 Katy Vandereyken
Katy Vandereyken Jelle Van Leene
Jelle Van Leene Barbara De Coninck
Barbara De Coninck Bruno P. A. Cammue
Bruno P. A. Cammue- 1Centre of Microbial and Plant Genetics, KU Leuven, Heverlee, Belgium
- 2VIB Center for Plant Systems Biology, Ghent, Belgium
- 3Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium
- 4Division of Crop Biotechnics, KU Leuven, Heverlee, Belgium
Plant stress responses involve numerous changes at the molecular and cellular level and are regulated by highly complex signaling pathways. Studying protein-protein interactions (PPIs) and the resulting networks is therefore becoming increasingly important in understanding these responses. Crucial in PPI networks are the so-called hubs or hub proteins, commonly defined as the most highly connected central proteins in scale-free PPI networks. However, despite their importance, a growing amount of confusion and controversy seems to exist regarding hub protein identification, characterization and classification. In order to highlight these inconsistencies and stimulate further clarification, this review critically analyses the current knowledge on hub proteins in the plant interactome field. We focus on current hub protein definitions, including the properties generally seen as hub-defining, and the challenges and approaches associated with hub protein identification. Furthermore, we give an overview of the most important large-scale plant PPI studies of the last decade that identified hub proteins, pointing out the lack of overlap between different studies. As such, it appears that although major advances are being made in the plant interactome field, defining hub proteins is still heavily dependent on the quality, origin and interpretation of the acquired PPI data. Nevertheless, many hub proteins seem to have a reported role in the plant stress response, including transcription factors, protein kinases and phosphatases, ubiquitin proteasome system related proteins, (co-)chaperones and redox signaling proteins. A significant number of identified plant stress hubs are however still functionally uncharacterized, making them interesting targets for future research. This review clearly shows the ongoing improvements in the plant interactome field but also calls attention to the need for a more comprehensive and precise identification of hub proteins, allowing a more efficient systems biology driven unraveling of complex processes, including those involved in stress responses.
Introduction
Plants are continuously exposed to different kinds of environmental stresses. These can be abiotic, such as drought, salinity, non-optimal light or temperature conditions, or biotic in origin, such as pathogen or herbivore attacks. The plant stress response is therefore highly complex and although the molecular mechanisms that occur upon stress detection have been extensively studied, much remains to be unraveled (Bari and Jones, 2009; Dodds and Rathjen, 2010; Atkinson and Urwin, 2012; Duque et al., 2013; Rejeb et al., 2014). In the current era of systems biology driven research it is important to analyze systems as a whole, rather than their individual constituents. Consequently, studying the plant protein interactome—i.e., the set of physical protein interactions in a cell—can provide valuable new insights into biological processes (Ideker et al., 2001; Garbutt et al., 2014; Mine et al., 2014; Sheth and Thaker, 2014).
In the past decade, advances in high-throughput proteomics and interactomics technologies resulted in an accumulation of increasingly accurate plant protein-protein interaction (PPI) data (Morsy et al., 2008; Braun et al., 2013; Xing et al., 2016). These data are often represented graphically in the form of PPI networks, not to be confused with genetic interactions or gene regulatory networks used to elucidate how genes function as a network in biological processes (Figure 1; Zhang et al., 2010; Krouk et al., 2013). Studies show that most biological networks including PPI networks, are scale-free, meaning that only a limited number of proteins interact with numerous others (Figure 2; Albert, 2005; Vallabhajosyula et al., 2009). Because of their high connectivity, these proteins are called hub proteins (or hubs) and they are of critical importance to PPI networks and whole biological systems (Higurashi et al., 2008). Indeed, when a hub is eliminated or interfered with, the structure of the biological networks will change drastically, having a vast impact on the organismal fitness (He and Zhang, 2006). Specifically in plant stress response-related networks, hubs are assumed to play an essential role in the signal transduction cascade following stress detection and loss-of-function mutants often show abnormal stress responses (Dietz et al., 2010). Still, despite their great importance and major advances in the interactome field, there is a lack of clarity regarding hub proteins, made evident by inconsistent definitions, characteristics, and classification systems. This review critically assesses the current knowledge in the plant interactome field, focusing specifically on plant stress response related hubs in large-scale plant PPI networks.
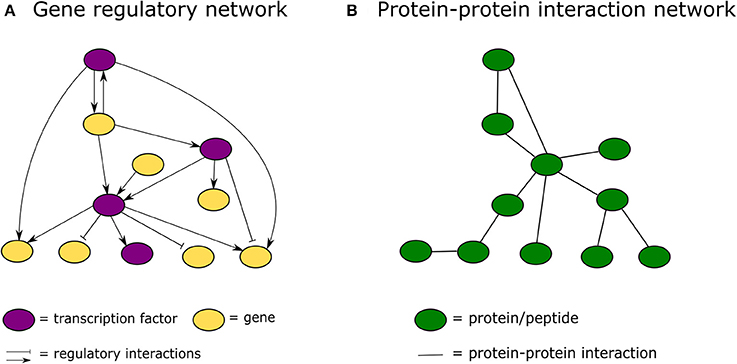
Figure 1. Graphical representation of a gene regulatory network vs. a protein-protein interaction network. (A) In a gene regulatory network nodes represent genes or proteins and lines between them regulatory interactions. (B) In a protein-protein interaction network nodes always represent proteins and the connecting lines physical protein-protein interactions.
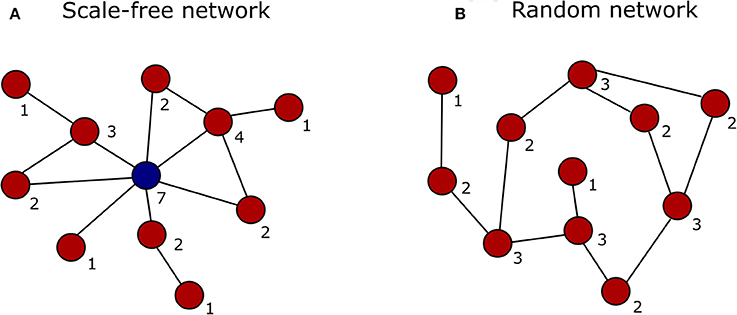
Figure 2. Graphical representation of a scale-free vs. a random network. The degree (number of interactions) of each node is indicated by the digit below the node. (A) In a scale-free network the majority of nodes interact with just a few other nodes (red), while only some interact with many other nodes and represent the network hubs (blue). (B) In a random network the nodes (red) are connected with a uniform probability, resulting in most nodes having the same number of connections.
Characterizing Hub Proteins: Definitions and Properties
Current Hub Definitions
While the original definition of the term “hub” refers to the solid central part of a wheel into which spokes are inserted, nowadays a hub refers to any kind of system's center around which various other system components revolve or from which they radiate, and as such are interconnected. This definition was translated to molecular biology, where hub proteins were defined as highly connected central nodes in a systematic scale-free PPI network, having numerous interaction partners and connecting many network modules (Han et al., 2004; Ekman et al., 2006; Haynes et al., 2006; Tsai et al., 2009; Patil et al., 2010; Bertolazzi et al., 2013; Ota et al., 2016). Biological networks such as PPI networks often adopt a scale-free architecture, meaning that the vast majority of network nodes have a low degree of interactions while relatively few have high connectivity, the latter termed hubs (Figure 2; Jeong et al., 2001; Rangarajan et al., 2015). However, the exact number of interactions needed for a protein to be labeled as a hub, also called the degree threshold, differs between PPI studies. Some sources cite a fixed degree cutoff of 5 (Jeong et al., 2001; Han et al., 2004; Patil and Nakamura, 2006), 8 (Ekman et al., 2006), 10 (Haynes et al., 2006), 20 (Aragues et al., 2007), or 50 interactors (Mukhtar et al., 2011). Others take a floating cutoff as a top percentage of high degree nodes, with hubs most often defined as the top 10% of proteins with the highest number of interactors (Batada et al., 2006; Dosztányi et al., 2006; Jin et al., 2007). Hence there seems to be no consensus on the degree threshold. At present, the term “hub” is increasingly used more loosely, and even often incorrectly, as it has become a fashionable label for any central signaling network protein of some importance, independent from the fact that the concerned protein may or may not participate in numerous protein interactions. This unavoidably leads to an increase of per definition incorrectly labeled hubs and a growing confusion.
For the transition between high-degree nodes and intermediate or low degree nodes in general scale-free PPI networks a threshold of more than 5 interactions has previously been stated (Vallabhajosyula et al., 2009). However, this seems very low for hubs defined as the most highly connected proteins in comprehensive PPI networks. Indeed when looking at large-scale plant PPI networks, many proteins have more than 5 interactors, indicating that this is not a justified threshold. Some studies also suggest to differentiate between small (6–10), intermediate (11–50), major (51–100), and super hubs (>100 interactions). However, as PPI networks become more extensive, more interactions will be found for a single protein, making any fixed cutoff seemingly less suitable. In the view that hub function is a property of the given interactome network and not something that can be defined at the level of individual proteins, a floating cutoff can seem more relevant and flexible (Dosztányi et al., 2006). Still, this last method can also be subjective as to what exact top percentage should be chosen and the minimum degree for hubs will also depend on the size and connectivity of the network of interest. The quality and origin of the PPI data presents additional challenges for hub protein identification based on interaction degree (Andorf et al., 2013; Bertolazzi et al., 2013). A general cutoff for plant hub proteins is therefore not straightforward.
Network Properties
Besides their high connectivity, hub proteins are also often described as being characterized by other network properties (Table 1), with centrality being the most essential and referring to their central position in relationship to other proteins in the network (McCormack et al., 2015). There are many different measures for centrality in biological networks (De Arruda et al., 2014; Vella et al., 2017), but the most commonly used measures are (i) “degree centrality”, simply defined by the number of interactions a protein has in a network, (ii) “betweenness centrality”, based on the shortest paths between network proteins and referring to a protein's frequency in mediating connections to other proteins, and (iii) “eigenvector centrality”, referring to the influence of the protein in a network not strictly through the number of connections but through the importance of these connections (Liseron-Monfils and Ware, 2015; McCormack et al., 2015).

Table 1. Network and structural properties often seen as hub-defining.
Hub proteins with a high degree of connectivity and centrality in a PPI network are also often said to be essential for PPI network topology and interactome functionality and stability (Jeong et al., 2001; Vallabhajosyula et al., 2009; Dietz et al., 2010). This also implies that hub proteins can be weak points in PPI networks, with the potential of disrupting an entire system when, for example, deleted or targeted by pathogen attacks (Mukhtar et al., 2011; Garbutt et al., 2014; Weβling et al., 2014; Ota et al., 2016). Still, the relationship between hub degree, centrality and essentiality has been heavily debated. At first, Jeong et al. (2001) concluded that highly connected proteins with a central role in networks are more likely to be encoded by essential genes, that when deleted cause lethal phenotypes, leading to the so-called lethality-centrality rule. However, more recently this conclusion was reported to be incorrect and a consequence of using heavily biased datasets available at the time of the original observation (Yu et al., 2008). When less systematically biased datasets are used, the correlation between centrality and lethality is replaced by one between centrality and pleiotropy. The number of physical interactions mediated by a protein thus seems to be better correlated with the number of cellular processes in which it participates than with its essentiality/lethality (Yu et al., 2008). Several studies indeed indicated that hubs often participate in essential complexes and processes and mediate essential interactions, therefore affecting many phenotypic traits when knocked-out, but not necessarily resulting in lethality (He and Zhang, 2006; Zotenko et al., 2008; Song and Singh, 2013).
Another property contributed to hub proteins in some studies is low interconnectivity as compared to non-hub proteins (Maslov and Sneppen, 2002; Vallabhajosyula et al., 2009). However paradoxically, an increasing amount of studies define PPI network hubs as highly (inter)connected proteins, indicating that hubs are often together in larger complexes (de Folter, 2005; Popescu et al., 2007; Hunt et al., 2008; Causier et al., 2012; Lumba et al., 2014; Piya et al., 2014; McCormack et al., 2015).
Lastly, co-expression between hub proteins and their interaction partners is often found to have a statistically significant bimodal distribution, termed co-expression bimodality (Vallabhajosyula et al., 2009). This implies the existence of two classes of hub proteins, one with low and another with high averaged interactor co-expression values, referred to as date and party hubs, respectively (Han et al., 2004). Party (or static) hubs are usually larger proteins that are part of functional protein complexes and interact with multiple partners simultaneously and continuously, assuming co-expression. Date (or dynamic) hubs on the other hand are usually smaller proteins that interact more transiently with one or two of their interaction partners at different time points and locations, therefore showing low co-expression. Party hubs generally act as central parts of functional protein complexes, while date hubs function as bridges between network modules (Han et al., 2004; Yu et al., 2008). However, the date/party hub classification is also quite controversial and argued not to be comprehensive enough (Batada et al., 2006, 2007; Agarwal et al., 2010; Chang et al., 2013).
Structural Properties
Concerning structural properties, it is somewhat unclear what is needed for a protein to evolve into a hub, making it able to recognize and bind many different partners. However, some structure-related properties are statistically more often ascribed to and thus suggested to be characteristic for hub proteins (Table 1). Evidently, the proportion of multi-domain proteins or long proteins with repeating domains, by which they can effectively bind different partners, is significantly larger amongst proteins labeled as hubs than in non-hubs. These protein binding domains are usually associated with ordered or globular regions of the proteins (Ekman et al., 2006). Conversely, several studies suggest that the presence of disordered regions is a common feature of eukaryotic hub proteins (Haynes et al., 2006; Vincent and Schnell, 2016). Intrinsically disordered proteins (IDPs) or protein regions (IDPRs) are (segments of) proteins that lack a unique 3D structure, they do not completely fold and remain flexible (Kim et al., 2008). IDPRs facilitate multiple protein recognition and binding by acting as adaptable extended interaction surfaces. Short linear motifs (SLiMs) are often central to IDPRs. These short stretches of protein sequence, typically 3–11 residues long, mediate interactions through inducing secondary structure formation upon binding with a structured partner or globular protein domain and are especially relevant in signaling networks with high connectivity proteins (O'Shea et al., 2017). Hence IDPRs allow for high specificity, obtained through adopting an ordered conformation upon partner binding, coupled with low affinity (Uversky, 2013, 2016; Sun et al., 2014). These regions usually evolve through the expansion of internal repeat regions and exhibit low amino acid complexity (Dosztányi et al., 2006). Some smaller hub proteins, however, are too short to have multiple binding domains and have very few or no disordered regions. Many of these small hubs instead have highly charged surfaces, including arginine, tyrosine, histidine, and methionine, facilitating binding with multiple partners (Patil and Nakamura, 2006, 2007; Patil et al., 2010).
These structural hub characteristics gave rise to a second commonly used classification for hub proteins, differentiating between single interface hubs (SIHs) and multiple interface hubs (MIHs), depending on the presence of a single or multiple interaction sites, respectively, to bind their partners. There is a strong but not complete overlap between the SIH/MIH and date/party classification of hub proteins, but even combined they are argued not the be comprehensive enough as general hub classification systems (Andorf et al., 2013).
Identifying Hubs by Unravelling the Plant Interactome: Overcoming Challenges Through the Use of Complementary Approaches
Challenges in the Interactome Field
During evolution, higher organisms have developed immensely complex mechanisms and signaling pathways that allow them to respond to a wide variety of changing environmental conditions, pathogen attacks, and developmental challenges (Verma et al., 2016). In order to respond optimally to specific challenges whilst wasting a minimum of energy, these systems are also tightly regulated (Tomé et al., 2014). Partly due to this inherent complexity, many genes and proteins still remain unknown, uncharacterized or the understanding of their function is incomplete. Using interactomics-based approaches in research could help fill in these gaps, not in the least in further unraveling plant stress responses. Plants harbor vast and dynamic protein networks, with a number of protein interaction pairs estimated to range from 75,000 to 299,000 in proteomes of 30,000–40,000 proteins (Arabidopsis Interactome Mapping Consortium, 2011; Sheth and Thaker, 2014). Moreover, all these proteins have diverse physicochemical properties, expression levels, subcellular localizations and reaction kinetics. As a result, no single PPI identification approach can at present build a complete map of the plant interactome (McCormack et al., 2015). Additionally, interactome studies in plants are somewhat lagging behind the leading studies done in yeast, animal and human models (Rolland et al., 2014; Van Leene et al., 2015; Mehta and Trinkle-Mulcahy, 2016). Large-scale plant PPI networks have mostly been reconstructed during the past decade, but the data are still far from depicting global PPI maps in a plant cell (Hao et al., 2016). Plant PPI maps will thus become increasingly accurate, reliable, and extensive in the upcoming years, thereby also allowing easier and more precise identification of hub proteins. To this end, several complementary approaches, including a range of computational prediction methods and various experimental wet lab techniques, are being used and developed.
