
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 27 November 2018
Sec. Plant Pathogen Interactions
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01694
This article is part of the Research TopicUnravelling Citrus Huanglongbing DiseaseView all 10 articles
 Ming Huang1
Ming Huang1 Mikeal L. Roose2
Mikeal L. Roose2 Qibin Yu1
Qibin Yu1 Dongliang Du1
Dongliang Du1 Yuan Yu1Yi Zhang1
Yuan Yu1Yi Zhang1 Zhanao Deng3Ed Stover4
Zhanao Deng3Ed Stover4 Frederick G. Gmitter Jr.1*
Frederick G. Gmitter Jr.1*Huanglongbing (HLB), or citrus greening, is the most devastating disease in citrus worldwide. Commercial citrus varieties including sweet orange (Citrus sinensis) are highly susceptible to HLB, and trifoliate orange (Poncirus trifoliata, a close Citrus relative) is widely considered resistant or highly tolerant to HLB. In this study, an intergeneric F1 population of sweet orange and trifoliate orange was genotyped by Genotyping-by-Sequencing, and high-density SNP-based genetic maps were constructed separately for trifoliate orange and sweet orange. The two genetic maps exhibited high synteny and high coverage of the citrus genome. Progenies of the F1 population and their parents were planted in a replicated field trial, exposed to intense HLB pressure for 3 years, and then evaluated for susceptibility to HLB over 2 years. The F1 population exhibited a wide range in severity of HLB foliar symptom and canopy damage. Genome-wide QTL analysis based on the phenotypic data of foliar symptom and canopy damage in 2 years identified three clusters of repeatable QTLs in trifoliate orange linkage groups LG-t6, LG-t8 and LG-t9. Co-localization of QTLs for two traits was observed within all three regions. Additionally, one cluster of QTLs in sweet orange (linkage group LG-s7) was also detected. The majority of the identified QTLs each explained 18–30% of the phenotypic variation, indicating their major role in determining HLB responses. These results show, for the first time, a quantitative genetic nature yet the presence of major loci for the HLB tolerance in trifoliate orange. The results suggest that sweet orange also contains useful genetic factor(s) for improving HLB tolerance in commercial citrus varieties. Findings from this study should be very valuable and timely to researchers worldwide as they are hastily searching for genetic solutions to the devastating HLB crisis through breeding, genetic engineering, or genome editing.
Huanglongbing (HLB), commonly known as citrus greening, is the most devastating disease in citrus plantations worldwide. Since the first identification of the disease in Florida in 2005, HLB has spread throughout the state, and is now found in most states in the United States where citrus is grown. It is rapidly spreading throughout many of the world’s production areas, as well. Due to widespread infection and lack of effective management strategy, Florida’s nearly $11 billion citrus industry has experienced a rapid and continuous decline. The production of sweet orange dropped from 240 million boxes in 2004 to 45 million in 2018 (Florida Citrus Commission1).
Huanglongbing-diseased trees develop leaves with characteristic blotchy mottle, the shoots are stunted and yellowing, and the canopy and branches gradually dieback as the disease progresses. These symptoms are associated with the host limitations for photoassimilate transport and nutrient uptake induced by the disease, and finally can lead to tree death (Blaustein et al., 2018). HLB is generally considered to be caused by three species of Candidatus Liberibacter, of which Candidatus Liberibacter asiaticus (CLas) is the most widespread and virulent species and is the only species reported in the citrus industry of United States. CLas is a heat-tolerant Gram-negative bacterium, resides only in the phloem of plant hosts, and is vectored by the sap-sucking Asian citrus psyllid (ACP, Diaphorina citri) (Wang N.A. et al., 2017). As an obligate and insect-transmitted plant pathogen, CLas attacks all species and hybrids in the genus of Citrus as well as some closely related genera (Ramadugu et al., 2016). Most commercial citrus cultivars are highly susceptible to HLB (Stover et al., 2016a,b). Within cultivated citrus, high levels of tolerance to HLB were mostly found in some types that are commonly used as rootstocks, such as trifoliate orange and some of its hybrids (Albrecht and Bowman, 2011, 2012; Ramadugu et al., 2016).
Until now, very little is known about the molecular mechanism of pathogenesis of CLas (Martinelli and Dandekar, 2017). As to understanding the genetic architecture of citrus resistance or susceptibility to HLB, there has been no compelling progress, and no QTL related to CLas infection or HLB tolerance responses has been reported. Moreover, so far there is no sustainable management and control of HLB where it is endemic. Breeding and development of HLB resistant or tolerant cultivars is widely regarded to be the most practical strategy to support long-term control of this severe disease in the field. The reports of variability for tolerance of or sensitivity to HLB within citrus and its relatives encourages breeding and selection for tolerant or possibly resistant genotypes (Richardson and Hall, 2013; Ramadugu et al., 2016; Killiny et al., 2017; Miles et al., 2017). Traditional breeding through crossing elite cultivars with resistant materials can achieve this objective. However, the introgression of resistant germplasm into elite cultivars will likely require multiple rounds of backcrossing to recover desirable commercial traits. In addition, the breeding cycle for citrus ranges from 5 to 10 years, and the rescue of the citrus industries through HLB-resistant or tolerant cultivars demands considerable urgency. Identification of QTLs associated with resistance or tolerance to HLB in citrus can facilitate more rapid development of resistant cultivars through marker-assisted selection or genome editing.
To identify QTLs associated with phenotypic traits, a genetic map with high resolution and fine accuracy is crucial. Nowadays, the wide application of high-throughput genome sequencing and efficient SNP genotyping allows the construction of genetic maps with numerous markers at an acceptable cost (Deschamps et al., 2012). While initially confined to annual herbaceous plants, high-density genetic mapping is increasingly extended to perennial woody plants. Using high-throughput genotyping, the saturation of genetic maps has been greatly improved for some citrus species, such as sweet orange, mandarin and pummelo (Lyon, 2008; Ollitrault et al., 2012a; Shimada et al., 2014; Guo et al., 2015; Yu et al., 2016; Curtolo et al., 2017; Imai et al., 2017). However, in comparison with well-studied model and agronomic plants, citrus lags in development of high-density, high-resolution genetic maps with fine accuracy and precision. Moreover, so far there is no high-density genetic map for trifoliate orange.
This study inaugurates investigation of HLB infection in citrus by repeated phenotyping of segregating populations and QTL mapping. The objectives of this study were: (1) to evaluate severity level of HLB disease among a field population exposed to intense HLB pressure using two phenotypic traits, foliar symptoms and canopy damage; (2) to construct high-density genetic maps separately for trifoliate orange and sweet orange in an F1 mapping population through Genotyping-by-Sequencing; (3) to identify QTLs associated with citrus HLB infection and responses separately in trifoliate orange and sweet orange genetic maps.
Genotyping was carried out in an F1 population of 170 individuals derived from mixed intergeneric crosses between two sweet oranges (Citrus sinensis ‘Sanford’ and ‘Succari’) and two trifoliate oranges (Poncirus trifoliata ‘Argentina’ and ‘Flying Dragon’), including 79 progenies of ‘Sanford’ × ‘Argentina’, 40 progenies of ‘Succari’ × ‘Flying Dragon’, and 51 progenies of ‘Flying Dragon’ × ‘Succari’. Of the genotyping population, 86 individuals were randomly chosen as a phenotyping population, including 47 progenies of ‘Sanford’ × ‘Argentina,’ 14 progenies of ‘Succari’ × ‘Flying Dragon’, and 25 progenies of ‘Flying Dragon’ × ‘Succari’. In addition, two sweet orange varieties (‘Navel’ grafted on ‘Swingle’ and ‘Hamlin’ grafted on ‘C-35’), six trifoliate orange varieties (‘Flying-Dragon’, ‘Argentina’, and ‘Pomeroy’ grafted on ‘Volkamer’ lemon; and seedlings of ‘Rubidoux’, ‘Rich 16-6’, and ‘Large-flower’), and seedlings of ‘Volkamer’ lemon (Citrus volkameriana) were included as controls in the phenotyping. All progenies were clonally propagated by grafting on ‘Volkamer’ lemon in the greenhouse of the USDA/ARS in Fort Pierce, FL in 2010. Except for ‘Volkamer’ lemon seedings that had 16 replicate trees, each of the progenies and control varieties had eight clonal replicate trees. A completely randomized experiment of 768 trees was established in a field trial at the USDA-ARS in Fort Pierce in 2011. The planting consisted of eight rows oriented south-north, with 4 m × 1.5 m planting distances. Guard citrus trees not analyzed in this study were planted at the ends of each row. The field population trees were irrigated and fertilized with professional practices, but pesticides were not applied during the study, to encourage psyllid population increase, feeding, colonization, and inoculation of CLas to the trees. Maintenance of the field trees was as described for a nearby experiment on the same farm (Richardson et al., 2011).
As reported in other concurrent studies (Lewis-Rosenblum et al., 2015; Ramadugu et al., 2016), the HLB disease pressure was high in the field trial at this location, which caused homogeneous inoculation of CLas naturally, providing excellent conditions to evaluate trees under natural disease spread conditions (Westbrook et al., 2011; Richardson and Hall, 2013). During the period of HLB disease evaluation, the status of CLas infection for each tree was diagnosed using the TaqMan label-based multiplex real-time PCR method (Li W.B. et al., 2006, 2008). At least four fully expanded mature leaves were randomly collected from different branches and different quadrants of each tree. The midribs were separated from the leaves and cut into small pieces for DNA extraction using the CTAB method (Aldrich and Cullis, 1993). Real-time qPCR was performed on an ABI 7500 thermocycler with probes specific to CLas 16S ribosomal gene and citrus cytochrome oxidase gene. The mean cycle threshold (Ct) values of qPCR for direct CLas detection were normalized with Ct values of the corresponding host plant gene. Trees with Ct value under 33 were considered to be HLB-positive (Albrecht and Bowman, 2012).
