 Kai-Ming Shih1
Kai-Ming Shih1 Chung-Te Chang
Chung-Te Chang Yu-Chung Chiang
Yu-Chung Chiang Shih-Ying Hwang
Shih-Ying Hwang- 1Department of Life Science, National Taiwan Normal University, Taipei, Taiwan
- 2Department of Geography, National Taiwan University, Taipei, Taiwan
- 3Division of Silviculture, Taiwan Forestry Research Institute, Taipei, Taiwan
- 4Department of Biological Sciences, National Sun Yat-Sen University, Kaohsiung, Taiwan
Double digest restriction site-associated DNA sequencing (ddRADseq) is a tool for delivering genome-wide single nucleotide polymorphism (SNP) markers for non-model organisms useful in resolving fine-scale population structure and detecting signatures of selection. This study performs population genetic analysis, based on ddRADseq data, of a coniferous species, Keteleeria davidiana var. formosana, disjunctly distributed in northern and southern Taiwan, for investigation of population adaptive divergence in response to environmental heterogeneity. A total of 13,914 SNPs were detected and used to assess genetic diversity, FST outlier detection, population genetic structure, and individual assignments of five populations (62 individuals) of K. davidiana var. formosana. Principal component analysis (PCA), individual assignments, and the neighbor-joining tree were successful in differentiating individuals between northern and southern populations of K. davidiana var. formosana, but apparent gene flow between the southern DW30 population and northern populations was also revealed. Fifteen of 23 highly differentiated SNPs identified were found to be strongly associated with environmental variables, suggesting isolation-by-environment (IBE). However, multiple matrix regression with randomization analysis revealed strong IBE as well as significant isolation-by-distance. Environmental impacts on divergence were found between populations of the North and South regions and also between the two southern neighboring populations. BLASTN annotation of the sequences flanking outlier SNPs gave significant hits for three of 23 markers that might have biological relevance to mitochondrial homeostasis involved in the survival of locally adapted lineages. Species delimitation between K. davidiana var. formosana and its ancestor, K. davidiana, was also examined (72 individuals). This study has produced highly informative population genomic data for the understanding of population attributes, such as diversity, connectivity, and adaptive divergence associated with large- and small-scale environmental heterogeneity in K. davidiana var. formosana.
Introduction
Conifers are reported to have slower evolutionary rate due to reduced levels of nucleotide mutation and large effective population size, but with higher ratio of non-synonymous to synonymous divergence, in comparison with angiosperms (Buschiazzo et al., 2012). Local adaptation in populations of coniferous species is not uncommon (e.g., Mimura and Aitken, 2010; Grivet et al., 2011; Chen et al., 2012; Fang et al., 2013). Limited dispersal shaping genetic structure of populations isolated geographically can result in a correlation between genetic and geographic distance known as isolation-by-distance (IBD) (Wright, 1943). However, adaptive divergence may occur between isolated populations because of topographical and ecological complexity known as isolation-by-environment (IBE), in which genetic distance is positively correlated with environmental distance (Wang et al., 2013; Sexton et al., 2014). The pattern of population divergence within a species can be either IBD, IBE, or both IBD and IBE, and IBD could be more prominent than IBE in plant species divergence (Sexton et al., 2014). Disentangling the effects of IBD from IBE is crucial to understanding their relative impact on population genetic structure, particularly because the relative contributions of IBD and IBE may vary among and within species (Wang et al., 2013; Sexton et al., 2014).
Most Taiwan endemic coniferous species are thought to be colonized from their ancestral species occurring in China with the exception of Chamaecyparis formosensis and Ch. taiwanensis (Wang et al., 2003; Chung et al., 2004; Chen et al., 2009; Chou et al., 2011). Genetic studies revealed congeneric sister species pair relationships of conifers occur in Taiwan and China, such as Cunninghamia konishii (Taiwan) and Cu. lanceolata (China) (Chung et al., 2004), Calocedrus macrolepis var. formosana (Taiwan) and Ca. macrolepis (China) (Chen et al., 2009), and Taiwania cryptomeriodes occurs in Taiwan colonized from China (Chou et al., 2011). Ch. formosensis and Ch. taiwanensis occur in Taiwan are known to be congeneric sister species pairs with Ch. pisifera and Ch. obtusa occur in Japan, respectively (Wang et al., 2003). Coniferous species endemic to Taiwan may display not only population adaptive divergence on island but also species level divergence with their ancestors. Genetic changes in response to local environments might have potential to keep pace with the rate of climate change (Robertson et al., 2014). Population structure of species may exhibit a pattern of IBE due to range expansion since colonization of their ancestral species and lead to locally adapted lineages associated with environmental heterogeneity (Holt, 2003; Schlotfeldt and Kleindorfer, 2006; Huang et al., 2015). Post-glacial expansion since the Last Glacial Maximum (LGM, 26.5–19.0 thousand years ago) (Lambeck and Chappell, 2001) may have also played a role in invoking locally adapted variants correlated with ecological complexity and climate change (Aitken et al., 2008; Chen et al., 2017).
Geographic isolation since the marine transgression following the LGM may induce higher probabilities of allopatric isolation between island-mainland sister species (Otte and Endler, 1989; Losos and Ricklefs, 2009). However, assumption of allopatric divergence between sister species of island and adjacent mainland since the isolation caused by the last marine transgression has been challenged (Li et al., 2010; Burridge et al., 2013). The level of allopatric divergence may be lower than expected because of prolonged gene flow between geographically isolated closely related species (Li et al., 2010) and also due to the recent colonization from mainland to island (Burridge et al., 2013). Species identification and characterization between closely related species have often been a challenge because of weak interspecific barriers. Low levels of genetic differentiation are frequently observed between Taiwan coniferous species and their adjacent mainland counterparts (Chung et al., 2004; Chou et al., 2011). In addition, multiple cycles of connection and isolation between Taiwan and adjacent large landmass would have led to high levels of interspecies gene flow causing difficulty in species delimitation (e.g., Chung et al., 2004; Worth et al., 2009; Strijk et al., 2012), and the differentiation of progenitor-derivative species pair may have restricted to some limited genomic hot-spots, while most of the genetic information are shared between sister species (Via, 2009; Wolf et al., 2010; Strasburg et al., 2012).
Populations of the warmth-loving Taiwan cow-tail fir (Keteleeria davidiana (Franchet) Beissner var. formosana) are presently disjunctly distributed on northern and southern rocky mountain ridges, respectively, at elevations of 300–600 m and 500–900 m (Li and Hsuan, 1994). The northern and southern populations of K. davidiana var. formosana occupy different environmental niches with varying floristic compositions (Chou et al., 2009). Taiwan cow-tail fir is thought to be derived from K. davidiana (Bertrand) Beissner that occurs in China (Farjon, 1989). The occurrence of Keteleeria since the Plio-Pleioscene boundary was revealed by a pollen record and in minor proportion during the early Pleistocene Praetiglian, late Pleistocene, and early Holocene around 10,000 years ago in central Taiwan (Tsukada, 1967). The disappearance of Keteleeria from areas other than northern and southern Taiwan may have resulted from its recalcitrant seed storage behavior (Yang et al., 2006) and low natural regeneration rate (Wang, 1987) that rendered Keteleeria incompetent in competition with rapidly growing subtropical species such as Machilus and Castanopsis (Su, 1984). In addition, human disturbance may also have contributed to the disappearance of K. davidiana var. formosana from parts of its former range.
Restriction site-associated DNA sequencing (RADseq) (Baird et al., 2008) and its related methodologies, such as genotyping-by-sequencing (Elshire et al., 2011) and double digest RADseq (ddRADseq) (Peterson et al., 2012) are powerful methods in genotyping thousands of genomic markers distributed randomly across genome. These techniques, share the common features of using one or more restriction enzymes to sample a subset of genomic loci, are applicable in population genetics study on species with no reference sequence information (Davey and Blaxter, 2010). RADseq and related techniques can sample genomic variation at reduced complexity from many individuals particularly for non-model organisms at reasonable cost, and are important technologies for ecological, evolutionary, and conservation genomics (Hoffmann et al., 2015; Andrews et al., 2016). RADseq related techniques can be useful in examining natural population adaptive divergence (Parchman et al., 2012; Pannell and Fields, 2014) and in revealing candidate genome regions that involved in speciation (Eaton and Ree, 2013; Sobel and Streisfeld, 2015).
In a previous study based on amplified fragment length polymorphism (AFLP), local adaptation was found in K. davidiana var. formosana (Fang et al., 2013). However, no clear genetic distinction between disjunctly distributed northern and southern populations was found except when the FST outliers potentially evolved under selection were used in the analysis. One of the main findings of the previous study was the associations of environmental variables, such as temperature, precipitation, and humidity, with FST outliers, indicating IBE between disjunctly distributed northern and southern populations of K. davidiana var. formosana. However, because of geographically distant distributions of the northern and southern populations, IBD can also be important influencing population genetic structure. In the present study, we employed ddRADseq in genotyping samples of K. davidiana var. formosana to obtain genome-wide single nucleotide polymorphism (SNP) markers for the purpose of investigating population adaptive divergence in K. davidiana var. formosana. We hypothesized the occurrence of IBE as well as IBD because of habitat heterogeneity and geographic isolation, in particular, between northern and southern populations of Taiwan cow-tail fir. The relative importance of geography and environment shaping the patterns of genetic variation was assessed to gain a deeper understanding of how environmental factors influence evolutionary processes. We also aimed to test the selection of SNPs closely associated with environmental variables across populations of K. davidiana var. formosana and to examine whether potential selective outliers link to specific gene functions that may have played significant roles underlying local adaptation. To examine the genetic relationships between populations of K. davidiana var. formosana and K. davidiana, samples of K. davidiana were also genotyped based on ddRADseq.
Materials and Methods
Sample Collections and DNA Extraction
Samples of endangered K. davidiana var. formosana were collected, including three northern and two southern populations (Table 1, Figure 1). The number of old growth trees, ages between 100 and 300 years, were ranged between 20 and 81 for Taiwan cow-tail fir in natural stands (Chung, J.-D., unpublished data). The distances between pairs of the three northern populations are within 1–3 km, and they are distantly separated from the two southern populations (263–290 km). Distance between the two southern populations is about 26 km. Approval of sample collection was granted by the Forestry Bureau, Council of Agriculture, Taiwan (permit number: 101-AgroScience-1.1.2-B-e1). Genomic DNA was extracted from K. davidiana (n = 10, Kunming Botanical Garden, Yunnan, China) and K. davidiana var. formosana (n = 62) using a modified cetyl trimethyl ammonium bromide (CTAB) protocol (Dehestani and Kazemitabar, 2007) and quantified with a Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
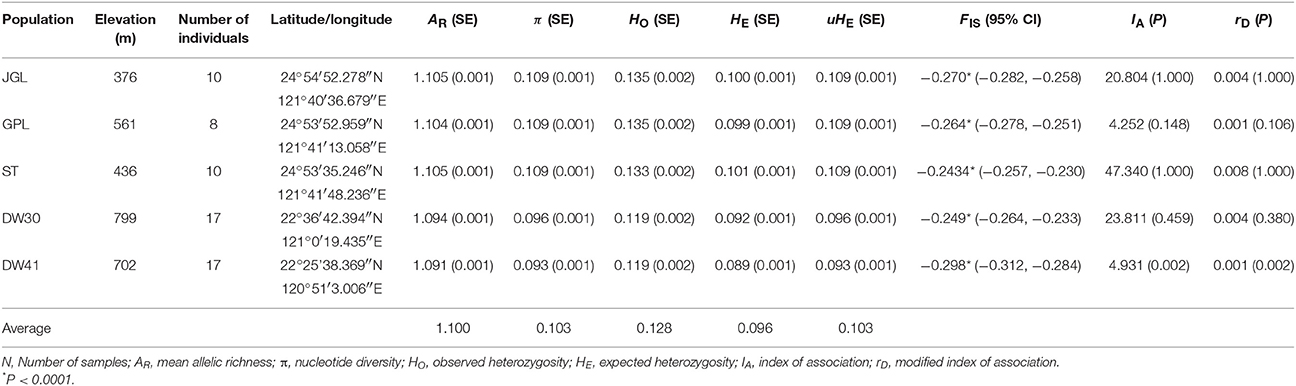
Table 1. Population genetic parameters of the five sampled populations of Taiwan cow-tail fir based on ddRADseq.
