
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Plant Sci. , 15 May 2018
Sec. Technical Advances in Plant Science
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.00639
 Yuchen Long1†
Yuchen Long1† Yvonne Stahl2
Yvonne Stahl2 Stefanie Weidtkamp-Peters3Wouter Smet1Yujuan Du1Theodorus W. J. Gadella Jr4Joachim Goedhart4Ben Scheres1
Stefanie Weidtkamp-Peters3Wouter Smet1Yujuan Du1Theodorus W. J. Gadella Jr4Joachim Goedhart4Ben Scheres1 Ikram Blilou1,5*
Ikram Blilou1,5*Protein complex formation has been extensively studied using Förster resonance energy transfer (FRET) measured by Fluorescence Lifetime Imaging Microscopy (FLIM). However, implementing this technology to detect protein interactions in living multicellular organism at single-cell resolution and under native condition is still difficult to achieve. Here we describe the optimization of the labeling conditions to detect FRET-FLIM in living plants. This study exemplifies optimization procedure involving the identification of the optimal position for the labels either at the N or C terminal region and the selection of the bright and suitable, fluorescent proteins as donor and acceptor labels for the FRET study. With an effective optimization strategy, we were able to detect the interaction between the stem cell regulators SHORT-ROOT and SCARECROW at endogenous expression levels in the root pole of living Arabidopsis embryos and developing lateral roots by FRET-FLIM. Using this approach we show that the spatial profile of interaction between two transcription factors can be highly modulated in reoccurring and structurally resembling organs, thus providing new information on the dynamic redistribution of nuclear protein complex configurations in different developmental stages. In principle, our optimization procedure for transcription factor complexes is applicable to any biological system.
In living organisms, many cellular functions are executed by protein complexes. Over the decades, the concept of “protein-protein interaction networks” has emerged: rather than working as monomeric entities, most cellular proteins are known to dynamically engage in binding events. To understand the dynamic nature of these protein complexes, it is crucial to correlate the in vivo spatiotemporal interactions between key proteins and their impact on different biological processes. This holds true especially in a multicellular context, where heterogeneity of protein complexes between cell populations can lead to different outcomes in distinct cells within an intact organism.
Protein interactions are frequently studied with biochemical methods. These methods can be arduous, especially for protein complexes of low abundance. Improvements of protein purification procedures and the increased sensitivity of mass spectrometers have dramatically enhanced protein complex detectability (Bensimon et al., 2012; Pardo and Choudhary, 2012; Young et al., 2012; Aryal et al., 2014; Jorge et al., 2016; Wendrich et al., 2017). In addition, automated methods have been developed to isolate specific cell populations, further allowing high throughput proteome-wide analysis of protein complexes in selected cellular environments (Bridgeman et al., 2010; Petricka et al., 2012). Despite these technical advances, biochemical methods remain challenging when dealing with dynamic interactions in transient protein complexes.
Alternatively, fluorescence-based microscopic techniques have been developed to study protein-protein interactions. Bimolecular fluorescence complementation (BiFC) assays are commonly employed to visualize protein interaction in living cells, where two non-fluorescent fragments of a fluorescent protein can form a bimolecular fluorescent complex upon interaction (Hu et al., 2002). Successful BiFC applications in intact living organisms have been reported (Zhang et al., 2004; Gohl et al., 2010; Hudry et al., 2011; Smaczniak et al., 2012). However, the irreversible formation of bimolecular complexes limits its use to follow dynamic protein interactions (Lalonde et al., 2008; Horstman et al., 2014; Xing et al., 2016). Conversely, other strategies such as employing Förster resonance energy transfer (FRET) can provide better means to visualize and quantify dynamic protein complexes in living cells (Weidtkamp-Peters and Stahl, 2017), with the spatial information presented as a microscopic lifetime image. FRET describes the phenomenon of energy transfer from an excited donor fluorophore to a non-excited acceptor chromophore in its direct vicinity through dipole-dipole coupling (Förster, 1948; Figure 1A). Since FRET only occurs when the two fluorophores are within a short radius (on the scale of several nanometers), direct protein-protein interaction can be detected by tagging candidate proteins with appropriate fluorophores, such as different green fluorescent protein (GFP) variants (Kremers and Goedhart, 2009). Upon interaction, FRET will lead to a decreased donor emission, relative to that measured in a non-FRET situation, and an elevated acceptor emission (Clegg, 2009). These changes in emission intensities can be used to reflect the level of protein interaction by directly monitoring donor-acceptor emission ratio changes or measuring donor emission recovery after acceptor photobleaching (Gu et al., 2004; Adjobo-Hermans et al., 2011). However, these emission level-based techniques are highly dependent on the concentrations and good signal-to-noise ratios of both donor and acceptor, which are often difficult to achieve for lowly expressed proteins at endogenous levels.
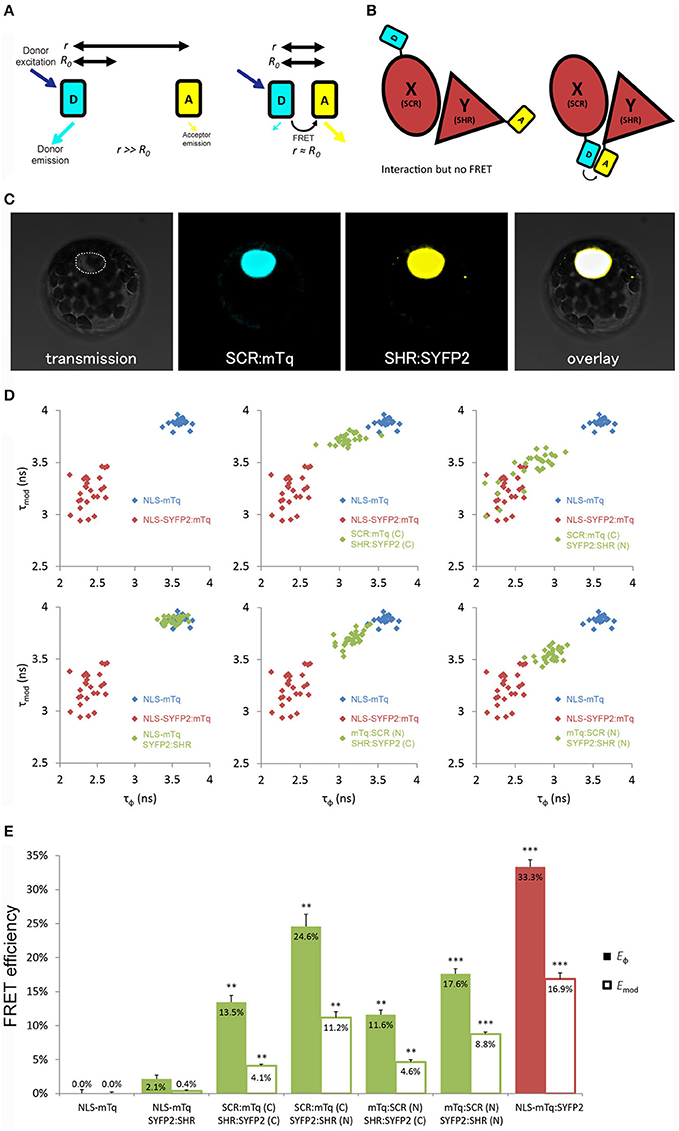
Figure 1. Optimization of tagging orientation for FRET-FLIM detection. (A) Illustration of FRET principle. D, donor fluorophore; A, acceptor fluorophore; r, distance between D and A; R0, Förster radius for D and A. (B) Illustration emphasizing the necessity to optimize tagging orientation for FRET. X and Y, two proteins of interest. Limited to no FRET might be observed when fluorophores are located at the distant ends of X and Y, yielding false negative result. (C) Arabidopsis mesophyll protoplast co-expressing SCR:mTq and SHR:SYFP2. Dotted line circles the nucleus. (D) Scatterplots showing distribution of phase lifetime τϕ against modulation lifetime τmod from protoplast measurements. Each FRET pair was plotted against the same positive and donor-only samples. n > 10 for each sample. (E) Bar chart showing FRET efficiency E derived from τϕ and τmod in (D), error bars represent standard errors within one set of experiment. * represent p-values, **, 10−20 < p < 10−2; ***p < 10−20, p-values calculated by Student's t-test compared to the donor-only samples.
FRET can also be quantified by measuring the fluorescence lifetime decrease of the donor molecules by fluorescence lifetime imaging microscopy (FLIM) (Gadella et al., 1993). Applications of FRET-FLIM have been mostly applied to analyze protein-protein interaction in living cells or as means to analyze biosensors (Tonaco et al., 2006; Crosby et al., 2011; Kardash et al., 2011; Bücherl et al., 2013; Stahl et al., 2013). Since accurate FRET-FLIM measurements are less dependent on emission intensity, it can be especially useful to detect interaction between proteins under native conditions without resorting to overexpression, which can alter cell states. Therefore, dynamic protein complex association at cellular resolution can be detected non-invasively using a microscopy-based approach (Bücherl et al., 2013). With these technical advantages, one would be able to follow and quantify such interactions in living multicellular organisms and determine their specificity in different cell types and developmental contexts.
