 Aleksandra Dimitrijevic
Aleksandra Dimitrijevic Renate Horn
Renate Horn
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci., 17 January 2018
Sec. Plant Breeding
Volume 8 - 2017 | https://doi.org/10.3389/fpls.2017.02238
This article is part of the Research TopicAdvances in Oil Crops Research – Classical and New Approaches to Achieve Sustainable ProductivityView all 24 articles
In sunflower, molecular markers for simple traits as, e.g., fertility restoration, high oleic acid content, herbicide tolerance or resistances to Plasmopara halstedii, Puccinia helianthi, or Orobanche cumana have been successfully used in marker-assisted breeding programs for years. However, agronomically important complex quantitative traits like yield, heterosis, drought tolerance, oil content or selection for disease resistance, e.g., against Sclerotinia sclerotiorum have been challenging and will require genome-wide approaches. Plant genetic resources for sunflower are being collected and conserved worldwide that represent valuable resources to study complex traits. Sunflower association panels provide the basis for genome-wide association studies, overcoming disadvantages of biparental populations. Advances in technologies and the availability of the sunflower genome sequence made novel approaches on the whole genome level possible. Genotype-by-sequencing, and whole genome sequencing based on next generation sequencing technologies facilitated the production of large amounts of SNP markers for high density maps as well as SNP arrays and allowed genome-wide association studies and genomic selection in sunflower. Genome wide or candidate gene based association studies have been performed for traits like branching, flowering time, resistance to Sclerotinia head and stalk rot. First steps in genomic selection with regard to hybrid performance and hybrid oil content have shown that genomic selection can successfully address complex quantitative traits in sunflower and will help to speed up sunflower breeding programs in the future. To make sunflower more competitive toward other oil crops higher levels of resistance against pathogens and better yield performance are required. In addition, optimizing plant architecture toward a more complex growth type for higher plant densities has the potential to considerably increase yields per hectare. Integrative approaches combining omic technologies (genomics, transcriptomics, proteomics, metabolomics and phenomics) using bioinformatic tools will facilitate the identification of target genes and markers for complex traits and will give a better insight into the mechanisms behind the traits.
Sunflower represents the second most important crop based on hybrid breeding, after maize (Seiler et al., 2017). It is mainly used for its seed oil, even though the seeds of confectionary sunflower also serve as snacks. With up to 12% of the global production of vegetable oils worldwide, sunflower takes position number four after palm oil, soybean and canola oil (Rauf et al., 2017). Apart from its use for human nutrition, sunflower oil has a number of industrial applications as, e.g., basic component for polymer synthesis, biofuel, emulsifier or lubricants (Dimitrijevic et al., 2017).
Up until the beginning of the 1970s of the last century sunflower production was based on open-pollinated varieties (Vear, 2016). Events that led to changing sunflower production to hybrid breeding were the discoveries of the first cytoplasmic male sterility (CMS) source (Leclercq, 1969) and the identification of corresponding restorer genes (Kinman, 1970; Leclercq, 1971). Soon after, in 1972, the first sunflower commercial hybrid was available for production in United States (Putt, 1978). Exploitation of heterosis for hybrid development enabled farmers to obtain higher seed and oil yields, as well as increased uniformity (Bohra et al., 2016). The development of sunflower hybrids set up sunflower as a major viable crop worldwide and encouraged the founding of numerous public and private breeding centers (Skoric, 2012; Seiler et al., 2017). In recent years, public and private sector contributed to assemble huge plant genetic resources in sunflower, to identify markers for marker assisted selection (MAS) and to establish the use of new high-throughput technologies in sunflower. Today, the estimated value of global sunflower production reaches $20 billion per year (FAO, 2016).
Basic directions in sunflower hybrid breeding include developing: (1) high seed and oil yield hybrids resistant to dominant diseases and tolerant to drought, (2) hybrids with changed oil properties, (3) confectionary hybrids, (4) herbicide resistant hybrids and (5) ornamental hybrids (Jocic et al., 2015). In addition, special markets have particular demands such as (1) achene and kernel properties as well as high protein content and lower oil content (lower than 40%) in confectionary sunflower production, (2) specific fatty acid and tocopherol composition in food and non-food industry or (3) plant height, ray and disk flower color, duration of flowering in ornamental sunflower hybrid breeding. The common needs for resistance against abiotic and biotic stress as well as the special needs of the various breeding purposes require the development of markers to facilitate the introduction of different traits.
Botanically, sunflower (Helianthus annuus L.) is a member of the Asteraceae family, one of the most diverse and largest families of flowering plants. Due to the economic importance of the cultivated sunflower and the ecophysiological variability within the genus Helianthus, sunflower became a model plant species for genome studies in the family (Bachlava et al., 2012). The sunflower genome with 3.6 Gb is quite large (Badouin et al., 2017), three times larger than the rapeseed genome (Chalhoub et al., 2014), or more than eight times larger than the one of rice (Arumuganathan and Earle, 1991). Due to its ability to grow in different agroecological conditions and its moderate drought tolerance, sunflower may become the oil crop of preference in the future, especially in the light of global environmental changes. Even though simulations showed an increase of sunflower yield for northern parts of Europe in view of predicted climate changes, negative effects on sunflower yield may occur in southern latitudes (Debaeke et al., 2017). Consequently, more attention should be paid to breeding for better adaptation with regard to climate changes. These traits should include not only improvement in drought tolerance, but also introduction of pest resistance, salt tolerance and changes of plant architecture for better adaptation. Exploitation of available plant genetic resources in combination with the use of modern molecular tools for genome-wide association studies (GWAS) and application of genomic selection (GS) could lead to considerable improvements in sunflower. However, only in the recent years plant and genomic resources have become available in sunflower comparable to other crops (Figure 1). In this review we will talk about the long way that sunflower breeders and biotechnologists have to go and the future perspectives of using modern molecular tools in sunflower breeding.
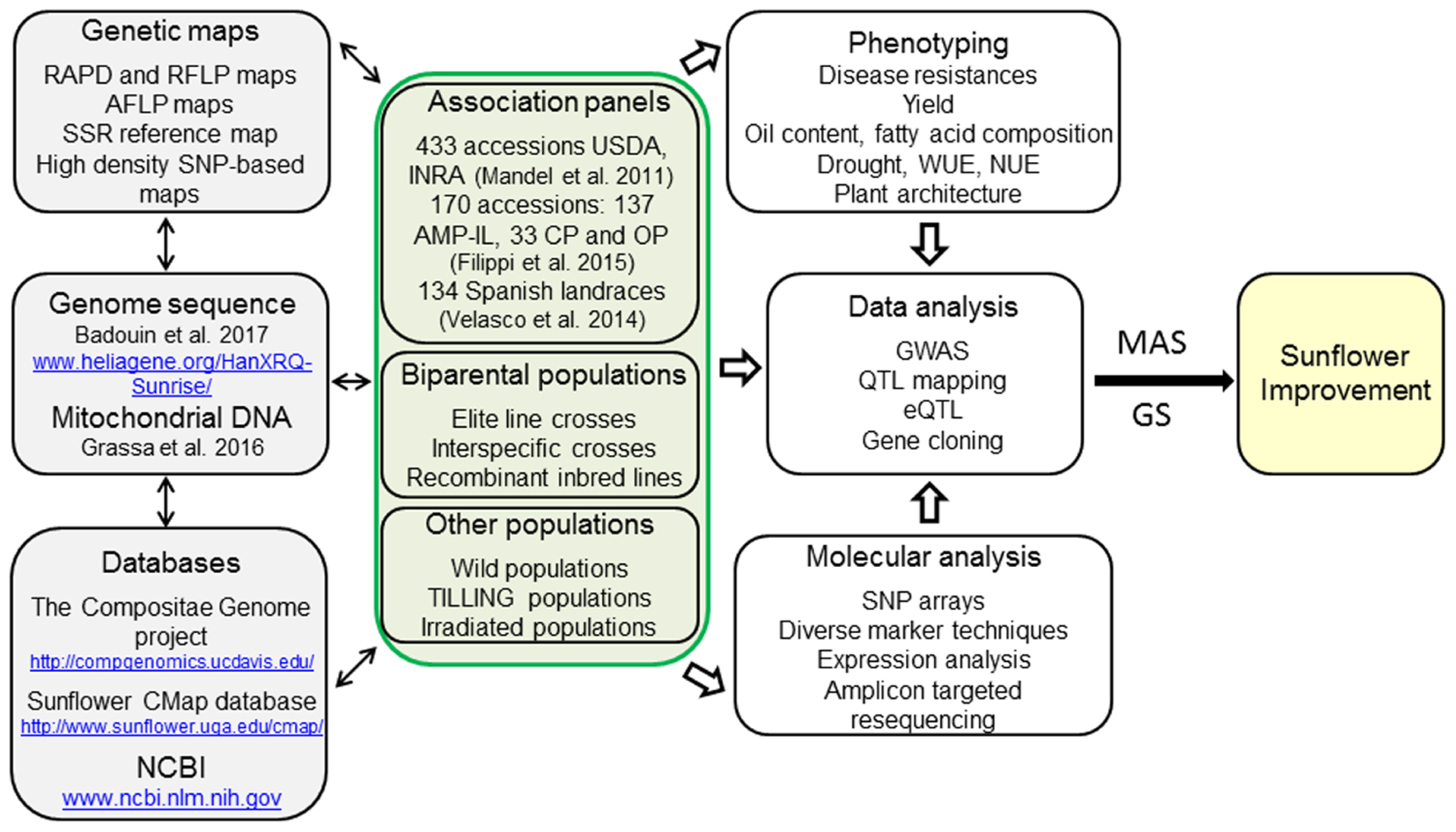
FIGURE 1. Schematic overview of the resources available in sunflower for marker-assisted selection (MAS) and future genomic selection (GS). Diverse plant genetic resources for sunflower breeding are available representing a large genetic diversity that can be exploited for sunflower improvement. The access to the sunflower genome sequences, the large resources of SNP, being part of high resolution maps or SNP arrays, and the huge amount of expression data will accelerate sunflower breeding by making the selection steps more efficient and precise. Future developments will move marker-assisted breeding toward genomic selection based on genomic estimated breeding values (GEBVs). WUE, water use efficiency; NUE, nitrogen use efficiency.
Biparental populations based on crosses between elite breeding, conventional, or introgressed lines (e.g., Berry et al., 1995; Horn et al., 2003; Vera-Ruiz et al., 2006; Kane et al., 2013; Livaja et al., 2016) as well as landraces and wild species (e.g., Quillet et al., 1995; Kim and Rieseberg, 1999; Brouillette et al., 2007; Ma et al., 2017) have been employed in sunflower for mapping of genes, marker detection, QTL analyses and gene cloning. In addition, recombinant inbred lines (RILs) have been developed that as immortals can be maintained forever by self-propagation (e.g., Berrios et al., 1999; Tang et al., 2002; Tang et al., 2006; Poormohammad Kiani et al., 2007a; Talukder et al., 2016). However, biparental populations have three major disadvantages: (1) these populations have to be individually established for each research project requiring time and resources, (2) only two alleles per locus can be evaluated and (3) due to missing recombination events populations show low resolutions in mapping (Bernardo, 2008; Patrick and Alfonso, 2013). The use of association panels overcomes these problems. To verify the usefulness of association panels the genetic diversity between wild populations and the genetic diversity fixed in association panels were compared (Mandel et al., 2011; Filippi et al., 2015). Even though alleles present only in the wild populations were detected, the majority of the alleles were present in the investigated association panels.
The largest sunflower collection is handled at the Institute of Field and Vegetable Crops, Novi Sad, Serbia consisting of over 7,000 sunflower inbred lines developed from different genetic sources and 21 perennial and 7 annual species (447 accessions in total)1 (Atlagic and Terzic, 2014). The next largest collection of more than 5000 cultivated and wild Helianthus accessions is held at the USDA-ARS NPGS in Ames (Marek, 2016). About half of these, 2,519 accessions, represent the world’s largest wild relatives sunflower collection, comprising 53 species – 39 perennial and 14 annual species (Seiler et al., 2017). Another large collection for sunflower (cultivated and wild) is maintained at the Vavilov Institute of Plant Industry, which consists of a total of 2,780 accessions from which 2,230 represent cultivated sunflower accessions and 550 wild sunflower accessions belonging to 24 species (19 perennials and 5 annuals) (Gavrilova et al., 2014). Some smaller numbers of 585 accessions of H. annuus are available via GRIN-CA, the Plant Gene Resources of Canada2 and additional 613 sunflower accessions of diverse origin are distributed by the IPK Gatersleben3. These resources represent mostly uncharacterized plant material. In contrast to these, a well-defined collection of 400 open-pollinated varieties, landraces and breeding pools has been assembled by INRA to reflect the worldwide diversity present in sunflower (Mangin et al., 2017b). However, conservation of population diversity of sunflower populations represents a challenge in the maintenance process due to the self-incompatibility of wild sunflowers (Gandhi et al., 2005) and the possibility of genetic drifts occurring during the propagation of seed stocks (Mangin et al., 2017b). To study the preservation of the genetic variability, a set of 114 cultivated sunflower populations of the INRA collection were genotyped using a 384 Golden Gate SNP Assay. In conclusion, multiplication in isolation fields or use of cages is recommended to reduce loss of genetic variability in cultivated genetic resources.
These worldwide available collections of sunflower represent a valuable resource for the sunflower community. It could be of interest to include some additional accessions of these large collections to the existing association panels described below.
Association panels have to be characterized by molecular markers like SSRs or SNPs to avoid false associations due to the population structure and family relationship. The review here focusses on association panels that are online available as the prior mentioned sunflower collections. To analyze the primary gene pool of sunflower an association panel consisting of 433 cultivated accessions from North America and Europe in addition to 24 wild sunflower populations distributed over the whole of United States were characterized by 34 selected EST-SSRs chosen on the presumptive neutrality toward domestication and breeding efforts (Chapman et al., 2008; Mandel et al., 2011). USDA cultivated accessions in this panel were assigned to the following categories: HA and RHA being either non-oil or oil, landrace, open-pollinated variety (OPV), non-oil introgressed, oil introgressed, other non-oil, and other oil. The INRA accessions could only be categorized into INRA-HA and INRA-RHA as the information on oil and non-oil was not always available. Analyses using the software STRUCTURE (Pritchard et al., 2000) and Principle Coordinates (PCO) analyses (Patterson et al., 2006) did not reveal deep genetic divisions within the germplasm (Mandel et al., 2011). The cultivated and the wild populations separated into two different groups and within the cultivated accessions the restorer-oil (RHA-oil) category stayed apart from the remaining gene pool (Mandel et al., 2011). This is not unexpected due to the hybrid history of sunflower in which the maintainer and restorer pools have been kept separate on purpose to maximize heterosis (Fick and Miller, 1997). A selection of 288 accessions still covers nearly 90% of the genetic diversity available in the original larger panel. This association panel was named UGA-SAM1 and consists of 259 accessions, which are distributed by the Germplasm Resources Information Network (GRIN4) of the USDA National Plant Germplasm System (NPGS) and 29 accessions available through the French National Institute for Agricultural Research (INRA, France). This UGA-SAM1 population, which has been successfully employed in association studies (Mandel et al., 2013; Nambeesan et al., 2015), represents a very valuable tool for future association studies for the whole sunflower community. A minimal core set of 12 accessions (representing HA, RHA, oil and non-oil accessions as well as INRA material) capturing nearly 50% of the total allelic diversity might be ideal to build up a MAGIC (Multi-parent advanced generation intercross) population for sunflower. The MAGIC strategy is interesting for studies of multiple alleles in order to exploit higher recombination frequencies and better mapping resolution (Cavanagh et al., 2008). Development of MAGIC populations is in progress for numerous plant species (Bandillo et al., 2013) and would be interesting for sunflower as well.
Argentinean germplasm also represents a valuable genetic resource due to the long history of sunflower breeding in Argentina (de Bertero, 2003; Moreno et al., 2013). Filippi et al. (2015) characterized the population structure und calculated the genetic diversity of the association mapping population (AMP-IL), representing 137 INTA (National Institute of Agricultural Technology, Argentina) accessions and 33 accessions of open-pollinated and composite populations. The plant material is maintained by The Active Germplasm Bank of INTA Manfredi (AGB-IM). Using 42 SSR markers and/or SNP markers, detected by a 384 Illumina SNP-oligo pool array, estimated and observed heterozygosity as well as clustering using STRUCTURE and Discriminant Analysis of Principle Components (DAPC) were compared for the two marker types. As in other studies (Mandel et al., 2011, 2013) the population structure was dominated by the maintainer/restorer trait (Filippi et al., 2015).
A germplasm collection of 196 Spanish confectionary sunflower accessions is maintained at the Centre of Plant Genetic Resources of the National Institute for Agricultural and Food Research and Technology (CRF-INIA)5. A large genetic variation was revealed regarding hundred-seed weight, kernel percentage, seed oil content, fatty acid and tocopherol composition, phytosterols and other traits (Velasco et al., 2014; Pérez Vich et al., 2017).
