 Xiaofei Yang
Xiaofei Yang Minglei Yang
Minglei Yang Hongjing Deng
Hongjing Deng Yiliang Ding
Yiliang Ding- 1Department of Cell and Developmental Biology, John Innes Centre, Norwich, United Kingdom
- 2State Key Laboratory of Plant Genomics and National Center for Plant Gene Research, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, China
- 3College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
The dynamic structure of RNA plays a central role in post-transcriptional regulation of gene expression such as RNA maturation, degradation, and translation. With the rise of next-generation sequencing, the study of RNA structure has been transformed from in vitro low-throughput RNA structure probing methods to in vivo high-throughput RNA structure profiling. The development of these methods enables incremental studies on the function of RNA structure to be performed, revealing new insights of novel regulatory mechanisms of RNA structure in plants. Genome-wide scale RNA structure profiling allows us to investigate general RNA structural features over 10s of 1000s of mRNAs and to compare RNA structuromes between plant species. Here, we provide a comprehensive and up-to-date overview of: (i) RNA structure probing methods; (ii) the biological functions of RNA structure; (iii) genome-wide RNA structural features corresponding to their regulatory mechanisms; and (iv) RNA structurome evolution in plants.
RNA secondary structure plays many essential roles in RNA synthesis, metabolism, and regulatory pathways (Bevilacqua et al., 2016; Vandivier et al., 2016). Previous efforts to determine RNA structure depended on classical and time-consuming techniques, such as nuclear magnetic resonance spectroscopy (NMR), X-ray crystallography, and cryo-electron microscopy (Table 1) (Lengyel et al., 2014). However, these methods yielded data limited to a few key RNAs with comparatively short length (less than 200 nt) and high abundance (∼1 μmol).
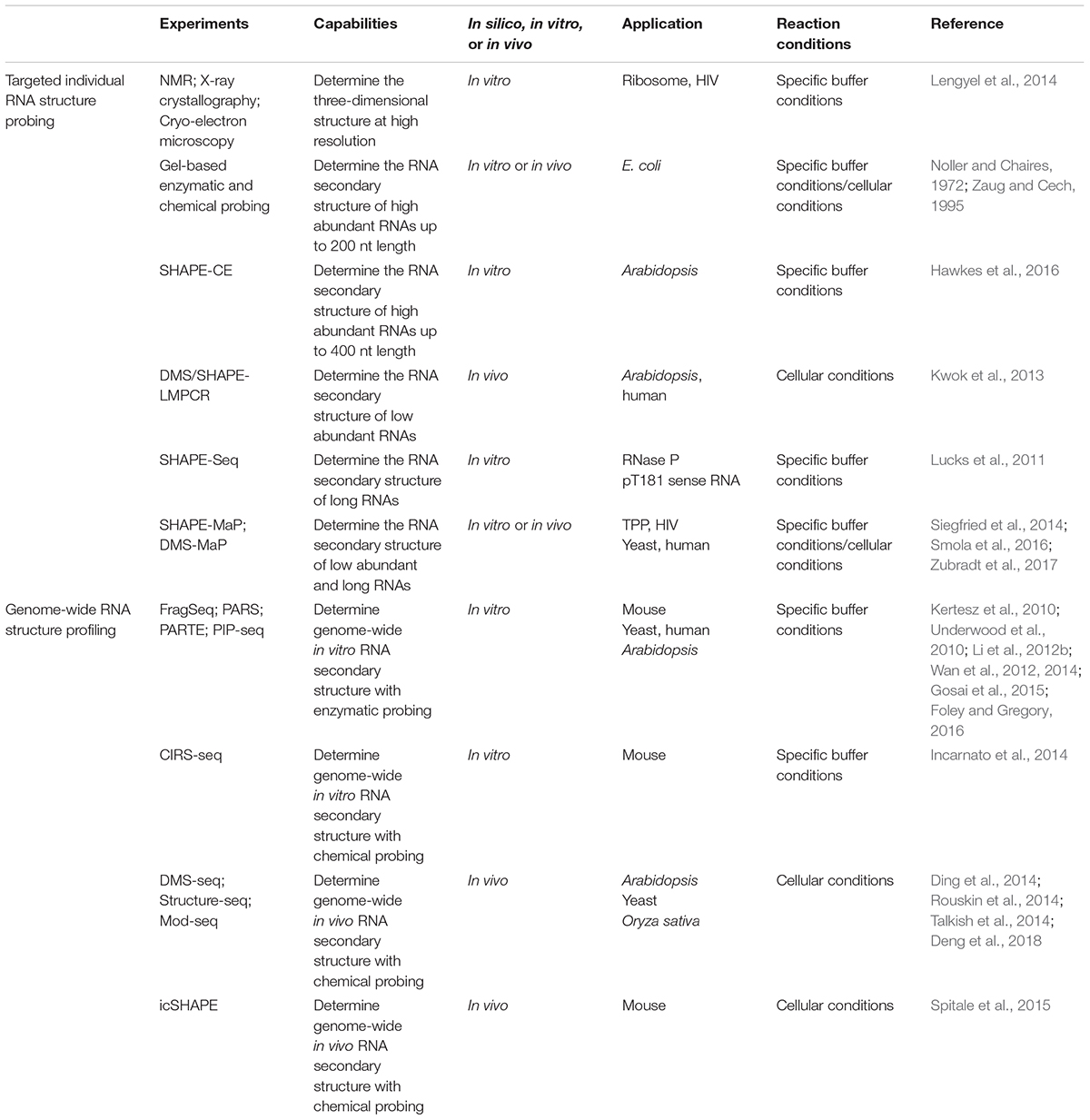
TABLE 1. RNA structure probing methods for both individual RNAs and at the genome-wide scale.
