, qClVIR-11, located on mini chromosome 11, thus explaining 85% of the variability in virulence of the C. lentis population (Bhadauria et al., 2019).
Cowpea is one of the highly cultivated legume crops and is susceptible to many biotic stresses caused by nematodes, bacteria, and fungi. Root-knot nematodes (RKN) are the most important pests of cowpea, resulting in huge losses due to their interference with the root architecture, which results in poor development of the plants (Santos et al., 2018). Previously, two resistance genes, Resistance to Root knot (Rk) and Rk2, were identified to confer resistance against RKN in cowpea (Das et al., 2010; Ndeve et al., 2019). A recent QTL mapping study using RIL population 524B x IT84S-2049 in cowpea resulted in the identification of a major QTL, QRk-vu9.1, associated with resistance to Meloidogyne javanica reproduction (Santos et al., 2018). This QTL was mapped on linkage group LG9 at position 13.37 cM using egg production data. Interestingly, the mapped intervals for this QTL corresponded with six TIR-NBS-LRR (TNL) genes that were identified using transcriptomic analysis between NILs resistant and susceptible to RKN (Santos et al., 2018). A majority of the examples quoted here are in early studies towards achieving economic benefits by developing disease-resistant cultivars.
In plant species, underlying variation with phenotypic data can be quantified, and genome-wide association mapping (GWAS) can be applied for identifying genes and for associating them with the phenotypes. This type of GWAS analysis for SNP discovery is made possible through the development of several target-enrichment or reduction-of-genome-complexity methods such as Genotyping-by-Sequencing (GBS) (Elshire et al., 2011; Glaubitz et al., 2014) or restriction site-associated DNA sequencing (RADSeq) (Davey and Blaxter, 2010). RADseq combines two simple molecular biology methods such as restriction digestion of DNA into fragments and then tagging them using identifier tags followed by NGS (Das et al., 2010). Recently, Diversity Arrays Technology (DArT) in combination with NGS platforms, known as DArTseq™, was used to develop a relatively large number of polymorphic markers to build dense genetic maps (Kilian et al., 2012; Kilian and Graner, 2012). In the GBS methodology, genome complexity is reduced through the use of methylation-sensitive restriction enzymes. Such a method helps avoid the sequencing of repetitive regions and can aid in the sequencing of low copy regions with high efficiency (Elshire et al., 2011). The majority of the GWAS studies described below in this section followed the GBS methodology to generate the genotyping data for SNP identification and exclusively used the GWAS pipelines developed for model and non-model plants (Lipka et al., 2012; Glaubitz et al., 2014; Tang et al., 2016). A variety of experiments have been conducted using GWAS to study the variations associated with the phenotypes in Arabidopsis (Atwell et al., 2010), Maize (Tian et al., 2011), rice (Huang et al., 2010), soybean (Lam et al., 2010), and M. truncatula (Branca et al., 2011). In this section, we will review GWAS studies of legumes in relation to disease resistance phenotypes. Alternatively, HapMap accessions that are sequenced by whole genome sequencing provide an excellent opportunity for the identification of SNPs across the HapMap populations (http://www.medicagohapmap.org/). Some of the M. truncatula HapMap accessions (288 accessions) were used to map flowering time traits linked to nitrogen fixation and for identifying other traits of interest (Stanton-Geddes et al., 2013; Kang et al., 2015; Kang et al., 2019). GWAS was used to estimate linkage disequilibrium levels and identify quantitative resistance loci (QRL) controlling resistance to both anthracnose and Angular leaf spot (ALS) diseases of 180 accessions of common bean. The study resulted in the identification of 21 and 17 statistically significant SNPs associated with anthracnose and ALS diseases of common bean, respectively (Perseguini et al., 2016). Bonhomme and his colleagues (Bonhomme et al., 2014) used high-density SNPs (∼5.1 million single nucleotide polymorphisms) to perform GWAS studies with Aphanomyces root rot resistance against 179 HapMap accessions of M. truncatula. With the use of GWAS, they were able to identify two QTL loci on chromosome 3, with candidate SNPs in the promoter and coding regions of an F-box protein coding gene (Bonhomme et al., 2014). GWAS was recently used in 175 Pisum sativum lines and were genotyped for resistance to A. euteiches using 13,204 SNPs from the GenoPea Infinium® BeadChip (Desgroux et al., 2016). The study resulted in the identification of 52 QTLs of small size intervals associated with resistance to A. euteiches and further validated six of the seven previously reported QTLs (Desgroux et al., 2016). A similar GWAS study was performed to find the association between the plant system architecture of pea and A. euteiches resistance by using 266 pea lines that varied in both of the traits (plant system architecture and disease resistance). Genotyping the lines with 14,157 SNP markers resulted in the identification of one significant SNP mapped to major QTL Ae-Ps7.6 associated with both resistance and root system architecture (RSA) traits (Desgroux et al., 2017).
Brown stem rot (BSR) of soybean, caused by the soilborne fungus Cadophora gregata, affects soybean production in the Northern United States, Canada, and Brazil. Using GWAS, a BSR resistance QTL has been identified in chromosome 16 and is located between 32.8 and 33.1 Mb based on the Glyma2.0 assembly (Rincker et al., 2016). More importantly, this region also maps to previously identified Resistance to Brown Stem Rot genes (Rbs) Rbs1, Rbs2, and Rbs3 (Rincker et al., 2016). This narrow range of resistance QTL could be useful for MAS breeding programs. A comprehensive global view of disease resistance loci in soybean against multiple plant pathogens was presented through the use of GWAS on public Germplasm Resources Information Network and public SNP data (SoySNP50K; (Chang et al., 2016). Using GWAS, the authors identified significant novel SNPs associated with resistance to: bacterial pustule caused by Xanthomonas axonopodis pv. glycines; BSR caused by fungus C. gregata; Diaporthe stem canker caused by Diaporthe phaseolorum var. caulivora and D. phaseolorum var. meridionalis; SDS caused by F. virguliforme; ASR caused by P. pachyrhizi; SCN caused by reniform nematode, Rotylenchulus reniformis; and bean pod mottle virus (Chang et al., 2016).
GWAS was applied to detect SNPs significantly associated with resistance to H. glycines in the core collection of the common bean. There were 84,416 SNPs identified in 363 common bean accessions (Wen et al., 2019). GWAS identified SNPs on chromosome 1 that were significantly associated with resistance to H. glycines type 2.5.7. These SNPs were in linkage disequilibrium with a gene cluster orthologous to the three genes at the Resistance to H. glycines (Rhg1) locus in soybean. A novel signal on chromosome 7 was detected and associated with resistance to H. glycines type 1.2.3.5.6.7. Genomic predictions for resistance to these two H. glycines types in common bean achieved prediction accuracy of 0.52 and 0.41, respectively (Wen et al., 2019).
The molecular markers developed in legume species such as M. truncatula, pea, lentil, faba bean, and lupin can be used in other legumes. Recently, the transferability of molecular markers was tested in legumes such as chickling pea (Lathyrus cicera) and grass pea (L. sativus) (Almeida et al., 2014). During this study, ∼130 markers were successfully cross-amplified in L. cicera and L. sativus with an efficiency of 55% for gene-based markers (Almeida et al., 2014). Such comparative mapping can greatly boost the use of resources and expand the knowledge base in other related species as well.
Gene introgression through breeding often comes with some undesirable trait inheritance that can perturb a desired outcome. Hence, the integration of plant breeding with precise editing of target genes can efficiently aid in the implementation of pathogen resistance in plants. This precision editing is recently made possible through the use of programmable sequence-specific nucleases such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and, more recently, the clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein9 (Cas9)-based genome editing tool (CRISPR/Cas9). These tools effectively generate target site mutations based on base-pairing of the engineered single-guide RNAs (sgRNAs) to the target DNA sites. More information on the development and applications of the CRISPR-Cas9 technologies in plant genomes can be found in previous reviews (Cong et al., 2013; Jiang et al., 2013; Perez-Pinera et al., 2013; Shan et al., 2013; Belhaj et al., 2015; Piatek et al., 2015; Kleinstiver et al., 2016; Ma et al., 2016; Tsai and Joung, 2016; Knott and Doudna, 2018). CRISPR/Cas9 or TALEN entry vectors were developed for gateway cloning in soybean and M. truncatula (Curtin et al., 2018). Several new web tools, such as E-CRISP (Heigwer et al., 2014) and CHOPCHOP (Montague et al., 2014), for the identification of CRISPR-Cas9 target sites are available both for target site identification and also for identification of off-target sites. In legumes, one such tool was developed for CRISPR/Cas9 design (Michno et al., 2015), and a methodology to perform gene editing in M. truncatula also is available (Curtin, 2018).
Using the CRISPR/cas9 technology, severe loss-of-function mutants were developed in the necrotrophic fungal pathogen Sclerotinia sclerotiorum. Using the previously characterized Ssoah1 gene as the gene target, insertional gene mutants were generated that were found to be less virulent on soybean, Brassica spp. and tomato (Li et al., 2018a). Similarly, gene editing was adapted in P. sojae to generate mutants of P. sojae by manipulating Avr4/6 genes of the pathogen (Fang and Tyler, 2016). These studies were important for determining the function of fungal or oomycete genes in pathogen virulence.
Recently, it has been demonstrated that, by using CRISPR/Cas9 genome editing of promoters, diverse cis-regulatory alleles can be generated and that quantitative variation can be an invaluable tool for breeding. A genetic scheme designed by Rodriguez-Leal et al. (2017) exploits transgenerational heritability of Cas9 activity in heterozygous loss-of-function mutant backgrounds. Such a system could also be used in the screening of QTLs for disease resistance if we knew the functions of the cis-regulatory alleles and could be a valuable tool for breeding (Rodriguez-Leal et al., 2017). This concept of generating variations was made possible through the use of epimutagenesis, a method that rapidly generates DNA methylation variation through random demethylation. This ability to manipulate plant methylomes to create epigenetically distinct individuals could be an invaluable breeding tool (Ji et al., 2018). Even though currently not many legume plants have been gene-edited to confer resistance against pathogens, in the future, we anticipate that gene editing will be used more frequently to engineer legume plants with yield-saving disease resistance.