In addition to well characterized association panels, considerable plant genetic resources are nowadays available in sunflower (cultivated as well as wild H. annuus accessions and accessions representing other species in the genus Helianthus).
To increase the naturally available genetic variability sunflower has been mutagenized (Zambelli et al., 2015). Mutant populations have been successfully developed and used to screen for mutant phenotypes interesting for breeding purposes with regard to flowering time, dwarf habitus, oil content, high oleic trait, herbicide resistance and branching (Soldatov, 1976; Gabard and Huby, 2001; Sala et al., 2008a; Cvejic et al., 2011b; Leon et al., 2013). Recently, a TILLING (Targeted Induced Local Lesion In Genomes) population for high throughput screening of EMS (ethyl methane sulfonate)-induced mutations in sunflower was established by Sabetta et al. (2011) and used for studies of genes involved in the fatty acid biosynthesis. Optimized mutagenesis using EMS was used to develop an additional sunflower TILLING platform (Kumar et al., 2013). Phenotypic characterization of 5,000 M2 lines was performed to estimate the mutation rates and to select interesting mutants. As seed oil biosynthesis is of major importance in sunflower, TILLING of FatA and SAD genes were investigated and revealed an overall mutation rate of one mutation every 480 kb (Kumar et al., 2013). Another possibility to develop mutant populations is to apply gamma irradiation or fast neutrons (Cvejic et al., 2011a). Optimal ranges for gamma irradiation and fast neutrons were explored in comparison to EMS concentrations.
Besides induced mutagenized populations, natural mutations that have occurred in wild sunflower populations have had significant impact on sunflower hybrid breeding, especially in the area of herbicide resistance. In the recent years intensive use of herbicides has led to the emergence of resistant wild sunflower populations. The first case was a population of common sunflower found in a soybean field in Rossville (KS, United States), in which imazethapyr that belongs the group of AHAS (acetohydroxy acid synthase) inhibitors was used over a time course of seven consecutive years for weed control. Thus, creating the first sunflower population, named ANN-PUR, resistant to one of the AHAS inhibitors (Al-Khatib et al., 1998). Resistance from this population was successfully introduced into commercial sunflower hybrids (Miller and Al-Khatib, 2000; Jocić et al., 2004). Sunflower production based on the use of this imidazolinone (IMI) resistance, which provides an efficient and easy control of post-emergence broadleaf weeds in Europe, is called Clearfield® technology. In addition to the discovery of IMI resistant sunflowers, another population of wild sunflowers (ANN-KAN), tolerant to another AHAS herbicide group called sulfonylurea, was discovered in Kansas (United States) (Al-Khatib et al., 1999). The same tolerance was also obtained by EMS mutagenesis (Gabard and Huby, 2001). Later, more populations of wild sunflowers resistant to AHAS herbicides were found (e.g., White et al., 2002, 2003; Jacob et al., 2017). In addition, a new tolerance for imidazoline called Clearfield Plus® was selected from an M2 population of 600,000 plants treated with EMS (Sala et al., 2008a).
Natural genetic diversity and naturally occurring or chemically/gamma-ray induced genetic variability represent a perquisite for selection in breeding. The wide range of accessions maintained and made available by the germplasm banks for the research community is an extremely valuable starting point for successful breeding programs in sunflower allowing association studies and introduction of new traits into existing commercial breeding material. However, mutagenesis can create additional new genetic variability in traits where the natural variability is not sufficient.
Different molecular markers, which have been applied in mapping genes and development of sunflower linkage maps (Figure 1), set the basis for the assessment of the genetic diversity present in the genus Helianthus as well as in cultivated and wild sunflower accessions. Positioning of desirable genes allowed the identification and development of more specific molecular markers. At the Sunflower CMap database6 genetic maps available for sunflower have been listed and can be compared with each other by using the program CMAP (Kane et al., 2013).
The first map was developed on wild sunflower using RAPD markers (Rieseberg et al., 1993). A couple of years later maps were generated and published by using non-PCR based RFLP markers in different crosses of cultivated sunflower (Berry et al., 1995; Gentzbittel et al., 1995; Jan et al., 1998). These maps were published several years later than RLFP maps, e.g., in wheat, maize, barley, rice, and oilseed rape due to companies being involved in construction of the sunflower map (Hu, 2010). Later on, AFLP markers were added to the maps (Peerbolte and Peleman, 1996; Gedil et al., 2001). Most sunflower linkage maps contained 17 linkage groups (LG), representing the number of haploid chromosomes in sunflower. These maps were followed by genetic maps based on SSR markers (Tang et al., 2003b; Yu et al., 2003). The first composite genetic SSR map consisted of 278 single-locus SSR markers as well as additional 379 markers (public and proprietary), covering 1423 cM. This map that nowadays serves as reference genetic map for sunflower (Tang et al., 2003b) was then further saturated with additional SSR markers exploring three new mapping populations (Yu et al., 2003). In between more than 2,000 SSR have been derived from genomic sequences (gSSR) and EST (EST-SSR) and are now available for mapping and genotyping (Brunel, 1994; Dehmer and Friedt, 1998; Paniego et al., 2002; Tang et al., 2003b; Yu et al., 2003; Poormohammad Kiani et al., 2007b; Chapman et al., 2008; Heesacker et al., 2008). Existing sunflower maps were further enriched by these gSSRs, EST-SSRs, INDELs, TRAPs markers (Hu et al., 2007; Heesacker et al., 2008). These SSR markers (sequences and primers available through NCBI) represent a very valuable tool as they allow the localization of genes on individual linkage groups (Tang et al., 2003b) as well as on the recently published sunflower genome sequence of HanXRQ7 (Badouin et al., 2017). About 3 gigabases (Gb) representing 80% of the whole genome size were assembled and represent an extremely useful tool for all different research programs that aim at the improvement of sunflower hybrids.
Finally, the step toward high-density maps was made possible by using SNP-based markers, starting with Lai et al. (2005) who derived SNPs from an EST database (as part of the Compositae Genome Project) and used them for mapping. An Infinium Beadchip including 9,480 SNPs based on transcriptome data was developed by Bachlava et al. (2012) and employed by Bowers et al. (2012) to obtain four high-density genetic maps. Each of these maps contained 3,500–5,500 loci. Even though the maps were highly colinear, gaps in individual maps were observed. To solve this issue a consensus map of 10,080 loci was constructed from these data (Bowers et al., 2012). Talukder et al. (2014a) developed a high density map of 5,019 SNP markers obtained via RAD-sequencing. The rust resistance gene R12 was fine-mapped using this SNP-based map. In addition, 118 SSR markers were included in the SNP map to address and orientate the linkage groups according to the sunflower reference genetic map. Celik et al. (2016) pioneered the use of genotyping-by-sequencing for large scale SNP detection in sunflower and developed a SNP-based linkage map of 817 SNP-markers covering all 17 LG by analyzing an F2 obtained from the cross RHA 436 × H08 M1. Using the newly developed 25 K SNP array in sunflower Livaja et al. (2016) were able to construct a linkage map based on 6,355 SNP markers for the RIL population NDBLOSsel × CM625. The connection between genetic linkage maps and the sunflower karyotype was finally made by developing a molecular cytogenetic map for H. annuus (Feng et al., 2013). BAC and BIBAC clones with known genetic locations were used in fluorescence in situ hybridization (FISH) experiments to address the individual chromosomes.
The high resolution of the recently developed high-density maps in sunflower facilitates to narrow down the regions of interest, which should allow identification and cloning of genes for various relevant traits in the near future. In addition, SNP-based maps deliver markers closely linked to, e.g., resistance genes that can be applied in large scale marker-assisted breeding programs or can be integrated in SNP arrays.
Developed linkage maps set a good basis for localization and mapping of simply inherited traits. Most of the downy mildew resistance genes, conferring resistance to the oomycete Plasmopara halstedii, have been found to be dominantly inherited and consequently, relatively easy to map by using molecular markers. Identification of closely linked markers also represents a good basis for map-based cloning of the genes.
Great efforts were put into examination of downy mildew resistance genes (R genes), designated as Pl genes, that are distributed throughout sunflower genome: Pl13, Pl14, Pl16, and PlArg on LG1; Pl1, Pl2, Pl6, Pl7, Pl15, and Pl20 on LG8; Pl5, Pl8, and Pl21 on LG13; Pl17 and Pl19 on LG4; while Pl18 is localized on LG2 of the sunflower SSR reference map (Mouzeyar et al., 1995; Roeckel-Drevet et al., 1996; Vear et al., 1997; Bert et al., 2001; Slabaugh et al., 2003; Yu et al., 2003; Mulpuri et al., 2009; Wieckhorst et al., 2010; Bachlava et al., 2011; Vincourt et al., 2012; Qi et al., 2015a; Qi L.L. et al., 2016; Zhang et al., 2016; Ma et al., 2017). Most of Pl genes are clustered, except for PlArg and Pl18.
The Pl cluster on LG8 was the first to be detected by molecular markers. Mouzeyar et al. (1995) used RAPD and RFLP markers for mapping the first downy mildew resistance gene, Pl1, which is a part of a large Pl cluster (Pl1, Pl2, Pl6, Pl7). Of all genes in the cluster, Pl6 gene was the most intensively examined since it conferred resistance to all the races present for a long time, except race 304. Different marker types were used for the introgression of Pl6 into susceptible sunflower material including STS (Sequence-Tagged Sites) markers belonging to the TIR-NBS-LRR class of RGA (Resistance-Gene Analog) (Bouzidi et al., 2002) and co-dominant CAPS (Cleaved Amplified Polymorphic Sequence) markers (Table 1). The developed markers have been successfully used to introduce Pl6 by MAS or to track the introduction of Pl6 in backcrosses during conversion of downy mildew susceptible lines into resistant ones (Dimitrijevic et al., 2010; Jocic et al., 2010).
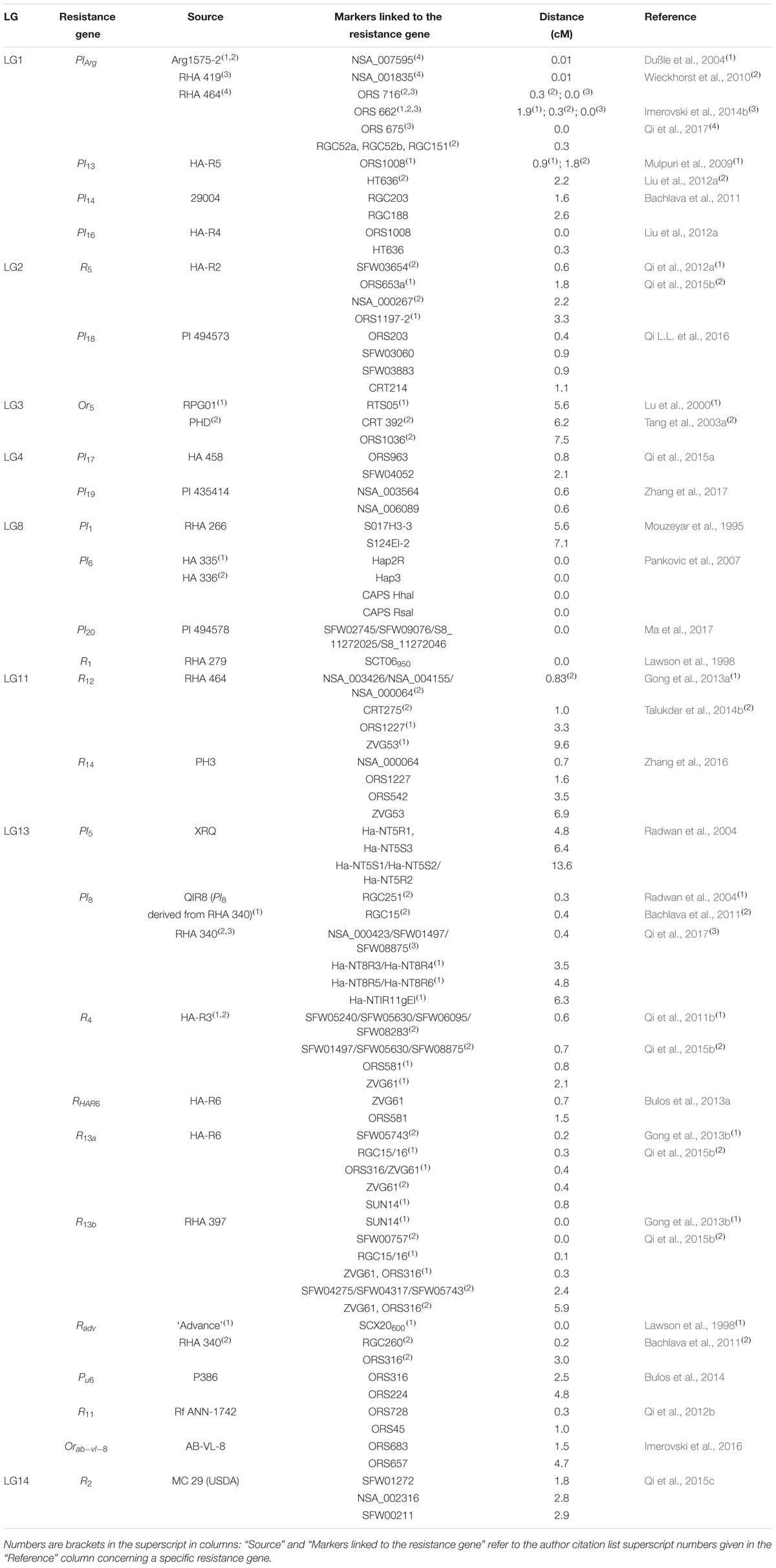
TABLE 1. Overview of resistance sources, locations, and markers of disease resistance genes in sunflower.
Two Pl genes originating from H. argophyllus, Pl8 and PlArg, were also subject of numerous studies. PlArg confers resistance to all present downy mildew races and Pl8 to 96% of all isolates collected in the north-central region of United States (Gilley et al., 2016). Radwan et al. (2004) developed STS markers for detection of the Pl5/Pl8 locus that were later on also explored in other sunflower genotypes (Dimitrijevic et al., 2011). Bachlava et al. (2011) developed two resistance gene candidate (RGC) markers, RGC251 and RGC15/16, closely linked to Pl8 that belong to the group of SSCP (Single-Strand Conformational Polymorphism) markers. However, SSCPs are labor-intensive and time-consuming in MAS. A comprehensive study of Pl8 by Qi et al. (2017) explored previously published SNP markers as well as two SSR markers (Bowers et al., 2012; Talukder et al., 2014a) to genotype a F2 population derived from the cross HA434 × RHA340. The three closest SNP markers, NSA_000423, NSA_002220, and NSA_002251, were then investigated to check the specificity of the identified markers, concluding that NSA_000423 and NSA_002220 could serve as diagnostic markers in 87% of the tested sunflower lines when RHA 340 is used as donor for the Pl8 gene. Validation of these markers across 548 sunflower lines proved their usefulness for MAS. However, a larger panel of sunflower lines should to be tested.
Unlike, the Pl8 gene, PlArg is not clustered. Several authors identified and developed different types of markers (SSRs, SNPs, RGCs) for MAS (Dußle et al., 2004; Wieckhorst et al., 2010; Imerovski et al., 2014b), some of these were also validated across a panel of sunflower lines. ORS716 was identified as the most useful marker in MAS (Table 1). Recently, Qi et al. (2017) combined available genomic data for the population obtained from the cross HA 89 × RHA 464 by use of SNP markers (Pegadaraju et al., 2013; Talukder et al., 2014a) with the phenotypic evaluation for resistance. The two nearest SNP markers (NSA_007595 and NSA_001835) narrowed the PlArg locus down to an area of 2.83 Mb. The nine identified SNP markers represent valuable diagnostic tools for introgression of PlArg into most genetic backgrounds in sunflower.
Other markers for use in MAS for downy mildew resistance include the identification of the tightly linked SSR marker ORS1008 to Pl13 gene (Mulpuri et al., 2009), development of RGC markers tightly linked to Pl14 gene (Bachlava et al., 2011) and the identification of one dominant co-segregating SSR marker (ORS1008) and one co-dominant tightly linked (EST)-SSR (HT636) to Pl16. Interestingly, HT636 and ORS1008 were reported to be linked to both, Pl13 and Pl16, indicating that these genes are in close vicinity to each other (Liu et al., 2012a). Qi et al. (2015a) used SSRs to place Pl17 onto LG4 and then used SNPs identified by the National Sunflower SNP Consortium (Talukder et al., 2014a) and by Bowers et al. (2012) to saturate the region surrounding the Pl17 gene. The authors identified SNP SFW04052 and ORS963 as the closest flanking markers linked to Pl17. A year later, Qi L.L. et al. (2016) used the same methodology to map Pl18 to LG2 and found two SSRs and 10 SNPs flanking the Pl18 gene. Pl18 represents the first gene mapped to LG2. In 2017, two new Pl genes, Pl19 and Pl20, were reported and mapped to LG4 and LG8, respectively (Ma et al., 2017; Zhang et al., 2017). Two SSRs and two SNPs were mapped in close vicinity to Pl19, while four SNP markers (SFW02745, SFW09076, S8_11272025, and S8_11272046) co-segregated with Pl20. All markers can be used in MAS and most importantly in pyramiding Pl genes in order to achieve long lasting resistance toward downy mildew. The development of SNP markers is of special interest because of the large number of markers generated that increase the likelihood to have markers available for any cross combination.
