
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 21 August 2018
Sec. Plant Breeding
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01146
This article is part of the Research Topic The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits View all 18 articles
 Jieqin Li1†Weijie Tang2,3†Ya-Wen Zhang4†Kai-Ning Chen2Chenchen Wang1Yanlong Liu1Qiuwen Zhan1Chunming Wang3Shi-Bo Wang2
Jieqin Li1†Weijie Tang2,3†Ya-Wen Zhang4†Kai-Ning Chen2Chenchen Wang1Yanlong Liu1Qiuwen Zhan1Chunming Wang3Shi-Bo Wang2 Shang-Qian Xie2*Lihua Wang1*
Shang-Qian Xie2*Lihua Wang1*Understanding the genetic function of the forage quality-related traits, including crude protein (CP), neutral detergent fiber (NDF), acid detergent fiber (ADF), hemicellulose (HC), and cellulose (CL) contents, is essential for the identification of forage quality genes and selection of effective molecular markers in sorghum. In this study, we genotyped 245 sorghum accessions by 85,585 single-nucleotide polymorphisms (SNPs) and obtained the phenotypic data from four environments. The SNPs and phenotypic data were applied to multi-locus genome-wide association studies (GWAS) with the mrMLM software. A total of 42 SNPs were identified to be associated with the five forage quality-related traits. Moreover, three and two quantitative trait nucleotides (QTNs) were simultaneously detected among them by three and two multi-locus methods, respectively. One QTN on chromosome 5 was found to be associated simultaneously with CP, NDF, and ADF. Furthermore, 3, 2, 2, 5, and 2 candidate genes were identified to be responsible for CP, NDF, ADF, HC, and CL contents, respectively. These results provided insightful information of the forage quality-related traits and would facilitate the genetic improvement of sorghum forage quality in the future.
Sorghum (Sorghum bicolor L.) is a popular crop worldwide, which is used a food source, animal fodder, and raw material for alcoholic beverages and biofuels in industries (Paterson et al., 2009). Most of the important agronomic traits are genetically controlled by quantitative trait loci (QTLs) (Zou et al., 2012; Boyles et al., 2017). For example, the forage quality is an important quantitative trait. Thus, understanding their genetic mechanism is essential for identifying the candidate genes and selecting effective molecular markers in sorghum breeding.
The forage digestibility and crude protein (CP) content are the main focus for forage sorghum breeding (Murray et al., 2008). Forage digestibility is mainly determined by the cellulose (CL), hemicellulose (HC), and lignin contents (Wang H. et al., 2016), which are important components of the neutral detergent fiber (NDF). On the other hand, acid detergent fiber (ADF) is a portion of sorghum fiber and is obtained from acid detergent-treated forage. The two types of fibers, NDF and ADF, are the two vital components of forage digestibility. Recently, the forage quality traits have been studied in sorghum and some related QTLs have been identified (Murray et al., 2008; Shiringani and Friedt, 2011; Li et al., 2015). However, these identified QTLs were observed to be less sensitive due to the limitation of linkage analysis based on bi-parental mapping populations.
Compared with the linkage analysis of bi-parental mapping populations, genome-wide association studies (GWAS), which is based on linkage disequilibrium (LD) and provided sufficient genetic background information, have become a powerful alternative for the investigation of quantitative traits. There are three main strategies for GWAS. Firstly, a generalized linear model (GLM) was proposed for the genetic analysis of the quantitative traits (Price et al., 2006), but it did not effectively control the polygenic background. Secondly, a mixed linear model (MLM) was elaborated to take into account the population structure and polygenic background using the pedigree relationship or marker information (Zhang et al., 2005; Yu et al., 2006). These methods involve a large calculation burden due to the tremendous number of existing markers. Therefore, a series of rapid detection methods were finally developed, such as EMMA (Kang et al., 2008), FaST-LMM (Lippert et al., 2011), GRAMMAR-Gamma (Svishcheva et al., 2012), ECMLM (Li et al., 2014), SUPER (Wang et al., 2014), BOLT-LMM (Loh et al., 2015), and FarmCPU (Liu et al., 2016). Although the above methods have been widely adopted, the complex traits controlled by multiple QTNs could not be effectively identified. To address this issue, Zhang's group has developed a series of multi-locus GWAS methods, including mrMLM (Wang S. B. et al., 2016), FASTmrMLM (Tamba et al., 2017), FASTmrEMMA (Wen et al., 2017), ISIS EM-BLASSO (Tamba et al., 2017), pLARmEB (Zhang et al., 2017), and pKWmEB (Ren et al., 2018).
In our study, we utilized the advantageous multi-locus GWAS to investigate the sorghum forage quality-related traits. We genotyped 245 sorghum accessions by using 85,585 single-nucleotide polymorphisms (SNPs) and phenotyped them in the four environments. The data were analyzed by the multi-locus GWAS software, mrMLM.
