 Stefan Burén
Stefan Burén Xi Jiang
Xi Jiang Gema López-Torrejón
Gema López-Torrejón Carlos Echavarri-Erasun
Carlos Echavarri-Erasun Luis M. Rubio
Luis M. Rubio- Centro de Biotecnología y Genómica de Plantas (CBGP), Universidad Politécnica de Madrid (UPM), Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Madrid, Spain
Active NifB is a milestone in the process of engineering nitrogen fixing plants. NifB is an extremely O2-sensitive S-adenosyl methionine (SAM)–radical enzyme that provides the key metal cluster intermediate (NifB-co) for the biosyntheses of the active-site cofactors of all three types of nitrogenases. NifB and NifB-co are unique to diazotrophic organisms. In this work, we have expressed synthetic codon-optimized versions of NifB from the γ-proteobacterium Azotobacter vinelandii and the thermophilic methanogen Methanocaldococcus infernus in Saccharomyces cerevisiae and in Nicotiana benthamiana. NifB proteins were targeted to the mitochondria, where O2 consumption is high and bacterial-like [Fe-S] cluster assembly operates. In yeast, NifB proteins were co-expressed with NifU, NifS, and FdxN proteins that are involved in NifB [Fe–S] cluster assembly and activity. The synthetic version of thermophilic NifB accumulated in soluble form within the yeast cell, while the A. vinelandii version appeared to form aggregates. Similarly, NifB from M. infernus was expressed at higher levels in leaves of Nicotiana benthamiana and accumulated as a soluble protein while A. vinelandii NifB was mainly associated with the non-soluble cell fraction. Soluble M. infernus NifB was purified from aerobically grown yeast and biochemically characterized. The purified protein was functional in the in vitro FeMo-co synthesis assay. This work presents the first active NifB protein purified from a eukaryotic cell, and highlights the importance of screening nif genes from different organisms in order to sort the best candidates to assemble a functional plant nitrogenase.
Introduction
Agricultural systems in developed countries are largely based on cereal crops, which provide most of the calories and proteins in the human diet (Borlaug, 2002). Nitrogen and water availability are the most important factors limiting cereal crop productivity. Over the last 100 years, cereal crop yields have been increased by addition of chemically synthesized nitrogen fertilizers (Smil, 2000; Galloway et al., 2008). However, the extensive use of these commercial nitrogen fertilizers in developed countries poses enormous and pressing environmental threats (Vitousek et al., 1997). On the other hand, the cost of chemical fertilizers is prohibitive for poor farmers, and they are scarcely used in most of Africa with consequence of poverty and hunger derived from extremely low crop yields (Sanchez and Swaminathan, 2005). During the last years, the scientific community has paid considerable attention to this problem and acknowledged the need of disruptive technological changes. One way to tackle this problem could be the generation of so-called nitrogen-fixing plants, aimed to solve the nitrogen problem (Stokstad, 2016; Vicente and Dean, 2017).
Nitrogen fixation is the conversion of inert atmospheric N2 into NH3, a biologically active form of nitrogen. Only a few bacteria are naturally able to fix nitrogen through the activity of an enzyme called nitrogenase; collectively known as diazotrophs (nitrogen eaters). In this regard, transferring bacterial nitrogen fixation (nif) genes into the plant genome could result in plants able to fix nitrogen, and therefore in crops less dependent of external nitrogen fertilization. This direct approach eliminates the need to generate or optimize interactions of cereals with specific symbiotic or associative nitrogen fixing bacteria (Oldroyd and Dixon, 2014). However, two main barriers are believed to impair the direct nif gene transfer approach (Curatti and Rubio, 2014): the known sensitivity of nitrogenase to O2 and the genetic complexity of nitrogenase biosynthesis. Two decades of genetic and biochemical analyses culminated with the unambiguous identification of the essential proteins required for nitrogenase cofactor biosynthesis (Curatti et al., 2007). On the contrary, overcoming the O2 sensitivity barrier in plants remains largely unexplored.
Nitrogenases have two O2-sensitive protein components: a dinitrogenase that catalyzes the nitrogen fixation reaction and a dinitrogenase reductase that serves as obligate electron donor to dinitrogenase (Bulen and LeComte, 1966). In the case of the widespread molybdenum nitrogenase these components are called Fe protein and MoFe protein. The Fe protein is a homodimer of the nifH gene product that contains a [4Fe–4S] cluster at the subunit interface. The MoFe protein is a heterotetramer of the nifD and nifK gene products that contains two complex iron–sulfur (Fe–S) clusters called iron-molybdenum cofactor (FeMo-co) and P-cluster. The type of [4Fe–4S] cluster found in NifH is ubiquitous in nature. In fact, plants carry [Fe–S] cluster assembly machineries in mitochondria, chloroplasts, and cytosol, which are all capable of synthesizing [4Fe–4S] clusters (Balk and Pilon, 2011). However, the P-cluster and FeMo-co are unique to diazotrophs. Their uniqueness implies that specialized cellular biosynthetic pathways, involving multiple nif gene products, are required for cofactor synthesis and NifDK maturation (Rubio and Ludden, 2008).