More recently, enzymatic and chemical structure probing methods have been developed to routinely and efficiently obtain structural information of individual RNAs. Ribonucleases (RNase) cleave either single-stranded (ss) RNA regions or double-stranded (ds) RNA regions to indicate RNA base-pairing status. The most commonly used enzymatic probing reagents include RNase V1 (for dsRNA), RNase S1 (for ssRNA), RNase A (for C/U in ssRNA), and RNase T1 (for G in ssRNA) (Knapp, 1989). The RNase-based RNA structure probing method has been used extensively in studying RNA structure with less toxicity, but with the limitation of cell permeability (Kwok, 2016). For chemical probing, two main types of chemical reagent can be used. One modifies the Watson-Crick base-pairing face on the nucleobase, as a direct measure of single-strandedness. Dimethyl sulfate (DMS) is one of the most commonly used nucleobase probing reagents as it easily penetrates the cell, a pre-requisite for in vivo chemical probing (Zaug and Cech, 1995). Another example is 1-cyclohexyl-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate (CMCT), which targets the unpaired N3 position of uracil and the unpaired N1 position of guanine (Incarnato et al., 2014); while 3-ethoxy-1,1-dihydroxy-2-butanone (kethoxal) attacks the unpaired N1 and unpaired exocyclic amine positions of guanine (Noller and Chaires, 1972). Among these reagents, DMS is predominantly used to probe RNA structures in different organisms (Ding et al., 2014; Rouskin et al., 2014; Talkish et al., 2014; Deng et al., 2018). The other type of chemical reagent modifies the ribose, by selective 2′-hydroxyl acylation and which can be analyzed by primer extension (SHAPE) (Mortimer and Weeks, 2007; Spitale et al., 2013). A particular advantage of SHAPE is that it generates structural information for all four nucleotides at the same time.
Polyacrylamide gel electrophoresis (PAGE) assays were traditionally used to measure the modified pattern of both enzymatic and chemical reactions (Noller and Chaires, 1972; Knapp, 1989; Zaug and Cech, 1995). However, these gel-based assays were limited to highly abundant and short (less than 200 nt) RNAs. The application of capillary electrophoresis (CE) improved the detection limits of both the length (up to 400 nt) and the abundance of RNA (Table 1) (Watts et al., 2009). A recent application of CE on Arabidopsis thaliana long non-coding RNA (lncRNA), COOLAIR, revealed the remarkable complexity of RNA structure up to 750 nt (Hawkes et al., 2016). A further improved method on probing sensitivity, DMS/SHAPE-LMPCR, was developed in Arabidopsis thaliana (Table 1). This method achieved “attomole” sensitivity allowing RNA structure probing of low abundance RNAs in living cells (Kwok et al., 2013). By subsequently combining the action of DMS with next-generation sequencing high-depth RNA structural information of very long RNAs was achieved (Lucks et al., 2011; Smola et al., 2016). For instance, the structural information of over 18 kb lncRNA, Xist, was fulfilled in a single experiment (Smola et al., 2016). The development of these approaches has significantly improved the sensitivity and resolution for probing individual RNA structure both in vitro and in vivo. The capability for single nucleotide-resolution quantitative measurements on any RNA down to 1 attomole and up to 18 kb enables efficient functional investigation of RNA structure in biological processes.