Recently we have shown that FRET-FLIM can be used to study transcription factor associations in the model organism Arabidopsis thaliana (Long et al., 2017), particularly in the root tip which is ideal for live imaging with confocal microscopy due to its transparency and its simple, organized structure. We exploited the intensively-studied interaction between the two GRAS domain transcription factors SHORT-ROOT (SHR) and SCARECROW (SCR) (Di Laurenzio et al., 1996; Helariutta et al., 2000). SHR and SCR control the radial pattern of the Arabidopsis root through generating formative cell divisions in the stem cell called the cortex-endodermis initial (CEI) (Di Laurenzio et al., 1996; Helariutta et al., 2000). SHR is also required for endodermal specification (Helariutta et al., 2000; Long et al., 2015a,b; Moreno-Risueno et al., 2015). SHR transcript is produced in the vasculature and its protein moves outward into the surrounding cell layer consisting the quiescent center (QC), CEI and endodermis, collectively called as the U-shaped domain (Nakajima et al., 2001; Supplementary Figure 1). SHR physically interacts with SCR in the U-shaped domain of the main root, and the interaction is more pronounced in the CEI to regulate downstream target expressions such as CYCLIN D6;1 (CYCD6;1) to promote formative divisions (Cui et al., 2007; Cruz-Ramírez et al., 2012; Long et al., 2015a, 2017).
Here, we provide a guideline for utilizing the FRET-FLIM technology to visualize and quantify protein interactions at physiological conditions in living Arabidopsis roots at cellular resolution, with SCR and SHR as the example protein pair. We extended our analysis to Arabidopsis lateral roots and embryos to show that in vivo FRET-FLIM can also be applied to visualize interactions in other organs. In this study, we addressed the key optimization steps for transcription factors as prerequisites for measurable FRET to occur in living Arabidopsis tissues. These include (1) testing position of fluorescent tags at amino- and carboxyl-termini of each tested protein, (2) evaluating fluorophores suitability, and (3) in vivo fusion protein functionality.
Our work demonstrates that in vivo FRET-FLIM can be used to visualize nuclear protein interactions in a living, intact organism and provides evidence that the level of interaction between transcription factors can be heterogeneous throughout their domain of co-localization, and their interaction pattern can change depending on the developmental stage. Our optimization set up and procedure to detect in vivo protein-protein interaction can in principle be applied to any protein pair in any biological system.
Coding sequences (CDS) of SCFP3A, mTurquoise, SYFP2, mCherry, mStrawberry and mRFP (Kremers et al., 2006; Goedhart et al., 2007) were subcloned into multiple Gateway cassettes with flanking attB sites. A general SV40 nuclear localizing signal (NLS) (Lassner et al., 1991) was attached to the N-terminal of mTq and SYFP2 to generate NLS-mTq and NLS-SYFP2. For C-terminal tagging, fluorescent protein sequences were recombined into pGEMTeasyR2R3 vector by Gateway BP reaction; while pGEMTeasyR1R2-derived entry clones were generated for N-terminal tagging. SHR and SCR coding sequence in pDONR221-derived entry clones (Welch et al., 2007) were used for C-terminal tagging clones; while for N-terminal tagging SHR and SCR were subcloned into pGEMTeasyR2R3. For protoplast transfection, 35S promoter-driven fusions of SHR and SCR with N- or C-terminal tagging were created in pB7m34GW or pH7m34GW binary vectors (Karimi et al., 2007) by multiple Gateway LR reactions (Invitrogen). Positive controls of 35S::NLS-SYFP2:mTq and 35S::NLS-SYFP2:SCFP3A were generated by combining previously described tags in entry clones. Root expression vectors of SHR and SCR were created similarly with endogenous pSHR and pSCR promoters (Long et al., 2015a). For better stem cell niche localization (see below), pSHR::SYFP2-SHRΔ1a was generated by site-directed mutagenesis (QuikChange II, Aligent) from pSHR::SYFP2:SHR vector. For HeLa cell expression, SYFP-Δ1a-SHR was generated by subcloning SHR CDS with flanking restriction sites (5′-BsrGI-SHR-BamHI-3′) into pSYFP2-C1 (Kremers et al., 2006) followed by site-directed mutagenesis as described. SCR-mCherry was generated by subcloning SCR CDS with flanking restriction sites (5′-KpnI-SCR-AgeI-3′) into pmTurquoise-N1 (Goedhart et al., 2010), followed by swapping mTurquoise with mCherry (5′-AgeI-mCherry-NotI-3′) (Goedhart et al., 2007). Primers for cloning are listed in Supplementary Table 1.
Arabidopsis thaliana ecotype Columbia (Col-0) plants containing SHR and SCR transgenes were grown as previously described (Long et al., 2017). Stably transformed lines were generated by Agrobacterium tumefaciens-mediated transformation via floral dip method (Clough and Bent, 1998).
A. thaliana Col-0 mesophyll protoplasts were prepared and transfected according to (Díaz-Triviño et al. (2017). A. thaliana Col-0 tissue culture protoplasts were prepared and transfected according to Axelos et al. (1992). Ten microgram donor vector and 20 μg acceptor vector were transfected.
HeLa cell culture and transfection were as described in Jiang et al. (2014), constructs were transfected using FuGENE 6 protocol (Promega).
Living transfected protoplasts were collected in LabTek chambered coverglass (Nunc) for frequency-domain FLIM measurements. Samples with cyan fluorescent donors were acquired according to Goedhart et al. (2010) and samples with yellow fluorescent donor were acquired according to Goedhart et al. (2007). Briefly, CFP-variants were excited with a 440 nm modulated diode laser (LDH-M-C-440; PicoQuant) at 75.1 MHz, the light was reflected by a 455DCLP dichroic mirror and emission was passed through a D480/40 band-pass emission filter (Chroma Technology). SYFP2 fluorescence was excited with a 514 nm Argon laser (Melles-Griot) intensity-modulated at a frequency of 75.1 MHz and the light was reflected by a 525DCXR dichroic mirror and emission was passed through a HQ545/30 band-pass emission filter (Chroma Technology). Emission was detected using a radio frequency (RF)-modulated image intensifier (Lambert Instruments II18MD) coupled to a charge-coupled device (CCD) camera (Photometrics HQ) as detector. FLIM stacks of 18 phase images were acquired in permutated recording order with an exposure time of 50-1000 ms per image depending on sample brightness. The average fluorescence lifetime of individual nuclei was quantified from which an average lifetime for the sample was determined. FRET efficiency was calculated as described in Goedhart et al. (2007) More than 10 cells were analyzed for each sample.
Protoplasts, Arabidopsis embryos and lateral roots were imaged with a LSM 710 laser-scanning confocal microscope (Carl Zeiss GmbH) with an Objective C-Apochromat 40x/1.2 W Corr M27. A 2 air unit (AU) pinhole was set for weak SHR expression. Cyan fluorescence was detected at 465–500 nm with 458 nm excitation and 458/514 beam splitter; yellow detected at 520–560 nm with 514 nm excitation and 458/514 beam splitter; and red detected at 600–660 nm with 543 nm excitation and 488/543/633 beam splitter, respectively. Images were taken with no offset, and signal-to-noise ratio (SNR) was calculated as follows:
where S is the nuclear fluorescence signal from imaged root endodermis, and N auto-fluorescence signal in the adjacent non-fluorescent area in the root to emphasize the challenge of measurement in Arabidopsis root with high background signal. More than 10 roots were analyzed for each SNR calculation, except for pSCR::SCR:mStrawberry (n = 8), pSCR::SCR:mCherry (n = 7) and pSHR::SHR:mRFP (n = 9).
Roots of 6 dpg seedlings were mounted in water for measurements in LRP. Late heart-/early torpedo-stage embryos were mounted in 5% glucose for measurements. FLIM was performed on a confocal laser scanning microscope (Zeiss LSM 780) additionally equipped with a single-photon counting device with picosecond time resolution (PicoQuant Hydra Harp 400). SYFP2 fluorescence was excited at 485 nm using a linearly polarized diode laser (LDH-D-C-485) operated at a repetition rate of 32 MHz. Excitation power was around 1 μW at the objective C-Apochromat 40x/1.2 W Corr M27). The emitted light was collected in the same objective and separated into its perpendicular and parallel polarization (Thorlabs PBS 101, Thorlabs GmbH, Germany). Fluorescence was then detected by Tau-SPADs (PicoQuant) in a narrow range of SYFP2's emission spectrum (band-pass filter: HC535/30 AHF). Images were taken with 12.6 μs pixel time and a resolution of 0.1 μm/pixel for roots and embryos and 0.21 μm/pixel for LRP (Zoom 4 and 2, 256 × 256). A series of 60 frames were merged into one image and further analyzed (Widengren et al., 2006).