Computational Approaches
In silico predictions usually yield large PPI networks that are often based on evolutionary conservation (e.g., sequence alignment), protein structural information (e.g., physicochemical properties, primary structures, 3D structures, docking ability, protein domain interactions), co-expression of the corresponding genes and/or integration of various available datasets (Cui et al., 2008; De Bodt et al., 2012; Braun et al., 2013; Zahiri et al., 2013; Vella et al., 2017). Many computationally predicted interactome maps are based on known sequence similarities between interacting proteins in other model species, called conserved interacting orthologs or interologs (Yu et al., 2004; Windram et al., 2014). Interolog mapping was used to build the first truly large-scale plant interactome map for Arabidopsis thaliana (Geisler-Lee et al., 2007) and later also for several other plant and crop species (Tables 2, 3). Genetic algorithms, mostly domain-based like the ENTS algorithm (Elucidating Network Topology with Sequence), are also used for PPI network reconstruction. This approach uses pairwise combination of conserved domains and predictions of subcellular protein localization as input features and have already predicted large PPI networks for several organisms, including A. thaliana and Populus trichocarpa (Rodgers-Melnick et al., 2013; Hao et al., 2016).
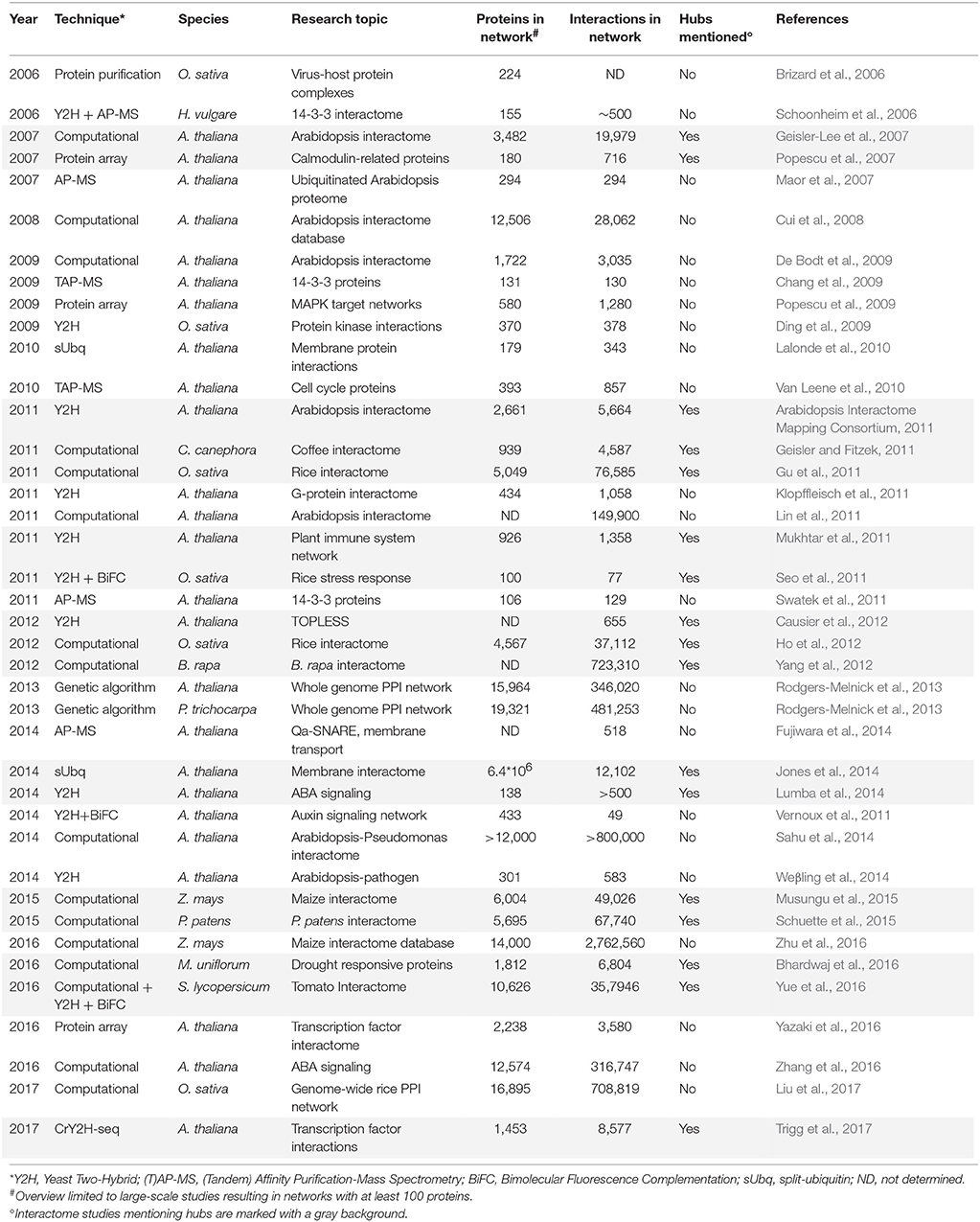
Table 2. Historical overview of large-scale plant interactome studies.
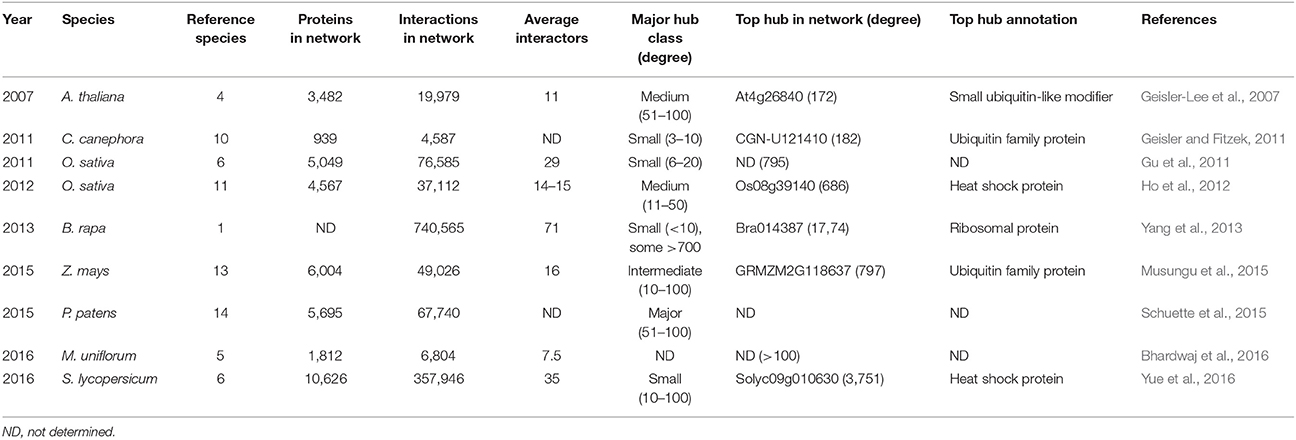
Table 3. Details of large-scale interolog-based computational plant interactome studies defining hub proteins.
Though these predicted PPIs are in many cases a necessary starting point in mapping large-scale PPI networks and provide very useful data, the approaches are not able to predict interactions between proteins without conserved domains or clear interacting domains. Also, computationally predicted networks that are based on literature data, like the interolog-based networks, are limited by what is known at a given time point and/or which data are included in the analyses and are therefore not really able to give a correct representation of the molecular connectivity. Hence, computational predictions are often seen as preliminary data that need to be verified experimentally and preferably by multiple complementary approaches to further increase their reliability. Predicted PPIs can thus give information on the interaction potential of proteins, but whether these interactions actually take place within a cell or tissue or at a given time can only be validated through experimental approaches, preferably in planta.
Experimental Approaches
The most common techniques for the production of experimental plant PPI data are high-throughput methods like in vivo yeast two-hybrid (Y2H) and split-ubiquitin (sUbq) systems, the latter specifically used for identifying membrane protein interactions (Brückner et al., 2009; Zhang et al., 2010). The first experimental so-called “proteome-scale” interactome map for A. thaliana (Arabidopsis Interactome I, AI-1) was composed using a systematic Y2H-based approach (Arabidopsis Interactome Mapping Consortium, 2011; Tables 2, 4). Improvements to these techniques are always ongoing, further increasing scalability and sensitivity while reducing costs (Ecker et al., 2016; Xing et al., 2016; Yachie et al., 2016). Recently, the CrY2H-seq method, a massively multiplexed Y2H method combining a Cre recombinase interaction reporter and next-generation DNA sequencing, allowing deep-coverage interactome mapping, has been presented. Using this technique to investigate A. thaliana transcription factor (TF) interactions, a deep-coverage A. thaliana TF interaction network was created (AtTFIN-1), greatly expanding the number of known plant TF interactions (Trigg et al., 2017). Other systematic approaches include classic in vitro protein arrays (Feilner et al., 2005; Popescu et al., 2007, 2009) or the nucleic acid programmable protein arrays (NAPPA), replacing immobilized purified proteins on the array surface by plasmid DNA and using cell-free expression systems to generate proteins (Miersch and LaBaer, 2011). The recent development of a new HaloTag-NAPPA method greatly increased protein array technology capacity, allowing proteome scale-screenings. These high-density protein arrays were also used to create an A. thaliana TF interactome network (Yazaki et al., 2016). Although all these experimental approaches usually give high quality results, they are based on yeast or in vitro systems and still require in planta confirmation of the interactions. Therefore, a range of targeted PPI techniques can be used for validation purposes, including fluorescence resonance energy transfer (FRET) assays (Bhat et al., 2006; Long et al., 2017), split-luciferase systems (SLS) (Fujikawa and Kato, 2007; Li et al., 2011), bimolecular fluorescence complementation (BiFC) (Miller et al., 2015), and co-immunoprecipitation (Co-IP) (Roux et al., 2011).
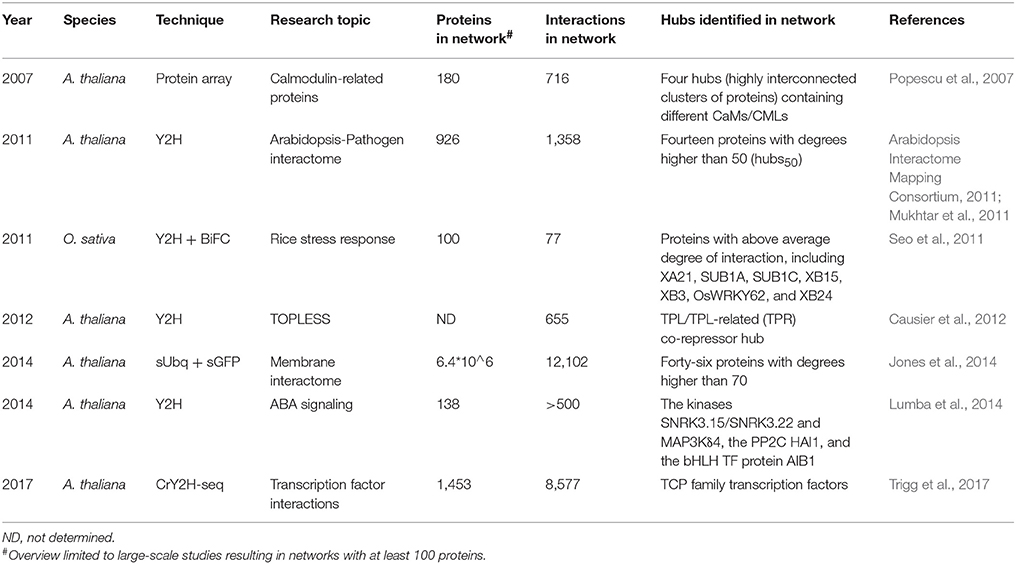
Table 4. Details of large-scale experimental plant interactome studies defining hub proteins.
Lastly, approaches specifically used for the identification of protein complexes in plants, include classic immunoprecipitation assays and tag-based affinity purification (AP) methods combined with mass spectrometry (MS) (Dedecker et al., 2015). Especially tandem affinity purification followed by mass spectrometry (TAP-MS) is a very powerful and often used method because of its high specificity. With this technique protein complexes have already been identified in A. thaliana and O. sativa, both in stably transformed cell suspension cultures and seedlings, and in Medicago truncatula hairy roots (Goossens et al., 2016). Combining TAP with quantitative MS also allowed mapping of dynamic PPI networks over the different growth zones of maize leaves (Nelissen et al., 2015). To filter out non-specific contaminants, a non-specific background interactions “black list” is often defined (Van Leene et al., 2015). However, nowadays quantitative AP-MS (q-AP-MS) methods are increasingly being used, comparing the quantity of proteins that co-purify with the bait to a negative control, in order to greatly improve confidence in identified interaction partners without losing possibly important interactors (Meyer and Selbach, 2015).
Validation, Tools, and Databases
The above-mentioned techniques are all characterized by their own inherent advantages and drawbacks, regarding throughput, background, false positive and negative rates, fusion tags, biochemistry, technical demand, costs, etc. As a result, it is important to realize that different techniques can lead to the detection of different subsets of interaction partners which are not always overlapping between the different approaches. This complicates the validation of interactions via multiple approaches (Van Leene et al., 2010, 2011; Gadeyne et al., 2014; Yazaki et al., 2016). For example, though Y2H and TAP-MS have been shown to be highly complementary, it is often challenging to validate interactions identified with one method, using the other (Yu et al., 2008; Brückner et al., 2009). Extensive knowledge on the properties and issues inherent to each method and the use of proper controls during experiments is therefore essential as the differences between methods, based on the used technology (i.e., tags, reporters, read out), the type of interactions identified (i.e., stable or transient) and the interaction environment (i.e., in vitro or in vivo, in yeast or in planta), can all cause diverse outcomes for the interaction analyses. For detailed information on the various plant PPI identification and validation techniques, we refer to some other excellent reviews (Morsy et al., 2008; Braun et al., 2013; McCormack et al., 2015).
Nevertheless, when a PPI can be identified through multiple high-quality approaches, its reliability increases significantly and this is an important requirement for developing networks that optimally reflect the biological reality. Therefore, increasing emphasis is also being put on creating better PPI networks through complementing them with data from various other sources, including co-expression, co-localization, co-evolution, and functional data (De Bodt et al., 2009; Zahiri et al., 2013; Vella et al., 2017). Several publically available databases, resources and tools exist to retrieve, analyze and visualize plant PPI and other related data (Table 5). Since PPI reliability is crucial in building PPI networks and maps, many of the databases also provide further details and trust values to their presented interactions and proteins.