The 245 sorghum accessions (Table S1) included 238 mini-core collection sorghum and 7 breeding varieties. These accessions were planted in the Fengyang campus of Anhui Science and Technology University (Fengyang, China, 32°52′ N, 177°33′ E) and Tengqiao town of Hainan Province (Tengqiao, China, 18°24′ N, 109°45′ E) in 2015 and 2016. All the experiments in the four environments used a completely randomized block design with three replicates. The aboveground parts were harvested when 70% accessions were at the heading stage. The harvested plants were dried at 75°C for three days. The plant material was then milled using a grinder and filtered using a 0.5 mm sieve.
Seven hundred and thirty-five sorghum samples (3 replicates) were measured for CP, CL, HC, NDF, and ADF using the traditional chemical methods, and simultaneously scanned for near-infrared (NIR) spectra with an Antaris™ II FT-NIR Analyzer (Thermo, USA). A model was established using TQ Analyst software based on the NIR spectra and the results of the chemical analysis. The samples were then scanned for NIR spectra, and their CP, CL, HC, NDF, and ADF were calculated using the model. The mean of the phenotypic data and the correlation coefficients were calculated using Microsoft Excel.
Total DNA was extracted using the DNAsecure Plant Kit (Qiagen, Cat.No. DP320). All the samples were standardized to 50 ng/μL, and 10 μL of each sample was digested with the enzymes, PstI (CTGCAG) and MspI (CCGG), at 37°C for 2 h and then at 65°C for 20 min. The digested samples were ligated with the adapters from Illumina (San Diego, CA, USA). The ligated samples were then pooled using the same volume (10 μL) for PCR-amplification in a single tube. The fragment length was analyzed using a Bioanalyzer (Agilent), and the PCR products were quantified by a Qubit3.0 fluorometer (Invitrogen). The GBS library was run on an Illumina Hiseq2500 (San Diego, CA, USA).
The sequencing reads of the 245 samples were extracted from the raw data of RAD-seq and filtered by using fastx_barcode_splitter and fastq_quality_filter with parameters (-q 20 -p 80 -Q 33) of fastx_toolkit-0.0.13.2 (http://hannonlab.cshl.edu/fastx_toolkit/). The high-quality sequencing data were aligned using BWA MEM (Li and Durbin, 2009). The software—samtools, mpileup, and bcftools (Li et al., 2009), were then used to call the SNPs from the alignment files of the 245 samples; these were kept as the genotype of the sorghum population. These genotypic data were used to calculate the population structure using the fastSTRUCTURE software (Raj et al., 2014).
The GWAS for the five forage quality-related traits (CP, CL, HC, NDF, and ADF) was performed using six methods, including mrMLM, FASTmrMLM, FASTmrEMMA, pLARmEB, pKWmEB, and ISIS EM-BLASSO in the mrMLM software. The main model used in this study in the mrMLM software is as follows :y = Wα+Xβ+Zu+ε, where y is an n × 1 phenotypic vector of quantitative traits, and n is the number of accessions. W = (ω1, ω2, ⋯ , ωc) is an n × c matrix of covariates (fixed effects), including a column vector of 1; the population structure or principal components can be incorporated intoW. Moving on, α is a c × 1 vector of fixed effects, including the intercept, and X is an n × 1 vector of marker genotypes. is the random effect of putative QTN. Z is an n × m design matrix, and is an m × 1 vector of polygenic effects. K is a known n × n relatedness matrix. is an n × 1 vector of residual errors, and is residual variance. In is an n × n identity matrix, and MVN denotes multivariate normal distribution. An LOD score of 3 was used as the critical threshold for significant QTNs for all the six methods.
Genes that were hit directly by the associated QTNs within a 50-kb stretch were selected to choose the candidate genes as described in Upadhyaya et al. (2016). The physical locations of the QTNs were recorded according to the assembly genome (Sorghum_bicolor_NCBIv3) and the annotation GFF file (https://www.ncbi.nlm.nih.gov/genome/108). The detailed functions of the corresponding genes were annotated by performing BLASTP search at the NCBI website, and the candidate genes were assigned to different biological processes based on the function of their homologs in other species in literature or with the help of data in the Conserved Domains Database. The selected candidate genes were associated with the main QTNs of the five traits if they made a contribution (r2) greater than 5%.
Extensive phenotypic variations of CP, CL, HC, NDF, and ADF were observed in the 245 sorghum samples in the four environments, including two locations in 2 years (Fengyang and Tengqiao in 2015 and 2016, Table 1). The variation range of the five traits was 1.5 to 3.5-fold: the phenotype values of the CP content were 3.80 to 13.24% with 2.5 to 3.5-fold variation. The NDF content varied from 0.38 to 0.75 g/g with 1.5 to 1.9-fold variation, while the ADF content varied from 0.18 to 0.52 g/g with a 1.8 to 2.2-fold variation. Lastly, the HC and CL contents varied from 0.14 to 0.42 g/g and 0.12 to 0.45 g/g with 1.6 to 2.2-fold and 1.8 to 2.8-fold variations, respectively.