Evaluations of HLB disease symptoms were performed twice per year from 2015 to 2016 in September and October, which is the optimal time for HLB disease evaluation considering both the growth condition of citrus (Poncirus and some of its hybrids are deciduous or semi-deciduous) and the period of the most evident disease symptoms. At each time of disease evaluation, the visual evaluation of foliar symptom and canopy damage was conducted twice per tree on each side along the tree rows. All evaluations were carried out by the same individual researcher. Foliar disease symptom severity was assessed on a 6-point scale visual rating for comprehensive typical HLB symptom including mottled rugose leaves and yellow shoots: 0 = no symptom for whole tree, 1 = isolated (less than 1/10 of tree) slight symptom, 2 = partly (approximately 1/3 of tree) slight symptom or isolated severe symptom, 3 = mostly (approximately 2/3 of tree) slight symptom or partly severe symptom, 4 = mostly severe symptom, 5 = severe symptom for whole tree. The slight foliar symptom rating refers to trees with leaves that are slightly mottled, pale or leathery, while the severe symptom rating refers to severely blotchy mottled, rugose or leathery. Canopy damage severity was also assessed on a 6-point scale visual rating basing on dieback, defoliation or stunting: 0 = full tree canopy without apparent dieback or defoliation or stunting, 1 = full tree canopy with isolated short dieback or very slight defoliation, 2 = partly short dieback or isolated long dieback or slight defoliation or stunting, 3 = mostly short dieback or partly long dieback or moderate defoliation or stunting, 4 = mostly long dieback or main branch dieback or severe defoliation or stunting, 5 = trunk dieback or whole tree dead. The mean rating score for each genotype in each year was the averaged rating score of all replicate trees at the two times of evaluation.
Descriptive statistics of all phenotypic data were calculated using IBM SPSS (Statistical Package for Social Sciences) Statistics 17.0. Significant differences between phenotypic data were declared with p ≤ 0.05 by Student’s t-test. To evaluate whether the data followed a normal distribution, a normality analysis by Kolmogorov–Smirnov and Shapiro–Wilk tests was performed separately for the dataset of each trait in each year. The Box-Cox transformation was performed before QTL analysis if data presented a non-normal distribution. Histograms for each trait were constructed using the mean rating score of the complete dataset. Pearson’s correlation coefficients were calculated between year-means for each phenotypic trait.
Genomic DNA of each citrange hybrid and parent was extracted using a modified CTAB method (Aldrich and Cullis, 1993), Genomic DNA was digested with PstI restriction endonuclease and then processed into restriction site associated DNA (RAD) libraries according to a previously described protocol (Baird et al., 2008). The constructed RAD libraries were sequenced on an Illumina HiSeq2000 platform following the manufacturer’s protocol. Library construction, sequencing and SNP calling were provided by Floragenex (Floragenex Inc., Portland, OR, United States). SNP calling of parents and progenies was performed using the Clementine mandarin genome v1.02 as a reference. Genotypes at each locus were determined using the VCF Popgen Pipeline version 4.0 to generate a customized VCF 4.1 (variant call format) database with parameters set as follows: minimum allele frequency for genotyping 0.075, minimum Phred score 15, minimum depth of sequencing coverage of 12, and minimum 75% of individuals with the specific genotype call. The marker configuration codes “lm × ll” and “nn × np” were used to represent markers that were heterozygous only in one of the parents, and code “hk × hk” to represent markers that heterozygous in both parents.
Due to asexual means of evolution (somatic mutations), molecular polymorphism among sweet orange cultivars is very rare (Ollitrault et al., 2012b); likewise very low polymorphism was found between the trifoliate orange varieties, so all F1 progenies from different crosses between the two genera were considered as a single family, after excluding the inconsistent marker loci between parental varieties. To ensure high quality of linkage mapping for each parent, SNP markers segregating from only one of the parents (with configuration codes “lm × ll” and “nn × np”) were selected to construct the linkage maps; markers with code “hk × hk” were not considered in this study. SNP markers matching the following criteria were excluded from linkage analysis: (1) had a missing genotype in more than 10% of progenies; (2) had a missing genotype for one of the parents; (3) had inconsistent genotypes among different parental sweet oranges or trifoliate oranges; (4) had homozygous genotypes for both parents; (5) had heterozygous genotype for both parents; (6) had genotypes in F1 progeny not expected for the parental genotypes; (7) had no segregation in F1 progeny. Segregation distortion was tested by χ2 conformity tests against the Mendelian segregation ratio of 1:1. Because the method of linkage analysis was based upon a test for independence of logarithm of the odds (LOD) scores that is not affected by segregation distortion, markers with certain skewed segregation were included in linkage analysis referring to previous studies on citrus (Bernet et al., 2010; Ollitrault et al., 2012a; Raga et al., 2012). Linkage analyses were performed using JoinMap 4.1 (Van Ooijen, 2011). Linkage mapping was performed under a two-way pseudo-testcross scheme (Grattapaglia and Sederoff, 1994) with two separate datasets, one with markers segregating from the trifoliate orange and the other one with markers segregating from sweet orange. Markers with identical segregation patterns or segregating similarity higher than 98% were excluded from the linkage groups. Phases of the linked marker loci were automatically detected by the software. Linked markers were grouped using the independence LOD with a threshold LOD score of 4.0 and a maximum recombination fraction (θ) of 0.4. Map distances were estimated in centiMorgans (cM) using the regression mapping algorithm and the Kosambi mapping function. The linkage groups were numbered according to the corresponding scaffold number of the reference Clementine mandarin genome, and marker names were composed of the scaffold number and SNP position on the reference genome. The genetic map was drawn using the MapChart 2.2 program (Voorrips, 2002).
QTL mapping was carried out on two parental maps separately using MapQTL 6 under the backcross model by composite interval mapping (CIM) (van Ooijen, 1992). Phenotypic data were analyzed separately for each trait in each year. The LOD thresholds to declare a significant QTL for each trait were determined via permutation tests using 1,000 permutations at a genome-wide significance level of 0.90. The CIM analysis was performed in 1 cM steps to detect significant QTLs with a LOD score higher than the threshold. The nearest marker to the likelihood peak of each significant QTL was selected as a cofactor to perform restricted multiple QTL mapping (rMQM). If the LOD value linked with a cofactor fell below the threshold during rMQM mapping, the cofactor was removed, and the analysis repeated. This process was continued until the cofactor list remained stable. Kruskal–Wallis (KW) test was also used to provide complementary validation for significant genotypic means. Each significant QTL was characterized by its LOD score, percentage of explained phenotypic variation, confidence interval (in cM) corresponding to threshold LOD score and extension region at either side of the likelihood peak until the LOD score dropped to 2.0. QTLs that showed clearly overlapping confidence intervals were considered as co-localized. The position of QTLs on the genetic map was drawn using the MapChart 2.2 program (Voorrips, 2002).
Four parental varieties (‘Argentina’ and ‘Flying Dragon’ trifoliate orange; ‘Sanford’ and ‘Succari’ sweet orange) and 170 F1 progenies were processed for RAD sequencing on the NGS Illumina platform. Except one progeny, all had acceptable quality of sequencing reads. The average number of reads was 8.14E6 and 7.37E6 for trifoliate orange and sweet orange, respectively. The read counts for the 169 F1 progenies ranged from 1.05E6 to 14.91E6, with an average of 4.53E6 per progeny. After alignment with the reference genome, the average number of read clusters was 1.83E5 and 1.47E5 for trifoliate oranges and sweet oranges, respectively, and the numbers of read clusters for the F1 progenies ranged from 0.32E5 to 2.79E5 and averaged to be 1.03E5 (the detailed data are available in Supplementary File S1).
In total, 51,687 putative SNP loci were determined, of which 55.6% were transitions and 44.4% were transversions. Excluding SNP loci without calls, 96.7% of the genotyped SNP loci were identical between ‘Argentina’ and ‘Flying Dragon’ trifoliate orange, and 98.2% were identical between ‘Sanford’ and ‘Succari’ sweet orange (the detailed data are available in Supplementary File S2). Considering the low levels of genetic diversity found between the trifoliate orange parents and between the sweet orange parents, all the F1 progenies from different crosses were combined as a single family. After stringent filtering, a total of 3,861 high quality SNP markers with configuration code “lm × ll” or “nn × np” were retained for genetic map construction, of which 1,408 SNP markers were heterozygous in trifoliate orange and 2,453 SNP markers were heterozygous in sweet orange.
After eliminating markers with identical or similar segregation patterns that accounted for 62% of the filtered markers, all the remaining SNP markers in each dataset were grouped under the threshold LOD score of 4.0 into nine linkage groups, which is consistent with the haploid number of chromosomes in citrus. Additionally, all nine linkage groups conserved their integrity up to LOD of 10 for both parents. Finally, for trifoliate orange, a total of 647 high-quality SNP markers were mapped on nine linkage groups with unique loci, spanning a total genetic length of 1030.8 cM, with an average inter-locus distance of 1.59 cM (Table 1 and Figure 1A). The number of markers within each linkage group ranged from 52 (for LG-t6) to 125 (for LG-t3), spanning a genetic distance ranging from 78.7 (for LG-t6) to 155.2 cM (for LG-t3). In total, 85.5% of the inter-locus gaps on the genome-wide genetic map were smaller than 3 cM and no gap was larger than 10 cM. For sweet orange, 754 high-quality SNP markers with unique loci were mapped into nine linkage groups, spanning a total genetic length of 760.2 cM with an average inter-locus distance of 1.01 cM (Table 1 and Figure 1B). The number of markers within each linkage group ranged from 57 (for LG-s1) to 118 (for LG-s3), spanning a genetic distance ranging from 60.5 (for LG-s9) to 105.3 cM (for LG-s3). Of the inter-locus gaps on the whole genetic map, 93.3% were smaller than 3 cM and only one gap was larger than 10 cM (11.2 cM on LG-s1).