Successful expression and maturation of the prokaryotic nitrogenase protein in a eukaryotic host, in order to develop N2 fixing cereal crops, could revolutionize agricultural systems worldwide. For this to succeed, a deeper understanding of the processes involved in the formation of active nitrogenase in a eukaryotic cell is required. In this regard, expression of nif genes in Saccharomyces cerevisiae have shown that: (1) active NifH can be achieved upon mitochondrial targeting (Lopez-Torrejon et al., 2016), providing a proof of concept that O2-sensitive Nif proteins can be assembled in an eukaryotic cell organelle, and (2) that expression and mitochondria targeting of nine Nif proteins (NifUSHMDKBEN) resulted in proper mitochondria targeting, processing and NifDK tetramer formation (Burén et al., 2017), an essential step of nitrogenase assembly. However, to obtain similar results in a plant cell background is likely to be more challenging, as the O2 generated during photosynthesis could create even a harsher environment for nitrogenase proteins, especially in the chloroplast. This was recently suggested from work by Ivleva et al. (2016), where Fe protein activity from transplastomic Nicotiana tabacum plants only could be detected at very low levels in plants previously incubated at sub-ambient O2 (Ivleva et al., 2016). Importantly, a recent study showed that 16 mitochondria targeted Nif proteins from Klebsiella pneumoniae (among them NifB) could be successfully expressed in leaves of Nicotiana benthamiana (Allen et al., 2017). Although no protein activities were reported, this work showed that most Nif proteins were well expressed and accumulated at their estimated sizes within the plant tissue, with the exception of NifD that appeared to be processed to a polypeptide of smaller size, as has also been observed in yeast (Burén et al., 2017).
A main hurdle to overcome in order to generate functional nitrogenase proteins is obtaining active NifB. NifB is an extremely O2-sensitive S-adenosyl methionine (SAM)–radical enzyme (Curatti et al., 2006), that provides the key intermediate metal cluster (called NifB-co) in the biosynthesis of FeMo-co (Shah et al., 1994; Guo et al., 2016). As NifB-co also serves as precursor for FeV-co in the vanadium nitrogenase and for FeFe-co in the iron-only nitrogenase, NifB is required for all biological nitrogen fixation activity in nature (Bishop and Joerger, 1990; Curatti et al., 2007; Dos Santos et al., 2012). Because of its O2 sensitivity and instability, and since it is unlikely that NifB or NifB-co can be replaced by components of plant origin, plant cell NifB-co accumulation is likely to be one of the main barriers in the generation of an active plant nitrogenase (Curatti and Rubio, 2014; Vicente and Dean, 2017).
In this work, two naturally occurring NifB proteins, from the model-diazotroph Azotobacter vinelandii and from the thermophilic methanogen Methanocaldococcus infernus (see accompanying paper by Arragain et al., submitted manuscript), were expressed in S. cerevisiae and targeted to mitochondria. Mitochondria was chosen due to the high rate of O2 consumption, and the plentiful ATP and reducing power generated by respiration (Curatti and Rubio, 2014), in addition to the bacterial-like [Fe–S] cluster assembly machinery available (Lill and Muhlenhoff, 2008). NifB proteins were co-expressed with NifU, NifS, and FdxN proteins, involved in NifB [Fe–S] cluster formation and activity (Yuvaniyama et al., 2000; Johnson et al., 2005; Zhao et al., 2007; Jiménez-Vicente et al., 2014). Surprisingly, only NifB from the thermophile was found to accumulate in a soluble form, while NifB from A. vinelandii appeared to form aggregates. The soluble M. infernus NifB was purified and proven functional in the in vitro FeMo-co synthesis assay. A. vinelandii and M. infernus NifB were also targeted to the mitochondria in leaf cells of N. benthamiana (tobacco). As in yeast, the synthetic version of NifB from M. infernus was better expressed and accumulated as a soluble protein while the A. vinelandii NifB was mainly associated with the non-soluble cell fraction. These results underline the importance of screening for functionality each one of the Nif proteins required to mature nitrogenase.
Results
Generation of Yeast Platform Strains for NifB Expression
Synthetic versions of A. vinelandii nifB (nifBAv) and M. infernus nifB (nifBMi) were codon-optimized for S. cerevisiae and cloned into expression vectors under the control of the galactose inducible promoters (Supplementary Figures S1, S2). As NifU, NifS, and FdxN participate in NifB maturation and activity (Zhao et al., 2007; Jiménez-Vicente et al., 2014), synthetic versions of the A. vinelandii nifU, nifS, and fdxN genes, codon-optimized for S. cerevisiae, were additionally cloned into expression vectors under the control of the galactose inducible promoters. To ensure mitochondria targeting of the expressed proteins, SU9 leader sequences, previously shown to deliver yeast expressed Nif proteins to the mitochondria (Burén et al., 2017) were added in-frame and upstream of each gene. To facilitate NifB purification by affinity chromatography, the coding sequence for 10 histidines were added 3′ of each nifB gene. Expression vectors were co-introduced into S. cerevisiae strain W303-1a, generating yeast strains SB09Y and SB10Y for the expression of mitochondria targeted NifU, NifS, and FdxN, together with NifBAv-His10 or NifBMi-His10 (hereafter called yNifBAv and yNifBMi), respectively (Table 1).