The advancements in cost-economic sequencing technologies have enabled global transcription profiling during plant–pathogen interactions in legumes and identified several pathways and candidate genes responsible for either disease susceptibility or resistance (Table 1). This progress has enabled a broader understanding of both plant and pathogen strategies during resistant and susceptible disease interactions. These studies have identified a repertoire of candidate genes that play key roles in resistance or disease processes. However, functional studies to evaluate their roles in plant–pathogen interactions are limited in some legume species, largely due to lack of mutant resources and appropriate methods for gene function validation. The Tnt1-mediated insertion mutagenesis in M. truncatula has generated ∼21,000 lines with ∼90% gene-tagging coverage in the genome (Tadege et al., 2008; Cheng et al., 2014; Sun et al., 2019). This genetic resource has been utilized to evaluate some candidate genes involved in plant–pathogen interactions. Similarly, several genetic resources are being developed for other legume species such as soybean and L. japonicus (Sato et al., 2007; Libault and Dickstein, 2014). Functional characterization of a few candidate genes has been achieved through RNAi methods and recombinant gene expression studies (Singh et al., 2013). Several genes identified in microarray analysis of SCN–soybean interactions have been characterized by overexpression studies and grouped into genes that enhance, reduce, or have no impact on disease susceptibility (Matthews et al., 2013). Such studies will augment the genomics data generated through whole-genome transcriptional studies. A variety of molecular markers, including AFLPs, SSRs, and SNPs, have been developed and then used to identify QTLs governing resistance to fungal and bacterial pathogens and to root-knot nematodes (Table 2). More recently, the underlying phenotypic variations combined with genotype information (SNPs) have been used for GWAS and are being used extensively in legume crops to identify the QTLs associated with the resistance loci against plant microbes. Precise genome editing technologies such as CRISPR-Cas9 have been employed to effectively knock out P. sojae effector Avr4/6 and uncover the functional role of the corresponding resistance gene RPS4/6 (Fang and Tyler, 2016). The utilization of these resources will help the biological function of genes identified through various genomic approaches to be better understood. Introgression of plant defense-related traits identified through genomics is in its early infancy and could lead to an economic success in the next few years. We predict that the use of the genomics tools in breeding mentioned in this review such as the use QTL introgression, GWAS, and CRISPR/cas9 editing of the genomes for generating plant variation will become increasingly popular in the next few years and will further advance our understanding as well as define our approaches to making improved cultivars in legumes. Genomic studies of plant–pathogen interaction will continue to provide us with novel disease resistance or defense-related genes that can be incorporated into elite legume cultivars, either by classical breeding or by biotechnological approaches.

Table 1 Summary of genomic methods and legume–pathogen interaction studies.
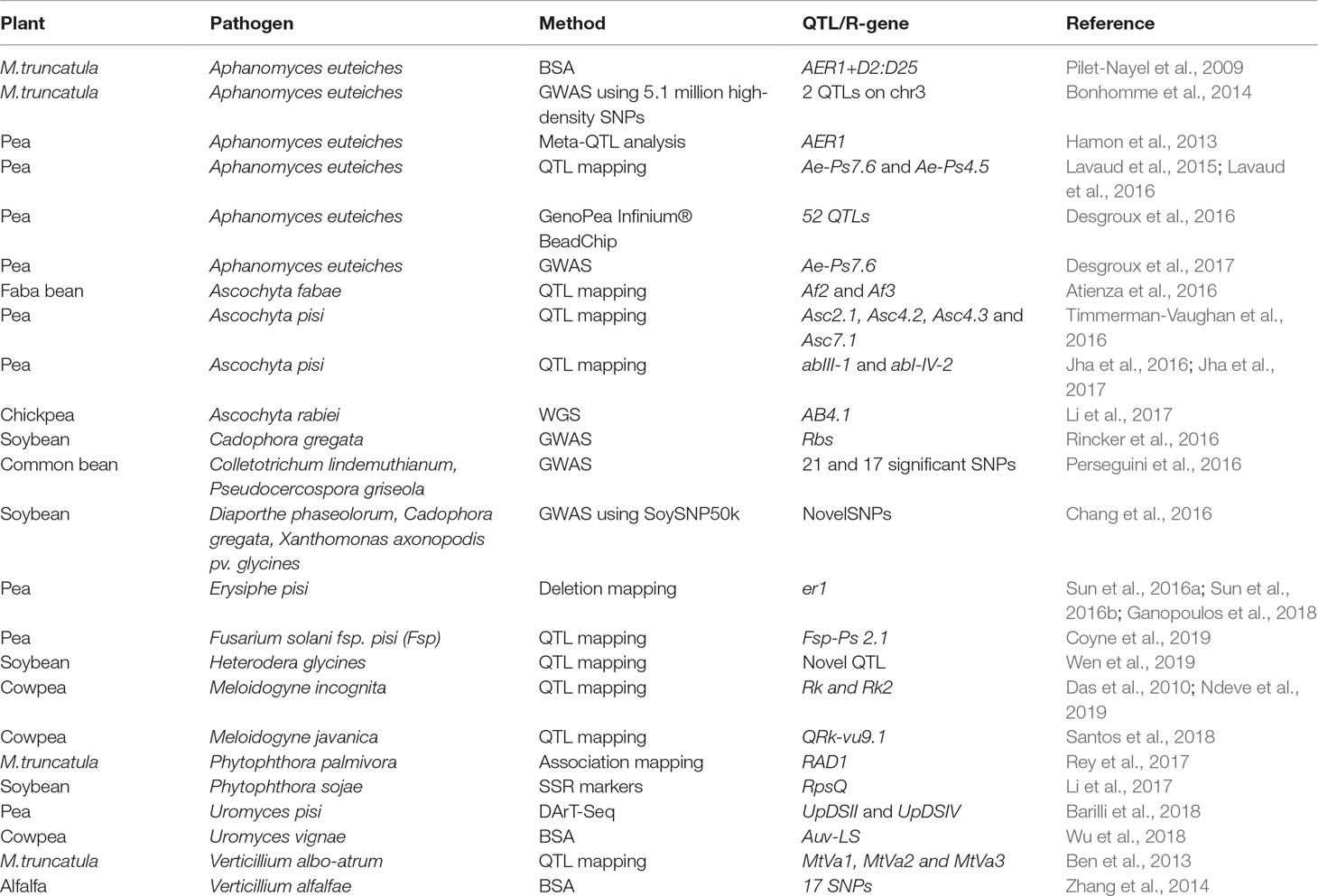
Table 2 Summary of QTL/marker analysis in legume–pathogen interaction studies.
PK and KM conceived the idea. PK and RN wrote the manuscript. KM edited the manuscript.