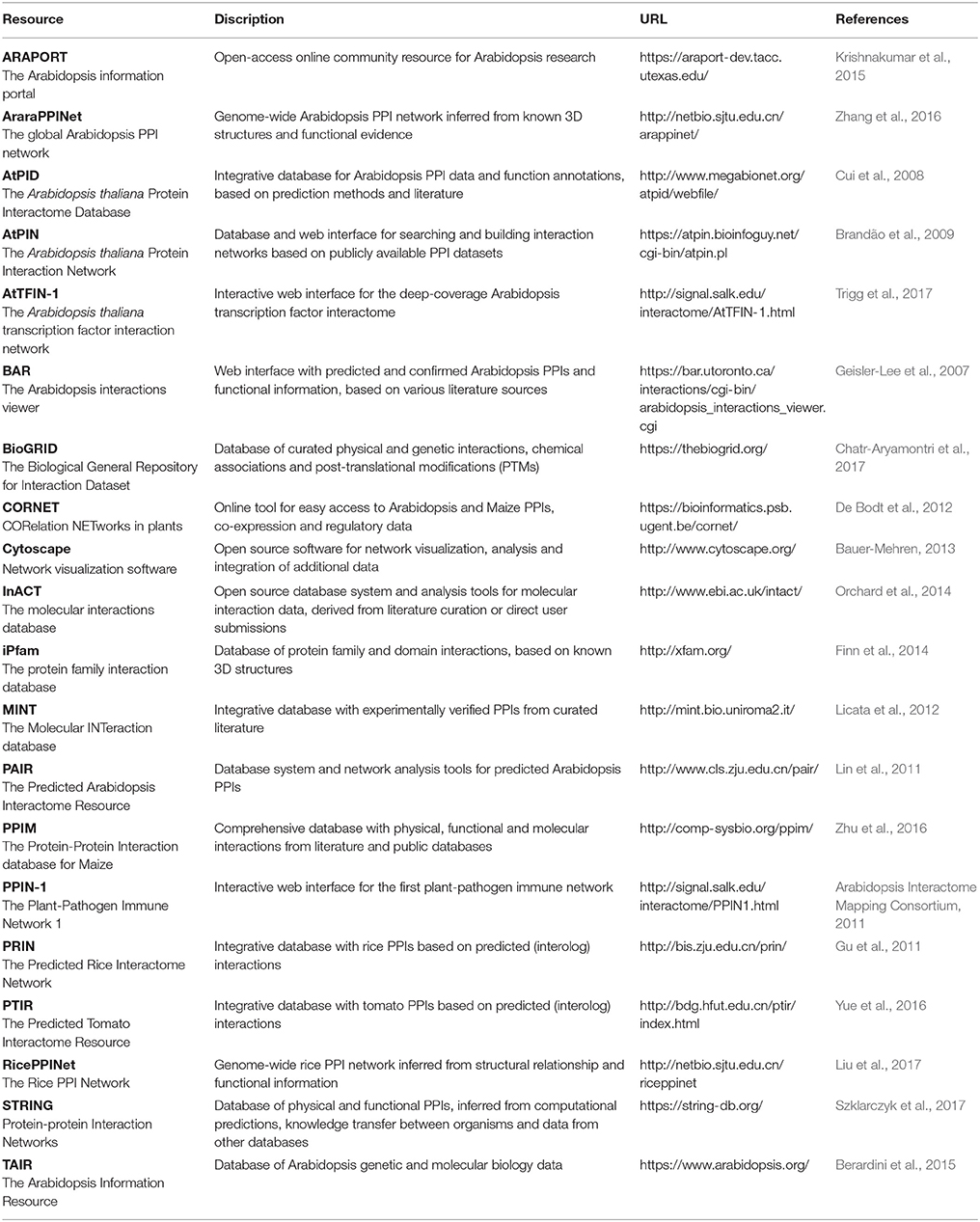
Table 5. Available resources, databases and tools to retrieve and analyze plant PPI data.
Hubs in Large-Scale Plant PPI Networks: The Lack of Overlap
In general, hub proteins are most often identified when studying large-scale protein networks. Table 2 gives a historical overview of large-scale plant PPI studies resulting in networks containing at least 100 proteins, with first reports starting in 2006. As illustrated, they consist of both broad-spectrum as well as more specific studies, mainly focusing on the A. thaliana interactome and primarily on signal transduction, stress responses, cell proliferation, protein ubiquitination, transcription factors, and membrane-protein interactions. The importance of hubs in these networks cannot be underestimated, however, less than half of the large-scale plant PPI studies focus on or even mention the identification of hub proteins (Table 2). Moreover, within those studies hubs are often defined differently and/or focus is restricted to hubs within a specific protein (sub)network of interest. Also apparent, both identified PPIs as well as the total amount of interactions (degree) found for specific proteins significantly differ between studies, especially when comparing computational and experimental networks. As a consequence, the overlap in plant proteins labeled as hubs in different studies is small and the differences in degree and characteristics between the identified hubs once more illustrate that there is no real unifying and binding definition for PPI network hubs (Tables 3, 4, Supplementary Tables 1, 2).
Computationally Predicted Plant Hubs
The first real large-scale plant PPI network was computationally predicted for A. thaliana through interolog mapping, predicting 19,979 interactions for 3,617 A. thaliana proteins (Geisler-Lee et al., 2007). Hub proteins of different degrees were identified, distinguishing between minor (3–5 interactions), small (6–10 interactions), medium (11–50 interactions), major (51–100 interactions), and super hubs (101+ interactions). In general, proteins had an average of 11 interactors with the largest class consisting of the medium hubs (Table 3). This same study showed that major and super hubs tend to have nuclear localization, with 11% of them annotated as indispensable. The most highly connected hubs appeared to be highly conserved proteins, involved in important signaling processes (Geisler-Lee et al., 2007). Later, hubs were also identified via similar interolog-based computational studies for Coffea canephora (Geisler and Fitzek, 2011), Oryza sativa (Gu et al., 2011; Ho et al., 2012), Brassica rapa (Yang et al., 2012), Zea mays (Musungu et al., 2015), Physcomitrella patens (Schuette et al., 2015), S. lycopersicum (Yue et al., 2016), and Macrotyloma uniflorum (Bhardwaj et al., 2016). In these studies a higher number of reference species was used as more interactome data became available (Table 3), including a diverse range of eukaryotic and prokaryotic organisms, allowing greater confidence (Schuette et al., 2015). An exception was the B. rapa interactome, inferred only from PPI data of A. thaliana, belonging to the same family of Brassicaceae (Yang et al., 2012).
Differences can be seen between the average amount of interaction partners and the largest hub class inferred from these interactomes (Table 3), but similar general conclusions regarding the hub proteins were made. As in the A. thaliana study, the highest degree hubs tend to be highly conserved and essential proteins, including heat shock proteins, nuclear DNA repair proteins, cytoskeleton-related proteins, ribosomal proteins and proteins associated with ubiquitin-mediated breakdown pathways (Supplementary Table 2; Geisler-Lee et al., 2007). However, conserved hub proteins with an unknown function were also always identified. More information on these computational plant interactome studies can be found in the corresponding publications (Geisler-Lee et al., 2007; Geisler and Fitzek, 2011; Gu et al., 2011; Ho et al., 2012; Yang et al., 2013; Musungu et al., 2015; Schuette et al., 2015; Bhardwaj et al., 2016; Yue et al., 2016).
Experimentally Determined Plant Hubs
The first experimentally constructed and comprehensive large-scale plant interactome was presented by the Arabidopsis Interactome Mapping Consortium (2011). This A. thaliana interactome (Arabidopsis Interactome 1, AI-1) was constructed using a systematic binary Y2H-based approach screening the pairwise interactions between 8,543 A. thaliana proteins (constructed from AtORFeome2.0) and resulted in the identification of 5,664 highly reliable interactions among 2,661 proteins (Arabidopsis Interactome Mapping Consortium, 2011). A subsequent study using the same Y2H-based approach determined the interactions between the 8,543 initially screened Arabidopsis proteins (in AI-1) and an extra 552 A. thaliana immunity proteins and effector proteins from the phytopathogens Pseudomonas syringae and Hyaloperonospora arabidopsidis. This resulted in an extended PPI network composed of 1,358 interactions among 926 proteins, including 83 pathogen effectors, 170 immune proteins, and 673 other A. thaliana proteins. Combining this network with AI-1 and literature-curated interactions for the 926 interacting proteins, resulted in a plant-pathogen immune network (PPIN-1), containing 3,148 interactions (Mukhtar et al., 2011). Additionally, 15 A. thaliana proteins with a degree higher than 50 in AI-1 were identified as hub proteins (hubs50), with 7 of these hubs50 targeted by effectors from both P. syringae and H. arabidopsidis (Supplementary Table 1). A third study defined the interactions between 12,000 A. thaliana proteins, including the 8,543 initially screened proteins, and pathogen effectors from Golovinomyces orontii, yielding a G. orontii effector-host interactome network (Gor_EHIn12k). Combining all P. syringae, H. arabidopsidis, and G. orontii effector host-protein interactions with interaction data from AI-1, PPIN-1, and literature, yielded the comprehensive Plant-Pathogen Immune Network 2 (PPIN-2; Weβling et al., 2014). When limited to the interactions of the 8,543 initially screened proteins, a network was generated with identical experimental parameters consisting of 178 A. thaliana host proteins and 123 effectors, connected by 421 effector host-proteins interactions and 162 interactions among host proteins (PPIN-28k_sys) (Weβling et al., 2014). These studies allowed the observation of a remarkable convergence of effectors onto a common set of host hub proteins, including members of the TCP (teosinte-branched/cycloidea/proliferating cell factor) TF family and the LSU (low sulfur upregulated) protein family (Supplementary Table 1). Similarly to the computational studies, also some proteins with an unknown function were identified as hubs.
Additionally, several more specific large-scale experimental studies identifying hub proteins in A. thaliana were published. These include a protein microarray study focusing on calmodulin targets (Popescu et al., 2007) and Y2H studies focusing specifically on the TOPLESS (TPL) transcriptional co-repressor (Causier et al., 2012) and ABA (abscisic acid) (Lumba et al., 2014; Table 4). Though multiple hubs were identified in these studies, they were defined as highly (inter)connected clusters of proteins in the unraveled networks and not as single hub proteins. More recently, the newly developed CrY2H-seq method, allowed the creation of a deep-coverage A. thaliana TF interaction network (AtTFIN-1), greatly expanding the number of known TF interactions and providing more evidence for the validation of TCP TFs as hub proteins (Trigg et al., 2017). One large-scale A. thaliana interactome study focused on membrane protein interactions using sUbq-approaches and identified several membrane hub proteins. A test-space of 6.4 × 10∧6 protein pairs was covered, determining 12,102 membrane/signaling PPIs, with more than 99% of the interactions previously unknown. Interactions were confirmed at a rate of 32% in in planta split–GFP interaction assays (Jones et al., 2014). A total of 46 hub proteins with a degree higher than 70 were identified, with functions consistent with roles that require interaction with a large number of target proteins (Table 4; Jones et al., 2014). However, as membrane PPIs require specific approaches to be identified and are generally not picked up by other methods, the unraveled plant membrane protein interactome is even more incomplete and many biologically relevant interactions are undoubtedly still being missed.
Aside from A. thaliana, several experimental large-scale interactome studies have also been published for economically important crops, but only one Y2H study for O. sativa focused on hub proteins, with several identified as having a key role in the stress response (Table 4; Seo et al., 2011).
Other Plant Hub-Defining Studies
To be complete, next to extended interactome studies, some small-scale PPI studies also define hubs in their limited (sub)networks, but these can hardly be seen as true hubs. Additionally, several other reports define plant hub proteins primarily based on their central role in signaling pathways. When it is indeed shown through PPI studies that these proteins engage in numerous physical interactions with other proteins, these can be correctly labeled as true hub proteins. By contrast, when they are solely termed hubs because of their importance in signaling, one could seriously doubt this labeling, based on the original hub protein definition. Although, this could also be interpreted as a slow emergence of a distinction between degree hubs (based on physical connectivity) and signaling hubs (based on a central signaling role). The fact that some hubs fall into both categories, however, and the distinction between the categories is not clear to all, often causes confusion for researchers from different fields, e.g., functional biologists vs. network theorists.
Proteins that are often labeled as signaling hubs include various transcription factors with a role in the plant hormone response, including ERFs (Ethylene response factors), ARFs (Auxine response factors), TPL (TOPLESS), and JAZ (Jasmonate ZIM-domain) proteins (Sheard et al., 2010; Mukhtar et al., 2011; Wager and Browse, 2012; Piya et al., 2014; Windram et al., 2014; Müller and Munné-Bosch, 2015), various PP2Cs (protein phosphatase 2C) and MAPKs (Mitogen-activated protein kinases) (Pitzschke et al., 2009; Dietz et al., 2010; Rodrigues et al., 2013), heat shock proteins (Taipale et al., 2010), and several ubiquitin ligase complex-associated proteins (Shabek and Zheng, 2014). Others identified in more focused studies include PIF (Phytochrome Interacting Factor) proteins (Leivar and Quail, 2011; Pfeiffer et al., 2014), peroxiredoxins (Muthuramalingam et al., 2009), calcium-dependent protein kinases (Schulz et al., 2013; Ranty et al., 2016), GTPases (Dietz et al., 2010), and specific proteins like Mediator 25 (Çevik et al., 2012), RPM1-interacting protein 4 (Sun et al., 2014), the miR156/SPL module (Wang and Wang, 2015), and the stress-associated plant protein RCD1 (radical-induced cell death 1; O'Shea et al., 2017).
Plant Stress Response Hubs: An Overview of the Major Classes
Many plant proteins labeled as hubs play a role in the plant stress response. Most of these are true hubs, participating in numerous PPIs and having an important central role in signaling pathways. They broadly fall into the functional categories of transcription factors, kinases and phosphatases, or components of ubiquitin ligase complexes. Other hubs include heat shock proteins, proteins involved in redox signaling, DNA repair, ribosomal proteins, and cytoskeleton-interacting proteins that play a role in protecting cells and cell components, making them essential and often conserved. There are, however, also proteins labeled as highly interacting hubs which are still uncharacterized or do not have a completely unraveled function, making them interesting targets for future research. In the next paragraphs we will discuss some of the best studied plant hub representatives with a role in the plant stress response, specifically focusing on hub protein characteristics. The resulting findings are summarized in Table 6.
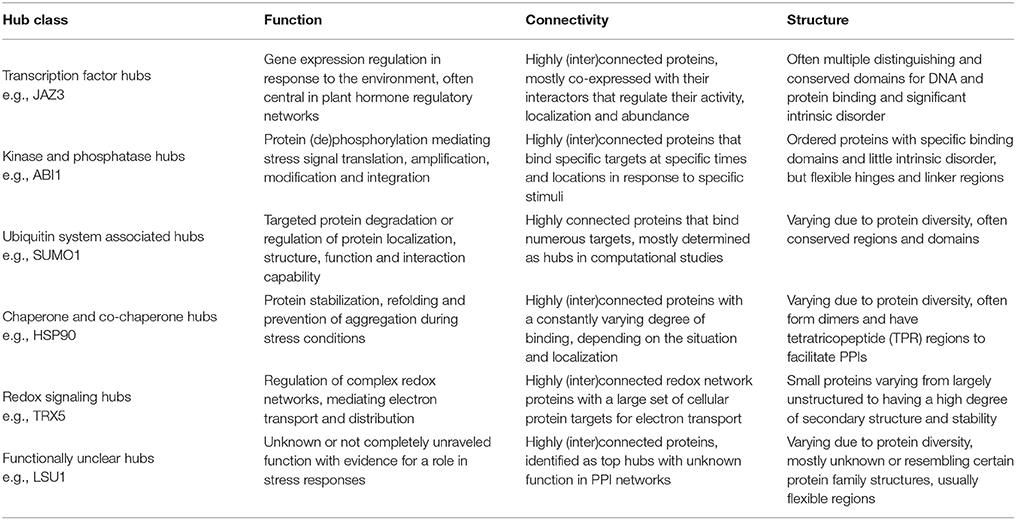
Table 6. Key features of the major classes of plant stress response-related hub proteins.
Transcription Factor Hubs
During evolution, DNA-binding proteins became prevalent in eukaryotic genomes and several families of transcription factors (TFs) arose. These proteins contain specific domains for DNA-binding, but their activity, localization, and abundance is often regulated through binding with other proteins. TFs usually have multiple conserved DNA and protein binding domains, but studies also show a significant prevalence of intrinsic disorder in eukaryotic TFs, making them more flexible and efficient (Liu et al., 2006). Recently published A. thaliana TF interactome networks show a large amount of interactions for most TF (Yazaki et al., 2016; Trigg et al., 2017). In the network created by Yazaki et al. (2016), on average 95 interactors were found, with a maximum of 499 interactors reported for TGA1, a bZIP TF important in auxin and SA signaling (Yazaki et al., 2016). Other studies specifically indicated several plant TFs as hubs regulating transcriptional cascades, including A. thaliana JAZ proteins, ERFs, ARFs, TCPs, and TPLs (Mukhtar et al., 2011; Pauwels and Goossens, 2011; Causier et al., 2012; Piya et al., 2014; Li et al., 2015; Müller and Munné-Bosch, 2015; Trigg et al., 2017). JAZ proteins, ERFs, and TCPs all have important roles in plant stress responses and defense response pathways. JAZ family proteins function as transcriptional repressors of jasmonate (JA) signaling (Pauwels and Goossens, 2011; Kazan and Manners, 2012; Wager and Browse, 2012), while ERFs regulate molecular responses to both pathogens and abiotic stresses (Mizoi et al., 2012; Müller and Munné-Bosch, 2015). TCPs regulate plant growth and development through control of cell proliferation and differentiation (Aguilar-Martínez and Sinha, 2013), but have recently also been linked to plant immunity (Sugio et al., 2011; Kim et al., 2014; Weβling et al., 2014). In comparison to JAZ and TCP TFs, however, only a limited number of interactions are currently known for ERFs (Figure 3).
