
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 May 2018
Sec. Evolutionary and Population Genetics
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.00604
This article is part of the Research Topic Genetics and Genomics of Polyploid Plants View all 18 articles
 Qing Lu1†
Qing Lu1† Haifen Li1†Yanbin Hong1†Guoqiang Zhang2Shijie Wen1Xingyu Li1Guiyuan Zhou1Shaoxiong Li1Hao Liu1Haiyan Liu1Zhongjian Liu2
Haifen Li1†Yanbin Hong1†Guoqiang Zhang2Shijie Wen1Xingyu Li1Guiyuan Zhou1Shaoxiong Li1Hao Liu1Haiyan Liu1Zhongjian Liu2 Rajeev K. Varshney3,4Xiaoping Chen1*Xuanqiang Liang1*
Rajeev K. Varshney3,4Xiaoping Chen1*Xuanqiang Liang1*Peanut (Arachis hypogaea L.), an important leguminous crop, is widely cultivated in tropical and subtropical regions. Peanut is an allotetraploid, having A and B subgenomes that maybe have originated in its diploid progenitors Arachis duranensis (A-genome) and Arachis ipaensis (B-genome), respectively. We previously sequenced the former and here present the draft genome of the latter, expanding our knowledge of the unique biology of Arachis. The assembled genome of A. ipaensis is ~1.39 Gb with 39,704 predicted protein-encoding genes. A gene family analysis revealed that the FAR1 family may be involved in regulating peanut special fruit development. Genomic evolutionary analyses estimated that the two progenitors diverged ~3.3 million years ago and suggested that A. ipaensis experienced a whole-genome duplication event after the divergence of Glycine max. We identified a set of disease resistance-related genes and candidate genes for biological nitrogen fixation. In particular, two and four homologous genes that may be involved in the regulation of nodule development were obtained from A. ipaensis and A. duranensis, respectively. We outline a comprehensive network involved in drought adaptation. Additionally, we analyzed the metabolic pathways involved in oil biosynthesis and found genes related to fatty acid and triacylglycerol synthesis. Importantly, three new FAD2 homologous genes were identified from A. ipaensis and one was completely homologous at the amino acid level with FAD2 from A. hypogaea. The availability of the A. ipaensis and A. duranensis genomic assemblies will advance our knowledge of the peanut genome.
Peanut (Arachis hypogaea L.) is a grain legume and oilseed crop that is an important source of vegetable oil and protein. It is widely cultivated in tropical and subtropical regions. In Africa and some Asia countries, peanut is more prevalent than any other leguminous crop, including soybean. With an annual production of ~46 million tons and a remarkable 45–56% oil content, it plays a key role in daily human nutrition. Moreover, peanut oil is important to human health owing to its rich nutritional elements, such as oleic acid, linoleic acid, resveratrol, fiber, and vitamins (Parthasarathy et al., 1990).
The Arachis genus originated in South America and is composed of about 80 diploid species that have been divided taxonomically into nine sections (Krapovickas and Gregory, 1994). Arachis species have an unusual reproductive biology in that all members have a geocarpic reproductive habit, with unique growth characteristics of aerial flowers and subterranean fruit (Smith, 1950), that allows them to adapt to particularly harsh environments (Tan et al., 2010). A. hypogaea, cultivated peanut or groundnut, is an allotetraploid (2n = 4x = 40), with an AABB genomic constitution (Temsch and Greilhuber, 2000), which was probably derived from a single recent hybridization of two diploid progenitors (Kochert et al., 1991, 1996; Moretzsohn et al., 2013). Molecular evidence indicates that Arachis duranensis and Arachis ipaensis are the two most likely progenitors that donated the A and B subgenomes, respectively (Kochert et al., 1996; Ramos et al., 2006; Grabiele et al., 2012; Moretzsohn et al., 2013). The genome sizes of the two species are ~1.25 and ~1.56 Gb, respectively (Samoluk et al., 2015), and their sum is close to the total genome size of A. hypogaea (~2.8 Gb) (Temsch and Greilhuber, 2000), indicating that no large changes that affected genome size have taken place since polyploidy. Moreover, researches indicated that the genomes of A. duranensis and A. ipaensis are similar to cultivated peanut's A and B subgenomes (Kochert et al., 1996; Seijo et al., 2007; Robledo et al., 2009; Robledo and Seijo, 2010; Moretzsohn et al., 2013). The high-DNA identity between the A. ipaensis genome and the B subgenome of cultivated peanut, along with biogeographic evidence, indicates that A. ipaensis may be the direct descendant of A. hypogaea that contributed the B subgenome (Bertioli et al., 2016).
The large genome size of A. hypogaea (~2.8 Gb) and highly repetitive content (64%) makes the assembly of the peanut genome sequence very challenging (Dhillon et al., 1980; Temsch and Greilhuber, 2000; Bertioli et al., 2016). Therefore, sequencing and analyzing the genomes of the two diploid ancestors to uncover the genome of cultivated peanut was considered a sensible initial strategy. Our previous sequencing of the peanut A-genome progenitor, A. duranensis, provided new insights into Arachis biology, evolution and genomic changes (Chen et al., 2016). To gain insights into the genomic evolution, as well as the divergence, of the peanut B subgenome and to provide candidate genes to enable a better understanding of the biology of leguminous species, we sequenced the suspected peanut B-genome progenitor, A. ipaensis, and re-sequenced two A-genome and three B-genome genotypes (Chen et al., 2016). The A. ipaensis genome sequencing will facilitate future research on the genome assembly of cultivated peanut and, has the potential to accelerate the molecular breeding of peanut varieties.
The genome of the peanut B-genome progenitor, A. ipaensis (ICG_8206), was sequenced using a shotgun approach on the Illumina HiSeq2500 platform (Supplementary File 1: Figure S1). We generated 250.40 Gb of high-quality reads, representing 149.53 × genome coverage, with fragment lengths ranging from 250 to 20 Kb (Supplementary File 1: Table S1). A total of ~1,391.70 Mb of the A. ipaensis genome sequence was assembled using SOAPdenovo2 (Luo et al., 2012) with a contig N50 of 8,067 bp and a scaffold N50 of 170,050 bp (Table 1; Supplementary File 1: Tables S2, S3). An assessment of the draft genome assembly using the core eukaryotic gene mapping approach method (Parra et al., 2007) revealed that >98% of conserved genes were present in the assembly (Supplementary File 1: Table S4). Over 98% of transcript sequences (>500 bp) were mapped to the assembled genome (Supplementary File 1: Table S5). Based on k-mer statistics, the A. ipaensis genome is estimated to be ~1,475.83 Mb, which is consistent with the total scaffold length (Supplementary File 1: Table S6 and Figure S2). The average GC content is 36.70% (Table 1; Supplementary File 1: Figure S3), which is equivalent to that of the A. duranensis genome (Chen et al., 2016), and its distribution is highly similar to previously reported Arachis genomes (Bertioli et al., 2016; Chen et al., 2016) but different from those of Glycine max, Arabidopsis thaliana, and Oryza sativa (Supplementary File 1: Figure S4).
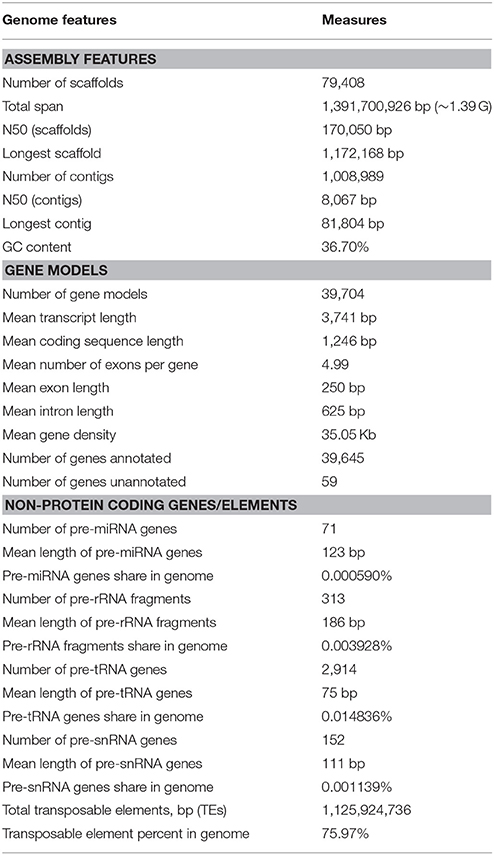
Table 1. Genome assembly and annotation of the A. ipaensis.
We predicted 39,704 genes with average transcript and coding sequence lengths of 3,741 and 1,246 bp, respectively (Table 1). The whole-genome's gene density is one gene per 35.05 Kb (Figure 1 and Table 1), and the mean exon and intron lengths per gene are 250 and 625 bp (Table 1), respectively, which were relatively longer than those in other leguminous species, such as Cicerarietinum (Varshney et al., 2013) and G. max (Schmutz et al., 2010). Compared with the gene sets of legumes, oilseeds, and other plant species (Supplementary File 1: Table S7), the distribution of the A. ipaensis gene features is most similar to those of A. duranensis and legumes, such as C. arietinum and G. max, but different from those of non-leguminous species, such as A. thaliana and O. sativa (Supplementary File 1: Table S8 and Figure S5). Moreover, the A. ipaensis gene number is comparable to those of Lotus japonicus (39,366) and Zea mays (39,498), greater than that of C. arietinum (24,819), and substantially lower than those of G. max (54,174) and Medicago truncatula (50,444) (Supplementary File 1: Table S9). Functions were tentatively assigned to 39,645 genes but not to 59 genes that may be peanut-specific (Table 1). Most of the A. ipaensis genes have homologous gene models in the TrEMBL (99.82%) and Interpro (71.29%) databases (Bairoch and Apweiler, 2000; Zdobnov and Apweiler, 2001), and ~99.85% of the gene models matched entries in publically available databases (Supplementary File 1: Table S10). Conservative analyses indicated that the predicted proteins of A. ipaensis were most similar to those of A. duranensis (88.10%), followed by Cajanus cajan (67.4%), and least similar to those of gramineous crops, such as Sorghum italica (33.53%) and S. bicolor (34.51%) (Supplementary File 1: Table S8).