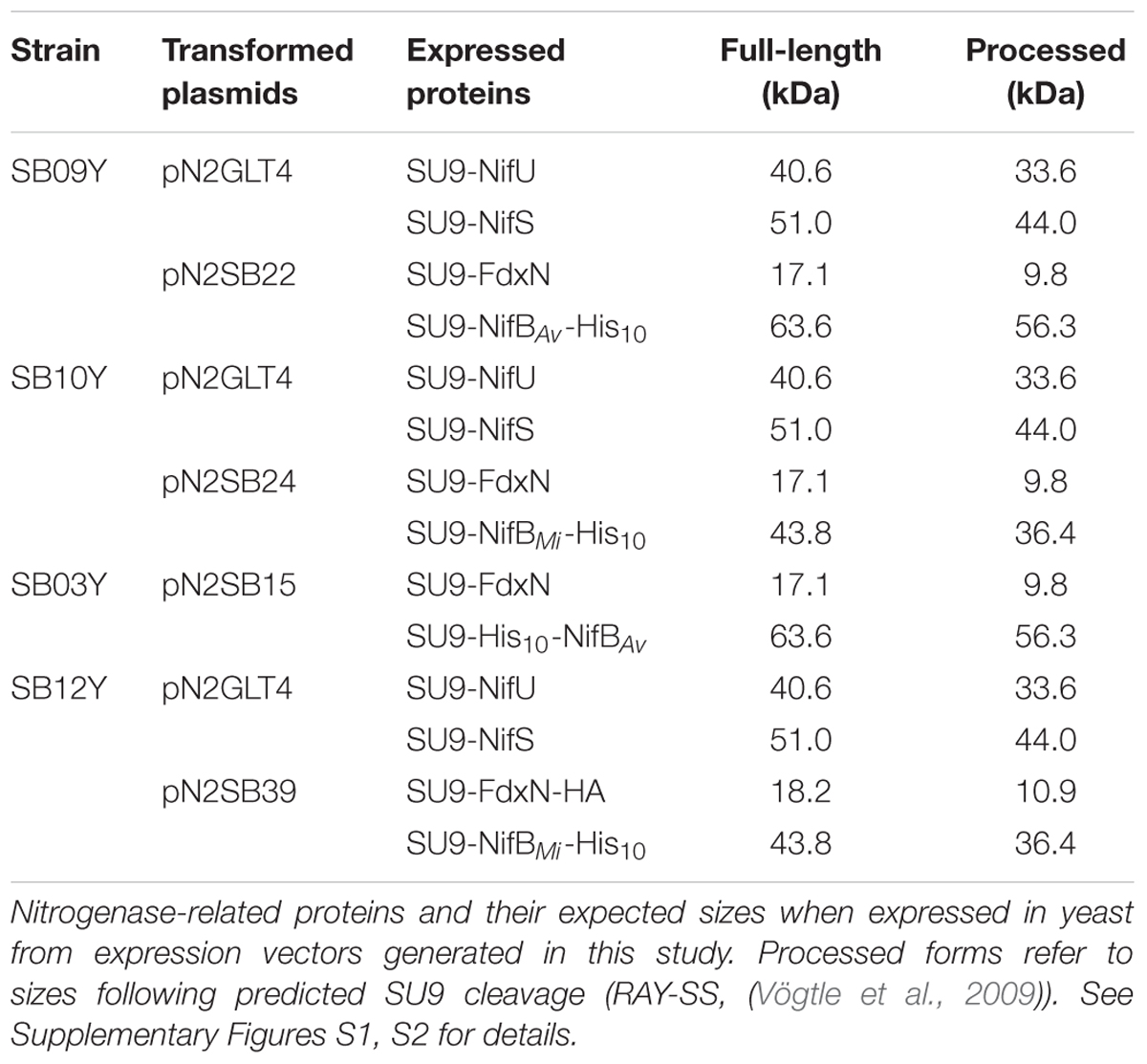
TABLE 1. Yeast expression plasmids and yeast strains used in this work.
NifB Expression, Mitochondria Targeting, and Solubility
Western blot analysis of yeast cell-free extracts using antibodies specifically recognizing NifU, NifS, and histidine-tag (for yNifBAv or yNifBMi) confirmed expression of all these proteins in SB09Y and SB10Y strains grown aerobically with galactose as inducer. Protein migrations in SDS-PAGE were consistent with efficient mitochondria leader sequence processing (Table 1 and Figure 1A). Detection of FdxN in SB09Y and SB10Y was difficult, presumably due to the small size of FdxN (10 kDa) and/or to weak binding of FdxN antibodies generated for this study. To confirm that FdxN was successfully expressed from the GAL10 promoter, presence of fdxN transcripts were verified in SB09Y and SB10Y strains (Supplementary Figure S3). In addition, an epitope-tagged version of the protein where a C-terminal HA-tag was added to the SU9-FdxN construct was generated and expressed in strain SB12Y (Figure 1B).
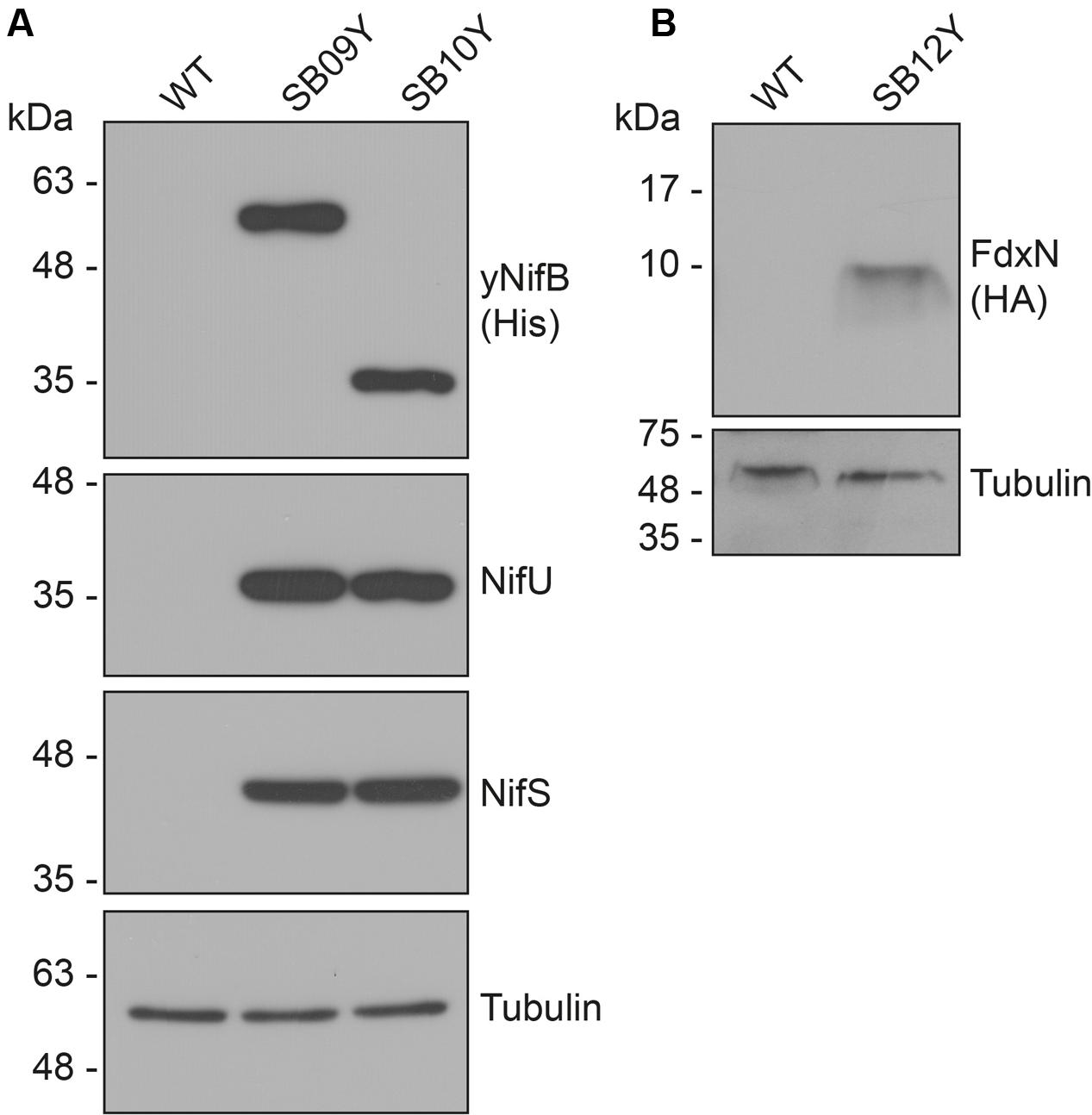
FIGURE 1. Expression of NifB, NifU, NifS and FdxN proteins in S. cerevisiae. (A) Western blot analysis of yNifBAv and yNifBMi, as well as NifU and NifS, in total protein extracts from strains SB09Y and SB10Y. (B) Western blot analysis of C-terminally HA-tagged FdxN in total protein extract from SB12Y strain. Extracts in (A) and (B) were prepared from aerobically grown cells following galactose induction, and proteins in the extract separated by SDS-PAGE before transferred to membranes. Antibodies recognizing NifUAv, NifSAv, His epitope, HA epitope, and tubulin were used. Tubulin immunoblot signal intensity is used as loading control. See Table 1 for details about recombinant yeast strains.