Infections of sunflower plants with Puccinia helianthi Schwein lead to the rust disease. This fungus, which is mostly spread in North America, Argentina, South Africa, and Australia, can cause significant damage and yield reduction in infected fields. Genetic control of the disease can be effective; however, due to fast emergence of new races either by sexual or asexual reproduction, resistance achieved is short-termed. Consequently, a significant effort has been made into discovering rust resistance genes and the introduction into commercial lines and hybrids with a final goal of pyramiding several resistance genes in order to achieve long-term resistance. Most of the rust resistance genes (R genes), described so far, are monogenic dominant. R genes are located on different LGs of the sunflower genome with the majority being located on the LG13 [R4, Ru6, R11, Radv, R13a (RHAR6), and R13b] (Bachlava et al., 2011; Qi et al., 2011b, 2012b; Gong et al., 2013b; Bulos et al., 2014).
First molecular studies were conducted on discovering markers for R1 and Radv genes by use of RAPD and SCAR markers (Lawson et al., 1996, 1998). While R1 gene was the first rust resistance gene present in a large number of sunflower lines, Radv is present in the line P2 owned by Pioneer Hi-Bred Australia (Lawson et al., 1998; Qi et al., 2011a). Radv is also present in the USDA line RHA 340, which Bachlava et al. (2011) used for mapping of the gene. Lawson et al. (1998) developed the SCAR marker SCT06950 linked to R1 gene, which proved to be useful for detection of R1 in different genetic backgrounds, except for the sunflower line MC29, which carries the R2 and R10 genes. For mapping of R2, Qi et al. (2015c) used a different MC29 line, called MC29 (USDA) as it was cultivated in the USDA-ARS Sunflower Research Unit, Fargo, North Dakota, which differs in term of resistance to NA race 6 in comparison to the MC29 line used by Lawson et al. (1998). Qi et al. (2015c) reported two SNP markers, NSA_002316 and SFW01272, flanking the R2 gene on LG14. Since, the closest marker, SFW01272, can only to a certain extent be used to detect the R2 gene across different genetic backgrounds; the authors recommend the use of two flanking SNP markers in order to minimize selection of false positives in MAS.
Further molecular studies of R genes include identification of molecular markers closely linked to R4, Radv, Pu6, R11, R13a (RHAR6), and R13b genes that are located on LG13. Qi et al. (2011b) identified two markers flanking R4 gene (ORS581 and ZVG61) in the cross HA 89 × HA-R3, which were later also reported to be linked to rust resistance genes R13a (RHAR6) and R13b located on the lower end of the LG13 (Bulos et al., 2013a; Gong et al., 2013b; Qi et al., 2015b) (Table 1). Further on, Gong et al. (2013b) saturated the region flanking the genes by analysis of RGC markers that were present in vicinity of downy mildew resistance gene Pl8, which was also mapped in the lower end of LG13. Another R gene that mapped in vicinity of Pl8 and fertility restorer gene Rf1 was Radv. A completely co-segregating SCAR marker (Lawson et al., 1998) as well as RGC and SSR markers tightly linked to Radv were identified (Bachlava et al., 2011) (Table 1). Recently, Bulos et al. (2014) mapped Pu6 gene and identified closely linked SSRs to this gene in the sunflower line P386 on lower end of LG13. However, these markers are too far away to be useful in MAS (Table 1). Pu6 and R4 map 6.25 cM apart from each other. Qi et al. (2012b) examined the R11 gene and mapped it 1.6 cM from fertility restoration gene Rf5 also on the lower end of LG13, hypothesizing the presence of a great rust R-gene cluster of Radv/R11/R4. SSR marker ORS45 was the closest to R11 gene and was mapped 1 cM proximal to the gene, while ORS728 was shown to be a common marker for R11 and Rf5 genes. The results allow the conclusion that the lower end of LG13 harbors the second largest cluster of NBS-LRR encoding genes: rust resistance and downy mildew resistance genes. Based on SSR and RGC markers used in this area, Gong et al. (2013b) proposed that this big cluster could be sub-divided into two clusters. Radv and R11 form sub-cluster I, while R4, R13a/b, Pl5, Pl8 form subcluster II. Pl21 that was also positioned on LG13 mapped 8 cM proximal to Pl5/Pl8 (Radwan et al., 2004; Vincourt et al., 2012).
Other rust resistance genes investigated by use of molecular markers include analysis of R5. This is to date the only R gene discovered on LG2. Qi et al. (2012a, 2015b) identified two SSR and two SNP markers flanking the gene, with the closest being 0.6 cM away (Table 1). On LG11 two rust resistance genes have been mapped so far: R12 and R14. Both genes were positioned in the middle of LG11, and were discovered in wild sunflower accessions, however, they have different origin: R12 from PI413047 and R14 from PI413038 (Gong et al., 2013a; Zhang et al., 2016). Both genes were mapped between the markers ORS1227 and ZVG53 (ORS1227 with 3.3 and 1.6 cM and ZVG53 with 9.6 and 6.9 cM from R12 and R14, respectively). Talukder et al. (2014b) performed fine mapping of the R12 gene region by using SNP markers. Five SNP markers (NSA_000064, NSA_008884, NSA_004155, NSA_003320, and NSA_003426) were linked with 0.83 cM to the gene, but only two markers (NSA_003426 and NSA_004155) proved to have diagnostic quality for R12 (Table 1). The nearest SNP marker to R14 was NSA_000064, which was mapped with 0.7 cM from the gene in the F2 mapping population obtained from the cross HA 343 × PH3 (Zhang et al., 2016). However, this marker amplified the same banding pattern in RHA 464 (R12) and PH3 (R14). Zhang et al. (2016) identified thirteen SSR/InDel and two SNP markers that amplified different profiles between the two donors of R12 and R14 indicating polymorphisms between these regions.
One of the latest efforts in saturation mapping of R genes was published by Qi et al. (2015b) who used previously developed SFW and NSA SNP markers in order to saturate the regions surrounding R4, R5, R13a, and R13b genes and succeeded in identifying markers that are under 1 cM distant from all analyzed genes thus raising the efficiency of introduction of rust resistance in to susceptible material (Table 1). The authors used previously developed SSR markers and newly developed SNP markers for identification of homozygous “double-resistant” F2 individuals in a population obtained from a cross combination between a BC3F2 plant harboring R5 and HA-R6 bearing R13a. The F4 progeny obtained from chosen plants showed improved resistance toward races 336 and 777 in comparison to lines that possess only one resistance gene. Qi et al. (2015c) also performed marker-assisted pyramiding of R2 and R13a in confectionary sunflower by use of SSR and SNP markers. Further pyramiding of R genes could lead to long-term improvements in sunflower rust resistance. The process of converting susceptible into resistant forms can be greatly facilitated and accelerated by use of the reported molecular markers.
Another constraint in sunflower production is broomrape (Orobanche cumana), a parasitic flowering plant, that can cause significant yield loss of up to 100%. Most of the genes that confer resistance to broomrape were found to be monogenic dominant for broomrape races A to E and G (Vranceanu et al., 1980; Velasco et al., 2012), while resistance to race F was either inherited by a monogenic dominant gene (Pacureanu-Joita et al., 1998; Pérez-Vich et al., 2004) or by two recessive genes (Rodríguez-Ojeda et al., 2001) depending on the genetic background. Broomrape resistance genes are denoted as Or genes. Imerovski et al. (2014a) reported a single recessive resistance gene in the sunflower line HA-267 that carried a resistance gene higher than Or6. The majority of molecular analyses were conducted in investigating and creating different types of molecular markers for detection of Or5 that conveys resistance to broomrape race E or lower (Lu et al., 2000; Tang et al., 2003a) (Table 1). The efficiency of RAPD and SSR primers in MAS for Or5 were tested by Iuoras et al. (2004), however, none of the primers proved to be efficient or accurate enough. Imerovski et al. (2013) identified SSR markers associated with Or2, Or4, and Or6 genes that could be used in converting broomrape susceptible sunflower genotypes into resistant ones. However, O. cumana populations belonging to race F have shown different aggressiveness (Molinero-Ruiz et al., 2009). Imerovski et al. (2016) mapped newly identified broomrape resistant gene conferring resistance to broomrape races overcoming race F from sunflower inbred line AB-VL-8 on LG3. The authors named the gene Orab-vl-8, which was shown to be recessive and ORS683 mapped 1.5 cM from the gene. Further molecular analysis are needed in order to develop co-segregating markers for some of the Or genes. In addition, finding novel resistance sources is essential since broomrape races are emerging at a high speed. Recent work of Louarn et al. (2016) involved using 586, 985 SNPs from SUNRISE project8 on GeneTitan® (Affymetrix) for identification of QTL for resistance to broomrape races F and G. The authors identified 17 QTL spread throughout 9 LGs. Among them was a stable QTL on LG13 that controlled the number of broomrape emergence that explained 15–30% of the phenotypic variability. This QTL was marked as the one that could be the most rapidly used. A molecular characterization of O. cumana populations in Europe using RAPD-PCR identified four groups (Molinero-Ruiz et al., 2014). These markers might be useful as molecular tools to detect first broomrape appearances in fields that had been free of virulent races (Molinero-Ruiz et al., 2014).
Different tolerances against herbicides inhibiting the large, catalytic subunit of the acetohydroxyacid synthase (AHASL) have become a very necessary tool in sunflower hybrid production and cultivation as these facilitate the application of either imidazolinones (IMIs) or sulfonylureas (SUs) against broadleaf weeds (Sala et al., 2012). It also allows a race independent control of broomrape (Skoric and Pacureanu, 2010). Three AHASL genes were isolated from sunflower: AHASL1 located on LG9, AHASL2 on LG6 and AHASL3 on LG2 (Kolkman et al., 2004). Only mutations in AHASL1 seem to be involved in the herbicide tolerance in sunflower. Four different mutated alleles have been explored for commercial use in sunflower hybrid breeding: Imisun/Clearfield®, Clearfield Plus®, Sures and ExpressSun® (Sala et al., 2012). Point mutations from C-T in codon 205 (Ahasl1-1) and in codon 197 (Ahasl1-2) (adopting the Arabidopsis nomenclature) confer moderate tolerance to IMIs and high tolerance to SUs, respectively. The allele Ahasl1-3 is characterized by a G-A mutation in codon 122 and results in high levels of IMI tolerance (Sala et al., 2008b). The broadest range of herbicide tolerance is shown by allele Ahasl1-4, which has a G-T mutation in codon 574 (Sala and Bulos, 2012). A first SNP marker based on the C-T change in codon 205 proved to be very useful as it cosegregated with partially dominant herbicide tolerance for the Imisun/Clearfield® system (Kolkman et al., 2004), even though an additional non-target gene is required for the tolerance (Bruniard and Miller, 2001; Miller and Al-Khatib, 2002). One SSR marker exploiting the differences in the (ACC) repeats present in the AHASL gene allows the differentiation between the wild type Ahasl1 allele and alleles Ahasl1-1 and Ahasl1-2 (Sures and ExpressSun®) (Kolkman et al., 2004; Bulos et al., 2013b). A CAPS marker developed by Bulos et al. (2013b) uses the A-T exchange to detect the Ahasl1-3 allele (Clearfield Plus®) by digesting the PCR product with the restriction enzyme BmgBI. The markers can now help to select for herbicide tolerance. Nevertheless, the development of efficient screening tests for herbicide tolerance is crucial (e.g., Breccia et al., 2011; Vega et al., 2012).
Several oil properties have been characterized as quantitative traits, however, some traits such as oleic acid content (OAC) could, to a certain extent, be considered a semi-qualitative trait since OAC is dependent not only on the environment, but also on the genetic background of the receiver line (Ferfuia et al., 2015; Regitano Neto et al., 2016). A partial duplication of the FAD2-1 allele caused by chemical mutation leads to an increase in OAC by silencing the FAD2-1 gene encoding FAD2 (oleoyl-phosphatidylcholine desaturase) (Lacombe et al., 2002; Schuppert et al., 2006). This enzyme catalyzes the synthesis of linoleic acid from oleic acid and by silencing its activity oleic acid is accumulated. Soldatov (1976) created the Pervenets cultivar with elevated OAC, which has become the main source of elevated OAC in sunflower breeding programs worldwide due to the beneficial properties of high oleic sunflower oil (Allman-Farinelli et al., 2005; Vannozzi, 2006). Inheritance of the OAC trait has been a subject of numerous studies and different results were reported from a single dominant gene to several genes influencing OAC (Urie, 1984; Lacombe et al., 2004; Joksimovic et al., 2006; Bervillé, 2010; Ferfuia and Vannozzi, 2015; Premnath et al., 2016; Dimitrijevic et al., 2017). Gene/genes involved in inheritance of OAC have been denoted as Ol genes. Different markers were employed in mapping and detecting the mutation (Ol mutation) in sunflower. The two RAPD markers, F15-690 and AC10-765, were linked with 7.0 and 7.2 cM to Ol1 gene, respectively (Dehmer and Friedt, 1998). Later on, the Ol1-FAD2-1 locus was placed onto LG14 (Pérez-Vich et al., 2002; Schuppert et al., 2006). One major QTL identified by Pérez-Vich et al. (2002) explained 84.5% of the variation in the OAC. Schuppert et al. (2006) provided dominant INDEL markers for tracking the Ol mutation in addition to identifying 49 SNPs and five INDELs in the 3′-region of FAD2-1. Three years later, a co-dominant SSR marker tightly linked to the Ol mutation and dominant markers specific for the mutation were published (Lacombe et al., 2009). Recently, Premnath et al. (2016) identified in addition to the QTL on LG14, two additional QTL for OAC on LG8 and LG9. The two markers HO_Fsp_b for the QTL on LG14 (Schuppert et al., 2006) and ORS762 for the QTL on LG8 explained about 60% of the phenotypic variation in OAC. Several of the markers have been used for validation across numerous sunflower lines (Nagarathna et al., 2011; Singchai et al., 2013; Bilgen, 2016; Dimitrijevic et al., 2016). Dimitrijevic et al. (2017) reported marker F4-R1 created by Schuppert et al. (2006) as the most efficient in MAS for OAC.
Development of reliable tools for detection of cytoplasmic male sterility (cms) and restorer of fertility (Rf) genes would significantly improve and accelerate the process of developing sunflower hybrids. In sunflower, CMS PET1 originating from an interspecific hybridization of H. petiolaris with H. annuus (Leclercq, 1969) is the only CMS cytoplasm worldwide used for hybrid breeding. Male sterility is caused by the co-transcription of the atpA gene with the new CMS-specific orfH522 leading to the expression of a 16-kDa-protein (Horn et al., 1991; Köhler et al., 1991). Fertility restoration suppresses the co-transcription anther-specific (Monéger et al., 1994). In sunflower, the restorer genes for the PET1 cytoplasm represent the best characterized due to the commercial use of this cytoplasm in sunflower hybrid breeding. The restorer gene Rf1, which was originally discovered by Kinman (1970) in the line T66006-2-1-B, has since then been integrated into a number of USDA/ARS RHA lines like RHA 271, RHA 272, RHA 273, and others (Korell et al., 1992; Serieys, 2005). A second major dominant restorer gene Rf2 was discovered in a test cross between T66006-2-1-B and MZ01398. However, this Rf2 gene seems to be ubiquitously present in almost all cultivated sunflower lines, along with maintainer lines of CMS PET1 (Serieys, 2005). Only Rf1 is responsible for restoring male fertility in sunflower hybrids (Leclercq, 1984). RAPD markers in combination with AFLP markers were very useful for mapping of the restorer gene Rf1 (Horn et al., 2003), which was first positioned on LG6 of the RFLP sunflower map (Gentzbittel et al., 1995). Two RAPD markers OPK13_454 and OPY10_740, which mapped 0.8 and 2.0 cM from Rf1, respectively, were converted into more reliable, easier to handle SCAR markers HRG01 and HRG02 (Horn et al., 2003). A recent study of these SCAR markers for breeding practice proved that HRG01 is more efficient for Rf1 detection in perennial species, whereas HRG02 gave better results for annual species (Markin et al., 2017). In addition a multiplex TaqMan assay was established that allowed the detection of HRG01 and orfH522 at the same time (Markin et al., 2017). Using the SSR markers ORS1030, Rf1 had been mapped to LG13 (Kusterer et al., 2005) of the sunflower reference map (Tang et al., 2003b). In addition, a CAPS marker H13, which mapped 7.7 cM from Rf1 gene, was developed from the RAPD marker OPH13_337 by digesting the PCR product with HinfI (Kusterer et al., 2005). The tight linkage between CAPS H13 and Rf1 was confirmed in Xenia hybrid combination (Port et al., 2013). An additional SSR marker ORS511 and a TRAP marker K11F05Sa12-160 were mapped to the Rf1 gene with distances of 3.7 and 0.4 cM, respectively (Yue et al., 2010). A fertility restorer gene Rf3, which could be shown to be different from Rf1 and Rf2, was identified in the confectionery restorer line RHA 280 (Jan and Vick, 2007). Rf3 could be linked with eight markers to LG7, including five known SSR markers (ORS328, ORS331, ORS928, ORS966, and ORS1092) and three new SSR markers HT-619-1, HT619-2, and HT1013 derived from expressed sequence tags (Liu et al., 2012b). SSR ORS328, which mapped 0.7 cM distant from Rf3, represents so far the closest co-dominant marker to the gene (Liu et al., 2012b). Another restorer gene Rf3 in RHA 340 has also been mapped to LG7 (Abratti et al., 2008). Rf ANN-1742, a restorer line derived from wild H. annuus showed resistance to rust (Qi et al., 2012b). The new rust resistance gene R11 mapped with 1.6 cM closely to a restorer gene on the lower end of LG13. The SSR marker ORS728 was mapped 1.3 cM proximal from this restorer gene and 0.3 cM distal to R11. Marker analyses using HRG01, HRG02, STS115, and ORS728 indicated that this restorer gene, now called Rf5, might not be allelic to Rf1 (Qi et al., 2012b).