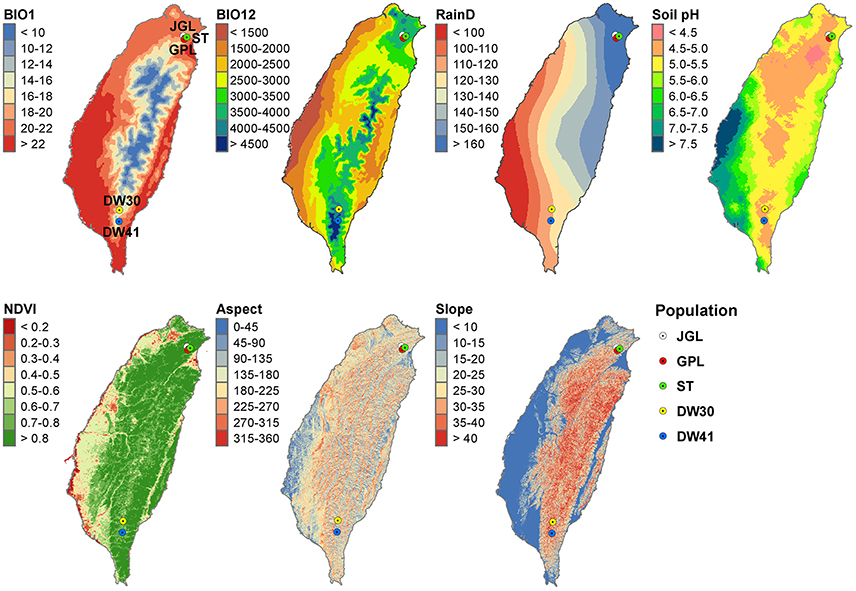
Figure 1. Geographic distribution of the five populations of Taiwan cow-tail fir and annual mean gradients of seven environmental variables. BIO1, annual mean temperature; BIO12, annual precipitation; RainD, number of rainfall days per year. Annual mean gradients were smoothed using a universal spherical model of the Kriging method in ArcGIS.
ddRADseq Library Construction and Sequencing
ddRADseq libraries were prepared following Peterson et al. (2012) with modifications. Genomic DNA was double digested with EcoRI (20 units) and MseI (20 units) and ligated to barcoded P1 and indexed P2 adapters binding to EcoRI and MseI overhangs, respectively. Fragments in the 250–450 bp size range were selected from an agarose gel. Amplified fragment libraries were quantified using quantitative real time polymerase chain reaction (qPCR) and pooled in equimolar amounts for sequencing on the Illumina HiSeq2000 platform (Illumina, San Diego, CA, USA) with 36 samples per lane. Sequence reads, 85 bp in read 1 and 90 bp in read 2, were assigned to individual samples based on barcode sequences. The ddRADseq library construction, sequencing, and the following processing of sequence reads were conducted at the Beijing Genomic Institute, China.
ddRAD Sequence Analysis
Restriction site sequences on both paired-end (PE) reads were removed using FASTX-Toolkit v.0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/). The two base pairs following EcoRI restriction sites were removed avoiding the effect of GC content asymmetry that may cause problem in the subsequent de novo assembly (Kozarewa et al., 2009). A 9-bp sequence following MseI restriction sites was also removed to generate equal length in the two PE reads. Moreover, a 5-bp C and a 5-bp T, respectively, was added to the end of read 1 and read 2, resulting in 78 bp in both reads, using a php script (Supplementary File 1) considering that the two PE reads with high sequence similarity will not be treated as same locus during de novo assembly.
STACKS software pipeline v.1.40 was used for read filtering and SNP genotyping (Catchen et al., 2011). Using FASTQC v.0.11.2 (Andrews, 2010), low quality sequence reads with a Phred33 quality score < 10 (as suggested by the authors of STACKS) were discarded and any reads with an uncalled base was also removed using the “process_radtags” module. The “ustacks,” “cstacks,” “sstacks,” and “populations” modules of STACKS were used to obtain final genotypes called at a Minor Allele Frequency of 5%. The number of SNP obtained was used to determine the values of parameters m, M, and N for “ustacks” module and of parameter n for “cstacks” module (Paris et al., 2017). The settings of these parameters are known to influence the number of SNP obtained, estimation of population genetic diversity measures, and downstream population genetic inference (Mastretta-Yanes et al., 2015; Paris et al., 2017; Shafer et al., 2017). Three individuals from each of the five populations of K. davidiana var. formosana and K. davidiana were used to compute number of SNP (in two of three samples in each population) obtained and percentage of polymorphic loci (Supplementary Table 1; Paris et al., 2017). Parameter m was evaluated from 3 to 5, and m = 3 had the highest number of SNP obtained. Parameter M was evaluated from 0 to 5. Parameter N was set to M+2 (Paris et al., 2017). The number of SNP obtained increased by increasing M and reached a plateau between M = 2 and M = 4, but dropped apparently when M = 5. When M and n were set to 2 and 1, respectively, no apparent change in percentage of polymorphic loci was found among different values of m. No apparent change in percentage of polymorphic loci was also found among different values of M when m and n were set to 3 and 1, respectively. Therefore, the values of parameters m, M, N, and n were set to 3, 2, 4, and 1, respectively, for STACKS pipeline. The “cstacks” module created a catalog of SNPs, which was used to genotype each individual with the “sstacks” module. In “populations” module, we obtained three data sets that allowed the minimum proportions of non-missing genotypes at 40, 50, and 60% of samples across populations with multiple SNPs per stack, respectively.
Environmental Variables
Environmental variables include 19 bioclimate, 10 ecological, and three topological variables. Bioclimate data at 30 s spatial resolution (~1 km) were downloaded from the WorldClim v.1.4 (http://www.worldclim.org/; Hijmans et al., 2005). Monthly mean values of ecological variables at spatial resolution of 1 km included relative humidity, cloud cover, time of sunshine, wet days (number of days with >0.1 mm of rain per month), number of rainfall days per year, and mean wind speed were obtained from the Data Bank for Atmospheric Research (DBAR, http://www.narlabs.org.tw/en/, recorded in 1990–2013) using a universal spherical model of the Kriging method in ArcGIS (Chang et al., 2014). Remote sensing data based on moderate resolution imaging spectroradiometer (MODIS) for ecological factors included normalized difference vegetation index (NDVI) and enhanced vegetation index were obtained from Land Process Distributed Active Archive Center (http://lpdaac.usgs.gov). Monthly MODIS images were generated based on a maximum values composite procedures (Huete et al., 2002). Soil pH values of sample sites were acquired from the Agriculture and Food Agency of Taiwan based on an island-wide soil investigation (n = 1,150) conducted in 1969–1986 (Chang et al., 2009). Annual moisture index (Thornthwaite, 1948) was also calculated for each sample site. Topographic variables, including aspect, elevation, and slope, were derived from a 40 m resolution digital terrain model, and monthly mean values for sampling sites were computed in ArcGIS (Chang et al., 2014). Variance inflation factor (VIF) was computed for environmental variables using the “vif” function of usdm package (Naimi et al., 2014) in the R environment (R Development Core Team, 2013). Correlation coefficient and VIF were calculated for the three sets of environmental variables (bioclimate, ecological, and topological variables) separately, and environmental variables with VIF > 20 (Borcard et al., 2011) and highly correlated with other variables (|r| > 0.8) were removed. Seven environmental variables were retained, including bioclimate: annual mean temperature and annual precipitation; ecology: number of rainfall days per year, Soil pH, and NDVI; and topology: aspect and slope (Supplementary Table 2).
Detection of FST Outliers and Association with Environmental Variables
BAYSECAN v.2.1 (Foll and Gaggiotti, 2008) was used to identify FST outliers. BAYESCAN estimates the posterior odds (PO), the ratio of posterior probabilities of selection over neutrality. The parameters for running BAYESCAN were a 100 pilot runs of 50,000 iterations and followed by a sample size of 50,000 with thinning interval of 20 among 106 iterations. Any SNP with log10(PO) > 0.5 was considered to have substantial evidence for selection (Jeffreys, 1961). FDIST within the LOSITAN workbench (Beaumont and Nichols, 1996; Antao et al., 2008) was also used to identify outliers potentially evolved under selection. In FDIST, outliers were identified by comparing observed distribution of FST conditioned on expected heterozygosity with neutral expectations at a 99.5% confidence interval (CI) and a false discovery rate (FDR) of 1% with each run comprising 106 simulations with both “neutral mean FST” and “force mean FST” selected, and removed FST outliers to increase the reliability when calculating the global distribution of FST. Loci that are detected as outliers by both BAYESCAN and FDIST were analyzed with Samβada (Stucki et al., 2014) for the associations between all possible pairs of allele frequencies of SNPs and environmental variables using multiple univariate logistic regression. The 23 outlier SNPs identified by BAYESCAN and FDIST (see Results) were coded for allelic presence (“1”) or absence (“0”), producing 69 SNP genotypes (i.e., “00,” “01,” and “11” for each of the 23 outlier SNPs). These genotypes were tested for associations with the seven environmental variables retained (Supplementary Table 2), resulting in 483 tests. Models including and excluding the environmental variables were compared, and significant fit was identified based on both Wald and G scores with an FDR cutoff of 0.01. Three data sets of genetic variation were generated, including total, neutral, and outlier data sets, after identification of FST outliers. Sequences containing outlier SNPs were searched (BLASTN) against the NCBI non-redundant nucleotide database for potential gene region identification. In BLASTN, an E-value of 0.001 was used as threshold for significant sequence similarity. Pairwise linkage disequilibrium (LD) between outlier SNPs was assessed using a two-locus exact test implemented in ARLEQUIN v.3.5, and significance determined by 10,000 permutations (Excoffier and Lischer, 2010).
Genetic Diversity, Structure, and Relationships
Data with missing values may influence the individual assignment and phylogenetic analysis using reduced representation of RADseq data (Chattopadhyay et al., 2014; Huang and Knowles, 2016). The number of SNPs retained by STACKS based on the extent of missing data vary dramatically (Supplementary Table 3). In the present study, different percentages of missing value (SNPs found in at least 40, 50, and 60% of samples across populations) data sets were generated. These data sets were evaluated for the potential effects on distributions of the levels of population genetic diversity measures and pairwise locus FST using Kolmogorov Smirnov (KS) test (the “ks.test” function of R). Data of 50% missing value across populations was adopted, based on the number of SNP obtained (Supplementary Table 3), and used for all the following analyses. We calculated nucleotide diversity (π), observed (HO), and expected (HE) heterozygosity using STACKS. Nei's unbiased HE (uHE) was also calculated (Nei, 1978). Pairwise locus FST was calculated with the “popgenreport” function of R package PopGenReport (Adamack and Gruber, 2014). CI (95%) of FIS for each population was calculated using “boot.ppfis” function of R package hierfstat with 999 bootstrap resampling (Goudet, 2005), and P-values calculated. Mean allelic richness (i.e., mean number of alleles per locus, AR) was calculated with the function “allel.rich” of R package PopGenReport. Proportion of shared alleles between species and between populations was calculated with the “pairwise.propShared” function of R package PopGenReport. Index of association (IA) (Brown et al., 1980) and modified index of association (rD) (Agapow and Burt, 2001) represent multilocus LD were calculated using the “ia” function of R poppr package (Kamvar et al., 2014), and significance of non-zero IA and rD values was tested with 999 permutations.
We used linear mixed-effects models with maximum likelihood (ML) estimation using the “lmer” function of R package lme4 (Bates et al., 2015) to assess whether genetic diversity measures (AR, π, HO, HE, and uHE) were significantly different between populations of K. davidiana var. formosana. Population and locus were used as fixed and random effects, respectively, in linear mixed-effects models. Overall difference was tested using the “Anova” function of R package car (Fox and Weisberg, 2011) based on the type-II Wald χ2-test, and Tukey's post-hoc pairwise comparisons were performed using the “lsmeans” function of R package lsmeans (Lenth, 2016).