Further analysis of the soluble fractions prepared from yeast cells lysed in absence of detergents indicated that most yNifBMi and nearly all yNifBAv were of poor solubility (Figure 2). This suggested that the proteins were either forming insoluble aggregates upon strong expression or interacting with membranes. A similar behavior was observed for NifBAv and NifBKo (Klebsiella oxytoca NifB) overexpressed in Escherichia coli (data not shown). Exchanging the C-terminal His-tag for an N-terminal variant, and addition of detergents during lysis (see Materials and Methods for details), did not improve solubility (Supplementary Figure S4). As NifB from the thermophile M. infernus previously showed heat-resistant properties (Wilcoxen et al., 2016), several distinct extraction conditions were tested (including different temperatures) to screen yNifBMi solubility and to find a protocol for extraction and enrichment of yNifBMi. While increased concentration of glycerol did not improve solubility, the pH of the extraction buffer was important (Supplementary Figure S5). In addition, exposing the total yeast lysate to elevated temperatures before centrifugation not only reduced the amount of total yeast proteins remaining in solution, but also increased the levels of yNifBMi in the soluble fraction of the extract. Unfortunately, no similar improvement could be obtained for yNifBAv (Figures 3A,B and Supplementary Figure S6) impairing yNifBAv purifications. Further optimization confirmed that maximum solubility yNifBMi was obtained at pH 8 upon treatment at 60–65°C (Figure 3C). Therefore 65°C was chosen for the following yNifBMi extractions in order to minimize the complexity of the yeast cell-free extracts used for affinity chromatography.