Projects in the KSM laboratory are funded by the Noble Research Institute, LLC, and the National Science Foundation. We thank Courtney Leeper (Noble Research Institute) for editing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alkharouf, N., Khan, R., Matthews, B. (2004). Analysis of expressed sequence tags from roots of resistant soybean infected by the soybean cyst nematode. Genome 47, 380–388. doi: 10.1139/g03-114
Alkharouf, N. W., Klink, V. P., Chouikha, I. B., Beard, H. S., Macdonald, M. H., Meyer, S., et al. (2006). Timecourse microarray analyses reveal global changes in gene expression of susceptible Glycine max (soybean) roots during infection by Heterodera glycines (soybean cyst nematode). Planta 224, 838–852. doi: 10.1007/s00425-006-0270-8
Almeida, N. F., Krezdorn, N., Rotter, B., Winter, P., Rubiales, D., Vaz Patto, M. C. (2015). Lathyrus sativus transcriptome resistance response to Ascochyta lathyri investigated by deepSuperSAGE analysis. Front. Plant Sci. 6, 178. doi: 10.3389/fpls.2015.00178
Almeida, N. F., Trindade Leitao, S., Caminero, C., Torres, A. M., Rubiales, D., Vaz Patto, M. C. (2014). Transferability of molecular markers from major legumes to Lathyrus spp. for their application in mapping and diversity studies. Mol. Biol. Rep. 41, 269–283. doi: 10.1007/s11033-013-2860-4
Alves, M. S., Soares, Z. G., Vidigal, P. M. P., Barros, E. G., Poddanosqui, A. M. P., Aoyagi, L. N., et al. (2015). Differential expression of four soybean bZIP genes during Phakopsora pachyrhizi infection. Funct. Integr. Genomics 15, 685–696. doi: 10.1007/s10142-015-0445-0
Aoyagi, L. N., Lopes-Caitar, V. S., De Carvalho, M. C., Darben, L. M., Polizel-Podanosqui, A., Kuwahara, M. K., et al. (2014). Genomic and transcriptomic characterization of the transcription factor family R2R3-MYB in soybean and its involvement in the resistance responses to Phakopsora pachyrhizi. Plant Sci. 229, 32–42. doi: 10.1016/j.plantsci.2014.08.005
Asamizu, E., Nakamura, Y., Sato, S., Tabata, S. (2004). Characteristics of the Lotus Japonicus Gene Repertoire Deduced from Large-Scale Expressed Sequence Tag (EST) Analysis. Plant Mol. Biol. 54, 405–414. doi: 10.1023/B:PLAN.0000036372.46942.b8
Ashraf, N., Basu, S., Narula, K., Ghosh, S., Tayal, R., Gangisetty, N., et al. (2018). Integrative network analyses of wilt transcriptome in chickpea reveal genotype dependent regulatory hubs in immunity and susceptibility. Sci. Rep. 8, 6528. doi: 10.1038/s41598-018-19919-5
Ashraf, N., Ghai, D., Barman, P., Basu, S., Gangisetty, N., Mandal, M. K., et al. (2009). Comparative analyses of genotype dependent expressed sequence tags and stress-responsive transcriptome of chickpea wilt illustrate predicted and unexpected genes and novel regulators of plant immunity. BMC Genomics 10, 415. doi: 10.1186/1471-2164-10-415
Atienza, S. G., Palomino, C., Gutiérrez, N., Alfaro, C. M., Rubiales, D., Torres, A. M., et al. (2016). QTLs for ascochyta blight resistance in faba bean (Vicia faba L.): validation in field and controlled conditions. Crop Pasture Sci. 67, 216–224. doi: 10.1071/CP15227
Atwell, S., Huang, Y. S., Vilhjalmsson, B. J., Willems, G., Horton, M., Li, Y., et al. (2010). Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465, 627–631. doi: 10.1038/nature08800
Babu, M., Gagarinova, A. G., Brandle, J. E., Wang, A. (2008). Association of the transcriptional response of soybean plants with soybean mosaic virus systemic infection. J. Gen. Virol. 89, 1069–1080. doi: 10.1099/vir.0.83531-0
Barilli, E., Cobos, M. J., Carrillo, E., Kilian, A., Carling, J., Rubiales, D. (2018). A High-Density Integrated DArTseq SNP-Based Genetic Map of Pisum fulvum and Identification of QTLs Controlling Rust Resistance. Front. Plant Sci. 9, 167. doi: 10.3389/fpls.2018.00167
Belhaj, K., Chaparro-Garcia, A., Kamoun, S., Patron, N. J., Nekrasov, V. (2015). Editing plant genomes with CRISPR/Cas9. Curr. Opin. Biotechnol. 32, 76–84. doi: 10.1016/j.copbio.2014.11.007
Ben, C., Toueni, M., Montanari, S., Tardin, M. C., Fervel, M., Negahi, A., et al. (2013). Natural diversity in the model legume Medicago truncatula allows identifying distinct genetic mechanisms conferring partial resistance to Verticillium wilt. J. Exp. Bot. 64, 317–332. doi: 10.1093/jxb/ers337
Bencke-Malato, M., Cabreira, C., Wiebke-Strohm, B., Bucker-Neto, L., Mancini, E., Osorio, M. B., et al. (2014). Genome-wide annotation of the soybean WRKY family and functional characterization of genes involved in response to Phakopsora pachyrhizi infection. BMC Plant Biol. 14, 236. doi: 10.1186/s12870-014-0236-0
Beneventi, M. A., Da Silva, O. B., De Sá, M. E. L., Firmino, A.a.P., De Amorim, R. M. S., Albuquerque, É.V.S., et al. (2013). Transcription profile of soybean-root-knot nematode interaction reveals a key role of phythormones in the resistance reaction. BMC Genomics 14, 322. doi: 10.1186/1471-2164-14-322
Bhadauria, V., Maclachlan, R., Pozniak, C., Cohen-Skalie, A., Li, L., Halliday, J., et al. (2019). Genetic map-guided genome assembly reveals a virulence-governing minichromosome in the lentil anthracnose pathogen Colletotrichum lentis. New Phytol. 221, 431–445. doi: 10.1111/nph.15369
Bhadauria, V., Vijayan, P., Wei, Y., Banniza, S. (2017). Transcriptome analysis reveals a complex interplay between resistance and effector genes during the compatible lentil-Colletotrichum lentis interaction. Sci. Rep. 7, 42338. doi: 10.1038/srep42338
Bilgin, D. D., Aldea, M., O'neill, B. F., Benitez, M., Li, M., Clough, S. J., et al. (2008). Elevated ozone alters soybean-virus interaction. Mol. Plant Microbe Interact. 21, 1297–1308. doi: 10.1094/MPMI-21-10-1297
Bonhomme, M., Andre, O., Badis, Y., Ronfort, J., Burgarella, C., Chantret, N., et al. (2014). High-density genome-wide association mapping implicates an F-box encoding gene in Medicago truncatula resistance to Aphanomyces euteiches. New Phytol. 201, 1328–1342. doi: 10.1111/nph.12611
Branca, A., Paape, T. D., Zhou, P., Briskine, R., Farmer, A. D., Mudge, J., et al. (2011). Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc. Natl. Acad. Sci. U. S. A. 108, E864–E870. doi: 10.1073/pnas.1104032108
Campe, R., Loehrer, M., Conrath, U., Goellner, K. (2014). Phakopsora pachyrhizi induces defense marker genes to necrotrophs in Arabidopsis thaliana. Physiol. Mol. Plant Pathol. 87, 1–8. doi: 10.1016/j.pmpp.2014.04.005
Chang, C., Tian, L., Ma, L., Li, W., Nasir, F., Li, X., et al. (2019). Differential responses of molecular mechanisms and physiochemical characters in wild and cultivated soybeans against invasion by the pathogenic Fusarium oxysporum Schltdl. Physiol. Plant 166, 1008–1025. doi: 10.1111/ppl.12870
Chang, H. X., Lipka, A. E., Domier, L. L., Hartman, G. L. (2016). Characterization of disease resistance loci in the USDA soybean germplasm collection using genome-wide association studies. Phytopathology 106, 1139–1151. doi: 10.1094/PHYTO-01-16-0042-FI
Chatnaparat, T., Prathuangwong, S., Lindow, S. E. (2016). Global Pattern of gene expression of xanthomonas axonopodis pv. glycines within soybean leaves. Mol. Plant Microbe Interact. 29, 508–522. doi: 10.1094/MPMI-01-16-0007-R
Chen, H., Adam Arsovski, A., Yu, K., Wang, A. (2017). Deep sequencing leads to the identification of eukaryotic translation initiation factor 5A as a key element in Rsv1-mediated lethal systemic hypersensitive response to Soybean mosaic virus infection in soybean. Mol. Plant Pathol. 18, 391–404. doi: 10.1111/mpp.12407
Chen, H., Arsovski, A. A., Yu, K., Wang, A. (2016). Genome-wide investigation using sRNA-Seq, Degradome-Seq and transcriptome-seq reveals regulatory networks of microRNAs and their target genes in soybean during soybean mosaic virus Infection. PLOS One 11, e0150582. doi: 10.1371/journal.pone.0150582
Chen, Y., Ren, X., Zhou, X., Huang, L., Yan, L., Lei, Y., et al.. (2014). Dynamics in the resistant and susceptible peanut (Arachis hypogaea L.) root transcriptome on infection with the Ralstonia solanacearum. BMC Genomics 15, 1078. doi: 10.1186/1471-2164-15-1078
Cheng, X., Wang, M., Lee, H. K., Tadege, M., Ratet, P., Udvardi, M., et al. (2014). An efficient reverse genetics platform in the model legume Medicago truncatula. New Phytol. 201, 1065–1076. doi: 10.1111/nph.12575
Choi, H. K., Kim, D., Uhm, T., Limpens, E., Lim, H., Mun, J. H., et al. (2004). A sequence-based genetic map of Medicago truncatula and comparison of marker colinearity with M. sativa. Genetics 166, 1463–1502. doi: 10.1534/genetics.166.3.1463
Choi, J. J., Alkharouf, N. W., Schneider, K. T., Matthews, B. F., Frederick, R. D. (2008). Expression patterns in soybean resistant to Phakopsora pachyrhizi reveal the importance of peroxidases and lipoxygenases. Funct. Integr. Genomics 8, 341–359. doi: 10.1007/s10142-008-0080-0
Cobb, J. N., Biswas, P. S., Platten, J. D. (2019). Back to the future: revisiting MAS as a tool for modern plant breeding. Theor. Appl. Genet. 132, 647–667. doi: 10.1007/s00122-018-3266-4