Genome-wide RNA structure profiling was initially achieved by coupling enzymatic probing with next-generation sequencing, PARS (parallel analysis of RNA structure) (Table 1). It was developed in yeast by measuring the catalytic activity of two enzymes, RNase V1 (for dsRNA) and S1 (for ssRNA) (Kertesz et al., 2010). This method was extended in Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, and Homo sapiens (Li et al., 2012a,b; Wan et al., 2014). An enhanced method, PIP-seq (protein interaction profiling sequencing), complements RNA–protein interaction information with in vitro RNA structure profiling (Table 1) (Foley et al., 2015; Gosai et al., 2015; Foley and Gregory, 2016). A further improvement on genome-wide scale RNA structure profiling extended to living cells and addressed native RNA folding status. By harnessing the cell permeability of DMS, the first genome-wide in vivo RNA structure profiling method, Structure-seq, was developed in Arabidopsis (Ding et al., 2014, 2015) in parallel with DMS-seq and Mod-seq in yeast (Rouskin et al., 2014; Talkish et al., 2014) (Table 1). Both methods reveal in vivo RNA structures are more single-stranded than in vitro and in silico computational predicted RNA structures. Use of the Structure-seq method was recently extended to rice (Deng et al., 2018). A follow-up genome-wide in vivo RNA structure profiling method, icSHAPE (in vivo click SHAPE), was developed in mouse by using the SHAPE chemical reagent with the power of four-nucleotide probing (Table 1) (Spitale et al., 2015). In addition to measuring reverse transcription stopping, chemical modification can also be determined by mutational profiling (Table 1) (Siegfried et al., 2014; Smola et al., 2016; Zubradt et al., 2017).
These powerful genome-wide methods can provide an accurate and quantitative in vivo RNA structure map over tens of thousands of RNA with single nucleotide-resolution. These technological advances create an unprecedented scale for the in-depth study of the global impact of RNA structure in gene regulation. For example, regulatory RNAs are able to act as a master regulator in gene expression. In general, these regulatory RNAs directly turn on or off gene expression by altering RNA secondary structure. A recent study of RNA structure characterization on a range of regulatory RNAs in Arabidopsis is illustrated below (Figure 1).
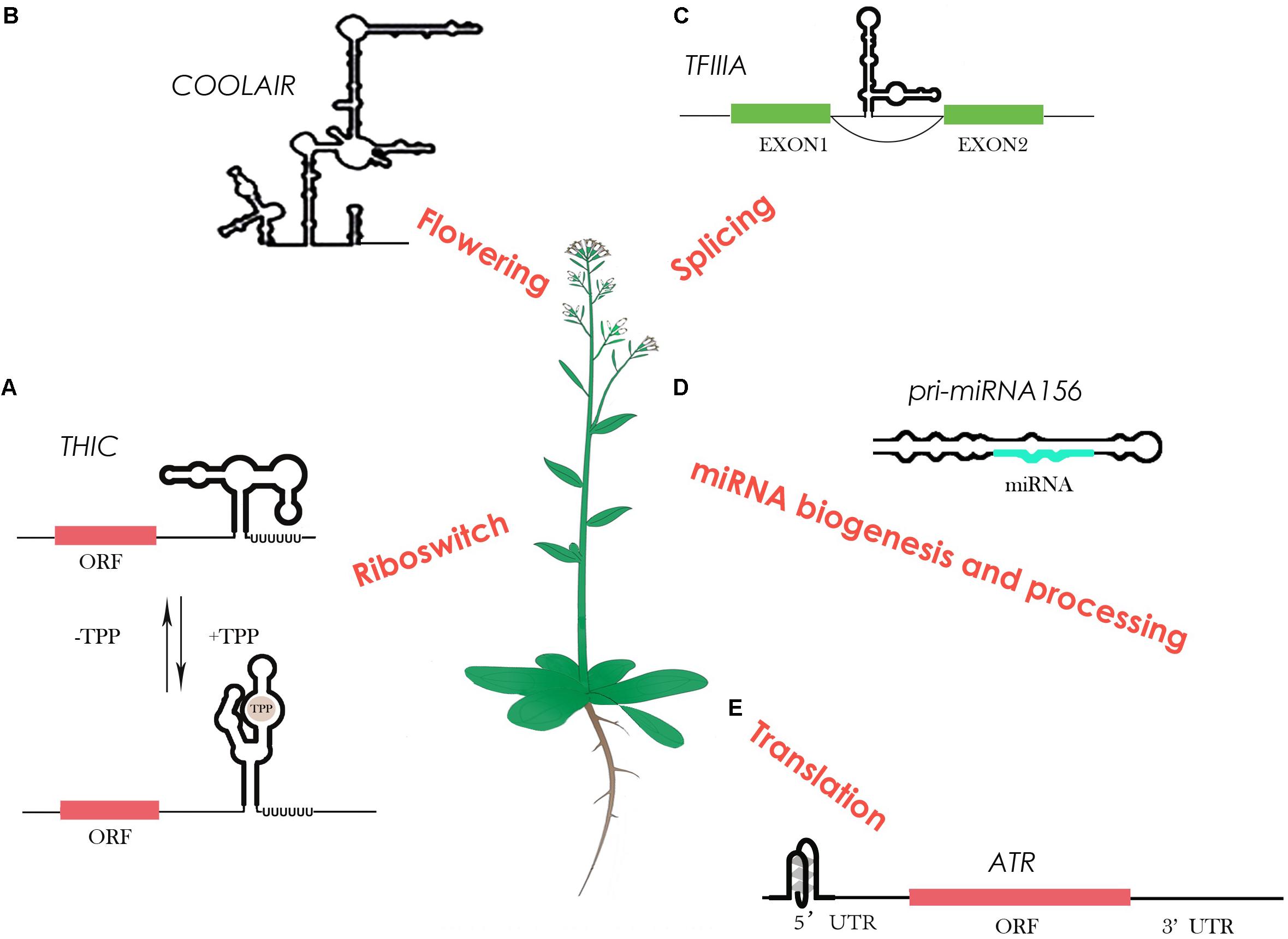
FIGURE 1. RNA structure characterization on regulatory RNAs in Arabidopsis. (A) The TPP (thiamin pyrophosphate) riboswitch in plants changes its own structure in response to different TPP concentrations, resulting in different 3′ end processing to control gene expression. (B) The highly-conserved plant lncRNA COOLAIR shows a highly complex structure that links to its biological function in flowering. (C) A 5S ribosomal RNA mimic regulates alternative splicing of transcription factor IIIA pre-mRNAs. (D) Several studies show that RNA structure determines miRNA biogenesis and processing. (E) An RNA G-quadruplex was reported to be able to regulate its own translation.