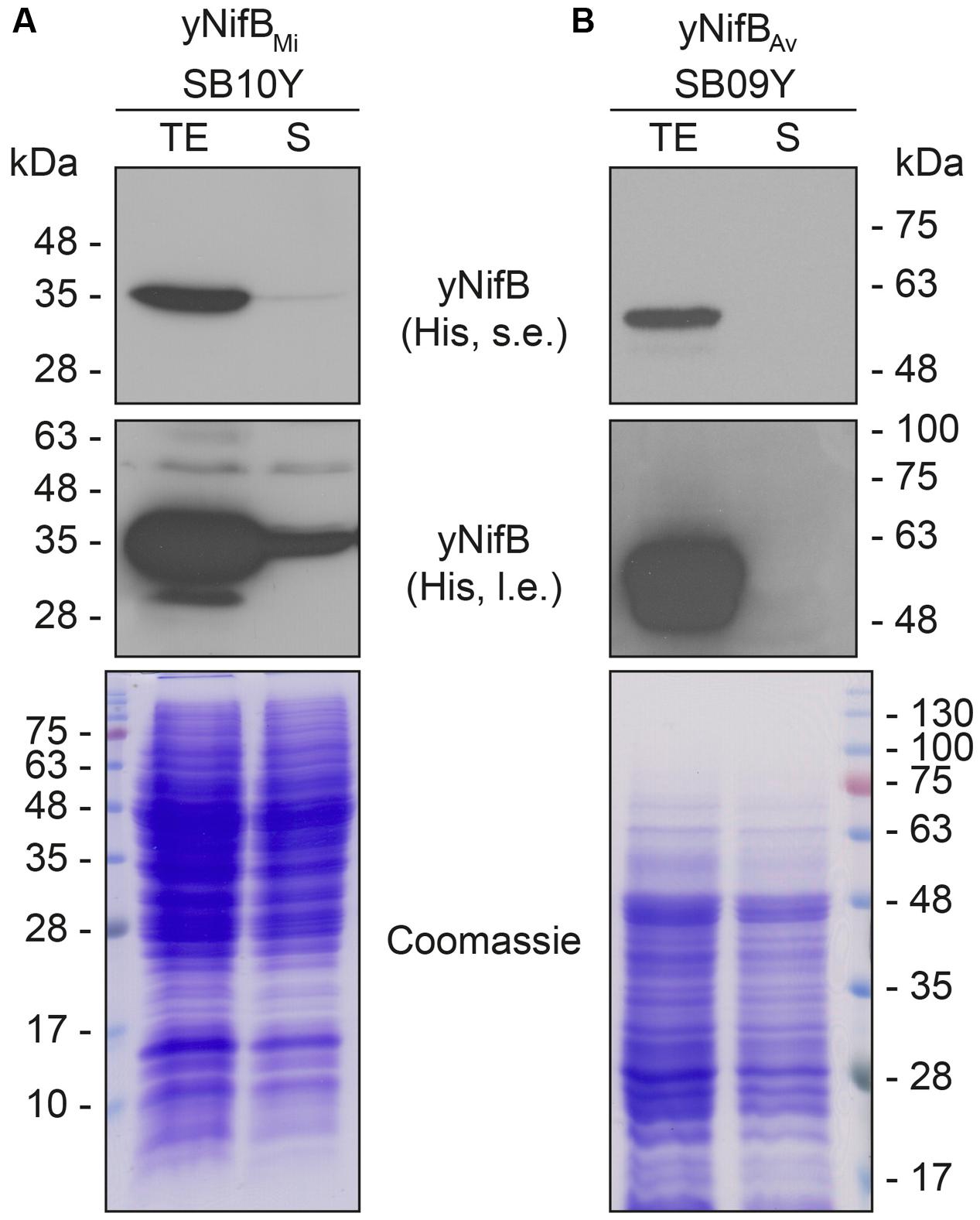
FIGURE 2. Solubility of A. vinelandii and M. infernus NifB proteins expressed in S. cerevisiae. (A,B) Western blot analysis of yNifBMi (A) and yNifBAv (B) present in total protein extracts (TE) and the soluble fraction (S) of yeast strains SB10Y and SB09Y. Conditions for strain growth, induction of protein expression, total extract preparation, and separation by SDS-PAGE are as in Figure 1. Antibodies recognizing the His epitope were used. Short (s.e.) and long (l.e.) film exposures are shown. Coomassie stained SDS gels (below) of the protein extracts are included as loading controls.
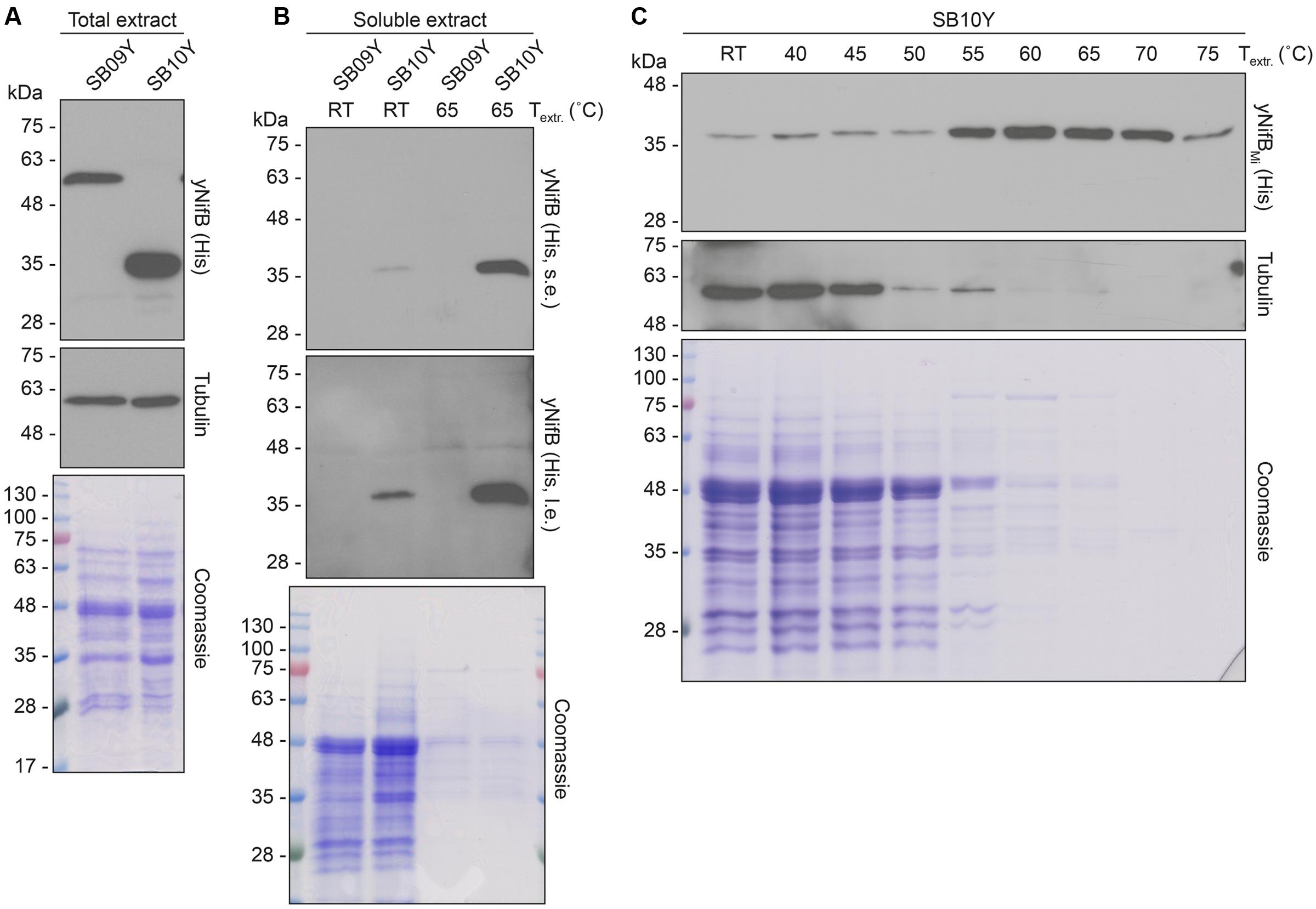
FIGURE 3. Levels of soluble yNifBAv and yNifBMi in heat-treated yeast extracts. (A,B) Total NifB levels (A) and levels of soluble NifB upon 65°C heat-treatment (B) of protein extracts from yeast expressing yNifBAv (SB09Y) or yNifBMi (SB10Y). Antibodies recognizing the His epitope were used. Short (s.e.) and long (l.e.) film exposures are shown. RT means room temperature. Tubulin and/or Coomassie stained SDS gels of the same protein extracts are included as loading controls. (C) Western blot analysis of soluble yNifBMi in SB10Y protein extracts upon heat-treatment at increasing temperatures. Heat-induced precipitation of yeast proteins in the extract at the different temperatures is shown using antibodies recognizing tubulin, as well as by Coomassie staining of proteins from the extract resolved by SDS-PAGE.
Yeast-Expressed NifBMi Is Active in the In Vitro FeMo-co Synthesis Assay
Typical yeast NifBMi purification yielded about 4 mg/100 g cell pellet (4.4 ± 1.1, mean and standard deviation from four individual purifications), and yNifBMi was at near purity as determined by SDS-PAGE analysis (Figure 4A). To confirm mitochondria import and functionality of the SU9 leader sequence, purified yNifBMi was subjected to N-terminal sequencing. Successful processing of the SU9 sequence was verified, and cleavage appeared at the site predicted from alignment of the SU9 peptide with a reported consensus sequence for yeast mitochondria proteins (Vögtle et al., 2009) (Figure 4B). While as isolated yNifBMi showed some color and UV-vis absorbance spectrum characteristic of Fe–S protein (3.3 ± 0.8 Fe atoms per