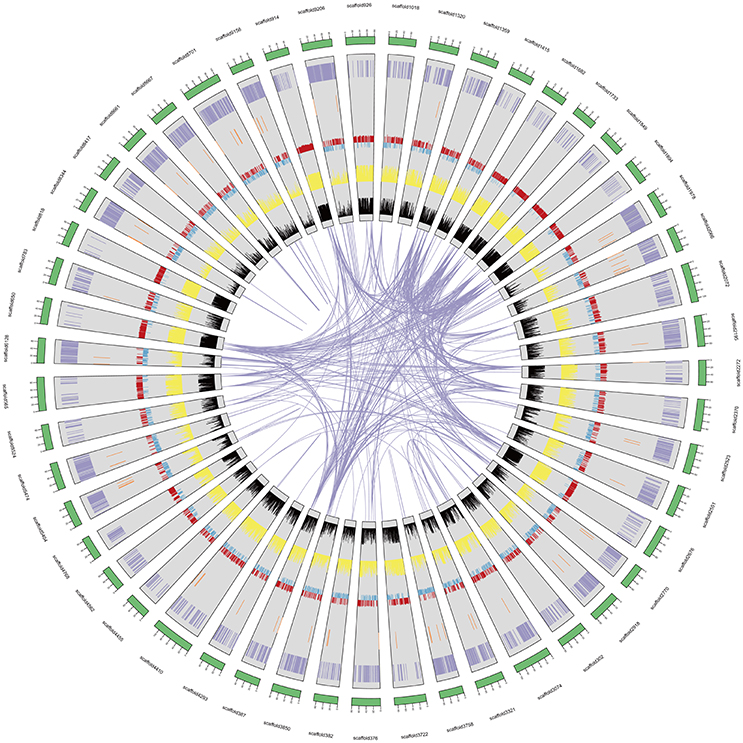
Figure 1. A. ipaensis genome overview. From the outer edge inward, circles represent the 50 largest DNA sequence scaffolds (green), the genes on each scaffold (purple), the non-coding RNA on each scaffolds (brown), GC content (red and blue), repeat density at 10 Kb (yellow), and transposable element density at 10 Kb (black).
A total of 2,530 putative A. ipaensis transcription factor (TF) genes were identified in 58 families, which was equal to or slightly higher than of the numbers found in O. sativa and A. thaliana, much higher than in L. japonicus but lower than in G. max and Glycine soja (Supplementary File 1: Figure S6). The distribution of the A. ipaensis TF genes among the families was highly similar to those of A. duranensis and G. max (Supplementary File 1: Figure S7). FAR1 was dominant in A. ipaensis (Figure 2A), as in the A-genome progenitor, A. duranensis (Chen et al., 2016). More importantly, the FAR1 TF families play pivotal roles in modulating phyA-signaling homeostasis (Lin et al., 2007), and phyA, together with phyB, regulate skotomorphogenesis and photomorphogenesis in higher plants (Medzihradszky et al., 2013). The FAR1 TF families identified in A. thaliana contained several conservative motifs (Supplementary File 1: Figure S8), and phyA and phyB were highly expressed in different tissues (shoot, seed, leaf, flower, and root) at different growth stages in A. thaliana (Supplementary File 1: Figure S9). In addition, previous non-synonymous substitutions per non-synonymous site (Ka)/synonymous substitutions per synonymous site (Ks) analyses of phyB in A. duranensis and G. max showed evidence of positive selection (Chen et al., 2016). These findings may enhance our understanding of peanut's unique fructification, having aerial flowers but subterranean fruit, as well as providing evidence for different regulators of biological functions in Arachis and other plants.
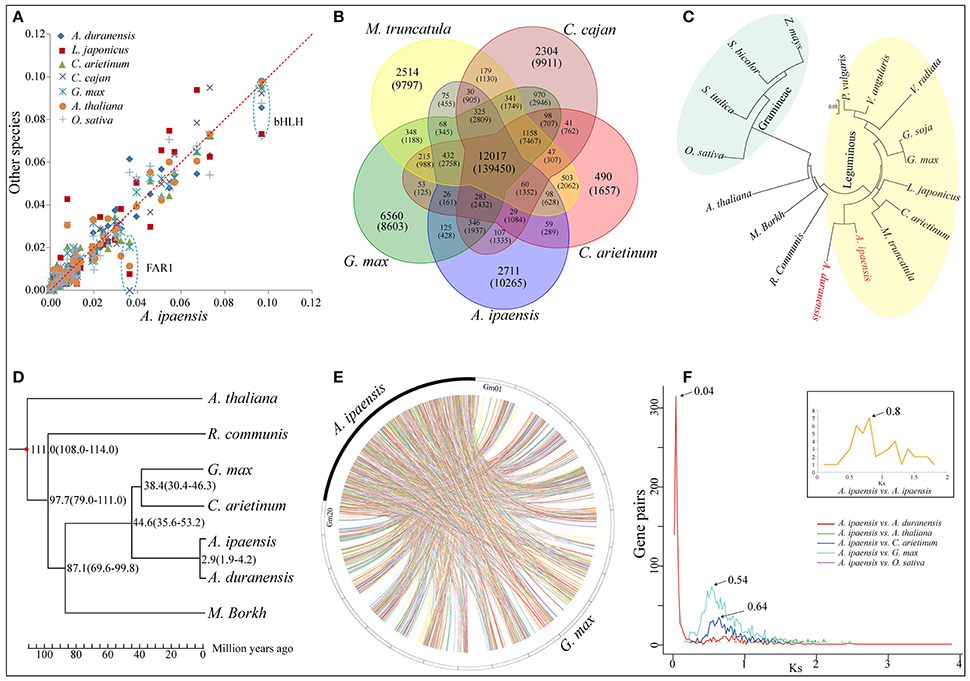
Figure 2. Comparative genomic and evolutionary analysis. (A) Scatter plot of percentage of A. ipaensis transcription factors in relation to L. japonicas, C. arietinum, C. cajan, G. max, A. thaliana and O. sativa. (B) Venn diagram showing distribution of gene families among A. ipaensis, G. max, M. truncatula, C. cajan and C. arietinum. (C) Cluster tree for 17 plant species including common leguminous and gramineous crops based on single copy orthologous genes. (D) Phylogenetic tree for 7 representative plant species. The numerical on each node represents the estimated differentiation time using the evolutionary time between A. thaliana and G. max (~108–114 Mya) as a correction. (E) Syntenic relationship between A. ipaensis scaffolds and G. max chromosomes. (F) Synonymous substitution rate (Ks) dating of duplication blocks in A. ipaensis and different combinations of orthologs of A. duranensis, A. thaliana, C. arietinum, G. max, and O. sativa. Different colored lines represent the distribution of Ks against orthologs gene pairs among different plant species. Inset shows the distribution of Ks between the gene pairs present in the duplicated blocks within the A. ipaensis genome.
We identified 71 Arachis pre-microRNAs (pre-miRNAs) (Supplementary File 2: Data S1) with an average length of 123 bp, 2,914 pre-transfer RNAs (pre-tRNAs) with an average length of 75 bp, 313 pre-ribosomal RNAs (pre-rRNAs) with an average length of 186 bp including 5S (108), 5.8S (55), 18S (82), and 28S (68), and 152 pre-small nuclear RNAs (pre-snRNAs) with an average length of 111 bp. These genes represent 0.000590, 0.014836, 0.003928, and 0.001139% of the A. ipaensis genome, respectively (Table 1; Supplementary File 1: Table S11).
Approximately 75.97% of the A. ipaensis genome is composed of transposable elements (Figure 1; Tables 1, 2), which was higher than other legumes, such as G. max (59.00%) (Schmutz et al., 2010), C. cajan (51.60%) (Varshney et al., 2011) and M. truncatula (30.50%) (Young et al., 2011). Long-terminal repeat (LTR) retrotransposons are the dominant transposable elements, covering 64.15% of the nuclear genome (Table 2). Sequence divergence analyses indicated that most of A. ipaensis transposable elements had a ~30% divergence rate (Supplementary File 1: Figure S10).
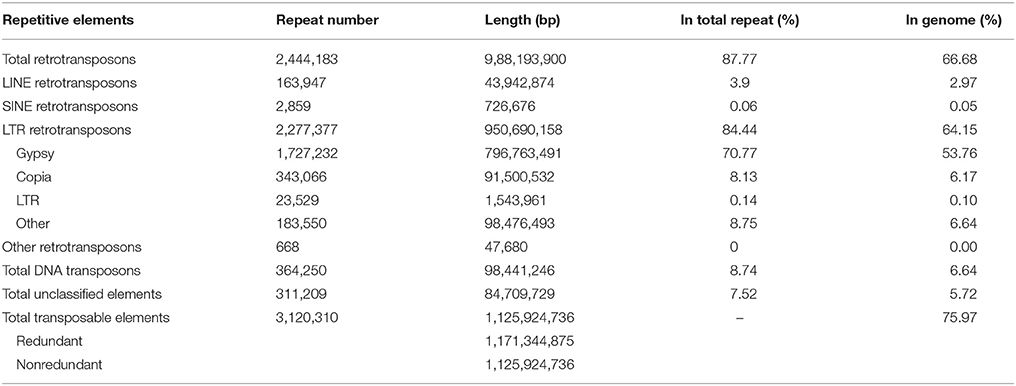
Table 2. Organization of repetitive sequences in A. ipaensis genome.
