 Chrysi Sergaki
Chrysi Sergaki Beatriz Lagunas
Beatriz Lagunas Ian Lidbury
Ian Lidbury Miriam L. Gifford
Miriam L. Gifford Patrick Schäfer
Patrick Schäfer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci. , 17 August 2018
Sec. Plant Pathogen Interactions
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01205
This article is part of the Research Topic Harnessing Useful Rhizosphere Microorganisms for Pathogen and Pest Biocontrol, Volume II View all 33 articles
We face major agricultural challenges that remain a threat for global food security. Soil microbes harbor enormous potentials to provide sustainable and economically favorable solutions that could introduce novel approaches to improve agricultural practices and, hence, crop productivity. In this review we give an overview regarding the current state-of-the-art of microbiome research by discussing new technologies and approaches. We also provide insights into fundamental microbiome research that aim to provide a deeper understanding of the dynamics within microbial communities, as well as their interactions with different plant hosts and the environment. We aim to connect all these approaches with potential applications and reflect how we can use microbial communities in modern agricultural systems to realize a more customized and sustainable use of valuable resources (e.g., soil).
Soil is considered as one of the most diverse habitats on Earth containing billions of bacteria and millions of fungi (comprising thousands of taxa), as well as larger organisms such as nematodes, ants, or moles (Bardgett and van der Putten, 2014). Recent advances in high throughput sequencing techniques and the increasing number of microbial culture libraries are contributing to show an expanded version of the tree of life dominated by bacterial diversification (Hug et al., 2016). This enormous diversity is driven by the ability of microbes to perform lateral gene transfer across disparate phylogenetic groups (McDonald and Currie, 2017). Moreover, microbial communities are built on high numbers of individuals for each species (Robbins et al., 2016), that can quickly proliferate and have high mutation rates (in the range of 10−4 in E. coli) (Kibota and Lynch, 1996; Boe et al., 2000; Denamur and Matic, 2006) as compared to higher organisms like humans [10−8] (Kuroki et al., 2006; Xue et al., 2015). These characteristics increase the diversification of microbes and microbial communities, where individual microbes of the same species could potentially bear different genetic endowments and thus functional characteristics.
Soil microbes play key roles in the cycling of nutrients such as nitrogen or phosphorus as well as providing plant protection against biotic and abiotic stress (Bender et al., 2016; Lladó et al., 2017). Intensive agriculture has contributed to increases in crop yields but at the same time it has had detrimental effects on the physical and biological properties of soils (Pimentel et al., 1995; Bouwman et al., 2009). In intensively managed agricultural systems, the application of fertilizers can compensate for a loss of soil fertility, while tillage disrupts microbial communities (Johnson et al., 1997). This is particularly relevant in the light of current crop production systems with the degradation of more than one half of the global agricultural land while we face massive challenges associated with the disturbance of nitrogen and phosphorous cycles. This situation is very likely to worsen under the prospect of the climate change (Rockström et al., 2009; Yuan et al., 2018). As a consequence the United Nations has suggested the re-introduction of sustainable land management practices to minimize land degradation (Sanz, 2017). These practices include crop diversification, use of local adapted species or intercropping in order to maintain soil fertility, carbon sequestration, and nutrient cycling as well as to control soil erosion (Sanz, 2017). Interestingly, these procedures also enhance general soil disease suppression (Weller et al., 2002; Bonilla et al., 2015). In addition, sustaining microbial community diversity, structure and composition can help to support ecosystem functions, e.g., by regulating nutrient cycles.
During the last decade, microbiome research has modified our perception on the complexity and structure of microbial communities. However, we are only just starting to understand the organization of such complex communities, the interdependencies among themselves and with the biotic (e.g., plant) and abiotic (e.g., edaphic) environment. The increasing need for alternative experimental approaches, as well as the development of new tools has provided new insights into our understanding of the dynamics that occur within the microbiomes and their interaction with host organisms (Goodrich et al., 2017). In studying the human microbiome, the complexity of microbial interactions and the importance of analyzing them separately for each individual has already resulted in novel therapies. Considering the unique microbiome signature of each host, we could move toward a personalized application of microbiome, where we would be able to handle each case independently and better tailor the microbiome to the host’s needs, thus increasing the efficiency of the treatment and the potential of the host (Human Microbiome Project Consortium, 2012). Such “personalized” microbiome approaches would be particularly facilitated by the genetic uniformity of host genotypes of a given crop plant species in the field. Similar to human microbiome studies, there have been efforts to understand the complexity of soil and plant microbiomes (Bulgarelli et al., 2012; Lundberg et al., 2012) and to fuel new innovations in sustainable crop production as part of the next green revolution (Jez et al., 2016). However, to exploit the full potential of microbiomes, we require the development of new analytical strategies to comprehend the array of functional capabilities of microbial communities (Bashiardes et al., 2018). The importance of maintaining a diverse and well-balanced microbiome at the plant–soil interface is vital in crop production. Any microbiome applications, however, have to focus on improving key determinants of crop production such as nutrient availability, soil fertility and soil health (Syed Ab Rahman et al., 2018). In this respect, the key challenge is to transfer lab-generated knowledge to the field. In addition to unraveling the structure of the plant/soil microbiome (Schlaeppi and Bulgarelli, 2015), it especially requires us to connect microbial community dynamics with microbiome functioning (Sánchez-Cañizares et al., 2017). In this review we present the challenges and latest efforts that have been made in order to advance our understanding of the different dimensions of microbiomes (e.g., structure, dynamics) and how it affects plants. We further introduce future approaches to access the full potential of the soil microbiome, including beneficial microbes, in improving crop production.
The reduction in sequencing costs in addition to advances in sequencing technologies and increased computational power has facilitated an overwhelming number of soil and rhizosphere-related microbiome studies (Prosser, 2015; Fierer, 2017). Researchers commonly employ three main types of sequencing: (1) metataxonomic primer-based amplicon sequencing, which focuses on the amplification of specific regions of ubiquitous genetic markers, usually 16S rRNA (Bacteria and Archaea) or the intergenic spacer (ITS) region (Eukaryotes), (2) shotgun sequencing of the entire genomic or transcriptomic information within a given sample (metagenomics and metatranscriptomics) (3) detection of separated and fragmented proteins (metaproteomics), usually by combining liquid chromatography mass spectrometry (LC MS-MS), and (4) detection of metabolites, normally through MS or nuclear magnetic resonance (NMR) (metabolomics). The technical limitations of such metaomics and amplicon sequencing, such as sampling errors, primer or processing biases, computational power and adequate analytical algorithms have been extensively discussed in several comprehensive reviews and will not be discussed here (Hirsch et al., 2010; Carvalhais et al., 2012; Pinto and Raskin, 2012; Scholz et al., 2012; Temperton and Giovannoni, 2012; Tkacz and Poole, 2015). Encouragingly, major progress has been made in alleviating these limitations, but even with “perfect” metaomics techniques, many conceptual limitations, such as extrapolating accurate information from metaomics datasets to draw meaningful conclusions, still exist and require careful experimental design (Prosser, 2015). For example, mis-annotation of genes in datasets is a major limitation on extrapolating data from omics datasets (Lidbury et al., 2014; Fierer, 2017). In addition, particularly with regards to metagenomics, we tend to focus on genes with known functions and ignore a whole suite of genetic information that harbors the potential to perform novel functions, particularly with regards to genes that encode ecologically important extracellular proteins (Christie-Oleza et al., 2015).
One clear benefit of amplicon sequencing is that multiple samples can be processed simultaneously in one sequencing experiment allowing for increased spatiotemporal resolution and the ability to test multivariate factors. To this end, amplicon sequencing has been invaluable in determining general patterns of microbial diversity within the plant microbiome (Bulgarelli et al., 2012; Lundberg et al., 2012; Peiffer et al., 2013; Tkacz et al., 2015). Amplicon sequencing is now frequently employed with more sophisticated techniques, for example exometabolic profiling of plant exudates (Badri et al., 2013; Zhalnina et al., 2018), multi-generation plant trait selection experiments (Haney et al., 2015; Panke-Buisse et al., 2015), crop mutant line experiments (Senga et al., 2017), and microscopy (Rybakova et al., 2017) to help answer specific questions about plant-mediated bacterial recruitment and functioning in the rhizosphere. Indeed, using 16S rRNA gene data has also revealed that members of the “rare” biosphere are actively recruited in the rhizosphere suggesting that they may play an important role despite their low abundance (Dawson et al., 2017). Furthermore, Bulgarelli et al. (2015) suggested the need to combine alternative markers to 16S rRNA, such as 18S rRNA or internal transcribed spacers (ITSs), in order to access and characterize a broader range of microbes and to get a more representative picture of the microbial diversity and structure. Indeed, metagenomics or metatranscriptomics can partially alleviate this problem by sequencing all the genomic content in a sample simultaneously (Chaparro et al., 2014; Bulgarelli et al., 2015).