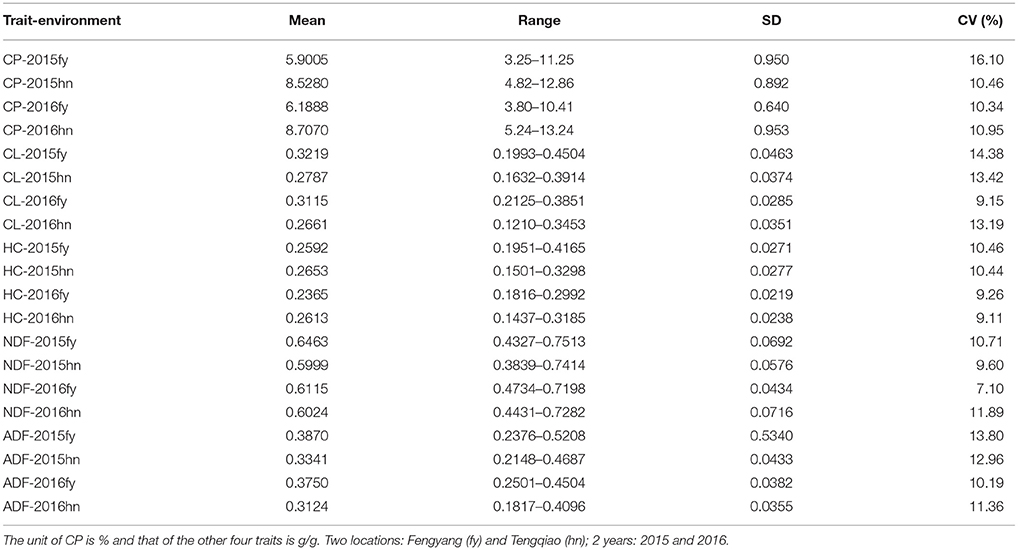
Table 1. The statistical description for CP, CL, HC, NDF, and ADF in 245 sorghum accessions in the four environments.
The correlation coefficients between a pair of traits were assessed. It was revealed that there were significant and positive correlations between ADF, NDF, CL, and HC. However, they correlated significantly but negatively with the CP phenotype, except for HC in 2015fy, 2015hn, and 2016fy and NDF in the 2016fy environments (Table S2). These results indicated that the four traits of ADF, NDF, HC, and CL could be genetically linked or that some genes could play pleiotropic roles in controlling these phenotypes.
A total of 85,585 SNPs were identified in the genotypes of the 245 accessions using RAD-seq (Table 2). Chromosome 1 had the most SNP markers (11,719), while chromosome 10 had the least (5,994). The highest SNP density was observed on chromosome 3 with 1.5 SNP markers per 10 kb, whereas the lowest density was on chromosome 7 with 0.9 SNP markers per 10 kb. The average density was 1.2 markers per 10 kb. Altogether, the genotyping results were of high quality in this research. The population structure was analyzed using the fastSTRUCTURE software. The results showed that the best value for the number of sub-populations was 5 (Figure 1), which was selected to perform further GWAS analysis.
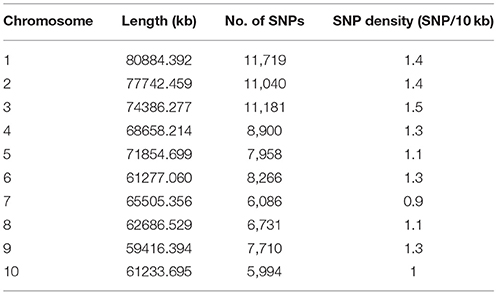
Table 2. Number of SNPs on the 10 chromosomes of sorghum.
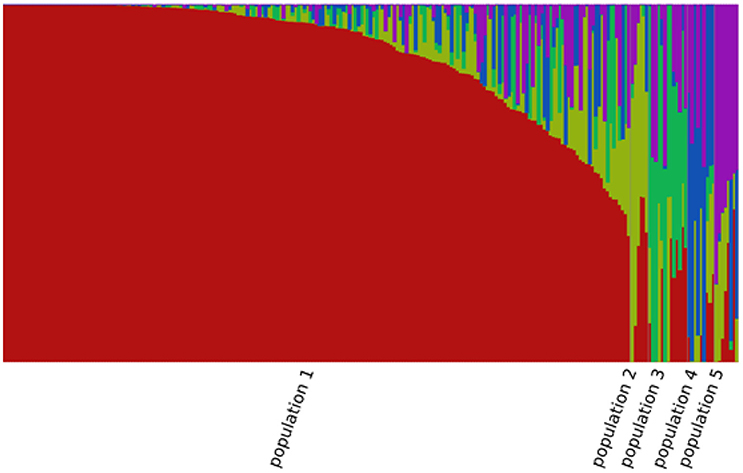
Figure 1. Population structure of the 245 sorghum accessions.