The fluorescence lifetime of SYFP2 was determined and analyzed pixel-wise in merged images to increase photon numbers for analysis using the software tools “AnI-3SF” and “Margarita” developed in Prof. C.A.M Seidel group [Software Package for Multiparameter Fluorescence Spectroscopy, Full Correlation and Multiparameter Fluorescence Imaging (http://www.mpc.uni-duesseldorf.de/en/software/software-package.html)] for Multiparameter Fluorescence Image Spectroscopy (MFIS) (Kudryavtsev et al., 2007; Weidtkamp-Peters et al., 2009). In fluorescence lifetime measurements, high spatial resolution microscopy and low excitation power prevent photo bleaching; the number of photons per pixel is exceptionally low, ranging from 100 to 2,000 photons per pixel. Therefore, a model to fit the data with a minimal number of parameters has to be applied in conjunction with a maximum likelihood estimator (MLE) (Schaffer et al., 1999; Eggeling et al., 2001; Widengren et al., 2006; Weidtkamp-Peters et al., 2009; Sisamakis et al., 2010). The decay of SYFP2 is approximated in the subsequent fluorescence lifetime analysis by an (fluorescence-weighted) average lifetime, τ. We therefore used a monoexponential model function with two variables (fluorescence lifetime τ and scatter contribution γ); as described elsewhere (Stahl et al., 2013), fitted with MLE. The instrument response function was measured using the dye erythrosine, which exhibits a very short fluorescence lifetime, which is additionally quenched in an aqueous, saturated potassium iodide solution.
Nuclear areas of no smaller than 25 pixels, based on the nuclei's appearances after the 100-photon-per-pixel background subtraction, were selected from independent cells. Cellular fluorescence lifetimes were computed by least-square fitting the Gaussian peaks of each cells' lifetime distributions. Fluorescence lifetimes at the same cell position were pooled from independent measurements without normalization, enabled by the robust FRET-FLIM acquisition between samples and between experiments. Reduction of fluorescence lifetime (Δτ) between donor-only and FRET samples were calculated from the means of donor-only and FRET samples at each cell position, with inclusion of fractional standard errors. Significances, between donor-only and FRET samples at specific cell positions in the same or different experiments, were resolved by Student's t-test with critical value of p < 0.01.
Our optimization procedure featured an ex vivo to in vivo pipeline, where we first employed the transient Arabidopsis protoplast expression system as a convenient tool to test a large number of FRET-FLIM pair combinations to select optimal positions of fluorescent tags and system-specific fluorophores, before evaluating protein functionality in Arabidopsis roots. For rapid data acquisition, we exploited widefield frequency-domain FLIM (Supplementary Figure 2A; Verveer and Hanley, 2009) measurements for protoplast samples with high transgene expression levels. Lifetime measurements in living Arabidopsis tissues were conducted with time-correlated single photon counting (TCSPC)-based time-domain FLIM (Supplementary Figure 2B; Gerritsen et al., 2009) with confocal imaging of lowly-expressed proteins at endogenous levels.
Close proximity between the donor and the acceptor is a prerequisite for achieving measureable FRET (Figure 1B). We first optimized the tagging position to detect FRET between SHR and SCR with a cyan-emitting mTurquoise (mTq) (Goedhart et al., 2010) as donor and a yellow-emitting SYFP2 (Kremers et al., 2006) as acceptor in Arabidopsis protoplasts. We fused mTq and SYFP2 to either the amino- or carboxyl-termini of the SHR and SCR proteins. We constructed SCR:mTq, mTq:SCR, SHR:SYFP2, and SYFP2:SHR under the constitutive promoter of Cauliflower Mosaic Virus 35S RNA (35S) by the Gateway cloning system, and introduced them into Arabidopsis protoplasts as pairs (example in Figure 1C). As a negative control, we co-transfected SYFP2:SHR with a nuclear-localizing mTq (NLS-mTq), while for positive control we constructed a nuclear-localizing fusion between SYFP2 and mTq (NLS-SYFP2:mTq), where constitutive FRET occurs. Upon paired co-transfection, we measured lifetimes for each SHR-SCR combination by frequency-domain FLIM measurements. Frequency domain FLIM measurements yield a fluorescence lifetime based on the phase shift (τϕ) and demodulation (τmod) of the fluorescence emission relative to the modulated excitation source (Supplementary Figure 2A; Verveer and Hanley, 2009). From these lifetimes and the lifetime of the donor-only sample, the average FRET efficiency was calculated, yielding Eϕ and Emod (Supplementary Figure 2A). As shown in Figures 1D,E, different combinations of tagging orientations gave varying levels of lifetime changes, i.e., different shifts of lifetimes in the scatterplots. This results in the unequal FRET efficiencies in the bar chart. The SCR:mTq SYFP2:SHR combination scored the highest FRET efficiency of Eϕ = 24.6% ± 1.8% and Emod = 11.2% ± 0.9% (Figures 1D,E; Long et al., 2017). These results suggest that the carboxyl-terminus of SCR and the amino-terminus of SHR are in close proximity. Up to 33.3% FRET efficiency was measured in the positive control NLS-SYFP2::mTq (Figure 1E), comparable to the previous reported value (Goedhart et al., 2010). The NLS-mTq SYFP2:SHR negative control gave near-ground level FRET (Figure 1E), indicating that FRET between each SHR-SCR combination reflects specific binding. To achieve the highest sensitivity, we selected carboxyl-terminal-tagged SCR and amino-terminal-tagged SHR for further optimizations and analyses.
The brightness and quantum yield of the fluorescent proteins depends on pH, temperature and other conditions introduced by different biological systems. To identify the optimal fluorophores suitable for FRET-FLIM measurement in Arabidopsis, we compared the performances of several fluorescent proteins in protoplasts and roots (Tables 1, 2).
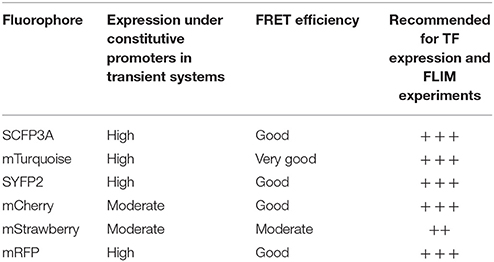
Table 1. Summary of fluorophores used in this study and their performance in transient systems.
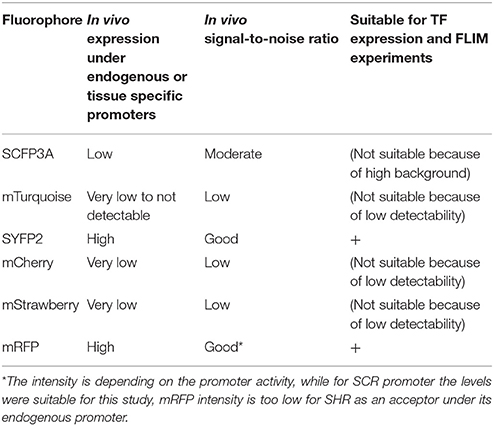
Table 2. Overview on the fluorophores performance when used under native promotors in living Arabidopsis roots.
First, we evaluated whether cyan fluorescent protein (CFP) variants SCFP3A and mTq, in the context of our FRET pair combination SCR and SHR, could be used in a common cyan-yellow FRET-FLIM setup in plant cells (Kremers et al., 2006; Hamers et al., 2014). As shown in Figure 2A, SCR:mTq yielded a higher FRET efficiency than SCR:SCFP3A in combination with SYFP2:SHR in protoplasts, most likely due to mTq's higher quantum yield. However, SCR:SCFP3A SYFP2:SHR measurements were more precise (Figure 2A, Supplementary Figure 3A). The reduced precision of mTq-SYFP2 measurements might reflect suboptimal mTq performance in plant nuclei (see Discussion).