The A. ipaensis genome contains 188,075 simple sequence repeats (SSRs), for which 80,218 SSR primers were designed (Supplementary File 1: Table S12; Supplementary File 3: Data S2). Of these SSRs, the di-nucleotide repeats are the most abundant, accounting for 48.38% of the total SSRs, followed by tri- nucleotide repeats (28.06%) (Supplementary File 1: Table S12). Among the di-nucleotide type, the AT/AT motif type had the greatest frequency (~21.9%). Among the tri-nucleotide type, the AAT/ATT is dominant (~11.4%) (Supplementary File 1: Figure S11). Using two A-genome genotypes (ICG_8123 and ICG_8138) and three B-genome genotypes (ICG_8960, ICG_8209, and ICG_13160) that were re-sequenced in our earlier study (Chen et al., 2016), we identified 26,050,150 variations, including 24,688,277 single nucleotide polymorphisms (SNPs) and 1,361,873 insertion-deletions (InDels) (Supplementary File 1: Table S13 and Figure S12). Among these variations, ~4 million SNPs were present in the two diploid A species (ICG_8123 and ICG_8138). By contrast, ~5 million SNPs were identified in the comparison of the three diploid B species (ICG_8960, ICG_8209, and ICG_13160) (Supplementary File 1: Table S13 and Figure S12). Thus, the diploid B species Arachis magna and Arachis batizocoi may have more abundant genetic diversity than the diploid A species A. duranensis when compared with the reference A. ipaensis (ICG_8206) genome assembly. The geographical origin of Arachis indicated that the distribution of A. duranensis is more extensive and also closer to that of A. ipaensis which has only one known location of origin, than A. magna (Bertioli et al., 2016). Another source of confusion among the variations may result from the two A-genome genotypes having fewer mapped reads than the three B-genome genotypes.
A total of 16,791 orthologous gene groups were identified among 18 species using OrthoMCL (Li et al., 2003; Supplementary File 4: Data S3), including 959 A. ipaensis-specific families containing 6,443 genes (Supplementary File 1: Table S14). A gene ontology (GO) annotation suggested differentially enriched functional categories in the peanut-specific families, indicating that these gene families may be closely related to the unique Arachis growth characteristics, such as aerial flowers but subterranean fruit, and lipid biosynthesis (Supplementary File 1: Figures S13–S15). Moreover, 1,624 of these orthologous groups were single-copy orthologs (Supplementary File 1: Table S15 and Figure S16). In addition, 6,443 unique paralogs of A. ipaensis genes occurred in species-specific homolog groups, indicating that these groups could arise from genomic structural rearrangements that obscured simple orthology, such as nonallelic recombination or gene conversion, followed by duplication (Supplementary File 1: Table S15 and Figure S16; Varshney et al., 2013). We identified 12,017 orthologous groups common to all five leguminous species (Figure 2B), 11,985 groups between A. ipaensis and Ricinus communis (oilseed crop) (Supplementary File 1: Figure S17), 9,099 groups between A. ipaensis and Gramineae/Poaceae crops (Supplementary File 1: Figure S18), and 10,501 orthologous groups are common to A. ipaensis and other distantly related plant species (Supplementary File 1: Figure S19). These results provide an important molecular foundation for comparative biology and for functional mechanistic inferences in A. ipaensis, as well as other species, because simple orthologous family genes often exhibit conserved molecular functions that were maintained during evolution process.
A polygenetic tree based on single-copy orthologous genes showed A. ipaensis and A. duranensis in the same clade, which did not include any other leguminous species, indicating their closer genetic distance and divergence time (Figure 2C). Furthermore, a special phylogenetic tree estimated that the divergence of the two species occurred ~2.9 million years ago (Mya) (Figure 2D), which was fairly consistent with a previous report (~2.16 Mya) (Bertioli et al., 2016). Syntenic blocks identified between A. ipaensis and other species was found to be extensively conserved (Supplementary File 1: Table S16). The largest number of syntenic blocks was identified between A. ipaensis and G. max (Figure 2E). The longest syntenic block (>10 Kb) was observed between A. ipaensis and A. duranensis (Supplementary File 1: Table S16). The numbers of syntenic blocks identified within the respective A. ipaensis and A. duranensis genomes were extremely lower than the number between the two genomes (Supplementary File 1: Figure S20) as well as the number between the A. ipaensis and G. max genomes (Supplementary File 1: Figure S21; Bertioli et al., 2016), indicating that few large-scale genome duplication events occurred in the A. ipaensis genome's evolution or that syntenic blocks were lost after genome duplication events.
The Ks values between paralogous or orthologous genes reveals a mechanism of molecular evolution (Lna, 1996). Distributions of Ks distances between paralogs within A. ipaensis and orthologs among A. ipaensis, leguminous crops and other species were plotted (Figure 2F). The A. ipaensis paralogs showed a peak at ~ 0.80, which is similar to those of M. truncatula (~0.80) and L. japonicus (~0.73) (Cannon et al., 2006) but lower than those of A. duranensis (~0.9) and A. ipaensis (~0.95) (Chen et al., 2016). Thus, the whole-genome duplication events of A. duranensis and A. ipaensis occurred around the time that corresponds to a Ks value range of 0.8–0.95. In addition, A. duranensis and A. ipaensis orthologs showed a prominent peak at ~0.04, which is consistent with a previous study (Bertioli et al., 2016). Assuming a synonymous substitution rate per synonymous site of 6.1 × 10−9 per year for eudicots (Lynch and Conery, 2000), the two species were estimated to have diverged ~3.28 Mya, which is close to the estimation based on the phylogenetic tree (Figure 2D). Furthermore, Ks dating suggested the divergence of A. ipaensis and G. max (Ks = ~0.54) at 44.3 Mya and that of A. ipaensis and C. arietinum (Ks = ~0.64) at 52.5 Mya.
The graphic trend of the Ka/Ks (ω) and Ks between the orthologs of A. duranensis and A. ipaensis formed three clusters, such as Ks = 0–0.3, 0.5–1.5, and >1.5, and the ω values decreased as the Ks values increased (Supplementary File 1: Figure S22). The genes with Ks ≥ 1.5 are attributed to pan-eudicot palaeoploidization, and the genes with lower ω ratios are considered to be under neutral selection. Here, the 45 A. ipaensis genes with ω ratios > 1 may be under positive selection pressure (Supplementary File 1: Figure S23).
Peanut is an allotetraploid species that may have originated from a single resent hybridization event between two diploid species, followed by polyploidization. Cytogenetic, phylogeographic and molecular evidence indicates that A. duranensis and A. ipaensis are the most likely donors of the A and B subgenomes, respectively (Kochert et al., 1996; Seijo et al., 2007; Robledo et al., 2009; Robledo and Seijo, 2010; Moretzsohn et al., 2013). A previous study estimated the divergence of the two species at ~2.88 Mya (Moretzsohn et al., 2013). The estimation using a comparative genomics analyses between them was ~2.9 Mya, which was fairly consistent with our report. Moreover, sequence comparisons with tetraploid cultivated peanut estimated the divergence times of A. duranensis and A. ipaensis from the A and B subgenomes of A. hypogaea as ~247,000 and ~9,400 years, respectively (Bertioli et al., 2016).
Comparative genomics analyses of chromosomal structure and synteny between A. duranensis and A. ipaensis indicated that some chromosomes shared a conservative linear structure that was partially in accordance with our other analyses (Supplementary File 1: Figure S20). Other analyses showed a large inversion in one or both arms of a chromosome (Bertioli et al., 2016). In contrast, chromosomes 07 and 08 have undergone complex rearrangements that were consistent with cytogenetic observations (Seijo et al., 2007; Nielen et al., 2010). Importantly, a genomic comparison showed a fundamentally one-to-one correspondence between the diploid chromosomes and cultivated peanut linkage groups. However, the A. duranensis chromosomes were less similar to A. hypogaea sequences than those of A. ipaensis (Bertioli et al., 2016). These results may help to uncover potential mechanisms of hybridization events in the future.
Plant NBS-LRR proteins encoded by resistance genes (R genes) play key roles in the responses of plants to various pathogens. The R genes can be classified into various subfamilies based on the present of different domain, such as CC-NB-LRR, TIR-NB-LRR, ser/thr-LRR, Kin-LRR, and others (e.g., Mol and Asc-1; Sanseverino et al., 2010). The A. ipaensis genomic assembly contains 1,437 putative disease R genes as assessed by a screening of the PRG database (Supplementary File 1: Table S17; Supplementary File 5: Data S4; Sanseverino et al., 2010). Compared with other legumes, the A. ipaensis genome possesses more R genes than the G. max and M. truncatula genomes but less than the A. duranensis and C. cajan genomes. Moreover, these R genes tend to cluster on different scaffolds. For example, several large clusters containing 6–10 R genes are located on six different scaffolds (Supplementary File 1: Figure S24). The NL subfamily of genes, which confers resistance against pests and diseases, is the second largest R gene-containing family, and these genes can be clustered reasonably into different individual clades in A. ipaensis, A. duranensis, and A. thaliana, indicating that gene divergence occurred during the evolution of the three species (Supplementary File 1: Figure S25). In addition, we analyzed protein motifs in the most homology of the top 20 R genes found in PRG database using MEME (Bailey et al., 2009), and the results showed highly conserved LRR motifs (Supplementary File 1: Figure S26). However, further investigation is required to determine the biological functions of these R genes.