monomer from four individual purifications, S not determined), in vitro reconstitution with Fe and S increased color intensity and the 320 and 420 nm features of the UV-vis spectrum indicative of [4Fe–4S] cluster formation (Figures 4C,D). Treatment with dithionite (DTH) reduced absorbance at 420 nm as expected for a redox responsive Fe–S protein. Fe and S content of reconstituted yNifBMi was consistent with the presence of, at minimum, two [Fe-S] clusters in addition to the SAM-binding [4Fe-4S] cluster (12.5 ± 2.8 Fe and 10.6 ± 3.1 S atoms per monomer; average ± standard deviation from four individual purifications). All these features are typical of NifB proteins (Curatti et al., 2006; Wilcoxen et al., 2016).
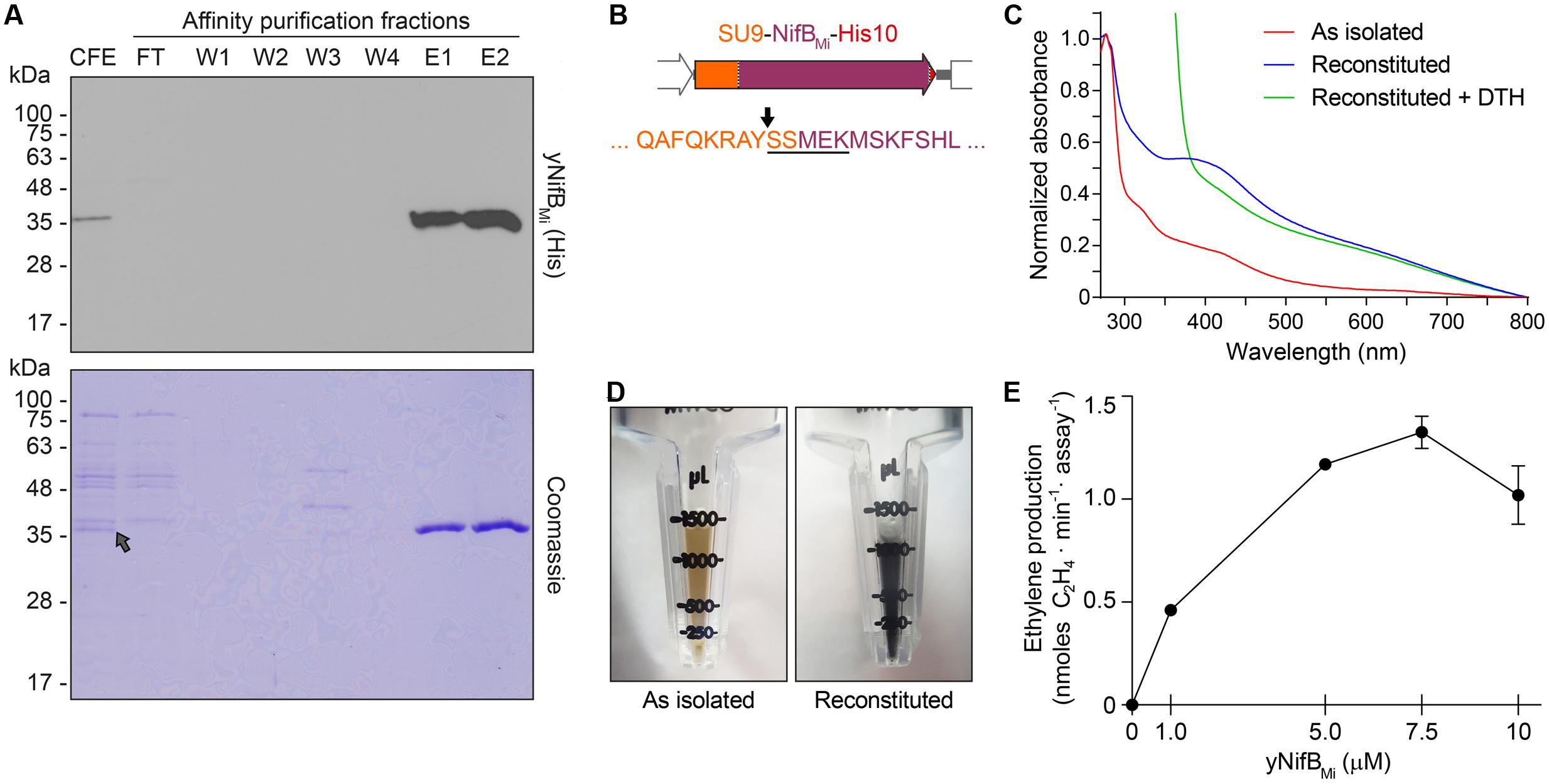
FIGURE 4. Purification and biochemical properties of yNifBMi. (A) SDS-PAGE and Western blot analysis of yNifBMi purification. CFE, 65°C heated SB10Y cell-free extract; FT, affinity chromatography flow through; W1-W4 and E1-E2, affinity chromatography wash and elution fractions containing increasing concentrations of imidazole (see Materials and Methods for details). Grey arrow in the Coomassie stained panel points to the position of yNifBMi in the gel. (B) SU9 processing site (black arrow) of yNifBMi. Underlined sequence indicates the N-terminal amino acids of yNifBMi identified by Edman degradation. (C) UV-visible spectra of as isolated, reconstituted, and dithionite (DTH)-reduced reconstituted yNifBMi. (D) Typical color of as isolated and reconstituted yNifBMi purified preparations. (E) Titration of FeMo-co synthesis and nitrogenase reconstitution assay with yNifBMi. The indicated concentrations of yNifBMi monomer were used. NifB activity was determined by acetylene reduction assay of reconstituted NifDK from ΔnifB A. vinelandii UW140 cell-free extracts. Data represent mean ± standard deviation (n = 2) at each yNifBMi concentration.
NifB can be used for in vitro FeMo-co synthesis and nitrogenase reconstitution assays using cell-free extracts of ΔnifB A. vinelandii, supplemented with ATP-regenerating mixture, molybdenum (Mo), and homocitrate (Curatti et al., 2006). When in vitro FeMo-co synthesis occurs, de novo-synthesized FeMo-co is incorporated into apo-MoFe nitrogenase present in the extract and activity