Cobos, J. M., Satovic, Z., Rubiales, D., Fondevilla, S. (2018). Er3 gene, conferring resistance to powdery mildew in pea, is located in pea LGIV. Euphytica 214, 203. doi: 10.1007/s10681-018-2292-2
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Copley, T. R., Aliferis, K. A., Kliebenstein, D. J., Jabaji, S. H. (2017). An integrated RNAseq-(1)H NMR metabolomics approach to understand soybean primary metabolism regulation in response to Rhizoctonia foliar blight disease. BMC Plant Biol. 17, 84. doi: 10.1186/s12870-017-1020-8
Coyne, C. J., Porter, L. D., Boutet, G., Ma, Y., Mcgee, R. J., Lesne, A., et al. (2019). Confirmation of Fusarium root rot resistance QTL Fsp-Ps 2.1 of pea under controlled conditions. BMC Plant Biol. 19, 98. doi: 10.1186/s12870-019-1699-9
Curtin, S. J. (2018). Editing the medicago truncatula genome: targeted mutagenesis using the CRISPR-Cas9 reagent. Methods Mol. Biol. 1822, 161–174. doi: 10.1007/978-1-4939-8633-0_12
Curtin, S. J., Xiong, Y., Michno, J. M., Campbell, B. W., Stec, A. O., Cermak, T., et al. (2018). CRISPR/Cas9 and TALENs generate heritable mutations for genes involved in small RNA processing of Glycine max and Medicago truncatula. Plant Biotechnol. J. 16, 1125–1137. doi: 10.1111/pbi.12857
Das, S., Ehlers, J. D., Close, T. J., Roberts, P. A. (2010). Transcriptional profiling of root-knot nematode induced feeding sites in cowpea (Vigna unguiculata L. Walp.) using a soybean genome array. BMC Genomics 11, 480. doi: 10.1186/1471-2164-11-480
Davey, J. W., Blaxter, M. L. (2010). RADSeq: next-generation population genetics. Brief. Funct. Genomics 9, 416–423. doi: 10.1093/bfgp/elq031
Desgroux, A., Baudais, V. N., Aubert, V., Le Roy, G., De Larambergue, H., Miteul, H., et al. (2017). Comparative genome-wide-association mapping identifies common loci controlling root system architecture and resistance to aphanomyces euteiches in pea. Front. Plant Sci. 8, 2195. doi: 10.3389/fpls.2017.02195
Desgroux, A., L'anthoene, V., Roux-Duparque, M., Riviere, J. P., Aubert, G., Tayeh, N., et al. (2016). Genome-wide association mapping of partial resistance to Aphanomyces euteiches in pea. BMC Genomics 17, 124. doi: 10.1186/s12864-016-2429-4
Ek, M., Eklund, M., Von Post, R., Dayteg, C., Henriksson, T., Weibull, P., et al.. (2005). Microsatellite markers for powdery mildew resistance in pea (Pisum sativum L.). Hereditas 142, 86–91. doi: 10.1111/j.1601-5223.2005.01906.x
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379. doi: 10.1371/journal.pone.0019379
Fan, S., Jiang, L., Wu, J., Dong, L., Cheng, Q., Xu, P., et al. (2015). A novel pathogenesis-related class 10 protein gly m 4l, increases resistance upon phytophthora sojae infection in soybean (Glycine max [L.] Merr.). PLoS One 10, e0140364. doi: 10.1371/journal.pone.0140364
Fang, Y., Tyler, B. M. (2016). Efficient disruption and replacement of an effector gene in the oomycete Phytophthora sojae using CRISPR/Cas9 . Mol. Plant Pathol. 17, 127–139. doi: 10.1111/mpp.12318
Feng, J., Chang, K. F., Hwang, S. F., Strelkov, S. E., Conner, R. L., Gossen, B. D., et al. (2012). Analysis of expressed sequence tags derived from pea leaves infected by Peronospora viciae f. sp. pisi. Annals of Applied Biology 161, 214–222. doi: 10.1111/j.1744-7348.2012.00566.x
Fondevilla, S., Kuster, H., Krajinski, F., Cubero, J. I., Rubiales, D. (2011). Identification of genes differentially expressed in a resistant reaction to Mycosphaerella pinodes in pea using microarray technology. BMC Genomics 12, 28. doi: 10.1186/1471-2164-12-28
Fondevilla, S., Rotter, B., Krezdorn, N., Jüngling, R., Winter, P., Rubiales, D. (2014). Identification of genes involved in resistance to didymella pinodes in pea by deepSuperSAGE transcriptome profiling. Plant Mol. Biol. Rep. 32, 258–269. doi: 10.1007/s11105-013-0644-6
Fondevilla, S., Rubiales, D. (2012). Powdery mildew control in pea. Rev. Agron. Sustainable Dev. 32, 401–409. doi: 10.1007/s13593-011-0033-1
Fonseca, J. P., Mysore, K. S. (2019). Genes involved in nonhost disease resistance as a key to engineer durable resistance in crops. Plant Sci. 279, 108–116. doi: 10.1016/j.plantsci.2018.07.002
Gamas, P., Debellé, F., Berges, H., Godiard, L., Niebel, A., Journet, E., et al. (2006). “Medicago truncatula cDNA and genomic libraries,” in The Medicago truncatula handbook. Eds. Mathesius, U., Journet, E., Sumner, L.W..
Ganopoulos, I., Mylona, P., Mellidou, I., Kalivas, A., Bosmali, I., Kontzidou, S., et al. (2018). Microsatellite genotyping and molecular screening of pea (Pisum sativum L.) germplasm with high-resolution melting analysis for resistance to powdery mildew. Plant Gene 15, 1–5. doi: 10.1016/j.plgene.2018.05.004
Gill, U. S., Lee, S., Mysore, K. S. (2015). Host versus nonhost resistance: distinct wars with similar arsenals. Phytopathology 105, 580–587. doi: 10.1094/PHYTO-11-14-0298-RVW
Glaubitz, J. C., Casstevens, T. M., Lu, F., Harriman, J., Elshire, R. J., Sun, Q., et al. (2014). TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9, e90346. doi: 10.1371/journal.pone.0090346
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Graham, P. H., Vance, C. P. (2003). Legumes: importance and constraints to greater use. Plant Physiol. 131, 872–877. doi: 10.1104/pp.017004
Guimaraes, P. M., Guimaraes, L. A., Morgante, C. V., Silva, O. B., Araujo, A. C., Martins, A. C., et al. (2015). Root transcriptome analysis of wild peanut reveals candidate genes for nematode resistance. PLoS One 10, e0140937. doi: 10.1371/journal.pone.0140937
Guo, N., Ye, W. W., Wu, X. L., Shen, D. Y., Wang, Y. C., Xing, H., et al. (2011). Microarray profiling reveals microRNAs involving soybean resistance to Phytophthora sojae. Genome 54, 954–958. doi: 10.1139/g11-050
Gupta, S., Bhar, A., Chatterjee, M., Ghosh, A., Das, S. (2017). Transcriptomic dissection reveals wide spread differential expression in chickpea during early time points of Fusarium oxysporum f. sp. ciceri Race 1 attack. PLoS One 12, e0178164. doi: 10.1371/journal.pone.0178164
Gupta, S., Chakraborti, D., Rangi, R. K., Basu, D., Das, S. (2009). A molecular insight into the early events of chickpea (Cicer arietinum) and Fusarium oxysporum f. sp. ciceri (race 1) interaction through cDNA-AFLP analysis. Phytopathology 99, 1245–1257. doi: 10.1094/PHYTO-99-11-1245
Gupta, S., Chakraborti, D., Sengupta, A., Basu, D., Das, S. (2010). Primary metabolism of chickpea is the initial target of wound inducing early sensed Fusarium oxysporum f. sp. ciceri race I. PLoS One 5, e9030. doi: 10.1371/journal.pone.0009030
Gurjar, G. S., Giri, A. P., Gupta, V. S. (2012). Gene Expression profiling during wilting in chickpea caused by fusarium oxysporum F. sp. Ciceri. Am. J. Plant Sci. 3, 190–201. doi: 10.4236/ajps.2012.32023
Hamon, C., Coyne, C. J., Mcgee, R. J., Lesne, A., Esnault, R., Mangin, P., et al. (2013). QTL meta-analysis provides a comprehensive view of loci controlling partial resistance to Aphanomyces euteiches in four sources of resistance in pea. BMC Plant Biol. 13, 45. doi: 10.1186/1471-2229-13-45
Heigwer, F., Kerr, G., Boutros, M. (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123. doi: 10.1038/nmeth.2812
Hosseini, S., Elfstrand, M., Heyman, F., Funck Jensen, D., Karlsson, M. (2015). Deciphering common and specific transcriptional immune responses in pea towards the oomycete pathogens Aphanomyces euteiches and Phytophthora pisi. BMC Genomics 16, 627. doi: 10.1186/s12864-015-1829-1
Huang, J., Yan, L., Lei, Y., Jiang, H., Ren, X., Liao, B. (2012). Expressed sequence tags in cultivated peanut (Arachis hypogaea): discovery of genes in seed development and response to Ralstonia solanacearum challenge. J. Plant Res. 125, 755–769. doi: 10.1007/s10265-012-0491-9
Huang, X., Wei, X., Sang, T., Zhao, Q., Feng, Q., Zhao, Y., et al. (2010). Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967. doi: 10.1038/ng.695
Humphry, M., Reinstadler, A., Ivanov, S., Bisseling, T., Panstruga, R. (2011). Durable broad-spectrum powdery mildew resistance in pea er1 plants is conferred by natural loss-of-function mutations in PsMLO1. Mol. Plant Pathol. 12, 866–878. doi: 10.1111/j.1364-3703.2011.00718.x
Ibrahim, H. M., Hosseini, P., Alkharouf, N. W., Hussein, E. H., Gamal El-Din Ael, K., Aly, M. A., et al. (2011). Analysis of gene expression in soybean (Glycine max) roots in response to the root knot nematode Meloidogyne incognita using microarrays and KEGG pathways. BMC Genomics 12, 220. doi: 10.1186/1471-2164-12-220
Ishiga, Y., Uppalapati, S. R., Gill, U. S., Huhman, D., Tang, Y., Mysore, K. S. (2015). Transcriptomic and metabolomic analyses identify a role for chlorophyll catabolism and phytoalexin during Medicago nonhost resistance against Asian soybean rust. Sci. Rep. 5, 13061. doi: 10.1038/srep13061
Ishiga, Y., Upplapapti, S., Mysore, K. S. (2013). Expression analysis reveals a role for hydrophobic or epicuticular wax signals in pre-penetration structure formation of Phakopsora pachyrhizi. Plant Signal Behav 8, e26959.