So far, 72 new CMS sources have been described for sunflower (Serieys, 2005). However, only for very few of these CMS sources markers have been detected linked to the corresponding restorer genes (Horn et al., 2016). Feng and Jan (2008) tagged an additional restorer gene Rf4 with molecular markers and assigned it to LG3 of the sunflower general reference map (Tang et al., 2003b). Rf4 is restoring male fertility to a newly identified CMS cytoplasm GIG2. Schnabel et al. (2008) identified AFLP markers that mapped in close vicinity of the restorer gene Rf_PEF1, which represent a major restorer gene for the PEF1 CMS cytoplasm, another potentially interesting CMS source for commercial sunflower hybrid breeding. In addition, markers were developed that allowed the distinction between the PET1 cytoplasm and the PEF1 cytoplasm. For CMS 514A, a H. tuberosus based male sterile cytoplasm, the restorer gene Rf6 was located on LG3 with eight markers (Liu et al., 2013). Two SSR markers, ORS13 and ORS1114, mapped as close as 1.6 cM to Rf6. GISH showed Rf6 to be present on a small translocation introgressed from H. angustifolius.
Further analyses are needed in order to develop more tightly linked molecular markers to Rf genes to locate them on the genetic map and to get an insight on the fertility restoration mechanisms in sunflower. In other species, most of the so far cloned restorer of fertility genes belong to the pentatricopeptide repeat gene family (PPR), however, also other types of restorer genes have been identified (Horn et al., 2014).
For association mapping two approaches have been explored: (1) genome-wide association studies (GWAS) and (2) candidate gene approaches. For most plant species, the last strategy was predominantly applied because whole genome sequences have only recently become available (Fusari et al., 2008). However, high-throughput marker systems nowadays give full genome coverage, which makes approaches as genome-wide association mapping, QTLSeq mapping and genomic selection possible (Mammadov et al., 2012). As the linkage disequilibrium (LD) in sunflower rapidly decays (Liu and Burke, 2006; Kolkman et al., 2007; Fusari et al., 2008) studies based on associations could result in resolution levels detecting genes underlying quantitative trait loci. However, it is important to analyze the population structure of the association mapping population to avoid false associations.
In sunflower, only one of the association mapping studies so far was performed genome-wide (Mandel et al., 2013), all others were candidate gene based (Fusari et al., 2012; Cadic et al., 2013; Talukder et al., 2014b; Nambeesan et al., 2015; McAssey et al., 2016). Genome-wide association mapping was performed in an association population of 271 lines (Mandel et al., 2011), using 5,359 SNP marker from the Illumina Infinium Beadchip (Mandel et al., 2013). Associations were studied regarding flowering time, branching and heterotic groups. LD showed considerable variability across the genome, but significant marker-trait associations were detected. Selection for disease resistance as well as initial domestication might be responsible for the genome-wide differences in the LD profile (Mandel et al., 2013). This first screen was followed by a more detailed, refined association mapping approach based on candidate genes for branching (Nambeesan et al., 2015). Shoot branching was differentiated in no branching, apical, mid-apical, mid, mid-basal, basal branching as well as whole plant branching or other phenotype. A total of 48 candidate genes described to be involved in branching in other plant species were used to detect homologs to 39 genes in sunflower. Up to eight of the highest BLAST hit for each gene were included in the analyses due to the recent triplication of the sunflower genome (Badouin et al., 2017). For 13 candidate genes for branching co-localization of SNPs associated with branching was observed (Nambeesan et al., 2015). Most of these were found on LG10, where previous QTL mapping had detected the B-Locus for recessive branching (Tang et al., 2006; Bachlava et al., 2009). With regard to flowering time, a SNP in HaFT2 was identified that co-localized with a flowering time QTL (McAssey et al., 2016).
Association mapping and linkage mapping were combined with QTL detection to identify mutations responsible for changes in flowering time (Cadic et al., 2013). Associations with flowering time could be demonstrated for 11 regions distributed over 10 LGs. In addition, QTL for flowering time were detected on 11 LGs in a RIL population by linkage mapping. This large number of QTL is consistent with the polygenic pattern of inheritance of flowering time reported before (Leon et al., 2000). SNPs detected by association mapping were then investigated with regard to positional overlaps with QTL identified in the RIL population. The remaining eight regions contained five candidate genes potentially associated with flowering time in other species that showed SNPs in sunflower, one of the genes was the gibberellin receptor GID1B (Cadic et al., 2013). Thirty genes, including this gene had before been investigated as candidate genes for flowering time with regard to domestication and improvement in sunflower (Blackman et al., 2011). One major QTL, which was detected on LG14 by linkage mapping (Poormohammad Kiani et al., 2009), was not detected by the association study (Cadic et al., 2013). This can happen if alleles are present in a low frequency in an association panel as one disadvantage of association mapping is that rare alleles are difficult to be associated with traits.
Sclerotinia sclerotiorum, a necrotrophic, fungal pathogen, is one of the most devastating diseases in sunflower. The fungus can cause three different types of diseases depending on which part of the plants gets infected and whether the infection occurs via ascospores or mycelia (Gulya et al., 1997). These are stalk rot, mid-stalk rot and head rot. In a panel consisting of 94 sunflower lines 16 candidate genes were screened for associations to Sclerotinia head rot using a Mixed Linear Model (MLM) that also considers family relationship as well as population structure (Fusari et al., 2012). These candidate genes had been derived from previous transcript profiling in sunflower (Peluffo, 2010) and Brassica (Zhao et al., 2007) after infecting the plants with S. sclerotiorum. Significant association of the haplotype 3 of the gene HaRIC_B, representing a truncated gene, was detected and accounted for 20% reduction in Sclerotinia head rot. Candidate gene association mapping for Sclerotinia stalk rot was also performed in another association panel of 260 cultivated sunflower lines (Talukder et al., 2014b). Eight genes, which had been identified in defense response against S. sclerotiorum in Arabidopsis (Guimaraes and Stotz, 2004; Guo and Stotz, 2007), served as basis to identify the orthologous genes in sunflower. The panel was divided in two groups representing either the best resistance response or the most susceptible lines. Association studies found strong association of HaCOI1-1 and HaCOI1-2 with resistance against Sclerotinia stalk rot, explaining 7.4% of the observed phenotypic variation (Talukder et al., 2014b).
Association mapping studies in the recent years have shown that this approach represents an interesting alternative to linkage mapping especially regarding quantitative inherited traits.
Genomic selection (GS) is so far mostly used in animals, e.g., dairy cattle (Van Raden et al., 2009). However, application of genomic selection got started as well in plant breeding, e.g., in maize (Massman et al., 2013; Bandeira e Sousa et al., 2017; Cantelmo et al., 2017; Lyra et al., 2017), potato (Habyarimana et al., 2017), soybean (de Azevedo Peixoto et al., 2017), sugar beet (Würschum et al., 2013), and wheat (Bassi et al., 2016). Genomic selection was regarded as promising in hybrid breeding of self-pollinating crops as wheat (Longin and Reif, 2014; Zhao et al., 2015), especially if little is known about the heterotic pools. To implement GS into sunflower breeding programs some general aspects of genomic selection need to be emphasized.
Genomic selection selects the individuals based on genomic breeding values (GEBVs) (Meuwissen et al., 2001). The idea of GS is to use genome-wide molecular data to effectively select for quantitative trait loci (Bernardo, 2008; Massman et al., 2013; Würschum et al., 2013). More than 10,000 QTL have been detected by traditional mapping approaches considering 12 major crop species, but only very few have been successfully applied in marker-assisted breeding programs (Bernardo, 2008). Genomic selection is a concept that becomes more attractive as high-throughput genotyping becomes feasible due to recent advances in genotyping platforms and to considerable price reductions in the last few years. As first step in GS, a training population has to be established that is genotyped and phenotyped. This training population is needed to adjust the statistical models, which are then applied to predict breeding and genotypic values of individuals that have not been phenotyped (Bassi et al., 2016). The breeding population consists of these not phenotyped individuals that are only genotyped. Selection is performed in the breeding population. Finally, a validation population serves to estimate the accuracy of the GS models (Bassi et al., 2016). Comparing traditional MAS and GS, three major differences are obvious: (1) within the training phase markers linked with a gene of interest and quantitative traits are identified in MAS, whereas in GS models are developed to predict GEBVs, (2) in the breeding phase only few markers are used in traditional MAS for genotyping, whereas in the GS genome-wide genome data are collected and (3) regarding the selection in the breeding phase traditional MAS uses only the identified markers to select the individuals by genotype, whereas selection in GS is performed based on the GEBV (Nakaya and Isobe, 2012). For the success of GS, the accuracy of the prediction of GEBV is the most important factor. The accuracy of prediction relies on the characteristics of the training population as size, marker density, trait heritability and kinship between training and breeding population as well as the ratio of training population : breeding population (Nakaya and Isobe, 2012; Bassi et al., 2016). In traditional MAS, markers tightly linked to a QTL could be applied in most other breeding population, so that the relationship between the mapping and the breeding population had not to be considered by the breeder. However, in GS the interrelationship between training and breeding population is crucial for the predictive power (Nakaya and Isobe, 2012).
In sunflower, prediction of hybrid performance was based on fingerprinting data in form of 572 AFLP markers (Reif et al., 2013). Intragroup (133) and intergroup hybrids and the parental lines were evaluated at two locations in 2 years for grain yield, oil content and oil yield. If no information on the General Combining Ability (GCA) of the parental lines was accessible, prediction of hybrid performance using genomic selection methods was accurate if the parents were closely related, but with genetically distant lines prediction proved challenging (Reif et al., 2013). However, prediction based on GCA could not be improved by genomic selection. In the recent years, large sets of markers were generated in sunflower by genotyping-by-sequencing (Baute et al., 2016; Celik et al., 2016; Talukder et al., 2016; Ma et al., 2017; Qi et al., 2017), application of the new 25 K SNP genotyping array (Livaja et al., 2016) and sequencing of parental lines (Mangin et al., 2017a). However, so far only SNP array data were used for genomic prediction of Sclerotinia resistance (Livaja et al., 2016) and sequencing data for the genomic prediction of sunflower hybrid oil content (Bonnafous et al., 2016; Mangin et al., 2017a). In the latter case, an incomplete factorial design consisting of 36 CMS lines and 36 restorer lines was used to compare prediction accuracy of GS and classical GCA modeling in sunflower. Multi-environmental field trials were performed to characterize 452 sunflower hybrids of the panel with regard to hybrid performance in oil content, which represents a primarily additive trait with high heritability. In addition, all 72 parental lines were sequenced to obtain genome-wide SNP markers (Mangin et al., 2017a). Genomic predictions were then made for missing hybrids and hybrid combinations lacking information about at least one parental line. In conclusion, GS led to considerable improvement in breeding efficiency compared to the conventional GCA modeling if little is known about one or both parental lines (Mangin et al., 2017a). For Sclerotinia midstalk rot, the prediction ability of a genome-based best linear unbiased prediction (GBLUB) model was evaluated in a biparental population genotyped with the 25 K SNP array (Livaja et al., 2016). High predictive abilities were obtained for “stem lesion length” and lower predictive abilities for “leaf lesion length” and “speed of fungal growth,” which represent traits with lower heritabilities. These first experimental trials for genomic predictions, using and comparing the results of different models, have shown the potential and the limitations for genomic selection in sunflower.
In this review the emphasis was given to plant genetic resources and molecular tools used to detect and exploit genetic diversity and to facilitate sunflower hybrid breeding. Traditional MAS has been successfully used to introduce monogenic traits into the breeding material, especially disease resistance as well as herbicide tolerance. Validation of identified molecular markers across different genotypes has also shown the limitation in markers to be used in different genetic backgrounds. However, sunflower researchers have put a lot of effort in the identification of markers linked to specific traits without gaining insight into the function of the involved genes, even though this would allow a better understanding of the metabolism and mechanisms behind traits. Breeding for complex polygenic traits is still challenging. With this regard, it is necessary to stress the importance of precise phenotypic evaluation, on which molecular biologists rely to correctly interpret the molecular and phenotypic data. High-throughput phenotyping as applied and tested in other crops would be also interesting for sunflower (Sankaran et al., 2015). There has been a first report on testing remote sensing on sunflower and maize in China with regard to future applications (Yu and Shang, 2017). In recent years, high throughput genotyping platforms, e.g., SNP arrays, GBS and whole genome sequencing have been established and successfully used in sunflower (Livaja et al., 2016; Qi L. et al., 2016; Talukder et al., 2016). GWAS (genome wide association study) and GS (genomic selection) using large amounts of markers across a wide range of genotypes provided by these techniques open up new possibilities to address complex traits in sunflower. However, GWAS is still expensive and unavailable for many researchers and breeders. Some initial steps have been made in order to create the most appropriate models for prediction of hybrid performance based on GWAS and GS data (Bonnafous et al., 2016; Mangin et al., 2017a), yet there is still a need for further improvement of prediction models, which mostly take additive effects into account, whereas for heterosis also dominance and epistasis play an important role. As in conventional breeding, species-specific strategies will have to be developed for GS taking into account reproduction system, generation time, genome structure, harvested organs and breeding purposes (Nakaya and Isobe, 2012). However, first empirical GS studies in plants showed the potential for GS also in plant breeding. It could be demonstrated that the correct choice of population allows successful performance of GS even with lower numbers of markers and reasonable sizes of populations (Nakaya and Isobe, 2012).
Access to the recently published sunflower genome sequence (Badouin et al., 2017) should allow researchers and breeders to make sunflower breeding more efficient in the coming years. However, exploring the sunflower genome on its own is not enough. Extensive transcriptomics, proteomics and metabolomics data are required as only the combination of all Omics data will enable us to get to the bottom of some important physiological and molecular mechanisms unique to sunflower. This is especially important for quantitative traits such as drought tolerance or biotic stress resistance (e.g., against Sclerotinia, Phoma, Phomopsis). First results in this direction have been published. Transcriptional profiling has been done with regard to disease reactions of resistant and sensitive genotypes to pathogens as S. sclerotiorum (Muellenborn et al., 2011), Plasmopara halstedii (Livaja et al., 2013) and Verticillium dahliae (Guo et al., 2017). Identification of the differentially expressed genes now allows a better understanding of the mechanisms behind pathogen attacks and plant reactions. This knowledge will be helpful with regard to developing resistant cultivars. Earlier metabolome data of head rot between genotypes with different reactions to S. sclerotiorum also gave an indication to 63 metabolites involved in the attack of the pathogen (Peluffo et al., 2010). To analyze the response of sunflower to drought transcriptome analyses of sunflower genotypes under water-limited conditions in comparison to well-water plants have been performed by RNASeq or microarray analyses (Liang et al., 2017; Moschen et al., 2017; Sarazin et al., 2017). Combination of the transcriptomic and metabolic data made the identification of drought relevant hubs for transcription possible (Moschen et al., 2017). Leaf senescence is a naturally occurring process, but the onset and progress of senescence plays a major role for yield. Integration of transcriptomic and metabolomics data identified metabolites and transcription factors as applicable biomarkers (Moschen et al., 2016a,b). To explore the potential of other species in the genus Helianthus for sunflower breeding, transcriptomics have also been performed to address populations of, e.g., perennial sunflowers as H. maximiliani (Kawakami et al., 2014) and H. tuberosus (Jung et al., 2014) as well as interspecific hybrids of annuals in the first generation (Rowe and Rieseberg, 2013). Proteomic analyses in sunflower have been performed with regard to drought stress (Castillejo et al., 2008; Fulda et al., 2011; Ghaffari et al., 2013, 2017), cold acclimation (Balbuena et al., 2011), response to metal-ion contamination (Garcia et al., 2006; Printz et al., 2013; Lopes Júnior et al., 2015), seed protein composition (De Sousa Barbosa et al., 2013), heterosis performance (Mohayeji et al., 2014), and resistance to O. cumana (Yang et al., 2017). In addition, the sunflower genome database represents a very valuable tool, which allows access to a wide range of transcriptome data, which have already been successfully used to address flowering time and oil metabolism (Badouin et al., 2017). However, further studies in sunflower are still needed in order to analyze in detail responses to different abiotic and biotic stress conditions and to prepare sunflower for future climatic challenges. Combining Omics data will allow system biology approaches to improve sunflower hybrids. Another aspect is the optimization of plant architecture to a more compact form, which would have an influence on photosynthesis, lodging, climatic adaptation and possible plant densities. This could also improve sunflower hybrid performance and increase yields per hectare by use of higher plant densities (Hall et al., 2010). Picheny et al. (2017) used the crop model SUNFLO to design sunflower ideotypes with optimized morphological and physiological traits for certain environments.