A riboswitch is a type of regulatory RNA that contains specific RNA structure segments, which can change conformation depending on specific ligand binding, e.g., metabolites. A well-studied example of a riboswitch is the vitamin B1 derivative thiamin pyrophosphate (TPP), which resides in the 3′ UTR region of the thiamin biosynthetic gene THIC (Wachter et al., 2007) (Figure 1A). With a low TPP concentration, the 3′ end processing of THIC mRNA results in a short 3′ UTR that permits high expression of the THIC gene. Conversely, with a high TPP concentration, TPP binds directly with the 3′ end of the RNA and induces a structural change that prevents splicing. This results in a long 3′ UTR inducing RNA degradation, subsequently reducing THIC gene expression (Wachter et al., 2007). Unlike riboswitches in bacteria that control translation through a structural change in the 5′ UTR, plants may have evolved a diverse and more complicated alternative 3′ end processing mechanism in order to cope with a large number of metabolites (Wachter et al., 2007).
Not only are some metabolites able to bind to specific RNA structures to regulate their synthesis pathways, but some are also able to regulate their own expression levels. A plant conserved pre-mRNA of transcription factor IIIA (TFIIIA) contains a 5S rRNA mimic structural element in one of its exons (Hammond et al., 2009) (Figure 1C). When ribosomal protein L5 binds to this 5S rRNA mimic, it triggers exon skipping in TFIIIA mRNA to control TFIIIA levels (Hammond et al., 2009). This ribosomal protein–mRNA interaction provides a new-found class of RNAs regulating alternative splicing to control the protein level.
Furthermore, specific RNA structural motifs such as G-quadruplexes (GQS) also play an important role in gene expression regulation. RNA GQSs are typically more stable in the presence of potassium or sodium. Tens of thousands of putative GQSs were identified in Arabidopsis and other plant species (Mullen et al., 2010). A recent study reported the first highly-conserved plant RNA GQS located in the 5′ UTR of ATAXIA TELANGIECTASIA-MUTATED AND RAD3-RELATED (ATR), inhibiting its translation when forming stable GQS structures (Kwok et al., 2015) (Figure 1E). Interestingly, potassium concentrations in plant cells can dramatically increase under drought stress (Mullen et al., 2010). Thus, GQS structural motifs in plants may specifically act as a regulator in response to abiotic stress, such as drought and salinity.
Long non-coding RNAs have also been shown as important regulatory RNAs involved in various biological processes. The study of lncRNA structures has been limited in the past due to their long length and low abundance. Advances in probing methods has enabled the highly-conserved plant lncRNA COOLAIR structure to be determined by chemical profiling with CE (Hawkes et al., 2016). COOLAIR is a key regulator of a major plant developmental gene FLC (FLOWERING LOCUS C), in response to vernalisation. The distal COOLAIR isoform in Arabidopsis (Figure 1) is highly-structured with numerous secondary structural motifs, an intricate multi-way junction, and two unusual asymmetric 5′ internal loops (Hawkes et al., 2016) (Figure 1B). Interestingly, a single nucleotide polymorphism (SNP) in the natural variation accession, Var2-6, is able to change the structure to affect the RNA stability, resulting in a late-flowering phenotype in Var2-6 (Hawkes et al., 2016). RNA secondary structure determination has progressed our understanding of the structure–function relationship of lncRNAs for the first time in plants.