Three data sets, including total, neutral, and outlier, were used for computation of genetic differentiation via analysis of molecular variance (AMOVA) and across population FST. We performed AMOVA using the “poppr.amova” function of R package poppr and significance tested with the “randtest” function of R package ade4 (Dray and Dufour, 2007). Across population FST was calculated using the “popStructTest” function of R package strataG (Archer et al., 2017) and tested the significance (999 permutations). For genetic assignment of individuals, only the total data was used. Estimation of individual ancestries was performed with ADMIXTURE v.1.3 based on ML method (Alexander et al., 2009). We ran ADMIXTURE for each K from K = 1 to K = 5 (K. davidiana var. formosana only) and from K = 1 to K = 6 (both K. davidiana and K. davidiana var. formosana) using the default settings, and the best K evaluated with 10-fold cross-validation (CV) procedure. Genetic assignment of individuals was also inferred based on sparse non-negative factorization (sNMF) and least-squares optimization with the “snmf” function of R package LEA (Frichot and Francois, 2015). The snmf settings were: regularization parameter = 100, iterations = 200, and repetitions = 10 with other arguments set to defaults, and the best K evaluated with the means of minimal cross-entropy (CE).
For principal component analysis (PCA), allelic frequency data was first generated with the “makefreq” function of R package adegenet (Jombart and Ahmed, 2011) with missing values replaced with the mean of the corresponding allele and analyzed using the “prcomp” function of R based on correlation matrix. A neighbor joining (NJ) tree was generated based on Nei's genetic distance (Nei, 1978), and bootstrap support value (BSV) was calculated using the “aboot” function of R package poppr with 1,000 bootstrap resampling, missing values were also replaced with the mean of the corresponding allele in the NJ tree construction.
Importance of Environmental Variables Explaining Genetic Variation
The most important environmental variables explaining genetic variation based on the total and outlier data sets were analyzed according to the double stopping criterion (Blanchet et al., 2008) using the “forward.sel” function of R package packfor (Dray, 2013). Significance was determined using 999 permutations. In the forward selection analysis, three categories (bioclimate, ecology, and topology) of explanatory variables were used separately.
Isolation-by-Environment and Isolation-by-Distance
Mantel correlation between geographic distance and environmental heterogeneity was assessed with the “mantel” function of R package vegan (Oksanen et al., 2017). Environmental and geographic distance matrices were generated using the “dist” function of R based on Euclidean distance. Nei's genetic distance matrix (Nei, 1978) was calculated using the “nei.dist” function of R package poppr. The relative role of environment and geography on population genetic distance was evaluated using multiple matrix regression with randomization (MMRR) implemented in the “MMRR” function of R (Wang, 2013) based on the total and outlier data sets. MMRR was used to quantify how dependent variable (genetic variation) responds to changes in explanatory variables (environmental, geographic, and environmental plus geographic). In MMRR, regression coefficients IBE (βE) and IBD (βD) were calculated and tested with 999 permutations.
Results
Genotyping and SNP Calling
A total of 582,985,540 PE reads were obtained following ddRADseq of 72 individuals of K. davidiana and K. davidiana var. formosana (Supplementary Table 4). After filtering using STACKS pipeline to remove low quality reads, ambiguous barcodes, and overrepresented sequences, 574,480,673 reads remained. A catalog containing 7,663,569 loci was created for generation of genotypes in all individuals. For each individual, an average number of 395,546 stacks were assembled with an average read depth of 5.29 per stack. Further filtering with the presence of SNPs in at least 50% of examined samples across populations resulted in a total of 17,982 SNPs genotyped for samples included both K. davidiana and K. davidiana var. formosana (Supplementary Table 3). When only samples of K. davidiana var. formosana were used, we obtained 13,914 SNPs.
We also obtained the 40 and 60% missing data sets and examined the effect of the levels of missing values on the distributions of the levels of population genetic diversity measures and pairwise locus FST. The distributions of the levels of genetic diversity measures across populations and distributions of pairwise locus FST for the three missing data sets are shown in Supplementary Figures 1, 2. KS test revealed that the distributions of population genetic diversity measures across populations and pairwise locus FST differed significantly between 40 and 60% missing data sets (Supplementary Figures 1, 2; Supplementary Table 5). Significant shape shifting between the distributions of pairwise locus FST of 40 and 50% missing data sets was revealed (P = 0.038). KS test showed no significant differences between 40 and 50% and between 50 and 60% missing data sets in the distributions of π, HO, HE, and uHE across populations. However, the distributions of AR across populations were significantly different between 50 and 60% missing data sets probably due to the significant increase in the number of different alleles per locus in 60% than in 50% missing data set (Supplementary Figure 1). In addition, 60% missing data set had much lower numbers of SNPs compared with 50% missing data set (Supplementary Table 3) and may cause the loss of valuable information in individual assignments (Huang et al., 2010; Chattopadhyay et al., 2014) and removing SNPs that with high mutation rate for recent divergence (Huang and Knowles, 2016). In contrast, 40% missing data set that with higher number of missing values may also influence individual assignments (Chattopadhyay et al., 2014; Huang and Knowles, 2016). We chose to adopt the 50% missing data set, based on number of SNP obtained (Supplementary Table 3), for further use in the present study. All raw sequences are available at NCBI SRA Bioproject PRJNA419582 and Biosample SAMN08093166–SAMN08093171. The 50% missing data sets included both investigated species and K. davidiana var. formosana only in STRUCURE format (Pritchard et al., 2000) are provided in Data Sheets 1, 2, respectively.
Population Genetic Diversity of Taiwan Cow-Tail Fir
We found average values of all population genetic diversity measures were smaller in K. davidiana var. formosana (AR = 1.078, π = 0.080, HO = 0.100, HE = 0.075, and uHE = 0.080) than the values in K. davidiana (AR = 1.096, π = 0.101, HO = 0.121, HE = 0.093, and uHE = 0.100) (Supplementary Table 6). Population measures of genetic diversity within K. davidiana var. formosana, including AR, π, HO, HE, and uHE, averaged 1.100 (1.091–1.105), 0.103 (0.093–0.109), 0.128 (0.119–0.135), 0.096 (0.089–0.101), and 0.103 (0.093–0.109), respectively (Table 1). Overall, genetic diversity measures within K. davidiana var. formosana differed significantly among populations (type-II Wald χ2-test, AR: χ2 = 334.25; π: χ2 = 436.85; HO: χ2 = 359.03; HE: χ2 = 254.86; and uHE: χ2 = 436.9, and all P < 0.0001). HE was lower compared to HO in all populations (25.4, 26.1, 24.1, 22.6, and 25.3% lower, respectively, for populations JGL, GPL, ST, DW30, and DW41). The levels of genetic diversity measures (AR, π, HO, HE, and uHE) were comparatively higher in the northern than in the southern populations (Table 1, Tukey's Ps < 0.0001). No difference was found between the three northern populations in all genetic diversity measures. The levels of all genetic diversity measures except HO were also significantly different between the two southern populations (AR: P = 0.026; π: P = 0.027; HO: P = 0.999; HE: P = 0.033; and uHE: P = 0.027). FIS-values were all negative and deviate significantly from zero for all populations either with data included only K. davidiana var. formosana (Table 1) or data included both K. davidiana var. formosana and K. davidiana (Supplementary Table 6). Multilocus LD assessed using IA and rD found only the DW41 population had significant non-zero values (IA = 4.931, P = 0.002; rD = 0.001, P = 0.002, Table 1) than expected under a null distribution, indicating significant non-random associations between alleles in the DW41 population.
Potential Selective Outliers in Taiwan Cow-Tail Fir
Twenty-three outlier SNPs (0.2%) were found using both BAYESCAN and FDIST in global and pairwise population comparisons (Table 2), and 15 of them were found to be associated strongly with environmental variables, including annual mean temperature, number of rainfall days per year, aspect, and soil pH using Samβada (Table 2). Most of these 15 outlier SNPs had log10(PO) > 1.0 in either global or between population comparisons indicating strong evidence for selection (Jeffreys, 1961), and outlier SNPs that had log10(PO) = 1,000 were observed when the southern DW41 population was compared to the northern populations. Samβada revealed four outlier SNPs (109734_46, 334591_7, 505960_78, and 522238_59) with log10(PO) = 1,000 in global comparison associated strongly with environmental variables: CC with aspect and TT with number of rainfall days per year for SNP 109734_46 (the minor C allele frequencies were 0.92, 0.86, 0.79, 0.20, and 0.00, respectively, for populations JGL, GPL, ST, DW30, and DW41), TT and GG with annual mean temperature for SNP 334591_7 (the minor T allele frequencies were 0.00, 0.00, 0.00, 0.70, and 1.00, respectively, for populations JGL, GPL, ST, DW30, and DW41), CC and AA with annual mean temperature for SNP 505960_78 (the minor C allele frequencies were 0.00, 0.00, 0.00, 0.71, and 1.00, respectively, for populations JGL, GPL, ST, DW30, and DW41), and GG with aspect and AA with number of rainfall days per year for SNP 522238_59 (the minor G allele frequencies were 0.92, 0.80, 0.64, 0.23, and 0.00, respectively, for populations JGL, GPL, ST, DW30, and DW41). No significant LD between these four outlier SNPs was found with two-locus exact test (Supplementary Table 7). Functional annotation of the sequences containing the outlier SNPs with BLASTN (Supplementary Table 8) found high sequence similarities for locus 227675 (outlier SNP found in global comparison) to the mitochondrial alternative oxidase 1 (AOX1) of Araucaria angustifolia (E = 5E-10), locus 334591 (outlier SNP found in global and all pairwise population comparisons) to the clone GQ03405_M22 mRNA of Picea glauca (E = 4E-23), and locus 521876 (outlier SNP found in comparison between the southern neighboring DW30 and DW41 populations) to the mitochondrial large subunit ribosomal RNA gene (LSU rRNA) of Abies homolepis (E = 3E-35). Clone GQ03405_M22 mRNA sequence of Pic. glauca is corresponded with the sequences of mitochondrial cytochrome oxidase subunit 1 (COX1) of larix gmelinii (EF053147.1)
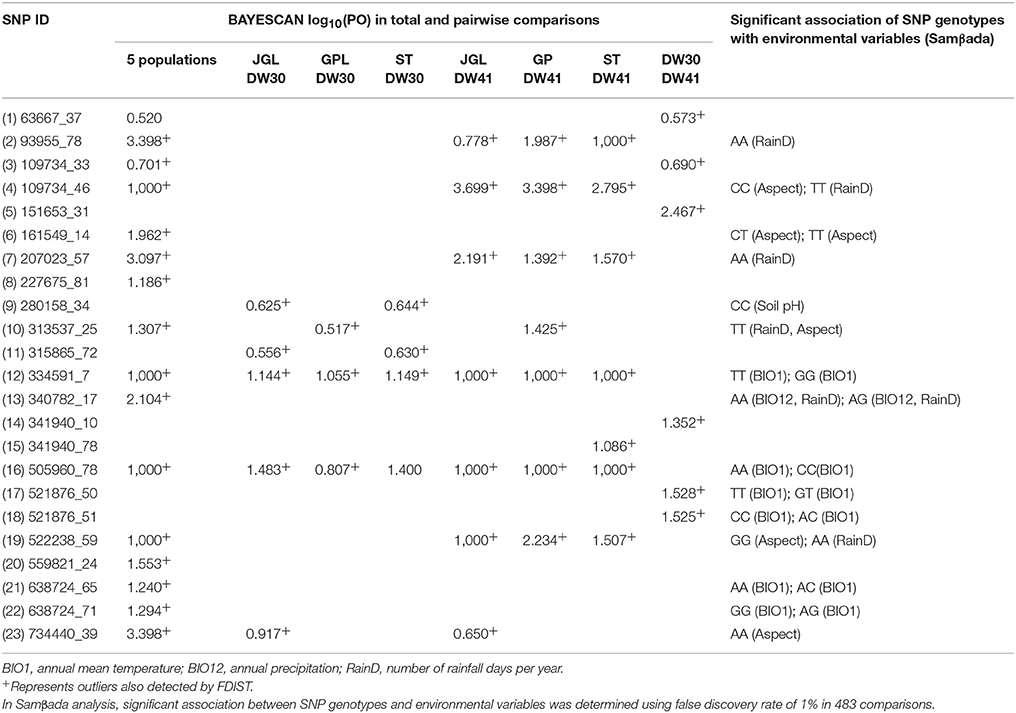
Table 2. Selective outliers identified by BAYESCAN and FDIST and their correlations with environmental variables analyzed with Samβada.