Nitrogen is one of the most important plants require nutrients, and in agriculture nitrogen availability influences both crop yield and quality. Leguminous plants, such as peanut, soybean, and Medicago, can transform molecular nitrogen into available ammonia nitrogen through the leguminous-root-nodule bacteria nitrogen-fixing system that results from the symbiotic interactions between leguminous plants and rhizobia (Figure 3A). In the A. ipaensis and A. duranensis genomic assemblies, 16 and 38 root-nodule developmental genes respectively, have been identified (Supplementary File 1: Table S18; Supplementary File 6: Data S5). As expected, there are greater numbers of nodulation-related genes present in leguminous plants than in non-leguminous plants, such as A. thaliana, O. sativa, and Z. mays (Supplementary File 1: Figure S27).
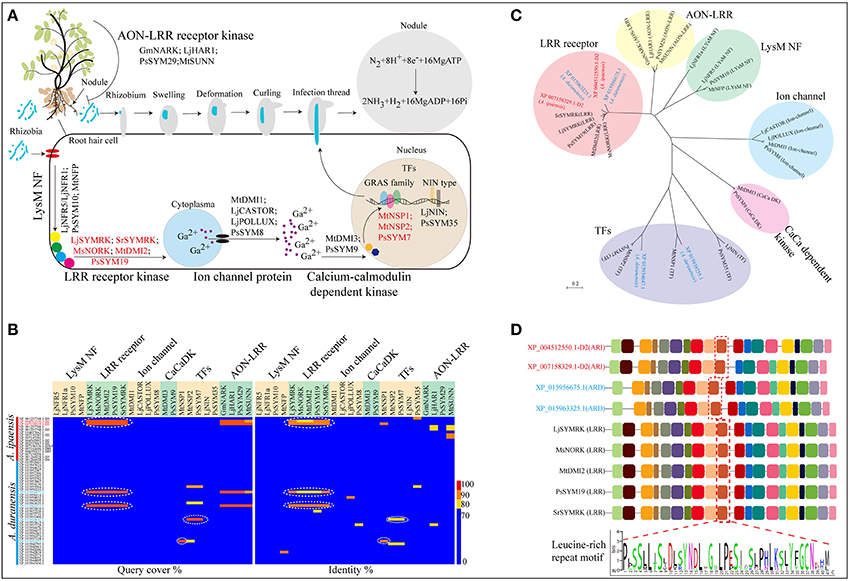
Figure 3. Biological nitrogen fixation in leguminous plants. (A) Genes involved in nodule initiation, development and signal recognition pathway. (B) Protein sequence alignment of Nod related genes identified in A. ipaensis and A. duranensis. (C) Phylogenetic tree of nodule development genes and their homologs from A. ipaensis and A. duranensis. (D) Identification of high conserved domains of leucine-rich repeat (LRR) receptor kinases. Red dashed boxes represent LRR conserved motif. (A) The rhizobium (blue) attach to the surface of root hair cell. After swelling, deformation, curling and infection thread, the bacteria are released into cells via endocytosis then a vacuole-like structures (symbiosomes), in which the bacteria convert N2 to NH3, formed. But how is the Nod signal transmitted? Initially, the rhizobia-derived signal is perceived by LysM-type protein receptor kinases, such as NRF1 and 5 (Radutoiu et al., 2003) and SYM10 (Schneider et al., 2002) identified in L. japonicus and P. sativuml, followed by a downstream leucine-rich receptor kinase, for example SYMRK (Stracke et al., 2002 and Capoen et al., 2005), NORK (Endre et al., 2002), DMI2 (Catoira et al., 2000), and SYM19 (Stracke et al., 2004) from L. japonicus, Sesbania rostrata, M. sativa, M. truncatula, and P. sativuml, respectively. Then, the Nod factor (NF) signal is processed through a signal transduction cascade involving proteins including ion channels [MDI1(Ané et al., 2004), CASTOR (Imaizumi-Anraku et al., 2005), POLLUX (Imaizumi-Anraku et al., 2005), and SYM8 (Edwards et al., 2007)], calcium-calmodulin-dependent kinase (CaCaDK) (MDI3 and SYM9) (Lévy et al., 2004) and transcription factors [NSP1 (Smit et al., 2005), NSP2 (Kaló et al., 2005), SYM7 (Kaló et al., 2005), NIN (Schauser et al., 1999), and SYM35 (Borisov et al., 2003)]. Finally, rhizobia infection occurred primarily through uncharacterized target genes that may be activated by these TFs.
Nitrogen-fixing root nodules are important symbiotic organs that provide an epiphytic site for rhizobia to convert atmospheric nitrogen to ammonia, and supply its host plant with fixed nitrogen. In return, the rhizobia gain photosynthates from the plant (Figure 3A). In leguminous plants, multiple genes are involved in the formation and development of root nodules, as well as in the autoregulation of the nodulation (AON) process, which is a systemic feedback loop used to avoid an excessive energy expenditure from “unwanted” nodulation (Figure 3A; Supplementary File 7: Data S6). Here, four homologous LRR receptor kinase genes were identified in A. ipaensis (XP_004512550.1-D2 and XP_007158329.1-D2) and A. duranensis (XP_015956675.1 and XP_015963325.1) (Figure 3B; Supplementary File 1: Figure S28; Supplementary File 7: Data S6). A phylogenetic tree showed that the four homologous genes were clustered into an independent clade, together with other LRR receptor kinase genes (Figure 3C). Interestingly, these four genes contain multiple common motifs, including a conserved LRR motif, indicating a similar biological function (Figure 3D). The GO analyses suggested that the four homologous genes are involved in ion binding and signal transducer activity (Supplementary File 1: Figures S29–S32). More importantly, the proteins encoded by the four genes showed similar three-dimensional structures and localized on the cell membrane (Supplementary File 1: Figures S29–S32).
We also identified two other nodule development-related genes (XP_015934647.1 and XP_015939255.1) that are homologous to the TF genes of the GRAS family in A. duranensis. One gene is homologous with MtNSP2 and PsSYM7 (Kaló et al., 2005), while the other is an ortholog of MtNSP1 (Imaizumi-Anraku et al., 2005) (Figures 3A,B; Supplementary File 1: Figure S33; Supplementary File 7: Data S6). The phylogenetic tree indicated that the two homologs were classified into the TF category but appeared in different branches (Figure 3C). In addition, the GO enrichment indicated that the two genes participate in the regulation of multiple biological processes, such as nucleic acid-binding TF and signal transducer activities (Supplementary File 1: Figures S34, S35). The three-dimensional structures of the two proteins were completely dissimilarity, and the two proteins localized in the nucleus (Supplementary File 1: Figures S34, S35). These results could provide candidate genes and basic bioinformation for further functional studies of nodule formation in leguminous crops.
Peanut (A. hypogaea L.) is a typical upland crop in tropical, subtropical, and warm temperate climates. Drought adaptation plays a central role in their growth and development. During drought stress, TFs, such as MYB, MYB-related, NAC, WRKY, bZIP, and ERF, are involved in numerous physiological responses (Shinozaki and Yamaguchi-Shinozaki, 2000) (Supplementary File 1: Figure S36). Here, the total number of TF genes identified in upland crops was greater than that found in hygrophilous plants (Supplementary File 1: Table S19 and Figure S37). Notably, in A. ipaensis we identified 185 MYB and 129 MYB-related TFs (Supplementary File 1: Table S19), most of which contain a highly conserved DNA-binding domain, and they are key factors in regulatory networks controlling development, metabolism and responses to biotic and abiotic stresses (Dubos et al., 2010). The second large number of drought tolerance-related TFs, with 170 members, is the ERF family (Supplementary File 1: Table S19). The ERF proteins, sharing a conserved 58–59 amino-acid domain, are key regulators linked to responses to plant stresses, such as cold, drought and pathogen attack (Supplementary File 1: Figure S38; Singh et al., 2002). In A. duranensis, A. ipaensis and A. hypogaea species, sets of 51, 57 and 53 ERF TF family proteins, respectively, were obtained from the Plant Transcription Factor Database (Jin et al., 2015, 2017; Supplementary File 1: Figures S39–S41). These TF proteins contained different DNA-binding domains and can be categorized into different branches based on different motif permutation structures, indicating the distinct functional and evolutionary features of ERF TFs in different Arachis species (Supplementary File 1: Figures S39–S41).
Heat-shock proteins (Hsps)/chaperones are important defense mechanism members against abiotic stresses, such as drought, salinity and extreme temperatures (Wang et al., 2004; Supplementary File 1: Figure S42). Drought stress is a common factors that induces Hsp expression (Kimpel et al., 1990; Sun et al., 2002). To elucidate the cause of drought tolerance, five major families of Hsps/chaperones were identified in upland crops and hygrophilous plants (Supplementary File 1: Table S20 and Figure S43). As expected, the total number of Hsps/chaperones obtained in upland crops was much great than in hygrophilous plants (Supplementary File 1: Figure S43).In particular, A. ipaensis and G. soja had 118 and 34 Hsp70 subfamilies, respectively, compared with only 1 in rice (Supplementary File 1: Table S20). The great number of Hsps/chaperones detected in A. ipaensis and G. soja indicates the nature of drought adaptation in upland crops.
The subtilisin-like protease (SDD1) gene family is involved in the regulation of stomatal density and distribution to adjust for drought stress by modulating the apertures of these pores flanked by two guard cells (Berger and Altmann, 2000). In the expanded gene families, 39 and 40 SDD1 genes were identified in A. ipaensis and A. duranensis, respectively (Supplementary File 8: Data S7). These gene families were divided into different clusters according to their related functions but showed a pattern of cross-distribution in each cluster based on their different genetic relationships (Supplementary File 1: Figure S44).