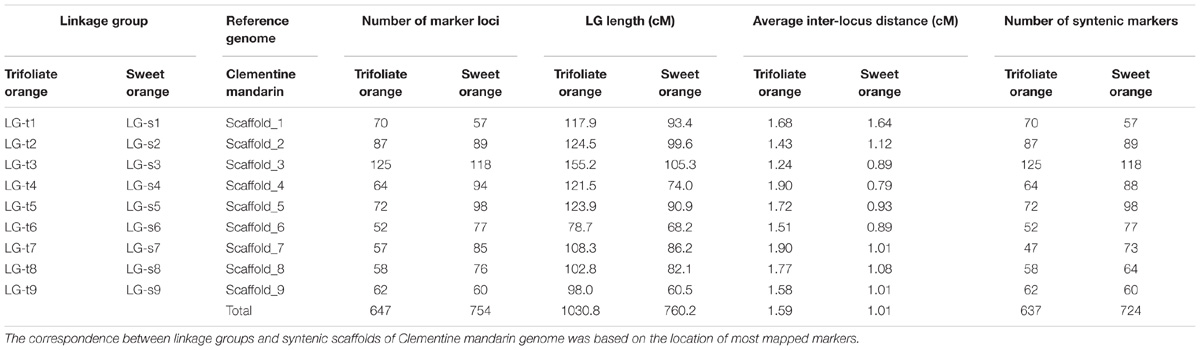
TABLE 1. Summary of the genetic linkage maps of trifoliate orange and sweet orange.

FIGURE 1. Distribution of markers with unique loci in the genetic linkage map of trifoliate orange (A) and sweet orange (B). The nine linkage groups correspond to the nine major scaffolds of the Clementine mandarin genome. Map distances in centiMorgans (cM) are indicated by the ruler at the left.
Through alignment, all SNP markers on the two genetic maps were successfully mapped onto nine major scaffolds of the Clementine mandarin genome. For trifoliate orange, except for 10 markers of LG-t7 which mapped on Scaffold_5, all other 637 markers were mapped onto syntenic scaffolds (Table 1 and Figure 2). The overall coverage ratio of mapped markers on the Clementine genome was 98.6%, and the coverage ratio on each scaffold ranged from 95.5 (for LG-t7) to 99.7% (for LG-t5). For sweet orange, except for a total of 30 markers on LG-s4, LG-s7 and LG-s8, all other 724 markers were mapped onto syntenic scaffolds (Table 1 and Figure 3). The overall coverage ratio of mapped markers on the Clementine genome was 95.0%, and the coverage ratio on each scaffold ranged from 87.7 (for LG-s9) to 99.3% (for LG-s8). Only some minor discrepancies were observed between the genetic maps and the Clementine genome, possibly due to genetic divergence among different citrus species, potential errors in the current linkage grouping, or erroneous assemblies in the reference genome. Collinear analysis of the consensus between the genetic map and the Clementine genome via a dot-plot diagram not only showed variations of the ratios between genetic distance to physical distance, but also clearly revealed high consensus between the genetic maps of trifoliate orange and sweet orange (Supplementary Figure S1).
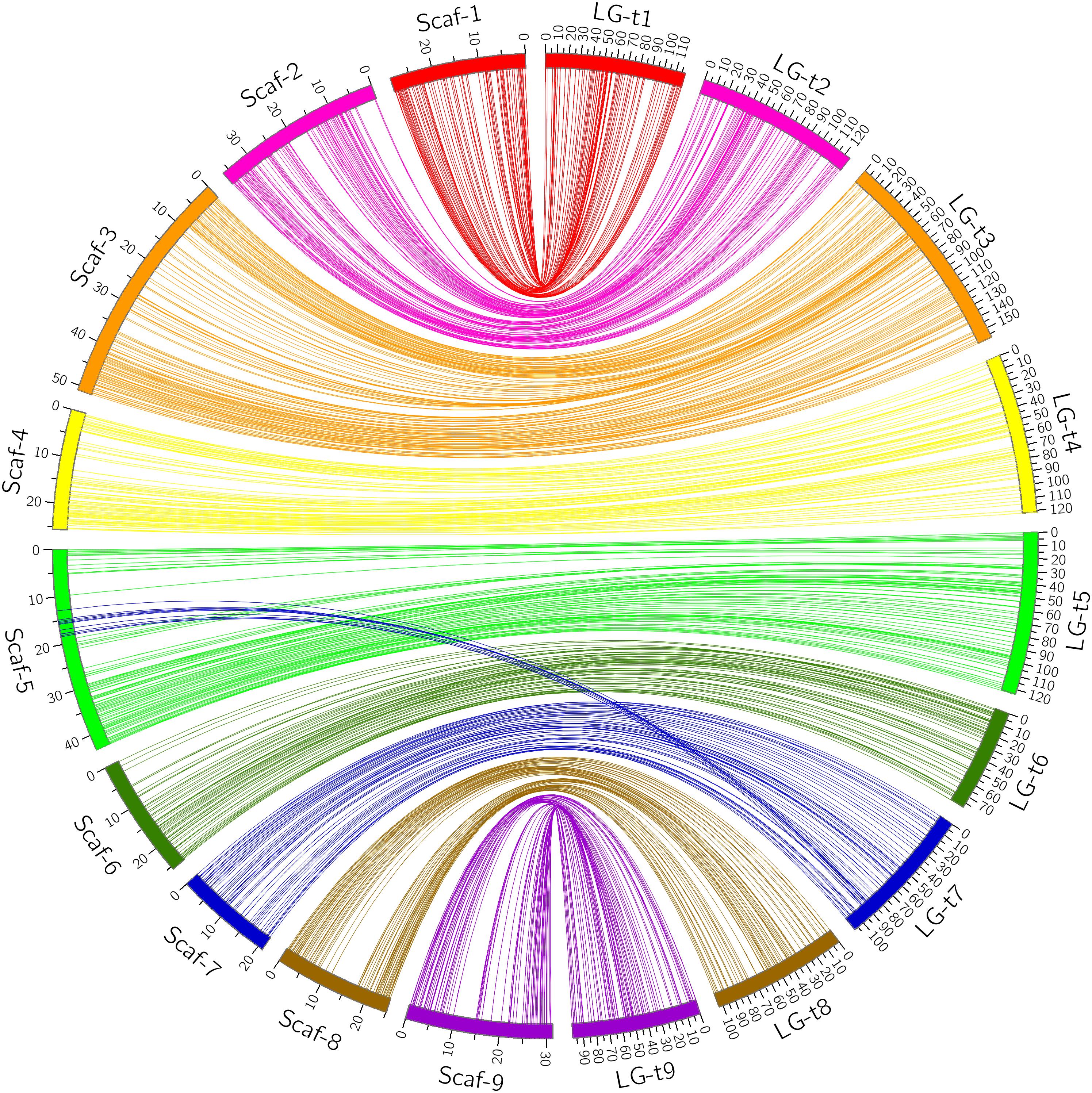
FIGURE 2. Conservation of synteny and linear order of markers between trifoliate orange genetic map and Clementine mandarin genome via circle diagram.
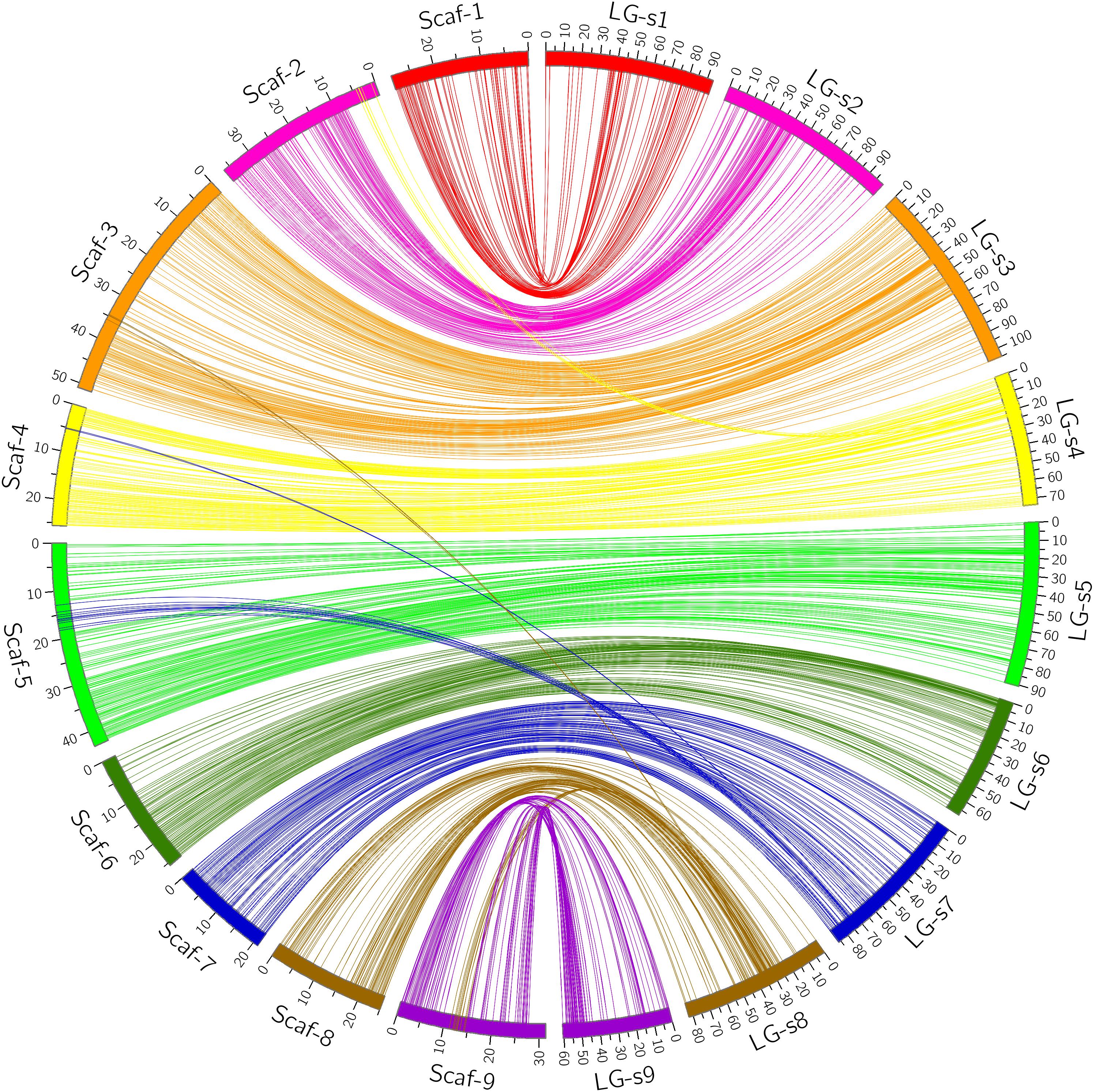
FIGURE 3. Conservation of synteny and linear order of markers between sweet orange genetic map and Clementine mandarin genome via circle diagram.