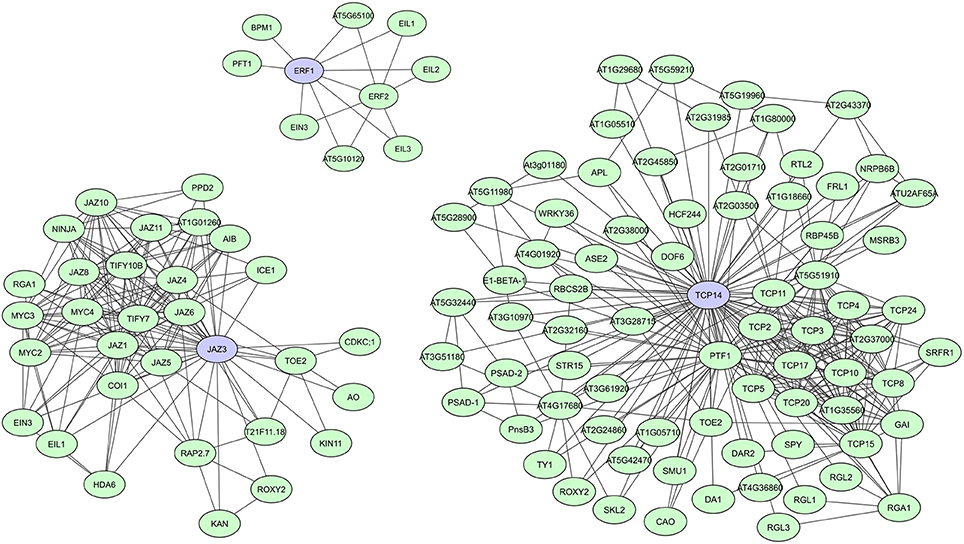
Figure 3. STRING-based networks for the A. thaliana ERF1, JAZ3, and TCP14 transcription factors. Interactors experimentally determined or from curated databases (medium confidence 0.400).
Most JAZ family proteins are highly connected and interconnected and share many of their interactors with which they are co-expressed (Pauwels and Goossens, 2011). Typical JAZ interactors are DELLA proteins, COI1 (CORONATINE INSENSITIVE1) and NINJA, but they also form homo- and heterodimers and interact with several other TFs (Figure 3) (Pauwels and Goossens, 2011; Wager and Browse, 2012). A. thaliana JAZ3, for example, was shown to have 23 interactors in AI-1 and is targeted by effectors of three distinct pathogens (Supplementary Table 1) (Arabidopsis Interactome Mapping Consortium, 2011; Mukhtar et al., 2011; Weβling et al., 2014). Currently over 30 interactors have been determined for JAZ3 according to the STRING database (Figure 3). JAZ mutants also show clear effects on pathogen susceptibility, metabolism, and development (Pauwels and Goossens, 2011; Pieterse et al., 2014; Weβling et al., 2014). For the TCP TF family it seemed at first that they could have highly connected and interconnected members, as well as much less connected members. As such, A. thaliana TCP14 and TCP13 were characterized as hub50 proteins in AI-1 and as targets of three distinct pathogens, while TCP15 and TCP19 were shown to have over 20 interactors and be targets of at least two pathogens, and TCP1 was only found to have two interactors (Supplementary Table 1; Arabidopsis Interactome Mapping Consortium, 2011; Mukhtar et al., 2011; Weβling et al., 2014). However, the most recently created A. thaliana TF interactome network (AtTFIN-1) expanded the number of known TCP TF interactions. TCPs were shown to interact with 18 other TF families and although differences in degree between TCP family members were still apparent, with over 200 interactors found for TCP14 compared to 30 interactors for TCP1, most were found to be highly interacting (Trigg et al., 2017). Additionally, several TCP mutants were shown to have altered disease susceptibility phenotypes, suggesting an important and universal role of this class of TFs during pathogen infection (Weβling et al., 2014).
Protein Kinase and Phosphatase Hubs
Plant signaling pathways are highly regulated through reversible protein phosphorylation, mediated by protein kinases and phosphatases (Chae et al., 2010). Some function in integrating signals triggered by a wide range of stresses and are thus said to function as key hubs. These include certain calcium-dependent protein kinases (CDPKs), calmoduline (CaM)-like proteins (CMLs), mitogen-activated protein kinases (MAPKs), and serine/threonine proteins phosphatases (including PP2Cs).
CDPKs comprise Ser/Thr protein kinases with a conserved structure and several ordered domains. They function as complex signaling date hubs, able to interact with many different proteins, but with specific targets at specific times and locations, in response to specific environmental stimuli (Schulz et al., 2013). Plant CaMs and CMLs have a wide range of diverse targets, including TFs, intracellular and receptor protein kinases, F-box proteins, RNA-binding proteins, and several proteins of unknown function. They have little disorder in their structure, but highly flexible hinges and linker regions that allow for the binding with multiple partners (Popescu et al., 2007). Several A. thaliana CaM/CML proteins, including CaM1, CaM6, CaM7, and CaM9, were shown to be very highly connected and form the main hubs of the CaM/CML network (Table 4, Popescu et al., 2007).
MAPK signaling cascades are often required for further translation, amplification and modification of environmental stimuli. All MAPKs share similar two lobed 3D structures, with a protein substrate binding on the surface of the C-terminal domain and the phosphorylation loop sequence influencing substrate specificity. However, MAPK signaling location, specificity and duration are regulated by interacting scaffold proteins and MAPK phosphatases (Taj et al., 2010). Both MPK3 and MPK6, well-known MAPKs in plant defense responses (Beckers et al., 2009), are said to function as date hubs (Dietz et al., 2010). Using a protein microarray-based method allowing high-throughput study of protein phosphorylation, 48 and 39 potential substrates could be identified for MPK3 and MPK6, respectively (Feilner et al., 2005). Later, a study of Popescu et al. (2009), focusing on MAPK targets (Table 2) found that each MAPK bound and phosphorylated an average of 128 other proteins, with 184 phosphorylated by MPK6 (Popescu et al., 2009).
PP2C-type protein phosphatases are involved in signaling regulation, cooperating with other phosphatases and kinases, often in a stress-induced manner (Rodriguez, 1998; Sheen, 1998; Merlot et al., 2001). A general characteristic of PP2C-type phosphatases is the presence of 11 subdomains in the catalytic part of the proteins. PP2Cs interact with both substrates as well as regulator proteins, via N-terminal extensions that function as binding sites to specific substrates (Schweighofer et al., 2004). In A. thaliana, clade A PP2Cs act as regulatory hubs for different abiotic stress responses via interaction with a wide array of targets. They function as negative regulators of the abscisic acid (ABA) signaling pathway through their interaction with SnRK2s [SNF1 (Sucrose Non-fermenting-1) -Related Protein Kinase 2]. The PP2Cs ABI1 and PP2CA, were shown to also function as SnRK1 phosphatases (Rodrigues et al., 2013). A STRING-based interaction network for ABI1 shows more than 90 determined interactors, including many other phosphatases and kinases, TFs, metabolic enzymes and proteins of unknown function (Figure 4). Lastly, the PP2C HAI1, was indicated as a ABA signaling interactome hub, highly correlated with the expression of many of its partners in both osmotic and salt stress data sets (Lumba et al., 2014).
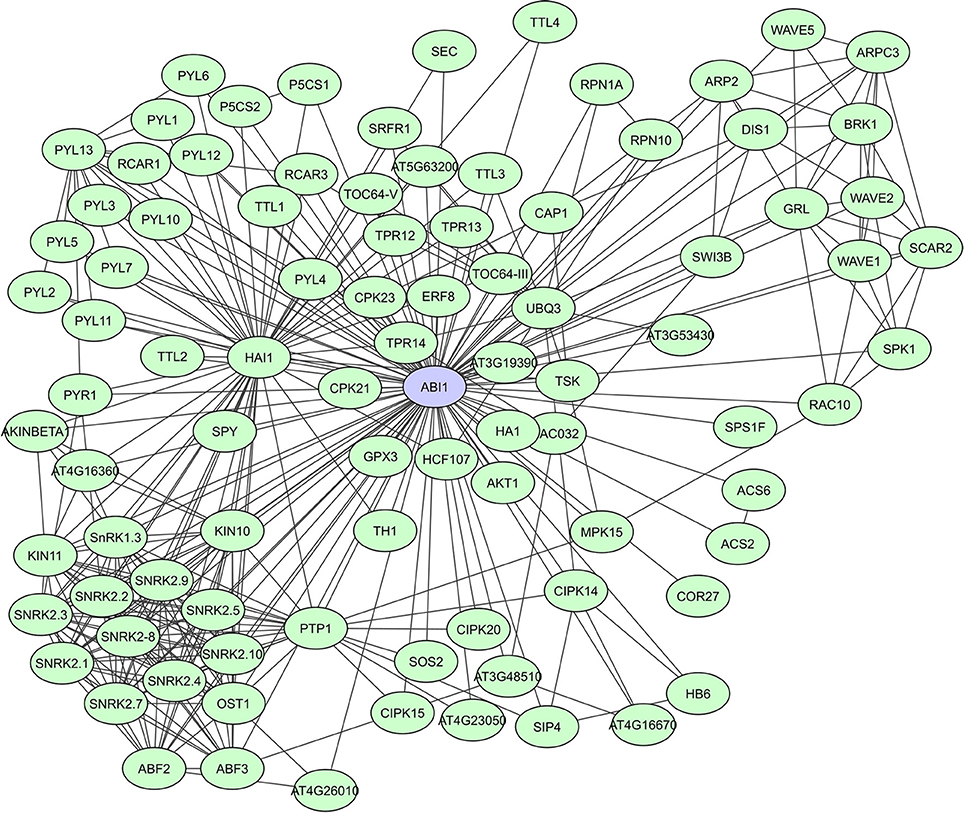
Figure 4. STRING-based network for the A. thaliana ABI1 protein phosphatase. Interactors experimentally determined or from curated databases (medium confidence 0.400).
Ubiquitin System Associated Hubs
The ubiquitin (Ub)-proteasome protein breakdown system has a fundamental role in plants, both in growth and development but also in plant immune signaling (Stone and Callis, 2007; Delauré et al., 2008; Marino et al., 2012). Ubiquitination or ubiquitin-like modifications, such as sumoylation, can regulate plant stress signaling via affecting protein localization, structure, function, and interaction capability (Miura and Hasegawa, 2010). About 6% of the plant proteome is said to be composed of Ub-proteasome system (UPS) related proteins (Hua and Vierstra, 2011).
When looking at large-scale plant PPI studies, UPS proteins are especially identified as highly interacting proteins in computational studies. In the Geisler-Lee study (2007) the small Ub-like modifier AtSUMO1 was identified as the top hub with 172 interactors, followed by the Ub carrier protein AtUBC1 with 119 interactors, and the multi-Ub chain binding 26S proteasome subunit protein ATMCB1 with 108 interactors (Supplementary Table 2). Many AtSUMO1 interactions have however also been confirmed via a Y2H screen (Figure 5, Elrouby and Coupland, 2010). In later computational PPI studies for C. canephora (Geisler and Fitzek, 2011) and Z. mays (Musungu et al., 2015) UPS proteins were also found as top hubs (Table 3). In the experimental membrane-linked interactome composed by Jones et al. (2014) several UPS proteins were identified as hub proteins with degrees higher than 70 (Jones et al., 2014). Additionally, the F-box proteins or substrate receptors of multisubunit E3 ligase complexes, like the plant Skp-cullin-F-box complexes (SCF) and the cullin-RING ligases (CRLs) are often considered as hub proteins (Shabek and Zheng, 2014). A typical example is the A. thaliana COI1 F-box protein that allows the SCFCOI1 complex to regulate JA-responsive gene expression through JAZ protein breakdown (Pauwels and Goossens, 2011). Still, UPS proteins are seemingly less prevalent in experimentally determined interactomes. This is probably a result of the more specific nature of some of these interactomes and because general UPS proteins are often put on non-specific interactors lists as they are almost always present and often found to interact with many proteins.
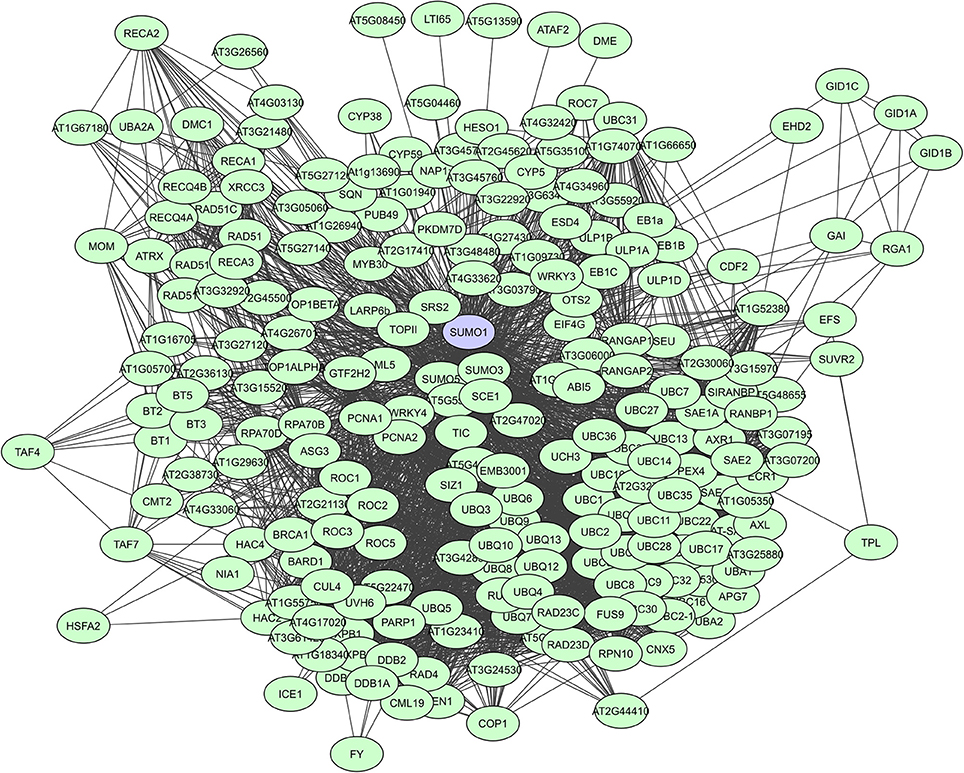
Figure 5. STRING-based network for the A. thaliana Ub-like modifier SUMO1. Interactors experimentally determined or from curated databases (medium confidence 0.400).
Chaperone and Co-chaperone Hubs
Chaperone proteins or heat shock proteins (HSPs) are essential for cellular homeostasis and are responsible for proper protein folding, localization, and degradation (Wang et al., 2004; Al-Whaibi, 2011). Five major families of plant chaperones are currently recognized according to their approximate molecular weight: the HSP70 family, the chaperonins, the HSP90 family, the HSP100 family and the small HSP (sHSP) family (Gupta et al., 2010). As a result of their stabilizing function they interact with numerous substrates, but also with several co-chaperones that regulate their activity, substrate recognition and refolding (Al-Whaibi, 2011; Breiman, 2014). As a result, both chaperones and co-chaperones are often identified as hubs.
As such, HSP90s, for example, are core components of many protein complexes and act as key regulators of plant growth and immunity, by directly interacting with R proteins and diverse proteins like kinases and TFs that activate defense responses (Liu et al., 2004; Sangster and Queitsch, 2005; Sangster et al., 2007; Breiman, 2014; Park and Seo, 2015). They are large dimeric proteins with each monomer having three characterizing domains i.e. a highly conserved N-terminal domain that binds and hydrolyzes ATP following substrate interaction, a middle domain thought to have an important role in substrate recognition, and a C-terminal domain mediating dimerization and interaction with many co-chaperones (Taipale et al., 2010). Chaperone proteins also often have tetratricopeptide regions (TPRs) which can form scaffolds to mediate PPIs or the assembly of large protein complexes (Blatch and Lässle, 1999). However, their binding degree varies continuously depending on the cell and tissue type, interactions with other proteins, alternative splicing, post-translational modifications and cell signaling events. A total of more than 380 interactors have currently been determined for HSP90.1, based on the STRING database (Figure 6).