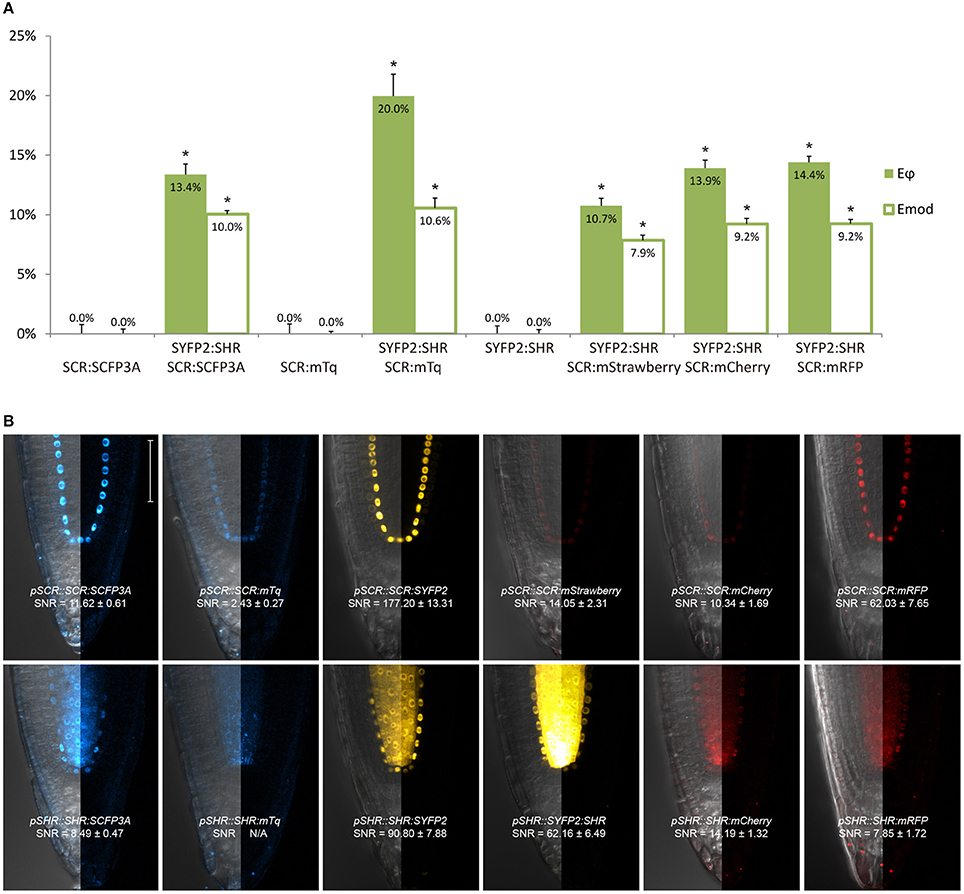
Figure 2. Selection of an appropriate fluorescent protein pair for FRET-FLIM analysis. (A) Bar chart of FRET efficiency Eϕ and Emod between SCR and SHR tagged with different fluorescent proteins, with error bars of standard error of mean, n > 10 for each sample. *p < 10−2, p-values calculated by Student's t-test compared to the donor-only samples. (B) Confocal images of roots expressing SCR and SHR tagged with different fluorescent proteins, with signal-to-noise ratio (SNR) calculated from endodermal nuclear fluorescence signal. Scale bar, 50 μm. Each image displays the overlay image of transmission and fluorescent channels in the left half and the fluorescence channel in the right half from the same root.
We next tested the performance of SCFP3A, mTq and SYFP2 in Arabidopsis roots. Since SHR and SCR co-localize in the U-shaped domain, it is essential to detect them in these cells to assess where they interact. Under endogenous promoters, both cyan-variant-tagged SCR and SHR transgenic lines displayed low fluorescence levels relative to the background: signal of pSCR::SCR:SCFP3A, pSCR::SCR:mTq, and pSHR::SHR:SCFP3A could be detected in the endodermis with low signal-to-noise ratios (SNR); while endodermal signal of pSHR::SHR:mTq was indistinguishable from background signal (Figure 2B).
Since FRET-FLIM is more dependent on donor fluorescence, the poor detection of these two cyan variants made them unsuitable as donor tags in this system. On the contrary, pSCR::SCR:SYFP2 and pSHR::SYFP2:SHR yielded readily detectable emissions supported by higher SNR (Figure 2B), hence we favored SYFP2 as donor tag. Since it has been previously shown that red fluorescent proteins are efficient FRET acceptors for SYFP2 with Förster radii > 5.6 nm (Goedhart et al., 2007), we proceeded to optimize the labeling conditions for yellow-red FRET pairs.
Three red-emitting variants, mStrawberry, mCherry and mRFP, were tested for their performance as mentioned above. In protoplasts, SHR and SCR tagged with all three red variants and SYFP2 gave comparable FRET efficiency, with SYFP2-mStrawberry pair slightly lower (Figure 2A, Supplementary Figure 3B). When expressed in roots, pSCR::SCR:mRFP exhibited higher detectability than pSCR::SCR:mStrawberry and pSCR::SCR:mCherry, making mRFP a better choice. In the case of SHR, all the red variants displayed low detectability correlating with low signal-to-noise ratios (SNR) (Figure 2B). Considering that sufficient FRET analysis requires more acceptor molecules than donors, or “donor saturation,” SCR is then more suitable as acceptor due to its higher endogenous expression level than SHR (Long et al., 2017, Table 3). Therefore, we selected SYFP2:SHR and SCR:mRFP for in vivo FRET-FLIM studies.

Table 3. Summary of the performance of fluorophore pairs used in this study.
Tagging proteins of interest with fluorescent proteins has a potential pitfall: the resulting fusions might reduce biological function due to undesired conformational changes or steric hindrance introduced by the tags. Measurements carried out with such non-functional or dysfunctional fusions might not accurately reflect their endogenous behaviors. Therefore, it is crucial to evaluate the functionality of fusion proteins before FRET-FLIM measurements.
The C terminal fusion pSCR::SCR:mRFP was reported to be functional (Long et al., 2015a, 2017). For SHR fusion, despite its high detectability in the endodermis, we noticed that only 11% of the roots harboring pSHR::SYFP2:SHR showed clearly visible signal in the stem cell niche (Figure 3b), while such signal was readily visible in 80% of roots harboring the carboxyl-terminal-tagged pSHR::SHR:SYFP2 (Figure 3a). This indicated that SYFP2:SHR might not move efficiently between certain cells. As previously shown, SHR movement from the vasculature is essential for root growth regulation, and altering its mobility can cause abnormal CEI division and disrupted root architecture (Cui et al., 2007; Vatén et al., 2011; Koizumi et al., 2012; Long et al., 2015a). Additionally, SHR and SCR co-localize in the endodermis and stem cell niche, it is thus essential to have sufficient SHR movement into the stem cell niche to measure SHR-SCR interaction. Since amino-terminal tagging on SHR was not reported to disrupt SHR movement (Heidstra et al., 2004), we reasoned that the Gateway linker between SYFP2 and SHR might cause an undesired conformational change to the fusion, and attempted to restore SYFP2:SHR mobility by linker alteration. A typical attB2 Gateway recombination site with flanking sequence is recommended to be 27 base pairs after recombination (Invitrogen), translating to a linker of 9 amino acids DPAFLYKVA between SYFP2 and SHR. Although longer, more flexible linkers are usually favored for functional tagging, farther tag displacement can potentially increase the distance and reduce the probability of spatial association between donor and acceptor fluorophores beyond the Förster radii, thereby reducing FRET. Thus, we shortened the linker using site-directed mutagenesis, and generated pSHR::SYFP2-SHRΔ1a by removing 5 amino acids, reducing it from DPAFLYKVA to DKVA, similar in length to the described functional N-terminal SHR fusion (Heidstra et al., 2004). Both linkers are estimated to be shorter than the 5.6 nm Förster radius for SYFP2-mRFP pair (Goedhart et al., 2007). As shown in Figure 3c, up to 71% of the roots harboring pSHR::SYFP2-SHRΔ1a showed significant improvement of SHR fusion signal in the stem cell niche. The linker alteration of SYFP2-SHRΔ1a did not change the FRET efficiency between SHR and SCR in protoplasts (Supplementary Figures 3A,B), indicating that neither fluorophore distance nor dipole orientation was disrupted. This enabled us to measure FRET-FLIM between SHR and SCR in their endogenous conditions.
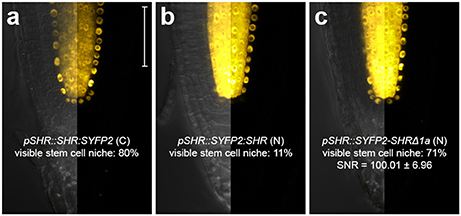
Figure 3. Improvement of SHR fusion protein mobility. Confocal images of roots expressing SHR fusion proteins differentially tagged with SYFP2, with signal-to-noise ratio (SNR) calculated from endodermal nuclear fluorescence signal. (a) pSHR::SHR:SYFP2, (b) pSHR::SYFP2:SHR, (c) pSHR::SYFP2-SHRΔ1a, n > 10 for each sample. Scale bar, 50 μm. For every image, the left half displays the overlay image and the right half fluorescence channel from the same root.
Our optimization procedure revealed that the combination of analysis in protoplasts (ex vivo) and intact plants (in vivo) is essential for the selection of the appropriate donor-acceptor pairs and protein fusions strategies for in vivo FRET-FLIM measurements. A summary of choosing the optimal fluorophores ex vivo and in vivo as well as additional considerations of using this technology can be found in Supplementary Materials.
In a previous study, we implemented in vivo FRET-FLIM measurements between SYFP2-SHRΔ1a and SCR:mRFP in the Arabidopsis primary root meristem, and showed that SHR and SCR interact in the QC, CEI and endodermis in Arabidopsis roots (Long et al., 2017). The primary root meristem is pre-established in the embryotic root pole (ten Hove et al., 2015), while de novo root meristems repetitively emerge in the forms of lateral roots, adventitious roots and during root regeneration (Verstraeten et al., 2014; Efroni et al., 2016). Although highly resembling in structure and sharing the transcriptional regulatory network, the precise regulatory mechanisms have been proposed to differ between these root meristems (Lucas et al., 2011; Verstraeten et al., 2014; Efroni et al., 2016; Du and Scheres, 2017). To explore the SHR-SCR interaction profile in other developmental contexts, we extend the application of in vivo FRET-FLIM measurements to Arabidopsis embryos and developing lateral roots.