However, only a combined effort of the sunflower research community can make sunflower more competitive to other oil crops. The new high-throughput technologies combined with new genomic-based breeding strategies give us the opportunity, as never before, to understand and mine genetic variation and to use it for improvement of sunflower hybrids.
Both authors AD and RH have made an equal substantial, direct and intellectual contribution to writing the review and its revision, and approved it for publication. The table was prepared by AD, the figure by RH. The authors complied to the ethical standards.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the University of Rostock and the DFG for funding part of the research included (HO 1593/5-1, HO 1593/5-2, and HO 1593/6-1). They are also grateful for the University of Rostock and the DFG for funding the open access publication. Another part of the research presented was supported by project TR31025 (Ministry of Education, Science and Technological Development, R. Serbia).
Abratti, G., Bazzalo, M. E., and León, A. (2008). “Mapping a novel fertility restoration gene in sunflower,” in Proceedings of the 17th International Sunflower Conference, Córdoba, 617–621.
Al-Khatib, K., Baumgartner, J. R., and Currie, R. S. (1999). “Survey of common sunflower (Helianthus annuus) resistance to ALS inhibiting herbicides in northeast Kansas,” in Proceedings of the 21st Sunflower Research Workshop, Bismarck, ND, 210–215.
Al-Khatib, K., Baumgartner, J. R., Peterson, D. E., and Currie, R. S. (1998). Imazethapyr resistance in common sunflower - Helianthus annuus L. Weed Sci. 46, 403–407.
Allman-Farinelli, M. A., Gomes, K., Favaloro, E. J., and Petocz, P. (2005). A diet rich in high-oleic-acid sunflower oil favorably alters low-density lipoprotein cholesterol, triglycerides, and factor VII coagulant activity. J. Am. Diet. Assoc. 105, 1071–1079. doi: 10.1016/j.jada.2005.04.008
Arumuganathan, K., and Earle, E. D. (1991). Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–219. doi: 10.1007/BF02672016
Atlagic, J., and Terzic, S. (2014). “Sunflower genetic resources – interspecific hybridization and cytogenetics in prebreeding,” in Sunflowers: Growth and Development, Environmental Influences and Pests/Diseases, ed. J. I. Arribas (New York, NY: Nova Science Publishers), 95–130.
Bachlava, E., Radwan, O. E., Abratti, G., Tang, S., Gao, W., Heesacker, A. F., et al. (2011). Downy mildew (Pl8 and Pl14) and rust (RAdv) resistance genes reside in close proximity to tandemly duplicated clusters of non-TIR-like NBS-LRR-encoding genes on sunflower chromosomes 1 and 13. Theor. Appl. Genet. 122, 1211–1221. doi: 10.1007/s00122-010-1525-0
Bachlava, E., Tang, S., Pizarro, G., and Knapp, S. J. (2009). Pleiotropy of the branching locus (B) masks and unlinked quantitative trait loci affecting seed traits in sunflower. Theor. Appl. Genet. 120, 829–842. doi: 10.1007/s00122-009-1212-1
Bachlava, E., Taylor, C. A., Tang, S., Bowers, J. E., Mandel, J. R., Burke, J. M., et al. (2012). SNP discovery and development of a high-density genotyping array for sunflower. PLOS ONE 7:e29814. doi: 10.1371/journal.pone.0029814
Badouin, H., Gouzy, J., Grassa, C. J., Murat, F., Staton, S. E., Cottret, L., et al. (2017). The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546, 148–152. doi: 10.1038/nature22380
Balbuena, T. S., Salsa, J. J., Martinez-Force, E., Garces, R., and Thelen, J. J. (2011). Proteome analysis of cold acclimation in sunflower. J. Proteome Res. 10, 2330–2346. doi: 10.1021/pr101137q
Bandeira e Sousa, M., Cuevas, J., de Oliveira Couto, E. G., Péres-Rodríguez, P., Jarquin, D., Fritsche-Neto, R., et al. (2017). Genomic-enabled prediction in maize using kernel models with genotype x environment interaction. G3 7, 1995–2014. doi: 10.1534/g3.117.042341
Bandillo, N., Raghvan, C., Muyco, P. A., Sevilla, M. A. L., Lobina, I. T., Dilla-Ermita, C., et al. (2013). Multi-parent advanced generation inter-cross (MAGIC) populations in rice: progress and potential for genetics research and breeding. Rice 6:11. doi: 10.1186/1939-8433-6-11
Bassi, F. M., Bentley, A. R., Charmet, G., Ortiz, R., and Crossa, J. (2016). Breeding schemes for the implementation of genomic selection in wheat (Triticum sp.). Plant Sci. 242, 23–36. doi: 10.1016/j.plantsci.2015.08.021
Baute, G. J., Owens, G. L., Bock, D. G., and Rieseberg, L. H. (2016). Genome-wide genotyping-by-sequencing data provide a high-resolution view of wild Helianthus diversity, genetic structure, and interspecies gene flow. Am. J. Bot. 103, 2170–2177. doi: 10.3732/ajb.1600295
Bernardo, R. (2008). Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci. 48, 1649–1664. doi: 10.2135/cropsci2008.03.0131
Berrios, E. F., Gentzbittel, L., Alibert, G., Griveau, Y., Bervillé, A., and Sarrafi, A. (1999). Genetic control of in vitro-organogenesis in recombinant inbred lines of sunflower (Helianthus annuus L.). Plant Breed. 118, 359–361. doi: 10.1046/j.1439-0523.1999.00370.x
Berry, S. T., León, A. J., Hanfrey, C. C., Challis, P., Burkholz, A., Barnes, S. R., et al. (1995). Molecular marker analysis of Helianthus annuus L. 2. Construction of an RFLP linkage map for cultivated sunflower. Theor. Appl. Genet. 91, 195–199. doi: 10.1007/BF00220877
Bert, P. F., Tourvieille de Labrouhe, D., Philippon, J., Mouzeyar, S., Jouan, I., Nicolas, P., et al. (2001). Identification of a second linkage group carrying genes controlling resistance to downy mildew (Plasmopara halstedii) in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 103, 992–997. doi: 10.1007/s001220100660
Bervillé, A. (2010). “Oil composition variations,” in Genetics, Genomics and Breeding of Sunflower, eds J. Hu, G. Seiler, and C. Kole (Boca Raton, FL: Science Publisher), 253–277.
Bilgen, B. (2016). “Characterization of sunflower inbred lines with high oleic acid content by DNA markers,” in Proceedings of the 19th International Sunflower Conference, Edirne, 662–668.
Blackman, B. K., Rasmussen, D. A., Strasburg, J. L., Raduski, A. R., Burke, J. M., Knapp, S. J., et al. (2011). Contribution of flowering time genes to sunflower domestication and improvement. Genetics 187, 271–287. doi: 10.1534/genetics.110.121327
Bohra, A., Jha, U. C., Adhimoolam, P., Bisht, D., and Singh, N. P. (2016). Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 35, 967–993. doi: 10.1007/s00299-016-1949-3
Bonnafous, F., Langdale, N., Sunrise Consortium and Mangin, B. (2016). “Inclusion of dominance effect in genomic selection model to improve predictive ability for sunflower hybrid performance,” in Proceedings of the 19th International Sunflower Conference, Edirne, 285.
Bouzidi, M. F., Badaoui, S., Cambon, F., Vear, F., De Labrouhe, D., Nicolas, P., et al. (2002). Molecular analysis of a major locus for resistance to downy mildew in sunflower with specific PCR-based markers. Theor. Appl. Genet. 104, 592–600. doi: 10.1007/s00122-001-0790-3
Bowers, J. E., Bachlava, E., Brunick, R. L., Knapp, S. J., and Burke, J. M. (2012). Development of a 10,000 locus genetic map of the sunflower genome based on multiple crosses. G3 2, 721–729. doi: 10.1534/g3.112.002659
Breccia, G., Gil, M., Vega, T., Zorzoli, R., Picardi, L., and Nestares, G. (2011). Rapid test for detection of imidazolinone resistance in sunflower (Helianthus annuus L.). Plant Breed. 130, 109–113. doi: 10.1111/j.1439-0523.2009.01756.x
Brouillette, L. C., Rosenthal, D. M., Rieseberg, L. H., Lexer, C., Malmberg, R. L., and Donovan, L. A. (2007). Genetic architecture of leaf ecophysiological traits in Helianthus. J. Hered. 98, 141–146. doi: 10.1093/jhered/esl063
Brunel, D. (1994). A microsatellite marker in Helianthus annuus L. Plant Mol. Biol. 24, 397–400. doi: 10.1007/BF00020177
Bruniard, J. M., and Miller, J. F. (2001). Inheritance of imidazolinone herbicide resistance in sunflower. Helia 24, 11–16.
Bulos, M., Ramos, M. L., Altieri, E., and Sala, C. A. (2013a). Molecular mapping of a sunflower rust resistance gene from HAR6. Breed. Sci. 63, 141–146. doi: 10.1270/jsbbs.63.141
Bulos, M., Sala, C. A., Altieri, E., and Ramos, M. L. (2013b). Marker assisted selection for herbicide resistance in sunflower. Helia 59, 1–16. doi: 10.2298/HEL1359001B
Bulos, M., Vergani, P. N., and Altieri, E. (2014). Genetic mapping, marker assisted selection and allelic relationships for the Pu6 gene conferring rust resistance in sunflower. Breed. Sci. 64, 206–212. doi: 10.1270/jsbbs.64.206
Cadic, E., Coque, M., Vear, F., Grezes-Besset, B., Puaquet, J., Piquemal, J., et al. (2013). Combined linkage and association mapping of flowering time in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 126, 1337–1356. doi: 10.1007/s00122-013-2056-2
Cantelmo, F. N., Von Pinho, R. G., and Balestre, M. (2017). Genome-wide prediction for maize single-cross hybrids using the GBLUB model and validation in different crop seasons. Mol. Breed. 37:51. doi: 10.1007/s11032-017-0651-7
Castillejo, M. A., Maldonado, A. M., Ogueta, S., and Jorrin, J. V. (2008). Proteomic analysis of responses to drought stress in sunflower (Helianthus annuus) leaves by 2DE gel electrophoresis and mass spectrometry. Open Proteomics J. 1, 59–71. doi: 10.2174/1875039700801010059
Cavanagh, C., Morell, M., Mackay, I., and Powell, W. (2008). From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 11, 215–221. doi: 10.1016/j.pbi.2008.01.002
Celik, I., Bodur, S., Frary, A., and Doganlar, S. (2016). Genome-wide SNP discovery and genetic linkage map construction in sunflower (Helianthus annuus L.) using a genotyping by sequencing (GBS) approach. Mol. Breed. 36:133. doi: 10.1007/s11032-016-0558-8
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.125343
Chapman, M. A., Pashley, C. H., Wenzler, J., Hvala, J., Tang, S., Knapp, S. J., et al. (2008). A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annuus L.). Plant Cell 20, 2931–2945. doi: 10.1105/tpc.108.059808
Cvejic, S., Afza, R., Jocic, S., Prodanovic, S., Miklic, V., Skoric, D., et al. (2011a). Radiosensitivity of sunflower inbred lines to mutagenesis. Helia 34, 99–106. doi: 10.2298/HEL1154099C
Cvejic, S., Jocic, S., Prodanovic, S., Terzic, S., Miladinovic, D., and Balalic, I. (2011b). Creating new genetic variability in sunflower using induced mutations. Helia 34, 47–55. doi: 10.2298/HEL1155047C
de Azevedo Peixoto, L., Moellers, T. C., Zhang, J., Lorenz, A. J., Bhering, L. L., Beavis, W. D., et al. (2017). Leveraging genomic prediction to scan germplasm collection crop improvement. PLOS ONE 12:e0179191. doi: 10.1371/journal.pone.0179191
de Bertero, R. A. (2003). Origin of the Argentine sunflower. Helia 26, 127–136. doi: 10.2298/HEL0338127d
De Sousa Barbosa, H., Quirino de Souza, D. L., Koolen, H. H. F., Gozzo, F. C., and Arruda, M. A. Z. (2013). Sample preparation focusing on plant proteomics: extraction, evaluation and identification of proteins from sunflower seeds. Anal. Methods 5, 116–123. doi: 10.1039/C2AY25503K
Debaeke, P., Casadebaig, P., Flenet, F., and Langlade, N. (2017). Sunflower crop and climate change: vulnerability, adaptation, and mitigation potential from case-studies in Europe. OCL 24:D102. doi: 10.1051/ocl/2016052
Dehmer, K. J., and Friedt, W. (1998). Development of molecular markers for high oleic acid content in sunflower (Helianthus annuus L.). Ind. Crops Prod. 7, 311–315. doi: 10.1016/S0926-6690(97)00063-0
Dimitrijevic, A., Imerovski, I., Miladinovic, D., Cvejic, S., Jocic, S., Zeremski, T., et al. (2017). Oleic acid variation and marker-assisted detection of Pervenets mutation in high- and low-oleic sunflower cross. Crop Breed. Appl. Biotechnol. 17, 229–235. doi: 10.1590/1984-70332017v17n3a36
Dimitrijevic, A., Imerovski, I., Miladinovic, D., Jocic, S., and Miklic, V. (2011). “Use of molecular markers in identification of non-TIR-NBS-LRR RGA linked to downy mildew resistance locus Pl5/Pl8 in sunflower (Helianthus annuus L.),” in Proceedings of the 19th Symposium of the Serbian Plant Physiology Society, Banja Vrujci, 80.
Dimitrijevic, A., Imerovski, I., Miladinovic, D., Jockovic, M., Cvejic, S., Jocic, S., et al. (2016). “Screening of the presence of ol gene in NS sunflower collection,” in Proceedings of the 19th International Sunflower Conference, Edirne, 661–667.
Dimitrijevic, A., Imerovski, I., Miladinovic, D., Tancic, S., Dušanic, N., Jocic, S., et al. (2010). Use of SSR markers in identification of sunflower isogenic lines in late generations of backcrossing. Helia 33, 191–198. doi: 10.2298/hel1053191d
Dußle, C. M., Hahn, V., Knapp, S. J., and Bauer, E. (2004). Plarg from Helianthus argophyllus is unlinked to other known downy mildew resistance genes in sunflower. Theor. Appl. Genet. 109, 1083–1086. doi: 10.1007/s00122-004-1722-9
FAO (2016). FAOSTAT. Rome: FAO. Available at: http://www.fao.org/faostat/en/#data/QC (accessed October 5, 2016).
Feng, J., and Jan, C.-C. (2008). Introgression and molecular tagging of Rf4, a new male fertility restoration gene from wild sunflower Helianthus maximiliani L. Theor. Appl. Genet. 117, 241–249. doi: 10.1007/s00122-008-0769-4
Feng, J., Liu, Z., Cai, X., and Jan, C.-C. (2013). Towards a molecular cytogenetic map for cultivated sunflower (Helianthus annuus L.) by landed BAC/BIBAC clones. G3 3, 31–40. doi: 10.1534/g3.112.004846
Ferfuia, C., Turi, M., and Vannozzi, G. P. (2015). Variability of seed fatty acid composition to growing degree-days in high oleic acid sunflower genotypes. Helia 38, 61–78. doi: 10.1515/helia-2014-0022
Ferfuia, C., and Vannozzi, G. P. (2015). Maternal effect on seed fatty acid composition in a reciprocal cross of high oleic sunflower (Helianthus annuus L.). Euphytica 205, 325–336. doi: 10.1007/s10681-015-1378-3
Fick, G. N., and Miller, J. F. (1997). “Sunflower breeding,” in Sunflower Production and Technology, ed. A. A. Scheiter (Madison, WI: ASA), 395–440.
Filippi, C. V., Aguirre, N., Rivas, J. G., Zubrzycki, J., Puebla, A., Cordes, D., et al. (2015). Population structure and genetic diversity characterization of a sunflower association mapping population using SSR and SNP markers. BMC Plant Biol. 15:52. doi: 10.1186/s12870-014-0360-x
Fulda, S., Mikkat, S., Stegmann, H., and Horn, R. (2011). Physiology and proteomics of drought stress acclimation in sunflower (Helianthus annuus L.). Plant Biol. 13, 632–642. doi: 10.1111/j.1438-8677.2010.00426
Fusari, C. M., Di Rienzo, J. A., Troglia, C., Nishinakamasu, V., Moreno, M. V., Maringolo, C., et al. (2012). Association mapping in sunflower for Sclerotinia head rot resistance. BMC Plant Biol. 12:93. doi: 10.1186/1471-2229-12-93
Fusari, C. M., Lia, V. V., Hopp, H. E., Heinz, R. A., and Paniego, N. B. (2008). Identification of single nucleotide polymorphisms and analysis of linkage disequilibrium in sunflower elite inbred lines using the candidate gene approach. BMC Plant Biol. 8:7. doi: 10.1186/147-2229.8-7
Gabard, J. M., and Huby, J. P. (2001). Sulfonylurea-tolerant sunflower plants. U.S. Patent No. 20050044587.