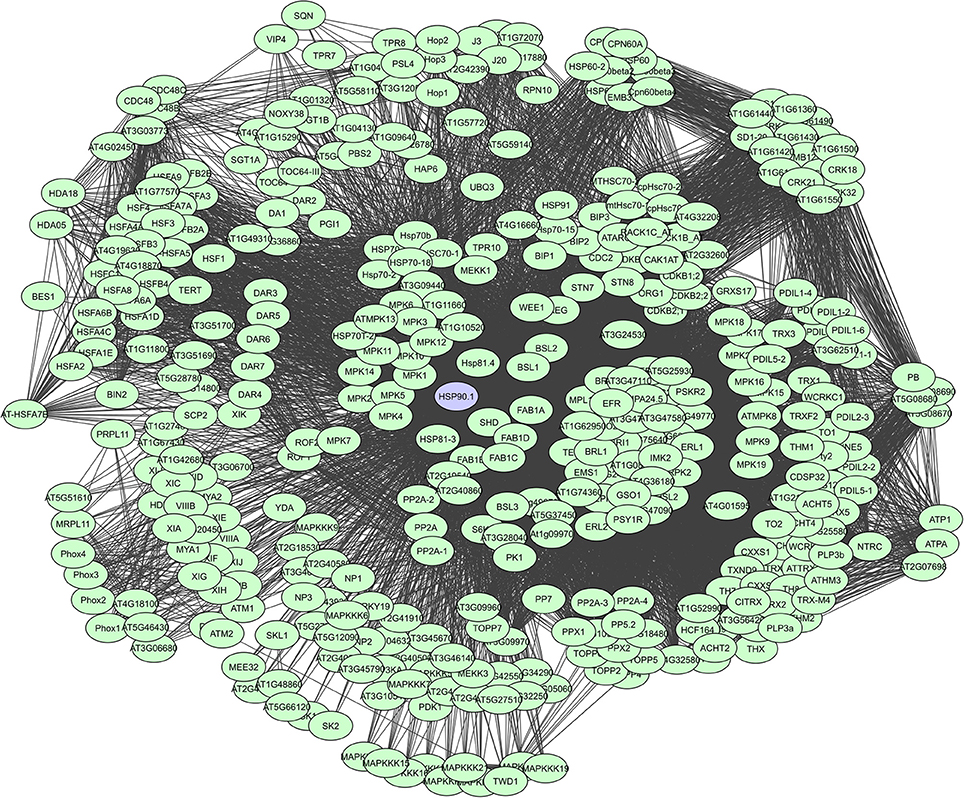
Figure 6. STRING-based network for the A. thaliana HSP90.1 chaperone. Interactors experimentally determined or from curated databases (medium confidence 0.400).
Another example is the mitochondrion-localized small heat shock protein AtHSP23.6 that was identified as one of the hub50 proteins in AI-1, with 87 interactors, and targeted by H. arabidopsidis effector proteins (Supplementary Table 1; Arabidopsis Interactome Mapping Consortium, 2011; Mukhtar et al., 2011). In the studies of Ho et al. (2012) and Yue et al. (2016) a rice HSP90 and a tomato HSP70 were found as top hub proteins with 686 and 3,7551 interactions, respectively (Table 3; Ho et al., 2012; Yue et al., 2016). In the computational study of Geisler-Lee et al. (2007) the prefoldin co-chaperone AtPDF6 was identified as one of the top 20 hub proteins with 93 predicted interactors. However, only five of these interactors were experimentally confirmed in AI-1 (Supplementary Table 2; Arabidopsis Interactome Mapping Consortium, 2011; Mukhtar et al., 2011). Like UPS proteins, chaperones, and co-chaperones, are seemingly less prevalent in datasets from experimentally determined plant interactomes. This can be the result of HSPs not covalently but transiently binding to their targets, not making them part of the final complex and thus being more difficult to pick up with certain experimental approaches. HSPs are also often not included in more specific screens or are often added to lists of false positive or non-specific interactors as they are inherently associated with many proteins as a result of their stabilizing function.
Redox Signaling Hubs
Reactive oxygen species (ROS) in plants are produced through mitochondrial respiration and photosynthetic electron transport in the chloroplasts (Foyer and Noctor, 2013; Schwarzländer and Finkemeier, 2013; Kalia et al., 2017). Regulating ROS and redox signals is essential for plant survival and requires complex redox networks. These networks contain several small hub proteins, including certain ferredoxins, thioredoxins, and peroxiredoxins, that play a central role in electron distribution by mediating electron transfer through interacting with many other redox network proteins (Dietz, 2008; Dietz et al., 2010).
Fe2S2 ferredoxins in chloroplasts function as electron carriers in the photosynthetic electron transport chain and as electron donors to various cellular proteins. They have a large proportion of unstructured regions with a high content of loops, but the residues necessary for PPIs are present in the α-helices (Dietz et al., 2010). Thioredoxins (Trx) also accept electrons through thiol-disulfide interchange and adjust the redox state of multiple target proteins. They have a high degree of secondary structure coupled to an extraordinary stability. In plants, the Trx system is particularly complex and many isoforms exist in various subcellular compartments, with a large set of plant Trx targets, exceeding 300 proteins, having been identified (Motohashi et al., 2001; Marchand et al., 2004; Lemaire et al., 2007; Meyer et al., 2009). In the experimental membrane-linked interactome composed by Jones et al. (2014) the A. thaliana H-type Trx ATH7 and ATH5 were identified as hub proteins with 97 and 71 interactors, respectively. ATH5 seems to be specifically involved in responses to pathogens and oxidative stress, and also exhibits antimicrobial activity (Laloi et al., 2004; Park et al., 2017). A STRING-based interaction network for ATH5 (TRX5) shows more than 120 interactors (Figure 7). Lastly, peroxiredoxins function as thioredoxin-dependent peroxidases and in plants 2-CysPyr functions to protect the photosynthetic membrane against photo-oxidative damage (Baier and Dietz, 1999). In the computational interactome of Geisler-Lee et al. (2007) 52 interactors were identified for 2-CysPyr. Multiple redox and non-redox interactions define 2-CysPyr as a regulatory hub in the chloroplast (Muthuramalingam et al., 2009; Dietz et al., 2010).
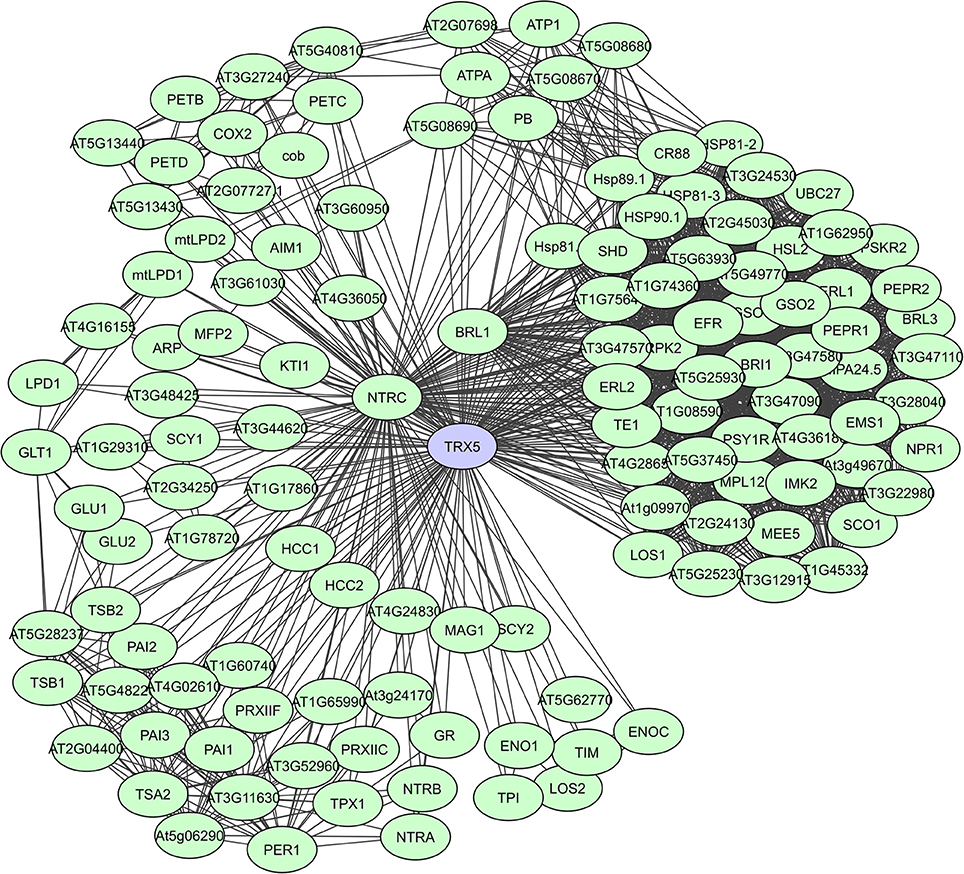
Figure 7. STRING-based networks for the A. thaliana TRX5 thioredoxin. Interactors experimentally determined or from curated databases (medium confidence 0.400).
Functionally Unclear Hubs
In many of the large-scale plant interactome studies, several putative proteins have been identified as hubs, like the unknown hub50 proteins in AI-1 (Supplementary Table 1), or proteins with a not yet completely unraveled function. A typical example of the latter is the family of LSU proteins, identified as hubs with a potentially important but not (fully) understood function in the plant stress response. These small proteins were initially identified as strongly induced during sulfur deficiencies (Maruyama-Nakashita et al., 2006; Lewandowska et al., 2010). A. thaliana has four members (LSU1-4), but homologs can be found in all higher land plants (Sirko et al., 2015; Garcia-Molina et al., 2017). LSU1 was identified as a hub50 protein with 80 interactors in AI-1, while 37 interactors were found for LSU2 (Supplementary Table 1) and more than 100 interactors were later identified for LSU3 in the expanded PPIN-1 network (Arabidopsis Interactome Mapping Consortium, 2011; Mukhtar et al., 2011). For LSU4 no interaction data are as yet available, but it is likely that all members of this protein family have hub potential. They have no specific binding domains, but their structure is predicted as a coiled-coil, allowing for flexibility and facilitating protein binding (Sirko et al., 2015).
Interestingly, identified LSU interactors were characterized as functionally very diverse and located in different cellular compartments, suggesting that LSU proteins have complex regulatory functions in various plant processes, including plant immunity and the abiotic stress response (Mukhtar et al., 2011; Sirko et al., 2015; Garcia-Molina et al., 2017). High sequence similarities and existing overlap in interactors between LSU1, LSU2, and LSU3 (Figure 8) suggest partial overlapping functions, while promotor analyses, unique interactors and differences in mutant phenotypes do assume some functional specificity (Sirko et al., 2015). A recent study suggests that LSU proteins can function as network hubs through integrating abiotic and biotic stress responses via interactions with the Fe superoxide dismutase FSD2 (Garcia-Molina et al., 2017). The putative RNA binding protein AtRAP was also recently shown to interacts with LSU2 in chloroplasts and suppress LSU2 under normal conditions (Wang et al., 2017). In general, different modes of action are expected for the LSU proteins due to their high connectivity and seeming involvement in many plant processes, making them highly interesting hubs and targets for further interactomics-based research.
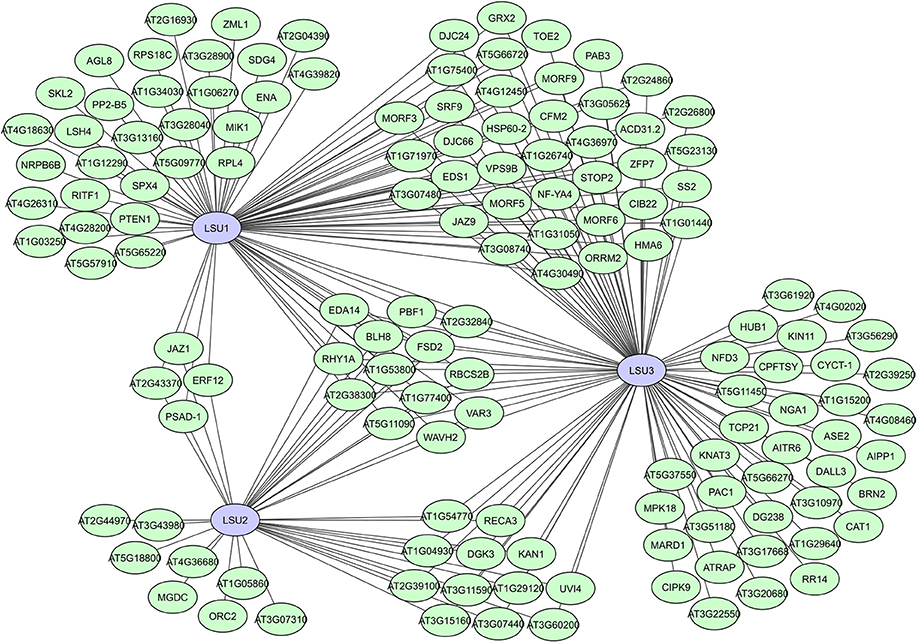
Figure 8. Local interactome for the A. thaliana LSU-like proteins LSU1, LSU2, and LSU3. Network based on data from the interactome studies of the Arabidopsis Interactome Mapping Consortium (2011) and Mukhtar et al. (2011).
Conclusions
Plant systems biology driven research is mainly supported by high-throughput -omics analyses with increasing performance, including proteomics and interactomics approaches. In order to represent this burst of data in a comprehensive way, graphical network presentations are used, with the most highly connected node proteins, being of great importance for network structure, stability and functionality. However, authors do not always follow the same definition to characterize these nodes in PPI networks as “hubs.”
The original definition of a hub in biology is that of an outlier in the degree distribution of a scale-free network, like a protein interaction network. Nowadays a hub protein is usually defined as a highly connected central node in a systematic PPI network. Several studies have resulted in a list of network and structural characteristics that are statistically more often attributed to proteins labeled as hubs compared to non-hubs (Table 1). However, not all hub proteins can be defined by one fixed set of such characteristics and depending on whether they are considered as part of large protein networks or as a separate entities, different properties appear to become more important and defining. Defining a protein as a hub is thus a complex matter, as rather the cumulative effect of having (some of) these properties is important to function as a hub in a given PPI network and be labeled as such. Even the intuitive setting of an interaction degree cutoff for hubs is challenging as either fixed or floating cutoffs can be preferred depending on the analyzed PPI networks. Moreover, the originally stated threshold of more than 5 interactions for defining high-degree nodes in general scale-free PPI networks (Vallabhajosyula et al., 2009), appears very low and thus inappropriate for comprehensive PPI networks, while alternatively choosing floating cutoffs also seems rather subjective and depending on the size and connectivity of the network. As such, the setting of a degree cutoff for hub proteins is complex and should be considered with care. There has also been some debate on correlations between degree, essentiality, centrality and pleiotropy, but currently the latter two are generally considered as inherent hub protein characteristics (Yu et al., 2008).
Nevertheless, the term “hub” has recently been used more frequently, and often incorrectly, to loosely label any central signaling network protein of some importance, independent of its connectivity, resulting in some justified confusion between researchers from different fields. However, as there is seemingly a growing emergence of proteins being labeled as hubs, a possible solution could be to make a distinction between signaling hubs (based on their involvement in numerous signaling pathways) and degree hubs (based on their high physical connectivity), potentially further dividing this last group in classes based on the number of interactions, like for example, small (6–10), intermediate (11–50), major (51–100), and super degree hubs (>100 interactions). Additionally, a clear choice should be made to define a PPI network hub as a single highly connected protein and not as a group of several different highly interconnected proteins, as stated by some studies. Here, the terms hub complex or hub module could be used. Nonetheless, new or improved hub classification systems should be considered and validated by network analysis experts, to cope with the boost of novel PPI network data. As hubs have great potential in further unraveling the intricacies of complex biological processes and networks, more clarity and consensus regarding their definition, characteristics and classifications will undoubtedly improve the growing systems biology based research.
In the plant interactome field, early breakthroughs were the first large-scale interolog-based computational A. thaliana interactome published by Geisler-Lee et al. (2007) and the high-throughput Y2H-based A. thaliana interactomes published by the Arabidopsis Interactome Mapping Consortium (2011) and Mukhtar et al. (2011). Later similar interolog-based studies were performed for other plant species (Tables 2, 3) and more large-scale experimental studies reported, both general and more specific, although only some defined or mentioned hub proteins (Tables 2, 4). In addition, during the last decade major technological advances have been made in the plant interactome field. These include improvements to computational predictions and experimental PPI identifications techniques, as well as improved literature curation of low-throughput experimental studies and increased data from high-quality and high-throughput experimental PPI studies deposited in public PPI databases. An array of databases, tools and resources have been developed to easily retrieve, analyze and visualize plant PPI and other related data (Table 5).