Because peanut is an important oilseed crop, 1,613 A. ipaensis genes related to the biosynthesis of fatty acids and triacylglycerols were identified, which was more than were identified in the nonoilseed plant Arabidopsis (1,380) and rice (1,419) (Supplementary File 1: Table S21). In addition, fatty acids and triacylglycerols synthesis involves many key enzymes, such as ACCase (Slabas and Fawcett, 1992), acyl-ACP thioesterase (A and B) (Dörmann et al., 2000; Bonaventure et al., 2003; Serrano-Vega et al., 2005), LACS (Zhao et al., 2010), DGAT (Yen et al., 2008), and FAD (Pham et al., 2012) (Supplementary File 1: Figure S45). When we manually investigated the homologous genes in the storage lipid biosynthesis pathway using the Arabidopsis Lipid Gene Database (Mekhedov) (http://lipids.plantbiology.msu.edu/), 116 nonredundant homologs potentially involve in lipid biosynthesis were obtained in A. ipaensis (Supplementary File 9: Data S8). Consistent with the lipids produced in peanut seeds, one, and nine homologous genes encoding acyl-ACP thioesterase A and B (FATA and FATB), respectively, the two key enzymes leading to the synthesis of fatty acid, were identified. Moreover, multiple copies or isoforms of some key genes, such as FAD2, LACS, and KAS, involved in triacylglycerol synthesis were also detected in the A. ipaensis genome (Supplementary File 9: Data S8).
FAD2, encoding δ-12 oleic acid desaturase, is the essential gene that controls linoleic acid biosynthesis (López et al., 2000). It converts oleic acid to linoleic acid by desaturating the δ-12 carbon and determines the multi-polyunsaturated fatty acid content and proportion in most oil seed plants (Figure 4A). In this study, three new FAD2 homologous genes (XP_004497897.1-D3, XP_007162321.1, and XP_007162321.1-D2) were identified in A. ipaensis (Supplementary File 1: Figure S46). The proteins of FAD2 and its homologs contain the highly conserved feature of three enzyme-specific histidine boxes (Figure 4B), which are considered to be essential for desaturase activity because they act as potential ligands for iron atoms (Sakai and Kajiwara, 2005). A phylogenetic tree showed that FAD2 clustered into five groups based on its genus, and the three homologous genes were more closely related to the evolutionary kinship of oil seed plants, especially A. hypogaea (Figure 4C). This result indicated that FAD2 is an extremely conserved gene in the fatty acid biosynthesis pathway. In addition, the GO terms revealed that the three homologous genes having δ-12 oleic acid dehydrogenase activities (ω-6 fatty acid desaturase activities) were involved in the fatty acid biosynthesis process and that the proteins encoded by the three genes were subcomponents of the endoplasmic reticulum membrane. They had similarity three-dimensional structures, which was supported by the predicted protein subcellular localization (Supplementary File 1: Figures S47–S49).
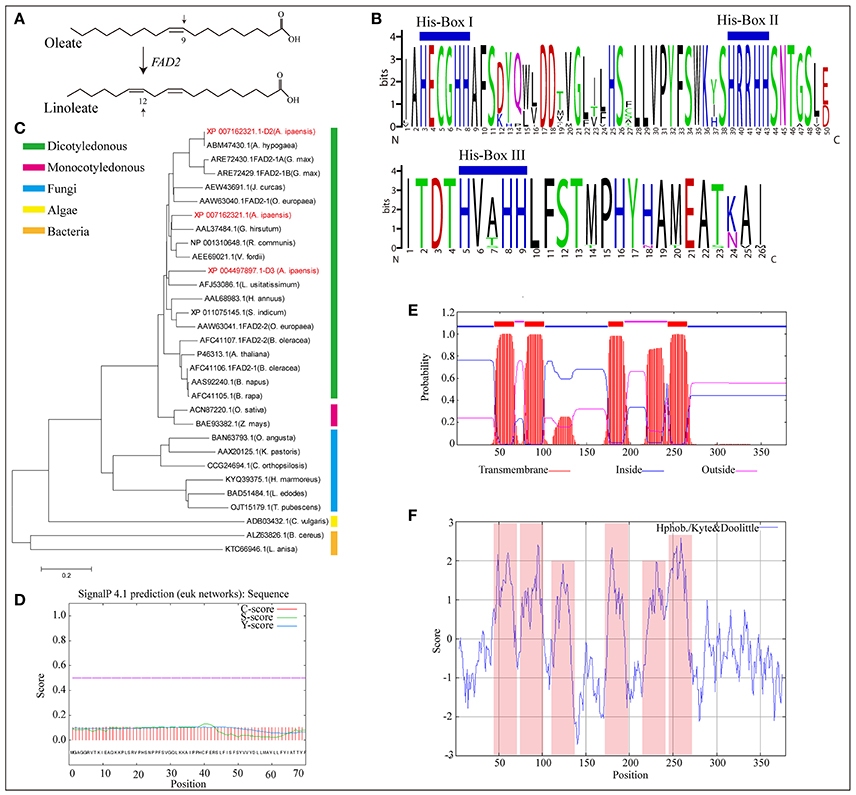
Figure 4. Homologous genes of δ-12 oleic acid desaturase (FAD2). (A) FAD2 catalyze oleate into linoleate. (B) Multiple alignment of amino acid sequence of substrate binding motif of FAD2 in oil seed plants and its homologous genes in A. ipaensis. (C) Phylogenetic tree of FAD2 and its homologous genes from different species. (D) Signal peptides analysis of FAD2 homologous gene (XP_007162321.1-D2) from A. ipaensis. (E) Tansmembrane region prediction of FAD2 homologous gene, XP_007162321.1-D2. Red, blue, and pink boxes represent transmembrane, inside, and outside domains. (F) Hydrophobicity and hydrophilicity prediction for the homologous gene XP_007162321.1-D2. Pink box represent protein hydrophobic region.
Pairwise comparisons of the amino acid sequences of XP_007162321.1-D2 from A. ipaensis with FAD2 from A. hypogaea revealed 100% sequence identities with no gaps (Supplementary File 1: Figure S50), which confirmed the ancestral origin of FAD2 as being the A. ipaensis genome. A signal peptide analysis showed a low level S-score, indicating a typical non-secretory protein with no leading peptide (Figure 4D). This was supported by the predicted protein subcellular localization (Supplementary File 1: Figure S47E). Moreover, four transmembrane domains were predicted in their amino acid sequence (Figure 4E). Importantly, the protein hydrophobicity/hydrophilicity prediction revealed four strong hydrophobic regions, which completely overlapped with the transmembrane regions (Figure 4F). These results provide information for exploring the origin of FAD2, and the homologous gene will be of service to peanut improvement for high oleic acid.
Among the key enzyme-encoding genes, 82 nonredundant homologous genes had high distributions of non-synonymous substitutions (Ka/Ks > 1.0) between A. ipaensis and A. thaliana as assessed by the branch-site likelihood ratio test, indicating positive selection during domestication (Supplementary File 1: Figure S51; Supplementary File 10: Data S9). Coincidentally, 21 fatty acid biosynthesis genes located in multiple improvement-selective sweeps regions were obtained through combined genome selective sweeps and GWAS analyses in soybean (Zhou et al., 2015). Thus, we hypothesize that these 82 genes, including FAD2 (2), KASIII (2), and FATB (6) homologs with high Ka/Ks values (Supplementary File 1: Figure S52) may also have undergone domestication.
TFs that regulate seed development play crucial roles in seed lipid accumulation. To date, the TFs regulating lipid metabolism mainly belong to the following 6 super gene families, AP2/EREBP, B3, NF-Y, Dof, MYB, and MYC (Song et al., 2016). The number of the TF families identified in oilseed crops is much great than in non-oilseed plants (Supplementary File 1: Figure S53). Information related to these genes involved in fatty acid and triacylglycerol metabolic pathways is important for modifying the oil quality of peanut as well as other oilseed crops.
The draft genome sequence of A. ipaensis, together with those of L. japonicus, M. truncatula, C. cajan, C. arietinum, and G. max, will provide new biological information for an important branch of the legume clade. The A. ipaensis genome sequence presented here, combined with our previous sequence of A. duranensis, will shed light on the genomic evolution and polyploidization mechanisms of polyploid species. In addition, the biological information of the A. ipaensis genome provides a fundamental resource for understanding disease resistance, symbiotic nitrogen fixation, environmental adaptation and oil biosynthesis in peanut. Moreover, high-density molecular markers, such as SSRs and SNPs, identified in the A. ipaensis draft genome can be used to investigate the genetic diversity and make genetic changes to improve important agronomic traits in peanut.
The Arachis genus is composed mostly of diploid species (2n = 2x = 20). Peanut (A. hypogaea L.) is an allotetraploid (AABB-type genome; 2n = 4x = 40), probably derived from a single recent hybridization event between A. duranensis (AA subgenome, 2n = 2x = 20) and A. ipaensis (BB subgenome, 2n = 2x = 20) (Supplementary File 1: Figure S1; Koppolu et al., 2010; Chen et al., 2016). In 2016, an accession of A. ipaensis K30076 has already been sequenced (Bertioli et al., 2016). The accession collected by A. Krapovickas, W.C. Gregory, D.J. Banks, J.R. Pietrarelli, A. Schinini, and C.E. Simpson in 1977 was maintained at Embrapa Genetic Resources and Biotechnology (Brasília, Brazil), which probably originated form Villa Montes near Camatindi or Tigüipa, Bolivia (https://www.peanutbase.org/; Bertioli et al., 2016). In this study, the accession of A. ipaensis ICG_8206 maintained at International Centre for Research in the Semi-Arid Tropics (India) then introduced to Crops Research Institute-Guangdong Academy of Agricultural Sciences (China) was used. Although cytogenetic, phylogeographic and molecular evidence showed that the accession of A. ipaensis K30076 was the most probable B-genome donor for A. hypogaea (Seijo et al., 2007; Robledo and Seijo, 2010; Bertioli et al., 2016), genetic relationship analyses indicated that the B-genome accession ICG 8206 (A. ipaensis) was the most closely related to A. hypogaea (Koppolu et al., 2010).