In heart stage embryos, SHR and SCR expression domains at the root pole resemble those in the postembryonic roots (Figure 4a). Similar to the observations in the primary root meristem (Long et al., 2017), we found that SYFP2-SHRΔ1a exhibited strong FRET with SCR:mRFP in QC, CEI and endodermis of late heart-/early torpedo-stage embryos (Figures 4b,c). Interestingly, FRET between SYFP2-SHRΔ1a and SCR:mRFP in the embryo was enhanced in QC and the first endodermal cell (endodermis 1), to similar levels occurring in the CEI (Figure 4c). This observation might reflect enhanced SHR-SCR interaction or closer SHR-SCR association in multimeric protein complexes in these embryonic cells. Alternatively, the contribution of high background signal (reduced SNR in Figure 4a) with generally shorter lifetimes in the embryos might have influenced FRET detections and resulted in a general lifetime reduction. To distinguish between these possibilities, detailed expression analysis of direct target genes of SHR-SCR complex like CYCD6;1 during embryogenesis, as well as creating mutations in the SHR-SCR interaction domain, will be necessary to fully understand these observations. Nevertheless, our in vivo FRET-FLIM results hint that, despite the structural resemblance and developmental similarity, the underlying molecular wiring regulating embryonic root can be different from the root tip.
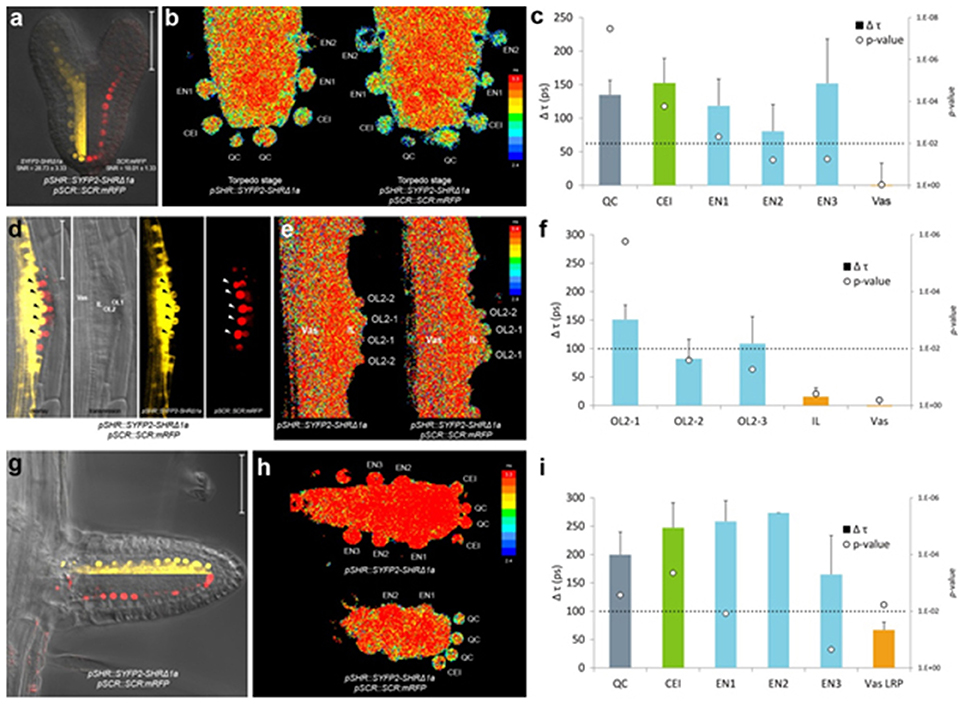
Figure 4. In vivo FRET-FLIM of SHR-SCR in embryos and lateral roots. (a) Early torpedo stage Arabidopsis embryo co-expressing pSHR::SYFP2-SHRΔ1a and pSCR::SCR:mRFP, with signal-to-noise ratio (SNR) calculated from endodermal nuclear fluorescence signal. Scale bar, 50 μm. Yellow fluorescence channel (left) and red fluorescence channel (right) were overlaid with transmission image from the same root. (b) Heatmaps of fluorescence lifetime in donor-only and sample embryo. (c) Quantification of lifetime change (Δτ) in single cells. Column color matches with tissue type illustrated in this figure. Circles indicate p-value calculated by Student's t-test of sample lifetimes comparing to donor-only lifetimes at each cell position, with the dotted line marking the 0.01 significant value. Donor embryos n = 18, FRET sample embryos n = 34. (d) Arabidopsis stage IV LRP co-expressing pSHR::SYFP2-SHRΔ1a and pSCR::SCR:mRFP. Scale bar, 50 μm. OL1 and OL2, outer layer 1 and 2; IL, inner layer; Vas, primary root vasculature. Arrowheads point to OL2 cells where SHR and SCR co-localize. (e) Fluorescence lifetime heatmaps of donor-only and sample LRP. OL2 cells were numbered with OL2-1 in the middle of the LRP and OL2-2 and−3 progressively further from LRP midline. (f) Quantification of FRET between SYFP2-SHRΔ1a and SCR:mRFP measured in (e). Donor LRP n = 13, FRET sample LRP n = 17. (g) Arabidopsis emerged lateral root co-expressing pSHR::SYFP2-SHRΔ1a and pSCR::SCR:mRFP. Scale bar, 50 μm. Yellow fluorescence channel (upper) and red fluorescence channel (lower) were overlaid with transmission image from the same root. (h) Fluorescence lifetime heatmaps of donor-only and sample emerged lateral root. (i) Quantification of FRET between SYFP2-SHRΔ1a and SCR:mRFP measured in (h). Donor lateral roots n = 11, FRET sample lateral roots n = 3. Vas LRP, vasculature of LRP.
New root meristems are formed from differentiated root tissue in a process called lateral root formation. Lateral root primordia (LRP) initiation is marked by a series of cell divisions originating from the vasculature, particularly the pericycle cells opposing the xylem pole (Malamy and Benfey, 1997). Using in vivo FRET-FLIM, we studied the interaction between SHR and SCR during lateral root formation. As shown in Figure 4d, SHR and SCR only co-localized in a subset of cells in the developing stage IV LRP: SCR:mRFP was detected in both of the two outer layers (OL1 and OL2), while SYFP2-SHRΔ1a resided in the OL2 nuclei and maintained nuclear-and-cytoplasmic localization in the inner layer (IL), similar to mature vasculature. Within OL2 where SYFP2-SHRΔ1a and SCR:mRFP co-localized, FRET was detected higher in the central cells (OL2-1, Figure 4e,f). In contrast, OL2 cells displaced from LRP midline (OL2-2 and OL2-3, Figure 4f) exhibited lower FRET levels similar to those in the endodermis in the primary root (Long et al., 2017). No FRET was detected in the IL or vasculature due to the absence of detectable SCR:mRFP (Figure 4f).
After emergence, the lateral root morphology resembles the primary root, with similar cellular organization and expression patterns of SHR and SCR (Figure 4g). However, the FRET levels between SYFP2-SHRΔ1a and SCR:mRFP in emerged lateral roots were generally higher with no significant difference between QC, CEI and endodermis (Figure 4i).
Analyses between SYFP2-SHRΔ1a and SCR:mRFP in Arabidopsis embryos and LRP show that in vivo FRET-FLIM can be utilized within different developmental contexts. The generally preserved but slightly altered interaction patterns further suggests that the transcriptional regulations of SHR and SCR may exhibit different network topology in different developmental stages.
Interaction between SHR and SCR has been shown by many approaches including assays in mammalian cells (Long et al., 2017). To assess whether this interaction can be detected by FRET-FLIM in a system devoided from plant specific transcriptional regulations, we measured FRET-FLIM between SYFP2-SHRΔ1a and SCR-mRFP the HeLa cells and we could detect interaction (Supplementary Figures 4C,D), albeit at a lower level. This demonstrates that plant protein interaction can be analyzed in heterologous systems like animal cells.
In the present study, we outline an optimization procedure of the labeling conditions for applying the FRET-FLIM technology to inspect nuclear protein interactions in living plants. We show that protein complex formation can be mapped to specific cells in different organs in vivo and that the interaction domain is spatially modulated during development. This technique therefore overcomes previous limitations to studying protein complex dynamics at cellular resolution.