Genetic Differentiation
Both AMOVA and FST measures revealed significant differentiation between K. davidiana and K. davidiana var. formosana (ΦCT = 0.233, P = 0.001; FST = 0.077, P = 0.001; Table 3). The levels of genetic differentiation were shallow but significant in all comparisons when the total and neutral data sets were used in the analyses. However, significantly high levels of genetic differentiation were found in all comparisons using the outlier data set (Table 3). Across populations of K. davidiana var. formosana, significant ΦST (= 0.422, P = 0.001) and across population FST (= 0.427, P = 0.001) were found based on the outlier data. Comparing between populations of the North and South regions, ΦST was 0.425 (P = 0.001) and FST was 0.437 (P = 0.001). Significant AMOVA and FST were also found between populations within the North (ΦST = 0.133, P = 0.001; FST = 0.125, P = 0.001) and within the South (ΦST = 0.326, P = 0.001; FST = 0.290, P = 0.001).
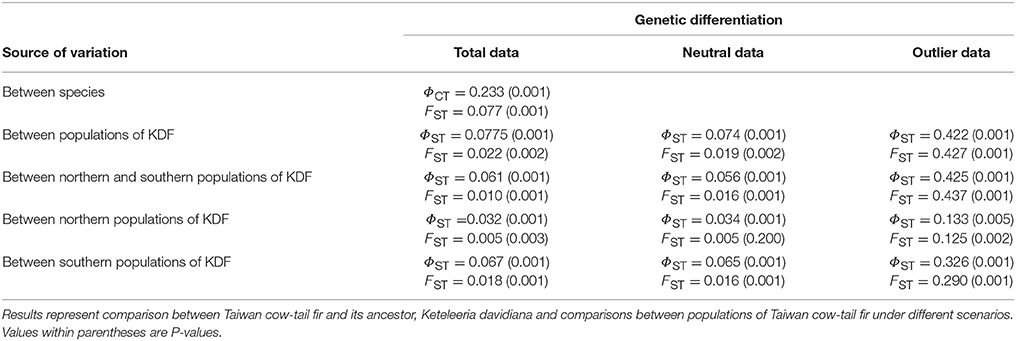
Table 3. Summary of the analysis of molecular variance (AMOVA) and across population FST.
Genetic Clustering
Using the total data, eigenvalues for the first two PCs were 33.43 and 14.00 and 16.32 and 9.84, when both species and K. davidiana var. formosana only were analyzed, respectively. However, only small amounts of genetic variation were explained by the first two PCs (both species: PC1 = 8.01%, PC2 = 3.35%; K. davidiana var. formosana only: PC1 = 4.43%, PC2 = 2.67%) (Figure 2), suggesting that only minor proportion of SNPs possessed the power of species delimitation and individual distinction, and most alleles were shared between ancestor and derivative species (95.0 ± 1.35%) and between populations of K. davidiana var. formosana (94.7 ± 0.4%), but effective genetic clustering was observed. PCA revealed clear distinction between K. davidiana and K. davidiana var. formosana (Figure 2A). In general, three distinct clusters of the northern, southern DW30, and southern DW41 populations of K. davidiana var. formosana were found (Figure 2B), however, with amalgamation of four DW30 individuals with the northern cluster. Individuals of the DW41 population were found to be distinct genetically.
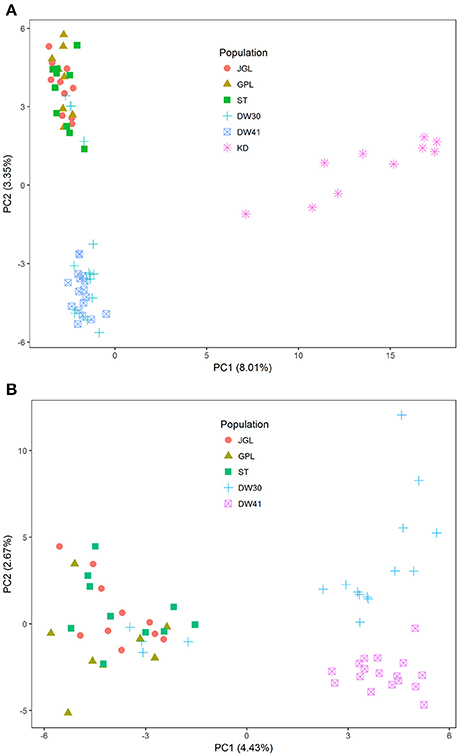
Figure 2. Scatter plots of the first two principal components (PCs) based on allelic frequencies of SNPs. (A) Samples of Taiwan cow-tail fir and Keteleeria davidiana (n = 72) and (B) samples of Taiwan cow-tail fir (n = 62). See Table 1 for population code abbreviations for Taiwan cow-tail fir. KD, K. davidiana.
CE was minimized at K = 2 and K = 1, respectively, when data included K. davidiana and K. davidiana var. formosana and data included only K. davidiana var. formosana, based on sNMF algorithm of LEA (Supplementary Figures 3A,B). Individual ancestry inferred with ADMIXTURE found CV error was minimized at K = 1 in both data sets, and no difference for 10 runs of a given K was found estimating CV (Supplementary Figures 3C,D). However, genetically homogeneous groups that resolved substructure with the highest biological meaning revealed otherwise. With LEA and ADMIXTURE, a clear phylogenetic break between K. davidiana and K. davidiana var. formosana was observed at K = 3 and 4 based on the total data (Figure 3A). Both LEA and ADMIXTURE showed distinction between northern and southern populations of K. davidiana var. formosana when K = 4 (Figures 3A,B). Admixtures between individuals of the southern DW30 population and northern populations were also observed. Individuals of the southern DW41 population were clearly separated from individuals of all other populations of K. davidiana var. formosana at K = 3 analyzed with LEA and at K = 4 analyzed with ADMIXTURE (Figure 3B). Similar pattern of genetic structuring was also found based on the NJ tree (Figure 4). The NJ tree revealed a close relationship between K. davidiana (KD clade) and the DW30 population of K. davidiana var. formosana (DW30 clade) forming a KD + DW30 clade, which can be easily collapsed to a KD + DW30 + DW41 clade (BSV > 90%) due to a low BSV of < 70%. The individuals of the three northern populations of K. davidiana var. formosana formed a well-supported separate clade with a BSV of >90%.
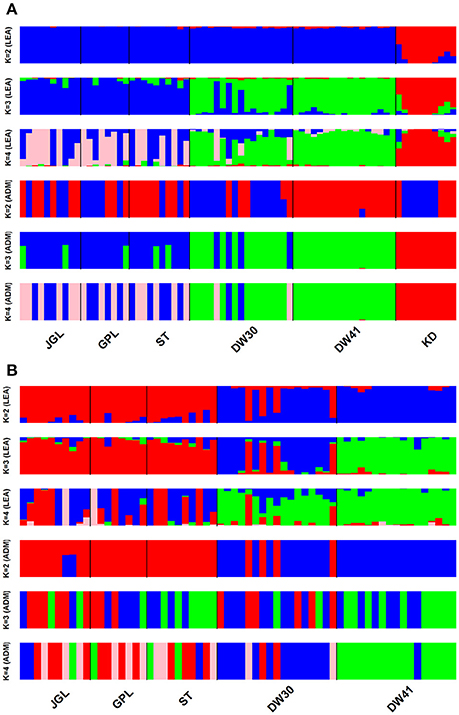
Figure 3. Barplots represent inference of individual assignments based on LEA and ADMIXTURE (ADM) for different clustering scenarios (K). (A) Samples of Taiwan cow-tail fir and Keteleeria davidiana (n = 72) and (B) samples of Taiwan cow-tail fir (n = 62). See Table 1 for population code abbreviations. KD, K. davidiana.
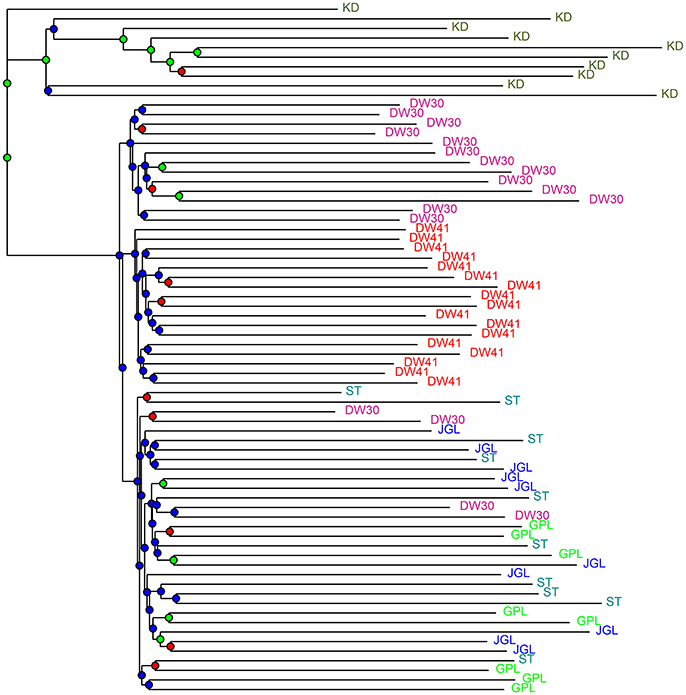
Figure 4. Neighbor-joining tree of samples of Taiwan cow-tail fir and Keteleeria davidiana based on Nei's genetic distance. Bootstrap support values (BSVs) were coded with colored nodes with BSVs ≥ 90% (green), 90% < BSVs ≥ 70% (red), and BSVs < 70% (blue), respectively. See Table 1 for population code abbreviations. KD, K. davidiana.
Importance of Environmental Variables Explaining Genetic Variation and the Effect of Environment and Geography on Genetic Variation of Taiwan Cow-Tail Fir
Because similar results were found for the forward selection and MMRR analyses based on the total and neutral data sets, only the results based on the total and outlier data sets are reported (Tables 4, 5). All environmental variables explained little genetic variation but was significant (F-test) when the total data was used (Table 4). When the outlier data was used in the forward selection, substantial amounts of outlier genetic variation were explained significantly by environmental variables. The most important environmental variables that explained outlier genetic variation were number of rainfall days per year (adjusted R2 = 0.311, F = 28.55, P = 0.001), aspect (adjusted R2 = 0.230, F = 19.18, P = 0.001), and annual mean temperature (adjusted R2 = 0.164, F = 12.95, P = 0.001). Soil pH (adjusted R2 = 0.090, F = 9.99, P = 0.001), annual precipitation (adjusted R2 = 0.070, F = 6.49, P = 0.001), and slope (adjusted R2 = 0.041, F = 4.33, P = 0.001) were also found to significantly explain the outlier genetic variation.
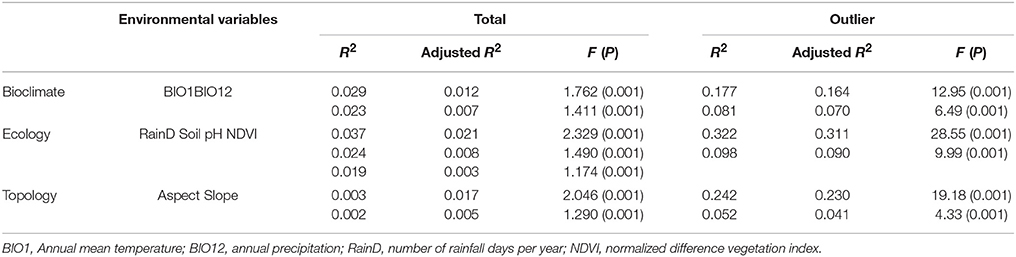
Table 4. Forward selection of environmental variables classified into three categories (bioclimate, ecology, and topology) explaining genetic variation within Taiwan cow-tail fir.