Ithal, N., Recknor, J., Nettleton, D., Hearne, L., Maier, T., Baum, T. J., et al. (2007). Parallel genome-wide expression profiling of host and pathogen during soybean cyst nematode infection of soybean. Mol. Plant Microbe Interact. 20, 293–305. doi: 10.1094/MPMI-20-3-0293
Jain, S., Chittem, K., Brueggeman, R., Osorno, J. M., Richards, J., Nelson, B. D., Jr. (2016). comparative transcriptome analysis of resistant and susceptible common bean genotypes in response to soybean cyst nematode infection. PLoS One 11, e0159338. doi: 10.1371/journal.pone.0159338
Jaulneau, V., Cazaux, M., Wong Sak Hoi, J., Fournier, S., Esquerre-Tugaye, M. T., Jacquet, C., et al. (2010). Host and nonhost resistance in Medicago-Colletotrichum interactions. Mol. Plant Microbe Interact. 23, 1107–1117. doi: 10.1094/MPMI-23-9-1107
Jha, A. B., Gali, K. K., Tar’an, B., Warkentin, T. D. (2017). Fine mapping of QTLs for ascochyta blight resistance in pea using heterogeneous inbred families. Front. Plant Sci. 8, 765. doi: 10.3389/fpls.2017.00765
Jha, A. B., Tar’an, B., Stonehouse, R., Warkentin, T. D. (2016). Identification of QTLs associated with improved resistance to ascochyta blight in an interspecific pea recombinant inbred line population. Crop Sci. 56, 2926–2939. doi: 10.2135/cropsci2016.01.0001
Ji, L., Jordan, W. T., Shi, X., Hu, L., He, C., Schmitz, R. J. (2018). TET-mediated epimutagenesis of the Arabidopsis thaliana methylome. Nat. Commun. 9, 895. doi: 10.1038/s41467-018-03289-7
Jiang, W., Zhou, H., Bi, H., Fromm, M., Yang, B., Weeks, D. P. (2013). Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41, e188. doi: 10.1093/nar/gkt780
Jones, J. D. G., Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kandoth, P. K., Ithal, N., Recknor, J., Maier, T., Nettleton, D., Baum, T. J., et al.. (2011). The Soybean Rhg1 locus for resistance to the soybean cyst nematode Heterodera glycines regulates the expression of a large number of stress- and defense-related genes in degenerating feeding cells. Plant Physiol. 155, 1960–1975. doi: 10.1104/pp.110.167536
Kang, Y., Sakiroglu, M., Krom, N., Stanton-Geddes, J., Wang, M., Lee, Y. C., et al. (2015). Genome-wide association of drought-related and biomass traits with HapMap SNPs in Medicago truncatula. Plant Cell Environ. 38, 1997–2011. doi: 10.1111/pce.12520
Kang, Y., Torres-Jerez, I., An, Z., Greve, V., Huhman, D., Krom, N., et al.. (2019). Genome-wide association analysis of salinity responsive traits in Medicago truncatula. Plant Cell Environ. 42, 1513–1531. doi: 10.1111/pce.13508
Khan, R., Alkharouf, N., Beard, H., Macdonald, M., Chouikha, I., Meyer, S., et al. (2004). Microarray analysis of gene expression in soybean roots susceptible to the soybean cyst nematode two days post invasion. J. Nematol. 36, 241–248.
Kido, E. A., Pandolfi, V., Houllou-Kido, L. M., Andrade, P. P., Marcelino, F. C., Nepomuceno, A. L., et al.. (2010). Plant antimicrobial peptides: an overview of SuperSAGE transcriptional profile and a functional review. Curr. Protein Pept. Sci. 11, 220–230. doi: 10.2174/138920310791112110
Kilian, A., Wenzl, P., Huttner, E., Carling, J., Xia, L., Blois, H., et al. (2012). Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol. Biol. 888, 67–89. doi: 10.1007/978-1-61779-870-2_5
Kilian, B., Graner, A. (2012). NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genomics 11, 38–50. doi: 10.1093/bfgp/elr046
Kim, K. H., Kang, Y. J., Kim, D. H., Yoon, M. Y., Moon, J. K., Kim, M. Y., et al. (2011). RNA-Seq analysis of a soybean near-isogenic line carrying bacterial leaf pustule-resistant and -susceptible alleles. DNA Res. 18, 483–497. doi: 10.1093/dnares/dsr033
Kleinstiver, B. P., Pattanayak, V., Prew, M. S., Tsai, S. Q., Nguyen, N. T., Zheng, Z., et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495. doi: 10.1038/nature16526
Klink, V. P., Hosseini, P., Macdonald, M. H., Alkharouf, N. W., Matthews, B. F. (2009a). Population-specific gene expression in the plant pathogenic nematode Heterodera glycines exists prior to infection and during the onset of a resistant or susceptible reaction in the roots of the Glycine max genotype Peking. BMC Genomics 10, 111. doi: 10.1186/1471-2164-10-111
Klink, V. P., Hosseini, P., Matsye, P., Alkharouf, N. W., Matthews, B. F. (2009b). A gene expression analysis of syncytia laser microdissected from the roots of the Glycine max (soybean) genotype PI 548402 (Peking) undergoing a resistant reaction after infection by Heterodera glycines (soybean cyst nematode). Plant Mol. Biol. 71, 525–567. doi: 10.1007/s11103-009-9539-1
Klink, V. P., Hosseini, P., Matsye, P. D., Alkharouf, N. W., Matthews, B. F. (2010). Syncytium gene expression in Glycine max([PI 88788]) roots undergoing a resistant reaction to the parasitic nematode Heterodera glycines. Plant Physiol. Biochem. 48, 176–193. doi: 10.1016/j.plaphy.2009.12.003
Klink, V. P., Overall, C. C., Alkharouf, N. W., Macdonald, M. H., Matthews, B. F. (2007a). Laser capture microdissection (LCM) and comparative microarray expression analysis of syncytial cells isolated from incompatible and compatible soybean (Glycine max) roots infected by the soybean cyst nematode (Heterodera glycines). Planta 226, 1389–1409. doi: 10.1007/s00425-007-0578-z
Klink, V. P., Overall, C. C., Alkharouf, N. W., Macdonald, M. H., Matthews, B. F. (2007b). A time-course comparative microarray analysis of an incompatible and compatible response by Glycine max (soybean) to Heterodera glycines (soybean cyst nematode) infection. Planta 226, 1423–1447. doi: 10.1007/s00425-007-0581-4
Knott, G. J., Doudna, J. A. (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. doi: 10.1126/science.aat5011
Kou, Y., Wang, S. (2010). Broad-spectrum and durability: understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 13, 181–185. doi: 10.1016/j.pbi.2009.12.010
Kou, Y., Wang, S. (2012). Toward an understanding of the molecular basis of quantitative disease resistance in rice. J. Biotechnol. 159, 283–290. doi: 10.1016/j.jbiotec.2011.07.002
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Langenbach, C., Campe, R., Beyer, S. F., Mueller, A. N., Conrath, U. (2016a). Fighting asian soybean rust. Front. Plant Sci. 7, 797. doi: 10.3389/fpls.2016.00797
Langenbach, C., Campe, R., Schaffrath, U., Goellner, K., Conrath, U. (2013). UDP-glucosyltransferase UGT84A2/BRT1 is required for Arabidopsis nonhost resistance to the Asian soybean rust pathogen Phakopsora pachyrhizi. New Phytol. 198, 536–545. doi: 10.1111/nph.12155
Langenbach, C., Schultheiss, H., Rosendahl, M., Tresch, N., Conrath, U., Goellner, K. (2016b). Interspecies gene transfer provides soybean resistance to a fungal pathogen. Plant Biotechnol. J. 14, 699–708. doi: 10.1111/pbi.12418
Lanubile, A., Muppirala, U. K., Severin, A. J., Marocco, A., Munkvold, G. P. (2015). Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genomics 16, 1089. doi: 10.1186/s12864-015-2318-2
Lavaud, C., Baviere, M., Le Roy, G., Herve, M. R., Moussart, A., Delourme, R., et al. (2016). Single and multiple resistance QTL delay symptom appearance and slow down root colonization by Aphanomyces euteiches in pea near isogenic lines. BMC Plant Biol. 16, 166. doi: 10.1186/s12870-016-0822-4
Lavaud, C., Lesne, A., Piriou, C., Le Roy, G., Boutet, G., Moussart, A., et al. (2015). Validation of QTL for resistance to Aphanomyces euteiches in different pea genetic backgrounds using near-isogenic lines. Theor. Appl. Genet. 128, 2273–2288. doi: 10.1007/s00122-015-2583-0
Lee, H. A., Lee, H. Y., Seo, E., Lee, J., Kim, S. B., Oh, S., et al. (2017). Current understandings of plant nonhost resistance. Mol. Plant Microbe Interact. 30, 5–15. doi: 10.1094/MPMI-10-16-0213-CR
Li, J., Zhang, Y., Zhang, Y., Yu, P. L., Pan, H., Rollins, J. A. (2018a). Introduction of large sequence inserts by crispr-cas9 to create pathogenicity mutants in the multinucleate filamentous pathogen sclerotinia sclerotiorum. Mbio 9. doi: 10.1128/mBio.00567-18
Li, S., Chen, Y., Zhu, X., Wang, Y., Jung, K. H., Chen, L., et al. (2018b). The transcriptomic changes of Huipizhi Heidou (Glycine max), a nematode-resistant black soybean during Heterodera glycines race 3 infection. J. Plant Physiol. 220, 96–104. doi: 10.1016/j.jplph.2017.11.001
Li, Y., Sun, S., Zhong, C., Wang, X., Wu, X., Zhu, Z. (2017). Genetic mapping and development of co-segregating markers of RpsQ, which provides resistance to Phytophthora sojae in soybean. Theor. Appl. Genet. 130, 1223–1233. doi: 10.1007/s00122-017-2883-7
Libault, M., Dickstein, R., (2014). “Advances in Functional Genomics in Legumes,” in Legumes in the Omic Era. Eds. Gupta, S., Nadarajan, N., Gupta, D. S. (New York, NY: Springer New York), 15–39. doi: 10.1007/978-1-4614-8370-0_2
Lin, F., Zhao, M., Baumann, D. D., Ping, J., Sun, L., Liu, Y., et al.. (2014). Molecular response to the pathogen Phytophthora sojae among ten soybean near isogenic lines revealed by comparative transcriptomics. BMC Genomics 15, 18. doi: 10.1186/1471-2164-15-18
Lipka, A. E., Tian, F., Wang, Q., Peiffer, J., Li, M., Bradbury, P. J., et al.. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399. doi: 10.1093/bioinformatics/bts444
Loehrer, M., Langenbach, C., Goellner, K., Conrath, U., Schaffrath, U. (2008). Characterization of nonhost resistance of Arabidopsis to the Asian soybean rust. Mol. Plant Microbe Interact. 21, 1421–1430. doi: 10.1094/MPMI-21-11-1421
Lygin, A. V., Li, S., Vittal, R., Widholm, J. M., Hartman, G. L., Lozovaya, V. V. (2009). The importance of phenolic metabolism to limit the growth of Phakopsora pachyrhizi. Phytopathology 99, 1412–1420. doi: 10.1094/PHYTO-99-12-1412
Ma, X., Zhu, Q., Chen, Y., Liu, Y. G. (2016). CRISPR/Cas9 Platforms for Genome Editing in Plants: Developments and Applications. Mol. Plant 9, 961–974. doi: 10.1016/j.molp.2016.04.009
Madrid, E., Gil, J., Rubiales, D., Krajinski, F., Schlereth, A., Millan, T. (2010). Transcription factor profiling leading to the identification of putative transcription factors involved in the Medicago truncatula-Uromyces striatus interaction. Theor. Appl. Genet. 121, 1311–1321. doi: 10.1007/s00122-010-1390-x
Madrid, E., Palomino, C., Plötner, A., Horres, R., Rotter, B., Winter, P., et al.. (2013). DeepSuperSage analysis of the Vicia faba transcriptome in response to Ascochyta fabae infection. Phytopathol. Mediterr.