We show that fluorophores exhibit different performances in plant cells when fused to two interacting transcription factors. For example, mTq is well recognized as a preferred CFP variant for use as a FRET donor (Goedhart et al., 2010). In the Arabidopsis root, endodermal signal was low for SCR:mTq and undetectable for SHR:mTq (Figure 2B) relative to autofluorescence. Such low mTq detectability, however, was not reported when expressed at high levels (Figure 1C; Hecker et al., 2015) or localized to cell membranes, cytoplasm or cytoskeleton in intact Arabidopsis plants (Roppolo et al., 2011; Peremyslov et al., 2012; Waadt et al., 2014). This is possibly due to high expression levels of these fusion proteins concentrated at different subcellular domains, or might suggest that mTq protein is sensitive to the plant nuclear microenvironment. Nevertheless, our optimization procedure highlights the importance of selecting appropriate fluorophores for different cellular and subcellular conditions (see Supplementary Materials). Linker optimization between the protein-of-interest and the fluorophore is also crucial for ensuring close proximity, favorable dipole orientation and fusion protein functionality. Our studies confirmed that the linker introduced by common Gateway recombination site is sufficiently short for FRET between SHR and SCR, although functionality of N-terminal SHR fusion was only restored with shortened linker without compromising FRET detection (Figure 3). It is therefore important to optimize fusion linkers for functional in vivo FRET studies.
Optimizing FRET-FLIM in living Arabidopsis roots allowed visualization of spatiotemporal bindings between endogenous SHR and SCR during different developmental stages, which cannot be addressed by in vivo over-expressions or cell lines (Long et al., 2017). We found that the FRET levels between SYFP2-SHRΔ1a and SCR:mRFP vary among different developmental contexts, and among different cell types within each developmental stage. The enhanced FRET-FLIM signals in CEI reflect a specific conformation of a multimeric complex modified by the presence of other binding partners (Long et al., 2017). We have recently shown that SHR and SCR interact with the BIRD protein JACKDAW which regulate SHR intercellular mobility and transcriptional activity, and that SHR-SCR-JKD complexes display distinct conformations within the U-shaped domain (Long et al., 2015a, 2017). The cell cycle regulator RETINOBLASTOMA-RELATED (RBR) also physically associates with the SHR-SCR complex to repress ectopic formative divisions in the endodermis (Cruz-Ramírez et al., 2012). The in vivo binding dynamics of RBR and other interacting BIRD proteins to the SHR-SCR complexes have not yet been tested. To this end, extending our optimized in vivo FRET-FLIM technique for proteins interacting with SHR-SCR complex to create a protein interaction map at cellular resolution will be a big step toward understanding the cell-specific protein complex dynamics in vivo and their functions during different stages of Arabidopsis development.
The discovery of SHR-SCR interaction heterogeneity highlights the spatiotemporal sensitivity of in vivo FRET-FLIM. However, FRET requires the donor and acceptor being within the stringent Förster radius and the fluorophore dipoles parallel to each other, making it especially sensitive to close-ranged protein associations but inefficient to detect interactions between far-end-tagged proteins due to functionality obligations or associations of proteins within big protein complexes that exceed Förster radii. Meanwhile, single molecule spectroscopy analyses such as fluorescence correlation spectroscopy (FCS)-based techniques, can detect protein-protein association without Förster radius requirement. While single molecule tracking of SHR-SCR complex using FCS was in line with our findings (Clark et al., 2016), however, it was proven impractical in the stem cell niche due to high background level, while in vivo FRET-FLIM succeeded in obtaining interaction information thanks to the stringently controlled fitting procedure. To sum up, one can obtain a broader spectrum of information regarding protein-protein interaction by combining FRET-FLIM and FCS-based techniques in vivo.
Nevertheless, our heterologous analyses forecast future applications of in vivo FRET-FLIM in studying protein-protein interactions in other biological systems. Indeed, attempts of applying FRET-FLIM measurements in living animals or intact tumors to study interactions between exogenous proteins or monitor biosensors have been reported (Kelleher et al., 2009; Kardash et al., 2011; Venugopal et al., 2012; Nobis et al., 2013), promising the possibility of in vivo FRET-FLIM usage. Multiphoton FRET-FLIM (Peter et al., 2005) may further enhance SNR, improve detection depth in thicker tissues and reduce photobleaching, although the near-infrared excitation will likely require additional optimizations to address potential cross-excitation and signal bleedthrough for the SYFP2-mRFP pair. Following our optimization procedure, endogenous protein interactions should be readily analyzable in living animals and other multicellular organisms.
In conclusion, optimization of FRET-FLIM allows detection of protein complexes in living tissue at cellular resolution. Our optimization procedure is, in principle, appropriate for any protein interaction pair and in various subcellular compartments (Stahl et al., 2013; Somssich et al., 2015; Weidtkamp-Peters and Stahl, 2017). Additionally, homo-FRET measured by fluorescence anisotropy can help in further deciphering protein complex compositions. Low abundance of certain proteins and potential limitations in engineering effective fusions without disrupting protein function still remain as major challenges for in vivo FRET-FLIM measurements. Technical advances will rely on continuous improvements of fluorescent tags and detection sensitivity. Characterizing and implementing mTurquoise2, mScarlet (Bindels et al., 2017) and other fluorophores with high quantum yield in future FRET-FLIM measurements, in addition to the application of other microscopic techniques such as single-molecule FRET-FLIM or FCS-based techniques in living organisms, will allow us to precisely monitor the composition of multiprotein complexes and their dynamics in vivo.
The scientific conception, is due to IB and YL. IB and YL designed and executed the experiments. YS, SW-P assisted in setting up, optimizing FRET-FLIM experiments and data analysis. JG helped with FRET-FLIM measurements in protoplast. TG helped with discussions related to FRET-FLIM quantification. IB and YL wrote the manuscript. All authors were involved in data analysis, interpretation and revision of the manuscript.
This work was supported by an NWO VIDI grant 015.003.003 for IB and YL. YL was further supported by ERC Advanced Grant SysArc no 232914 and NWO Spinoza Grant OND1352967 to BS. SW-P was supported by DFG-project WE 5343/1-1. Publication fee was supported by King Abdullah University of Science and Technology (KAUST).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Prof Anna Akhmanova for providing mammalian cell line and lab facilities to conduct transfections in Hela cells and to Prof Rudiguer Simon for critical reading of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00639/full#supplementary-material
Adjobo-Hermans, M. J., Goedhart, J., van Weeren, L., Nijmeijer, S., Manders, E. M., Offermanns, S., et al. (2011). Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol. 9:32. doi: 10.1186/1741-7007-9-32
Aryal, U. K., Xiong, Y., McBride, Z., Kihara, D., Xie, J., Hall, M. C., et al. (2014). A proteomic strategy for global analysis of plant protein complexes. Plant Cell 26, 3867–3882. doi: 10.1105/tpc.114.127563
Axelos, M., Curie, C., Mazzolini, L., Bardet, C., and Lescure, B. (1992). A protocol for transient gene expression in Arabidopsis thaliana protoplasts isolated from cell suspension cultures. Plant Physiol. Biochem. 30, 123–128.
Bensimon, A., Heck, A. J., and Aebersold, R. (2012). Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405. doi: 10.1146/annurev-biochem-072909-100424
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56. doi: 10.1038/nmeth.4074
Bridgeman, J. S., Blaylock, M., Hawkins, R. E., and Gilham, D. E. (2010). Development of a flow cytometric co-immunoprecipitation technique for the study of multiple protein-protein interactions and its application to T-cell receptor analysis. Cytom. Part J. Int. Soc. Anal. Cytol. 77, 338–346. doi: 10.1002/cyto.a.20840
Bücherl, C. A., van Esse, G. W., van Kruis, A., Luchtenberg, J., Westphal, A. H., Aker, J. de, et al. (2013). Visualization of BRI1 and BAK1(SERK3) membrane receptor heterooligomers during brassinosteroid signaling. Plant Physiol. 162, 1911–1925. doi: 10.1104/pp.113.220152
Clark, N. M., Hinde, E., Winter, C. M., Fisher, A. P., Crosti, G., Blilou, I., et al. (2016). Tracking transcription factor mobility and interaction in Arabidopsis roots with fluorescence correlation spectroscopy. eLife 5:e14770. doi: 10.7554/eLife.14770
Clegg, R. M. (2009). “Förster resonance energy transfer—FRET what is it, why do it, and how it's done,” in Laboratory Techniques in Biochemistry and Molecular Biology, ed T. W. J. Gadella (Amsterdam; Oxford, UK: Elsevier), 1–57.