Beyond determining microbiome structures under different environments, it is particularly challenging to assign a function to individual microbiome members or groups and we tend to consider each microbial group as a functional group. However, even species within a particular genus can have completely different lifestyles - from pathogen to mutualist depending on the environment (Hacquard et al., 2016; Hiruma et al., 2016), or due to horizontal transfer of specific functional genes (Qiu et al., 2009). This variability can lead to dramatic changes in microbial phenotypes of desired traits, such as phosphorus mobilization (Lidbury et al., 2016). More sensitive methods to characterize the microbiome beyond the genus level are needed, along with a better functional characterization of each species, which would require large scale/high throughput techniques (Schlaeppi and Bulgarelli, 2015). With the rate at which technology advances, combining, e.g., computational/modeling methods are very promising. For instance, there is a transition from metagenomics to metaphenomics, which combine the product of the metagenome (or “expressed functions encoded in microbial genomes”) with the environment, taking all the parameters into account that may influence the dynamics of the interactions within the community and the environment (Jansson and Hofmockel, 2018). In this respect, metagenomics can be potentially powerful and provide information about the broad functional capabilities (e.g., secondary metabolite production or carbohydrate utilization) (Bulgarelli et al., 2015) or about specific gene sets (e.g., metabolic pathways) of the microbiome (Wang et al., 2015; Ofaim et al., 2017). With new analytical methods, we can also gain deeper insights into the specific taxa responsible for harboring key functional traits (Prosser, 2015). With the development of easy-to-use commercial kits, extracting DNA from a given sample is straightforward. However, DNA only gives the functional “potential” of a microbial community. Furthermore, particularly in soils, the vast majority (>90%) of microbial biomass (or genetic information) is inactive or dormant (for a comprehensive review, see Fierer, 2017). Nevertheless, this number is likely to drop significantly in the rhizosphere and root, due to the step-wise selection of microbes by plant-mediated factors meaning more microbes are metabolically active in these niches (Bulgarelli et al., 2013). Efforts have been made to extract RNA from rhizosphere samples to look at those microbes that are metabolically active and reveal which genetic pathways they are inducing in response to plant and microbial stimuli (Turner et al., 2013b; Chaparro et al., 2014; Yergeau et al., 2014). Therefore, many studies combine the enrichment of 13C-labeled CO2 with metatranscriptomics to identify microbes responding to plant exudates to improve our understanding on the interactions between microbiome and plant host (Haichar et al., 2016; Lueders et al., 2016).
Unlike, the shorter turnover times of RNA, which reduces the simplicity and robustness of sampling efficiency (Prosser, 2015), proteins, especially exoproteins, are more stable in the environment (Armengaud et al., 2012). Thus, sampling is methodologically easier prone to sampling errors/artifacts. Metaproteomics also provides an exciting opportunity in omics research as it gives a profile of expressed proteins, and hence, (metabolic) activities in a given sample (Heyer et al., 2015). In turn, exoproteomics or exometaproteomics, enriches for the more ecologically important proteins that are involved in nutrient acquisition and microbial–microbial and microbial–host interactions (e.g., extracellular hydrolytic enzymes and transporter systems) (Armengaud et al., 2012; Lidbury et al., 2016). However, there are several drawbacks to metaproteomics, particularly a requirement for sufficient starting material (sometimes up to 100 g of soil is needed) (Johnson-Rollings et al., 2014), as well as the accurately assignment of peptides detected to the correct proteins, which relies on a comprehensive databases (metagenome) and sufficient computational power (Muth et al., 2013, 2016; Timmins-Schiffman et al., 2017). Perhaps this is why meta(exo)proteomics has not been extensively utilized in rhizosphere research, in comparison to studies of other less complex microbial niches, e.g., seawater and anaerobic digesters (Sowell et al., 2009; Williams et al., 2012; Heyer et al., 2015), and enriched chitin-degrading sandy soil samples (Johnson-Rollings et al., 2014). Similarly, this approach can be very powerful for identifying the major extracellular enzymes involved in phosphate mobilization within the rhizosphere (Lidbury et al., 2016; Lidbury et al., unpublished results).
As the major rhizobacterial phyla (Actinobacteria, Proteobacteria, Bacteroidetes, and Firmicutes) are amenable to cultivation, a number of new studies have reverted to extensive isolation efforts followed by genome sequencing and phenotypic characterisation (Bai et al., 2015; Mauchline et al., 2015; Levy et al., 2018). These are often combined with the reconstruction of synthetic communities to determine keystone species and patterns of recruitment in the rhizosphere (Bai et al., 2015; Niu et al., 2017). Reconstruction of microbial communities can help to identify microbe–microbe interactions that have an effect on plant growth (Hartman et al., 2017). The advantage of the isolation approach is that sequencing and assembling of individual genomes is much simpler and usually provides a higher resolution of data than assembling metagenomes collected in situ. More, the isolation approach is a sophisticated tool to functionally validate isolates within a community and/or their interaction with host plants (Levy et al., 2018). Any isolates exhibiting either novel or improved functionality can easily be deployed for further investigation to identify the precise molecular mechanisms and associated rate kinetics of key enzymes. For example, studying the transcriptomic or proteomics response of individual bacterial or fungal isolates to the plant microbiome or associated nutrient stresses has provided useful information on the genes involved in potentially important plant growth promotion (PGP) processes and recruitment of beneficial microbes (Mauchline et al., 2006; Fernández et al., 2013; Lidbury et al., 2016; Martino et al., 2018). This approach can also be particularly useful for the discovery of novel traits associated with PGP activities mediated by microbes (Bruto et al., 2014). Since the in vitro screening methods for PGP traits do not necessarily reveal phenotypes associated with plants, the use of genomic screening tools could provide a fast, large scale screening while encouraging the discovery of novel PGP traits/genes (Finkel et al., 2017). Combining these methods with complementary molecular approaches, such as mutagenic and bio-reporter expression systems (Wetmore et al., 2015; Cheng et al., 2016; Pini et al., 2017) will uncover the role of these PGP traits/genes and improve our predictions about the mechanisms driving interactions within plant microbiomes.
Plants can affect the structure of their root microbiome in favor of beneficial microbes and against pathogens or other deleterious microbes (Berendsen et al., 2012; Bulgarelli et al., 2015). In turn, microbes can also manipulate the host for their own benefit, e.g., altering host metabolism (Jones and Dangl, 2006; Engelstädter and Hurst, 2009; Hacquard et al., 2015; Levy et al., 2018). In addition, the abiotic environment (e.g., edaphic factors) influences both the plant and microbial communities, further enhancing the complexity of ecosystems (Bakker et al., 2014). Network analyses have shown the importance of “microbial hubs,” which are strongly interconnected microbial taxa that severely influence communities and that are thought to be a key to understand microbiome dynamics, and the effect of single microbes on the structure of microbial communities (Agler et al., 2016). For instance, Niu et al. (2017) created synthetic communities using seven representative bacterial strains from the three most abundant phyla obtained from maize roots. Employing this simplified community, they aimed to uncover mechanisms that determine the dynamics of this system. Interestingly, the removal of one strain led to the complete collapse of the community, highlighting the importance of individual members of the microbiome (Niu et al., 2017). It further suggests that small or even subtle changes can lead to significant effects on microbiome structures. Therefore, deciphering underlying inter-microbial dynamics driving community structures can be key in validating stable synthetic communities. There are new efforts to analyse those complex interactions and establish reliable systems that can (i) overcome the soil ecosystem complexity and (ii) build our fundamental knowledge of microbial and plant–microbe interactions.
Since the revelation of the Arabidopsis thaliana core root microbiome, which gave more detailed insights into plant microbiome structures (Bulgarelli et al., 2012), numerous studies have collectively highlighted the importance of microbiomes in ecosystem functioning (Agler et al., 2016). Furthermore, the isolation and characterisation of microbial species together with the development of defined gnotobiotic systems (Lebeis et al., 2015; Finkel et al., 2017) allows the targeted functional characterisation of individual members of plant microbiomes (Bai et al., 2015). Gnotobiotic systems, in particular, have been recognized as being essential for microbiome research as they allow to distinguish between the effect of microbes or microbial combinations and the environment (by applying defined conditions) on plant phenotypes. By revealing individual processes in multicomponent plant–microbe–environment interactions, it gives the possibility to associate genotypes with phenotypes. However, the reduced complexity of such systems as well as the “artificial” or de novo assembly of microbiomes can prevent the recapitulation and, hence, full functionality of natural systems (Vorholt et al., 2017).
Hartman et al. (2017) presented a different way of microbe application as an effort to understand complex interactions within microbiomes. They isolated microbes from the roots of Trifolium pratense and chose one representative strain from each of the four most abundant microbial groups (OTUs) to inoculate sterile microcosms. They reported a negative effect of Flavobacterium on the growth of Trifolium, which was alleviated in the presence of either of the three other bacterial representatives from Pseudomonas, Janthinobacterium, and Microbacterium. Interestingly, none of the three bacteria affected the abundance of the Flavobacterium in the synthetic community. Therefore, the negative activity of Flavobacterium was somehow “buffered” by Pseudomonas, Janthinobacterium, and Microbacterium (Hartman et al., 2017). Such a reductionist approach can reveal new “keystone” players in regulating microbiome function and its interaction with the plant.
All these analytical approaches have greatly benefitted from the advancements in computational analyses and machine learning and their significance for the development of microbiome research (Knight et al., 2018). COREMIC, for example, is a bioinformatics tool that allows the generation and confirmation of hypothetical models, by associating microbes with certain plants or habitants using existing databases (Rodrigues et al., 2018). As for other omics-based analyses, the need for reproducibility as well as the development of “golden standards” to improve consistency and comparability of experiments have been particularly highlighted (Knight et al., 2018). Furthermore, network analyses have equipped microbiome research with sophisticated tools that can analyse and explain the complexity of microbial communities (Adair and Douglas, 2017; Wang H. et al., 2017). While network analyses often build the basis in revealing the function of microbial taxa and the nature of microbial interactions (Poudel et al., 2016), it has certain limitations in identifying synergistic, additive and antagonistic effects. As a result, key functions of certain low abundant microbes might be underestimated or not even recognized (Shade et al., 2014; Shi et al., 2016). The ultimate aim of these efforts, the identification of interactions within microbial communities on plants as a result of inter–microbial communication, therefore requires sensitive tools uncovering correlative interactions that can be verified in biological assays.