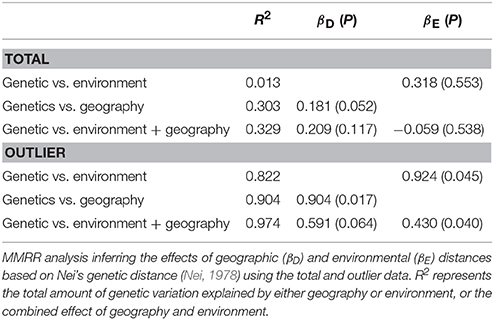
Table 5. Results of multiple matrix regression with randomization (MMRR) analysis.
Strong correlation between environmental and geographic distance matrices was found (Mantel r = 0.465, P = 0.025). Essentially no correlation was found between genetic variation and environment, between genetic variation and geography, and between genetic variation and combined effect of environment and geography, based on the total data (Table 5). Using the outlier data, significant IBE was found between genetic variation and environment (R2 = 0.822, βE = 0.924, P = 0.045). Significant IBD (between genetic variation and geography) was also found (R2 = 0.904, βD = 0.904, P = 0.017). When considering both environment and geography, results showed a pattern of strong IBE based on the outlier data (R2 = 0.974; IBD: βD = 0.591, P = 0.064; IBE: βE = 0.430, P = 0.040).
Discussion
Species Delimitation between K. Davidiana and K. davidiana var. formosana
Species delimitation using genome-wide markers has been demonstrated in plants (e.g., Eaton and Ree, 2013; Paun et al., 2016). In the present study, AMOVA showed significantly high differentiation at species level (ΦCT = 0.233, P = 0.001) though between species FST was low but also significant (FST = 0.077, P = 0.001). Nevertheless, distinction between the closely related island and mainland Keteleeria species pair were elucidated using ddRADseq. Three lines of evidence, including PCA (Figure 2A), individual assignments (LEA and ADMIXTURE, Figure 3A), and the NJ tree (Figure 4), suggest that each of species studied is distinct. Although the NJ tree revealed that K. davidiana (KD clade) was most closely related to the DW30 population of K. davidiana var. formosana, Keteleeria colonization from China into southern Taiwan cannot be inferred based solely on the NJ tree. Moreover, the disappearance of K. davidiana var. formosana from its historically occupied habitats in central Taiwan (Tsukada, 1967) and other areas hinders the investigation of Keteleeria colonization route.
Population Genetic Diversity and Outbreeding of Taiwan Cow-Tail Fir
Similar trends in AR, π, HO, HE, and uHE were observed across populations of K. davidiana var. formosana (Supplementary Figure 1). The average levels of population genetic diversity measures, including AR, π, HO, HE, and uHE (Table 1), were lower in restricted and disjunctly distributed K. davidiana var. formosana compared with widespread species of Pinaceae, e.g., Pic. glauca (white spruce) (AR = 1.920, HO = 0.276, uHE = 0.270, Namroud et al., 2008), Pic. mariana (black spruce) (AR = 1.850, HO = 0.241, HE = 0.247, Prunier et al., 2011), Pinus albicaulis (whitebark pine) (AR = 1.93, HO = 0.32, HE = 0.35, uHE = 0.36, Liu et al., 2016), and Pic. abies (Norway spruce) (π = 0.0893, HO = 0.138, HE = 0.258, Fagernäs, 2017). In the present study, we found that populations of K. davidiana var. formosana appear to harbor substantial amount of variation relative to K. davidiana (AR: 98.3%, π: 80.0%, HO: 82.4%, HE: 81.1%, and uHE: 80.0%) and may have potential for adaptive evolution under natural selection (Petit and Hampe, 2006; Barrett and Schluter, 2008).
Within K. davidiana var. formosana, the northern populations had comparatively higher levels of genetic diversity measures than those in the southern populations (Table 1). The levels of HE were lower than the level of HO in all populations, resulting in negative FIS-values. A negative value of FIS indicates heterozygote excess and is typical of conifers that are outcrossing (Hamrick et al., 1992; Hamrick and Godt, 1996). An excess of heterozygosity could indicate negative assortative mating or higher fitness of heterozygotes (Lachance, 2008). Moreover, significant non-zero IA and rD values were only found in the DW41 population, indicating significant non-random associations between alleles, which might have related to the higher degree of local adaptation in the DW41 than in other populations of Taiwan cow-tail fir (Bürger and Akerman, 2011). However, all K. davidiana var. formosana populations showed significant non-zero IA and rD values based on AFLP (Fang et al., 2013). The discrepancy could be due to the probable excess of homozygous genotypes resulted from the loss of restriction sites in AFLP, however, the presence/absence of restriction sites is not the primary source of information in ddRADseq (Cariou et al., 2016). We used the same restriction enzymes in the present study as that used in the AFLP study, standard errors of the estimates of all genetic diversity measures in all populations were similar and smaller based on ddRADseq, while dissimilar and larger standard errors of population uHE were observed based on AFLP (Fang et al., 2013), suggesting more reliable estimation of IA and rD within populations based on ddRADseq data. It is likely that errors produced during the PCR and sequencing can be filtered away and producing a large number of reliable high quality SNPs for population genetic analysis.
Multilocus LD based on ddRADseq revealed only one population (DW41) had significant non-zero IA and rD values, indicating rapid decay of LD over time in most examined populations of K. davidiana var. formosana conforming to studies of other Pinaceae species (Brown et al., 2004; Neale and Savolainen, 2004; Heuertz et al., 2006). Retention of significant LD in the DW41 population may have been caused by recent bottlenecks (Petit and Hampe, 2006) and/or natural selection (Barrett and Schluter, 2008; Eckert et al., 2010). Nonetheless, we cannot exclude the possibility of past bottlenecks occurred in all populations since the levels of genetic diversity measures were low across populations compared with widespread Pinaceae species (Namroud et al., 2008; Prunier et al., 2011; Liu et al., 2016; Fagernäs, 2017).
Population Differentiation and Structure of Taiwan Cow-Tail Fir
The levels of genetic differentiation within K. davidiana var. formosana based on the total and neutral data sets are in accordance with the general realization that conifers typically exhibit low population differentiation due to long distance wind pollination (Hamrick et al., 1992; Hamrick and Godt, 1996). Based on the total data, significant differentiation between K. davidiana and K. davidiana var. formosana was found (Table 3), suggesting apparent distinction between ancestor-derivative species pair. Only moderate level of genetic differentiation was found when compared between northern populations based on the outlier data (Table 3), and we did not detect any outlier SNPs when compared between the northern populations based on BAYESCAN and FDIST. High levels of genetic differentiation were found when comparisons involved the southern DW30 and DW41 populations based on the outlier data. These results are only partially concordant to the results of previous AFLP study (Fang et al., 2013), in which moderate level of differentiation based on FST outliers was found, probably because of high level of stringency applied in the identification of FST outliers in the present study.
Genetic clustering, based on the total data, using ddRADseq data in the present study provided a prominent phylogeographic break between northern and southern populations within K. davidiana var. formosana compared with no clear northern-southern distinction based on the total AFLP data (Fang et al., 2013). The admixtures of individuals between the southern DW30 population and northern populations, based on PCA (Figure 2B), LEA (Figure 3), ADMIXTURE (Figure 3), and the NJ tree (Figure 4), might have resulted from incomplete lineage sorting of ancestral variation or recent hybridization. Long distance seed dispersal between the southern DW30 population and northern populations may be less likely because of the recalcitrant seed storage behavior (Yang et al., 2006) and low rate of regeneration in natural stands (Wang, 1987) in K. davidiana var. formosana. Moreover, the NJ tree showed close relationship between K. davidiana and the southern populations of K. davidiana var. formosana. This result suggests that recent hybridization between the southern DW30 population and northern populations could be probable via effective pollen dispersal, in agreement with the ages of old growth trees between 100 and 300 years. Viable long distance pollen migration in Pin. taeda of at least 41 km was found (Williams, 2010). Pine pollens can travel up to 600–1,000 km (Dyakowska, 1948), whereas later Dyakowska (1959) suggested a pollen traveling range of 74.7 km. Szczepanek et al. (2017) reported a pine pollen travel distance of 500–750 km from Ukraine and Slovakia to southern Poland. In addition, viable long distance pollen dispersal can be aided by tropical cyclones and seasonal monsoons (Williams, 2009). Hence, recent hybridization between the southern DW30 population and northern populations of K. davidiana var. formosana via effective pollen migration could be possible.
Factors such as spatial context of selection and the balance between the strength of divergent selection and the between-population migration rates are important influencing population divergence (Endler, 1973; Lenormand, 2002). Individuals of the three northern populations were not clearly distinguished from each other probably because of the high rate of effective pollen dispersal among populations that are in geographical proximity, particularly in wind-pollinated conifers (Hamrick et al., 1992; Hamrick and Godt, 1996). Spatial environmental heterogeneity at overall scale among K. davidiana var. formosana populations was suggested by significant Mantel correlation between geographic and environmental distances (Mantel r = 0.465, P = 0.025). Spatial environmental heterogeneity and strong IBE based on the outlier data (Table 5) suggest that gene flow, particularly between the two southern neighboring populations (DW30 and DW41), could have been restricted due probably to habitat isolation or immigrant inviability arisen from local optimal for the environment and reduced survival and reproduction of migrants (Nosil et al., 2005; Jump and Peñuelas, 2006).
Adaptive Divergence in Association with Environmental Heterogeneity in Taiwan Cow-Tail Fir Despite Significant Role of Geography on Population Differentiation
Evolutionary theory predicts that population genetic divergence should be correlated with both geographic distance and environmental heterogeneity. Gene flow among populations could follow both IBD and IBE patterns, corresponding to geographic distance and environmental context (Sexton et al., 2014). Of the 26 studies related to plants summarized by Sexton et al. (2014), 38.5% found both IBD and IBE patterns, and 30.8 and 11.5% found IBD and IBE, respectively. In the present study, geographic distance was strongly correlated with environmental distance (Mantel r = 0.465, P = 0.025) suggesting that both IBD and IBE may have influenced population divergence within K. davidiana var. formosana. No significant correlation was found between genetic and environmental distances and between genetic and combined effect of environmental and geographic distances based on the total data using MMRR (Table 5). However, marginal significance between genetic and geographic distances (βD = 0.181, P = 0.052) was found based on the total data, suggesting that IBD could have played an important role in shaping the population genetic divergence of Taiwan cow-tail fir. When the outlier data was used, genetic variation was significantly correlated with either geographic or environmental (βD = 0.904, P = 0.017; βE = 0.924, P = 0.045) distances. In addition, strong IBE was found when considering combined effect of geography and environment (βD = 0.591, P = 0.064; βE = 0.430, P = 0.040). Our results suggest adaptive divergence corresponding to environmental heterogeneity despite strong IBD within K. davidiana var. formosana.
With the seven environmental variables retained that were separated into three categories: bioclimate, ecology, and topology, annual mean temperature, number of rainfall days per year, and aspect were found to be the most important environmental factors that explained substantial amounts of the outlier genetic variation using forward selection (Table 4). Results of forward selection are conformed to the results of Samβada analysis that 15 outlier SNPs were found to be strongly correlated with environmental variables using multiple univariate logistic regression (Table 2). Environmental gradients of annual temperature, number of rainfall days per year, and aspect are the most important environmental factors influencing adaptive variation in K. davidiana var. formosana. However, the second most important environmental factors, including annual precipitation, soil pH, and slope could also be important environmental factors that may have played key roles in driving adaptive divergence in K. davidiana var. formosana.