The other well-known regulatory RNAs, miRNAs, also heavily rely on RNA structure for their regulatory functions (Herr and Baulcombe, 2004). The double-stranded region of miRNA precursors (pri-miRNAs) are recognized and processed by Dicer protein, an RNase III-like enzyme (Herr and Baulcombe, 2004). Previous studies in plants on both individual miRNA precursors and genome-wide assessment of pri-miRNA processing products confirmed that different structure determinants within pri-miRNAs compete for the processing machinery (Song et al., 2010; Bologna et al., 2013) (Figure 1D). A recent RNA structure characterization study by NMR shows the upper stem of a double-stranded region of pri-miR156 is important for Dicer processing at different temperatures, that substantiates the structure-determined Dicer processing feature (Kim et al., 2017). After Dicer processing, an Argonaute (AGO) protein will recognize the processed duplex miRNA to target mRNA containing complementary sequence for either RNA cleavage or translational inhibition (Valencia-Sanchez et al., 2006). Genome-wide in vitro RNA structure profiling in Arabidopsis revealed a less structured pattern in miRNA target sites that indicates the relationship between miRNA target efficiency and the single-stranded structural feature (Li et al., 2012b).
Apart from these studies on regulatory RNAs, recent genome-wide research also reveals the general role of mRNA structure in a variety of post-transcriptional regulations such as RNA maturation, RNA stability, RNA location and translation.
Alternative splicing is an important process in RNA maturation. More than 40% of Arabidopsis genes possess alternative spliced isoforms (Filichkin et al., 2010). The first in vivo RNA structure profiling in Arabidopsis revealed a significantly less structural pattern in the 40 nt region upstream of the 5′ splice site for unspliced events (including exon skipping and intron retention) (Ding et al., 2014). PIP-seq further revealed that this kind of structural pattern results in more RNA-protein interactions in Arabidopsis nuclei (Gosai et al., 2015; Foley and Gregory, 2016). Interestingly, PIP-seq also found the robust structure at the 3′ splice site is responsible for more protein interactions (Gosai et al., 2015; Foley and Gregory, 2016). Thus, these RNA structural features indicate an important role of RNA structure in regulating alternative splicing.
Another RNA maturation process is alternative polyadenylation (APA) that is found in over 60% of mRNAs in Arabidopsis (Loke et al., 2005). In vivo RNA structure profiling shows a strong structural pattern in the U- and A-rich upstream region of the cleavage site as well as a single-stranded region at the cleavage site (Ding et al., 2014). These patterns may correlate with the recognition of endonucleases for regulating APA. Further study using PIP-seq shows more protein bound up- and downstream of the APA cleavage site as compared to constitutive polyadenylation events (Gosai et al., 2015). However, APA sites do not exhibit altered in vitro RNA secondary structure compared to constitutive sites (Gosai et al., 2015). This suggests there may be different effects of RNA structure on both protein binding and cleavage activity, that warrant closer investigation.
In addition to RNA maturation, the relationship between RNA structure and RNA degradation has also been uncovered by in vitro RNA structurome analysis in Arabidopsis. Unlike yeast, highly-structured mRNAs are more likely to be degraded in Arabidopsis, probably via specific siRNA processing (Li et al., 2012b).
An interesting study on RNA mobility in plants shows that a stem-bulge-stem-loop tRNA-derived structural motif is sufficient to mediate mRNA transport. A large number of mRNAs containing this motif can be moved through graft junction (Zhang et al., 2016). Thus, RNA structure might also affect intercellular communication across plants.
Another major impact of RNA structure is its regulatory role in translation. Both in vitro and in vivo RNA structure profiling show a single-stranded region upstream of the start codon that might facilitate ribosome initiation (Li et al., 2012b; Ding et al., 2014). Moreover, a triplet periodic trend is observed in the CDS region but not in UTRs. These structure patterns are obvious in mRNAs with high translation efficiency and are absent in those with low translation efficiency (Ding et al., 2014). This implies that ribosomes may recognize RNA secondary structure as an additional layer of information alongside sequence content.
Additionally, RNA structure is also strongly associated with RNA methylation sites and RNA binding protein (RBP) sites. For example, N6-methylation of adenosine alters the stability of the A⋅U pair (Harcourt et al., 2017). Cellular RNAs show a decrease in base pairing around sites of m6A when they were methylated (Harcourt et al., 2017). Recent genome-wide studies indicate that the N6-methyladenosine (m6A) prefers single-stranded conformations rather than double-stranded structures (Zhao et al., 2017). A genome-wide study in Arabidopsis shows an enrichment of m6A around the start codon, stop codon and 3′ UTR region (Luo et al., 2014). Interestingly, this enrichment region of m6A is well-correlated with the single-stranded region identified in RNA structure profiling (Luo et al., 2014; Zhao et al., 2017). A study of the RNA structurome in rice also confirmed that higher m6A modification sites tend to have less RNA structure (Deng et al., 2018). This indicates that m6A association may alter RNA structure to more single-strandedness to facilitate gene regulation.
Another key player in post-transcriptional regulations is RBP. Unlike DNA binding protein, RBP associates not only with the primary sequence motifs, but also RNA structural patterns. A recent study combining genome-wide RBP profiling and RNA secondary structure profiling shows that RBP binding sites tend to be more single-stranded (Gosai et al., 2015). Interestingly, a nuclear PIP-seq study confirms that both RNA secondary structure and RBP binding sites show quite different patterns between hair and non-hair cells in plants (Foley et al., 2017). This suggests that cell-type-specific RNA structure and RBP binding may be a new regulatory mechanism during plant development.