Six methods in the mrMLM software were used for the detection of QTNs. A total of 42 significant QTNs were detected for the five forage quality-related traits (CP, CL, HC, NDF, and ADF) across the four environments using six methods (Table 3). There were 5, 3, 3, 24, and 7 QTNs that were associated with CP, CL, HC, NDF, and ADF, respectively. Each trait was controlled by multiple QTNs. The 5 SNPs associated with the CP content were identified on chromosomes 2, 5, 7, and 9. The 3 SNPs associated with the CL content were present on chromosomes 2, 5, and 8, while the 3 SNPs associated with the HC content were located on chromosomes 1 and 9. The 24 SNPs associated with NDF were present on chromosomes 1, 2, 6, 7, 8, 9, and 10. Lastly, the 7 SNPs associated with the ADF content were present on chromosomes 3, 4, 5, 8, and 10. Among these QTNs, there were 4 significant QTNs, each of which was responsive for more than one trait. The three traits of ADF, CL, and NDF were associated with one QTN on chromosome 5 (RSS50197); both CL and NDF were associated with two QTNs (RSS21890 and RSS76122); ADF and CL were associated with one QTN (RSS68908) on chromosome 8.
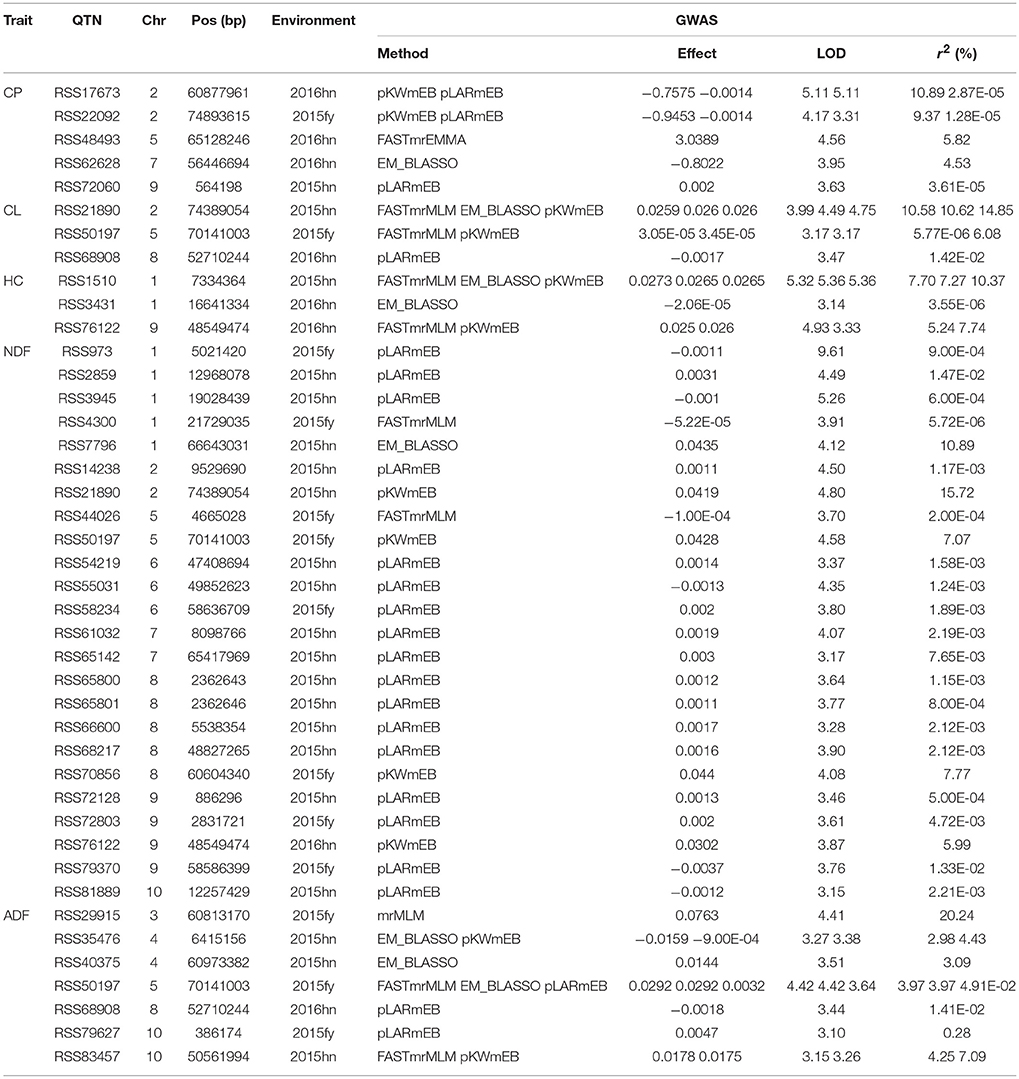
Table 3. QTNs for CP, CL, HC, NDF, and ADF in the four environments using six multi-locus GWAS methods.