Gandhi, S. D., Heesacker, A. F., Freeman, C. A., Argyris, J., Bradford, K., and Knapp, S. J. (2005). The self-incompatibility locus (S) and quantitative trait loci for self-pollination and seed dormancy in sunflower. Theor. Appl. Genet. 111, 619–629. doi: 10.1007/s00122-005-1934-7
Garcia, J. S., Gratao, P. L., Azevedo, R. A., and Arruda, M. A. Z. (2006). Metal contamination effects on sunflower (Helianthus annuus L.) growth and protein expression in leaves during development. J. Agric. Food Chem. 54, 8623–8630. doi: 10.1012/jf061593
Gavrilova, V. A., Rozhkova, V. T., and Anisimova, I. N. (2014). Sunflower genetic collection at the Vavilov institute of plant industry. Helia 37, 1–16. doi: 10.1515/helia-2014-0001
Gedil, M. A., Wye, C., Berry, S. T., Seger, B., Peleman, J., Jones, R., et al. (2001). An integrated RFLP-AFLP linkage map for cultivated sunflower. Genome 44, 213–221. doi: 10.1139/g00-111
Gentzbittel, L., Vear, F., Zhang, Y. X., and Berville, A. (1995). Development of a consensus linkage RFLP map of cultivated sunflower (Helianthus annuus L.). Theor. Appl. Genet. 90, 1079–1086. doi: 10.1007/BF00222925
Ghaffari, M., Toorchi, M., Valizadeh, M., and Komatsu, S. (2013). Differential response of root proteome to drought stress in drought sensitive and tolerant sunflower inbred lines. Funct. Plant Biol. 40, 609–617. doi: 10.1071/FP12251
Ghaffari, M., Toorchi, M., Valizadeh, M., and Shakiba, M. (2017). Proteomic prospects for tolerance of sunflower (Helianthus annuus) to drought stress during the flowering stage. Crop Pasture Sci. 68, 457–465. doi: 10.1071/CP17105
Gilley, M. A., Misar, C. G., Gulya, T. J., and Markell, S. G. (2016). “Prevalence and virulence of Plasmopara halstedii (downy mildew) in sunflowers,” in Proceedings of the 38th Sunflower Research Forum, Fargo, ND.
Gong, L., Guyla, T. J., Markell, S. G., Hulke, B. S., and Qi, L. L. (2013a). Genetic mapping of rust resistance genes in confection sunflower line HAR6 and oilseed line RHA 397. Theor. Appl. Genet. 126, 2039–2049. doi: 10.1007/s00122-013-2116-7
Gong, L., Hulke, B. S., Gulya, T. J., Markell, S. G., and Qi, L. L. (2013b). Molecular tagging of a novel rust resistance gene R12 in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 126, 93–99. doi: 10.1007/s00122-012-1962-z
Grassa, C. J., Ebert, D. P., Kane, N. C., and Rieseberg, L. H. (2016). Complete mitochondrial genome sequence of sunflower (Helianthus annuus L.). Genome Announc. 4:e00981-16. doi: 10.1128/genomeA.00981-16
Guimaraes, R. L., and Stotz, H. U. (2004). Oxalate production by Sclerotinia sclerotiorum deregulates guard cells during infection. Plant Physiol. 136, 3703–3711. doi: 10.1104/pp.104.049650
Gulya, T., Rashid, K. Y., and Masirevic, S. N. (1997). “Sunflower diseases,” in Sunflower Technology and Production, ed. A. A. Schneiter (Madison, WI: ASA), 263–379.
Guo, S., Zuo, Y., Zhang, Y., Wu, C., Su, C., Jin, W., et al. (2017). Large-scale transcriptome comparison of sunflower genes responsive to Verticillium dahliae. BMS Genomics 18:42. doi: 10.1186/s12864-016-3386-7
Guo, X., and Stotz, H. U. (2007). Defense against Sclerotinia sclerotiorum in Arabidopsis is dependent on jasmonic acid (JA), salicylic acid (SA), and ethylene (ET) signaling. Mol. Plant Microbe Interact. 20, 1384–1395. doi: 10.1094/MPMI-20-11-1384
Habyarimana, E., Parisi, B., and Mandolino, G. (2017). Genomic prediction for yields, processing and nutritional quality traits in cultivated potato (Solanum tuberosum L.). Plant Breed. 136, 245–252. doi: 10.1111/pbr.12461
Hall, A. J., Sporsaro, M. M., and Chimenti, C. A. (2010). Stem lodging in sunflower: variations in stem failure moment of force and structure across crop population densities and post-anthesis developmental stages in two genotypes of contrasting susceptibility to lodging. Field Crops Res. 116, 46–51. doi: 10.1016/j.fcr.2009.11.008
Heesacker, A., Kishore, V. K., Gao, W., Tang, S., Kolkman, J. M., Gingle, A., et al. (2008). SSRs and INDELs mined from the sunflower EST database: abundance, polymorphisms, and cross-taxa utility. Theor. Appl. Genet. 117, 1021–1029. doi: 10.1007/s00122-008-0841-0
Horn, R., Gupta, J. K., and Colombo, N. (2014). Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 19, 198–205. doi: 10.1016/j.mito.2014.04.004
Horn, R., Köhler, R., and Zetsche, K. (1991). A mitochondrial 16 kDa protein is associated with cytoplasmic male sterility in sunflower. Plant Mol. Biol. 17, 29–36. doi: 10.1007/BF00036803
Horn, R., Kusterer, B., Lazarescu, E., Prüfe, M., and Friedt, W. (2003). Molecular mapping of the Rf1 gene restoring pollen fertility in PET1-based F1-hybrids in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 106, 599–606. doi: 10.1007/s00122-002-1078-y
Horn, R., Reddemann, A., and Drumeva, M. (2016). “Comparison of cytoplasmic male sterility based on PET1 and PET2 cytoplasm in sunflower (Helianthus annuus L.),” in Proceedings of the 19th International Sunflower Conference, Edirne, 620–629.
Hu, J. (2010). “Genetic linkage maps: strategies, resources and achievements genetics,” in Genomics and Breeding of Sunflower, eds J. Hu, G. Seiler, and C. Kole (Enfield, NH: Science Publishers), 79–109. doi: 10.1201/b10192-4
Hu, J., Yue, B., and Vick, B. A. (2007). Integration of trap markers onto a sunflower SSR marker linkage map constructed from 92 recombinant inbred lines. Helia 30, 25–36. doi: 10.2298/HEL0746025H
Imerovski, I., Dimitrijevic, A., Miladinovic, D., Dedic, B., Jocic, S., and Cvejic, S. (2014a). “Preliminary SSR analysis of a novel broomrape resistance source,” in Proceedings of the Third International Symposium on Broomrape in Sunflower, Cordoba, 217–221.
Imerovski, I., Dimitrijevic, A., Miladinovic, D., Dedic, B., Jocic, S., Kovacevic, B., et al. (2013). Identification of PCR markers linked to different Or genes in sunflower. Plant Breed. 132, 115–120. doi: 10.1111/pbr.12022
Imerovski, I., Dimitrijevic, A., Miladinovic, D., Dedic, B., Jocic, S., Tubic, N. K., et al. (2016). Mapping of a new gene for resistance to broomrape races higher than F. Euphytica 209, 281–289. doi: 10.1007/s10681-015-1597-7
Imerovski, I., Dimitrijevic, A., Miladinovic, D., Jocic, S., Dedic, B., Cvejic, S., et al. (2014b). Identification and validation of breeder-friendly DNA markers for Plarg gene in sunflower. Mol. Breed. 34, 779–788. doi: 10.1007/s11032-014-0074-7
Iuoras, M., Stanciu, D., Ciuca, M., Nastase, D., and Geronzi, F. (2004). Preliminary studies related to the use of marker assisted selection for resistance of Orobanche cumana Wallr. in sunflower. Rom. Agric. Res. 21, 33–37.
Jacob, J., Mulpuri, S., and Sivannarayana Kodeboyina, V. (2017). Screening of cultivated and wild Helianthus species reveals herbicide tolerance in wild sunflower and allelic variation at Ahas11 (acetohydroxyacid synthase 1 large subunit) locus. Plant Genet. Res. C 15, 421–429. doi: 10.1017/S14792622116000095
Jan, C. C., and Vick, B. A. (2007). Inheritance and allelic relationship of fertility restoration genes for seven new sources of male-sterile cytoplasm in sunflower. Plant Breed. 126, 213–217. doi: 10.1111/j.1439-0523.2007.01350.x
Jan, C. C., Vick, B. A., Miller, J. K., Kahler, A. L., and Butler, E. T. I. (1998). Construction of a RFLP linkage map for cultivated sunflower. Theor. Appl. Genet. 96, 15–22. doi: 10.1007/s001220050703
Jocic, S., Cvejic, S., Hladni, N., Miladinovic, D., and Miklic, V. (2010). Development of sunflower genotypes resistant to downy mildew. Helia 33, 173–180. doi: 10.2298/hel1053173j
Jocić, S., Malidža, G., and Škorić, D. (2004). Sunflower tolerant to imidazoline herbicides. J. Sci. Agric. Res. 229, 81–89.
Jocic, S., Miladinovic, D., and Kaya, Y. (2015). “Breeding and genetics of sunflower,” in Sunflower: Chemistry, Production, Processing, and Utilization, eds E. Martinez-Force, N. T. Dunford, and J. J. Salas (Urbana, IL: AOCS), 1–26. doi: 10.2298/HEL0644033J
Joksimovic, J., Atlagic, J., Marinkovic, R., and Jovanovic, D. (2006). Genetic control of oleic and linoleic acid contents in sunflower. Helia 29, 33–40. doi: 10.2298/hel0644033j
Jung, W. Y., Lee, S. S., Kim, C. W., Kim, H. S., Min, S. R., Moon, J. S., et al. (2014). RNA-seq analysis and de novo transcriptome assembly of Jerusalem artichoke (Helianthus tuberosus Linne). PLOS ONE 9:e111982. doi: 10.1371/journal.pone.0111982
Kane, N. C., Burke, J. M., Marek, L., Seiler, G., Vear, F., Baute, G., et al. (2013). Sunflower genetic, genomic and ecological resources. Mol. Ecol. Resour. 13, 10–20. doi: 10.11111/1755-0998.12023
Kawakami, T., Darby, B. J., and Ungerer, M. C. (2014). Transcriptome resources for the perennial sunflower Helianthus maximiliani obtained from ecologically divergent populations. Mol. Ecol. Resour. 14, 812–819. doi: 10.1111/1755-0998.12227
Kim, S.-C., and Rieseberg, L. H. (1999). Genetic architecture of species differences in annual sunflowers: implication for adaptive trait introgression. Genetics 153, 965–977.
Kinman, M. L. (1970). “New developments in the USDA and state experiment station sunflower breeding programs,” in Proceedings of the 4th International Sunflower Conference, Memphis, TN, 181–183.
Köhler, R. H., Horn, R., Lössl, A., and Zetsche, K. (1991). Cytoplasmic male sterility in sunflower is correlated with the co-transcription of a new open reading frame with the atpA gene. Mol. Gen. Genet. 227, 369–376. doi: 10.1534/genetics.107.074054
Kolkman, J. M., Slabaugh, M. B., Bruniard, J. M., Berry, S., Bushman, B. S., Olungu, C., et al. (2004). Acetohydroxyacid synthase mutations conferring resistance to imidazolinone or sulfonylurea herbicides in sunflower. Theor. Appl. Genet. 109, 1147–1159. doi: 10.1007/s00122-004-1716-7
Kolkman, J. M., Berry, S. T., Leon, A. J., Slabaugh, M. B., Tang, S., Gao, W., et al. (2007). Single nucleotide polymorphisms and linkage disequilibrium in sunflower. Genetics 177, 457–468. doi: 10.1534/genetics.107.074054
Korell, M., Mösges, G., and Friedt, W. (1992). Construction of a sunflower pedigree map. Helia 15, 7–16.
Kumar, A. P. K., Boualem, A., Bhattacharya, A., Parikh, S., Desai, N., Zambelli, A., et al. (2013). SMART – Sunflower mutant population and reverse genetic tools for crop improvement. BMC Plant Biol. 13:38. doi: 10.1186/1471-2229-13-38
Kusterer, B., Horn, R., and Friedt, W. (2005). Molecular mapping of the fertility restoration locus Rf1 in sunflower and development of diagnostic markers for the restorer gene. Euphytica 143, 35–43. doi: 10.1007/s10681-005-1795-9
Lacombe, S., Kaan, F., Griveau, Y., and Bervillé, A. (2004). The Pervenets high oleic mutation: methodological studies. Helia 27, 41–54. doi: 10.2298/hel0440041l
Lacombe, S., Leger, S., Kaan, F., Bervillé, A., and Sas, M. (2002). Genetic, molecular and expression features of the Pervenets mutant leading to high oleic acid content of seed oil in sunflower. OCL 9, 17–23. doi: 10.1051/ocl.2002.0017
Lacombe, S., Souyris, I., and Bervillé, A. J. (2009). An insertion of oleate desaturase homologous sequence silences via siRNA the functional gene leading to high oleic acid content in sunflower seed oil. Mol. Genet. Genomics 281, 43–54. doi: 10.1007/s00438-008-0391-9
Lai, Z., Livingstone, K., Zou, Y., Church, S. A., Knapp, S. J., Andrews, J., et al. (2005). Identification and mapping of SNPs from ESTs in sunflower. Theor. Appl. Genet. 111, 1532–1544. doi: 10.1007/s00122-005-0082-4
Lawson, W. R., Goulter, K. C., Henry, R. J., Kong, G. A., and Kochman, J. K. (1996). RAPD markers for a sunflower rust resistance gene. Aust. J. Agric. Res. 47, 395–401. doi: 10.1071/AR9960395
Lawson, W. R., Goulter, K. C., Henry, R. J., Kong, G. A., and Kochman, J. K. (1998). Marker-assisted selection for two rust resistance genes in sunflower. Mol. Breed. 4, 227–234. doi: 10.1023/A:1009667112088
Leclercq, P. (1969). Une sterilité mâle cytoplasmique chez le tournesol. Ann. Amelior. Plant. 19, 99–106.
Leclercq, P. (1971). La stérilité mâle cytoplasmique du tournesol. 1. Premières études sur la restauration de la fertilité. Ann. Amelior. Plant. 21, 45–54.
Leclercq, P. (1984). Identification de gènes de restauration de fertilité sur cytoplasmic stérilisants chez le tournesol. Agronomie 4, 573–576. doi: 10.1051/agro:19840611
Leon, A. J., Andrade, F. H., and Lee, M. (2000). Genetic mapping of factors affecting quantitative variation for flowering in sunflower. Crop Sci. 40, 404–407. doi: 10.2135/cropsci2000.402404x
Leon, A. J., Zambelli, A. D., Reid, R. J., Morat, M. M., and Kaspar, M. (2013). Nucleotide sequences mutated by insertion that encode a truncated oleate desaturase protein, proteins, methods and uses. Patent No. WO 2013/004281 A1.