From an evolutionary perspective, the conservation and diversity of RNA structurome between species remains poorly understood. A recent study compared, for the first time, the conservation and divergence of in vivo RNA structurome between plant species, to assess the evolutionary adaptation of RNA structure (Deng et al., 2018). This study found that in vivo RNA secondary structure conservation does not correlate with sequence conservation between rice and Arabidopsis. The conservation and divergence in both sequence and RNA secondary structure are highly relevant with specific biological processes (Deng et al., 2018). This indicates evolutionary selection not only modifies sequence, but also alters RNA structure to regulate gene expression. This in turn suggests that RNA secondary structure may serve a different layer of selection to sequence in plants.
Recent methodology advances have overcome previous limitations of both low-throughput and in vitro conditions for studying RNA secondary structure. These new methods, with single-nucleotide resolution, genome-wide scale and high sensitivity, significantly accelerate the study of in vivo RNA structure and associated biological functions. Plants are more sensitive than animals to varying environmental conditions, such as changes in temperature, salinity, acidity, and heavy metal concentrations (Schleuning et al., 2016). These factors are able to affect RNA folding (Tan and Chen, 2011; Wan et al., 2012; Sun et al., 2017). By applying these new RNA structure analysis methods under a range of environmental conditions, we will be able to determine how RNA structure alters in response to these changes. By integrating RNA structure profiling with mutagenesis assays and phenotypic analysis, the relationship between RNA structure and biological function can be investigated in greater details. Extending analyses to other plant species provides scope for exploring evolutionary selection at the RNA structure level. It is notable that this new era of studying RNA secondary structure provides unprecedented opportunity for discovering novel regulatory mechanisms of gene expression in plants.
Author Contributions
XY, MY, HD, and YD wrote the manuscript. MY produced the figure. XY, HD, and YD made the corrections. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This study was supported by the Biotechnology and Biological Sciences Research Council (Grant No. BB/L025000/1 to XY, HD, and YD) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant No. 680324 to MY and YD).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to authors whose work has not been cited here, because of the limitations associated with the review format.
References
Bevilacqua, P. C., Ritchey, L. E., Su, Z., and Assmann, S. M. (2016). Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 50, 235–266. doi: 10.1146/annurev-genet-120215-035034
Bologna, N. G., Schapire, A. L., Zhai, J., Chorostecki, U., Boisbouvier, J., Meyers, B. C., et al. (2013). Multiple RNA recognition patterns during microRNA biogenesis in plants. Genome Res. 23, 1675–1689. doi: 10.1101/gr.153387.112
Deng, H., Cheema, J., Zhang, H., Woolfenden, H., Norris, M., Liu, Z., et al. (2018). Rice in vivo RNA structurome reveals RNA secondary structure conservation and divergence in plants. Mol. Plant 11, 607–622. doi: 10.1016/j.molp.2018.01.008
Ding, Y., Kwok, C. K., Tang, Y., Bevilacqua, P. C., and Assmann, S. M. (2015). Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nat. Protoc. 10, 1050–1066. doi: 10.1038/nprot.2015.064
Ding, Y., Tang, Y., Kwok, C. K., Zhang, Y., Bevilacqua, P. C., and Assmann, S. M. (2014). In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700. doi: 10.1038/nature12756
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58. doi: 10.1101/gr.093302.109
Foley, S. W., Gosai, S. J., Wang, D., Selamoglu, N., Sollitti, A. C., Koster, T., et al. (2017). A global view of RNA-protein interactions identifies post-transcriptional regulators of root hair cell fate. Dev. Cell 41, 204.e5–220.e5. doi: 10.1016/j.devcel.2017.03.018
Foley, S. W., and Gregory, B. D. (2016). Protein interaction profile sequencing (PIP-seq). Curr. Protoc. Mol. Biol. 116, 27.5.1–27.5.15. doi: 10.1002/cpmb.21
Foley, S. W., Vandivier, L. E., Kuksa, P. P., and Gregory, B. D. (2015). Transcriptome-wide measurement of plant RNA secondary structure. Curr. Opin. Plant Biol. 27, 36–43. doi: 10.1016/j.pbi.2015.05.021