Among the above six methods, pLARmEB was the most powerful and accountable for the identification of the 24 QTNs that mainly contributed to the NDF content trait (17 QTNs); however, their contributions were less than what were detected by other methods, except for one major QTN (RSS17673), whose contribution was greater than 5% (Table 3). The other methods of PKWmEB, ISIS EM-BLASSO, FASTmrMLM, mrMLM, and FASTmrEMA identified 12, 8, 8, 1, and 1 QTNs, respectively. About 43% (13 of 30) of these SNPs included the major QTNs (r2 > 5%). Besides, 3 QTNs (RSS50197, RSS21890, and RSS1510) were detected simultaneously by 3 methods, and another 5 QTNs (RSS35476, RSS83457, RSS76122, RSS22092, and RSS17673) were identified simultaneously by 2 methods. The remaining QTNs were detected by a single method, but most of them were considered as reliable because of the high thresholds at which they were detected.
The assembled sorghum genome and the annotation file from NCBI were used to annotate the genes associated with the significant QTNs. There were 14 candidate genes for five forage quality-related traits. The NDF and CP content traits were associated with five and three candidate genes, respectively. The remaining 6 genes were related to the CL, HC, and ADF content traits with each trait being associated with two genes (Table 4).
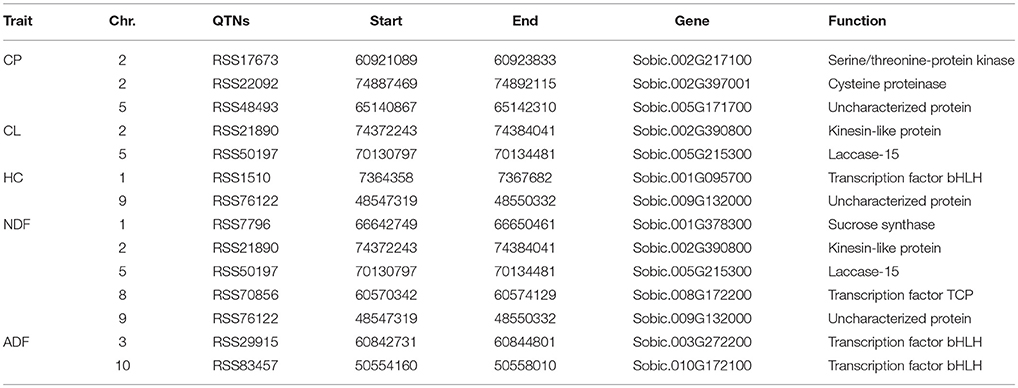
Table 4. Candidate genes for CP, CL, HC, NDF, and ADF traits.
For the CP content trait, one candidate gene that was associated with the major QTN (RSS17673) encoded a serine/threonine-protein kinase (Sobic.002G217100), which was consistent with a previous study that concluded that serine/threonine-protein kinases are involved in signal cascade for nitrogen metabolism in plants (Champigny, 1995). Besides, two candidate genes were identified for the CP content on chromosomes 2 and 5 with one gene encoding a cysteine proteinase and the other encoding an uncharacterized protein. In addition, one main QTN associated with the CL content trait on chromosome 2 was identified, and the associated candidate gene encoded a kinesin-like protein. The kinesin protein is reported to be involved in the deposition of CL during secondary growth of fiber cells in Arabidopsis (Kong et al., 2015). Furthermore, 5 main QTNs were detected in association with the NDF content; two of these (RSS21890 and RSS50197) were co-localized with those for the CL content trait. Therefore, the same two candidate genes were identified for the NDF and CL content (Sobic.005G215300 and Sobic.002G390800). For the ADF content trait, 2 main QTNs were detected on chromosomes 3 and 10, where both candidate genes encoded a bHLH transcription factor (Sobic.003G272200 and Sobic.010G172100).
Genome-wide association study is an important alternative for mapping quantitative traits. It has been applied rapidly and extensively in plant research. These methods have been widely adopted, but only a few QTNs for each complex trait have been identified. In this study, we implemented the latest multi-locus GWAS methods available in mrMLM (Wang S. B. et al., 2016; Tamba et al., 2017; Wen et al., 2017; Zhang et al., 2017; Ren et al., 2018), which can effectively overcome the above issue and actively detect the QTNs associated with the quantitative traits. Six methods in the mrMLM software were used to identify the QTNs of five forage quality-related traits in sorghum. Of these methods, pLARmEB detected the most significant QTNs, but most of them contributed insignificantly to heritability (Table 3). Most of the significant QTNs associated with the NDF content, detected using pLARmEB, were observed to be in the 2015hn (13 QTNs) and 2015fy (4 QTNs) environments (Table 3). This result might be associated with the range of values for this phenotypic trait (Table 1) and the difference of environments between Hainan Tengqiao (18°24′ N, 109°45′ E) and Anhui Fengyang (32°52′ N, 177°33′ E). The range of NDF-2015hn and NDF-2015fy was 0.36 and 0.32, which was higher than that in 2016hn (0.28) and 2016fy (0.25), respectively (Table 1). Similar conclusions can be drawn for other traits. It means that the greater the difference in phenotype, the more favorable it is for the detection of the associated QTNs. Hainan and Anhui are located in the tropics and subtropics, respectively, where the environment is particularly different in different climatic zones. The previous study has revealed that the climatic conditions, including temperature, water availability, and soil, are important factors which affect the forage quality of sorghum (Hussin et al., 2007). In our study, the QTN RSS50197 associated with the ADF, CL, and NDF traits was uniquely detected in the same environment of 2015fy by using three GWAS methods. The above results revealed the influence of environment in QTN detection. However, the latest methods of multi-locus GWAS applied in our study are currently unable to detect the QTN-by-environment interaction. Thus, we hope that in the future new methods can be developed by the theoretical researchers.