Besides comprehending the complexity of microbial communities and interactions, there is the need to uncover basic regulatory (communicative) principles of interactions that can inform experimental design. Recently, cytology-based systems have been developed to study microbial interactions. Hennessy et al. (2017) established a microplate reader-based system, to quantify the activities and interactions between living microbes. This method represents a potential high-throughput screen by using live imaging of fluorescing metabolites and microbial growth to identify and trace the expression of certain genes in defined microbial communities. In their study, they measured the rate of the production of fluorescent metabolites of Pseudomonas fluorescens in response to the presence of Fusarium graminearum, as an indicator for their interaction (Hennessy et al., 2017). Alternatively, Massalha et al. (2017) published recently a microfluidics-based system for in vivo imaging of plant root–microbe interactions. Using a transparent chamber, they could record root zone preferably colonized by a fluorophore-tagged microbe. By adding a second microbe to the system they were able to study microbe–microbe interactions in real time. Despite their minimalistic set-up, such studies reveal fundamental insights into basic principles that shape microbe–microbe and root–microbe interactions. In this respect, the application of transparent soil represents an innovative approach to study and live image microbes on plant roots in an environment which mimics different soil textures (Downie et al., 2012). It allows to detect processes driving the distribution of microbes in bulk substrate along the root (Downie et al., 2014) and study the effects of major root pests such as nematodes on microbe/community behavior (O’Callaghan et al., 2018).
In addition to bacterial and fungal microbiomes, soil and plant processes are directly influenced by other organisms including viruses, archaea, nematodes, and insects. Viruses play a very important role in soil biochemical processes and act as gene reservoirs for horizontal gene transfer, although their function is not completely understood (Pratama and van Elsas, 2018). Similarly, Archaea and nematodes significantly contribute to microbiome diversity and in interaction with other microbes to soil-plant processes and ecosystem functioning (Adam et al., 2017; Castillo et al., 2017; Elhady et al., 2017). In this respect, Benítez et al. (2017) has given a very interesting insight into plant–microbe–insect interactions. They reported that soil microorganisms can affect aboveground interactions between plants and insects, by modulating the release of plant volatiles (Pineda et al., 2015; Beck and Vannette, 2017; Benítez et al., 2017). These studies indicate that we need more comprehensive, holistic studies on multitrophic interactions in order to understand which edaphic and biotic factors determine the structure and, hence, function of soil and plant microbiomes structures.
Exploiting the full potential of microbes and microbial communities will depend on expertise from different fields. In addition to improving our understanding of complex plant–microbe and multitrophic interactions using plant biology and microbiology-based approaches, we need to develop new ecological systems with growing complexity. Most critically, in order for this knowledge to be successfully transferred to agriculture it is essential to understand the impact of various farming practices on the microbiome and how this is translated to plant health and, thus, crop productivity. In addition, it is necessary to test microbial community function in a highly complex and diverse system (e.g., field), bridging the gap between the lab and the farm.
Increasing evidence is showing that plant–microbe interactions can be beneficial or detrimental for either the host or microbial symbiont depending on the balance of associated biotic and abiotic factors. Whilst, experiments involving pairwise interactions under controlled conditions have increased our knowledge about gene and metabolite expression profiles involved in plant–microbe interactions, these experiments give us little information about microbial function in a natural ecosystem (de Boer, 2017). Although this was stated by de Boer (2017) for fungal–bacterial interactions it is applicable to many other interactions (even to those such as plant–rhizobia or plant–mycorrhizal interactions). For example, even some species of plant-growth promoting arbuscular mycorrhizae fungi (AMF) have been shown to inhibit plant growth under certain conditions, e.g., low light, low temperature or phosphorous (P) availability (Smith and Smith, 1996; Johnson et al., 1997). In addition, AMF activity can also be suppressed by the soil microbiota (Svenningsen et al., 2018) highlighting the practical need for field experiments to fully understand microbe behavior.
The main reason for the existence of this lab-farm gap is that lab studies generally do not capture the complexity of microbe–microbe interactions that occur in a natural setting. However, it is widely known now that microbial communities and plant–microbe interactions are highly dependent on the entire ecosystem (Bulgarelli et al., 2013; de Boer, 2017; Lewis et al., 2018). For instance, host genotypes have been shown to shape plant microbial communities (Bulgarelli et al., 2013; Horton et al., 2014) and a genome wide association study (GWAS) analysis revealed that both bacterial and fungal communities are structured by the same host biological processes (such as defense response or signal transduction). However, different genes seem to be involved in the interaction (Horton et al., 2014) and microbial communities are further fine-tuned during plant development according to host requirements (Chaparro et al., 2014). Host-dependent control of the microbial community is likely controlled by the flow of organic compounds from the root to the rhizosphere (rhizodeposition) (Chaparro et al., 2014; Baran et al., 2015) which has been shown to attract beneficial microbes and refrain pathogen attack. The legume-rhizobia symbiosis is an elegant example of rhizodeposits selecting for beneficial microbes. In response to low nitrogen, the host releases flavonoid compounds that initiate the molecular dialog with nitrogen-fixing rhizobia, resulting in root nodulation and nitrogen fixation (Oldroyd, 2013). In addition, rhizodeposition also functions as a chemical signal for the establishment of inter–root or root–microbe interactions (Jones et al., 2009). Therefore, different hosts, holding different gene sets, will trigger different responses to the same inoculant. Moreover, the same host will release different root exudates depending on the soil nutrient and microbial environment. All these examples represent the cyclic feedback between all the components of this ecosystem (plant–soil–microbes). This likely explains why field microbial inoculants fail to persist for long periods (Finkel et al., 2017). Moreover, the soil ecosystem plays a key role on the establishment of root microbiome (Edwards et al., 2015; Zarraonaindia et al., 2015), which means, that even if the inoculant survives within the soil community, it is not guaranteed that it would colonize the plant host.
Any benefits mediated by microbes observed under controlled conditions will ultimately need to be operative in the field. This implies their persistence in the field over time and successful plant colonization over a wide range of varying biotic, abiotic, and climate conditions. Therefore, finding single inoculants that can perform in such a variety of scenarios will be highly unlikely, which increases the need for the development of microbial precision agriculture mirroring the concept of human personalized medicine (Hamburg and Collins, 2010). In fact, the abundance of similarities between human gut and plant root microbiomes is striking and reveals the importance of the root microbiome in controlling plant fitness (Berendsen et al., 2012). Specifically, it has been shown that complex microbial inoculums can improve plant disease resistance and promote growth better than individual inoculums (reviewed in Finkel et al., 2017), highlighting the synergistic effects of a community. However, these findings still need to consider the soil context to address their potential use as soil amendments. In addition, a deeper understanding of a microbe’s function within a community and within a host would require functional studies where the ecosystem is challenged with different conditions (e.g., temperature, light, humidity). Those studies would validate their community interactions and their beneficial or detrimental outcome for plants as a prerequisite to justify for further field experiments (Figure 1).