Gosai, S. J., Foley, S. W., Wang, D., Silverman, I. M., Selamoglu, N., Nelson, A. D., et al. (2015). Global analysis of the RNA-protein interaction and RNA secondary structure landscapes of the Arabidopsis nucleus. Mol. Cell 57, 376–388. doi: 10.1016/j.molcel.2014.12.004
Hammond, M. C., Wachter, A., and Breaker, R. R. (2009). A plant 5S ribosomal RNA mimic regulates alternative splicing of transcription factor IIIA pre-mRNAs. Nat. Struct. Mol. Biol. 16, 541–549. doi: 10.1038/nsmb.1588
Harcourt, E. M., Kietrys, A. M., and Kool, E. T. (2017). Chemical and structural effects of base modifications in messenger RNA. Nature 541, 339–346. doi: 10.1038/nature21351
Hawkes, E. J., Hennelly, S. P., Novikova, I. V., Irwin, J. A., Dean, C., and Sanbonmatsu, K. Y. (2016). COOLAIR antisense RNAs form evolutionarily conserved elaborate secondary structures. Cell Rep. 16, 3087–3096. doi: 10.1016/j.celrep.2016.08.045
Herr, A. J., and Baulcombe, D. C. (2004). RNA silencing pathways in plants. Cold Spring Harb. Symp. Quant. Biol. 69, 363–370. doi: 10.1101/sqb.2004.69.363
Incarnato, D., Neri, F., Anselmi, F., and Oliviero, S. (2014). Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol. 15:491. doi: 10.1186/s13059-014-0491-2
Kertesz, M., Wan, Y., Mazor, E., Rinn, J. L., Nutter, R. C., Chang, H. Y., et al. (2010). Genome-wide measurement of RNA secondary structure in yeast. Nature 467, 103–107. doi: 10.1038/nature09322
Kim, W., Kim, H. E., Lee, A. R., Jun, A. R., Jung, M. G., Ahn, J. H., et al. (2017). Base-pair opening dynamics of primary miR156a using NMR elucidates structural determinants important for its processing level and leaf number phenotype in Arabidopsis. Nucleic Acids Res. 45, 875–885. doi: 10.1093/nar/gkw747
Knapp, G. (1989). Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 180, 192–212. doi: 10.1016/0076-6879(89)80102-8
Kwok, C. K. (2016). Dawn of the in vivo RNA structurome and interactome. Biochem. Soc. Trans. 44, 1395–1410. doi: 10.1042/BST20160075
Kwok, C. K., Ding, Y., Shahid, S., Assmann, S. M., and Bevilacqua, P. C. (2015). A stable RNA G-quadruplex within the 5′-UTR of Arabidopsis thaliana ATR mRNA inhibits translation. Biochem. J. 467, 91–102. doi: 10.1042/BJ20141063
Kwok, C. K., Ding, Y., Tang, Y., Assmann, S. M., and Bevilacqua, P. C. (2013). Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun. 4:2971. doi: 10.1038/ncomms3971
Lengyel, J., Hnath, E., Storms, M., and Wohlfarth, T. (2014). Towards an integrative structural biology approach: combining Cryo-TEM, X-ray crystallography, and NMR. J. Struct. Funct. Genomics 15, 117–124. doi: 10.1007/s10969-014-9179-9
Li, F., Zheng, Q., Ryvkin, P., Dragomir, I., Desai, Y., Aiyer, S., et al. (2012a). Global analysis of RNA secondary structure in two metazoans. Cell Rep. 1, 69–82. doi: 10.1016/j.celrep.2011.10.002
Li, F., Zheng, Q., Vandivier, L. E., Willmann, M. R., Chen, Y., and Gregory, B. D. (2012b). Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell 24, 4346–4359. doi: 10.1105/tpc.112.104232
Loke, J. C., Stahlberg, E. A., Strenski, D. G., Haas, B. J., Wood, P. C., and Li, Q. Q. (2005). Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiol. 138, 1457–1468. doi: 10.1104/pp.105.060541
Lucks, J. B., Mortimer, S. A., Trapnell, C., Luo, S., Aviran, S., Schroth, G. P., et al. (2011). Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc. Natl. Acad. Sci. U.S.A. 108, 11063–11068. doi: 10.1073/pnas.1106501108
Luo, G. Z., MacQueen, A., Zheng, G., Duan, H., Dore, L. C., Lu, Z., et al. (2014). Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5:5630. doi: 10.1038/ncomms6630
Mortimer, S. A., and Weeks, K. M. (2007). A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 129, 4144–4145. doi: 10.1021/ja0704028
Mullen, M. A., Olson, K. J., Dallaire, P., Major, F., Assmann, S. M., and Bevilacqua, P. C. (2010). RNA G-Quadruplexes in the model plant species Arabidopsis thaliana: prevalence and possible functional roles. Nucleic Acids Res. 38, 8149–8163. doi: 10.1093/nar/gkq804
Noller, H. F., and Chaires, J. B. (1972). Functional modification of 16S ribosomal RNA by kethoxal. Proc. Natl. Acad. Sci. U.S.A. 69, 3115–3118. doi: 10.1073/pnas.69.11.3115
Rouskin, S., Zubradt, M., Washietl, S., Kellis, M., and Weissman, J. S. (2014). Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705. doi: 10.1038/nature12894