The phenotyping population consisted of 86 F1 progenies randomly chosen from the above-mentioned mapping population, as well as six trifoliate oranges, two sweet oranges, and the rootstock seedings, with eight clonal replicate trees for each of individuals. After exposure to intense HLB pressure for 3 years in a replicated field trial, all phenotyped trees were diagnosed for CLas infection by the TaqMan label-based multiplex real-time PCR during the period of disease evaluation. As shown in Table 2, relatively few trees of trifoliate orange varieties were determined to be HLB-positive; in contrast, most trees of sweet orange varieties were HLB-positive. Meanwhile, F1 progenies were mostly infected by CLas in both 2015 and 2016. For the HLB-negative genotypes, it should be noted that these trees were not confirmed to be immune to CLas, but they mostly had low titers of CLas (Ct value of CLas diagnosis between 33 and 39), which might indicate partial resistance to CLas infection. The results indicate that all trees of the phenotyping population were adequately inoculated with CLas under continuous reinfection (the detailed data are available in Supplementary File S3).
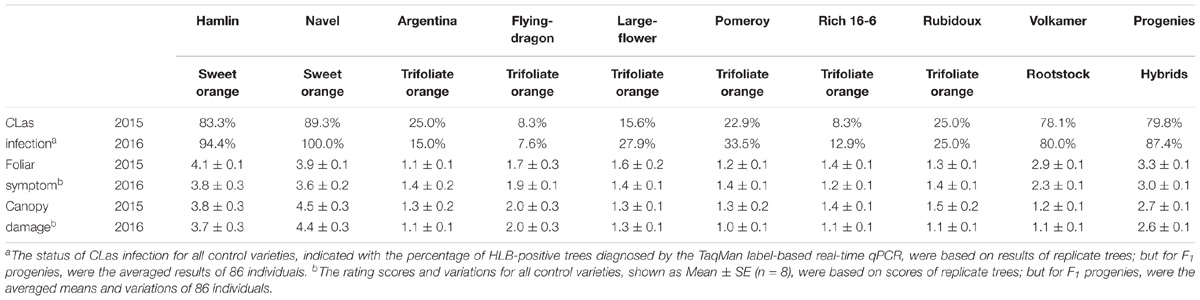
TABLE 2. Diagnosis of CLas infection and evaluation of severity level of HLB disease among control varieties and F1 progenies.
The evaluation of HLB disease among the phenotyping population was conducted in 2015 and 2016 through repeated rating of the foliar symptom and canopy damage separately (Table 2). The ratings of foliar symptom in trifoliate oranges (ranged from 1.1 to 1.9) were significantly lower than those in sweet oranges (ranged from 3.6 to 4.1). For F1 progenies, the ratings of foliar symptom ranged from 1.3 to 4.7, and the average rating was 3.3 in 2015 and 3.0 in 2016. The ratings of canopy damage in trifoliate oranges (ranged from 1.0 to 2.0) were also significantly lower than those in sweet oranges (ranged from 3.7 to 4.5). For F1 progenies, the ratings of canopy damage ranged from 0.9 to 4.1, and the average rating was 2.7 in 2015 and 2.6 in 2016. The frequency distributions of foliar symptom and canopy damage ratings among the F1 progenies are illustrated in Figure 4. As shown, obvious quantitative variation of the two traits were observed in both years. The correlation coefficient of phenotypic data between 2 years was 0.77 for foliar symptom rating and 0.91 for canopy damage rating. The correlation coefficients between two traits ranged from 0.47 to 0.74 and 0.58 in average, reflecting low consistency between the traits of disease in the progenies (the detailed data are available in Supplementary File S4).
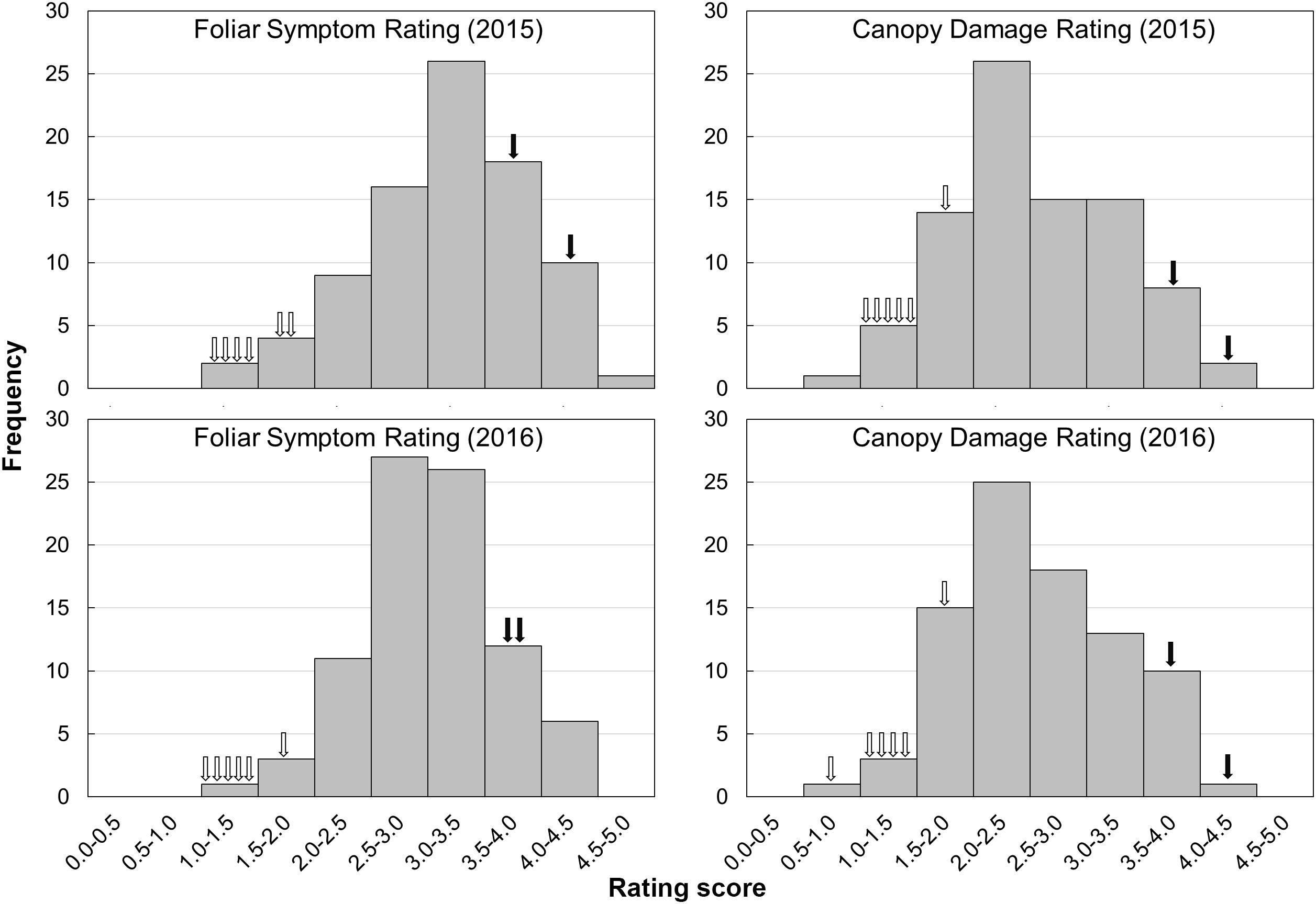
FIGURE 4. Frequency distributions of foliar symptoms rating and canopy damage rating in F1 progenies and parental varieties in 2 years. The hollow arrows indicate each of the trifoliate orange varieties, while the solid arrows indicate each of the sweet orange varieties.
QTLs associated with the two phenotypic traits were detected separately in two parental genetic maps for each year (Table 3). In trifoliate orange, six and three QTLs associated with foliar symptom, and three and two QTLs associated with canopy damage, were identified in 2015 and 2016, respectively. The maximum LOD score for each of the QTLs ranged from 2.7 to 5.5, explaining an estimated phenotypic variation (R2) ranging from 13.9 to 29.9%. In sweet orange, two QTLs associated with foliar symptom were identified in both 2015 and 2016, but no significant QTL associated with canopy damage was identified in either of the 2 years. The maximum LOD score for each of the QTLs ranged from 3.1 to 5.5, explaining an estimated phenotypic variation (R2) ranging from 17.3 to 29.1%. None of the QTLs alone could explain a majority of the phenotypic variation, but they collectively explained a major part. The graphics of QTL mapping are available in Supplementary Figure S2.
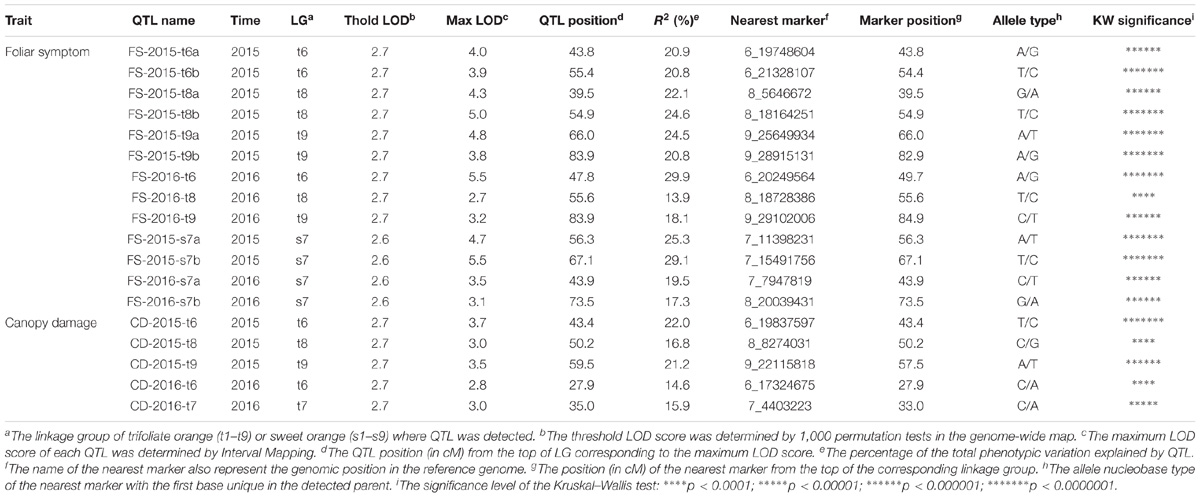
TABLE 3. QTLs detected separately in trifoliate orange and sweet orange genetic maps.