of reconstituted nitrogenase can be determined by the acetylene reduction assay. To test whether reconstituted yNifBMi was functional, 5 μM protein was added to UW140 extracts lacking NifB-co activity, but providing the rest of the protein components required for FeMo-co synthesis and activatable apo-MoFe nitrogenase. While extract without yNifBMi only showed negligible acetylene reduction, addition of yNifBMi resulted in 40-fold increase in ethylene formation (Table 2). Importantly, yNifBMi showed similar concentration-dependent activity as purified and reconstituted NifB from A. vinelandii (Curatti et al., 2006) (Figure 4E). The maximum activity appeared to occur at slightly higher concentration (5 μM vs. 1 μM), which could result from slight incompatibility between the yNifBMi and the other Nif components in the UW140 A. vinelandii extract, as has been shown for NifH (Emerich and Burris, 1976, 1978), or from the suboptimal reaction temperature for the thermophile M. infernus (optimal growth at 85°C) NifB protein (Jeanthon et al., 1998).

TABLE 2. yNifBMi-dependent in vitro FeMo-co synthesis and nitrogenase reconstitution assays.
In summary, yNifBMi exhibits the spectroscopic and catalytic properties of active NifB proteins. Further studies will aim to determine whether the yNifBMi protein can support NifB-co synthesis in vivo in mitochondria of yeast.
Expression and Mitochondria Targeting of NifB in Plant Leaves
In a work by Allen et al. (2017), 16 HA-tagged nitrogenase proteins from K. oxytoca were separately expressed in Agrobacterium tumefaciens infiltrated N. benthamiana leaves, and targeted to the mitochondria. K. oxytoca NifB was one of the Nif proteins that resulted in highest protein expression level (Allen et al., 2017). However, as leaf proteins were extracted in SDS buffer upon heating, the level of soluble NifB protein is difficult to estimate. In order to test whether differences in solubility of plant expressed and mitochondria targeted NifB proteins could be observed, as in yeast, NifBAv and NifBMi were cloned into plant expression vectors under the control of the constitutive 35S promoter (Supplementary Figures S7, S8). As yeast and tobacco codon usage is similar, no further sequence optimization and gene synthesis was performed.