Liang, C., Wang, W., Wang, J., Ma, J., Li, C., Zhou, F., et al. (2017). Identification of differentially expressed genes in sunflower (Helianthus annuus) leaves and roots under drought stress by RNA sequencing. Bot. Stud. 58:42. doi: 10.1186/s40529-017-0197-3
Liu, A., and Burke, J. M. (2006). Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173, 321–330. doi: 10.1534/genetics.105.051110
Liu, Z., Gulya, T. J., Seiler, G. J., Vick, B. A., and Jan, C. C. (2012a). Molecular mapping of the Pl(16) downy mildew resistance gene from HA-R4 to facilitate marker-assisted election in sunflower. Theor. Appl. Genet. 125, 121–131. doi: 10.1007/s00122-012-1820-z
Liu, Z., Mulpuri, S., Feng, J., Vick, B. A., and Jan, C. C. (2012b). Molecular mapping of the Rf3 fertility restoration gene to facilitate its utilization in breeding confection sunflower. Mol. Breed. 29, 275–284. doi: 10.1007/s11032-011-9563-0
Liu, Z., Wang, D. M., Feng, J., Seiler, G. J., Cai, X., and Jan, C. C. (2013). Diversifying sunflower germplasm by integration and mapping of a novel male fertility restoration gene. Genetics 193, 727–737. doi: 10.1534/genetics.112.146092/-/DC1
Livaja, M., Unterseer, S., Erath, W., Lehermeier, C., Wieseke, R., Plieske, J., et al. (2016). Diversity analysis and genomic prediction of Sclerotinia resistance in sunflower using a new 25 K SNP genotyping array. Theor. Appl. Genet. 129, 317–329. doi: 10.1007/s00122-015-2629-3
Livaja, M., Wang, Y., Wieckhorst, S., Haseneyer, G., Seidel, M., Hahn, V., et al. (2013). BSTA: a targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genomics 14:628. doi: 10.1186/1471-2164-14-628
Longin, C. F. H., and Reif, J. C. (2014). Redesigning the exploitation of wheat genetic resources. Trends Plant Sci. 19, 631–636. doi: 10.1016/j.tplants.2014.06.012
Lopes Júnior, C. A., Barbosa Hde, S., Moretto Galazzi, R., Ferreira Koolen, H. H., Gozzo, F. C., and Arruda, M. A. (2015). Evaluation of proteome alterations induced by cadmium stress in sunflower (Helianthus annuus L.) cultures. Ecotoxicol. Environ. Saf. 119, 170–177. doi: 10.1016/jecoenv.2015.05.016
Louarn, J., Boniface, M.-C., Pouilly, N., Velasco, L., Pérez-Vich, B., Vincourt, P., et al. (2016). Sunflower resistance to broomrape (Orobanche cumana) is controlled by specific QTLs for different parasitism stages. Front. Plant Sci. 7:590. doi: 10.3389/fpls.2016.00590
Lu, Y. H., Melero-Vara, J. M., Garcia-Tejada, J. A., and Blanchard, P. (2000). Development of SCAR markers linked to the gene Or5 conferring resistance to broomrape (Orobanche cumana Wallr.) in sunflower. Theor. Appl. Genet. 100, 625–632. doi: 10.1007/s001220050083
Lyra, D. H., Mendonça, L. F., Galli, G., Alves, F. C., Granato, I. S. C., and Fritsche-Neto, R. (2017). Multi-trait genomic prediction for nitrogen response indices in tropical maize hybrids. Mol. Breed. 37:80. doi: 10.1007/s11032-017-0681-1
Ma, G. J., Markell, S. G., Song, Q. J., and Qi, L. L. (2017). Genotyping-by-sequencing targeting of a novel downy mildew resistance gene Pl20 from wild Helianthus argophyllus for sunflower (Helianthus annuus L.). Theor. Appl. Genet. 130, 1519–1529. doi: 10.1007/s00122-017-2906-4
Mammadov, J., Aggarwal, R., Buyyarapu, R., and Kumpatla, S. (2012). SNP markers and their impact on plant breeding. Int. J. Plant Genomics 2012:728398. doi: 10.1155/2012/728398
Mandel, J. R., Dechaine, J. M., Marek, L. F., and Burke, J. M. (2011). Genetic diversity and population structure in cultivated sunflower and a comparison to its wild progenitor, Helianthus annuus L. Theor. Appl. Genet. 123, 693–704. doi: 10.1007/s00122-011-1619-3
Mandel, J. R., Nambeesan, S., Bowers, J. E., Marek, L., Ebert, D., Rieseberg, L. H., et al. (2013). Association mapping and the genomic consequences of selection in sunflower. PLOS Genet. 9:e1003378. doi: 10.1371/journal.pgen.1003378
Mangin, B., Bonnafous, F., Blanchet, N., Boniface, M.-C., Bret-Mestries, E., Carrère, S., et al. (2017a). Genomic prediction of sunflower hybrids oil content. Front. Plant Sci. 8:1633. doi: 10.3389/fpls.2017.01633
Mangin, B., Pouilly, N., Boniface, M.-C., Langdale, N. B., Vincourt, P., Vear, F., et al. (2017b). Molecular diversity of sunflower populations maintained as genetic resources is affected by multiplication processes and breeding for major traits. Theor. Appl. Genet. 130, 1099–1112. doi: 10.1007/s00122-017-2872-x
Marek, L. F. (2016). “Sunflower genetic resources,” in Proceedings of the 19th International Sunflower Conference, Edirne, 31–44.
Markin, N., Usatov, A., Makarenko, M., Azarin, K., Gorbachenko, O., Kolokolova, N., et al. (2017). Study of informative DNA markers of the Rf1 gene in sunflower for breeding practice. Czech J. Plant Breed. 53, 69–75. doi: 10.17221/108/2016-CJGPB
Massman, J. M., Jung, H.-J. G., and Bernardo, R. (2013). Genome wide selection versus marker-assisted recurrent selection to improve grain yield and stover-quality traits for cellulosic ethanol in maize. Crop Sci. 53, 58–66. doi: 10.2135/cropsci2012.02.0112
McAssey, E., Corbi, J., and Burke, J. M. (2016). Range-wide phenotypic and genetic differentiation in wild sunflower. BMC Plant Biol. 16:249. doi: 10.1186/s12870-016-0937-7
Meuwissen, T. H., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic values using genome-wide dense marker maps. Genetics 1157, 1819–1829.
Miller, F. J., and Al-Khatib, K. (2000). “Development of herbicide resistant germplasm in sunflower,” in Proceedings of the 15th International Sunflower Conference, Vol. 2, Toulouse, 12–15.
Miller, J. F., and Al-Khatib, K. (2002). Registration of imidazolinone herbicide-resistant sunflower maintainer (HA 425) and fertility restorer (RHA 426 and RHA 427) germplasms. Crop Sci. 42, 988–989. doi: 10.2135/cropsci2002.988a
Mohayeji, M., Capriotti, A. L., Cavaliere, C., Piovesana, S., Samperi, R., Stampachiacchiere, S., et al. (2014). Heterosis profile of sunflower leaves: a label free proteomics approach. J. Proteomics 99, 101–110. doi: 10.1016/j.jprot.2014.01.028
Molinero-Ruiz, L., García-Carneros, A. B., Collado-Romero, M., Raranciuc, S., Domínguez, J., and Melero-Vara, J. M. (2014). Pathogenic and molecular diversity in highly virulent populations of the parasitic weed Orobanche cumana (sunflower broomrape) from Europe. Weed Res. 54, 87–96. doi: 10.1111/wre.12056
Molinero-Ruiz, M. L., García-Ruiz, R., Melero-Vara, J. M., and Dominguez, J. (2009). Orobanche cumana race F: performance of resistant sunflower hybrids and aggressiveness of populations of the parasitic weed. Weed Res. 49, 469–478. doi: 10.1111/j.1365-3180.2009.00708.x
Monéger, F., Smart, C. J., and Leaver, C. J. (1994). Nuclear restoration of cytoplasmic male sterility in sunflower is associated with the tissue-specific regulation of a novel mitochondrial gene. EMBO J. 13, 8–17. doi: 10.1102/j.1460-2075.1994.tb06230x
Moreno, M. V., Nishinakamasu, V., Loray, M. A., Alvarez, D., Gieco, J., Vicario, A., et al. (2013). Genetic characterization of sunflower breeding resources from Argentina: assessing diversity in key open-pollinated and composite populations. Plant Genet. Resour. 11, 238–249. doi: 10.1017/S1479262113000075
Moschen, S., Bengoa Luoni, S., Di Rienzo, J. A., Caro Mdel, P., Watanabe, M., Hollmann, J., et al. (2016a). Integrating transcriptomic and metabolomic analysis to understand natural leaf senescence in sunflower. Plant Biotechnol. J. 14, 719–734. doi: 10.1111/pbi.12422
Moschen, S., Di Rienzo, J. A., Higgins, J., Tohge, T., Watanabe, M., Gonzalez, S., et al. (2017). Integration of transcriptomic and metabolic data reveals hub transcription factors involved in drought stress response in sunflower (Helianthus annuus L.). Plant Mol. Biol. 94, 549–564. doi: 10.1007/s11103-017-0625-5
Moschen, S., Higgins, J., Di Rienzo, J. A., Heinz, R. A., Paniego, N., and Fernandez, P. (2016b). Integration of sunflower transcriptomic and metabolic data by network and biosignature analysis. BMC Bioinformatics 17(Suppl. 5):174. doi: 10.1186/s12859-016-1045-2
Mouzeyar, S., Roeckel-Drevet, P., Gentzbittel, L., Philippon, J., De Labrouhe, D. T., Vear, F., et al. (1995). RFLP and RAPD mapping of the sunflower Pl1 locus for resistance to Plasmopara halstedii race 1. Theor. Appl. Genet. 91, 733–737. doi: 10.1007/BF00220951
Muellenborn, C., Krause, J.-H., and Cerboncini, C. (2011). Analysis of differential transcript expression reveals time-dependent leaf responses to Sclerotinia sclerotiorum in wild and cultivated sunflower. Plant Mol. Biol. Rep. 29, 597–608. doi: 10.1007/s11105-010-0265-2
Mulpuri, S., Liu, Z., Feng, J., Gulya, T. J., and Jan, C. C. (2009). Inheritance and molecular mapping of a downy mildew resistance gene, Pl13 in cultivated sunflower (Helianthus annuus L.). Theor. Appl. Genet. 119, 795–803. doi: 10.1007/s00122-009-1089-z
Nagarathna, T. K., Shadakshari, Y. G., and Ramanappa, T. M. (2011). Molecular analysis of sunflower (Helianthus annuus L.) genotypes for high oleic acid using microsatellite markers. Helia 34, 63–68. doi: 10.2298/hel1155063n
Nakaya, A., and Isobe, S. N. (2012). Will genomic selection be a practical method for plant breeding? Ann. Bot. 110, 1303–1316. doi: 10.1093/aob/mcs109
Nambeesan, S. U., Mandel, J. R., Bowers, J. E., Marek, L. F., Ebert, D., Corbi, J., et al. (2015). Association mapping in sunflower (Helianthus annuus L.) reveals independent control of apical vs. basal branching. BMC Plant Biol. 15:84. doi: 10.1186/s12870-015-0458-9
Pacureanu-Joita, M., Vranceanu, A. V., Soare, G., Marinescu, A., and Sandu, I. (1998). “The evaluation of the parasite-host interaction in the system Helianthus annuus L. -Orobanche cumana Wallr. in Romania,” in Proceedings of the 2nd Balkan Symposium in Field Crops, Novi Sad, 153–158.
Paniego, N., Echaide, M., Munoz, M., Fernandez, L., Torales, S., Faccio, P., et al. (2002). Microsatellite isolation and characterization in sunflower (Helianthus annuus L.). Genome 45, 34–43. doi: 10.1139/g01-120
Pankovic, D., Radovanovic, N., Jocic, S., Satovic, Z., and Skoric, D. (2007). Development of co-dominant amplified polymorphic sequence markers for resistance of sunflower to downy mildew race 730. Plant Breed. 126, 440–444. doi: 10.1111/j.1439-0523.2007.01376.x
Patrick, H., and Alfonso, C.-M. (2013). “New and renewed breeding methodology,” in Advance in Barley Science, eds G. Zhang, C. Li, and X. Liu (Dordrecht: Springer), 349–357. doi: 10.1007/978-94-007-4682-4_29
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and Eigen analysis. PLOS Genet. 2:e190. doi: 10.1371/journal.pgen.0020190
Peerbolte, R. P., and Peleman, J. (1996). “The CARTISOL sunflower RFLP map (146 loci) extended with 291 AFLP markers,” in Proceedings of the 18th Sunflower Research Forum, Jan 1996, Fargo, ND, USA (Bismarck, ND: National Sunflower Association), 174–178.
Pegadaraju, V., Nipper, R., Hulke, B. S., Qi, L. L., and Schultz, Q. (2013). De novo sequencing of the sunflower genome for SNP discovery using the RAD (Restriction site Associated DNA) approach. BMC Genomics 14:556. doi: 10.1186/1471-2164-14-556
Peluffo, L. (2010). Characterization of the Defense Mechanisms against Sclerotinia sclerotiorum, Causal Agent of Sunflower Head Rot, through Metabolic and Transcriptional Profiles Analyses. Ph.D. thesis, University of Buenos Aires, Buenos Aires.
Peluffo, L., Lia, V., Troglia, C., Maringolo, C., Norma, P., Escande, A., et al. (2010). Metabolic profiles of sunflower genotypes with contrasting response to Sclerotinia sclerotiorum infection. Phytochemistry 71, 70–80. doi: 10.1016/phytochem.2009.09.018
Pérez Vich, B., Aguirre, M. R., Guta, B., and Fernandez-Martinez, J. M. (2017). Genetic diversity of a germplasm collection of confectionery sunflower landraces from Spain. Front. Plant Sci.
Pérez-Vich, B., Fernández-Martıínez, J. M., Grondona, M., Knapp, S. J., and Berry, S. T. (2002). Stearoyl-ACP and oleoyl-PC desaturase genes cosegregate with quantitative trait loci underlying high stearic and high oleic acid mutant phenotypes in sunflower. Theor. Appl. Genet. 104, 338–349. doi: 10.1007/s001220100712
Pérez-Vich, B., Velasco, L., and Fernandez-Martinez, J. M. (2004). “QTL mapping of resistance to races E and F of broomrape (Orobanche cumana Wallr.) in sunflower,” in Proceedings of the Parasitic Plant Management in Sustainable Agriculture Meeting on Breeding for Orobanche Resistance in Sunflower, Bucharest, 6.
Picheny, V., Trépos, R., and Casadebaig, P. (2017). Optimization of black-box models with uncertain climatic inputs – Application to sunflower ideotype design. PLOS ONE 12:e0176815. doi: 10.1371/journal.pone.0176815
Poormohammad Kiani, S., Grieu, P., Maury, P., Hewezi, T., Gentzbittel, L., and Sarrafi, A. (2007a). Genetic variability for physiological traits under drought conditions and differential expression of water stress-associated genes in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 114, 193–207. doi: 10.1007/s00122-006-0419-7
Poormohammad Kiani, S., Maury, P., Nouri, L., Ykhef, N., Grieu, P., and Sarrafi, A. (2009). QTL analysis of yield-related traits in sunflower under different water treatments. Plant Breed. 128, 363–373. doi: 10.1111/j.1439-0523.2009.01628.x
Poormohammad Kiani, S., Talia, P., Maury, P., Grieu, R., Heinz, R., Perrault, A., et al. (2007b). Genetic analysis of plant water status and osmotic adjustment in recombinant inbred lines of sunflower under two water treatments. Plant Sci. 172, 773–787. doi: 10.1016/jplantsci.2006.12.007
Port, A., Nechifor, V., Midoni, A., and Duca, M. (2013). Usefulness of the diagnostic markers for the restorer gene Rf1 in inheritance studies at sunflower. Anal. Sci. 14, 11–17. doi: 10.1007/s11032-016-0527-2
Premnath, A., Narayana, M., Ramakrsihnan, C., Kuppusamy, S., and Chockalingam, V. (2016). Mapping quantitative trait loci controlling oil content, oleic acid and linoleic acid content in sunflower (Helianthus annuus L.). Mol. Breed. 36:106. doi: 10.1007/s11032-016-0527-2
Printz, B., Sergeant, K., Guignard, C., and Renaut, J. (2013). Physiological proteome study of sunflowers exposed to a polymetallic constraint. Proteomics 13, 1993–2015. doi: 10.1002/pmic.201200400
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Putt, E. D. (1978). “History and present world status,” in Sunflower Science and Technology, ed. J. F. Carter (Madison, WI: ASA), 1–25.