Plant PPI data can thus be obtained from several (complementary) sources, but it is important to realize that the potential of these data in defining hub proteins is strongly dependent on the quality and origin of the data. For instance, networks derived from computational approaches are mostly limited by what is already known in literature (e.g., interolog mapping) and therefore often biased and not accurately representing the biological reality. On the other hand, in networks resulting from experimental approaches, correct hub identification is strongly dependent both on the properties of the applied identification techniques and the experimental scale. Regarding the latter, for example, conclusions drawn from small-scale PPI studies might suffer from a research bias to the protein(s) of interest, resulting in a relatively higher number of identified interaction partners. Hubs are therefore preferably identified in large-scale PPI networks combined with comprehensive PPI networks assembled from curated databases. Still, the degree of proteins identified as hubs can also vary significantly between studies and resulting databases, mainly due to differences in applied techniques, their achieved interaction reliabilities and the risk of focusing on only a subset of the available data. It is also essential to keep in mind that every PPI identification technique has its own strengths and weaknesses. For example, approaches such as AP-MS, used for the identification of plant protein complexes, are very powerful methods but the resulting networks can hardly be used to define general hub proteins as protein degree is affected by preferred investigation and complex size, resulting in a network that is not scale-free. The advantages and disadvantages of different PPI identification techniques and their complementarity have been extensively reviewed (Morsy et al., 2008; Braun et al., 2013; McCormack et al., 2015) and a combination of techniques remains highly recommended for a more accurate PPI validation.
Though at present, various plant proteins have specifically been identified as hub proteins in plant PPI studies, the overlap in identified hubs and their interaction degree is often remarkably small. This becomes especially apparent when comparing hub proteins identified in large-scale computational and experimental PPI studies (Supplementary Tables 1, 2). However, as our knowledge grows and technology improves, it is expected that the gaps between the computationally and experimentally defined hubs will narrow. When looking at all the currently available plant PPI data, already similar (classes of) plant proteins are increasingly being characterized as important plant hub proteins, many of which with a reported role in the plant response to various types of stresses. They represent functional categories of transcription factors, kinases and phosphatases, ubiquitin system associated proteins, (co-)chaperones and redox signaling related proteins (Table 6). Furthermore, a significant number of identified plant hubs are still uncharacterized, making them most interesting targets for future research on (novel) plant stress signaling pathways. As interactomics approaches keep improving, this will undoubtedly lead to a more comprehensive identification of hub proteins and a more efficient system biology driven unraveling of complex biological processes, including those underlying the plant stress response.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
KV and BC acknowledge the receipt of a Ph.D. grant from the Flanders Innovation and Entrepreneurship (IWT) agency (IWT 141190) and a project grant from the Research Foundation—Flanders (FWO) (G0C6418N).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00694/full#supplementary-material
References
Agarwal, S., Deane, C. M., Porter, M. A., and Jones, N. S. (2010). Revisiting date and party hubs: novel approaches to role assignment in protein interaction networks. PLoS Comput. Biol. 6:e1000817. doi: 10.1371/journal.pcbi.1000817
Aguilar-Martínez, J. A., and Sinha, N. (2013). Analysis of the role of Arabidopsis class I TCP genes AtTCP7, AtTCP8, AtTCP22, and AtTCP23 in leaf development. Front. Plant Sci. 4:406. doi: 10.3389/fpls.2013.00406
Albert, R. (2005). Scale-free networks in cell biology. J. Cell Sci. 118, 4947–4957. doi: 10.1242/jcs.02714
Al-Whaibi, M. H. (2011). Plant heat-shock proteins: a mini review. J. King Saud Univ. Sci. 23, 139–150. doi: 10.1016/j.jksus.2010.06.022
Andorf, C. M., Honavar, V., Sen, T. Z., Eisenberg, D., Marcotte, E., Xenarios, I., et al. (2013). Predicting the binding patterns of hub proteins: a study using yeast protein interaction networks. PLoS ONE 8:e56833. doi: 10.1371/journal.pone.0056833
Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. doi: 10.1126/science.1203877
Aragues, R., Sali, A., Bonet, J., Marti-Renom, M. A., and Oliva, B. (2007). Characterization of protein hubs by inferring interacting motifs from protein interactions. PLoS Comput. Biol. 3:e30178. doi: 10.1371/journal.pcbi.0030178
Atkinson, N. J., and Urwin, P. E. (2012). The interaction of plant biotic and abiotic stresses: from genes to the field. J. Exp. Bot. 63, 3523–3544. doi: 10.1093/jxb/ers100
Baier, M., and Dietz, K. J. (1999). Protective function of chloroplast 2-cysteine peroxiredoxin in photosynthesis. Evid. Trans. Arabidop. Plant Physiol. 119, 1407–1414. doi: 10.1104/pp.119.4.1407
Bari, R., and Jones, J. D. G. (2009). Role of plant hormones in plant defence responses. Plant Mol. Biol. 69, 473–488. doi: 10.1007/s11103-008-9435-0
Batada, N. N., Reguly, T., Breitkreutz, A., Boucher, L., Breitkreutz, B.-J., Hurst, L. D., et al. (2007). Still stratus not altocumulus: further evidence against the date/party hub distinction. PLoS Biol. 5:e154. doi: 10.1371/journal.pbio.0050154
Batada, N. N., Reguly, T., Breitkreutz, A., Boucher, L., Breitkreutz, B. J., Hurst, L. D., et al. (2006). Stratus not altocumulus: a new view of the yeast protein interaction network. PLoS Biol. 4:e40317. doi: 10.1371/journal.pbio.0040317
Bauer-Mehren, A. (2013). Integration of genomic information with biological networks using Cytoscape. Methods Mol. Biol. 1021, 37–61. doi: 10.1007/978-1-62703-450-0_3
Beckers, G. J., Jaskiewicz, M., Liu, Y., Underwood, W. R., He, S. Y., Zhang, S., et al. (2009). Mitogen-activated protein kinases 3 and 6 are required for full priming of stress responses in Arabidopsis thaliana. Plant Cell 21, 944–953. doi: 10.1105/tpc.108.062158
Berardini, T. Z., Reiser, L., Li, D., Mezheritsky, Y., Muller, R., Strait, E., et al. (2015). The arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome. Genesis 53, 474–485. doi: 10.1002/dvg.22877
Bertolazzi, P., Bock, M. E., and Guerra, C. (2013). On the functional and structural characterization of hubs in protein-protein interaction networks. Biotechnol. Adv. 31, 274–286. doi: 10.1016/j.biotechadv.2012.12.002
Bhardwaj, J., Gangwar, I., Panzade, G., Shankar, R., and Yadav, S. K. (2016). Global de novo protein-protein interactome elucidates interactions of drought-responsive proteins in horse gram (Macrotyloma uniflorum). J. Proteome Res. 15, 1794–1809. doi: 10.1021/acs.jproteome.5b01114
Bhat, R. A., Lahaye, T., Panstruga, R., Goff, S., Ricke, D., Lan, T., et al. (2006). The visible touch: in planta visualization of protein-protein interactions by fluorophore-based methods. Plant Methods 2:12. doi: 10.1186/1746-4811-2-12
Blatch, G. L., and Lässle, M. (1999). The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21, 932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N
Brandão, M. M., Dantas, L. L., Silva-Filho, M. C., Giot, L., Bader, J., Brouwer, C., et al. (2009). AtPIN: Arabidopsis thaliana protein interaction network. BMC Bioinformatics 10:454. doi: 10.1186/1471-2105-10-454
Braun, P., Aubourg, S., Van Leene, J., De Jaeger, G., and Lurin, C. (2013). Plant protein interactomes. Annu. Rev. Plant Biol. 64, 161–187. doi: 10.1146/annurev-arplant-050312-120140
Breiman, A. (2014). Plant Hsp90 and its co-chaperones. Curr. Protein Pept. Sci. 15, 232–244. doi: 10.2174/1389203715666140331115603
Brizard, J. P., Carapito, C., Delalande, F., Van Dorsselaer, A., and Brugidou, C. (2006). Proteome analysis of plant-virus interactome: comprehensive data for virus multiplication inside their hosts. Mol. Cell. Proteomics 5, 2279–2297. doi: 10.1074/mcp.M600173-MCP200
Brückner, A., Polge, C., Lentze, N., Auerbach, D., and Schlattner, U. (2009). Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 10, 2763–2788. doi: 10.3390/ijms10062763
Causier, B., Ashworth, M., Guo, W., and Davies, B. (2012). The TOPLESS interactome: a framework for gene repression in Arabidopsis. Plant Physiol. 158, 423–438. doi: 10.1104/pp.111.186999
Çevik, V., Kidd, B. N., Zhang, P., Hill, C., Kiddle, S., Denby, K. J., et al. (2012). MEDIATOR25 acts as an integrative hub for the regulation of jasmonate-responsive gene expression in Arabidopsis. Plant Physiol. 160, 541–555. doi: 10.1104/pp.112.202697
Chae, L., Pandey, G. K., Luan, S., Cheong, Y. H., and Kim, K. N. (2010). “Protein kinases and phosphatases for stress signal transduction in plants,” in Abiotic Stress Adaptation in Plants: Physiological, Molecular and Genomic Foundation, eds A. Pareek, S. K. Sopory, H. J. Bohnert, and Govindjee (Dordrecht: Springer Netherlands), 123–163.
Chang, I. F., Curran, A., Woolsey, R., Quilici, D., Cushman, J. C., Mittler, R., et al. (2009). Proteomic profiling of tandem affinity purified 14-3-3 protein complexes in Arabidopsis thaliana. Proteomics 9, 2967–2985. doi: 10.1002/pmic.200800445
Chang, X., Xu, T., Li, Y., and Wang, K. (2013). Dynamic modular architecture of protein-protein interaction networks beyond the dichotomy of “date” and “party” hubs. Sci. Rep. 3:1691. doi: 10.1038/srep01691
Chatr-Aryamontri, A., Oughtred, R., Boucher, L., Rust, J., Chang, C., Kolas, N. K., et al. (2017). The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379. doi: 10.1093/nar/gkw1102
Cui, J., Li, P., Li, G., Xu, F., Zhao, C., Li, Y., et al. (2008). AtPID: Arabidopsis thaliana protein interactome database - An integrative platform for plant systems biology. Nucleic Acids Res. 36, D999–D1008. doi: 10.1093/nar/gkm844
De Arruda, G. F., Barbieri, A. L., Rodríguez, P. M., Rodrigues, F. A., Moreno, Y., and da Costa, L. F. (2014). Role of centrality for the identification of influential spreaders in complex networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 90:032812. doi: 10.1103/PhysRevE.90.032812
De Bodt, S., Hollunder, J., Nelissen, H., Meulemeester, N., and Inzé, D. (2012). CORNET 2.0: Integrating plant coexpression, protein-protein interactions, regulatory interactions, gene associations and functional annotations. New Phytol. 195, 707–720. doi: 10.1111/j.1469-8137.2012.04184.x
De Bodt, S., Proost, S., Vandepoele, K., Rouzé, P., and Van de Peer, Y. (2009). Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics 10:288. doi: 10.1186/1471-2164-10-288
de Folter, S. (2005). Comprehensive interaction map of the Arabidopsis MADS box transcription factors. Plant Cell Online 17, 1424–1433. doi: 10.1105/tpc.105.031831
Dedecker, M., Van Leene, J., and De Jaeger, G. (2015). Unravelling plant molecular machineries through affinity purification coupled to mass spectrometry. Curr. Opin. Plant Biol. 24, 1–9. doi: 10.1016/j.pbi.2015.01.001
Delauré, S. L., Van Hemelrijck, W., De Bolle, M. F. C., Cammue, B. P. A., and De Coninck, B. M. A. (2008). Building up plant defenses by breaking down proteins. Plant Sci. 174, 375–385. doi: 10.1016/j.plantsci.2008.01.008
Dietz, K. J. (2008). Redox signal integration: from stimulus to networks and genes. Physiol. Plant. 133, 459–468. doi: 10.1111/j.1399-3054.2008.01120.x
Dietz, K. J., Jacquot, J. P., and Harris, G. (2010). Hubs and bottlenecks in plant molecular signalling networks. New Phytol. 188, 919–938. doi: 10.1111/j.1469-8137.2010.03502.x
Ding, X., Richter, T., Chen, M., Fujii, H., Seo, Y. S., Xie, M., et al. (2009). A rice kinase-protein interaction map. Plant Physiol. 149, 1478–1492. doi: 10.1104/pp.108.128298
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Dosztányi, Z., Chen, J., Dunker, A. K., Simon, I., and Tompa, P. (2006). Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 5, 2985–2995. doi: 10.1021/pr060171o
Duque, A. S., de Almeida, A. M., da Silva, A. B., da Silva, J. M., Paula, A., Santos, D., et al. (2013). “Abiotic stress responses in plants: unraveling the complexity of genes and networks to survive,” in Abiotic Stress - Plant Responses and Applications in Agriculture, ed K. Vahdati (London: InTech), 49–101.
Ecker, J. R., Investigator, P., Trigg, S., Garza, R., Song, H., MacWilliams, A., et al. (2016). Next Generation Protein Interactomes for Plant Systems Biology and Biomass. Argonne, IL.
Ekman, D., Light, S., Björklund, Å. K., Elofsson, A., Gavin, A., Bosche, M., et al. (2006). What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 7:R45. doi: 10.1186/gb-2006-7-6-r45
Elrouby, N., and Coupland, G. (2010). Proteome-wide screens for small ubiquitin-like modifier (SUMO) substrates identify Arabidopsis proteins implicated in diverse biological processes. Proc. Natl. Acad. Sci. U.S.A. 107, 17415–17420. doi: 10.1073/pnas.1005452107
Feilner, T., Hultschig, C., Lee, J., Meyer, S., Immink, R. G. H., Koenig, A., et al. (2005). High throughput identification of potential arabidopsis mitogen-activated protein kinases substrates. Mol. Cell. Proteomics 4, 1558–1568. doi: 10.1074/mcp.M500007-MCP200
Finn, R. D., Miller, B. L., Clements, J., and Bateman, A. (2014). IPfam: a database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 42, D364–D373. doi: 10.1093/nar/gkt1210
Foyer, C. H., and Noctor, G. (2013). Redox signaling in plants. Antioxid. Redox Signal. 18, 2087–2090. doi: 10.1089/ars.2013.5278
Fujikawa, Y., and Kato, N. (2007). Split luciferase complementation assay to study protein-protein interactions in Arabidopsis protoplasts. Plant J. 52, 185–195. doi: 10.1111/j.1365-313X.2007.03214.x
Fujiwara, M., Uemura, T., Ebine, K., Nishimori, Y., Ueda, T., Nakano, A., et al. (2014). Interactomics of Qa-SNARE in Arabidopsis thaliana. Plant Cell Physiol. 55, 781–789. doi: 10.1093/pcp/pcu038
Gadeyne, A., Sánchez-Rodríguez, C., Vanneste, S., Di Rubbo, S., Zauber, H., Vanneste, K., et al. (2014). The TPLATE adaptor complex drives clathrin-mediated endocytosis in plants. Cell 156, 691–704. doi: 10.1016/j.cell.2014.01.039
Garbutt, C. C., Bangalore, P. V., Kannar, P., and Mukhtar, M. S. (2014). Getting to the edge: protein dynamical networks as a new frontier in plant-microbe interactions. Front. Plant Sci. 5:312. doi: 10.3389/fpls.2014.00312
Garcia-Molina, A., Altmann, M., Alkofer, A., Epple, P. M., Dangl, J. L., and Falter-Braun, P. (2017). LSU network hubs integrate abiotic and biotic stress responses via interaction with the superoxide dismutase FSD2. J. Exp. Bot. 68, 1185–1197. doi: 10.1093/jxb/erw498
Geisler, M., and Fitzek, E. (2011). A predicted interactome for coffee (Coffea canephora var robusta). J. Plant Mol. Biol. Biotechnol. 2, 34–46.