Temperature and precipitation are commonly found to play prominent roles as selective drivers for adaptive variation in various plant species (Manel et al., 2010, 2012; Bothwell et al., 2013; Fang et al., 2013; Hsieh et al., 2013; Huang et al., 2015). The topographic factor aspect is an important predictor of forest attributed to differences in radiation exposure and has a strong influence on the microclimate (Rosenberg et al., 1983; Bennie et al., 2008), and was found to be associated with genetic variation within (Manel et al., 2010, 2012; Bothwell et al., 2013) and between species (Nakazato et al., 2010; Huang et al., 2015). Slope is also a factor that may influence habitat microclimate (Brousseau et al., 2015) and contributed to intra- and inter-species adaptive divergence (Monahan et al., 2012; Brousseau et al., 2015). Small-scale habitat variation in soil alkali content has been found to be involved in intraspecific adaptive divergence of photosynthetic traits in a grass species, Phragmites australis (Qiu et al., 2017). In addition, Pease et al. (2016) found non-synonymous mutations in 43 genes strongly associated with soil pH in rapidly diverged wild Solanum species. In K. davidiana var. formosana, the aspect of the southern DW30 (34.7°) and DW41 (61.5°) (Supplementary Table 2) populations facing northeast, thereby, causing periodical drier conditions implying water stress or desiccation due to foehn winds induced by tropical cyclones (Chen et al., 2010). Tropical cyclones and the seasonal monsoon rainfall may have dramatically raised the amount of precipitation (Chen and Chen, 2011) in the southern DW41 population that had the highest annual precipitation (4,810 mm/year) compared with other populations (Supplementary Table 2). The more alkali condition of the southern DW30 population (soil pH = 5.5) compared with other populations may have played an important role in the divergence with the neighboring DW41 population. Therefore, the large-scale (temperature, number of rainfall days per year, precipitation) and the small-scale (aspect, slope, and soil properties) habitat variations could have played critical roles in shaping the population divergence between geographically distant and neighboring populations of K. davidiana var. formosana.
Sequences flanking three outlier SNPs that associated strongly with environmental variables were found to have high sequence similarities with low E-values to specific genes of mitochondrial AOX, COX, and LSU rRNA based on BLASTN search (Supplementary Table 8). The finding of the three annotated outlier SNPs associated with mitochondrial genes is interesting because mitochondrial genome is maternally inherited in Pinaceae (Hipkins et al., 1994) and displays higher subdivision among populations than paternally or biparentally inherited genes (Petit et al., 2005), and mitochondrial genes are known to play critical roles in plant local adaptation (Bock et al., 2014; Sloan, 2015). At the end of mitochondrial electron transport chain, oxygen can be reduced to water by either COX or AOX (Millar et al., 2008; Kühn et al., 2015). AOX and COX can relax the highly coupled and tensed electron transport process of mitochondria hence providing and maintaining metabolic homeostasis by reducing oxygen to water (Vanlerberghe, 2013). AOX and COX are also found to be important in stress signaling and plant stress response (Bartoli et al., 2005; Vacca et al., 2006; Costa et al., 2007; Dahan et al., 2014; Kühn et al., 2015). Mitochondrial ribosomal RNA genes were found to be involved in robustness of cell growth, proliferation, and therefore the whole plant survival (Greber et al., 2014). Our results suggest that these three outlier SNPs potentially evolved under selection may have involved in the growth and survival of locally adapted lineages in K. davidiana var. formosana.
Conclusions
Genome-wide SNPs obtained using ddRADseq has permitted for the evaluation of species delimitation between K. davidiana and K. davidiana var. formosana, and assessment of genetic diversity and fine-scale population genetic structure in K. davidiana var. formosana. Unlike AFLP data (Fang et al., 2013), genome-wide SNPs provided distinct regional substructuring within K. davidiana var. formosana based on the total data. Significant negative FIS values estimated with SNP data in the present study reflected outbreeding of K. davidiana var. formosana typical of conifers. The present study highlights the separation of environmental variables into separate categories for investigation of the impact on genetic variation. We identified outlier SNPs potentially evolved under selection strongly associated with specific environmental variables that might have played important roles in maintaining metabolic homeostasis and survival underlying local adaptation despite significant IBD. The present study emphasizes the importance of examining population isolation using ddRADseq to understand the spatial distribution of genetic variation across a species range. Our analyses suggest adaptive divergence between allopatrically distributed northern and southern populations and between the geographically neighboring DW30 and DW41 populations attributed to environmental heterogeneity. In addition, differential contributions of seed dispersal and pollen migration might have played crucial roles in shaping the population structure and spatial distribution of genetic diversity across species range of Taiwan cow-tail fir.
Author Contributions
S-YH proposed, funded, and designed the research; J-DC, K-MS, S-YH, and Y-CC collected samples; K-MS performed research; S-YH, K-MS, C-TC, and J-DC analyzed data; S-YH and K-MS wrote the paper. All authors have read and approved the final manuscript.
Funding
This work was supported by the Taiwan Ministry of Science and Technology under grant number of MOST 103-2313-B-003-001-MY3 and also endowed by the National Taiwan Normal University (NTNU), Taiwan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Jui-Hung Chen for writing a php script in adding a 5-bp C and a 5-bp T, respectively, to the end of PE read 1 and read 2, avoiding the combination of the two PE reads during de novo assembly. The authors would also like to thank the reviewers and associate editor whose insightful comments and suggestions have greatly improved the presentation of this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00092/full#supplementary-material
Supplementary Table 1. Determination of the values of parameters in the discovery of SNPs in STACKS pipeline.
Supplementary Table 2. Site environmental variables, including site names, annual mean temperature (BIO1), annual precipitation (BIO12), number of rainfall days per year (RainD), normalized difference vegetation index (NDVI), Soil pH, aspect (0–360°), and slope (0–90°), of the five populations of Taiwan cow-tail fir.
Supplementary Table 3. Number of SNP in data sets containing non-missing genotypes in at least 40, 50, and 60% of samples across populations.
Supplementary Table 4. Overview of STACKS pipeline analyses for ddRADseq.
Supplementary Table 5. P-values of pairwise Kolmogorov Smirnov test for distributions of genetic diversity measures across populations and pairwise locus FST between data sets of non-missing genotypes in at least 40, 50, and 60% of samples across populations.
Supplementary Table 6. Summary of population genetic parameters in Keteleeria davidiana and Taiwan cow-tail fir based on ddRADseq. KD, K. davidiana.
Supplementary Table 7. Test of two-locus linkage disequilibrium between the outlier SNPs identified by FST-based methods.
Supplementary Table 8. Summary of the sequences containing outlier SNPs matches to GenBank gene sequences using BLASTN.
Supplementary Figure 1. Distributions of population genetic diversity measures, including allelic richness, nucleotide diversity, observed heterozygosity, expected heterozygosity, and unbiased expected heterozygosity in data sets of non-missing genotypes in at least 40, 50, and 60% of samples across populations.
Supplementary Figure 2. Distributions of pairwise locus FST in data sets of non-missing genotypes in at least 40, 50, and 60% of samples across populations.
Supplementary Figure 3. Minimal cross-entropy and cross validation error analyzed using LEA and ADMIXTURE. (A,B) LEA and (C,D) ADMIXTURE using samples included both Taiwan cow-tail fir (KDF) and Keteleeria davidiana (KD) or samples of Taiwan cow-tail fir.
Supplementary File 1. A php script for the addition of a 5-bp C and a 5-bp T, respectively, to the end of PE read 1 and read 2.
Data Sheet 1. Fifty percent missing data set for the five populations of Keteleeria davidiana var. formosana in STRUCTUR format (Pritchard et al., 2000).
Data Sheet 2. Fifty percent missing data set for samples include Keteleeria davidiana and K. davidiana var. formosana in STRUCTUR format (Pritchard et al., 2000).
References
Adamack, A. T., and Gruber, B. (2014). PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol. Evol. 5, 384–387. doi: 10.1111/2041-210X.12158
Agapow, P.-M., and Burt, A. (2001). Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 1, 101–102. doi: 10.1046/j.1471-8278.2000.00014.x
Aitken, S. N., Yeaman, S., Holliday, J. A., Wang, T., and Curtis-McLane, S. (2008). Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol. Appl. 1, 95–111. doi: 10.1111/j.1752-4571.2007.00013.x
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G., and Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi: 10.1038/nrg.2015.28
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A., and Luikart, G. (2008). LOSITAN: a workbench to detect molecular adaptation based on a FST-outlier method. BMC Bioinformatics 9:323. doi: 10.1186/1471-2105-9-323
Archer, F. I., Adams, P. E., and Schneiders, B. B. (2017). strataG: an R package for manipulating, summarizing and analysing population genetic data. Mol. Ecol. Resour. 17, 5–11. doi: 10.1111/1755-0998.12559
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376. doi: 10.1371/journal.pone.0003376
Barrett, R. D. H., and Schluter, D. (2008). Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44. doi: 10.1016/j.tree.2007.09.008
Bartoli, C. G., Gomez, F., Gergoff, G., Guiamét, J. J., and Puntarulo, S. (2005). Up-regulation of the mitochondrial alternative oxidase pathway enhances photosynthetic electron transport under drought conditions. J. Exp. Bot. 56, 1269–1276. doi: 10.1093/jxb/eri111
Bates, D., Maechler, M., Bolker, B., and Walker, S. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Soft. 67, 1–48. doi: 10.18637/jss.v067.i01
Beaumont, M. A., and Nichols, R. A. (1996). Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. B 263, 1619–1626. doi: 10.1098/rspb.1996.0237
Bennie, J., Huntley, B., Wiltshire, A., Hill, M. O., and Baxter, R. (2008). Slope, aspect and climate: spatially explicit and implicit models of topographic microclimate in chalk grassland. Ecol. Model. 216, 47–59. doi: 10.1016/j.ecolmodel.2008.04.010
Blanchet, F. G., Legendre, P., and Borcard, D. (2008). Forward selection of explanatory variables. Ecology 89, 2623–2632. doi: 10.1890/07-0986.1
Bock, D. G., Andrew, R. L., and Rieseberg, L. H. (2014). On the adaptive value of cytoplasmic genomes in plants. Mol. Ecol. 23, 4899–4911. doi: 10.1111/mec.12920
Bothwell, H., Bisbing, S., Therkildsen, N. O., Crawford, L., Alvarez, N., Holderegger, R., et al. (2013). Identifying genetic signatures of selection in a non-model species, alpine gentian (Gentiana nivalis L.), using a landscape genetic approach. Conser Genet. 14, 467–481. doi: 10.1007/s10592-012-0411-5
Brousseau, L., Foll, M., Scotti-Saintagne, C., and Scotti, I. (2015). Neutral and adaptive drivers of microgeographic genetic divergence within continuous populations: the case of the neotropical tree Eperua falcata (Aubl.). PLoS ONE 10:e0121394. doi: 10.1371/journal.pone.0121394
Brown, A. H., Feldman, M. W., and Nevo, E. (1980). Multilocus structure of natural populations of Hordeum spontaneum. Genetics 96, 523–536.
Brown, G. R., Gill, G. P., Kuntz, R. J., Langley, C. H., and Neale, D. B. (2004). Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. U.S.A. 101, 15255–15260. doi: 10.1073/pnas.0404231101
Bürger, R., and Akerman, A. (2011). The effects of linkage and gene flow on local adaptation: a two-locus continent-island model. Theor. Popul. Biol. 80, 272–280. doi: 10.1016/j.tpb.2011.07.002
Burridge, C. P., Brown, W. E., Wadley, J., Nankervis, D. L., Olivier, L., Gardner, M. G., et al. (2013). Did postglacial sea-level changes initiate the evolutionary divergence of a Tasmanian endemic raptor from its mainland relative? Proc. R. Soc. B. 280:20132448. doi: 10.1098/rspb.2013.2448
Buschiazzo, E., Ritland, C., Bohlmann, J., and Ritland, K. (2012). Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 12:8. doi: 10.1186/1471-2148-12-8
Cariou, M., Duret, L., and Charlat, S. (2016). How and how much does RAD-seq bias genetic diversity estimates? BMC Evol. Biol. 16:240. doi: 10.1186/s12862-016-0791-0
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W., and Postlethwait, J. H. (2011). Stacks: building and genotyping loci de novo from short-read sequences. G3 (Bethesda) 1, 3171–3182. doi: 10.1534/g3.111.000240
Chang, C. T., Lin, T. C., and Lin, N. H. (2009). Estimating the critical load and the environmental and economic impact of acid deposition in Taiwan. J. Geogr. Sci. 56, 39–58.