Based on the confidence intervals of the QTLs on the genetic maps, most of the QTLs are repeatable between the 2 years (Figures 5, 6). For the QTLs associated with foliar symptoms, seven QTLs were co-localized in three locations of the trifoliate orange map, on LG-t6, LG-t8 and LG-t9, while four QTLs were co-localized in two locations of LG-s7 on the sweet orange map. For the QTLs associated with canopy damage, only two QTLs on LG-t6 showed certain co-localization. It is noteworthy that co-localization of QTLs between the two traits were also observed in three locations of the trifoliate orange map, respectively, on LG-t6, LG-t8 and LG-t9. Overall, all the locations of QTLs associated with the two traits could be grouped into four main clusters, respectively, located on LG-t6, LG-t8, LG-t9 and LG-s7. The approximate genomic size of corresponding regions on the reference genome for these QTL clusters were Scaffold_6, Scaffold_8, Scaffold_9 and Scaffold_7. Among the QTL clusters, the one on LG-t6 was the most repeatable QTL and explained the largest part of the phenotypic variation.
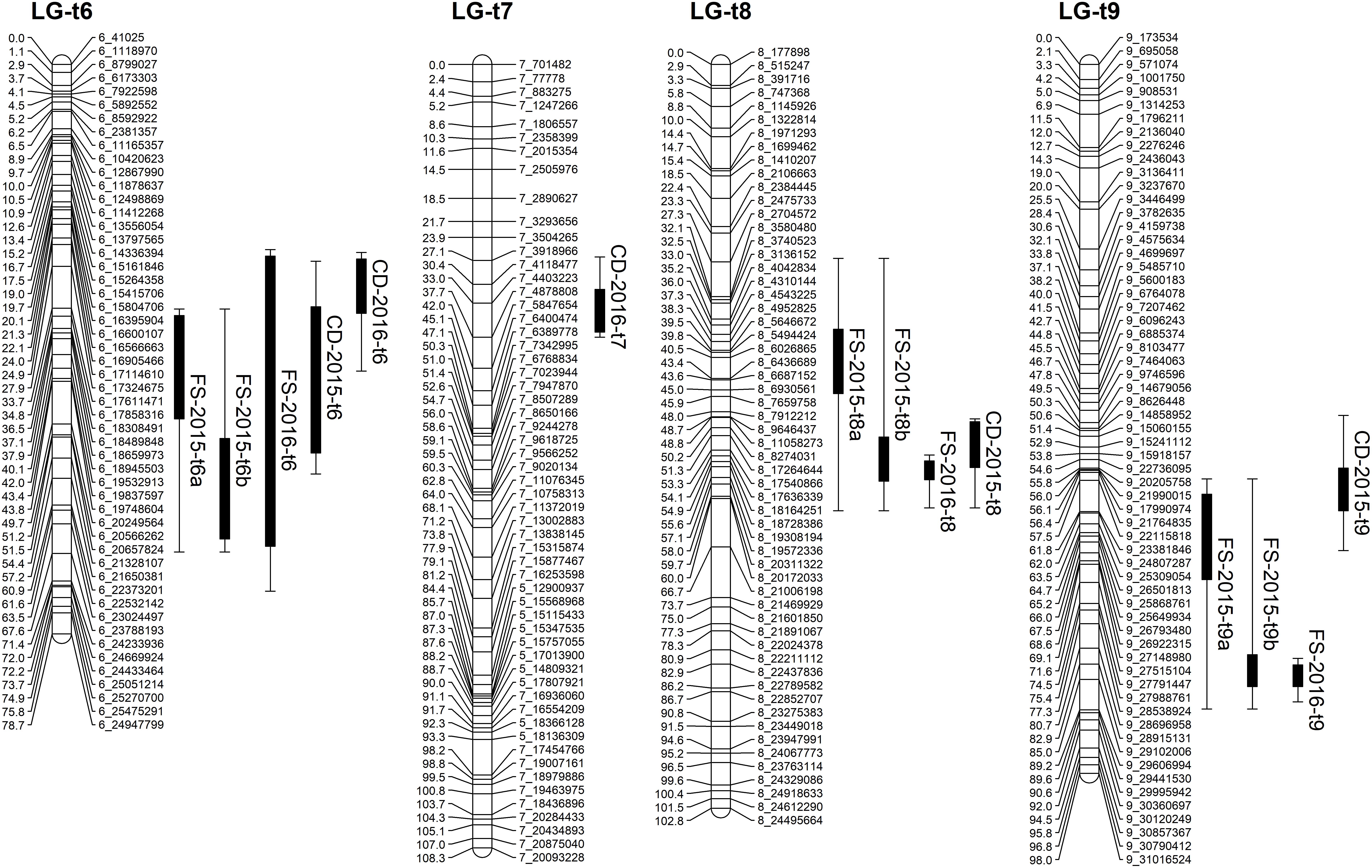
FIGURE 5. Mapping QTLs with foliar symptoms and canopy damage on the trifoliate orange genetic linkage map. Thick bars on the right side of each LG indicate confidence interval of QTLs with LOD score above threshold and flanking error bars indicate extension of QTL region at LOD score 2.0.
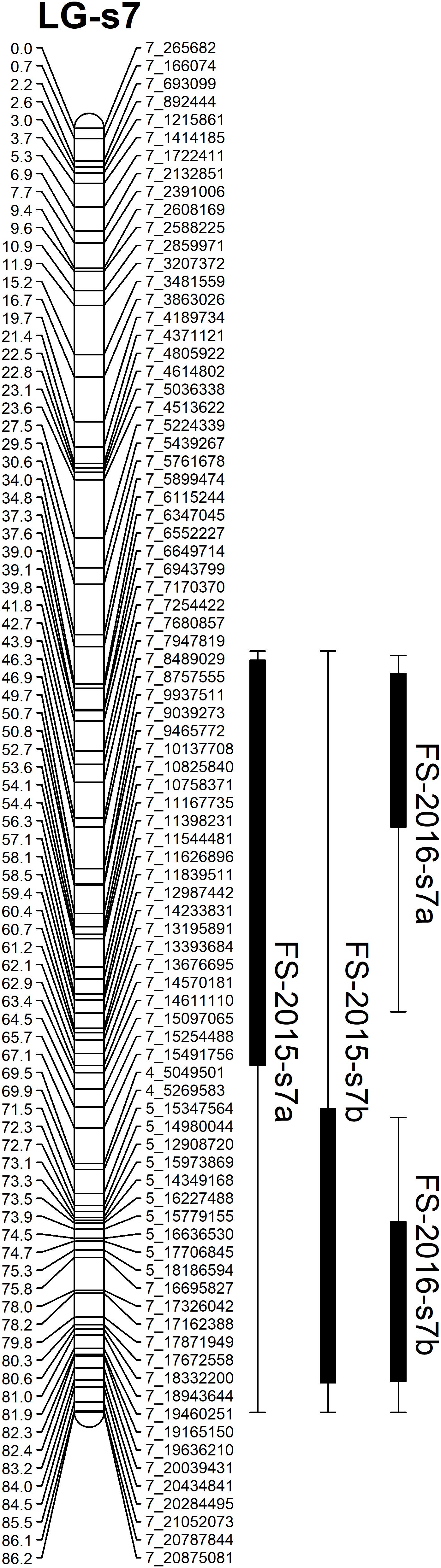
FIGURE 6. Mapping QTLs with foliar symptoms and canopy damage on the sweet orange genetic linkage map. Thick bars on the right side of each LG indicate confidence interval of QTLs with LOD score above threshold and flanking error bars indicate extension of QTL region at LOD score 2.0.
In this study, we constructed two separate parental genetic maps, which, to our knowledge, are the highest density genetic maps to date for trifoliate orange and sweet orange. The former highest density genetic map of trifoliate orange consisted of 146 SNP markers and 74 SSR markers, spanning a total genetic length of 937.1 cM and having an average density of 0.23 marker/cM (Lyon, 2008). Our trifoliate orange map consisted of 647 unique-loci SNP markers with an average density of 0.63 marker/cM and had quite high coverage of the citrus genome. Thus, our genetic mapping effort has greatly improved the saturation of the genetic map of trifoliate orange. This new linkage map can be utilized as a reference map in trifoliate orange genome assembly in the future. For sweet orange, the previously reported highest density genetic map consisted of 799 SNP markers and 189 SSR markers spanning a total genetic length of 1026.6 cM with an average density of 0.95 marker/cM (Lyon, 2008). The map was used as a reference map for the assembly of a sweet orange genome (Xu et al., 2013). We achieved a slightly higher density in our genetic map (0.99 marker/cM), though the map consisted of less markers. It is probably attributed to better quality of the genotyping and linkage mapping, resulting in smaller size of the linkage groups for our sweet orange map.