Here, the A. ipaensis (ICG_8206) was sequenced by Illumina HiSeq2500 platform. Total genomic DNA was isolated from the etiolated unopened young leaves of 20-day-old plants cultivated in dark chamber according to a modified CTAB procedure (Doyle and Doyle, 1990). This work will also be of great importance to guide cultivated peanut's genome assembly as a necessary complement in future.
Whole-genome shotgun sequencing was performed under the HiSeq2500 Sequencing System with 11 paired-end sequencing libraries, including 250, 500, 800 bp, 2, 5, 10, and 20 Kb using the standard protocol provided by Illumina (San Diego, USA).
SOAPdenovo2 (version 2.04.4) (Luo et al., 2012) was employed with optimized parameters to construct contigs and original scaffolds as previous described (Chen et al., 2016). Subsequently, SSPACE (version 2.0) (Boetzer et al., 2011) was used to link the scaffolds constructed by the SOAPdenovo2 as previous described (Chen et al., 2016).
The genome size was estimated based on the 17 K-mer distribution using the total length of sequence reads divided by sequencing depth, and the frequency of each 17-mer were calculated from the whole genome sequenced reads to evaluate the sequencing depth. Subsequently, the A. ipaensis genome size was calculated by following the algorithm: Genome size = K-mer number/Peak depth (Bertioli et al., 2016).
The gene coverage of the assembled genome was comprehensively evaluated using available public transcript sequence tags or expressed sequence tags. Core eukaryotic genes identified by CEGMA v.2.3 (Parra et al., 2007) were remapped to the A. ipaensis genome assembly by BLAT (Kent, 2002) to evaluate the quality of the assembly. CEGMA data were downloaded from the Korf Lab research group at the Genome Center, UC Davis (http://korflab.ucdavis.edu/datasets/cegma/#SCT6).
To annotate the A. ipaensis genome, an automated genome annotation pipeline MAKER was performed to produce de novo gene prediction, infer 5′ and 3′ UTR, and integrate these data to generate final downstream gene models with quality control statistics (Cantarel et al., 2008). All predicted genes were functionally annotated as previous described (Chen et al., 2016). The annotation was conducted using the BLAST+ (version 2.2.27) with 1e-5 as the E-value threshold to against the SwissProt and TrEMBL databases (Bairoch and Apweiler, 2000). To infer functions for the predicted genes, InterProScan (version 4.7) (Zdobnov and Apweiler, 2001) was used to search the predicted genes against the protein signature from InterPro with default parameters. All genes were also aligned against to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Kanehisa et al., 2004).
In order to evaluate the conservation of A. ipaensis ICG_8206 gene model, the BLASTP was used to query the A. ipaensis ICG_8206 proteome against the proteomes of other plant species (Supplementary File 1: Table S7) with an E value of 1e-10 as cut-off (Supplementary File 1: Table S8).
All the predicted gene models were analyzed using OrthoMCL (Li et al., 2003) to identify shared and specific gene families among 17 species (Supplementary File 1: Table S7). In the first step, inter and intra species BLASTP with an E-value cutoff of 1e-10 was performed to detect reciprocal best hit pairs between species (putative orthologs), as well as sets of genes within species (putative co-orthologs or in-paralogs). The reciprocal best hit matrix served as the basis for ortholog definition using OrthoMCL. Subsequently, orthologous groups were organized into species-specific and higher taxonomic level groups by requiring that at least one sequence from each clade under comparison be present in the intersecting set. Finally, based on fourfold degenerate sites of single-copy ortholog genes in all species, a phylogenetic tree was constructed using MEGA v6.0 (Tamura et al., 2013) and PhyML v3.0 (Guindon et al., 2010).
To identify TFs in A. ipaensis, the PlantTFDB database was used to search TFs in other plant species (http://planttfdb.cbi.pku.edu.cn/). The predicted genes were used to BLAST search against the PlantTFDB (E-value: 1e-10). The FAR1 motif was predicted using the Multiple Expectation Maximization for Motif Elicitation (MEME)/Motif Alignment and Search Tool (MAST) system (http://meme-suite.org/) (Bailey et al., 2009) and visualized using the TBtools (version 0.4999) (https://github.com/CJ-Chen/TBtools).
Non-coding RNAs were predicted by aligned A. ipaensis genome assembly to against the Rfam databese (version 12.1) (Nawrocki et al., 2015). The pre-tRNAs were identified using tRNAscan-SE (Lowe and Eddy, 1997), pre-rRNAs were predicted using RNAmmer (Lagesen et al., 2007), pre-snRNAs were annotated using INFERNAL (Nawrocki et al., 2009) and others were also obtained by BLAST search against the Rfam database.
The RepeatMasker (Chen, 2004), RepeatProteinMask (http://repeatmasker.org/), Tendem Repeats Finder (TRF) (Benson, 1999) and RepeatModeler (Smith and Hubley, 2014) were performed to identify repetitive sequences through homolog and de novo prediction. The RepeatMasker and RepeatProteinMask were used to screen the A. ipaensis genome against the RepBase database (http://www.girinst.org/). The transposable elements (TEs) were classified as described without consideration of the gaps in the genome assembly (Wicker et al., 2007).
MIcroSAtellite (http://pgrc.ipk-gatersleben.de/misa/) was used to mine SSRs in A. ipaensis genome, and primer 3 v3.0 was used for primer design (Thiel et al., 2003; Untergasser et al., 2012). A SSR was defined with at least 6 repeats for di-nucleotide motifs or 4 repeats for tri-, tetra-, penta-, and hexa-nucleotide motifs. The maximum number of interrupting nucleotides in a compound SSR was set as 100.
Reads from five re-sequenced genotypes including two A-genome genotypes (ICG_8123 and ICG_8138) and three B-genome genotypes (ICG_8960, ICG_8209, and ICG_13160) were used to identify genome SNP and InDel variations (Chen et al., 2016). Total of these sequenced reads were aligned to the reference genome (ICG_8026) using the Burrows Wheeler Aligner program (BWA) (Li and Durbin, 2009). Subsequently, SNPs and InDels were identified using GATK v3.5 (http://www.broadinstitute.org/gatk) with default parameters, respectively.
The phylogenetic tree was constructed based on single-copy orthologous genes shared in A. ipaensis and other 17 plants (Supplementary File 1: Table S7) using MEGA v6.0 with the maximum-liklihood algorithm (Tamura et al., 2013).
Syntenic blocks between the genomes of A. ipaensis and other plants were identified using the MCScanX with default parameters (Wang et al., 2012) and visualized on the genome using Circos (Krzywinski et al., 2009). Genomic sequences were first aligned annotated genes based on amino acid sequence using Promer package of Mummer (version 3.22) (Delcher et al., 2002). Whole genome dot plots were generated using Mummerplot (Delcher et al., 2002) and Gunplot 5.0 (www.gnuplot.info/). Ks values of the homologs within collinearity blocks were calculated using the perl script, add_ka_and_ks_to_collinearity.pl included in MCScanX package, and the median of Ks values was considered to be a representative of the collinearity blocks.
All the disease R genes were identified using the genome assembly of A. ipaensis and other plant species as a TBLASTN query to against the PRG datebase with an E-value of 1e-10 as cut-off. Amino acid sequences of all NBS-LRR genes from A. ipaensis, A. duranensis, and A. thaliana were aligned to construct phylogenetic tree using MEGA v6.0 with automatic bootstrap criteria (Maximum Likelihood) (Tamura et al., 2013). The conserved motifs of top 20 homologies NBS-LRR were identified using MEME suite (Bailey et al., 2009; Supplementary File 1: Figure S26).
Nodulation regulatory and nodulin genes were identified based on GO analyses. The GO IDs for each gene were obtained through BLAST search against KEGG proteins (E-value: 1e-5). Genes involved in symbiotic nitrogen fixation associated with nodule development and AON process were obtained by comparison with orthologous genes in other legumes using multiple protein sequence alignment in COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/). The PredictProtein was used to perform GO terms, protein-protein and protein-DNA binding sites and sub-cellular localization (Yachdav et al., 2014). The SWISS-MODEL was used to predict protein tertiary structure (Biasini et al., 2014).
Genes involved in oil biosynthesis for Arabidopsis were obtained from the Arabidopsis Lipid Gene Database (Mekhedov) (http://lipids.plantbiology.msu.edu/). All the Arabidopsis lipid genes (81) in the database were used to TBLASTN search against the A. ipaensis genome with a cutoff E-value of 1e-50. Finally, a total of 116 non-redundant oil biosynthesis genes were obtained in A. ipaensis. Multiple amino acid sequence alignment of FAD2 homologs was performed using the COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/). The PredictProtein and SWISS-MODEL was used to integrate GO terms, protein binding sites, sub-cellular localization and protein tertiary structure, respectively (Biasini et al., 2014; Yachdav et al., 2014).
Signal peptide analysis of the XP_007162321.1-D2 was predicted using SignalP 4.1 Server with default parameter (Petersen et al., 2011). Prediction of transmembrane helices was performed using TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Hydrophobicity and hydrophilicity regions were predicted using ProtScale (Gasteiger et al., 2005).