Geisler-Lee, J., O'Toole, N., Ammar, R., Provart, N. J., Millar, A. H., and Geisler, M. (2007). A predicted interactome for arabidopsis. Plant Physiol. 145, 317–329. doi: 10.1104/pp.107.103465
Goossens, J., De Geyter, N., Walton, A., Eeckhout, D., Mertens, J., Pollier, J., et al. (2016). Isolation of protein complexes from the model legume Medicago truncatula by tandem affinity purification in hairy root cultures. Plant J. 88, 476–489. doi: 10.1111/tpj.13258
Gu, H., Zhu, P., Jiao, Y., Meng, Y., and Chen, M. (2011). PRIN: a predicted rice interactome network. BMC Bioinformatics 12:161. doi: 10.1186/1471-2105-12-161
Gupta, S. C., Sharma, A., Mishra, M., Mishra, R. K., and Chowdhuri, D. K. (2010). Heat shock proteins in toxicology: how close and how far? Life Sci. 86, 377–384. doi: 10.1016/j.lfs.2009.12.015
Han, J.-D. J., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., Zhang, L. V., et al. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93. doi: 10.1038/nature02555
Hao, T., Peng, W., Wang, Q., Wang, B., and Sun, J. (2016). Reconstruction and application of protein-protein interaction network. Int. J. Mol. Sci. 17:907. doi: 10.3390/ijms17060907
Haynes, C., Oldfield, C. J., Ji, F., Klitgord, N., Cusick, M. E., Radivojac, P., et al. (2006). Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2:e20100. doi: 10.1371/journal.pcbi.0020100
He, X., and Zhang, J. (2006). Why do hubs tend to be essential in protein networks? PLoS Genet. 2:e88. doi: 10.1371/journal.pgen.0020088
Higurashi, M., Ishida, T., and Kinoshita, K. (2008). Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 17, 72–78. doi: 10.1110/ps.073196308
Ho, C.-L., Wu, Y., Shen, H. H., Provart, N. N. J., Geisler, M., Alon, U., et al. (2012). A predicted protein interactome for rice. Rice 5:15. doi: 10.1186/1939-8433-5-15
Hua, Z., and Vierstra, R. D. (2011). The cullin-RING ubiquitin-protein ligases. Annu. Rev. Plant Biol. 62, 299–334. doi: 10.1146/annurev-arplant-042809-112256
Hunt, A. G., Xu, R., Addepalli, B., Rao, S., Forbes, K. P., Meeks, L. R., et al. (2008). Arabidopsis mRNA polyadenylation machinery: comprehensive analysis of protein-protein interactions and gene expression profiling. BMC Genomics 9:220. doi: 10.1186/1471-2164-9-220
Ideker, T., Galitski, T., and Hood, L. (2001). A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372. doi: 10.1146/annurev.genom.2.1.343
Jeong, H., Mason, S. P., Barabási, A. L., and Oltvai, Z. N. (2001). Lethality and centrality in protein networks. Nature 411, 41–42. doi: 10.1038/35075138
Jin, G., Zhang, S., Zhang, X. S., and Chen, L. (2007). Hubs with network motifs organize modularity dynamically in the protein-protein interaction network of yeast. PLoS ONE 2:e1207. doi: 10.1371/journal.pone.0001207
Jones, A. M., Xuan, Y., Xu, M., Wang, R.-S., Ho, C.-H., Lalonde, S., et al. (2014). Border control—a membrane-linked interactome of Arabidopsis. Science 344, 711–716. doi: 10.1126/science.1251358
Kalia, R., Sareen, S., Nagpal, A., Katnoria, J., and Bhardwaj, R. (2017). “ROS-induced transcription factors during oxidative stress in plants: a tabulated review,” in Reactive Oxygen Species and Antioxidant Systems in Plants: Role and Regulation under Abiotic Stress, eds M. I. R. Khan and N. A. Khan (Singapore: Springer Singapore), 129–158.
Kazan, K., and Manners, J. M. (2012). JAZ repressors and the orchestration of phytohormone crosstalk. Trends Plant Sci. 17, 22–31. doi: 10.1016/j.tplants.2011.10.006
Kim, P. M., Sboner, A., Xia, Y., and Gerstein, M. (2008). The role of disorder in interaction networks: a structural analysis. Mol. Syst. Biol. 4:179. doi: 10.1038/msb.2008.16
Kim, S. H., Son, G. H., Bhattacharjee, S., Kim, H. J., Nam, J. C., Nguyen, P. D. T., et al. (2014). The Arabidopsis immune adaptor SRFR1 interacts with TCP transcription factors that redundantly contribute to effector-triggered immunity. Plant J. 78, 978–989. doi: 10.1111/tpj.12527
Klopffleisch, K., Phan, N., Augustin, K., Bayne, R. S., Booker, K. S., Botella, J. R., et al. (2011). Arabidopsis G-protein interactome reveals connections to cell wall carbohydrates and morphogenesis. Mol Syst Biol 7:532. doi: 10.1038/msb.2011.66
Krishnakumar, V., Hanlon, M. R., Contrino, S., Ferlanti, E. S., Karamycheva, S., Kim, M., et al. (2015). Araport: the arabidopsis information portal. Nucleic Acids Res. 43, D1003–D1009. doi: 10.1093/nar/gku1200
Krouk, G., Lingeman, J., Colon, A., Coruzzi, G., and Shasha, D. (2013). Gene regulatory networks in plants: learning causality from time and perturbation. Genome Biol. 14:123. doi: 10.1186/gb-2013-14-6-123
Laloi, C., Mestres-Ortega, D., Marco, Y., Meyer, Y., and Reichheld, J.-P. (2004). The Arabidopsis Cytosolic Thioredoxin-h5 gene induction by oxidative stress and its W-box mediated response to pathogen elicitor. Plant Physiol. 134, 1006–1016. doi: 10.1104/pp.103.035782
Lalonde, S., Sero, A., Pratelli, R., Pilot, G., Chen, J., Sardi, M. I., et al. (2010). A membrane protein/signaling protein interaction network for arabidopsis version AMPv2. Front. Physiol. 1:24. doi: 10.3389/fphys.2010.00024
Leivar, P., and Quail, P. H. (2011). PIFs: pivotal components in a cellular signaling hub. Trends Plant Sci. 16, 19–28. doi: 10.1016/j.tplants.2010.08.003
Lemaire, S. D., Michelet, L., Zaffagnini, M., Massot, V., and Issakidis-Bourguet, E. (2007). Thioredoxins in chloroplasts. Curr. Genet. 51, 343–365. doi: 10.1007/s00294-007-0128-z
Lewandowska, M., Wawrzynska, A., Moniuszko, G., Łukomska, J., Zientara, K., Piecho, M., et al. (2010). A contribution to identification of novel regulators of plant response to sulfur deficiency: characteristics of a tobacco gene UP9C, its protein product and the effects of UP9C silencing. Mol. Plant 3, 347–360. doi: 10.1093/mp/ssq007
Li, J. F., Bush, J., Xiong, Y., Li, L., and McCormack, M. (2011). Large-scale protein-protein interaction analysis in Arabidopsis mesophyll protoplasts by split firefly luciferase complementation. PLoS ONE 6:e27364. doi: 10.1371/journal.pone.0027364
Li, S., Xie, Z.-Z., Hu, C.-G., and Zhang, J.-Z. (2015). The Arabidopsis thaliana TCP transcription factors: a broadening horizon beyond development. Plant Signal. Behav. 10:e1044192. doi: 10.1080/15592324.2015.1044192
Licata, L., Briganti, L., Peluso, D., Perfetto, L., Iannuccelli, M., Galeota, E., et al. (2012). MINT, the molecular interaction database: 2012 Update. Nucleic Acids Res. 40, D857–D861. doi: 10.1093/nar/gkr930
Lin, M., Zhou, X., Shen, X., Mao, C., and Chen, X. (2011). The predicted Arabidopsis interactome resource and network topology-based systems biology analyses. Plant Cell 23, 911–922. doi: 10.1105/tpc.110.082529
Liseron-Monfils, C., and Ware, D. (2015). Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 3–4, 30–39. doi: 10.1016/j.cpb.2015.11.001
Liu, J., Perumal, N. B., Oldfield, C. J., Su, E. W., Uversky, V. N., and Dunker, A. K. (2006). Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888. doi: 10.1021/bi0602718
Liu, S., Liu, Y., Zhao, J., Cai, S., Qian, H., Zuo, K., et al. (2017). A computational interactome for prioritizing genes associated with complex agronomic traits in rice (Oryza sativa). Plant J. 90, 177–188. doi: 10.1111/tpj.13475
Liu, Y., Burch-Smith, T., Schiff, M., Feng, S., and Dinesh-Kumar, S. P. (2004). Molecular chaperone Hsp90 associates with resistance protein N and its signaling proteins SGT1 and Rar1 to modulate an innate immune response in plants. J. Biol. Chem. 279, 2101–2108. doi: 10.1074/jbc.M310029200
Long, Y., Stahl, Y., Weidtkamp-Peters, S., Postma, M., Zhou, W., Goedhart, J., et al. (2017). In vivo FRET-FLIM reveals cell-type-specific protein interactions in Arabidopsis roots. Nature 548, 97–102. doi: 10.1038/nature23317
Lumba, S., Toh, S., Handfield, L.-F. F., Swan, M., Liu, R., Youn, J.-Y. Y., et al. (2014). A mesoscale abscisic acid hormone interactome reveals a dynamic signaling landscape in arabidopsis. Dev. Cell 29, 360–372. doi: 10.1016/j.devcel.2014.04.004
Maor, R., Jones, A., Nühse, T. S., Studholme, D. J., Peck, S. C., and Shirasu, K. (2007). Multidimensional Protein Identification Technology (MudPIT) analysis of ubiquitinated proteins in plants. Mol. Cell. Proteomics 6, 601–610. doi: 10.1074/mcp.M600408-MCP200
Marchand, C., Le Maréchal, P., Meyer, Y., Miginiac-Maslow, M., Issakidis-Bourguet, E., and Decottignies, P. (2004). New targets of Arabidopsis thioredoxins revealed by proteomic analysis. Proteomics 4, 2696–2706. doi: 10.1002/pmic.200400805
Marino, D., Peeters, N., and Rivas, S. (2012). Ubiquitination during plant immune signaling. Plant Physiol. 160, 15–27. doi: 10.1104/pp.112.199281
Maruyama-Nakashita, A., Nakamura, Y., Tohge, T., Saito, K., and Takahashi, H. (2006). Arabidopsis SLIM1 is a central transcriptional regulator of plant sulfur response and metabolism. Plant Cell 18, 3235–3251. doi: 10.1105/tpc.106.046458
Maslov, S., and Sneppen, K. (2002). Specificity and stability in topology of protein networks. Science 296, 910–913. doi: 10.1126/science.1065103
McCormack, M. E., Lopez, J. A., Crocker, T. H., and Mukhtar, M. S. (2015). Making the right connections: network biology and plant immune system dynamics. Curr. Plant Biol. 5, 2–12. doi: 10.1016/j.cpb.2015.10.002
Mehta, V., and Trinkle-Mulcahy, L. (2016). Recent advances in large-scale protein interactome mapping. F1000Research 5:782. doi: 10.12688/f1000research.7629.1
Merlot, S., Gosti, F., Guerrier, D., Vavasseur, A., and Giraudat, J. (2001). The ABI1 and ABI2 protein phosphatases 2C act in a negative feedback regulatory loop of the abscisic acid signalling pathway. Plant J. 25, 295–303. doi: 10.1046/j.1365-313x.2001.00965.x
Meyer, K., and Selbach, M. (2015). Quantitative affinity purification mass spectrometry: a versatile technology to study protein-protein interactions. Front. Genet. 6:237. doi: 10.3389/fgene.2015.00237
Meyer, Y., Buchanan, B. B., Vignols, F., and Reichheld, J.-P. (2009). Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu. Rev. Genet. 43, 335–367. doi: 10.1146/annurev-genet-102108-134201
Miersch, S., and LaBaer, J. (2011). Nucleic acid programmable protein arrays: versatile tools for array-based functional protein studies. Curr. Protoc. Protein Sci. Chapter 27:Unit27.2. doi: 10.1002/0471140864.ps2702s64
Miller, K. E., Kim, Y., Huh, W. K., and Park, H. O. (2015). Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for Genome-Wide interaction studies. J. Mol. Biol. 427, 2039–2055. doi: 10.1016/j.jmb.2015.03.005
Mine, A., Sato, M., and Tsuda, K. (2014). Toward a systems understanding of plant-microbe interactions. Front. Plant Sci. 5:423. doi: 10.3389/fpls.2014.00423
Miura, K., and Hasegawa, P. M. (2010). Sumoylation and other ubiquitin-like post-translational modifications in plants. Trends Cell Biol. 20, 223–232. doi: 10.1016/j.tcb.2010.01.007
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Morsy, M., Gouthu, S., Orchard, S., Thorneycroft, D., Harper, J. F., Mittler, R., et al. (2008). Charting plant interactomes: possibilities and challenges. Trends Plant Sci. 13, 183–191. doi: 10.1016/j.tplants.2008.01.006
Motohashi, K., Kondoh, A., Stumpp, M. T., and Hisabori, T. (2001). Comprehensive survey of proteins targeted by chloroplast thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 98, 11224–11229. doi: 10.1073/pnas.191282098
Mukhtar, M. S., Carvunis, A.-R., Dreze, M., Epple, P., Steinbrenner, J., Moore, J., et al. (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601. doi: 10.1126/science.1203659
Müller, M., and Munné-Bosch, S. (2015). Ethylene response factors: a key regulatory hub in hormone and stress signaling. Plant Physiol. 169, 32–41. doi: 10.1104/pp.15.00677
Musungu, B., Bhatnagar, D., Brown, R. L., Fakhoury, A. M., and Geisler, M. (2015). A predicted protein interactome identifies conserved global networks and disease resistance subnetworks in maize. Front. Genet. 6:201. doi: 10.3389/fgene.2015.00201
Muthuramalingam, M., Seidel, T., Laxa, M., Nunes De Miranda, S. M., Gärtner, F., Ströher, E., et al. (2009). Multiple redox and non-redox interactions define 2-cys peroxiredoxin as a regulatory hub in the chloroplast. Mol. Plant 2, 1273–1288. doi: 10.1093/mp/ssp089
Nelissen, H., Eeckhout, D., Demuynck, K., Persiau, G., Walton, A., van Bel, M., et al. (2015). Dynamic changes in ANGUSTIFOLIA3 complex composition reveal a growth regulatory mechanism in the maize leaf. Plant Cell 27, 1605–1619. doi: 10.1105/tpc.15.00269
O'Shea, C., Staby, L., Bendsen, S. K., Tidemand, F. G., Redsted, A., Willemoës, M., et al. (2017). Structures and short linear motif of disordered transcription factor regions provide clues to the interactome of the cellular hub protein radical-induced cell death 1. J. Biol. Chem. 292, 512–527. doi: 10.1074/jbc.M116.753426
Orchard, S., Ammari, M., Aranda, B., Breuza, L., Briganti, L., Broackes-Carter, F., et al. (2014). The MIntAct project - IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363. doi: 10.1093/nar/gkt1115
Ota, M., Gonja, H., Koike, R., and Fukuchi, S. (2016). Multiple-localization and hub proteins. PLoS ONE 11:e0156455. doi: 10.1371/journal.pone.0156455
Park, C.-J., and Seo, Y.-S. (2015). Heat shock proteins: a review of the molecular chaperones for plant immunity. Plant Pathol. J. 31, 323–333. doi: 10.5423/PPJ.RW.08.2015.0150
Park, S.-C., Jung, Y. J., Kim, I. R., Lee, Y., Kim, Y.-M., Jang, M.-K., et al. (2017). Functional characterization of thioredoxin h type 5 with antimicrobial activity from Arabidopsis thaliana. Biotechnol. Bioprocess Eng. 22, 129–135. doi: 10.1007/s12257-017-0074-7
Patil, A., and Nakamura, H. (2006). Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 580, 2041–2045. doi: 10.1016/j.febslet.2006.03.003
Patil, A., and Nakamura, H. (2007). The role of charged surface residues in the binding ability of small hubs in protein-protein interaction networks. Biophysics 3, 27–35. doi: 10.2142/biophysics.3.27
Patil, A., Kinoshita, K., and Nakamura, H. (2010). Hub promiscuity in protein-protein interaction networks. Int. J. Mol. Sci. 11, 1930–1943. doi: 10.3390/ijms11041930
Pauwels, L., and Goossens, A. (2011). The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell Online 23, 3089–3100. doi: 10.1105/tpc.111.089300
Pfeiffer, A., Shi, H., Tepperman, J. M., Zhang, Y., Quail, P. H., Al-Sady, B., et al. (2014). Combinatorial complexity in a transcriptionally centered signaling hub in Arabidopsis. Mol. Plant 7, 1598–1618. doi: 10.1093/mp/ssu087