Chang, C. T., Wang, S. F., Vadeboncoeur, M. A., and Lin, T. C. (2014). Relating vegetation dynamics to temperature and precipitation at monthly and annual timescales in Taiwan using MODIS vegetation indices. Int. J. Remote Sens. 35, 598–620. doi: 10.1080/01431161.2013.871593
Chattopadhyay, B., Garg, K. M., and Ramakrishnan, U. (2014). Effect of diversity and missing data on genetic assignment with RAD-Seq markers. BMC Res. Notes 7:841. doi: 10.1186/1756-0500-7-841
Chen, C.-H., Huang, J.-P., Tsai, C.-C., and Chaw, S.-M. (2009). Phylogeny of Calocedrus (Cupressaceae), an eastern Asian and western North American disjunct gymnosperm genus, inferred from nuclear ribosomal nrITS sequences. Bot. Stud. 50, 425–433.
Chen, J.-H., Huang, C.-L., Lai, Y.-L., Chang, C.-T., Liao, P.-C., Hwang, S.-Y., et al. (2017). Postglacial range expansion and the role of ecological factors in driving adaptive evolution of Musa basjoo var. formosana. Sci. Rep. 7:5341. doi: 10.1038/s41598-017-05256-6
Chen, J., Källman, T., Ma, X., Gyllenstrand, N., Zaina, G., Morgante, M., et al. (2012). Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 191, 865–881. doi: 10.1534/genetics.112.140749
Chen, J.-M., and Chen, H.-S. (2011). Inter-decadal variability of summer rainfall in Taiwan associated with tropical cyclones and monsoon. J. Climate. 24, 5786–5798. doi: 10.1175/2011JCLI4043.1
Chen, T.-C., Wang, S.-Y., Yen, M.-C., Clark, A. J., and Tsay, J.-D. (2010). Sudden surface warming-drying events caused by typhoon passages across Taiwan. J. App. Meteorol. Clim. 49, 234–252. doi: 10.1175/2009JAMC2070.1
Chou, F.-S., Yang, C.-K., Lin, W.-L., Chen, T.-Y., Yang, Y.-P., and Liao, C.-K. (2009). Syntaxonomic and gradient analysis of Keteleeria davidiana var. formosana forests in Taiwan. Taiwan J. For. Sci. 24, 257–269.
Chou, Y.-W., Thomas, P. I., Ge, X.-J., LePage, B. A., and Wang, C.-N. (2011). Refugia and phylogeography of Taiwania in East Asia. J. Biogeogr. 38, 1992–2005. doi: 10.1111/j.1365-2699.2011.02537.x
Chung, J.-D., Lin, T.-P., Tan, Y.-C., Lin, M.-Y., and Hwang, S.-Y. (2004). Genetic diversity and biogeography of Cunninghamia konishii (Cupressaceae), an island species in Taiwan: a comparison with Cunninghamia lanceolata, a mainland species in China. Mol. Phylogenet. Evol. 33, 791–801. doi: 10.1016/j.ympev.2004.08.011
Costa, J. H., Jolivet, Y., Hasenfratz-Sauder, M.-P., Orellano, E. G., Da Guia Silva Lima, M., Dizengremel, P., et al. (2007). Alternative oxidase regulation in roots of Vigna unguiculata cultivars differing in drought/salt tolerance. J. Plant Physiol. 164, 718–727. doi: 10.1016/j.jplph.2006.04.001
Dahan, J., Tcherkez, G., Macherel, D., Benamar, A., Belcram, K., Quadrado, M., et al. (2014). Disruption of the CYTOCHROME C OXIDASE DEFICIENT1 gene leads to cytochrome c oxidase depletion and reorchestrated respiratory metabolism in Arabidopsis. Plant Physiol. 166, 1788–1802. doi: 10.1104/pp.114.248526
Davey, J. W., and Blaxter, M. L. (2010). RADSeq: next-generation population genetics. Brief. Funct. Genomics 9, 416–423. doi: 10.1093/bfgp/elq031
Dehestani, A., and Kazemitabar, S. K. (2007). A rapid efficient method for DNA isolation from plants with high levels of secondary metabolites. Asian J. Plant Sci. 6, 977–981. doi: 10.3923/ajps.2007.977.981
Dray, S. (2013). Packfor: Forward Selection with Permutation (Canoco p.46). R package version0.0-8. Available online at: http://r-forge.rproject.org/R/?group_id=195
Dray, S., and Dufour, A.-B. (2007). The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20. doi: 10.18637/jss.v022.i04
Dyakowska, J. (1948). The pollen rain on the sea and on the coasts of Greenland. Bull. Acad. Polon. Sci. Lettr. Ser. B 1, 25–33.
Dyakowska, J. (1959). Podrecznik Palynologii: Metody i Problemy. Warszawa: Wydawnictwa Geograficzne.
Eaton, D. A., and Ree, R. H. (2013). Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62, 689–706. doi: 10.1093/sysbio/syt032
Eckert, A. J., Bower, A. D., González-Martínez, S. C., Wegrzyn, J. L., Coop, G., and Neale, D. B. (2010). Back to nature: ecological genomics of loblolly pine (Pinus taeda, Pinaceae). Mol. Ecol. 19, 3789–3805. doi: 10.1111/j.1365-294X.2010.04698.x
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
Endler, J. A. (1973). Gene flow and population differentiation. Science 179, 243–250. doi: 10.1126/science.179.4070.243
Excoffier, L., and Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fagernäs, Z. (2017). Biogeography of Norway Spruce (Picea abies (L.) Karst.): Insights from a Genome-Wide Study. Dissertation Ph.D. thesis, University of Umeå, Sweden.
Fang, J.-Y., Chung, J.-D., Chiang, Y.-C., Chang, C.-T., Chen, C.-Y., and Hwang, S.-Y. (2013). Divergent selection and local adaptation in disjunct populations of an endangered conifer, Keteleeria davidiana var. formosana (Pinaceae). PLoS ONE 8:e70162. doi: 10.1371/journal.pone.0070162
Farjon, A. (1989). A second revision of the genus Keteleeria Carrière. Notes Roy. Bot. Gard. Edinburgh 46, 81–99.
Foll, M., and Gaggiotti, O. (2008). A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Fox, J., and Weisberg, S. (2011). An {R} Companion to Applied Regression, 2nd Edn. Thousand Oaks CA: Sage. Available online at: http://socserv.socsci.mcmaster.ca/jfox/Books/Companion
Frichot, E., and Francois, O. (2015). LEA: an R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929. doi: 10.1111/2041-210X.12382
Goudet, J. (2005). Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186. doi: 10.1111/j.1471-8286.2004.00828.x
Greber, B. J., Boehringer, D., Leitner, A., Bieri, P., Voigts-Hoffmann, F., Erzberger, J. P., et al. (2014). Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–519. doi: 10.1038/nature12890
Grivet, D., Sebastiani, F., Alía, R., Bataillon, T., Torre, S., Zabal-Aguirre, M., et al. (2011). Molecular footprints of local adaptation in two mediterranean conifers. Mol. Biol. Evol. 28, 101–116. doi: 10.1093/molbev/msq190
Hamrick, J. L., and Godt, M. (1996). Effects of life history traits on genetic diversity in plant species. Philos. Trans. Biol. Sci. 351, 1291–1298. doi: 10.1098/rstb.1996.0112
Hamrick, J. L., Godt, M. J. W., and Sherman-Broyles, S. L. (1992). Factors influencing levels of genetic diversity in woody plant species. New Forests 6, 95–124. doi: 10.1007/BF00120641
Heuertz, M., De Paoli, E., Källman, T., Larsson, H., Jurman, I., Morgante, M., et al. (2006). Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174, 2095–2105. doi: 10.1534/genetics.106.065102
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G., and Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978. doi: 10.1002/joc.1276
Hipkins, V. D., Krutovskii, K. V., and Strauss, S. H. (1994). Organelle genomes in conifers: structure, evolution, and diversity. For. Genet. 1:179–189.
Hoffmann, A., Griffin, P., Dillon, S., Catullo, R., Rane, R., Byrne, M., et al. (2015). A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim. Change Responses 2:1. doi: 10.1186/s40665-014-0009-x
Hsieh, Y.-C., Chung, J.-D., Wang, C.-N., Chang, C.-T., Chen, C.-Y., et al. (2013). Historical connectivity, contemporary isolation and local adaptation in a widespread but discontinuously distributed species endemic to Taiwan, Rhododendron oldhamii (Ericaceae). Heredity 111, 147–156. doi: 10.1038/hdy.2013.31
Huang, C.-L., Chang, C.-T., Huang, B.-H., Chung, J.-D., Chen, J.-H., Chiang, Y.-C., et al. (2015). Genetic relationships and ecological divergence in Salix species and populations in Taiwan. Tree Genet. Genom. 11, 39. doi: 10.1007/s11295-015-0862-1
Huang, H., He, Q., Kubatko, L. S., and Knowles, L. L. (2010). Sources of error inherent in species-tree estimation: impact of mutational and coalescent effects on accuracy and implications for choosing among different methods. Syst. Biol. 59, 573–583. doi: 10.1093/sysbio/syq047
Huang, H., and Knowles, L. L. (2016). Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst. Biol. 65, 357–365. doi: 10.1093/sysbio/syu046
Huete, A. R., Didan, K., Miura, T., Rodriguez, E. P., Gao, X., and Ferreira, L. G. (2002). Overview of the radiometric and biophysical performance of the MODIS vegetation indices. Remote Sens. Environ. 83, 195–213. doi: 10.1016/S0034-4257(02)00096-2
Jombart, T., and Ahmed, I. (2011). adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Jump, A. S., and Peñuelas, J. (2006). Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc. Natl. Acad. Sci. U.S.A. 103, 8096–8100. doi: 10.1073/pnas.0510127103
Kamvar, Z. N., Tabima, J. F., and Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281
Kozarewa, I., Ning, Z., Quail, M. A., Sanders, M. J., Berriman, M., and Turner, D. J. (2009). Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 6, 291–295. doi: 10.1038/nmeth.1311
Kühn, K., Yin, G., Duncan, O., Law, S. R., Kubiszewski-Jakubiak, S., Kaur, P., et al. (2015). Decreasing electron flux through the cytochrome and/or alternative respiratory pathways triggers common and distinct cellular responses dependent on growth conditions. Plant Physiol. 167, 228–250. doi: 10.1104/pp.114.249946
Lachance, J. (2008). A fundamental relationship between genotype frequencies and fitness. Genetics 180, 1087–1093. doi: 10.1534/genetics.108.093518
Lambeck, K., and Chappell, J. (2001). Sea level change through the last glacial cycle. Science 292, 679–686. doi: 10.1126/science.1059549
Lenormand, T. (2002). Gene flow and the limits to natural selection. Trends Ecol. Evol. 17, 183–189. doi: 10.1016/S0169-5347(02)02497-7
Lenth, R. V. (2016). Least-Squares Means: The R Package lsmeans. J. Stat. Soft. 69, 1–33. doi: 10.18637/jss.v069.i01
Li, H., and Hsuan, K. (1994). “Pinaceae,” in Flora of Taiwan, 2nd Edn, ed Editorial Committee of the Flora of Taiwan (Taipei: Epoch Publishing Co. Ltd.), 568–569.