XQL, and XC designed the experiments and managed the project. QL, HFL, and YH performed the research. QL, SW, XYL, GYZ, SL, HL, and HYL analyzed the data. QL wrote the manuscript with the help of GQZ, ZL, and RV. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (31771841 and 31501246); the Science and Technology Planning Project of Guangdong Province (2013B050800021, 2015A030313565, 2015B020231006, 2016B020201003, 2013B020301014, 2017A030311007); the Modern Agro-industry Technology Research System (CARS-13); the Research and Demonstration of Agricultural Technology Demand in Guangdong (2016LM3161, 2016LM3164); the Key Discipline Construction of the Guangdong Academy of Agricultural Sciences (201609); and the Special Foundation of President of the Guangdong Academy of Agricultural Sciences (201831).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank all the participants. In particular, we thank Jianan Zhang and Haofa Lan for helping us to analysis and upload the sequencing data. We thank Lesley Benyon, Ph.D., from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00604/full#supplementary-material
The genome assembly and annotation data were deposited in the DRYAD digital repository (https://datadryad.org/). All the data can be downloaded from DRYAD by searching for doi: 10.5061/dryad.hm5vs13 or by directly accessing the link: https://doi.org/10.5061/dryad.hm5vs13.
Supplementary File 1. Supplementary Tables S1–S21 and Figures S1–S53.
Supplementary File 2: Data S1. Details on the 71 miRNA identified in A. ipaensis.
Supplementary File 3: Data S2. Summary of primer sequences for SSR markers.
Supplementary File 4: Data S3. Summary of orthologous gene groups.
Supplementary File 5: Data S4. Detils on the 1,437 putative disease resistance genes in A. ipaensis.
Supplementary File 6: Data S5. Summary of 16 nodulin and nodulation associated genes in A. ipaensis.
Supplementary File 7: Data S6. Summary of multiple genes associated with nodulation development and nodule autoregulation (AON) signal pathway.
Supplementary File 8: Data S7. Summary of 39 subtilisin-like protease (SDD1) genes in A. ipaensis.
Supplementary File 9: Data S8. List of A. ipaensis genes orthologous to encoding key enzymes in the lipid biosynthesis pathways.
Supplementary File 10: Data S9. Details on the 82 oil sythesis genes with high Ka/Ks values (>1) in A. ipaensis.
Ané, J. M., Kiss, G. B., Riely, B. K., Penmetsa, R. V., Oldroyd, G. E., Ayax, C., et al. (2004). Medicago truncatula DMI1 required for bacterial and fungal symbioses in legumes. Science 303, 1364–1367. doi: 10.1126/science.1092986
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Benson, G. (1999). Tandem repeats finder, a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Berger, D., and Altmann, T. (2000). A subtilisin-like serine protease involved in the regulation of stomatal density and distribution in Arabidopsis thaliana. Genes Dev. 14, 1119–1131.
Bertioli, D. J., Cannon, S. B., Froenicke, L., Huang, G., Farmer, A. D., Cannon, E. K., et al. (2016). The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 48, 438–446. doi: 10.1038/ng.3517
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258. doi: 10.1093/nar/gku340
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bonaventure, G., Salas, J. J., Pollard, M. R., and Ohlrogge, J. B. (2003). Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell. 15, 1020–1033. doi: 10.1105/tpc.008946
Borisov, A. Y., Madsen, L. H., Tsyganov, V. E., Umehara, Y., Voroshilova, V. A., Batagov, A. O., et al. (2003). The Sym35 gene required for root nodule development in pea is an ortholog of Nin from Lotus japonicus. Plant Physiol. 131, 1009–1017. doi: 10.1104/pp.102.016071
Cannon, S. B., Sterck, L., Rombauts, S., Sato, S., Cheung, F., Gouzy, J., et al. (2006). Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. U.S.A. 103, 14959–14964. doi: 10.1073/pnas.0603228103
Cantarel, B. L., Korf, I., Robb, S. M., Parra, G., Ross, E., Moore, B., et al. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Capoen, W., Goormachtig, S., de Rycke, R., Schroeyers, K., and Holsters, M. (2005). SrSymRK, a plant receptor essential for symbiosome formation. Proc. Natl. Acad. Sci. U.S.A. 102, 10369–10374. doi: 10.1073/pnas.0504250102
Catoira, R., Galera, C., de Billy, F., Penmetsa, R. V., Journet, E. P., Maillet, F., et al. (2000). Four genes of Medicago truncatula controlling components of a nod factor transduction pathway. Plant Cell. 12, 1647–1666. doi: 10.1105/tpc.12.9.1647
Chen, N. (2004). Using repeatmasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4: Unit 4.10. doi: 10.1002/0471250953.bi0410s05
Chen, X., Li, H., Pandey, M. K., Yang, Q., Wang, X., Garg, V., et al. (2016). Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. U.S.A. 113, 6785–6790. doi: 10.1073/pnas.1600899113
Delcher, A. L., Phillippy, A., Carlton, J., and Salzberg, S. L. (2002). Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30, 2478–2483. doi: 10.1093/nar/30.11.2478
Dhillon, S. S., Rake, A. V., and Miksche, J. P. (1980). Reassociation kinetics and cytophotometric characterization of peanut (Arachis hypogaea L.) DNA. Plant Physiol. 65, 1121–1127. doi: 10.1104/pp.65.6.1121
Dörmann, P., Voelker, T. A., and Ohlrogge, J. B. (2000). Accumulation of palmitate in Arabidopsis mediated by the acyl-acyl carrier protein thioesterase FATB1. Plant Physiol. 123, 637–644. doi: 10.1104/pp.123.2.637
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., and Lepiniec, L. (2010). MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. doi: 10.1016/j.tplants.2010.06.005
Edwards, A., Heckmann, A. B., Yousafzai, F., Duc, G., and Downie, J. A. (2007). Structural implications of mutations in the pea SYM8 symbiosis gene, the DMI1 ortholog, encoding a predicted ion channel. Mol. Plant Microbe Interact. 20, 1183–1191. doi: 10.1094/MPMI-20-10-1183
Endre, G., Kereszt, A., Kevei, Z., Mihacea, S., Kaló, P., and Kiss, G. B. (2002). A receptor kinase gene regulating symbiotic nodule development. Nature 417, 962–966. doi: 10.1038/nature00842
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed J. M. Walker (Totowa, NJ: Humana press), 571–607. doi: 10.1385/1-59259-890-0:571
Grabiele, M., Chalup, L., Robledo, G., and Seijo, G. (2012). Genetic and geographic origin of domesticated peanut as evidenced by 5S rDNA and chloroplast DNA sequences. Plant Syst. Evol. 298, 1151–1165. doi: 10.1007/s00606-012-0627-3
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Imaizumi-Anraku, H., Takeda, N., Charpentier, M., Perry, J., Miwa, H., Umehara, Y., et al. (2005). Plastid proteins crucial for symbiotic fungal and bacterial entry into plant roots. Nature 433, 527–531. doi: 10.1038/nature03237
Jin, J., He, K., Tang, X., Li, Z., Lv, L., Zhao, Y., et al. (2015). An Arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol. Biol. Evol. 32, 1767–1773. doi: 10.1093/molbev/msv058
Jin, J., Tian, F., Yang, D. C., Meng, Y.Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D104–D105. doi: 10.1093/nar/gkw982
Kaló, P., Gleason, C., Edwards, A., Marsh, J., Mitra, R. M., Hirsch, S., et al. (2005). Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 308, 1786–1789. doi: 10.1126/science.1110951
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Kent, W. J. (2002). BLAT-The BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kimpel, J. A., Nagao, R. T., Goekjian, V., and Key, J. L. (1990). Regulation of the heat shock response in soybean seedlings. Plant Physiol. 94, 988–995. doi: 10.1104/pp.94.3.988
Kochert, G., Halward, T., Branch, W. D., and Simpson, C. E. (1991). RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor. Appl. Genet. 81, 565–570. doi: 10.1007/BF00226719
Kochert, G., Stalker, H. T., Gimenes, M., Galgaro, L., Lopes, C. R., and Moore, K. (1996). RFLP and cytogenetic evidence on the origin and evolution of allotetraploid domesticated peanut, Arachis hypogaea (leguminosae). Am. J. Bot. 83, 1282–1291. doi: 10.1002/j.1537-2197.1996.tb13912.x
Koppolu, R., Upadhyaya, H. D., Dwivedi, S. L., Hoisington, D. A., and Varshney, R. K. (2010). Genetic relationships among seven sections of genus Arachis studied by using SSR markers. BMC Plant Biol. 10:15. doi: 10.1186/1471-2229-10-15
Krapovickas, A., and Gregory, W. C. (1994). Taxonomia del genero “Arachis (leguminosae)”. Bonplandia 8, 1–186.