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. Cell Mol. Biol. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Crosby, K. C., Pietraszewska-Bogiel, A., Gadella, T. W. J. Jr., and Winkel, B. S. J. (2011). Förster resonance energy transfer demonstrates a flavonoid metabolon in living plant cells that displays competitive interactions between enzymes. FEBS Lett. 585, 2193–2198. doi: 10.1016/j.febslet.2011.05.066
Cruz-Ramírez, A., Díaz-Triviño, S., Blilou, I., Grieneisen, V. A., Sozzani, R., Zamioudis, C., et al. (2012). A bistable circuit involving SCARECROW-RETINOBLASTOMA integrates cues to inform asymmetric stem cell division. Cell 150, 1002–1015. doi: 10.1016/j.cell.2012.07.017
Cui, H., Levesque, M. P., Vernoux, T., Jung, J. W., Paquette, A. J., Gallagher, K. L., et al. (2007). An evolutionarily conserved mechanism delimiting SHR movement defines a single layer of endodermis in plants. Science 316, 421–425. doi: 10.1126/science.1139531
Díaz-Triviño, S., Long, Y., Scheres, B., and Blilou, I. (2017). Analysis of a plant transcriptional regulatory network using transient expression systems. Methods Mol. Biol. 1629, 83–103. doi: 10.1007/978-1-4939-7125-1_7
Di Laurenzio, L., Wysocka-Diller, J., Malamy, J. E., Pysh, L., Helariutta, Y., Freshour, G., et al. (1996). The SCARECROW gene regulates an asymmetric cell division that is essential for generating the radial organization of the Arabidopsis root. Cell 86, 423–433. doi: 10.1016/S0092-8674(00)80115-4
Du, Y., and Scheres, B. (2017). PLETHORA transcription factors orchestrate de novo organ patterning during Arabidopsis lateral root outgrowth. Proc. Natl. Acad. Sci. U.S.A. 114, 11709–11714. doi: 10.1073/pnas.1714410114
Efroni, I., Mello, A., Nawy, T., Ip, P.-L., Rahni, R., DelRose, N., et al. (2016). Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 165, 1721–1733. doi: 10.1016/j.cell.2016.04.046
Eggeling, C., Berger, S., Brand, L., Fries, J. R., Schaffer, J., Volkmer, A., et al. (2001). Data registration and selective single-molecule analysis using multi-parameter fluorescence detection. J. Biotechnol. 86, 163–180. doi: 10.1016/S0168-1656(00)00412-0
Gadella, T. W. J. Jr., Jovin, T. M., and Clegg, R.M. (1993). Fluorescence lifetime imaging microscopy (FLIM): spatial resolution of microstructures on the nanosecond time scale. Biophys. Chem. 48, 221–239. doi: 10.1016/0301-4622(93)85012-7
Gerritsen, H. C., Agronskaia, A. V., Bader, A. N., and Esposito, A. (2009). “Time domain FLIM: theory, instrumentation, and data analysis,” in Laboratory Techniques in Biochemistry and Molecular Biology, ed T. W. J. Gadella (Amsterdam; Oxford, UK: Elsevier), 95–132.
Goedhart, J., van Weeren, L., Hink, M. A., Vischer, N. O. E., Jalink, K., and Gadella, T. W. J. (2010). Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 7, 137–139. doi: 10.1038/nmeth.1415
Goedhart, J., Vermeer, J. E., Adjobo-Hermans, M. J., van Weeren, L., and Gadella, T. W. Jr. (2007). Sensitive detection of p65 homodimers using red-shifted and fluorescent protein-based FRET couples. PLoS ONE 2:e1011. doi: 10.1371/journal.pone.0001011
Gohl, C., Banovic, D., Grevelhörster, A., and Bogdan, S. (2010). WAVE forms hetero- and homo-oligomeric complexes at integrin junctions in Drosophila visualized by bimolecular fluorescence complementation. J. Biol. Chem. 285, 40171–40179. doi: 10.1074/jbc.M110.139337
Gu, Y., Di, W. L., Kelsell, D. P., and Zicha, D. (2004). Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing. J. Microsc. 215, 162–173. doi: 10.1111/j.0022-2720.2004.01365.x
Hamers, D., van Voorst Vader, L., Borst, J. W., and Goedhart, J. (2014). Development of FRET biosensors for mammalian and plant systems. Protoplasma 251, 333–347. doi: 10.1007/s00709-013-0590-z
Hecker, A., Wallmeroth, N., Peter, S., Blatt, M. R., Harter, K., and Grefen, C. (2015). Binary 2in1 vectors improve in planta (Co)localization and dynamic protein interaction studies. Plant Physiol. 168, 776–787. doi: 10.1104/pp.15.00533
Heidstra, R., Welch, D., and Scheres, B. (2004). Mosaic analyses using marked activation and deletion clones dissect Arabidopsis SCARECROW action in asymmetric cell division. Genes Dev. 18, 1964–1969. doi: 10.1101/gad.305504
Helariutta, Y., Fukaki, H., Wysocka-Diller, J., Nakajima, K., Jung, J., Sena, G., et al. (2000). The SHORT-ROOT gene controls radial patterning of the Arabidopsis root through radial signaling. Cell 101, 555–567. doi: 10.1016/S0092-8674(00)80865-X
Horstman, A., Tonaco, I. A., Boutilier, K., and Immink, R. G. H. (2014). A cautionary note on the use of split-YFP/BiFC in plant protein-protein interaction studies. Int. J. Mol. Sci. 15, 9628–9643. doi: 10.3390/ijms15069628
Hu, C.-D., Chinenov, Y., and Kerppola, T. K. (2002). Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798. doi: 10.1016/S1097-2765(02)00496-3
Hudry, B., Viala, S., Graba, Y., and Merabet, S. (2011). Visualization of protein interactions in living Drosophila embryos by the bimolecular fluorescence complementation assay. BMC Biol. 9:5. doi: 10.1186/1741-7007-9-5
Jiang, K., Hua, S., Mohan, R., Grigoriev, I., Yau, K. W., Liu, Q., et al. (2014). Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev. Cell 28, 295–309. doi: 10.1016/j.devcel.2014.01.001
Jorge, T. F., Rodrigues, J. A., Caldana, C., Schmidt, R., van Dongen, J. T., Thomas-Oates, J., et al. (2016). Mass spectrometry-based plant metabolomics: metabolite responses to abiotic stress. Mass Spectrom. Rev. 35, 620–649. doi: 10.1002/mas.21449
Kardash, E., Bandemer, J., and Raz, E. (2011). Imaging protein activity in live embryos using fluorescence resonance energy transfer biosensors. Nat. Protoc. 6, 1835–1846. doi: 10.1038/nprot.2011.395
Karimi, M., Bleys, A., Vanderhaeghen, R., and Hilson, P. (2007). Building blocks for plant gene assembly. Plant Physiol. 145, 1183–1191. doi: 10.1104/pp.107.110411
Kelleher, M. T., Fruhwirth, G., Patel, G., Ofo, E., Festy, F., Barber, P. R., et al. (2009). The potential of optical proteomic technologies to individualize prognosis and guide rational treatment for cancer patients. Target. Oncol. 4, 235–252. doi: 10.1007/s11523-009-0116-y
Koizumi, K., Hayashi, T., and Gallagher, K. L. (2012). SCARECROW reinforces SHORT-ROOT signaling and inhibits periclinal cell divisions in the ground tissue by maintaining SHR at high levels in the endodermis. Plant Signal. Behav. 7, 1573–1577. doi: 10.4161/psb.22437
Kremers, G., and Goedhart, J. (2009). “Visible fluorescent proteins for FRET,” in Laboratory Techniques in Biochemistry and Molecular Biology, ed T. W. J. Gadella (Amsterdam; Oxford, UK: Elsevier), 171–223.
Kremers, G.-J., Goedhart, J., van Munster, E. B., and Gadella, T. W. J. (2006). Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster Radius. Biochemistry (Mosc.) 45, 6570–6580. doi: 10.1021/bi0516273
Kudryavtsev, V., Felekyan, S., Wozniak, A. K., König, M., Sandhagen, C., Kühnemuth, R., et al. (2007). Monitoring dynamic systems with multiparameter fluorescence imaging. Anal. Bioanal. Chem. 387, 71–82. doi: 10.1007/s00216-006-0917-0
Lalonde, S., Ehrhardt, D. W., Loqué, D., Chen, J., Rhee, S. Y., and Frommer, W. B. (2008). Molecular and cellular approaches for the detection of protein-protein interactions: latest techniques and current limitations. Plant J. Cell Mol. Biol. 53, 610–635. doi: 10.1111/j.1365-313X.2007.03332.x
Lassner, M. W., Jones, A., Daubert, S., and Comai, L. (1991). Targeting of T7 RNA polymerase to tobacco nuclei mediated by an SV40 nuclear location signal. Plant Mol. Biol. 17, 229–234. doi: 10.1007/BF00039497
Long, Y., Goedhart, J., Schneijderberg, M., Terpstra, I., Shimotohno, A., Bouchet, B. P., et al. (2015b). SCARECROW-LIKE23 and SCARECROW jointly specify endodermal cell fate but distinctly control SHORT-ROOT movement. Plant J. Cell Mol. Biol. 84, 773–784. doi: 10.1111/tpj.13038
Long, Y., Smet, W., Cruz-Ramírez, A., Castelijns, B., de Jonge, W., Mähönen, A. P., et al. (2015a). Arabidopsis BIRD zinc finger proteins jointly stabilize tissue boundaries by confining the cell fate regulator SHORT-ROOT and contributing to fate specification. Plant Cell. 27, 1185–1199. doi: 10.1105/tpc.114.132407
Long, Y., Stahl, Y., Weidtkamp-Peters, S., Postma, M., Zhou, W., Goedhart, J., et al. (2017). In vivo FRET-FLIM reveals cell-type-specific protein interactions in Arabidopsis roots. Nature 548, 97–102. doi: 10.1038/nature23317
Lucas, M., Swarup, R., Paponov, I. A., Swarup, K., Casimiro, I., Lake, D., et al. (2011). Short-Root regulates primary, lateral, and adventitious root development in Arabidopsis. Plant Physiol. 155, 384–398. doi: 10.1104/pp.110.165126
Malamy, J. E., and Benfey, P. N. (1997). Organization and cell differentiation in lateral roots of Arabidopsis thaliana. Dev. Camb. Engl. 124, 33–44.