According to the GWAS analysis, 5, 3, 3, 7, and 24 QTNs were identified for CP, CL, HC, ADF, and NDF content, respectively. Of the 5 candidate loci for the CP content, 2 were already identified in the previous studies. The locus on chromosome 9 was mapped in the same region by Murray et al. (2008) and Li et al. (2015) in sorghum as well. Of the 3 candidate loci for the CL content, 2 were identified in the same region on chromosomes 2 and 8 by Murray et al. (2008) and Shiringani and Friedt (2011). Similarly, of the 7 loci for the ADF content, 2 were mapped on chromosome 4, which was in agreement with the report of Shiringani and Friedt (2011). As for the 24 loci for the NDF content, the 2 loci on chromosome 6 and 1 loci on chromosome 8 were also identified by Shiringani and Friedt (2011). More importantly, several QTNs that were detected by the six methods in this study were novel identifications for forage quality-related traits in sorghum.
The QTLs for the NDF or ADF content co-localized with those for the CL or HC content, which has been reported previously in sorghum. Cardinal et al. (2003) reported colocalization of QTLs that are associated with the cell wall components, such as lignin, NDF, and ADF in stalks of maize. Murray et al. (2008) and Shiringani and Friedt (2011) also found colocalization of QTLs associated with the CL, HC, NDF, and ADF content traits in sorghum by QTL mapping. In this study, we detected 4 co-localized QTNs: 1 for three traits and 3 for two traits. All of these QTNs were associated with NDF or ADF and with CL or HC. NDF is mainly composed of CL, HC, and lignin, while ADF is composed of CL and lignin. The difference between NDF and ADF is whether they have HC as a component or not. Furthermore, we found that NDF and ADF significantly correlated with CL or HC. It is reasonable that these QTNs were co-localized.
Both NDF and ADF include CL and lignin. There are a series of reports about the biosynthesis and signaling pathways of CL and lignin in plants (Kim et al., 2013; McNamara et al., 2015; Yoon et al., 2015; Chezem and Clay, 2016). In this study, we identified 5 and 2 candidate genes for the NDF and ADF content traits, respectively. Of these candidate genes, 1 gene (Sobic.001G378300) encoded a sucrose synthase, which is an integral component of the CL synthesis mechanism. Gerber et al. (2014) reported that deficient sucrose synthase activity in developing wood does not specifically affect the CL biosynthesis but causes an overall decrease in the cell wall polymers. Furthermore, Poovaiah et al. (2014) reported that the lignin content increases in all the transgenic switchgrass lines, where sucrose synthase (PvSUS1) was overexpressed.
Lignin, CL, and HC are the main components of secondary cell walls (Zhong et al., 2011). Secondary cell wall biosynthesis is positively regulated by NAD and MYB transcription factors (Zhong and Ye, 2014; Chezem and Clay, 2016). Moreover, studies have also identified several transcription factors (e.g., WRKY, ERF, and bHLH) that regulate the biosynthesis of secondary walls (Kim et al., 2013; Taylor-Teeples et al., 2015; Chezem and Clay, 2016). In this study, we identified a candidate gene encoding a bHLH transcription factor for CL and two bHLH genes for ADF. These transcription factors might also be involved in the regulation of CL or lignin biosynthesis. The function of the candidate genes identified in this work needs to be studied further by transformation experiments in the future.
JL, LW, and S-QX designed and conceived the experiments. Y-WZ, K-NC, and S-BW performed the computational analysis. WT and JL extracted the DNA and performed the experimental analysis. CCW and YL assisted with experiments in data collection and analysis. QZ and CW participated in the design and supervised the study. S-QX and JL discussed the results and interpretation of the final data. S-QX and JL drafted the manuscript. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (31671753, 31600667, 31760316), the key project of natural science research of Anhui provincial education department (KJ2016A177), the Key-construction Subject Plan of Anhui Province (WanJiaoMiKe[2014]28), the youth talent support program of Anhui Science and Technology University (XiaoRenFa[2015]69), Priming Scientific Research Foundation of Hainan University [KYQD(ZR)1721], and Science Foundation for The Youth Teachers of Hainan University in 2017 (hdkyxj201702).