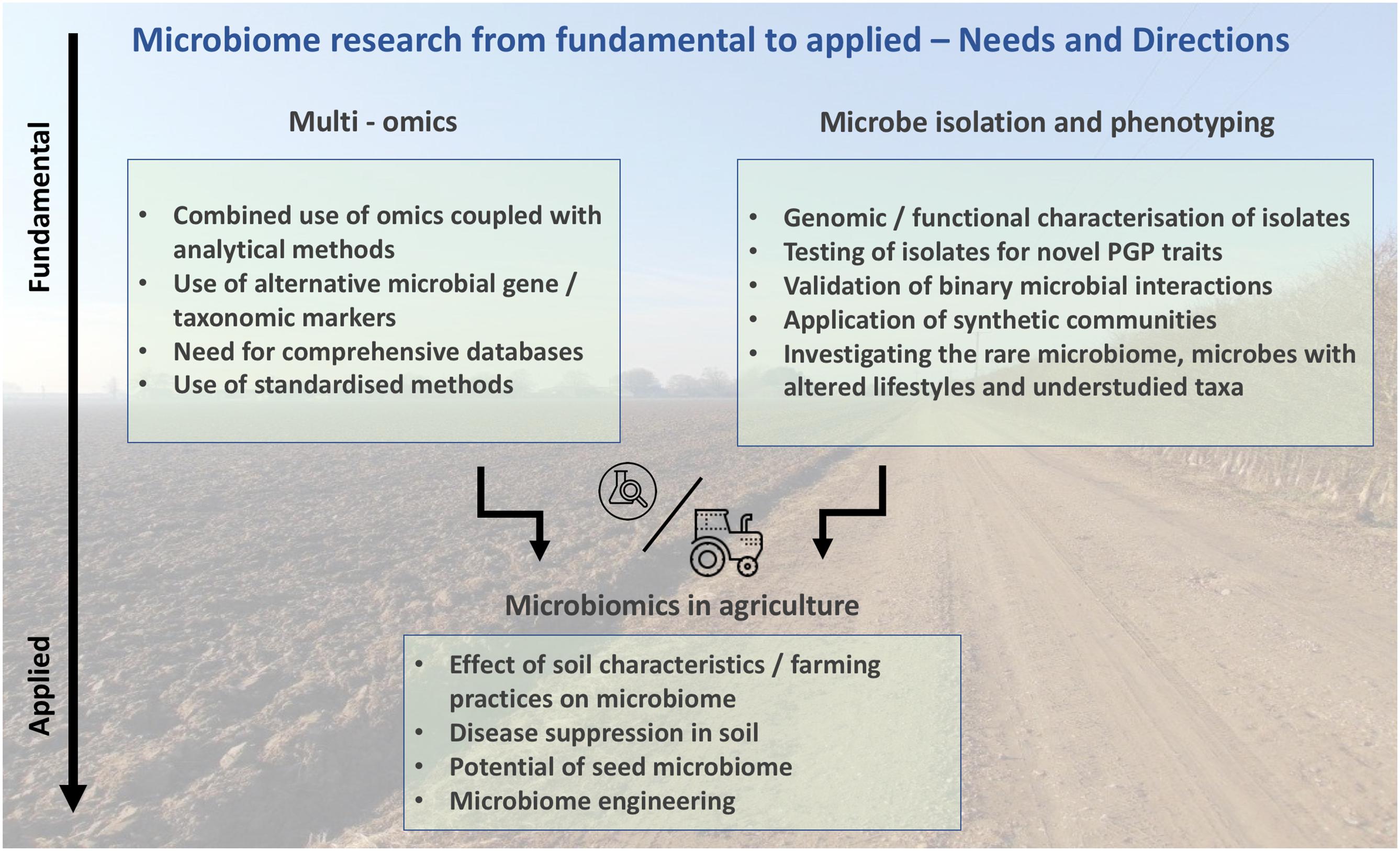
FIGURE 1. Potential of microbiome research; connecting fundamental microbiomics with applied microbiomics in agriculture.
The immense microbial biodiversity in soil is regulated at very different scales, for example changes in soil texture, biotic interactions or plant root exudates have enormous effects down to the smallest (microbial) scales. Variations in the physical and chemical properties of soil, such as pH, nutrient distribution or water retention, have effects on soil biodiversity on the medium (field) scales with soil pH as a suggested major driver of microbial diversity (Fierer and Jackson, 2006) linking microbial community structure with soil nutrient availability and cycling (Li et al., 2017). Finally, at larger scales, geo-localization and climate might play more relevant roles on controlling soil biodiversity (Bardgett and van der Putten, 2014).
The existence of disease suppressive soils is living proof that microbial communities cannot just promote plant growth (Chaparro et al., 2012; Van Der Heijden et al., 2008) but also provide protection against plant pathogens (Alabouvette, 1986; Andrivon, 1994; Shiomi et al., 1999). Disease suppression can be due to competition with native soil fauna (general suppression) or to the presence of specific subsets of microbes (specific suppression). These soil protection strategies are comparable to immunity strategies of animals (Raaijmakers and Mazzola, 2016). As common to all complex ecosystems, general suppression is also common to all soils. Specific suppression, in turn, is removed by soil pasteurization and can be transferred to other soils via soil transplants. Soils can lose their suppressiveness if non-host plants are grown and can be recovered if the susceptible host and pathogen are grown back in them (Wiseman et al., 1996; Berendsen et al., 2012; Raaijmakers and Mazzola, 2016).
The ability of plants to attract beneficial root microbes might represent a crucial strategy to survive under unfavorable environmental conditions. Several studies point out to the possibility of engineering microbiomes to control plant traits that can be used to increase and sustain plant production (Mueller and Sachs, 2015; Panke-Buisse et al., 2015; Herrera Paredes et al., 2018; Orozco-Mosqueda et al., 2018). Plants challenged with pathogens can recruit protective microbes in the rhizosphere and endosphere that can modulate the host immune responses (Berendsen et al., 2018). This strategy has been exploited to formulate bioorganic fertilizers that manipulate banana rhizosphere microbial structure and subsequently decrease the incidence of Panama disease (Xue et al., 2015). All these studies have demonstrated the feasibility to engineer plant microbiomes as a sustainable solution to increase yields as well as biotic and abiotic stress resistance.
Another recent breakthrough discovery highly relevant for microbiome research is the analysis of seed microbiomes. It is supposed to have been co-selected and evolved with the plant providing valuable traits that have driven and still drive plant evolution (Puente et al., 2009; Johnston-Monje and Raizada, 2011; Turner et al., 2013a; Bouffaud et al., 2014; Delaux et al., 2014; Hardoim et al., 2015). Seed microbiomes seem to consist of a limited range of microbial species and this restricted number is probably due to the requirement of these species to survive all seed developmental processes, even the most extreme such as desiccation (Truyens et al., 2015). These studies are in accordance with recent experiments that have shown the possibility to transfer the plant microbiota to the next generation (Mitter et al., 2017). All these findings have put plant microbial engineering and breeding at the forefront of sustainable agriculture (Wei and Jousset, 2017).
To ultimately bridge the lab-field gap, we need to take into account that field experiments (in contrast to glasshouse experiments) are subject to agricultural practices, and these have a significant influence on microbiomes and microbial diversity. According to the UN, sustainable land management practices such as (i) crop rotation, intercropping and use of local plant varieties, (ii) tillage and organic farming should be re-introduced to minimize land degradation (Sanz, 2017). However, it is not clear how these practices can be re-introduced in agricultural systems of the developed world whilst still sustaining or increasing crop production. These practices also have a major contribution to microbiome community structure and function (Oberson et al., 1993; Mäder et al., 2002; Dossa et al., 2012; Debenport et al., 2015; Reganold and Wachter, 2016; Hartman et al., 2017, 2018; Wang Y. et al., 2017). Therefore, they might be important to consider when designing microbiome field experiments or testing commercial field applications of microbial inoculants.
Some ancient agricultural practices started to become less important around the 1940s, since monocropping and synthetic fertilizer applications significantly increased crop yields. These massive agricultural changes were part of the Green Revolution with the intention of feeding an increasingly growing human population. Together with the development of input-intensive agricultural systems for various single cash crops, other agricultural practices were no longer practical in developed countries. However, under the current global scenario of land degradation, fertilizer shortage or global warming, developing sustainable agriculture solutions face the challenge of feeding the still growing human population with minimal ecological and economic impact.
Land management has significant impacts on soil and root microbial community structure and stability and consequently on microbiome-associated functions (Hartman et al., 2017, 2018). Crops grown in monoculture or short rotations often suffer yield decline, due to an enrichment of pathogenic relative to beneficial microbes (Bennett et al., 2012; Hilton et al., 2013; Santhanam et al., 2015). In response, in a field setting, inoculation with native root-associated bacterial isolates can significantly decrease the emergence of diseases associated with continuous cropping (Santhanam et al., 2015) illustrating the potential for employing local microbial resources to increase plant yield and fitness in sustainable agriculture. As mentioned earlier, since soil disease suppression is lost when a different host plant is grown, this property also seems to be directly related to continuous cropping of the same species. These two opposite outcomes for agricultural production do not only reflect the enormous impact that plant hosts have on the soil microbiome, but also how the latter can impact on plant species that can successfully colonize an environment in natural conditions.
In addition to general edaphic factors, different plant species (Rovira, 1969), plant ecotypes (Micallef et al., 2009), or even different locations (microenvironment) of a root system (Pinton et al., 2007) result in the release of distinct root exudates. Therefore, soil microbial communities are shaped differently depending on the plant species grown. Intercropping was an ancient agricultural practice that was abandoned due to the development of modern intensive agricultural systems. However, intercropping is still a common practice in developing countries, where different plant species are grown in close proximity. Intercropping experiments performed in the Sahel region (Africa) have shown that this practice increases crop yield, soil organic carbon levels and community diversity of both bacteria and fungi (Debenport et al., 2015). Moreover, the co-cultivation with indigenous shrubs improves soil quality and N conservation (Dossa et al., 2012) highlighting the importance of using local species that have already adapted strategies to exploit the natural resources of an ecosystem. Intercropping has been suggested as an alternative for sustainable agriculture production. However, for it to become a common practice in developed countries, multiple challenges would need to be addressed, such as the development of cropping systems adapted to this agricultural practice.
In terms of microbiome research, more studies are required to comprehend how different cropping practices have such a relevant impact in the soil microbiota and whether both cropping practices and microbiome engineering could contribute to sustainable agriculture in the long term.
Land tilling is extended in modern agriculture since it minimizes weed growth and creates a seedbed that is adapted to the machinery commonly used in the field. Since the introduction of plant growth regulators in the 1940s (Bagavathiannan and Davis, 2018), no-tillage systems have been explored as a practice in conservation agriculture. However, no tilling systems require the use of cover crops and especially higher amounts of herbicides, which puts off many consumers and farmers. In turn, this practice minimizes soil particle disturbance, increases organic carbon soil content and enhances soil aggregation as well as water infiltration (Álvaro-Fuentes et al., 2008; Hobbs et al., 2008; Li et al., 2017; Wang Y. et al., 2017). Long-term no tillage and organic input management practices impact soil pH (being slightly higher in organic systems) (Mäder et al., 2002) and nutrient flux from the soil matrix to the soil solution. In terms of microbiome research, no-tilling and organic farming practices correlate with increases in soil microbial diversity, biomass and microbial community stability (Oberson et al., 1993; Mäder et al., 2002; Reganold and Wachter, 2016; Wang Y. et al., 2017). These positive effects on the soil microbiota are likely due to the increase of organic matter (acting as food resources for the microbial community), the decrease of physical perturbations (Wang Y. et al., 2017) and the increase in soil aggregate stability (Siegrist et al., 1998).