Martin, K., Singh, J., Hill, J. H., Whitham, S. A., Cannon, S. B. (2016). Dynamic transcriptome profiling of Bean Common Mosaic Virus (BCMV) infection in Common Bean (Phaseolus vulgaris L.). BMC Genomics 17, 613. doi: 10.1186/s12864-016-2976-8
Matsye, P. D., Kumar, R., Hosseini, P., Jones, C. M., Tremblay, A., Alkharouf, N. W., et al. (2011). Mapping cell fate decisions that occur during soybean defense responses. Plant Mol. Biol. 77, 513–528. doi: 10.1007/s11103-011-9828-3
Matthews, B. F., Beard, H., Macdonald, M. H., Kabir, S., Youssef, R. M., Hosseini, P., et al.. (2013). Engineered resistance and hypersusceptibility through functional metabolic studies of 100 genes in soybean to its major pathogen, the soybean cyst nematode. Planta 237, 1337–1357. doi: 10.1007/s00425-013-1840-1
Mazarei, M., Liu, W., Al-Ahmad, H., Arelli, P. R., Pantalone, V. R., Stewart, C. N., Jr. (2011). Gene expression profiling of resistant and susceptible soybean lines infected with soybean cyst nematode. Theor. Appl. Genet. 123, 1193–1206. doi: 10.1007/s00122-011-1659-8
Mehrtens, F., Kranz, H., Bednarek, P., Weisshaar, B. (2005). The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 138, 1083–1096. doi: 10.1104/pp.104.058032
Michno, J. M., Wang, X., Liu, J., Curtin, S. J., Kono, T. J., Stupar, R. M. (2015). CRISPR/Cas mutagenesis of soybean and Medicago truncatula using a new web-tool and a modified Cas9 enzyme. GM Crops Food 6, 243–252. doi: 10.1080/21645698.2015.1106063
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M., Valen, E. (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407. doi: 10.1093/nar/gku410
Morales, A.M.a.P., O'rourke, J. A., Van De Mortel, M., Scheider, K. T., Bancroft, T. J., Borém, A., et al. (2013). Transcriptome analyses and virus induced gene silencing identify genes in the Rpp4-mediated Asian soybean rust resistance pathway. Funct. Plant Biol. 40, 1029–1047. doi: 10.1071/FP12296
Moy, P., Qutob, D., Chapman, B. P., Atkinson, I., Gijzen, M. (2004). Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Mol. Plant Microbe Interact. 17, 1051–1062. doi: 10.1094/MPMI.2004.17.10.1051
Narayanan, N. N., Grosic, S., Tasma, I. M., Grant, D., Shoemaker, R., Bhattacharyya, M. K. (2009). Identification of candidate signaling genes including regulators of chromosome condensation 1 protein family differentially expressed in the soybean-Phytophthora sojae interaction. Theor. Appl. Genet. 118, 399–412. doi: 10.1007/s00122-008-0895-z
Navarro, L., Jay, F., Nomura, K., He, S. Y., Voinnet, O. (2008). Suppression of the microRNA pathway by bacterial effector proteins. Science 321, 964–967. doi: 10.1126/science.1159505
Ndeve, A. D., Santos, J. R. P., Matthews, W. C., Huynh, B. L., Guo, Y. N., Lo, S., et al. (2019). A novel root-knot nematode resistance QTL on chromosome Vu01 in Cowpea. G3: Genes, Genomes, Genet. 9, 1199–1209. doi: 10.1534/g3.118.200881
Negahi, A., Ben, C., Gentzbittel, L., Maury, P., Nabipour, A., Ebrahimi, A., et al. (2014). Quantitative trait loci associated with resistance to a potato isolate of Verticillium albo-atrum in Medicago truncatula. Plant Pathol. 63, 308–315. doi: 10.1111/ppa.12100
Nemchinov, L. G., Shao, J., Lee, M. N., Postnikova, O. A., Samac, D. A. (2017). Resistant and susceptible responses in alfalfa (Medicago sativa) to bacterial stem blight caused by Pseudomonas syringae pv. syringae. PLoS One 12, e0189781. doi: 10.1371/journal.pone.0189781
Nimbalkar, S. B., Harsulkar, A. M., Giri, A. P., Sainani, M. N., Franceschi, V., Gupta, V. S. (2006). Differentially expressed gene transcripts in roots of resistant and susceptible chickpea plant (Cicer arietinum L.) upon Fusarium oxysporum infection. Physiol. Mol. Plant Pathol. 68, 176–188. doi: 10.1016/j.pmpp.2006.10.003
Nyamsuren, O., Colditz, F., Rosendahl, S., Tamasloukht, M. B., Bekel, T., Meyer, F., et al. (2003). Transcriptional profiling of Medicago truncatula roots after infection with Aphanomyces euteiches (oomycota) identifies novel genes upregulated during this pathogenic interaction. Physiol. Mol. Plant Pathol. 63, 17–26. doi: 10.1016/j.pmpp.2003.09.001
Oblessuc, P. R., Borges, A., Chowdhury, B., Caldas, D. G., Tsai, S. M., Camargo, L. E., et al. (2012). Dissecting Phaseolus vulgaris innate immune system against Colletotrichum lindemuthianum infection. PLoS One 7, e43161. doi: 10.1371/journal.pone.0043161
Ocaña, S., Seoane, P., Bautista, R., Palomino, C., Claros, G. M., Torres, A. M., et al. (2015). Large-scale transcriptome analysis in faba bean (Vicia faba L.) under Ascochyta fabae Infection. PLOS One 10, e0135143. doi: 10.1371/journal.pone.0135143
Ohmido, N., Ishimaru, A., Kato, S., Sato, S., Tabata, S., Fukui, K. (2010). Integration of cytogenetic and genetic linkage maps of Lotus japonicus, a model plant for legumes. Chromosome Res. 18, 287–299. doi: 10.1007/s10577-009-9103-5
Padder, B. A., Kamfwa, K., Awale, H. E., Kelly, J. D. (2016). Transcriptome Profiling of the Phaseolus vulgaris - Colletotrichum lindemuthianum Pathosystem. PLoS One 11, e0165823. doi: 10.1371/journal.pone.0165823
Pandey, A. K., Yang, C., Zhang, C., Graham, M. A., Horstman, H. D., Lee, Y., et al. (2011). Functional analysis of the Asian soybean rust resistance pathway mediated by Rpp2. Mol. Plant Microbe Interact. 24, 194–206. doi: 10.1094/MPMI-08-10-0187
Panthee, D. R., Marois, J. J., Wright, D. L., Narvaez, D., Yuan, J. S., Stewart, C. N. Jr. (2009). Differential expression of genes in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) is soybean growth stage-specific. Theor. Appl. Genet. 118, 359–370. doi: 10.1007/s00122-008-0905-1
Patwa, N., Nithin, C., Bahadur, R. P., Basak, J. (2018). Identification and characterization of differentially expressed Phaseolus vulgaris miRNAs and their targets during mungbean yellow mosaic India virus infection reveals new insight into Phaseolus-MYMIV interaction. Genomics. doi: 10.1016/j.ygeno.2018.09.005
Pecrix, Y., Staton, S. E., Sallet, E., Lelandais-Briere, C., Moreau, S., Carrere, S., et al. (2018). Whole-genome landscape of Medicago truncatula symbiotic genes. Nat. Plants 4, 1017–1025. doi: 10.1038/s41477-018-0286-7
Perez-Nadales, E., Almeida Nogueira, M. F., Baldin, C., Castanheira, S., El Ghalid, M., Grund, E., et al. (2014). Fungal model systems and the elucidation of pathogenicity determinants. Fungal Genet. Biol. 70, 42–67. doi: 10.1016/j.fgb.2014.06.011
Perez-Pinera, P., Kocak, D. D., Vockley, C. M., Adler, A. F., Kabadi, A. M., Polstein, L. R., et al. (2013). RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976. doi: 10.1038/nmeth.2600
Perseguini, J. M., Oblessuc, P. R., Rosa, J. R., Gomes, K. A., Chiorato, A. F., Carbonell, S. A., et al. (2016). Genome-wide association studies of anthracnose and angular leaf spot resistance in common bean (Phaseolus vulgaris L.). PLoS One 11, e0150506. doi: 10.1371/journal.pone.0150506
Piatek, A., Ali, Z., Baazim, H., Li, L., Abulfaraj, A., Al-Shareef, S., et al. (2015). RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol. J. 13, 578–589. doi: 10.1111/pbi.12284
Pilet-Nayel, M. L., Prosperi, J. M., Hamon, C., Lesne, A., Lecointe, R., Le Goff, I., et al. (2009). AER1, a major gene conferring resistance to Aphanomyces euteiches in Medicago truncatula. Phytopathology 99, 203–208. doi: 10.1094/PHYTO-99-2-0203
Poland, J., Rutkoski, J. (2016). Advances and challenges in genomic selection for disease resistance. Annu. Rev. Phytopathol. 54, 79–98. doi: 10.1146/annurev-phyto-080615-100056
Polhill, R. M., Raven, P. H., Stirton, C. H., (1981). “Evolution and systematics of the Leguminosae,” in Advances in Legume Systematics. Eds. Polhill, R. M., Raven, P. H. (Royal Botanical Gardens, Kew), 1–26.