The reviewer ML declared a shared affiliation, though no other collaboration, with several of the authors WT, CW to the handling Editor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Hari D Upadhyaya (International Crops Research Institute for the Semi-Arid Tropics) for providing sorghum mini-core accessions and the two reviewers for their constructive comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01146/full#supplementary-material
Boyles, R. E., Pfeiffer, B. K., Cooper, E. A., Rauh, B. L., Zielinski, K. J., Myers, M. T., et al. (2017). Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor. Appl. Genet. 130, 697–716. doi: 10.1007/s00122-016-2844-6
Cardinal, A. J., Lee, M., and Moore, K. J. (2003). Genetic mapping and analysis of quantitative trait loci affecting fiber and lignin content in maize. Theor. Appl. Genet. 106, 866–874. doi: 10.1007/s00122-002-1136-5
Champigny, M. L. (1995). Integration of photosynthetic carbon and nitrogen metabolism in higher plants. Photosynth. Res. 46, 117–127. doi: 10.1007/BF00020422
Chezem, W. R., and Clay, N. K. (2016). Regulation of plant secondary metabolism and associated specialized cell development by MYBs and bHLHs. Phytochemistry 131, 26–43. doi: 10.1016/j.phytochem.2016.08.006
Gerber, L., Zhang, B., Roach, M., Rende, U., Gorzsas, A., Kumar, M., et al. (2014). Deficient sucrose synthase activity in developing wood does not specifically affect cellulose biosynthesis, but causes an overall decrease in cell wall polymers. New Phytol. 203, 1220–1230. doi: 10.1111/nph.12888
Hussin, A., Khan, S., Sulatani, M. I., and Mohammad, D. (2007). Locational variation in green fodder yield, dry matter yield, and forage quality of sorghum. Pakistan J. Agric. Res. 20, 1–2.
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckerman, D., Daly, M. J., et al. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. doi: 10.1534/genetics.107.080101
Kim, W. C., Ko, J. H., Kim, J. Y., Kim, J., Bae, H. J., and Han, K. H. (2013). MYB46 directly regulates the gene expression of secondary wall-associated cellulose synthases in Arabidopsis. Plant J. 73, 26–36. doi: 10.1111/j.1365-313x.2012.05124.x
Kong, Z., Ioki, M., Braybrook, S., Li, S., Ye, Z., Julie Lee, Y., et al. (2015). Kinesin-4 functions in vesicular transport on cortical microtubules and regulates cell wall mechanics during cell elongation in plants. Mol. Plant 8, 1011–1023. doi: 10.1016/j.molp.2015.01.004
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J. Q., Wang, L. H., Zhan, Q. W., Liu, Y. L., Zhang, Q., Li, J. F., et al. (2015). Mapping quantitative trait loci for five forage quality traits in a sorghum-sudangrass hybrid. Genet. Mol. Res. 14, 13266–13273. doi: 10.4238/2015.October.26.23
Li, M., Liu, X., Bradbury, P., Yu, J., Zhang, Y. M., Todhunter, R. J., et al. (2014). Enrichment of statistical power for genome-wide association studies. BMC Biol. 12:73. doi: 10.1186/s12915-014-0073-5
Lippert, C., Listgarten, J., Liu, Y., Kadie, C. M., Davidson, R. I., and Heckerman, D. (2011). FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835. doi: 10.1038/nmeth.1681
Liu, X., Huang, M., Fan, B., Buckler, E. S., and Zhang, Z. (2016). Iterative usage of fixed and random effect models for powerful and efficient Genome-wide association studies. PLoS Genet. 12:e1005767. doi: 10.1371/journal.pgen.1005767
Loh, P. R., Tucker, G., Bulik-Sullivan, B. K., Vilhjalmsson, B. J., Finucane, H. K., Salem, R. M., et al. (2015). Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290. doi: 10.1038/ng.3190
McNamara, J. T., Morgan, J. L., and Zimmer, J. (2015). A molecular description of cellulose biosynthesis. Annu. Rev. Biochem. 84, 895–921. doi: 10.1146/annurev-biochem-060614-033930
Murray, S. C., Rooney, W. L., Mitchell, S. E., Sharma, A., Klein, P. E., Mullet, J. E., et al. (2008). Genetic improvement of sorghum as a biofuel feedstock: II. QTL for stem and leaf structural carbohydrates. Crop Sci. 48, 2180–2193. doi: 10.2135/cropsci2008.01.0068
Paterson, A. H., Bowers, J. E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., et al. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556. doi: 10.1038/nature07723