However, citrus genetic maps require substantial additional effort to achieve the high resolution found in model species genetic maps, which include thousands of markers with high accuracy and precision. Although we initially developed thousands of SNP markers for our linkage mapping, only a small portion of these markers were successfully mapped as unique loci. This has been observed in all previous SNP-based genetic maps of citrus, where many markers were located in “zero recombination clusters”, indicating nearly no meiotic recombination occurs between markers within those regions (Lyon, 2008; Shimada et al., 2014; Guo et al., 2015; Yu et al., 2016; Curtolo et al., 2017; Imai et al., 2017). As shown in Figures 3 and 4, many markers in some small regions (<2 cM) of the linkage groups fell into clusters, which all correspond to very large genomic regions (5–10 Mb) on citrus chromosomes. High redundancy of markers is commonly attributed to small population size, closely adjacent physical marker location, and regions with low recombination, which could be significantly improved by increasing the size of mapping populations and the evenness of marker distribution. These solutions have been applied to mapping of many plant species but are difficult for citrus species. Biological characteristics of citrus, such as long juvenility, seedlessness, polyembryony, apomixis, heterozygosis, gametophytic incompatibility, zygotic selection and gametic selection (Shimada et al., 2014), not only seriously hamper the development of numerous uniquely segregating markers and production of large full-sib populations, but also remarkably influence allelic segregation and recombination ratio (Bernet et al., 2010; Ollitrault et al., 2012a). In citrus hybridizations, parental genotype and homology have obvious effects on genetic map density. Guo et al. (2015) successfully constructed an integrated genetic map of pummelo with 1543 SNP markers using an intraspecific full-sib F1 population with only 124 individuals. By contrast, Curtolo et al. (2017) attempted to construct a genetic map using an interspecific full-sib F1 population with 278 individuals from crossing of tangor and sweet orange, but only 661 non-redundant SNP-based DArTseq markers were finally mapped on the integrated map. The differences indicate that the use of intraspecific populations tends to achieve much higher marker density in the construction of genetic maps than interspecific populations, even though the population size is much smaller. In our study, the mapping population is neither interspecific nor intraspecific hybrids, but intergeneric hybrids, which had made it more difficult to achieve high-density genetic maps. The potential karyotypic and genomic divergences between trifoliate orange and sweet orange can prevent normal meiotic pairing and homology recombination during meiosis. Some genomic divergences between the two species can be obviously observed from the conservation of synteny of the genetic linkage maps and the reference genome, e.g., the divergences on LG-4, LG-7 and LG-8. In addition to parental genotype, differential fitness of gamete genotypes, crossing direction, and regulatory gene interactions likely also contribute to the high level of segregation distortion in citrus (Bernet et al., 2010; Curtolo et al., 2017). It should be noted that our mapping population was not a full-sib family, but mixed progenies from three different crosses between two trifoliate orange varieties and two sweet orange varieties. Our genotyping results show that only 3.4 and 1.8% of genotyped SNP loci are inconsistent, respectively, between the trifoliate oranges and sweet oranges. However, when compared to the identified segregating SNP markers, the percentages rise up to 29.6 and 6.1%, respectively (Supplementary File S2). This is a substantial percentage of inconsistent segregating markers in trifoliate orange. By analyzing the distribution of these inconsistent SNP loci in trifoliate orange on the reference genome, we found that they are mostly concentrated in certain regions of the genome, and the two trifoliate oranges almost share all of these regions. Therefore, elimination of these polymorphic loci not only reduced the number of available segregating SNP markers, especially for the trifoliate orange map, but also resulted in several large regions with fewer markers on the genetic maps. Actually, many large gaps on the genetic maps were rightly located in the positions of the eliminated polymorphic SNP loci. In addition, in comparison to sweet orange, many fewer segregating markers were identified in trifoliate orange (Supplementary File S2). This problem was reported in all previous genetic maps of trifoliate orange (Chen et al., 2008; Lyon, 2008; Ollitrault et al., 2012a). The low availability of segregating markers in trifoliate orange is due to its lower heterozygosity. As the current sequence coverage only accounts for 2.3% of the citrus genome, to maximize detection of segregating SNP markers, greater sequence coverage is needed for the progenies and parents.
Significant differences in linkage group sizes were observed between trifoliate orange and sweet orange genetic maps. Each of the linkage groups of trifoliate orange is larger in genetic length than the corresponding one of sweet orange, even though there were less markers mapped on the trifoliate orange genetic map. Similar differences in genetic distances were evident in the previously reported EST-SSR genetic maps based on codominant markers segregating in both parents (Chen et al., 2008). Variation in map length could also be observed between SNP-based genetic maps of mandarin, pummelo and sweet orange (Ollitrault et al., 2012a). Due to the utilization of a reference genome, SNP-based genetic linkage maps are comparable if referred to physical distance. The variation in genetic map length is unrelated to genome size among different citrus species, which range from 360 to 398 Mb (Gmitter et al., 2012; Wu et al., 2014, 2018). Extensively distributed in our maps, nearly half of the markers exhibited significant segregation distortion, and such segregation distortion in citrus was proposed to result from gametic selection rather than zygotic selection (Ollitrault et al., 2012a). However, in a simulation study on factors affecting linkage map construction, segregation distortion from gametic selection had little influence on marker order and genetic distance (Hackett and Broadfoot, 2003). Thus, variation of genetic size between linkage group maps of trifoliate orange and sweet orange probably is not due to segregation distortion, but reflects differential recombination rates between the species. Studies on model plants amply demonstrate the impact of genome sequence divergence on recombination rate, and lower recombination rate is related to higher levels of genome divergence (Chetelat et al., 2000; Opperman et al., 2004; Li L. et al., 2006). For different citrus-related genera and species, the degree of genome heterozygosity differs dramatically. Sweet orange, known to be highly heterozygous, was demonstrated to be a complex interspecific hybrid derived from pummelo and mandarin (Xu et al., 2013; Wu et al., 2014). However, trifoliate orange, a genus related to Citrus, was found to be lower in heterozygosity (Chen and Gmitter, 2013). Therefore, in comparison to trifoliate orange, higher heterozygosity in the sweet orange genome probably suppresses recombination frequency, resulting in a smaller genetic size. This is in agreement with the difference in genetic distance between shared markers on genetic maps of Clementine mandarin and pummelo (Ollitrault et al., 2012a). Clementine mandarin is an interspecific hybrid of C. reticulata × C. sinensis with high genome heterozygosity (Wu et al., 2014), while pummelo is a progenitor species in Citrus with low genome heterozygosity (Wang X. et al., 2017). In addition, recombination rates are known to differ between sexes in both plants and animals (Lorch, 2005). The size of the male genetic map of Clementine mandarin was notably larger than its female genetic map (Ollitrault et al., 2012a). Our mapping population was a mix of several crosses between different varieties of trifoliate orange and sweet orange with taxa serving as male and female parents in different crosses. However, most of the progenies were generated with trifoliate orange as the male parent, which may also contribute to the larger size of the trifoliate orange genetic map.
This is the first report on identification of QTLs related to HLB disease and tolerance. Most of the recently reported QTLs in citrus are related to morphological and physiological traits (Kepiro and Roose, 2010; Sugiyama et al., 2011; Sahin-Cevik and Moore, 2012; Raga et al., 2014; Asins et al., 2015; Raga et al., 2016; Yu et al., 2016; Curtolo et al., 2017; Imai et al., 2017; Yu et al., 2017). Only a few reports focused on disease-related QTLs in citrus, such as resistance to citrus tristeza virus (CTV) (Asins et al., 2012; Ohta et al., 2015), Alternaria brown spot (ABS) (Cuenca et al., 2013, 2016), citrus leprosis virus (CiLV) (Bastianel et al., 2009), and citrus nematode (Ling et al., 2000). In genetic studies, estimated effects of each QTL for disease resistance in plants usually ranges from a few percent to 50% or more, and a QTL accounting for phenotypic variation of more than 20% is commonly described as a major QTL, or more than 50% as a dominant QTL (Davey et al., 2006; St Clair, 2010). Interestingly, only one QTL was found with a dominant effect on the phenotypic resistance for each of the diseases, suggesting that the inheritance of resistance for these diseases is mainly controlled by a single dominant allele. In our study, four clusters of QTLs associated with HLB tolerance were identified on the two parental genetic maps. Although all of them could be considered as major QTLs, none of them alone could explain a majority of the phenotypic variation. Our results indicate that the high degree of tolerance to HLB in trifoliate orange cannot be monogenic, because at least four genomic regions are involved. However, the HLB pathogenesis mechanism is still unknown and is likely complicated (Martinelli and Dandekar, 2017;Wang N.A. et al., 2017). Based on the most recent progress in studies on HLB, it was proposed that at least three main molecular mechanisms occur in citrus in response to HLB, and these mechanisms involve many pathways and genes (Martinelli and Dandekar, 2017). This is in agreement with our results that citrus tolerance to HLB is polygenic.
Unlike CTV, CiLV, ABS and citrus nematode for which strong resistance is available within the citrus gene pool, the suppression of HLB in trifoliate orange may be best described as tolerance. Trifoliate orange had been reported as resistant to CLas infection (Folimonova et al., 2009), but recently more studies suggested that it is not true resistant but highly tolerant (Ramadugu et al., 2016; Miles et al., 2017). In our study, though the trifoliate oranges were generally determined to be HLB-negative, most of the replicate trees had marginal results for CLas diagnosis (Ct value of qPCR ranging from 33 to 39), and a few trees were even HLB-positive (Ct value of qPCR under 33). It is important to note that at least eight replicate trees were clonally propagated for each genotype and they all were used throughout the phenotyping, which ensured high accuracy and reliability of the phenotypic results for each genotype. Thus, we believed that the trifoliate orange was probably infected by CLas, but the CLas titer was held at relatively low levels. Our results on HLB disease evaluation also support this interpretation. The trifoliate oranges were not completely healthy under the intense HLB pressure, but mostly displayed slight foliar disease symptoms and slight canopy damage. The results further suggest that trifoliate oranges are not immune to CLas infection, but can inhibit growth of CLas and show low symptom levels when they are infected. It is noteworthy that obviously different degrees of tolerance to HLB was observed among the F1 progenies, and a few progenies showed similar results of CLas diagnosis and disease evaluation as trifoliate oranges, indicating the HLB tolerance is inheritable. Moreover, some other hybrids containing the germplasm of trifoliate orange were also found with certain tolerance to HLB (Albrecht and Bowman, 2011, 2012; Ramadugu et al., 2016), probably due to the partial inheritances of HLB tolerance from trifoliate orange.