Pieterse, C. M. J., Pierik, R., and Van Wees, S. C. M. (2014). Different shades of JAZ during plant growth and defense. New Phytol. 204, 261–264. doi: 10.1111/nph.13029
Pitzschke, A., Schikora, A., and Hirt, H. (2009). MAPK cascade signalling networks in plant defence. Curr. Opin. Plant Biol. 12, 421–426. doi: 10.1016/j.pbi.2009.06.008
Piya, S., Shrestha, S. K., Binder, B., Stewart, C. N., and Hewezi, T. (2014). Protein-protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant Sci. 5:744. doi: 10.3389/fpls.2014.00744
Popescu, S. C., Popescu, G. V., Bachan, S., Zhang, Z., Gerstein, M., Snyder, M., et al. (2009). MAPK target networks in Arabidopsis thaliana revealed using functional protein microarrays. Genes Dev. 23, 80–92. doi: 10.1101/gad.1740009
Popescu, S. C., Popescu, G. V., Bachan, S., Zhang, Z., Seay, M., Gerstein, M., et al. (2007). Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays. Proc. Natl. Acad. Sci. U.S.A. 104, 4730–4735. doi: 10.1073/pnas.0611615104
Rangarajan, N., Kulkarni, P., and Hannenhalli, S. (2015). Evolutionarily conserved network properties of intrinsically disordered proteins. PLoS ONE 10:e0126729. doi: 10.1371/journal.pone.0126729
Ranty, B., Aldon, D., Cotelle, V., Galaud, J.-P., Thuleau, P., and Mazars, C. (2016). Calcium sensors as key hubs in plant responses to biotic and abiotic stresses. Front. Plant Sci. 7:327. doi: 10.3389/fpls.2016.00327
Rejeb, I., Pastor, V., and Mauch-Mani, B. (2014). Plant responses to simultaneous biotic and abiotic stress: molecular mechanisms. Plants 3, 458–475. doi: 10.3390/plants3040458
Rodgers-Melnick, E., Culp, M., and DiFazio, S. P. (2013). Predicting whole genome protein interaction networks from primary sequence data in model and non-model organisms using ENTS. BMC Genomics 14:608. doi: 10.1186/1471-2164-14-608
Rodrigues, A., Adamo, M., Crozet, P., Margalha, L., Confraria, A., Martinho, C., et al. (2013). ABI1 and PP2CA phosphatases are negative regulators of Snf1-related protein kinase1 signaling in Arabidopsis. Plant Cell 25, 3871–3884. doi: 10.1105/tpc.113.114066
Rodriguez, P. L. (1998). Protein phosphatase 2C (PP2C) function in higher plants. Plant Mol. Biol. 38, 919–927. doi: 10.1023/A:1006054607850
Rolland, T., Taşan, M., Charloteaux, B., Pevzner, S. J., Zhong, Q., Sahni, N., et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. doi: 10.1016/j.cell.2014.10.050
Roux, M., Schwessinger, B., Albrecht, C., Chinchilla, D., Jones, A., Holton, N., et al. (2011). The arabidopsis leucine-rich repeat receptor-like kinases BAK1/SERK3 and BKK1/SERK4 are required for innate immunity to Hemibiotrophic and Biotrophic pathogens. Plant Cell 23, 2440–2455. doi: 10.1105/tpc.111.084301
Sahu, S. S., Weirick, T., and Kaundal, R. (2014). Predicting genome-scale Arabidopsis-Pseudomonas syringae interactome using domain and interolog-based approaches. BMC Bioinformatics 15:8. doi: 10.1186/1471-2105-15-S11-S13
Sangster, T. A., and Queitsch, C. (2005). The HSP90 chaperone complex, an emerging force in plant development and phenotypic plasticity. Curr. Opin. Plant Biol. 8, 86–92. doi: 10.1016/j.pbi.2004.11.012
Sangster, T. A., Bahrami, A., Wilczek, A., Watanabe, E., Schellenberg, K., McLellan, C., et al. (2007). Phenotypic diversity and altered environmental plasticity in Arabidopsis thaliana with reduced Hsp90 levels. PLoS ONE 2:e648. doi: 10.1371/journal.pone.0000648
Schoonheim, P. J., Veiga, H., da Costa Pereira, D., Friso, G., van Wijk, K. J., and de Boer, A. H. (2006). A comprehensive analysis of the 14-3-3 interactome in barley leaves using a complementary proteomics and two-hybrid approach. PLANT Physiol. 143, 670–683. doi: 10.1104/pp.106.090159
Schuette, S., Piatkowski, B., Corley, A., Lang, D., and Geisler, M. (2015). Predicted protein-protein interactions in the moss Physcomitrella patens: a new bioinformatic resource. BMC Bioinformatics 16:89. doi: 10.1186/s12859-015-0524-1
Schulz, P., Herde, M., and Romeis, T. (2013). Calcium-dependent protein kinases: hubs in plant stress signaling and development. Plant Physiol. 163, 523–530. doi: 10.1104/pp.113.222539
Schwarzländer, M., and Finkemeier, I. (2013). Mitochondrial energy and redox signaling in plants. Antioxid. Redox Signal. 18, 2122–2144. doi: 10.1089/ars.2012.5104
Schweighofer, A., Hirt, H., and Meskiene, I. (2004). Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 9, 236–243. doi: 10.1016/j.tplants.2004.03.007
Seo, Y.-S., Chern, M., Bartley, L. E., Han, M., Jung, K.-H., Lee, I., et al. (2011). Towards establishment of a rice stress response interactome. PLoS Genet. 7:e1002020. doi: 10.1371/journal.pgen.1002020
Shabek, N., and Zheng, N. (2014). Plant ubiquitin ligases as signaling hubs. Nat. Struct. Mol. Biol. 21, 293–296. doi: 10.1038/nsmb.2804
Sheard, L. B., Tan, X., Mao, H., Withers, J., Ben-Nissan, G., Hinds, T. R., et al. (2010). Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 468, 400–405. doi: 10.1038/nature09430
Sheen, J. (1998). Mutational analysis of protein phosphatase 2C involved in abscisic acid signal transduction in higher plants. Proc. Natl. Acad. Sci. U.S.A. 95, 975–980. doi: 10.1073/pnas.95.3.975
Sheth, B. P., and Thaker, V. S. (2014). Plant systems biology: insights, advances and challenges. Planta 240, 33–54. doi: 10.1007/s00425-014-2059-5
Sirko, A., Wawrzynska, A., Rodrí-guez, M. C., and Sektas, P. (2015). The family of LSU-like proteins. Front. Plant Sci. 5:774. doi: 10.3389/fpls.2014.00774
Song, J., and Singh, M. (2013). From hub proteins to hub modules: the relationship between essentiality and centrality in the yeast interactome at different scales of Organization. PLoS Comput. Biol. 9:e1002910. doi: 10.1371/journal.pcbi.1002910
Stone, S. L., and Callis, J. (2007). Ubiquitin ligases mediate growth and development by promoting protein death. Curr. Opin. Plant Biol. 10, 624–632. doi: 10.1016/j.pbi.2007.07.010
Sugio, A., Kingdom, H. N., MacLean, A. M., Grieve, V. M., and Hogenhout, S. A. (2011). Phytoplasma protein effector SAP11 enhances insect vector reproduction by manipulating plant development and defense hormone biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 108, E1254–E1263. doi: 10.1073/pnas.1105664108
Sun, X., Greenwood, D. R., Templeton, M. D., Libich, D. S., McGhie, T. K., Xue, B., et al. (2014). The intrinsically disordered structural platform of the plant defence hub protein RPM1-interacting protein 4 provides insights into its mode of action in the host-pathogen interface and evolution of the nitrate-induced domain protein family. FEBS J. 281, 3955–3979. doi: 10.1111/febs.12937
Swatek, K. N., Graham, K., Agrawal, G. K., and Thelen, J. J. (2011). The 14-3-3 isoforms chi and epsilon differentially bind client proteins from developing Arabidopsis seed. J. Proteome Res. 10, 4076–4087. doi: 10.1021/pr200263m
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. doi: 10.1093/nar/gkw937
Taipale, M., Jarosz, D. F., and Lindquist, S. (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. doi: 10.1038/nrm2918
Taj, G., Agarwal, P., Grant, M., and Kumar, A. (2010). MAPK machinery in plants. Plant Signal. Behav. 5, 1370–1378. doi: 10.4161/psb.5.11.13020
Tomé, F., Nägele, T., Adamo, M., Garg, A., Marco-Llorca, C., Nukarinen, E., et al. (2014). The low energy signaling network. Front. Plant Sci. 5:353. doi: 10.3389/fpls.2014.00353
Trigg, S. A., Garza, R. M., MacWilliams, A., Nery, J. R., Bartlett, A., Castanon, R., et al. (2017). CrY2H-seq: a massively multiplexed assay for deep-coverage interactome mapping. Nat. Methods 14, 819–825. doi: 10.1038/nmeth.4343
Tsai, C. J., Ma, B., and Nussinov, R. (2009). Protein-protein interaction networks: how can a hub protein bind so many different partners? Trends Biochem. Sci. 34, 594–600. doi: 10.1016/j.tibs.2009.07.007
Uversky, V. N. (2013). A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 22, 693–724. doi: 10.1002/pro.2261
Uversky, V. N. (2016). Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J. Biol. Chem. 291, 6681–6688. doi: 10.1074/jbc.R115.685859
Vallabhajosyula, R. R., Chakravarti, D., Lutfeali, S., Ray, A., and Raval, A. (2009). Identifying Hubs in protein interaction networks. PLoS ONE 4:e5344. doi: 10.1371/journal.pone.0005344
Van Leene, J., Boruc, J., De Jaeger, G., Russinova, E., and De Veylder, L. (2011). A kaleidoscopic view of the Arabidopsis core cell cycle interactome. Trends Plant Sci. 16, 141–150. doi: 10.1016/j.tplants.2010.12.004
Van Leene, J., Eeckhout, D., Cannoot, B., De Winne, N., Persiau, G., Van De Slijke, E., et al. (2015). An improved toolbox to unravel the plant cellular machinery by tandem affinity purification of Arabidopsis protein complexes. Nat. Protoc. 10, 169–187. doi: 10.1038/nprot.2014.199
Van Leene, J., Hollunder, J., Eeckhout, D., Persiau, G., Van De Slijke, E., Stals, H., et al. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol. Syst. Biol. 6:397. doi: 10.1038/msb.2010.53
Vella, D., Zoppis, I., Mauri, G., Mauri, P., and Di Silvestre, D. (2017). From protein-protein interactions to protein co-expression networks: a new perspective to evaluate large-scale proteomic data. EURASIP J. Bioinform. Syst. Biol. 2017:6. doi: 10.1186/s13637-017-0059-z
Verma, V., Ravindran, P., Kumar, P. P., Kudla, J., Batistic, O., Hashimoto, K., et al. (2016). Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16:86. doi: 10.1186/s12870-016-0771-y
Vernoux, T., Brunoud, G., Farcot, E., Morin, V., Van Den Daele, H., Legrand, J., et al. (2011). The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol. Syst. Biol. 7, 508–508. doi: 10.1038/msb.2011.39
Vincent, M., and Schnell, S. (2016). A collection of intrinsic disorder characterizations from eukaryotic proteomes. Sci. Data 3:160045. doi: 10.1038/sdata.2016.45
Wager, A., and Browse, J. (2012). Social network: JAZ protein interactions expand our knowledge of jasmonate signaling. Front. Plant Sci. 3:41. doi: 10.3389/fpls.2012.00041
Wang, H., and Wang, H. (2015). The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant 8, 677–688. doi: 10.1016/j.molp.2015.01.008
Wang, H., Seo, J. K., Gao, S., Cui, X., and Jin, H. (2017). Silencing of AtRAP, a target gene of a bacteria-induced small RNA, triggers antibacterial defense responses through activation of LSU2 and down-regulation of GLK1. New Phytol. 215, 1144–1155. doi: 10.1111/nph.14654
Wang, W., Vinocur, B., Shoseyov, O., and Altman, A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9, 244–252. doi: 10.1016/j.tplants.2004.03.006
Weβling, R., Epple, P., Altmann, S., He, Y., Yang, L., Henz, S. R., et al. (2014). Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 16, 364–375. doi: 10.1016/j.chom.2014.08.004
Windram, O., Penfold, C. A., and Denby, K. J. (2014). Network modeling to understand plant immunity. Annu. Rev. Phytopathol. 52, 93–111. doi: 10.1146/annurev-phyto-102313-050103
Xing, S., Wallmeroth, N., Berendzen, K. W., and Grefen, C. (2016). Techniques for the analysis of protein-protein interactions in vivo. Plant Physiol. 171, 727–758. doi: 10.1104/pp.16.00470
Yachie, N., Petsalaki, E., Mellor, J. C., Weile, J., Jacob, Y., Verby, M., et al. (2016). Pooled-matrix protein interaction screens using Barcode Fusion Genetics. Mol. Syst. Biol. 12, 863–863. doi: 10.15252/msb.20156660
Yang, B., Ruan, R., Cantu, D., Wang, X., Ji, W., Ronald, P. C., et al. (2013). A comparative approach expands the protein–protein interaction node of the immune receptor XA21 in wheat and rice. Genome 56, 315–326. doi: 10.1139/gen-2013-0032
Yang, J., Osman, K., Iqbal, M., Stekel, D. J., Luo, Z., Armstrong, S. J., et al. (2012). Inferring the Brassica rapa interactome using protein-protein interaction data from Arabidopsis thaliana. Front. Plant Sci. 3:297. doi: 10.3389/fpls.2012.00297
Yazaki, J., Galli, M., Kim, A. Y., Nito, K., Aleman, F., Chang, K. N., et al. (2016). Mapping transcription factor interactome networks using HaloTag protein arrays. Proc. Natl. Acad. Sci. U.S.A. 113, E4238–E4247. doi: 10.1073/pnas.1603229113
Yu, H., Braun, P., Yildirim, M. A., Lemmens, I., Venkatesan, K., Sahalie, J., et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110. doi: 10.1126/science.1158684
Yu, H., Luscombe, N. M., Lu, H. X., Zhu, X., Xia, Y., Han, J. D. J., et al. (2004). Annotation transfer between genomes: Protein-protein interrologs and protein-DNA regulogs. Genome Res. 14, 1107–1118. doi: 10.1101/gr.1774904
Yue, J., Xu, W., Ban, R., Huang, S., Miao, M., Tang, X., et al. (2016). PTIR: Predicted Tomato Interactome Resource. Sci. Rep. 6:25047. doi: 10.1038/srep25047
Zahiri, J., Bozorgmehr, J. H., and Masoudi-Nejad, A. (2013). Computational prediction of protein–protein interaction networks: algorithms and resources. Curr. Genomics 14, 397–414. doi: 10.2174/1389202911314060004
Zhang, F., Liu, S., Li, L., Zuo, K., Zhao, L., and Zhang, L. (2016). Genome-wide inference of protein interaction network and its application to the study of crosstalk in Arabidopsis abscisic acid signaling. Plant Physiol. 171, 1511–1522. doi: 10.1104/pp.16.00057
Zhang, Y., Gao, P., and Yuan, J. S. (2010). Plant protein-protein interaction network and interactome. Curr. Genomics 11, 40–46. doi: 10.2174/138920210790218016
Zhu, G., Wu, A., Xu, X.-J., Xiao, P.-P., Lu, L., Liu, J., et al. (2016). PPIM: a Protein-Protein Interaction Database for Maize. Plant Physiol. 170, 618–626. doi: 10.1104/pp.15.01821
Keywords: hub proteins, protein-protein interaction networks, plant interactome, plant stress response, hub protein identification
Citation: Vandereyken K, Van Leene J, De Coninck B and Cammue BPA (2018) Hub Protein Controversy: Taking a Closer Look at Plant Stress Response Hubs. Front. Plant Sci. 9:694. doi: 10.3389/fpls.2018.00694
Received: 19 December 2017; Accepted: 07 May 2018;
Published: 05 June 2018.
Edited by:
Katja Baerenfaller, Swiss Institute of Allergy and Asthma Research (SIAF), SwitzerlandReviewed by:
Pascal Falter-Braun, Helmholtz Zentrum München—Deutsches Forschungszentrum für Gesundheit und Umwelt, GermanyTong Zhang, Pacific Northwest National Laboratory (DOE), United States
Copyright © 2018 Vandereyken, Van Leene, De Coninck and Cammue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno P. A. Cammue, YnJ1bm8uY2FtbXVlQGt1bGV1dmVuLnZpYi5iZQ==
†These authors have contributed equally to this work.