Schleuning, M., Frund, J., Schweiger, O., Welk, E., Albrecht, J., Albrecht, M., et al. (2016). Ecological networks are more sensitive to plant than to animal extinction under climate change. Nat. Commun. 7:13965. doi: 10.1038/ncomms13965
Siegfried, N. A., Busan, S., Rice, G. M., Nelson, J. A., and Weeks, K. M. (2014). RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965. doi: 10.1038/nmeth.3029
Smola, M. J., Christy, T. W., Inoue, K., Nicholson, C. O., Friedersdorf, M., Keene, J. D., et al. (2016). SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. U.S.A. 113, 10322–10327. doi: 10.1073/pnas.1600008113
Song, L., Axtell, M. J., and Fedoroff, N. V. (2010). RNA secondary structural determinants of miRNA precursor processing in Arabidopsis. Curr. Biol. 20, 37–41. doi: 10.1016/j.cub.2009.10.076
Spitale, R. C., Crisalli, P., Flynn, R. A., Torre, E. A., Kool, E. T., and Chang, H. Y. (2013). RNA SHAPE analysis in living cells. Nat. Chem. Biol. 9, 18–20. doi: 10.1038/nchembio.1131
Spitale, R. C., Flynn, R. A., Zhang, Q. C., Crisalli, P., Lee, B., Jung, J. W., et al. (2015). Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490. doi: 10.1038/nature14263
Sun, L. Z., Zhang, D., and Chen, S. J. (2017). Theory and modeling of RNA structure and interactions with metal ions and small molecules. Annu. Rev. Biophys. 46, 227–246. doi: 10.1146/annurev-biophys-070816-033920
Talkish, J., May, G., Lin, Y., Woolford, J. L. Jr., and McManus, C. J. (2014). Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA 20, 713–720. doi: 10.1261/rna.042218.113
Tan, Z. J., and Chen, S. J. (2011). Salt contribution to RNA tertiary structure folding stability. Biophys. J. 101, 176–187. doi: 10.1016/j.bpj.2011.05.050
Underwood, J. G., Uzilov, A. V., Katzman, S., Onodera, C. S., Mainzer, J. E., Mathews, D. H., et al. (2010). FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat. Methods 7, 995–1001. doi: 10.1038/nmeth.1529
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., and Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515–524. doi: 10.1101/gad.1399806
Vandivier, L. E., Anderson, S. J., Foley, S. W., and Gregory, B. D. (2016). The conservation and function of RNA secondary structure in plants. Annu. Rev. Plant Biol. 67, 463–488. doi: 10.1146/annurev-arplant-043015-111754
Wachter, A., Tunc-Ozdemir, M., Grove, B. C., Green, P. J., Shintani, D. K., and Breaker, R. R. (2007). Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell 19, 3437–3450. doi: 10.1105/tpc.107.053645
Wan, Y., Qu, K., Ouyang, Z., Kertesz, M., Li, J., Tibshirani, R., et al. (2012). Genome-wide measurement of RNA folding energies. Mol. Cell 48, 169–181. doi: 10.1016/j.molcel.2012.08.008
Wan, Y., Qu, K., Zhang, Q. C., Flynn, R. A., Manor, O., Ouyang, Z., et al. (2014). Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505, 706–709. doi: 10.1038/nature12946
Watts, J. M., Dang, K. K., Gorelick, R. J., Leonard, C. W., Bess, J. W. Jr., Swanstrom, R., et al. (2009). Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716. doi: 10.1038/nature08237
Zaug, A. J., and Cech, T. R. (1995). Analysis of the structure of Tetrahymena nuclear RNAs in vivo: telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. RNA 1, 363–374.
Zhang, W., Thieme, C. J., Kollwig, G., Apelt, F., Yang, L., Winter, N., et al. (2016). tRNA-related sequences trigger systemic mRNA transport in plants. Plant Cell 28, 1237–1249. doi: 10.1105/tpc.15.01056
Zhao, B. S., Roundtree, I. A., and He, C. (2017). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42. doi: 10.1038/nrm.2016.132
Keywords: RNA structurome, gene regulation, regulatory RNAs, RNA structure and function, plant RNA biology
Citation: Yang X, Yang M, Deng H and Ding Y (2018) New Era of Studying RNA Secondary Structure and Its Influence on Gene Regulation in Plants. Front. Plant Sci. 9:671. doi: 10.3389/fpls.2018.00671
Received: 28 February 2018; Accepted: 02 May 2018;
Published: 22 May 2018.
Edited by:
Dora Szakonyi, Instituto Gulbenkian de Ciência (IGC), PortugalReviewed by:
Cuncong Zhong, University of Kansas, United StatesCarmen Hernandez, Instituto de Biología Molecular y Celular de Plantas (IBMCP), Spain
Aleksandar Spasic, University of Rochester, United States
Copyright © 2018 Yang, Yang, Deng and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiliang Ding, eWlsaWFuZy5kaW5nQGppYy5hYy51aw==
†These authors have contributed equally to this work.