The generation of microbial communities with customized (beneficial) activities has the potential to serve as a powerful approach to enhance sustainable agricultural production by increasing crop health, through combatting plant diseases and reducing the application of fertilizers. To reach this goal a fundamental understanding regarding the functioning of the plant microbiome through microbe–microbe and plant–microbe interaction is required, as well as a deeper understanding of the soil microbial community structure over time (long-term studies) and its plasticity and response to the environmental changes. Also, since individual microbes are key for the regulation of microbial community structure and stability, more comprehensive studies investigating community dynamics using these individual microbes and their soil microbial communities would assist in advancing the field. This knowledge could help to fully understand the impact that these keystone microbes have on crop yields, disease resistance and global nutrient cycles, but also to reveal strategies for microbiome engineering.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Work in the PS and MG labs is funded by the EPSRC/BBSRC-funded Warwick Integrative Synthetic Biology Centre (BB/M017982/1 to PS), BBSRC (BB/P00-2145/1 to MG and PS) and a NERC-CENTA studentship (NE/L002493/1 to CS).
Adair, K. L., and Douglas, A. E. (2017). Making a microbiome: the many determinants of host-associated microbial community composition. Curr. Opin. Microbiol. 35, 23–29. doi: 10.1016/j.mib.2016.11.002
Adam, P. S., Borrel, G., Brochier-Armanet, C., and Gribaldo, S. (2017). The growing tree of Archaea: new perspectives on their diversity, evolution and ecology. ISME J. 11, 2407–2425. doi: 10.1038/ismej.2017.122
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub Taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14:e1002352. doi: 10.1371/journal.pbio.1002352
Alabouvette, C. (1986). Fusarium-wilt suppressive soils from the Châteaurenard region: review of a 10-year study. Agronomie 6, 273–284. doi: 10.1051/agro
Álvaro-Fuentes, J., Lopez, M. V., Cantero-Martinez, C., and Arrue, J. L. (2008). Tillage effects on soil organic carbon fractions in Mediterranean dryland agroecosystems. Soil Sci. Soc. Am. J. 72, 541–547. doi: 10.2136/sssaj2007.0164
Andrivon, D. (1994). Dynamics of the survival and infectivity to potato tubers of sporangia of Phytophthora infestans in three different soils. Soil Biol. Biochem. 26, 945–952. doi: 10.1016/0038-0717(94)90107-4
Armengaud, J., Christie-Oleza, J. A., Clair, G., Malard, V., and Duport, C. (2012). Exoproteomics: exploring the world around biological systems. Expert Rev. Proteomics 9, 561–575. doi: 10.1586/epr.12.52
Badri, D. V., Chaparro, J. M., Zhang, R., Shen, Q., and Vivanco, J. M. (2013). Application of natural blends of phytochemicals derived from the root exudates of arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem. 288, 4502–4512. doi: 10.1074/jbc.M112.433300
Bagavathiannan, M. V., and Davis, A. S. (2018). An ecological perspective on managing weeds during the great selection for herbicide resistance. Pest Manag. Sci. doi: 10.1002/ps.4920 [Epub ahead of print].
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Bakker, M. G., Schlatter, D. C., Otto-Hanson, L., and Kinkel, L. L. (2014). Diffuse symbioses: roles of plant-plant, plant-microbe and microbe-microbe interactions in structuring the soil microbiome. Mol. Ecol. 23, 1571–1583. doi: 10.1111/mec.12571
Baran, R., Brodie, E. L., Mayberry-Lewis, J., Hummel, E., Da Rocha, U. N., Chakraborty, R., et al. (2015). Exometabolite niche partitioning among sympatric soil bacteria. Nat. Commun. 6:8289. doi: 10.1038/ncomms9289
Bardgett, R. D., and van der Putten, W. H. (2014). Belowground biodiversity and ecosystem functioning. Nature 515, 505–511. doi: 10.1038/nature13855
Bashiardes, S., Godneva, A., Elinav, E., and Segal, E. (2018). Towards utilization of the human genome and microbiome for personalized nutrition. Curr. Opin. Biotechnol. 51, 57–63. doi: 10.1016/j.copbio.2017.11.013
Beck, J. J., and Vannette, R. L. (2017). Harnessing insect-microbe chemical communications to control insect pests of agricultural systems. J. Agric. Food Chem. 65, 23–28. doi: 10.1021/acs.jafc.6b04298
Bender, S. F., Wagg, C., and van der Heijden, M. G. A. (2016). An underground revolution: biodiversity and soil ecological engineering for agricultural sustainability. Trends Ecol. Evol. 31, 440–452. doi: 10.1016/j.tree.2016.02.016
Benítez, E., Paredes, D., Rodríguez, E., Aldana, D., González, M., Nogales, R., et al. (2017). Bottom-up effects on herbivore-induced plant defences: a case study based on compositional patterns of rhizosphere microbial communities. Sci. Rep. 7:6251. doi: 10.1038/s41598-017-06714-x
Bennett, A. J., Bending, G. D., Chandler, D., Hilton, S., and Mills, P. (2012). Meeting the demand for crop production: the challenge of yield decline in crops grown in short rotations. Biol. Rev. 87, 52–71. doi: 10.1111/j.1469-185X.2011.00184.x
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berendsen, R. L., Vismans, G., Yu, K., Song, Y., De Jonge, R., Burgman, W. P., et al. (2018). Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 12, 1496–1507. doi: 10.1038/s41396-018-0093-1
Boe, L., Danielsen, M., Knudsen, S., Petersen, J. B., Maymann, J., and Jensen, P. R. (2000). The frequency of mutators in populations of Escherichia coli. Mutat. Res. 448, 47–55. doi: 10.1016/S0027-5107(99)00239-0
Bonilla, N., Vida, C., Martínez-Alonso, M., Landa, B. B., Gaju, N., Cazorla, F. M., et al. (2015). Organic amendments to avocado crops induce suppressiveness and influence the composition and activity of soil microbial communities. Appl. Environ. Microbiol. 81, 3405–3418. doi: 10.1128/AEM.03787-14
Bouffaud, M. L., Poirier, M. A., Muller, D., and Moënne-Loccoz, Y. (2014). Root microbiome relates to plant host evolution in maize and other Poaceae. Environ. Microbiol. 16, 2804–2814. doi: 10.1111/1462-2920.12442
Bouwman, A. F., Beusen, A. H. W., and Billen, G. (2009). Human alteration of the global nitrogen and phosphorus soil balances for the period 1970-2050. Glob. Biogeochem. Cycles 23, 1–16. doi: 10.1029/2009GB003576
Bruto, M., Prigent-Combaret, C., Muller, D., and Moënne-Loccoz, Y. (2014). Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci. Rep. 4:6261. doi: 10.1038/srep06261
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren van Themaat, E., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Bulgarelli, D., Schlaeppi, K., Spaepen, S., van Themaat, E. V. L., and Schulze-Lefert, P. (2013). Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Carvalhais, L. C., Dennis, P. G., Tyson, G. W., and Schenk, P. M. (2012). Application of metatranscriptomics to soil environments. J. Microbiol. Methods 91, 246–251. doi: 10.1016/j.mimet.2012.08.011
Castillo, J. D., Vivanco, J. M., and Manter, D. K. (2017). Bacterial microbiome and nematode occurrence in different potato agricultural soils. Microb. Ecol. 74, 888–900. doi: 10.1007/s00248-017-0990-2
Chaparro, J. M., Badri, D. V., and Vivanco, J. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J. 8, 790–803. doi: 10.1038/ismej.2013.196
Chaparro, J. M., Sheflin, A. M., Manter, D. K., and Vivanco, J. M. (2012). Manipulating the soil microbiome to increase soil health and plant fertility. Biol. Fertil. Soils 48, 489–499. doi: 10.1007/s00374-012-0691-4
Cheng, G., Karunakaran, R., East, A. K., and Poole, P. S. (2016). Multiplicity of sulfate and molybdate transporters and their role in nitrogen fixation in Rhizobium leguminosarum bv. viciae Rlv3841. Mol. Plant Microbe Interact. 29, 143–152. doi: 10.1094/MPMI-09-15-0215-R
Christie-Oleza, J. A., Armengaud, J., Guerin, P., and Scanlan, D. J. (2015). Functional distinctness in the exoproteomes of marine Synechococcus. Environ. Microbiol. 17, 3781–3794. doi: 10.1111/1462-2920.12822
Dawson, W., Hör, J., Egert, M., van Kleunen, M., and Peste, M. (2017). A small number of low-abundance bacteria dominate plant species-specific responses during rhizosphere colonization. Front. Microbiol. 8:975. doi: 10.3389/fmicb.2017.00975
de Boer, W. (2017). Upscaling of fungal–bacterial interactions: from the lab to the field. Curr. Opin. Microbiol. 37, 35–41. doi: 10.1016/j.mib.2017.03.007
Debenport, S. J., Assigbetse, K., Bayala, R., Chapuis-Lardy, L., Dick, R. P., and McSpadden Gardener, B. B. (2015). Association of shifting populations in the root zone microbiome of millet with enhanced crop productivity in the Sahel Region (Africa). Appl. Environ. Microbiol. 81, 2841–2851. doi: 10.1128/AEM.04122-14