Postnikova, O. A., Hult, M., Shao, J., Skantar, A., Nemchinov, L. G. (2015). Transcriptome analysis of resistant and susceptible alfalfa cultivars infected with root-knot nematode Meloidogyne incognita. PLoS One 10, e0123157. doi: 10.1371/journal.pone.0118269
Potenza, C., Thomas, S. H., Sengupta-Gopalan, C. (2001). Genes induced during early response to Meloidogyne incognita in roots of resistant and susceptible alfalfa cultivars. Plant Sci. 161, 289–299. doi: 10.1016/S0168-9452(01)00415-0
Rey, T., Bonhomme, M., Chatterjee, A., Gavrin, A., Toulotte, J., Yang, W., et al. (2017). The Medicago truncatula GRAS protein RAD1 supports arbuscular mycorrhiza symbiosis and Phytophthora palmivora susceptibility. J. Exp. Bot. 68, 5871–5881. doi: 10.1093/jxb/erx398
Rincker, K., Lipka, A. E., Diers, B. W. (2016). Genome-Wide Association Study of Brown Stem Rot Resistance in Soybean across Multiple Populations. Plant Genome 9. doi: 10.3835/plantgenome2015.08.0064
Rodriguez-Leal, D., Lemmon, Z. H., Man, J., Bartlett, M. E., Lippman, Z. B. (2017). Engineering quantitative trait variation for crop improvement by genome editing. Cell 171, 470–480 e478. doi: 10.1016/j.cell.2017.08.030
Rubiales, D., Fondevilla, S., Chen, W., Gentzbittel, L., Higgins, T. J. V., Castillejo, M. A., et al. (2015). Achievements and challenges in legume breeding for pest and disease resistance. Crit. Rev. Plant Sci. 34, 195–236. doi: 10.1080/07352689.2014.898445
Samac, D. A., Penuela, S., Schnurr, J. A., Hunt, E. N., Foster-Hartnett, D., Vandenbosch, K. A., et al. (2011). Expression of coordinately regulated defence response genes and analysis of their role in disease resistance in Medicago truncatula. Mol. Plant Pathol. 12, 786–798. doi: 10.1111/j.1364-3703.2011.00712.x
Santini, L., Munhoz Cde, F., Bonfim, M. F., Brandao, M. M., Inomoto, M. M., Vieira, M. L. (2016). Host transcriptional profiling at early and later stages of the compatible interaction between phaseolus vulgaris and meloidogyne incognita. Phytopathology 106, 282–294. doi: 10.1094/PHYTO-07-15-0160-R
Santos, J. R. P., Ndeve, A. D., Huynh, B. L., Matthews, W. C., Roberts, P. A. (2018). QTL mapping and transcriptome analysis of cowpea reveals candidate genes for root-knot nematode resistance. PLoS One 13, e0189185. doi: 10.1371/journal.pone.0189185
Sato, S., Nakamura, Y., Asamizu, E., Isobe, S., Tabata, S. (2007). Genome sequencing and genome resources in model legumes. Plant Physiol. 144, 588–593. doi: 10.1104/pp.107.097493
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Kato, T., Nakao, M., et al. (2008). Genome Structure of the Legume, Lotus japonicus. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 15, 227–239. doi: 10.1093/dnares/dsn008
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Senthil-Kumar, M., Mysore, K. S. (2013). Nonhost resistance against bacterial pathogens: retrospectives and prospects. Annu. Rev. Phytopathol. 51, 407–427. doi: 10.1146/annurev-phyto-082712-102319
Shah, N., Hirakawa, H., Kusakabe, S., Sandal, N., Stougaard, J., Schierup, M. H., et al. (2016). High-resolution genetic maps of Lotus japonicus and L. burttii based on re-sequencing of recombinant inbred lines. DNA Res. doi: 10.1093/dnares/dsw033
Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K., Liang, Z., et al. (2013). Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 31, 686–688. doi: 10.1038/nbt.2650
Singh, N. K., Kumar, K. R., Kumar, D., Shukla, P., Kirti, P. B. (2013). Characterization of a pathogen induced thaumatin-like protein gene AdTLP from Arachis diogoi, a wild peanut. PLoS One 8, e83963. doi: 10.1371/journal.pone.0083963
Soria-Guerra, R. E., Rosales-Mendoza, S., Chang, S., Haudenshield, J. S., Padmanaban, A., Rodriguez-Zas, S., et al. (2010a). Transcriptome analysis of resistant and susceptible genotypes of Glycine tomentella during Phakopsora pachyrhizi infection reveals novel rust resistance genes. Theor. Appl. Genet. 120, 1315–1333. doi: 10.1007/s00122-009-1258-0
Soria-Guerra, R. E., Rosales-Mendoza, S., Chang, S., Haudenshield, J. S., Zheng, D., Rao, S. S., et al. (2010b). Identifying differentially expressed genes in leaves of Glycine tomentella in the presence of the fungal pathogen Phakopsora pachyrhizi. Planta 232, 1181–1189. doi: 10.1007/s00425-010-1251-5
Stanton-Geddes, J., Paape, T., Epstein, B., Briskine, R., Yoder, J., Mudge, J., et al.. (2013). Candidate genes and genetic architecture of symbiotic and agronomic traits revealed by whole-genome, sequence-based association genetics in Medicago truncatula. PLoS One 8, e65688. doi: 10.1371/journal.pone.0065688
Sudheesh, S., Lombardi, M., Leonforte, A., Cogan, N. O. I., Materne, M., Forste, J. W. (2015). Consensus genetic map construction for field pea (Pisum sativum L.), trait dissection of biotic and abiotic stress tolerance and development of a diagnostic marker for the er1 powdery mildew resistance gene. Plant Mol. Biol. Rep. 33, 1391–1403. doi: 10.1007/s11105-014-0837-7
Sumit, R., Sahu, B. B., Xu, M., Sandhu, D., Bhattacharyya, M. K. (2012). Arabidopsis nonhost resistance gene PSS1 confers immunity against an oomycete and a fungal pathogen but not a bacterial pathogen that cause diseases in soybean. BMC Plant Biol. 12, 87. doi: 10.1186/1471-2229-12-87
Sun, L., Gill, U. S., Nandety, R. S., Kwon, S., Mehta, P., Dickstein, R., et al. (2019). Genome-wide analysis of flanking sequences reveals that Tnt1 insertion is positively correlated with gene methylation in Medicago truncatula. Plant J. doi: 10.1111/tpj.14291
Sun, S., Deng, D., Wang, Z., Duan, C., Wu, X., Wang, X., et al. (2016a). A novel er1 allele and the development and validation of its functional marker for breeding pea (Pisum sativum L.) resistance to powdery mildew. Theor. Appl. Genet. 129, 909–919. doi: 10.1007/s00122-016-2671-9
Sun, S., Fu, H., Wang, Z., Duan, C., Zong, X., Zhu, Z. (2016b). Discovery of a novel er1 allele conferring powdery mildew resistance in chinese pea (Pisum sativum L.) Landraces. PLoS One 11, e0147624. doi: 10.1371/journal.pone.0147624
Swaminathan, S., Abeysekara, N. S., Knight, J. M., Liu, M., Dong, J., Hudson, M. E., et al. (2018). Mapping of new quantitative trait loci for sudden death syndrome and soybean cyst nematode resistance in two soybean populations. Theor. Appl. Genet. 131, 1047–1062. doi: 10.1007/s00122-018-3057-y
Tadege, M., Wen, J., He, J., Tu, H., Kwak, Y., Eschstruth, A., et al. (2008). Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula. Plant J. 54, 335–347. doi: 10.1111/j.1365-313X.2008.03418.x
Tang, Y., Liu, X., Wang, J., Li, M., Wang, Q., Tian, F., et al. (2016). GAPIT Version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9. doi: 10.3835/plantgenome2015.11.0120
Tian, B., Wang, S., Todd, T. C., Johnson, C. D., Tang, G., Trick, H. N. (2017). Genome-wide identification of soybean microRNA responsive to soybean cyst nematodes infection by deep sequencing. BMC Genomics 18, 572. doi: 10.1186/s12864-017-3963-4
Tian, F., Bradbury, P. J., Brown, P. J., Hung, H., Sun, Q., Flint-Garcia, S., et al. (2011). Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43, 159–162. doi: 10.1038/ng.746
Timmerman-Vaughan, G. M., Moya, L., Frew, T. J., Murray, S. R., Crowhurst, R. (2016). Ascochyta blight disease of pea (Pisum sativum L.): defence-related candidate genes associated with QTL regions and identification of epistatic QTL. Theor. Appl. Genet. 129, 879–896. doi: 10.1007/s00122-016-2669-3
Tiwari, K. R., Penner, G. A., Warkentin, T. (1997). Inheritance of powdery mildew resistance in pea. Can. J. Plant Sci. 77, 307–310. doi: 10.4141/P96-157
Toueni, M., Ben, C., Le Ru, A., Gentzbittel, L., Rickauer, M. (2016). Quantitative resistance to verticillium wilt in medicago truncatula involves eradication of the fungus from roots and is associated with transcriptional responses related to innate immunity. Front. Plant Sci. 7, 1431. doi: 10.3389/fpls.2016.01431
Tsai, S. Q., Joung, J. K. (2016). Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 17, 300–312. doi: 10.1038/nrg.2016.28
Upasani, M. L., Limaye, B. M., Gurjar, G. S., Kasibhatla, S. M., Joshi, R. R., Kadoo, N. Y., et al. (2017). Chickpea-Fusarium oxysporum interaction transcriptome reveals differential modulation of plant defense strategies. Sci. Rep. 7, 7746. doi: 10.1038/s41598-017-07114-x
Uppalapati, S. R., Marek, S. M., Lee, H. K., Nakashima, J., Tang, Y., Sledge, M. K., et al. (2009). Global gene expression profiling during Medicago truncatula-Phymatotrichopsis omnivora interaction reveals a role for jasmonic acid, ethylene, and the flavonoid pathway in disease development. Mol. Plant Microbe Interact. 22, 7–17. doi: 10.1094/MPMI-22-1-0007
Uppalapati, S. R., Young, C. A., Marek, S. M., Mysore, K. S. (2010). Phymatotrichum (cotton) root rot caused by Phymatotrichopsis omnivora: retrospects and prospects. Mol Plant Pathol 11, 325–334.