Qi, L., Long, Y., Talukder, Z. I., Seiler, G., Block, C. C., and Gulya, T. J. (2016). Genotyping-by-Sequencing uncovers the introgression alien segments associated with Sclerotinia basal stalk rot resistance from species – I. Helianthus argophyllus and H. petiolaris. Front. Genet. 7:219. doi: 10.3389/fgene.2016.00219
Qi, L. L., Foley, M. E., Cai, X. W., and Gulya, T. J. (2016). Genetics and mapping of a novel downy mildew resistance gene, Pl18, introgressed from wild Helianthus argophyllus into cultivated sunflower (Helianthus annuus L.). Theor. Appl. Genet. 129, 741–752. doi: 10.1007/s00122-015-2662-2
Qi, L. L., Gulya, T. J., Hulke, B. S., and Vick, B. A. (2012a). Chromosome location, DNA markers and rust resistance of the sunflower gene R5. Mol. Breed. 30, 745–756. doi: 10.1007/s11032-011-9659-6
Qi, L. L., Gulya, T. J., Seiler, G. J., Hulke, B. S., and Vick, B. A. (2011a). Identification of resistance to new virulent races of rust in sunflowers and validation of DNA markers in the gene pool. Phytopathology 101, 241–249. doi: 10.1094/PHYTO-06-10-0162
Qi, L. L., Hulke, B. S., Vick, B. A., and Gulya, T. J. (2011b). Molecular mapping of the rust resistance gene R4 to a large NBS-LRR cluster on linkage group 13 of sunflower. Theor. Appl. Genet. 123, 351–358. doi: 10.1007/s00122-011-1588-6
Qi, L. L., Long, Y. M., Jan, C. C., Ma, G. J., and Gulya, T. J. (2015a). Pl17 is a novel gene independent of known downy mildew resistance genes in the cultivated sunflower (Helianthus annuus L.). Theor. Appl. Genet. 128, 757–767. doi: 10.1007/s00122-015-2470-8
Qi, L. L., Long, Y. M., Ma, G. J., and Markell, S. G. (2015b). Map saturation and SNP marker development for the rust resistance genes (R4, R5, R13a, and R13b) in sunflower (Helianthus annuus L.). Mol. Breed. 35:196. doi: 10.1007/s11032-015-0380-8
Qi, L. L., Ma, G. J., Long, Y. M., Hulke, B. S., Gong, L., and Markell, S. G. (2015c). Relocation of a rust resistance gene R2 and its marker-assisted gene pyramiding in confection sunflower (Helianthus annuus L.). Theor. Appl. Genet. 128, 477–488. doi: 10.1007/s00122-014-2446-0
Qi, L. L., Seiler, G. J., Vick, B. A., and Gulya, T. J. (2012b). Genetics and mapping of the R11 gene conferring resistance to recently emerged rust races, tightly linked to male fertility restoration, in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 125, 921–932. doi: 10.1007/s00122-012-1883-x
Qi, L. L., Talukder, Z. I., Hulke, B. S., and Foley, M. E. (2017). Development and dissection of diagnostic SNP markers for the downy mildew resistance genes PlArg and Pl8 and marker-assisted gene pyramiding in sunflower (Helianthus annuus L.). Mol. Genet. Genomics 292, 1–13. doi: 10.1007/s00438-017-1290-8
Quillet, M. C., Madjidian, N., Griveau, Y., Serieys, H., Tersac, M., Lorieux, M., et al. (1995). Mapping genetic factors controlling pollen viability in an interspecific cross in Helianthus section Helianthus. Theor. Appl. Genet. 91, 1195–1202. doi: 10.1007/BF00220929
Radwan, O., Bouzidi, M. F., Nicolas, P., and Mouzeyar, S. (2004). Development of PCR markers for the Pl5/Pl8 locus for resistance to Plasmopara halstedii in sunflower, Helianthus annuus L. from complete CC-NBS-LRR sequences. Theor. Appl. Genet. 109, 176–185. doi: 10.1007/s00122-004-1613-0
Rauf, S., Jamil, N., Ali Tariq, S., Khan, M., and Kausar, M. (2017). Progress in modification of sunflower oil to expand its industrial value. J. Sci. Food Agric. 97, 1997–2006. doi: 10.1002/jsfa.8214
Regitano Neto, A., de Oliveira Miguel, A. M. R., Mourad, A. L., Henriques, E. A., and Alves, R. M. V. (2016). Environmental effect on sunflower oil quality. Crop Breed. Appl. Biotechnol. 16, 197–204. doi: 10.1590/1984-70332016v16n3a30
Reif, J. C., Zhao, Y., Würschum, T., Gowda, M., and Hahn, V. (2013). Genomic prediction of sunflower hybrid performance. Plant Breed. 132, 107–114. doi: 10.1111/pbr.12007
Rieseberg, L. H., Choi, H., Chan, R., and Spore, C. (1993). Genomic map of a diploid hybrid species. Heredity 70, 285–285. doi: 10.1038/hdy.1993.41
Rodríguez-Ojeda, M. I., Fernández-Escobar, J., and Alonso, L. C. (2001). “Sunflower inbred line (KI-374) carrying two recessive genes for resistance against a highly virulent Spanish population of Orobanche cernua Loelf/ O. cumana Wallr. race F,” in Proceedings of the 7th International Parasitic Weed Symposium, eds L. J. Muselman, C. Parker, and J. A. C. Verkleij (Nantes: Université de Nantes), 208–211.
Roeckel-Drevet, P., Gagne, G., Mouzeyar, S., Gentzbittel, L., Philippon, J., Nicolas, P., et al. (1996). Colocation of downy mildew (Plasmopara halstedii) resistance genes in sunflower (Helianthus annuus L.). Euphytica 91, 225–228. doi: 10.1007/BF00021074
Rowe, H. C., and Rieseberg, L. H. (2013). Genome-scale transcriptional analyses of first-generation interspecific sunflower hybrids reveals broad regulatory compatibility. BMC Genomics 14:342. doi: 10.1186/1471-2164-14-342
Sabetta, W., Alba, V., Blanco, A., and Montemurro, C. (2011). sunTILL: a TILLING resource for gene function analysis in sunflower. Plant Methods 7:20. doi: 10.1186/1746-4811-7-20
Sala, C. A., and Bulos, M. (2012). Inheritance and molecular characterization of broad range tolerance to herbicides targeting acetohydroxyacid synthase in sunflower. Theor. Appl. Genet. 124, 355–364. doi: 10.1007/s00122-011-1710-9
Sala, C. A., Bulos, M., Alteri, E., and Ramos, M. L. (2012). Genetics and breeding of herbicide tolerance in sunflower. Helia 35, 57–70. doi: 10.2298/HEL1257057S
Sala, C. A., Bulos, M., and Echarte, A. M. (2008a). Genetic analysis of an induced mutation conferring imidazolinone resistance in sunflower. Crop Sci. 48, 1817–1822. doi: 10.2135/cropsci2007.11.0625
Sala, C. A., Bulos, M., Echarte, A. M., Whitt, S. R., and Ascenzi, R. (2008b). Molecular and biochemical characterization of an induced mutation conferring imidazolinone resistance in sunflower. Theor. Appl. Genet. 108, 105–112. doi: 10.1007/s00122-008-0880-6
Sankaran, S., Khot, L. R., Espinoza, C. Z., Jarolmasjed, S., Sathuvalli, V. R., Vandemark, G. J., et al. (2015). Low-altitude, high-resolution aerial imaging systems for row and field crop phenotyping: a review. Eur. J. Agron. 70, 112–123. doi: 10.1016/j.eja.2015.07.004
Sarazin, V., Duclercq, J., Guillot, X., Sangwan, B., and Sangwan, R. S. (2017). Water-stressed sunflower transcriptome analysis revealed important molecular markers involved in drought stress response and tolerance. Environ. Exp. Bot. 142, 45–63. doi: 10.1016/jenvexpbot.2017.08.005
Schnabel, U., Engelmann, U., and Horn, R. (2008). Development of markers for the use of the PEF1-cytoplasm in sunflower hybrid breeding. Plant Breed. 127, 587–591. doi: 10.1111/j.1439-0532.2008.01516
Schuppert, G. F., Tang, S., Slabaugh, M. B., and Knapp, S. J. (2006). The sunflower high-oleic mutant Ol carries variable tandem repeats of FAD2-1, a seed-specific oleoyl-phosphatidyl choline desaturase. Mol. Breed. 17, 241–256. doi: 10.1007/s11032-005-5680-y
Seiler, G. J., Qi, L. L., and Marek, L. F. (2017). Utilization of sunflower crop wild relatives for cultivated sunflower improvement. Crop Sci. 57, 1083–1101. doi: 10.2135/cropsci2016.10.0856
Serieys, H. (2005). “Identification, study, utilization in breeding programs of new CMS sources in the FAO Subnetwork,” in Proceedings of the Sunflower Subnetwork Progress Report, Rome, 47–53.
Singchai, A., Muangsan, N., and Machikowa, T. (2013). Evaluation of SSR markers associated with high oleic acid in sunflower. Int. J. Biol. Food Vet. Agric. Eng. 7, 631–634.
Skoric, D. (2012). “Sunflower breeding,” in Sunflower Genetics and Breeding, ed. Z. Kovacevic (Novi Sad: Serbian Academy of Sciences), 165–354.
Skoric, D., and Pacureanu, M. (2010). “Sunflower breeding for resistance to broomrape (Orobanche cumana Wallr.),” in Proceedings of the International Symposium “Breeding of Sunflower on Resistance to Diseases”, Krasnodar, 19–29.
Slabaugh, M. B., Yu, J.-K., Tang, S., Heesacker, A., Hu, X., Lu, G., et al. (2003). Haplotyping and mapping a large cluster of downy mildew resistance gene candidates in sunflower using multilocus intron fragment length polymorphisms. Plant Biotechnol. J. 1, 167–185. doi: 10.1046/j.1467-7652.2003.00016.x
Soldatov, K. I. (1976). “Chemical mutagenesis in sunflower breeding,” in Proceedings of the 7th International Sunflower Conference, Krasnodar, 352–357.
Talukder, Z. I., Gong, L., Hulke, B. S., Pegadaraju, V., Song, Q., Schultz, Q., et al. (2014a). A high-density SNP map of sunflower derived from RAD-sequencing facilitating fine-mapping of the rust resistance gene R12. PLOS ONE 9:e98628. doi: 10.1371/journal.pone.0098628
Talukder, Z. I., Hulke, B. S., Qi, L., Scheffler, B. E., Pegadaraju, V., McPhee, K., et al. (2014b). Candidate gene association of Sclerotinia stalk rot resistance in sunflower (Helianthus annuus L.) uncovers the importance of COI1 homologs. Theor. Appl. Genet. 127, 193–209. doi: 10.1007/s00122-013-2210
Talukder, Z. I., Seiler, G. J., Song, Q., Ma, G., and Qi, L. (2016). SNP discovery and QTL mapping of Sclerotinia basal stalk rot resistance in sunflower using genotyping-by sequencing. Plant Genome 9, 1–16. doi: 10.3835/plantgenome2016.03.0035
Tang, S., Heesacker, A., Kishore, V. K., Fernandez, A., Sadik, E. S., Cole, G., et al. (2003a). Genetic mapping of the Or 5 gene for resistance to race E in sunflower. Crop Sci. 43, 1021–1028. doi: 10.2135/cropsci2003.1021
Tang, S., Kishore, V. K., and Knapp, S. J. (2003b). PCR-multiplexes for a genome-wide framework of simple sequence repeat marker loci in cultivated sunflower. Theor. Appl. Genet. 107, 6–19. doi: 10.1007/s00122-003-1233-0
Tang, S., Leon, A., Bridges, W. C., and Knapp, S. J. (2006). Quantitative trait loci for genetically correlated seed traits are tightly linked to branching and pericarp pigment loci in sunflower. Crop Sci. 46, 721–734. doi: 10.2135/cropsci2005.0006-7
Tang, S., Yu, J. K., Slabaugh, M. B., Shintani, D. K., and Knapp, S. J. (2002). Simple sequence repeat map of the sunflower genome. Theor. Appl. Genet. 105, 1124–1136. doi: 10.1007/s00122-002-0989-y
Urie, A. L. (1984). “Inheritance of very high oleic acid content in sunflower,” in Proceedings of the 6th Sunflower Research Workshop Sunflower Association, Bismarck, ND, 8–9.
Van Raden, P. M., Van Tassell, C. P., Wiggans, G. R., Sonstenard, T. S., Schnabel, R. D., Taylor, J. F., et al. (2009). Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92, 16–24. doi: 10.3168/jds.2008-1514
Vannozzi, G. P. (2006). The perspectives of use of high oleic sunflower for oleochemistry and energy raws. Helia 29, 1–24. doi: 10.2298/hel0644001v
Vear, F., Gentzbittel, L., Philippon, J., Mouzeyar, S., Mestries, E., Roeckel-Drevet, P., et al. (1997). The genetics of resistance to five races of downy mildew (Plasmopara halstedii) in sunflower (Helianthus annuus L.). Theor. Appl. Genet. 95, 584–589. doi: 10.1007/s001220050599
Vega, T., Breccia, G., Gil, M., and Nestares, G. (2012). Acetohydroxyacid synthase (AHAS) in vivo assay for screening imidazolinone-resistance in sunflower (Helianthus annuus L.). Plant Physiol. Biochem. 61, 103–107. doi: 10.1016/j.plaphy.2012.09.013
Velasco, L., Fernandez-Cuesta, A., and Fernandez-Martinez, J. M. (2014). Variability of seed quality traits in a collection of Spanish landraces of confectionary sunflower. Crop Pasture Sci. 65, 242–249. doi: 10.1071/CP13390
Velasco, L., Pérez-Vich, B., Yassein, A. A., Jan, C. C., and Fernandez- Martınez, J. M. (2012). Inheritance of resistance to sunflower broomrape (Orobanche cumana Wallr.) in an interspecific cross between Helianthus annuus and Helianthus debilis subsp. tardiflorus. Plant Breed. 131, 220–221. doi: 10.1111/j.1439-0523.2011.01915.x
Vera-Ruiz, E. M., Velasco, L., Leon, A. J., Fernández-Martínez, J. M., and Pérez-Vich, B. (2006). Genetic mapping of the Tph1 gene controlling beta-tocopherol accumulation in sunflower seeds. Mol. Breed. 17, 291–296. doi: 10.1007/s11032-005-5678-5
Vincourt, P., As-sadi, F., Bordat, A., Langlade, N. B., Gouzy, J., Pouilly, N., et al. (2012). Consensus mapping of major resistance genes and independent QTL for quantitative resistance to sunflower downy mildew. Theor. Appl. Genet. 125, 909–920. doi: 10.1007/s00122-012-1882-y
Vranceanu, A. V., Tudor, V. A., Stoenescu, F. M., and Pirvu, N. (1980). “Virulence groups of Orobanche cumana Wallr. different hosts and resistance sources and genes in sunflower,” in Proceedings of the 9th International Sunflower Conference, Torremolinos, 74–82.
White, A. D., Graham, M. A., and Owen, M. D. K. (2003). Isolation of acetolactate synthase homologs in common sunflower. Weed Sci. 51, 845–853. doi: 10.1614/P2002-136
White, A. D., Owen, M. D., Hartzler, R. G., and Cardina, J. (2002). Common sunflower resistance to acetolactate-inhibiting herbicides. Weed Sci. 50, 432–437. doi: 10.1614/0043-1745 (2002)050[0432:CSRTAS]2.0.CO;2
Wieckhorst, S., Bachlava, E., Dußle, C. M., Tang, S., Gao, W., Saski, C., et al. (2010). Fine mapping of the sunflower resistance locus Plarg introduced from the wild species Helianthus argophyllus. Theor. Appl. Genet. 121, 1633–1644. doi: 10.1007/s00122-010-1416-4
Würschum, T., Reif, J. C., Kraft, G., Janssen, G., and Zha, Y. (2013). Genomic selection in sugar beet breeding populations. BMC Genet. 14:85. doi: 10.1186/1471-2156-14-85
Yang, C., Xu, L., Zhang, N., Islam, F., Song, W., Hu, L., et al. (2017). iTRAQ-based proteomics of sunflower cultivars differing in resistance to parasitic weed Orobanche cumana. Proteomics 17, 13–14. doi: 10.1002/pmic.201700009
Yu, B., and Shang, S. (2017). Multi-year mapping of maize and sunflower in Hetao irrigation district of China with high spatial and temporal resolution vegetation index series. Remote Sens. 9:855. doi: 10.3390/rs9080855
Yu, J. K., Tang, S., Slabaugh, M. B., Heesacker, A., Cole, G., Herring, M., et al. (2003). Towards a saturated molecular genetic linkage map for sunflower. Crop Sci. 43, 367–387. doi: 10.2135/cropsci2003.3670
Yue, B., Vick, B. A., Cai, X., and Hu, J. (2010). Genetic mapping for the Rf1 (fertility restoration) gene in sunflower (Helianthus annuus L.) by SSR and TRAP markers. Plant Breed. 129, 24–28. doi: 10.1111/j.1439-0523.2009.01661.x
Zambelli, A., Leon, A., and Garcés, R. (2015). “Mutagenesis in sunflower,” in Sunflower Oilseed, Chemistry, Production, Processing and Utilization, eds E. Martínez Force, N. T. Dunford, and J. J. Salsa (Urbana, IL: AOCS), 27–52. doi: 10.1016/B978-1-893997-94-3.50008-8
Zhang, M., Liu, Z., and Jan, C. C. (2016). Molecular mapping of a rust resistance gene R14 in cultivated sunflower line PH 3. Mol. Breed. 36:32. doi: 10.1007/s11032-016-0456-0
Zhang, Z. W., Ma, G. J., Zhao, J., Markell, S. G., and Qi, L. L. (2017). Discovery and introgression of the wild sunflower-derived novel downy mildew resistance gene Pl19 in confection sunflower (Helianthus annuus L.). Theor. Appl. Genet. 130, 29–39. doi: 10.1007/s00122-016-2786-z
Zhao, J., Wang, J., An, L., Doerge, R. W., Chen, Z. J., Grau, C. R., et al. (2007). Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 227, 13–24. doi: 10.1007/s00425-007-0586-z
Keywords: association panel, genome-wide association studies, genomic estimated breeding value, genomic selection, genome sequence, marker-assisted selection, sunflower, traits
Citation: Dimitrijevic A and Horn R (2018) Sunflower Hybrid Breeding: From Markers to Genomic Selection. Front. Plant Sci. 8:2238. doi: 10.3389/fpls.2017.02238
Received: 15 October 2017; Accepted: 20 December 2017;
Published: 17 January 2018.
Edited by:
Leire Molinero-Ruiz, Instituto de Agricultura Sostenible (CSIC), SpainReviewed by:
Felicity Vear, INRA – Auvergne Rhône-Alpes Centre, FranceCopyright © 2018 Dimitrijevic and Horn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renate Horn, cmVuYXRlLmhvcm5AdW5pLXJvc3RvY2suZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.