Delaux, P. M., Varala, K., Edger, P. P., Coruzzi, G. M., Pires, J. C., and Ané, J. M. (2014). Comparative phylogenomics uncovers the impact of symbiotic associations on host genome evolution. PLoS Genet. 10: e1004487. doi: 10.1371/journal.pgen.1004487
Denamur, E., and Matic, I. (2006). Evolution of mutation rates in bacteria. Mol. Microbiol. 60, 820–827. doi: 10.1111/j.1365-2958.2006.05150.x
Dossa, E. L., Diedhiou, I., Khouma, M., Sene, M., Lufafa, A., Kizito, F., et al. (2012). Crop productivity and nutrient dynamics in a shrub (Guiera senegalensis)-based farming system of the sahel. Agron. J. 104:1255. doi: 10.2134/agronj2011.0399
Downie, H., Holden, N., Otten, W., Spiers, A. J., Valentine, T. A., and Dupuy, L. X. (2012). Transparent soil for imaging the rhizosphere. PLoS One 7: e44276. doi: 10.1371/journal.pone.0044276
Downie, H. F., Valentine, T. A., Otten, W., Spiers, A. J., and Dupuy, L. X. (2014). Transparent soil microcosms allow 3D spatial quantification of soil microbiological processes in vivo. Plant Signal. Behav. 9: e970421. doi: 10.4161/15592316.2014.970421
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Elhady, A., Giné, A., Topalovic, O., Jacquiod, S., Sørensen, S. J., Sorribas, F. J., et al. (2017). Microbiomes associated with infective stages of root-knot and lesion nematodes in soil. PLoS One 12: e0177145. doi: 10.1371/journal.pone.0177145
Engelstädter, J., and Hurst, G. D. D. (2009). The Ecology and Evolution of Microbes that Manipulate Host Reproduction. Annu. Rev. Ecol. Evol. Syst. 40, 127–149. doi: 10.1146/annurev.ecolsys.110308.120206
Fernández, M., Conde, S., Duque, E., and Ramos, J. L. (2013). In vivo gene expression of Pseudomonas putida KT2440 in the rhizosphere of different plants. Microb. Biotechnol. 6, 307–313. doi: 10.1111/1751-7915.12037
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. doi: 10.1073/pnas.0507535103
Finkel, O. M., Castrillo, G., Herrera Paredes, S., Salas González, I., and Dangl, J. L. (2017). Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 38, 155–163. doi: 10.1016/j.pbi.2017.04.018
Goodrich, J. K., Davenport, E. R., Clark, A. G., and Ley, R. E. (2017). The relationship between the human genome and microbiome comes into view. Annu. Rev. Genet. 51, 413–433. doi: 10.1146/annurev-genet-110711-155532
Hacquard, S., Garrido-Oter, R., González, A., Spaepen, S., Ackermann, G., Lebeis, S., et al. (2015). Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616. doi: 10.1016/j.chom.2015.04.009
Hacquard, S., Kracher, B., Hiruma, K., Münch, P. C., Garrido-Oter, R., Thon, M. R., et al. (2016). Survival trade-offs in plant roots during colonization by closely related beneficial and pathogenic fungi. Nat. Commun. 7:11362. doi: 10.1038/ncomms11362
Haichar, F. E.Z., Heulin, T., Guyonnet, J. P., and Achouak, W. (2016). Stable isotope probing of carbon flow in the plant holobiont. Curr. Opin. Biotechnol. 41, 9–13. doi: 10.1016/j.copbio.2016.02.023
Hamburg, M. A., and Collins, F. S. (2010). The path to personalized medicine-perspective. N. Engl. J. Med. 363, 301–304. doi: 10.1056/NEJMp1006304
Haney, C. H., Samuel, B. S., Bush, J., and Ausubel, F. M. (2015). Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat. Plants 1:15051. doi: 10.1038/nplants.2015.51
Hardoim, P. R., van Overbeek, L. S., Berg, G., Pirttilä, A. M., Compant, S., Campisano, A., et al. (2015). The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 79, 293–320. doi: 10.1128/MMBR.00050-14
Hartman, K., van der Heijden, M. G. A., Roussely-Provent, V., Walser, J. C., and Schlaeppi, K. (2017). Deciphering composition and function of the root microbiome of a legume plant. Microbiome 5:2. doi: 10.1186/s40168-016-0220-z
Hartman, K., van der Heijden, M. G. A., Wittwer, R. A., Banerjee, S., Walser, J. C., and Schlaeppi, K. (2018). Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 6:14. doi: 10.1186/s40168-017-0389-9
Hennessy, R. C., Stougaard, P., and Olsson, S. (2017). A microplate reader-based system for visualizing transcriptional activity during in vivo microbial interactions in space and time. Sci. Rep. 7:281. doi: 10.1038/s41598-017-00296-4
Herrera Paredes, S., Gao, T., Law, T. F., Finkel, O. M., Mucyn, T., Teixeira, P. J. P. L., et al. (2018). Design of synthetic bacterial communities for predictable plant phenotypes. PLoS Biol. 16:e2003962. doi: 10.1371/journal.pbio.2003962
Heyer, R., Kohrs, F., Reichl, U., and Benndorf, D. (2015). Metaproteomics of complex microbial communities in biogas plants. Microb. Biotechnol. 8, 749–763. doi: 10.1111/1751-7915.12276
Hilton, S., Bennett, A. J., Keane, G., Bending, G. D., Chandler, D., Stobart, R., et al. (2013). Impact of shortened crop rotation of oilseed rape on soil and rhizosphere microbial diversity in relation to yield decline. PLoS One 8:e59859. doi: 10.1371/journal.pone.0059859
Hirsch, P. R., Mauchline, T. H., and Clark, I. M. (2010). Culture-independent molecular techniques for soil microbial ecology. Soil Biol. Biochem. 42, 878–887. doi: 10.1016/j.soilbio.2010.02.019
Hiruma, K., Gerlach, N., Sacristán, S., Nakano, R. T., Hacquard, S., Kracher, B., et al. (2016). Root endophyte colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent. Cell 165, 464–474. doi: 10.1016/j.cell.2016.02.028
Hobbs, P. R., Sayre, K., and Gupta, R. (2008). The role of conservation agriculture in sustainable agriculture. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 543–555. doi: 10.1098/rstb.2007.2169
Horton, M. W., Bodenhausen, N., Beilsmith, K., Meng, D., Muegge, B. D., Subramanian, S., et al. (2014). Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5:5320. doi: 10.1038/ncomms6320
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Jansson, J. K., and Hofmockel, K. S. (2018). The soil microbiome — from metagenomics to metaphenomics. Curr. Opin. Microbiol. 43, 162–168. doi: 10.1016/j.mib.2018.01.013
Jez, J. M., Lee, S. G., and Sherp, A. M. (2016). The next green movement: plant biology for the environment and sustainability. Science 353, 1241–1244. doi: 10.1126/science.aag1698
Johnson, N. C., Graham, J. H., and Smith, F. A. (1997). Functioning of mycorrhizal associations along the mutualism-parasitism continuum. New Phytol. 135, 575–585. doi: 10.1046/j.1469-8137.1997.00729.x
Johnson-Rollings, A. S., Wright, H., Masciandaro, G., Macci, C., Doni, S., Calvo-Bado, L. A., et al. (2014). Exploring the functional soil-microbe interface and exoenzymes through soil metaexoproteomics. ISME J. 8, 2148–2150. doi: 10.1038/ismej.2014.130
Johnston-Monje, D., and Raizada, M. N. (2011). Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS One 6:e20396. doi: 10.1371/journal.pone.0020396
Jones, D. L., Nguyen, C., and Finlay, R. D. (2009). Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil 321, 5–33. doi: 10.1007/s11104-009-9925-0
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kibota, T. T., and Lynch, M. (1996). Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature .381, 694–696. doi: 10.1038/381694a0
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422. doi: 10.1038/s41579-018-0029-9
Kuroki, Y., Toyoda, A., Noguchi, H., Taylor, T. D., Itoh, T., Kim, D. S., et al. (2006). Comparative analysis of chimpanzee and human y chromosomes unveils complex evolutionary pathway. Nat. Genet. 38, 158–167. doi: 10.1038/ng1729
Lebeis, S. L., Paredes, S. H., Lundberg, D. S., Breakfield, N., Gehring, J., McDonald, M., et al. (2015). Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 349, 860–864. doi: 10.1126/science.aaa8764
Levy, A., Salas Gonzalez, I., Mittelviefhaus, M., Clingenpeel, S., Herrera Paredes, S., Miao, J., et al. (2018). Genomic features of bacterial adaptation to plants. Nat. Genet. 50, 138–150. doi: 10.1038/s41588-017-0012-9
Lewis, R., Barth, V., Coffey, T., McFarland, C., Huggins, D., and Sullivan, T. (2018). Altered bacterial communities in long-term no-till soils associated with stratification of soluble aluminum and soil pH. Soils 2:7. doi: 10.3390/soils2010007
Li, Y., Sun, Q., Zhan, J., Yang, Y., and Wang, D. (2017). Soil-covered strategy for ecological restoration alters the bacterial community structure and predictive energy metabolic functions in mine tailings profiles. Appl. Microbiol. Biotechnol. 101, 2549–2561. doi: 10.1007/s00253-016-7969-7
Lidbury, I., Murrell, J. C., and Chen, Y. (2014). Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc. Natl. Acad. Sci. U.S.A. 111, 2710–2715. doi: 10.1073/pnas.1317834111