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lévy, J., Bres, C., Geurts, R., Chalhoub, B., Kulikova, O., Duc, G., et al. (2004). A putative Ca2+ and calmodulin-dependent protein kinase required for bacterial and fungal symbioses. Science 303, 1361–1364. doi: 10.1126/science.1093038
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lin, R., Ding, L., Casola, C., Ripoll, D. R., Feschotte, C., and Wang, H. (2007). Transposase-derived transcription factors regulate light signaling in Arabidopsis. Science 18, 1302–1305. doi: 10.1126/science.1146281
Lna, Y. (1996). Pattern of synonymous and nonsynonymous substitutions: an indicator of mechanisms of molecular evolution. J. Genet. 75, 91. doi: 10.1007/BF02931754
López, Y., Nadaf, H. L., Smith, O. D., Connell, J. P., Reddy, A. S., and Fritz, A. K. (2000). Isolation and characterization of the Δ12-fatty acid desaturase in peanut (Arachis hypogaea L.) and search for polymorphisms for the high oleate trait in Spanish market-type lines. Theor. Appl. Genet. 101, 1131–1138. doi: 10.1007/s001220051589
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1:18. doi: 10.1186/2047-217X-1-18
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science. 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Medzihradszky, M., Bindics, J., Ádám, É., Viczián, A., Klement, É., Lorrain, S., et al. (2013). Phosphorylation of phytochrome B inhibits light-induced signaling via accelerated dark reversion in Arabidopsis. Plant Cell. 25, 535–544. doi: 10.1105/tpc.112.106898
Moretzsohn, M. C., Gouvea, E. G., Inglis, P. W., Leal-Bertioli, S. C., Valls, J. F., and Bertioli, D. J. (2013). A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann. Bot. 111, 113–126. doi: 10.1093/aob/mcs237
Nawrocki, E. P., Burge, S. W., Bateman, A., Daub, J., Eberhardt, R. Y., Eddy, S. R., et al. (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137. doi: 10.1093/nar/gku1063
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Nielen, S., Campos-Fonseca, F., Leal-Bertioli, S., Guimarães, P., Seijo, G., Town, C., et al. (2010). FIDEL–a retrovirus-like retrotransposon and its distinct evolutionary histories in the A- and B-genome components of cultivated peanut. Chromosome Res. 18, 227–246. doi: 10.1007/s10577-009-9109-z
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Parthasarathy, S., Parthasarathy, S., Khoo, J. C., Miller, E., Barnett, J., Witztum, J. L., et al. (1990). Low density lipoprotein rich in oleic acid is protected against oxidative modification: implication for dietary prevention of atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 87, 3894–3898. doi: 10.1073/pnas.87.10.3894
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pham, A. T., Shannon, J. G., and Bilyeu, K. D. (2012). Combinations of mutant FAD2 and FAD3 genes to produce high oleic acid and low linolenic acid soybean oil. Theor. Appl. Genet. 125, 503–515. doi: 10.1007/s00122-012-1849-z
Radutoiu, S., Madsen, L. H., Madsen, E. B., Felle, H. H., Umehara, Y., Grønlund, M., et al. (2003). Plant recognition of symbiotic bacteria requires two LysM receptor-like kinases. Nature 425, 585–592. doi: 10.1038/nature02039
Ramos, M. L., Fleming, G., Chu, Y., Akiyama, Y., Gallo, M., and Ozias-Akins, P. (2006). Chromosomal and phylogenetic context for conglutin genes in Arachis based on genomic sequence. Mol. Gen. Genomics. 275, 578–592. doi: 10.1007/s00438-006-0114-z
Robledo, G. and Seijo, G. (2010). Species relationships among the wild B genome of Arachis species (section Arachis) based on FISH mapping of rDNA loci and heterochromatin detection: a new proposal for genome arrangement. Theor. Appl. Genet. 121, 1033–1046. doi: 10.1007/s00122-010-1369-7
Robledo, G., Lavia, G. I., and Seijo, G. (2009). Species relations among wild Arachis species with the A genome as revealed by FISH mapping of rDNA loci and heterochromatin detection. Theor. Appl. Genet. 118, 1295–1307. doi: 10.1007/s00122-009-0981-x
Sakai, K., and Kajiwara, S. (2005). Cloning and functional characterization of a Δ12 fatty acid desaturase gene from the basidiomycete Lentinula edodes. Mol. Gen. Genomics 273, 336–341. doi: 10.1007/s00438-005-1138-5
Samoluk, S. S., Robledo, G., Podio, M., Chalup, L., Ortiz, J. P., Pessino, S. C., et al. (2015). First insight into divergence, representation and chromosome distribution of reverse transcriptase fragments from L1 retrotransposons in peanut and wild relative species. Genetica 143, 113–125. doi: 10.1007/s10709-015-9820-y
Sanseverino, W., Roma, G., De Simone, M., Faino, L., Melito, S., Stupka, E., et al. (2010). PRGdb, a bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res. 38, D814–D821. doi: 10.1093/nar/gkp978
Schauser, L., Roussis, A., Stiller, J., and Stougaard, J. (1999). A plant regulator controlling development of symbiotic root nodules. Nature 402, 191–195. doi: 10.1038/46058
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Schneider, A., Walker, A., Sagan, M., Duc, G., Ellis, N., and Downie, A. (2002). Mapping of the nodulation loci sym9 and sym10 of pea (Pisum sativum L.). Theor. Appl. Genet. 104, 1312–1316. doi: 10.1007/s00122-002-0896-2
Seijo, G., Lavia, G. I., Fernández, A., Krapovickas, A., Ducasse, D. A., and Bertioli, D. J. (2007). Genomic relationships between the cultivated peanut (Arachis hypogaea, Leguminosae) and its close relatives revealed by double GISH. Am. J. Bot. 94, 1963–1971. doi: 10.3732/ajb.94.12.1963
Serrano-Vega, M. J., Garcés, R., and Martínez-Force, E. (2005). Cloning, characterization and structural model of a FatA-type thioesterase from sunflower seeds (Helianthus annuus L.). Planta 221, 868–880. doi: 10.1007/s00425-005-1502-z
Shinozaki, K., and Yamaguchi-Shinozaki, K. (2000). Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol. 3, 217–223. doi: 10.1016/S1369-5266(00)00067-4
Singh, K., Foley, R. C., and O-ate-Sánchez, L. (2002). Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 5, 430–436. doi: 10.1016/S1369-5266(02)00289-3
Slabas, A. R., and Fawcett, T. (1992). The biochemistry and molecular biology of plant lipid biosynthesis. Plant Mol. Biol. 19, 169–191. doi: 10.1007/BF00015613
Smit, P., Raedts, J., Portyanko, V., Debellé, F., Gough, C., Bisseling, T., et al. (2005). NSP1 of the GRAS protein family is essential for rhizobial Nod factor-induced transcription. Science 308, 1789–1791. doi: 10.1126/science.1111025
Smith, A. F. A., and Hubley, R. (2014). Repeatmodeler open-1.0. [Internet]. Available online at: http://www.repeatmasker.org
Smith, B. W. (1950). Arachis hypogaea. Aerial flower and subterranean fruit. Am. J. Bot. 37, 802–815. doi: 10.1002/j.1537-2197.1950.tb11073.x
Song, L., Fan, C., Chen, Y., Zhang, X., and Hu, Z. (2016). The molecular regulation mechanism of the plant lipid biosynthesis. Mol. Plant Breed. 14, 2178–2187. doi: 10.13271/j.mpb.014.002178
Stracke, S., Kistner, C., Yoshida, S., Mulder, L., Sato, S., Kaneko, T., et al. (2002). A plant receptor-like kinase required for both bacterial and fungal symbiosis. Nature 417, 959–962. doi: 10.1038/nature00841
Stracke, S., Sato, S., Sandal, N., Koyama, M., Kaneko, T., Tabata, S., et al. (2004). Exploitation of colinear relationships between the genomes of Lotus japonicus, Pisum sativum and Arabidopsis thaliana, for positional cloning of a legume symbiosis gene. Theor Appl Genet. 108, 442–449. doi: 10.1007/s00122-003-1438-2
Sun, W., Van Montagu, M., and Verbruggen, N. (2002). Small heat shock proteins and stress tolerance in plants. Biochim. Biophys. Acta 1577, 1–9. doi: 10.1016/S0167-4781(02)00417-7
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tan, D. Y., Zhang, Y., and Wang, A. B. (2010). A review of geocarpy and amphicarpy in angiosperms, with special reference to their ecological adaptive significance. Chin. J. Plant Ecol. 34, 72–88. doi: 10.3773/j.issn.1005-264x.2010.01.011
Temsch, E. M., and Greilhuber, J. (2000). Genome size variation in Arachis hypogaea and A. monticola re-evaluated. Genome 43, 449–451. doi: 10.1139/g99-130
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3-new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Varshney, R. K., Chen, W., Li, Y., Bharti, A. K., Saxena, R. K., Schlueter, J. A., et al. (2011). Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89. doi: 10.1038/nbt.2022
Varshney, R. K., Song, C., Saxena, R. K., Azam, S., Yu, S., Sharpe, A. G., et al. (2013). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246. doi: 10.1038/nbt.2491
Wang, W., Vinocur, B., Shoseyov, O., and Altman, A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9, 244–252. doi: 10.1016/j.tplants.2004.03.006
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Yachdav, G., Kloppmann, E., Kajan, L., Hecht, M., Goldberg, T., Hamp, T., et al. (2014). PredictProtein-an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 42, W337–W343. doi: 10.1093/nar/gku366
Yen, C. L., Stone, S. J., Koliwad, S., and Harris, C. Jr. (2008). DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49, 2283–2301. doi: 10.1194/jlr.R800018-JLR200
Young, N. D., Debellé, F., Oldroyd, G. E., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 480, 520–524. doi: 10.1038/nature10625
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhao, L., Katavic, V., Li, F., Haughn, G. W., and Kunst, L. (2010). Insertional mutant analysis reveals that long-chain acyl-CoA synthetase 1 (LACS1), but not LACS8, functionally overlaps with LACS9 in Arabidopsis seed oil biosynthesis. Plant J. 64, 1048–1058. doi: 10.1111/j.1365-313X.2010.04396.x
Keywords: Arachis ipaensis, genome sequence, genome evolution, polyploidizations, whole genome duplication
Citation: Lu Q, Li H, Hong Y, Zhang G, Wen S, Li X, Zhou G, Li S, Liu H, Liu H, Liu Z, Varshney RK, Chen X and Liang X (2018) Genome Sequencing and Analysis of the Peanut B-Genome Progenitor (Arachis ipaensis). Front. Plant Sci. 9:604. doi: 10.3389/fpls.2018.00604
Received: 06 February 2018; Accepted: 16 April 2018;
Published: 03 May 2018.
Edited by:
Jun Yang, Shanghai Chenshan Plant Science Research Center (CAS), ChinaReviewed by:
Xuehui Huang, Shanghai Normal University, ChinaCopyright © 2018 Lu, Li, Hong, Zhang, Wen, Li, Zhou, Li, Liu, Liu, Liu, Varshney, Chen and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoping Chen, Y2hlbnhpYW9waW5nQGdkYWFzLmNu
Xuanqiang Liang, bGlhbmd4dWFucWlhbmdAZ2RhYXMuY24=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.