Moreno-Risueno, M. A., Sozzani, R., Yardimci, G. G., Petricka, J. J., Vernoux, T., Blilou, I., et al. (2015). Transcriptional control of tissue formation throughout root development. Science 350, 426–430. doi: 10.1126/science.aad1171
Nakajima, K., Sena, G., Nawy, T., and Benfey, P. N. (2001). Intercellular movement of the putative transcription factor SHR in root patterning. Nature 413, 307–311. doi: 10.1038/35095061
Nobis, M., McGhee, E. J., Morton, J. P., Schwarz, J. P., Karim, S. A., Quinn, J., et al. (2013). Intravital FLIM-FRET imaging reveals dasatinib-induced spatial control of src in pancreatic cancer. Cancer Res. 73, 4674–4686. doi: 10.1158/0008-5472.CAN-12-4545
Pardo, M., and Choudhary, J. S. (2012). Assignment of protein interactions from affinity purification/mass spectrometry data. J. Proteome Res. 11, 1462–1474. doi: 10.1021/pr2011632
Peremyslov, V. V., Klocko, A. L., Fowler, J. E., and Dolja, V. V. (2012). Arabidopsis myosin XI-K localizes to the motile endomembrane vesicles associated with F-actin. Plant Cell Biol. 3:184. doi: 10.3389/fpls.2012.00184
Peter, M., Ameer-Beg, S. M., Hughes, M. K. Y., Keppler, M. D., Prag, S., Marsh, M., et al. (2005). Multiphoton-FLIM quantification of the EGFP-mRFP1 FRET pair for localization of membrane receptor-kinase interactions. Biophys. J. 88, 1224–1237. doi: 10.1529/biophysj.104.050153
Petricka, J. J., Schauer, M. A., Megraw, M., Breakfield, N. W., Thompson, J. W., Georgiev, S., et al. (2012). The protein expression landscape of the Arabidopsis root. Proc. Natl. Acad. Sci. U.S.A. 109, 6811–6818. doi: 10.1073/pnas.1202546109
Roppolo, D., De Rybel, B., Dénervaud Tendon, V. D., Pfister, A., Alassimone, J., Vermeer, J. E. M., et al. (2011). A novel protein family mediates Casparian strip formation in the endodermis. Nature 473, 380–383. doi: 10.1038/nature10070
Schaffer, J., Volkmer, A., Eggeling, C., Subramaniam, V., Striker, G., and Seidel, C. A. M. (1999). Identification of single molecules in aqueous solution by time-resolved fluorescence anisotropy. J. Phys. Chem. A 103, 331–336. doi: 10.1021/jp9833597
Sisamakis, E., Valeri, A., Kalinin, S., Rothwell, P. J., and Seidel, C. A. M. (2010). Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 475, 455–514. doi: 10.1016/S0076-6879(10)75018-7
Smaczniak, C., Immink, R. G. H., Muiño, J. M., Blanvillain, R., Busscher, M., Busscher-Lange, J., et al. (2012). Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. U.S.A. 109, 1560–1565. doi: 10.1073/pnas.1112871109
Somssich, M., Ma, Q., Weidtkamp-Peters, S., Stahl, Y., Felekyan, S., Bleckmann, A., et al. (2015). Real-time dynamics of peptide ligand-dependent receptor complex formation in planta. Sci. Signal. 8:ra76. doi: 10.1126/scisignal.aab0598
Stahl, Y., Grabowski, S., Bleckmann, A., Kühnemuth, R., Weidtkamp-Peters, S., Pinto, K. G., et al. (2013). Moderation of Arabidopsis root stemness by CLAVATA1 and ARABIDOPSIS CRINKLY4 receptor kinase complexes. Curr. Biol. CB 23, 362–371. doi: 10.1016/j.cub.2013.01.045
ten Hove, C. A., Lu, K.-J., and Weijers, D. (2015). Building a plant: cell fate specification in the early Arabidopsis embryo. Development 142, 420–430. doi: 10.1242/dev.111500
Tonaco, I. A., Borst, J. W., de Vries, S. C., Angenent, G. C., and Immink, R. G. (2006). In vivo imaging of MADS-box transcription factor interactions. J. Exp. Bot. 57, 33–42. doi: 10.1093/jxb/erj011
Vatén, A., Dettmer, J., Wu, S., Stierhof, Y.-D., Miyashima, S., Yadav, S. R., et al. (2011). Callose biosynthesis regulates symplastic trafficking during root development. Dev. Cell 21, 1144–1155. doi: 10.1016/j.devcel.2011.10.006
Venugopal, V., Chen, J., Barroso, M., and Intes, X. (2012). Quantitative tomographic imaging of intermolecular FRET in small animals. Biomed. Opt. Express 3, 3161–3175. doi: 10.1364/BOE.3.003161
Verstraeten, I., Schotte, S., and Geelen, D. (2014). Hypocotyl adventitious root organogenesis differs from lateral root development. Front. Plant Sci. 5:495. doi: 10.3389/fpls.2014.00495
Verveer, P. J., and Hanley, Q. S. (2009). “Frequency domain FLIM theory, instrumentation, and data analysis,” in Laboratory Techniques in Biochemistry and Molecular Biology, ed T. W. J. Gadella (Amsterdam; Oxford, UK: Elsevier), 59–94.
Waadt, R., Hitomi, K., Nishimura, N., Hitomi, C., Adams, S. R., Getzoff, E. D., et al. (2014). FRET-based reporters for the direct visualization of abscisic acid concentration changes and distribution in Arabidopsis. eLife 3:e01739. doi: 10.7554/eLife.01739
Weidtkamp-Peters, S., Felekyan, S., Bleckmann, A., Simon, R., Becker, W., Kühnemuth, R., et al. (2009). Multiparameter fluorescence image spectroscopy to study molecular interactions. Photochem. Photobiol. Sci. 8, 470–480. doi: 10.1039/b903245m
Weidtkamp-Peters, S., and Stahl, Y. (2017). The use of FRET/FLIM to study proteins interacting with plant receptor kinases. Methods Mol. Biol. 1621, 163–175. doi: 10.1007/978-1-4939-7063-6_16
Welch, D., Hassan, H., Blilou, I., Immink, R., Heidstra, R., and Scheres, B. (2007). Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Genes Dev. 21, 2196–2204. doi: 10.1101/gad.440307
Wendrich, J. R., Boeren, S., Möller, B. K., Weijers, D., and De Rybel, B. (2017). In vivo identification of plant protein complexes using IP-MS/MS. Methods Mol. Biol. 1497, 147–158. doi: 10.1007/978-1-4939-6469-7_14
Widengren, J., Kudryavtsev, V., Antonik, M., Berger, S., Gerken, M., and Seidel, C. A. M. (2006). Single-molecule detection and identification of multiple species by multiparameter fluorescence detection. Anal. Chem. 78, 2039–2050. doi: 10.1021/ac0522759
Xing, S., Wallmeroth, N., Berendzen, K. W., and Grefen, C. (2016). Techniques for the analysis of protein-protein interactions in vivo. Plant Physiol. 171, 727–758. doi: 10.1104/pp.16.00470
Young, C. L., Britton, Z. T., and Robinson, A. S. (2012). Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol. J. 7, 620–634. doi: 10.1002/biot.201100155
Keywords: protein complexes, protein-protein interaction, fluorescent proteins, in vivo FRET-FLIM, SHORT-ROOT, SCARECROW
Citation: Long Y, Stahl Y, Weidtkamp-Peters S, Smet W, Du Y, Gadella TWJ Jr, Goedhart J, Scheres B and Blilou I (2018) Optimizing FRET-FLIM Labeling Conditions to Detect Nuclear Protein Interactions at Native Expression Levels in Living Arabidopsis Roots. Front. Plant Sci. 9:639. doi: 10.3389/fpls.2018.00639
Received: 27 November 2017; Accepted: 25 April 2018;
Published: 15 May 2018.
Edited by:
Roger Deal, Emory University, United StatesReviewed by:
Lei Li, Centre of Excellence in Plant Energy Biology (ARC), AustraliaCopyright © 2018 Long, Stahl, Weidtkamp-Peters, Smet, Du, Gadella, Goedhart, Scheres and Blilou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ikram Blilou, aWtyYW0uYmxpbG91QGthdXN0LmVkdS5zYQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.