Lidbury, I. D., Murphy, A. R. J., Scanlan, D. J., Bending, G. D., Jones, A. M. E., Moore, J. D., et al. (2016). Comparative genomic, proteomic and exoproteomic analyses of three Pseudomonas strains reveals novel insights into the phosphorus scavenging capabilities of soil bacteria. Environ. Microbiol. 18, 3535–3549. doi: 10.1111/1462-2920.13390
Lladó, S., López-Mondéjar, R., and Baldrian, P. (2017). Forest soil bacteria: diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 81:e00063-16. doi: 10.1128/MMBR.00063-16
Lueders, T., Dumont, M. G., Bradford, L., and Manefield, M. (2016). RNA-stable isotope probing: from carbon flow within key microbiota to targeted transcriptomes. Curr. Opin. Biotechnol. 41, 83–89. doi: 10.1016/j.copbio.2016.05.001
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Mäder, P., Fließbach, A., Dubois, D., Gunst, L., Fried, P., and Niggli, U. (2002). Soil fertility and biodiversity in organic farming. Science 296, 1694–1697. doi: 10.1126/science.1071148
Martino, E., Morin, E., Grelet, G. A., Kuo, A., Kohler, A., Daghino, S., et al. (2018). Comparative genomics and transcriptomics depict ericoid mycorrhizal fungi as versatile saprotrophs and plant mutualists. New Phytol. 217, 1213–1229. doi: 10.1111/nph.14974
Massalha, H., Korenblum, E., Malitsky, S., Shapiro, O. H., and Aharoni, A. (2017). Live imaging of root–bacteria interactions in a microfluidics setup. Proc. Natl. Acad. Sci. U.S.A. 114, 4549–4554. doi: 10.1073/pnas.1618584114
Mauchline, T. H., Chedom-Fotso, D., Chandra, G., Samuels, T., Greenaway, N., Backhaus, A., et al. (2015). An analysis of Pseudomonas genomic diversity in take-all infected wheat fields reveals the lasting impact of wheat cultivars on the soil microbiota. Environ. Microbiol. 4764–4778. doi: 10.1111/1462-2920.13038
Mauchline, T. H., Fowler, J. E., East, A. K., Sartor, A. L., Zaheer, R., Hosie, A. H. F., et al. (2006). Mapping the Sinorhizobium meliloti 1021 solute-binding protein-dependent transportome. Proc. Natl. Acad. Sci. U.S.A. 103, 17933–17938. doi: 10.1073/pnas.0606673103
McDonald, B. R., and Currie, C. R. (2017). Lateral gene transfer dynamics in the ancient bacterial genus Streptomyces. mBio 8:e00644-17. doi: 10.1128/mBio.00644-17
Micallef, S. A., Shiaris, M. P., and Colón-Carmona, A. (2009). Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J. Exp. Bot. 60, 1729–1742. doi: 10.1093/jxb/erp053
Mitter, B., Pfaffenbichler, N., Flavell, R., Compant, S., Antonielli, L., Petric, A., et al. (2017). A new approach to modify plant microbiomes and traits by introducing beneficial bacteria at flowering into progeny seeds. Front. Microbiol. 8:11. doi: 10.3389/fmicb.2017.00011
Mueller, U. G., and Sachs, J. L. (2015). Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617. doi: 10.1016/j.tim.2015.07.009
Muth, T., Benndorf, D., Reichl, U., Rapp, E., and Martens, L. (2013). Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol. Biosyst. 9, 578–585. doi: 10.1039/c2mb25415h
Muth, T., Renard, B. Y., and Martens, L. (2016). Metaproteomic data analysis at a glance: advances in computational microbial community proteomics. Expert Rev. Proteomics 13, 757–769. doi: 10.1080/14789450.2016.1209418
Niu, B., Paulson, J. N., Zheng, X., and Kolter, R. (2017). Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. U.S.A. 114, E2450–E2459. doi: 10.1073/pnas.1616148114
O’Callaghan, F. E., Braga, R. A., Neilson, R., MacFarlane, S. A., and Dupuy, L. X. (2018). New live screening of plant-nematode interactions in the rhizosphere. Sci. Rep. 8:1440. doi: 10.1038/s41598-017-18797-7
Oberson, A., Fardeau, J. C., Besson, J. M., and Sticher, H. (1993). Soil phosphorus dynamics in cropping systems managed according to conventional and biological agricultural methods. Biol. Fertil. Soils 16, 111–117. doi: 10.1007/BF00369411
Ofaim, S., Ofek-Lalzar, M., Sela, N., Jinag, J., Kashi, Y., Minz, D., et al. (2017). Analysis of microbial functions in the rhizosphere using a metabolic-network based framework for metagenomics interpretation. Front. Microbiol. 8:1606. doi: 10.3389/fmicb.2017.01606
Oldroyd, G. E. D. (2013). Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 11, 252–263. doi: 10.1038/nrmicro2990
Orozco-Mosqueda, M. D C., Rocha-Granados, M. D C., Glick, B. R., and Santoyo, G. (2018). Microbiome engineering to improve biocontrol and plant growth-promoting mechanisms. Microbiol. Res. 208, 25–31. doi: 10.1016/j.micres.2018.01.005
Panke-Buisse, K., Poole, A. C., Goodrich, J. K., Ley, R. E., and Kao-Kniffin, J. (2015). Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 9, 980–989. doi: 10.1038/ismej.2014.196
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110.
Pimentel, D., Harvey, C., Resosudarmo, P., Sinclair, K., Kurz, D., McNair, M., et al. (1995). Environmental and economic costs of soil erosion and conservation benefits. Science 267, 1117–1123. doi: 10.1126/science.267.5201.1117
Pineda, A., Soler, R., Pozo, M. J., Rasmann, S., and Turlings, T. C. J. (2015). Editorial: above-belowground interactions involving plants, microbes and insects. Front. Plant Sci. 6:318. doi: 10.3389/fpls.2015.00318
Pini, F., East, A. K., Appia-Ayme, C., Tomek, J., Karunakaran, R., Mendoza-Suárez, M., et al. (2017). Bacterial biosensors for in vivo spatiotemporal mapping of root secretion. Plant Physiol. 174, 1289–1306. doi: 10.1104/pp.16.01302
Pinto, A. J., and Raskin, L. (2012). PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7:e43093. doi: 10.1371/journal.pone.0043093
Pinton, R., Varanini, Z., Nannipieri, P., Darrah, P., Simonet, P., Neumann, G., et al. (2007). The Rhizosphere. Boca Raton, FL: CRC Press. doi: 10.1201/9781420005585
Poudel, R., Jumpponen, A., Schlatter, D. C., Paulitz, T. C., Gardener, B. B. M., Kinkel, L. L., et al. (2016). Microbiome networks: a systems framework for identifying candidate microbial assemblages for disease management. Phytopathology 106, 1083–1096. doi: 10.1094/PHYTO-02-16-0058-FI
Pratama, A. A., and van Elsas, J. D. (2018). The “Neglected” soil virome - potential role and impact. Trends Microbiol. 26, 649–662. doi: 10.1016/j.tim.2017.12.004
Prosser, J. I. (2015). Dispersing misconceptions and identifying opportunities for the use of “omics” in soil microbial ecology. Nat. Rev. Microbiol. 13, 439–446. doi: 10.1038/nrmicro3468
Puente, M. E., Li, C. Y., and Bashan, Y. (2009). Endophytic bacteria in cacti seeds can improve the development of cactus seedlings. Environ. Exp. Bot. 66, 402–408. doi: 10.1016/j.envexpbot.2009.04.007
Qiu, X., Kulasekara, B. R., and Lory, S. (2009). Role of horizontal gene transfer in the evolution of Pseudomonas aeruginosa virulence. Genome Dyn. 6, 126–139. doi: 10.1159/000235767
Raaijmakers, J. M., and Mazzola, M. (2016). Soil immune responses. Science 352, 1392–1393. doi: 10.1126/science.aaf3252
Reganold, J. P., and Wachter, J. M. (2016). Organic agriculture in the twenty-first century. Nat. Plants 2:15221. doi: 10.1038/nplants.2015.221
Robbins, R. J., Krishtalka, L., and Wooley, J. C. (2016). Advances in biodiversity: metagenomics and the unveiling of biological dark matter. Stand. Genomic Sci. 11:69. doi: 10.1186/s40793-016-0180-8
Rockström, J., Steffen, W., Noone, K., Persson,Å., Chapin, F. S., Lambin, E., et al. (2009). Planetary boundaries: exploring the safe operating space for humanity. Ecol. Soc. 14:32. doi: 10.5751/ES-03180-140232
Rodrigues, R. R., Rodgers, N. C., Wu, X., and Williams, M. A. (2018). COREMIC: a web-tool to search for a niche associated CORE MICrobiome. PeerJ 6:e4395. doi: 10.7717/peerj.4395
Rybakova, D., Mancinelli, R., Wikström, M., Birch-Jensen, A. S., Postma, J., Ehlers, R. U., et al. (2017). The structure of the Brassica napus seed microbiome is cultivar-dependent and affects the interactions of symbionts and pathogens. Microbiome 5:104. doi: 10.1186/s40168-017-0310-6
Sánchez-Cañizares, C., Jorrín, B., Poole, P. S., and Tkacz, A. (2017). Understanding the holobiont: the interdependence of plants and their microbiome. Curr. Opin. Microbiol. 38, 188–196. doi: 10.1016/j.mib.2017.07.001
Santhanam, R., Luu, V. T., Weinhold, A., Goldberg, J., Oh, Y., and Baldwin, I. T. (2015). Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. Proc. Natl. Acad. Sci. U.S.A. 112, E5013–E5020. doi: 10.1073/pnas.1505765112
Sanz M. J., de Vente J., Chotte J. L., Bernoux M., Kust G., Ruiz I., (eds). et al. (2017). “Sustainable land management contribution to successful land-based climate change adaptation and mitigation,” in A Report of the Science-Policy Interface. (Bonn: UNCCD).