Although high-density genetic maps were constructed for both trifoliate orange and sweet orange, the outcome of QTL mapping was still not fine enough for anchoring specific genomic regions or specifying candidate genes associated with citrus responses to HLB. The four anchored genomic regions spanned a total genomic length of approximately 44.6 Mb, consisting of thousands of genes. These genomic regions involved 57 markers from three regions of the trifoliate orange map and 54 markers from one region of the sweet orange map, indicating the marker density of our genetic maps is sufficient for fine mapping. One possible explanation for such phenomenon could be that the quality of the constructed genetic maps is still insufficient for fine mapping of QTLs. The characteristics of plant materials themselves and the utilization of mixed intergeneric populations for genotyping likewise may restrict the construction of genetic maps with sufficient resolution and accuracy. All the linkage groups in trifoliate orange where the QTLs were detected were constructed with fewer markers, especially within the regions of some QTLs. Another possible explanation could be that the size of the phenotyping population is not large enough to yield fine phenotypic data with wide variation. Due to the space requirements for trees and test costs, only 86 of the progenies were selected for phenotyping in our study, accounting for only a half of the genotyping population. Moreover, it is also possible that each of these regions consist of multiple QTLs and they generally co-segregated in the population. As presented in the graphs of QTLs, many QTLs have more than one peak within the LOD score profile (Supplementary Figure S2). Increasing the size of the phenotyping population, to equal that of the genotyping population, should greatly improve the quality of QTL mapping and minimize the number of anchored genomic regions associated with HLB tolerance; however, such larger experiments with tree crops can become cost-prohibitive, in comparison with similar experimental designs with annual or model plant species.
Based on Genotyping-by-Sequencing of an intergeneric F1 population of 170 progenies, we constructed two high-density genome-wide genetic maps for trifoliate orange and sweet orange. Each of the genetic maps contained nine firm linkage groups corresponding to the haploid chromosome number of the species and exhibited high synteny and high coverage of the reported citrus genome. The minor discrepancies among the genetic maps and genome assemblies may represent possible structural rearrangements among citrus species, or alternatively errors in the previous genome assembly. In the replicated field evaluation over 2 years, trifoliate orange and sweet orange showed significant differences in response to CLas infection and HLB, and their progenies exhibited an obvious continuous distribution for two phenotypic traits. Four clusters of QTLs were identified for HLB-incited foliar and canopy responses, respectively, located on LG-t6, LG-t8 and LG-t9 of the trifoliate orange genetic map and LG-s7 of the sweet orange genetic map. These QTLs collectively explained a major part of the phenotypic variation in response to HLB disease. Our results suggest that multiple QTLs are involved in the genetic control of HLB response in citrus. This work provides a starting point for future studies of the underlying genetic architecture of resistance or tolerance to HLB. These QTLs need to be confirmed further to facilitate breeding for resistance or tolerance to HLB in citrus. In addition, the corresponding genomic regions need to be refined if the objective is to discover and characterize candidate genes related to the host response to disease. The final identified QTLs and genes could be good targets for citrus breeding to support long-term solutions to this devastating disease.
FG, ES, MR, ZD, and MH conceived the study. FG and MR developed the mapping population. MR, MH, and QY conducted the work of genotyping. MH, QY, DD, YY, and YZ conducted the work of phenotyping. MH and MR analyzed the genotypic data. MH analyzed the phenotypic data, performed the QTL mapping, and drafted the manuscript. All authors read and approved the final manuscript.
This work was partly supported by grants from the Citrus Research and Development Foundation Inc. (CRDF #536, #15-010) and the New Varieties Development and Management Corporation (NVDMC), on behalf of the Florida citrus industry, and from USDA-NIFA-SCRI #2015-70016-23027.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with several of the authors MH, QY, DD, YY, YZ, and FG.
The authors thank Michael Irey for testing the samples; Randy Driggers for propagating and planting the trees and Sean Reif for maintenance of the field trees; Peng Ling, Yanbo Wang, Misty Holt, Fabieli Irizarry and Hui Wen for their assistance in sampling.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01694/full#supplementary-material
FIGURE S1 | Collinear analysis of consensus between genetic linkage maps and reference genome by dot-plot diagram.
FIGURE S2 | Full genetic linkage maps and genome-wide LOD score graphics of QTL mapping of two phenotypic traits.
FILE S1 | Summary of Genotyping by Sequencing for 170 F1 progenies and four parental varieties.
FILE S2 | Analysis of parental genotypes and distribution of SNP loci on reference genome in four parental varieties.
FILE S3 | Diagnosis of CLas infection by real-time qPCR in 86 F1 progenies and nine control varieties.
FILE S4 | Phenotypic data of two traits for the evaluation of HLB disease in 86 F1 progenies and nine control varieties.
Albrecht, U., and Bowman, K. D. (2011). Tolerance of the trifoliate citrus hybrid US-897 (Citrus reticulata Blanco x Poncirus trifoliata L. Raf.) to Huanglongbing. Hortscience 46, 16–22. doi: 10.1016/j.plantsci.2011.09.008
Albrecht, U., and Bowman, K. D. (2012). Tolerance of trifoliate citrus rootstock hybrids to Candidatus Liberibacter asiaticus. Sci. Hortic. 147, 71–80. doi: 10.1016/j.scienta.2012.08.036
Aldrich, J., and Cullis, C. A. (1993). RAPD analysis in flax: optimization of yield and reproducibility using klenTaq 1 DNA polymerase, chelex 100, and gel purification of genomic DNA. Plant Mol. Biol. Rep. 11, 128–141. doi: 10.1007/bf02670471
Asins, M. J., Fernandez-Ribacoba, J., Bernet, G. P., Gadea, J., Cambra, M., Gorris, M. T., et al. (2012). The position of the major QTL for Citrus tristeza virus resistance is conserved among Citrus grandis, C. aurantium and Poncirus trifoliata. Mol. Breed. 29, 575–587. doi: 10.1007/s11032-011-9574-x
Asins, M. J., Raga, V., Bernet, G. P., and Carbonell, E. A. (2015). Genetic analysis of reproductive, vegetative and fruit quality traits to improve Citrus varieties. Tree Genet. Genomes 11:117. doi: 10.1007/s11295-015-0949-8
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376. doi: 10.1371/journal.pone.0003376
Bastianel, M., Cristofani-Yaly, M., de Oliveira, A. C., Freitas-Astua, J., Garcia, A. A. F., de Resende, M. D. V., et al. (2009). Quantitative trait loci analysis of citrus leprosis resistance in an interspecific backcross family of (Citrus reticulata Blanco x C. sinensis L. Osbeck) x C. sinensis L. Osb. Euphytica 169, 101–111. doi: 10.1007/s10681-009-9950-3
Bernet, G. P., Fernandez-Ribacoba, J., Carbonell, E. A., and Asins, M. J. (2010). Comparative genome-wide segregation analysis and map construction using a reciprocal cross design to facilitate citrus germplasm utilization. Mol. Breed. 25, 659–673. doi: 10.1007/s11032-009-9363-y
Blaustein, R. A., Lorca, G. L., and Teplitski, M. (2018). Challenges for managing Candidatus Liberibacter spp. (Huanglongbing disease pathogen): current control measures and future directions. Phytopathology 108, 424–435. doi: 10.1094/Phyto-07-17-0260-Rvw
Chen, C. X., Bowman, K. D., Choi, Y. A., Dang, P. M., Rao, M. N., Huang, S., et al. (2008). EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet. Genomes 4, 1–10. doi: 10.1007/s11295-007-0083-3
Chen, C. X., and Gmitter, F. G. (2013). Mining of haplotype-based expressed sequence tag single nucleotide polymorphisms in citrus. BMC Genomics 14:746. doi: 10.1186/1471-2164-14-746
Chetelat, R. T., Meglic, V., and Cisneros, P. (2000). A genetic map of tomato based on BC1 Lycopersicon esculentum x Solanum lycopersicoides reveals overall synteny but suppressed recombination between these homeologous genomes. Genetics 154, 857–867.
Cuenca, J., Aleza, P., Garcia-Lor, A., Ollitrault, P., and Navarro, L. (2016). Fine mapping for identification of citrus alternaria brown spot candidate resistance genes and development of new SNP markers for marker-assisted selection. Front. Plant Sci. 7:1948. doi: 10.3389/fpls.2016.01948
Cuenca, J., Aleza, P., Vicent, A., Brunel, D., Ollitrault, P., and Navarro, L. (2013). Genetically based location from triploid populations and gene ontology of a 3.3-Mb genome region linked to Alternaria brown spot resistance in citrus reveal clusters of resistance genes. PLoS One 8:e76755. doi: 10.1371/journal.pone.0076755
Curtolo, M., Cristofani-Yaly, M., Gazaffi, R., Takita, M. A., Figueira, A., and Machado, M. A. (2017). QTL mapping for fruit quality in Citrus using DArTseq markers. BMC Genomics 18:289. doi: 10.1186/s12864-017-3629-2
Davey, M. W., Kenis, K., and Keulemans, J. (2006). Genetic control of fruit vitamin C contents. Plant Physiol. 142, 343–351. doi: 10.1104/pp.106.083279
Deschamps, S., Llaca, V., and May, G. D. (2012). Genotyping-by-Sequencing in plants. Biology 1, 460–483. doi: 10.3390/biology1030460
Folimonova, S. Y., Robertson, C. J., Garnsey, S. M., Gowda, S., and Dawson, W. O. (2009). Examination of the responses of different genotypes of citrus to Huanglongbing (citrus greening) under different conditions. Phytopathology 99, 1346–1354. doi: 10.1094/Phyto-99-12-1346
Gmitter, F. G., Chen, C., Machado, M. A., de Souza, A. A., Ollitrault, P., Froehlicher, Y., et al. (2012). Citrus genomics. Tree Genet. Genomes 8, 611–626. doi: 10.1007/s11295-012-0499-2
Grattapaglia, D., and Sederoff, R. (1994). Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and rapd markers. Genetics 137, 1121–1137.