Li, J.-W., Yeung, C. K. L., Tsai, P.-W., Lin, R.-C., Yeh, C.-F., Yao, C.-T., et al. (2010). Rejecting strictly allopatric speciation on a continental island: prolonged postdivergence gene flow between Taiwan (Leucodioptron taewanus, Passeriformes Timaliidae) and Chinese (L. canorum canorum) hwameis. Mol. Ecol. 19, 494–507. doi: 10.1111/j.1365-294X.2009.04494.x
Liu, J.-J., Sniezko, R., Murray, M., Wang, N., Chen, H., Zamany, A., et al. (2016). Genetic diversity and population structure of whitebark pine (Pinus albicaulis Engelm.) in western North America. PLoS ONE 11:e0167986. doi: 10.1371/journal.pone.0167986
Losos, J. B., and Ricklefs, R. E. (2009). Adaptation and diversification on islands. Nature 457, 830–836. doi: 10.1038/nature07893
Manel, S., Gugerli, F., Thuiller, W., Alvarez, N., Legendre, P., Holderegger, R., et al. (2012). Broad scale adaptive genetic variation in alpine plants is driven by temperature and precipitation. Mol. Ecol. 21, 3729–3738. doi: 10.1111/j.1365-294X.2012.05656.x
Manel, S., Poncet, B. N., Legendre, P., Gugerli, F., and Holderegger, R. (2010). Common factors drive adaptive genetic variation at different spatial scales in Arabis alpina. Mol. Ecol. 19, 3824–3835. doi: 10.1111/j.1365-294X.2010.04716.x
Mastretta-Yanes, A., Arrigo, N., Alvarez, N., Jorgensen, T. H., Piñero, D., and Emerson, B. C. (2015). Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Res. 15, 28–41. doi: 10.1111/1755-0998.12291
Millar, A. H., Small, I. D., Day, D. A., and Whelan, J. (2008). Mitochondrial biogenesis and function in Arabidopsis. Arabidopsis Book 6:e0111. doi: 10.1199/tab.0111
Mimura, M., and Aitken, S. N. (2010). Local adaptation at the range peripheries of Sitka spruce. J. Evolution. Biol. 23, 249–258. doi: 10.1111/j.1420-9101.2009.01910.x
Monahan, W. B., Pereira, R. J., and Wake, D. B. (2012). Ring distributions leading to species formation: a global topographic analysis of geographic barriers associated with ring species. BMC Biol. 10:20. doi: 10.1186/1741-7007-10-20
Naimi, B., Hamm, N. a. S., Groen, T. A., Skidmore, A. K., and Toxopeus, A. G. (2014). Where is positional uncertainty a problem for species distribution modelling? Ecography 37, 191–203. doi: 10.1111/j.1600-0587.2013.00205.x
Nakazato, T., Warren, D. L., and Moyle, L. C. (2010). Ecological and geographic modes of species divergence in wild tomatoes. Am. J. Bot. 97, 680–693. doi: 10.3732/ajb.0900216
Namroud, M.-C., Beaulieu, J., Juge, N., Laroche, J., and Bousquet, J. (2008). Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol. Ecol. 2008, 3599–3613. doi: 10.1111/j.1365-294X.2008.03840.x
Neale, D. B., and Savolainen, O. (2004). Association genetics of complex traits in conifers. Trends Plant Sci. 9, 325–330. doi: 10.1016/j.tplants.2004.05.006
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590.
Nosil, P., Vines, T. H., and Funk, D. J. (2005). Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution 59, 705–719. doi: 10.1111/j.0014-3820.2005.tb01747.x
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2017). Vegan: Community Ecology Package. R package version 2.4-2. Available online at: https://CRAN.R-project.org/package=vegan
Otte, D., and Endler, J. A. (1989). Speciation and Its Consequences. Sunderland: Sinauer Associates.
Pannell, J. R., and Fields, P. D. (2014). Evolution in subdivided plant populations: concepts, recent advances and future directions. New Phytol. 201, 417–432. doi: 10.1111/nph.12495
Parchman, T. L., Gompert, Z., Mudge, J., Schilkey, F. D., Benkman, C. W., and Buerkle, C. A. (2012). Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 21, 2991–3005. doi: 10.1111/j.1365-294X.2012.05513.x
Paris, J. R., Stevens, J. R., and Catchen, J. M. (2017). Lost in parameter space: a road map for stacks. Methods Ecol. Evol. 8, 1360–1373. doi: 10.1111/2041-210X.12775
Paun, O., Turner, B., Trucchi, E., Munzinger, J., Chase, M. W., and Samuel, R. (2016). Processes driving the adaptive radiation of a tropical tree (Diospyros, Ebenaceae) in New Caledonia, a biodiversity hotspot. Syst. Biol. 65, 212–227. doi: 10.1093/sysbio/syv076
Pease, J. B., Haak, D. C., Hahn, M. W., and Moyle, L. C. (2016). Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biol. 14:e1002379. doi: 10.1371/journal.pbio.1002379
Petit, R. J., Duminil, J., Fineschi, S., Hampe, A., Salvini, D., and Vendramin, G. G. (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 14, 689–701. doi: 10.1111/j.1365-294X.2004.02410.x
Petit, R. J., and Hampe, A. (2006). Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. 37, 187–214. doi: 10.1146/annurev.ecolsys.37.091305.110215
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., and Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. doi: 10.1371/journal.pone.0037135
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Prunier, J., Laroche, J., Beaulieu, J., and Bousquet, J. (2011). Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol. Ecol. 20, 1702–1716. doi: 10.1111/j.1365-294X.2011.05045.x
Qiu, T., Jiang, L., Li, S., and Yang, Y. (2017). Small-scale habitat-specific variation and adaptive divergence of photosynthetic pigments in different alkali soils in reed identified by common garden and genetic tests. Front. Plant Sci. 7:2016. doi: 10.3389/fpls.2016.02016
R Development Core Team (2013). R: A Language and Environment for Statistical Computing, version 3.0.0. Vienna: R Foundation for Statistical Computing. Available online at: http://www.Rproject.org/
Robertson, J. M., Langin, K. M., Sillett, T. S., Morrison, S. A., Ghalambor, C. K., and Funk, W. C. (2014). Identifying evolutionarily significant units and prioritizing populations for management on islands. Monogr. West. N. Am. Nat. 7, 397–411. doi: 10.3398/042.007.0130
Rosenberg, N. J., Blat, B. L., and Verma, S. B. (1983). Microclimate: The Biological Environment. New York, NY: Wiley.
Schlotfeldt, B. E., and Kleindorfer, S. (2006). Adaptive divergence in the superb fairy-wren (Malurus cyaneus): a mainland versus island comparison of morphology and foraging behaviour. Emu Aust. Ornithol. 106, 309–319. doi: 10.1071/MU06004
Sexton, J. P., Hangartner, S. B., and Hoffmann, A. A. (2014). Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68, 1–15. doi: 10.1111/evo.12258
Shafer, A. B. A., Peart, C. R., Tusso, S., Maayan, I., Bresfor, A., Wheat, C. W., et al. (2017). Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol. 8, 907–917. doi: 10.1111/2041-210X.12700
Sloan, D. B. (2015). Using plants to elucidate the mechnisms of cytonuclear co-evolution. New Phytol. 205, 1040–1046. doi: 10.1111/nph.12835
Sobel, J. M., and Streisfeld, M. A. (2015). Strong premating reproductive isolation drives incipient speciation in Mimulus aurantiacus. Evolution 69, 447–461. doi: 10.1111/evo.12589
Strasburg, J. L., Sherman, N. A., Wright, K. M., Moyle, L. C., Willis, J. H., and Rieseberg, L. H. (2012). What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Philos. Trans. Biol. Sci. 367, 364–373. doi: 10.1098/rstb.2011.0199
Strijk, J. S., Noyes, R. D., Strasberg, D., Cruaud, C., Gavory, F., Chase, M. W., et al. (2012). In and out of Madagascar: dispersal to peripheral islands, insular speciation and diversification of Indian Ocean daisy trees (Psiadia, Asteraceae). PLoS ONE 7:e42932. doi: 10.1371/journal.pone.0042932
Stucki, S., Orozco-terWengel, P., Bruford, M. W., Colli, L., Masembe, C., Negrini, R., et al. (2014). High performance computation of landscape genomic models integrating local indices of spatial association. arXiv:1405.7658v1 [q-bio.PE].
Su, H. J. (1984). Studies on the climate and vegetation types of the natural forest in Taiwan. (II). Altitudinal vegetation zones in relation to temperature gradient. Q. J. Chin. For. 17, 57–73.
Szczepanek, K., Myszkowska, D., Worobiec, E., Piotrowicz, K., Ziemianin, M., and Bielec-Bakowska, Z. (2017). The long-range transport of Pinaceae pollen: an example in Kraków (southern Poland). Aerobiologia 33, 109–125. doi: 10.1007/s10453-016-9454-2
Thornthwaite, C. W. (1948). An approach toward a rational classification of climate. Geogr. Rev. 38, 55–94. doi: 10.2307/210739
Tsukada, M. (1967). Vegetation in subtropical formosa during the pleistocene glaciations and the holocene. Palaeogeogr. Palaeocl. Paleoecol. 3, 49–64. doi: 10.1016/0031-0182(67)90005-3
Vacca, R. A., Valenti, D., Bobba, A., Merafina, R. S., Passarella, S., and Marra, E. (2006). Cytochrome c is released in a reactive oxygen species-dependent manner and is degraded via caspase-like proteases in tobacco Bright-Yellow 2 cells en route to heat shock-induced cell death. Plant Physiol. 141, 208–219. doi: 10.1104/pp.106.078683
Vanlerberghe, G. (2013). Alternative oxidase: a mitochondrial respiratory pathway to maintain metabolic and signaling homeostasis during abiotic and biotic stress in Plants. Int. J. Mol. Sci. 14:6805. doi: 10.3390/ijms14046805
Via, S. (2009). Natural selection in action during speciation. Proc. Natl. Acad. Sci. U.S.A. 106, 9939–9946. doi: 10.1073/pnas.0901397106
Wang, I. J. (2013). Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution 67, 3403–3411. doi: 10.1111/evo.12134
Wang, I. J., Glor, R. E., and Losos, J. B. (2013). Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol. Lett. 16, 175–182. doi: 10.1111/ele.12025
Wang, W.-P., Hwang, C.-Y., Lin, T.-P., and Hwang, S.-Y. (2003). Historical biogeography and phylogenetic relationships of the genus Chamaecyparis (Cupressaceae) inferred from chloroplast DNA polymorphism. Plant Syst. Evol. 241, 13–28. doi: 10.1007/s00606-003-0031-0
Wang, Y. (1987). Reproductive Cycle and Some Anatomical Studies on Keteleeria fomosana Hay. Dissertation, Ph.D. thesis, National Taiwan University, Taipei.
Williams, C. G. (2009). Conifer reproductive biology. Int. For. Rev. 11, 534–543. doi: 10.1007/978-1-4020-9602-0
Williams, C. G. (2010). Long-distance pine pollen still germinates after meso-scale dispersal. Am. J. Bot. 97, 846–855. doi: 10.3732/ajb.0900255
Wolf, J. B., Lindell, J., and Backström, N. (2010). Speciation genetics: current status and evolving approaches. Philos. Trans. Biol. Sci. 365, 1717–1733. doi: 10.1098/rstb.2010.0023
Worth, J. R., Jordan, G. J., McKinnon, G. E., and Vaillancourt, R. E. (2009). The major Australian cool temperate rainforest tree Nothofagus cunninghamii withstood Pleistocene glacial aridity within multiple regions: evidence from the chloroplast. New Phytol. 182, 519–532. doi: 10.1111/j.1469-8137.2008.02761.x
Keywords: adaptive divergence, fine-scale differentiation, Keteleeria davidiana, K. davidiana var. formosana, population genetics, SNP, species delimitation
Citation: Shih K-M, Chang C-T, Chung J-D, Chiang Y-C and Hwang S-Y (2018) Adaptive Genetic Divergence Despite Significant Isolation-by-Distance in Populations of Taiwan Cow-Tail Fir (Keteleeria davidiana var. formosana). Front. Plant Sci. 9:92. doi: 10.3389/fpls.2018.00092
Received: 02 October 2017; Accepted: 17 January 2018;
Published: 01 February 2018.
Edited by:
Octavio Salgueiro Paulo, Universidade de Lisboa, PortugalReviewed by:
Gonzalo Gajardo, University of Los Lagos, ChileJoshua Moses Miller, Yale University, United States
Copyright © 2018 Shih, Chang, Chung, Chiang and Hwang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shih-Ying Hwang, aHN5OTM0N0BudG51LmVkdS50dw==