Schlaeppi, K., and Bulgarelli, D. (2015). The plant microbiome at work. Mol. Plant Microbe Interact. 28, 212–217. doi: 10.1094/MPMI-10-14-0334-FI
Scholz, M. B., Lo, C. C., and Chain, P. S. G. (2012). Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr. Opin. Biotechnol. 23, 9–15. doi: 10.1016/j.copbio.2011.11.013
Senga, R. A., Alegria Terrazas, S., Balbirnie, K., Blank, M., Janiak, A., Szarejko, I., et al. (2017). Root hair mutations displace the barley rhizosphere microbiota. Front. Plant Sci. 8:1094. doi: 10.3389/fpls.2017.01094
Shade, A., Jones, S. E., Gregory Caporaso, J., Handelsman, J., Knight, R., Fierer, N., et al. (2014). Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5:e01371-14. doi: 10.1128/mBio.01371-14
Shi, S., Nuccio, E. E., Shi, Z. J., He, Z., Zhou, J., and Firestone, M. K. (2016). The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol. Lett. 19, 926–936. doi: 10.1111/ele.12630
Shiomi, Y., Nishiyama, M., Onizuka, T., and Marumoto, T. (1999). Comparison of bacterial community structures in the rhizoplane of tomato plants grown in soils suppressive and conducive towards bacterial wilt. Appl. Environ. Microbiol. 65, 3996–4001.
Siegrist, S., Schaub, D., Pfiffner, L., and Mäder, P. (1998). Does organic agriculture reduce soil erodibility? The results of a long-term field study on loess in Switzerland. Agric. Ecosyst. Environ. 69, 253–264. doi: 10.1016/S0167-8809(98)00113-3
Smith, F. A., and Smith, S. E. (1996). Mutualism and parasitism: diversity in function and structure in the “arbuscular” (VA) mycorrhizal symbiosis. Adv. Bot. Res. 22, 1–43. doi: 10.1016/S0065-2296(08)60055-5
Sowell, S. M., Wilhelm, L. J., Norbeck, A. D., Lipton, M. S., Nicora, C. D., Barofsky, D. F., et al. (2009). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 3, 93–105. doi: 10.1038/ismej.2008.83
Svenningsen, N. B., Watts-Williams, S. J., Joner, E. J., Battini, F., Efthymiou, A., Cruz-Paredes, C., et al. (2018). Suppression of the activity of arbuscular mycorrhizal fungi by the soil microbiota. ISME J. 12, 1296–1307. doi: 10.1038/s41396-018-0059-3
Syed Ab Rahman, S. F., Singh, E., Pieterse, C. M. J., and Schenk, P. M. (2018). Emerging microbial biocontrol strategies for plant pathogens. Plant Sci. 267, 102–111. doi: 10.1016/j.plantsci.2017.11.012
Temperton, B., and Giovannoni, S. J. (2012). Metagenomics: microbial diversity through a scratched lens. Curr. Opin. Microbiol. 15, 605–612. doi: 10.1016/j.mib.2012.07.001
Timmins-Schiffman, E., May, D. H., Mikan, M., Riffle, M., Frazar, C., Harvey, H. R., et al. (2017). Critical decisions in metaproteomics: achieving high confidence protein annotations in a sea of unknowns. ISME J. 11, 309–314. doi: 10.1038/ismej.2016.132
Tkacz, A., Cheema, J., Chandra, G., Grant, A., and Poole, P. S. (2015). Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J. 9, 2349–2359. doi: 10.1038/ismej.2015.41
Tkacz, A., and Poole, P. (2015). Role of root microbiota in plant productivity. J. Exp. Bot. 66, 2167–2175. doi: 10.1093/jxb/erv157
Truyens, S., Weyens, N., Cuypers, A., and Vangronsveld, J. (2015). Bacterial seed endophytes: genera, vertical transmission and interaction with plants. Environ. Microbiol. Rep. 7, 40–50. doi: 10.1111/1758-2229.12181
Turner, T. R., James, E. K., Poole, P. S., Gilbert, J., Meyer, F., Jansson, J., et al. (2013a). The plant microbiome. Genome Biol. 14:209. doi: 10.1186/gb-2013-14-6-209
Turner, T. R., Ramakrishnan, K., Walshaw, J., Heavens, D., Alston, M., Swarbreck, D., et al. (2013b). Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 7, 2248–2258. doi: 10.1038/ismej.2013.119
Van Der Heijden, M. G. A., Bardgett, R. D., and Van Straalen, N. M. (2008). The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11, 296–310. doi: 10.1111/j.1461-0248.2007.01139.x
Vorholt, J. A., Vogel, C., Carlström, C. I., and Müller, D. B. (2017). Establishing causality: opportunities of synthetic communities for plant microbiome research. Cell Host Microbe 22, 142–155. doi: 10.1016/j.chom.2017.07.004
Wang, H., Wei, Z., Mei, L., Gu, J., Yin, S., Faust, K., et al. (2017). Combined use of network inference tools identifies ecologically meaningful bacterial associations in a paddy soil. Soil Biol. Biochem. 105, 227–235. doi: 10.1016/j.soilbio.2016.11.029
Wang, Q., Fish, J. A., Gilman, M., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2015). Xander: employing a novel method for efficient gene-targeted metagenomic assembly. Microbiome 3:32. doi: 10.1186/s40168-015-0093-6
Wang, Y., Li, C., Tu, C., Hoyt, G. D., DeForest, J. L., and Hu, S. (2017). Long-term no-tillage and organic input management enhanced the diversity and stability of soil microbial community. Sci. Total Environ. 609, 341–347. doi: 10.1016/j.scitotenv.2017.07.053
Wei, Z., and Jousset, A. (2017). Plant Breeding Goes Microbial. Trends Plant Sci. 22, 555–558. doi: 10.1016/j.tplants.2017.05.009
Weller, D. M., Raaijmakers, J. M., Mcspadden Gardener, B. B., and Thomashow, L. S. (2002). Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 40, 309–348. doi: 10.1146/annurev.phyto.40.030402.110010
Wetmore, K. M., Price, M. N., Waters, R. J., Lamson, J. S., He, J., Hoover, C. A., et al. (2015). Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6:e00306-15. doi: 10.1128/mBio.00306-15
Williams, T. J., Long, E., Evans, F., Demaere, M. Z., Lauro, F. M., Raftery, M. J., et al. (2012). A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 6, 1883–1900. doi: 10.1038/ismej.2012.28
Wiseman, B. M., Neate, S. M., Keller, K. O., and Smith, S. E. (1996). Suppression of Rhizoctonia solani anastomosis group 8 in Australia and its biological nature. Soil Biol. Biochem. 28, 727–732. doi: 10.1016/0038-0717(95)00178-6
Xue, C., Penton, C. R., Shen, Z., Zhang, R., Huang, Q., Li, R., et al. (2015). Manipulating the banana rhizosphere microbiome for biological control of Panama disease. Sci. Rep. 5:11124. doi: 10.1038/srep11124
Yergeau, E., Sanschagrin, S., Maynard, C., St-Arnaud, M., and Greer, C. W. (2014). Microbial expression profiles in the rhizosphere of willows depend on soil contamination. ISME J. 8, 344–358. doi: 10.1038/ismej.2013.163
Yuan, Z., Jiang, S., Sheng, H., Liu, X., Hua, H., Liu, X., et al. (2018). Human perturbation of the global phosphorus cycle: changes and consequences. Environ. Sci. Technol. 52, 2438–2450. doi: 10.1021/acs.est.7b03910
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. mBio 6:e02527-14. doi: 10.1128/mBio.02527-14
Keywords: microbial community, root interactions, cropping systems, gnotobiotic, omics, microbial function
Citation: Sergaki C, Lagunas B, Lidbury I, Gifford ML and Schäfer P (2018) Challenges and Approaches in Microbiome Research: From Fundamental to Applied. Front. Plant Sci. 9:1205. doi: 10.3389/fpls.2018.01205
Received: 20 April 2018; Accepted: 26 July 2018;
Published: 17 August 2018.
Edited by:
Jesús Mercado-Blanco, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Giovanni Bubici, Istituto per la Protezione Sostenibile delle Piante (IPSP), ItalyCopyright © 2018 Sergaki, Lagunas, Lidbury, Gifford and Schäfer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chrysi Sergaki, U2VyZ2FraS5jaHJ5c2lAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.