Uppalapati, S. R., Ishiga, Y., Doraiswamy, V., Bedair, M., Mittal, S., Chen, J., et al., (2012). Loss of abaxial leaf epicuticular wax in Medicago truncatula irg1/palm1 mutants results in reduced spore differentiation of anthracnose and nonhost rust pathogens. Plant Cell 24, 353–370.
Urbanski, D. F., Malolepszy, A., Stougaard, J., Andersen, S. U. (2013). High-throughput and targeted genotyping of Lotus japonicus LORE1 insertion mutants. Methods Mol. Biol. 1069, 119–146. doi: 10.1007/978-1-62703-613-9_10
Van De Mortel, M., Recknor, J. C., Graham, M. A., Nettleton, D., Dittman, J. D., Nelson, R. T., et al. (2007). Distinct Biphasic mRNA Changes in Response to Asian Soybean Rust Infection. Mol. Plant-Microbe Interact. 20, 887–899. doi: 10.1094/MPMI-20-8-0887
Van Der Linden, C. G., Wouters, D. C., Mihalka, V., Kochieva, E. Z., Smulders, M. J., Vosman, B. (2004). Efficient targeting of plant disease resistance loci using NBS profiling. Theor. Appl. Genet. 109, 384–393. doi: 10.1007/s00122-004-1642-8
Vance, C. P., Graham, P. H., Allan, D. L., (2000). “Biological Nitrogen Fixation: Phosphorus - A Critical Future Need?,” in Nitrogen Fixation: From Molecules to Crop Productivity. Eds. Pedrosa, F. O., Hungria, M., Yates, G., Newton, W. E. (Dordrecht: Springer Netherlands), 509–514. doi: 10.1007/0-306-47615-0_291
Varshney, R. K., Chen, W., Li, Y., Bharti, A. K., Saxena, R. K., Schlueter, J. A., et al. (2012). Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89. doi: 10.1038/nbt.2022
Varshney, R. K., Song, C., Saxena, R. K., Azam, S., Yu, S., Sharpe, A. G., et al. (2013). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246. doi: 10.1038/nbt.2491
Villegas-Fernández, Á.M., Krajinski, F., Schlereth, A., Madrid, E., Rubiales, D. (2014). Characterization of transcription factors following expression profiling of medicago truncatula-botrytis spp. Interact. Plant Mol. Biol. Rep. 32, 1030–1040. doi: 10.1007/s11105-014-0710-8
Wan, J., Vuong, T., Jiao, Y., Joshi, T., Zhang, H., Xu, D., et al. (2015). Whole-genome gene expression profiling revealed genes and pathways potentially involved in regulating interactions of soybean with cyst nematode (Heterodera glycines Ichinohe). BMC Genomics 16, 148. doi: 10.1186/s12864-015-1316-8
Wang, B., Sumit, R., Sahu, B. B., Ngaki, M. N., Srivastava, S. K., Yang, Y., et al. (2018). Arabidopsis novel glycine-rich plasma membrane PSS1 protein enhances disease resistance in transgenic soybean plants. Plant Physiol. 176, 865–878. doi: 10.1104/pp.16.01982
Wang, X., Sato, S., Tabata, S., Kawasaki, S. (2008). A high-density linkage map of Lotus japonicus based on AFLP and SSR markers. DNA Res. 15, 323–332. doi: 10.1093/dnares/dsn022
Wang, Y., Fan, G., Liu, Y., Sun, F., Shi, C., Liu, X., et al. (2013). The sacred lotus genome provides insights into the evolution of flowering plants. Plant J. 76, 557–567. doi: 10.1111/tpj.12313
Wen, L., Chang, H. X., Brown, P. J., Domier, L. L., Hartman, G. L. (2019). Genome-wide association and genomic prediction identifies soybean cyst nematode resistance in common bean including a syntenic region to soybean Rhg1 locus. Hortic. Res. 6, 9. doi: 10.1038/s41438-018-0085-3
Wille, L., Messmer, M. M., Studer, B., Hohmann, P. (2019). Insights to plant-microbe interactions provide opportunities to improve resistance breeding against root diseases in grain legumes. Plant Cell Environ. 42, 20–40. doi: 10.1111/pce.13214
Wu, X., Li, G., Wang, B., Hu, Y., Wu, X., Wang, Y., et al. (2018). Fine mapping Ruv2, a new rust resistance gene in cowpea (Vigna unguiculata), to a 193-kb region enriched with NBS-type genes. Theor. Appl. Genet. 131, 2709–2718. doi: 10.1007/s00122-018-3185-4
Xu, P.-F., Wu, J.-J., Xue, A., Li, W.-B., Chen, W.-Y., Wei, L., et al. (2012). Differentially expressed genes of soybean during infection by phytophthora sojae. J. Integr. Agric. 11, 368–377. doi: 10.1016/S2095-3119(12)60021-5
Xue, R., Wu, J., Zhu, Z., Wang, L., Wang, X., Wang, S., et al. (2015). Differentially expressed genes in resistant and susceptible common bean (Phaseolus vulgaris L.) Genotypes in Response to Fusarium oxysporum f. sp. phaseoli. PLoS One 10, e0127698. doi: 10.1371/journal.pone.0127698
Yan, H. H., Mudge, J., Kim, D. J., Shoemaker, R. C., Cook, D. R., Young, N. D. (2004). Comparative physical mapping reveals features of microsynteny between Glycine max, Medicago truncatula, and Arabidopsis thaliana. Genome 47, 141–155. doi: 10.1139/g03-106
Young, N. D., Cannon, S. B., Sato, S., Kim, D., Cook, D. R., Town, C. D., et al. (2005). Sequencing the Genespaces of Medicago truncatula and Lotus japonicus. Plant Physiol. 137, 1174–1181. doi: 10.1104/pp.104.057034
Young, N. D., Debelle, F., Oldroyd, G. E. D., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524. doi: 10.1038/nature10625
Zhang, T., Yu, L. X., Mccord, P., Miller, D., Bhamidimarri, S., Johnson, D., et al. (2014). Identification of molecular markers associated with Verticillium wilt resistance in alfalfa (Medicago sativa L.) using high-resolution melting. PLoS One 9, e115953. doi: 10.1371/journal.pone.0115953
Zhao, Q., Li, H., Sun, H., Li, A., Liu, S., Yu, R., et al. (2018). Salicylic acid and broad spectrum of NBS-LRR family genes are involved in SMV-soybean interactions. Plant Physiol. Biochem. 123, 132–140. doi: 10.1016/j.plaphy.2017.12.011
Zhou, L., Mideros, S. X., Bao, L., Hanlon, R., Arredondo, F. D., Tripathy, S., et al. (2009). Infection and genotype remodel the entire soybean transcriptome. BMC Genomics 10, 49. doi: 10.1186/1471-2164-10-49
Zhou, X., Liao, H., Chern, M., Yin, J., Chen, Y., Wang, J., et al. (2018). Loss of function of a rice TPR-domain RNA-binding protein confers broad-spectrum disease resistance. Proc. Natl. Acad. Sci. U. S. A. 115, 3174–3179. doi: 10.1073/pnas.1705927115
Zhu, H., Choi, H.-K., Cook, D. R., Shoemaker, R. C. (2005). Bridging Model and crop legumes through comparative genomics. Plant Physiol. 137, 1189–1196. doi: 10.1104/pp.104.058891
Zou, J., Rodriguez-Zas, S., Aldea, M., Li, M., Zhu, J., Gonzalez, D. O., et al. (2005). Expression profiling soybean response to pseudomonas syringae reveals new defense-related genes and rapid HR-Specific downregulation of Photosynthesis. Mol. Plant-Microbe Interact. 18, 1161–1174. doi: 10.1094/MPMI-18-1161
Keywords: genomics, legumes, plant–pathogen interactions, transcriptome analysis, GWAS, QTLs, markers, CRISPR/Cas9
Citation: Kankanala P, Nandety RS and Mysore KS (2019) Genomics of Plant Disease Resistance in Legumes. Front. Plant Sci. 10:1345. doi: 10.3389/fpls.2019.01345
Received: 30 April 2019; Accepted: 27 September 2019;
Published: 30 October 2019.
Edited by:
Karam B. Singh, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaReviewed by:
Nicolas Rispail, Institute for Sustainable Agriculture, (CSIC), SpainCopyright © 2019 Kankanala, Nandety and Mysore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirankumar S. Mysore, a3NteXNvcmVAbm9ibGUub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.