Guo, F., Yu, H. W., Tang, Z., Jiang, X. L., Wang, L., Wang, X., et al. (2015). Construction of a SNP-based high-density genetic map for pummelo using RAD sequencing. Tree Genet. Genomes 11:2. doi: 10.1007/S11295-014-0831-0
Hackett, C. A., and Broadfoot, L. B. (2003). Effects of genotyping errors, missing values and segregation distortion in molecular marker data on the construction of linkage maps. Heredity 90, 33–38. doi: 10.1038/sj.hdy.6800173
Imai, A., Yoshioka, T., and Hayashi, T. (2017). Quantitative trait locus (QTL) analysis of fruit-quality traits for mandarin breeding in Japan. Tree Genet. Genomes 13:79. doi: 10.1007/s11295-017-1162-8
Kepiro, J. L., and Roose, M. L. (2010). AFLP markers closely linked to a major gene essential for nucellar embryony (apomixis) in Citrus maxima x Poncirus trifoliata. Tree Genet. Genomes 6, 1–11. doi: 10.1007/s11295-009-0223-z
Killiny, N., Valim, M. F., Jones, S. E., Omar, A. A., Hijaz, F., Gmitter, F. G. Jr., et al. (2017). Metabolically speaking: possible reasons behind the tolerance of ‘Sugar Belle’ mandarin hybrid to huanglongbing. Plant Physiol. Biochem. 116, 36–47. doi: 10.1016/j.plaphy.2017.05.001
Lewis-Rosenblum, H., Martini, X., Tiwari, S., and Stelinski, L. L. (2015). Seasonal movement patterns and long-range dispersal of Asian citrus psyllid in Florida citrus. J. Econ. Entomol. 108, 3–10. doi: 10.1093/jee/tou008
Li, L., Jean, M., and Belzile, F. (2006). The impact of sequence divergence and DNA mismatch repair on homeologous recombination in Arabidopsis. Plant J. 45, 908–916. doi: 10.1111/j.1365-313X.2006.02657.x
Li, W. B., Hartung, J. S., and Levy, L. (2006). Quantitative real-time PCR for detection and identification of Candidatus Liberibacter species associated with citrus huanglongbing. J. Microbiol. Methods 66, 104–115. doi: 10.1016/j.mimet.2005.10.018
Li, W. B., Li, D. Y., Twieg, E., Hartung, J. S., and Levy, L. (2008). Optimized quantification of unculturable Candidatus Liberibacter spp. in host plants using real-time PCR. Plant Dis. 92, 854–861. doi: 10.1094/PDIS-92-6-0854
Ling, P., Duncan, L. W., Deng, Z., Dunn, D., Hu, X., Huang, S., et al. (2000). Inheritance of citrus nematode resistance and its linkage with molecular markers. Theor. Appl. Genet. 100, 1010–1017. doi: 10.1007/s001220051382
Lorch, P. D. (2005). Sex differences in recombination and mapping adaptations. Genetica 123, 39–47. doi: 10.1007/s10709-003-2706-4
Lyon, M. P. (2008). A Genomic Genetic Map of the Common Sweet Orange and Poncirus trifoliata. Ph.D. dissertation, University of California, Riverside, CA.
Martinelli, F., and Dandekar, A. M. (2017). Genetic mechanisms of the devious intruder Candidatus Liberibacter in Citrus. Front. Plant Sci. 8:904. doi: 10.3389/fpls.2017.00904
Miles, G. P., Stover, E., Ramadugu, C., Keremane, M. L., and Lee, R. F. (2017). Apparent tolerance to Huanglongbing in Citrus and Citrus-related germplasm. HortScience 52, 31–39. doi: 10.21273/Hortsci11374-16
Ohta, S., Endo, T., Shimada, T., Fujii, H., Shimizu, T., Kita, M., et al. (2015). Construction of genetic linkage map and graphical genotyping of pseudo-backcrossed F2 (BC’2) progeny to introduce a CTV resistance from Poncirus trifoliata (L.) Raf. into Citrus by introgression breeding. Tree Genet. Genomes 11:797. doi: 10.1007/S11295-014-0797-Y
Ollitrault, P., Terol, J., Chen, C. X., Federici, C. T., Lotfy, S., Hippolyte, I., et al. (2012a). A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping. BMC Genomics 13:593. doi: 10.1186/1471-2164-13-593
Ollitrault, P., Terol, J., Garcia-Lor, A., Berard, A., Chauveau, A., Froelicher, Y., et al. (2012b). SNP mining in C. clementina BAC end sequences; transferability in the Citrus genus (Rutaceae), phylogenetic inferences and perspectives for genetic mapping. BMC Genomics 13:13. doi: 10.1186/1471-2164-13-13
Opperman, R., Emmanuel, E., and Levy, A. A. (2004). The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics 168, 2207–2215. doi: 10.1534/genetics.104.032896
Raga, V., Bernet, G. P., Carbonell, E. A., and Asins, M. J. (2012). Segregation and linkage analyses in two complex populations derived from the citrus rootstock Cleopatra mandarin. Inheritance of seed reproductive traits. Tree Genet. Genomes 8, 1061–1071. doi: 10.1007/s11295-012-0486-7
Raga, V., Bernet, G. P., Carbonell, E. A., and Asins, M. J. (2014). Inheritance of rootstock effects and their association with salt tolerance candidate genes in a progeny derived from ‘Volkamer’ lemon. J. Am. Soc. Hortic. Sci. 139, 518–528.
Raga, V., Intrigliolo, D. S., Bernet, G. P., Carbonell, E. A., and Asins, M. J. (2016). Genetic analysis of salt tolerance in a progeny derived from the citrus rootstocks Cleopatra mandarin and trifoliate orange. Tree Genet. Genomes 12:34. doi: 10.1007/s11295-016-0991-1
Ramadugu, C., Keremane, M. L., Halbert, S. E., Duan, Y. P., Roose, M. L., Stover, E., et al. (2016). Long-term field evaluation reveals Huanglongbing resistance in Citrus relatives. Plant Dis. 100, 1858–1869. doi: 10.1094/PDIS-03-16-0271-RE
Richardson, M. L., and Hall, D. G. (2013). Resistance of Poncirus and Citrus x Poncirus germplasm to the Asian citrus psyllid. Crop Sci. 53, 183–188. doi: 10.2135/cropsci2012.02.0091
Richardson, M. L., Westbrook, C. J., Hall, D. G., Stover, E., Duan, Y. P., and Lee, R. F. (2011). Abundance of citrus leafminer larvae on Citrus and Citrus-related germplasm. Hortscience 46, 1260–1264.
Sahin-Cevik, M., and Moore, G. A. (2012). Quantitative trait loci analysis of morphological traits in Citrus. Plant Biotech. Rep. 6, 47–57. doi: 10.1007/s11816-011-0194-z
Shimada, T., Fujii, H., Endo, T., Ueda, T., Sugiyama, A., Nakano, M., et al. (2014). Construction of a citrus framework genetic map anchored by 708 gene-based markers. Tree Genet. Genomes 10, 1001–1013. doi: 10.1007/s11295-014-0738-9
St Clair, D. A. (2010). Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 48, 247–268. doi: 10.1146/annurev-phyto-080508-081904
Stover, E., Hall, D. G., Shatters, R. G., and Moore, G. A. (2016a). Influence of citrus source and test genotypes on inoculations with Candidatus Liberibacter asiaticus. HortScience 51, 805–809.
Stover, E., Inch, S., Richardson, M. L., and Hall, D. G. (2016b). Conventional citrus of some scion/rootstock combinations show field tolerance under high Huanglongbing disease pressure. HortScience 51, 127–132.
Sugiyama, A., Omura, M., Matsumoto, H., Shimada, T., Fujii, H., Endo, T., et al. (2011). Quantitative trait loci (QTL) analysis of carotenoid content in citrus fruit. J. Jpn. Soc. Hortic. Sci. 80, 136–144. doi: 10.2503/jjshs1.80.136
van Ooijen, J. W. (1992). Accuracy of mapping quantitative trait loci in autogamous species. Theor. Appl. Genet. 84, 803–811. doi: 10.1007/bf00227388
Van Ooijen, J. W. (2011). Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet. Res. 93, 343–349. doi: 10.1017/S0016672311000279
Voorrips, R.E. (2002). Map Chart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, N. A., Stelinski, L. L., Pelz-Stelinski, K. S., Graham, J. H., and Zhang, Y. Z. (2017). Tale of the Huanglongbing disease pyramid in the context of the citrus microbiome. Phytopathology 107, 380–387. doi: 10.1094/Phyto-12-16-0426-Rvw
Wang, X., Xu, Y. T., Zhang, S. Q., Cao, L., Huang, Y., Cheng, J. F., et al. (2017). Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49, 765–772. doi: 10.1038/ng.3839
Westbrook, C. J., Hall, D. G., Stover, E., Duan, Y. P., and Lee, R. F. (2011). Colonization of Citrus and Citrus-related germplasm by Diaphorina citri (Hemiptera: Psyllidae). Hortscience 46, 997–1005.
Wu, G. A., Prochnik, S., Jenkins, J., Salse, J., Hellsten, U., Murat, F., et al. (2014). Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 32, 656–662. doi: 10.1038/nbt.2906
Wu, G. A., Terol, J., Ibanez, V., López-García, A., Pérez-Román, E., Borredá, C., et al. (2018). Genomics of the origin and evolution of Citrus. Nature 554, 311–316. doi: 10.1038/nature25447
Xu, Q., Chen, L. L., Ruan, X., Chen, D., Zhu, A., Chen, C., et al. (2013). The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45, 59–66. doi: 10.1038/ng.2472
Yu, Y., Bai, J., Chen, C., Plotto, A., Yu, Q., Baldwin, E. A., et al. (2017). Identification of QTLs controlling aroma volatiles using a ‘Fortune’ x ‘Murcott’ (Citrus reticulata) population. BMC Genomics 18:646. doi: 10.1186/s12864-017-4043-5
Keywords: Candidatus Liberibacter, genetic map, F1 population, genotyping-by-sequencing, Poncirus, QTL mapping, SNP
Citation: Huang M, Roose ML, Yu Q, Du D, Yu Y, Zhang Y, Deng Z, Stover E and Gmitter FG Jr (2018) Construction of High-Density Genetic Maps and Detection of QTLs Associated With Huanglongbing Tolerance in Citrus. Front. Plant Sci. 9:1694. doi: 10.3389/fpls.2018.01694
Received: 31 July 2018; Accepted: 31 October 2018;
Published: 27 November 2018.
Edited by:
Christopher Isaac Vincent, University of Florida, United StatesReviewed by:
Antonio Figueira, Universidade de São Paulo, BrazilCopyright © 2018 Huang, Roose, Yu, Du, Yu, Zhang, Deng, Stover and Gmitter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederick G. Gmitter Jr., ZmdtaXR0ZXJAdWZsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.