Poovaiah, C. R., Nageswara-Rao, M., Soneji, J. R., Baxter, H. L., Stewart, C. N. (2014), Altered lignin biosynthesis using biotechnology to improve lignocellulosic biofuel feedstocks. Plant Biotechnol. J. 12, 1163–1173. doi: 10.1111/pbi.12225, et al.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Raj, A., Stephens, M., and Pritchard, J. K. (2014). fastSTRUCTURE: variational Inference of population structure in large SNP data sets. Genetics 197, 573–589. doi: 10.1534/genetics.114.164350
Ren, W. L., Wen, Y. J., Dunwell, J. M., and Zhang, Y. M. (2018). pKWmEB: integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 120, 208–218. doi: 10.1038/s41437-017-0007-4
Shiringani, A. L., and Friedt, W. (2011). QTL for fibre-related traits in grain x sweet sorghum as a tool for the enhancement of sorghum as a biomass crop. Theor. Appl. Genet. 123, 999–1011. doi: 10.1007/s00122-011-1642-4
Svishcheva, G. R., Axenovich, T. I., Belonogova, N. M., van Duijn, C. M., and Aulchenko, Y. S. (2012). Rapid variance components-based method for whole-genome association analysis. Nat. Genet. 44, 1166–1170. doi: 10.1038/ng.2410
Tamba, C. L., Ni, Y. L., and Zhang, Y. M. (2017). Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 13:e1005357. doi: 10.1371/journal.pcbi.1005357
Taylor-Teeples, M., Lin, L., de Lucas, M., Turco, G., Toal, T. W., Gaudinier, A., et al. (2015). An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517, 571–175. doi: 10.1038/nature14099
Upadhyaya, H. D., Wang, Y. H., Sastry, D. V., Dwivedi, S. L., Prasad, P. V., Burrell, A. M., et al. (2016). Association mapping of germinability and seedling vigor in sorghum under controlled low-temperature conditions. Genome 59, 137–145. doi: 10.1139/gen-2015-0122
Wang, H., Li, K., Hu, X., Liu, Z., Wu, Y., and Huang, C. (2016). Genome-wide association analysis of forage quality in maize mature stalk. BMC Plant Biol. 16:227. doi: 10.1186/s12870-016-0919-9
Wang, Q., Tian, F., Pan, Y., Buckler, E. S., and Zhang, Z. (2014). A SUPER powerful method for genome wide association study. PLoS ONE 9:e107684. doi: 10.1371/journal.pone.0107684
Wang, S. B., Feng, J. Y., Ren, W. L., Huang, B., Zhou, L., Wen, Y. J., et al. (2016). Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 6:19444. doi: 10.1038/srep19444
Wen, Y. J., Zhang, H., Ni, Y. L., Huang, B., Zhang, J., Feng, J. Y., et al. (2017). Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinformatics 19, 700–712. doi: 10.1093/bib/bbx028
Yoon, J., Choi, H., and An, G. (2015). Roles of lignin biosynthesis and regulatory genes in plant development. J. Integr. Plant Biol. 57, 902–912. doi: 10.1111/jipb.12422
Yu, J., Pressoir, G., Briggs, W. H., Vroh, B. I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, J., Feng, J. Y., Ni, Y. L., Wen, Y. J., Niu, Y., Tamba, C. L., et al. (2017). pLARmEB: integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 118, 517–524. doi: 10.1038/hdy.2017.8
Zhang, Y. M., Mao, Y., Xie, C., Smith, H., Luo, L., and Xu, S. (2005). Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 169, 2267–2275. doi: 10.1534/genetics.104.033217
Zhong, R., Lee, C., McCarthy, R. L., Reeves, C. K., Jones, E. G., and Ye, Z. H. (2011). Transcriptional activation of secondary wall biosynthesis by rice and maize NAC and MYB transcription factors. Plant Cell Physiol. 52, 1856–1871. doi: 10.1093/pcp/pcr123
Zhong, R., and Ye, Z. H. (2014). Complexity of the transcriptional network controlling secondary wall biosynthesis. Plant Sci. 229, 193–207. doi: 10.1016/j.plantsci.2014.09.009
Zou, G., Zhai, G., Feng, Q., Yan, S., Wang, A., Zhao, Q., et al. (2012). Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J. Exp. Bot. 63, 5451–5462. doi: 10.1093/jxb/ers205
Keywords: sorghum, GWAS, forage quality-related traits, mrMLM, QTNs
Citation: Li J, Tang W, Zhang Y-W, Chen K-N, Wang C, Liu Y, Zhan Q, Wang C, Wang S-B, Xie S-Q and Wang L (2018) Genome-Wide Association Studies for Five Forage Quality-Related Traits in Sorghum (Sorghum bicolor L.). Front. Plant Sci. 9:1146. doi: 10.3389/fpls.2018.01146
Received: 15 May 2018; Accepted: 18 July 2018;
Published: 21 August 2018.
Edited by:
Zhenyu Jia, University of California, United StatesReviewed by:
Suhong Bu, Fujian Agriculture and Forestry University, ChinaCopyright © 2018 Li, Tang, Zhang, Chen, Wang, Liu, Zhan, Wang, Wang, Xie and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shang-Qian Xie, c3FpYW54aWVAaGFpbnUuZWR1LmNu
Lihua Wang, d2FuZ2xpaHVhZXJyQDEyNi5jb20=
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.