As SU9 is a mitochondria leader sequence from fungi without obvious plant homolog, the C-terminal His-tag of SU9-NifBAv and SU9-NifBMi was replaced with GFP to track SU9 functionality in N. benthamiana cells. Confocal microscopy analysis showed that SU9 successfully targeted the two NifB variants to the mitochondria of N. benthamiana, as seen from co-localization with a red fluorescent mitochondria marker (Candat et al., 2014) (Figures 5A–D). Specific and individual detection of the fluorescent signals was verified from adjacent cells expressing only each one of the constructs (Figure 5E). Confocal microscopy indicated that the expression level of SU9-NifBAv-GFP was lower than SU9-NifBMi-GFP, which was confirmed by Western blot analysis (Figure 6A). Importantly, SU9-NifBMi-GFP was only detected in the soluble fraction of the extract, in contrast to SU9-NifBAv-GFP that could also be seen in the pellet fraction (data not shown). Migration of the expressed fusion proteins was consistent with correct SU9 leader sequence processing in N. benthamiana cells (Figure 6A and Table 3).
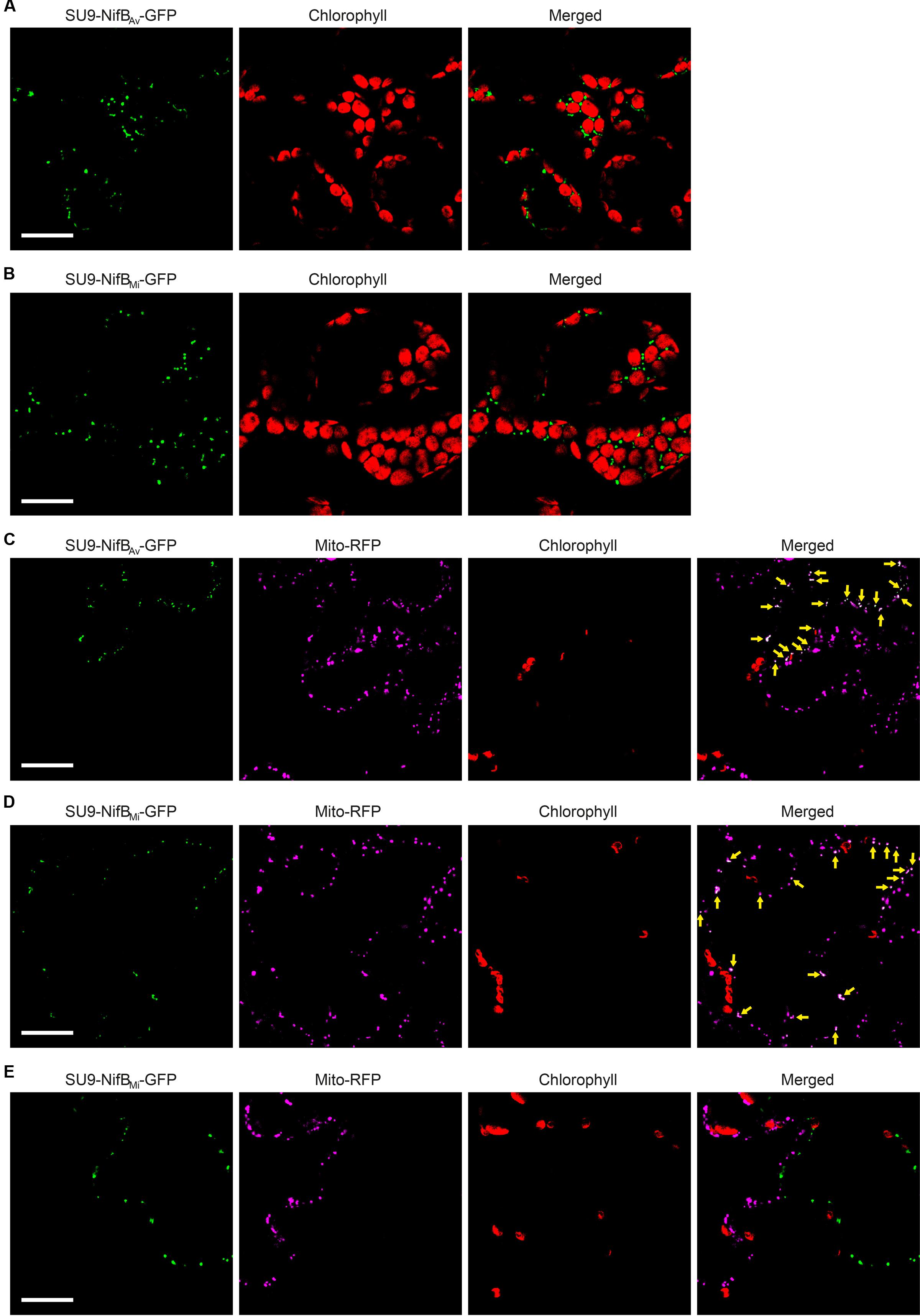
FIGURE 5. Expression of mitochondria targeted (SU9) NifBAv and NifBMi GFP fusions in N. benthamiana leaves. (A,B) Mesophyll cells expressing SU9-NifBAv-GFP (A) or SU9-NifBMi-GFP (B). GFP (green) and chlorophyll autofluorescence (red) of chloroplasts is shown. (C–E) Epidermal cells expressing SU9-NifBAv-GFP (C) and SU9-NifBMi-GFP (D,E), together with a fluorescent mitochondria marker (Mito-RFP). GFP (green), Mito-RFP (magenta) and chlorophyll autofluorescence (red) of chloroplasts is shown. Co-localization of SU9-NifBAv-GFP or SU9-NifBMi-GFP constructs with Mito-RFP labeled structures is shown as white in the merged images, and highlighted with yellow arrows. Adjacent cells expressing SU9-NifBMi-GFP or Mito-RFP are shown as control to verify the specificity of the signal recorded in each channel (E). Scale bars show 30 μm. Confocal Microscopy conditions are specified in Materials and Methods.
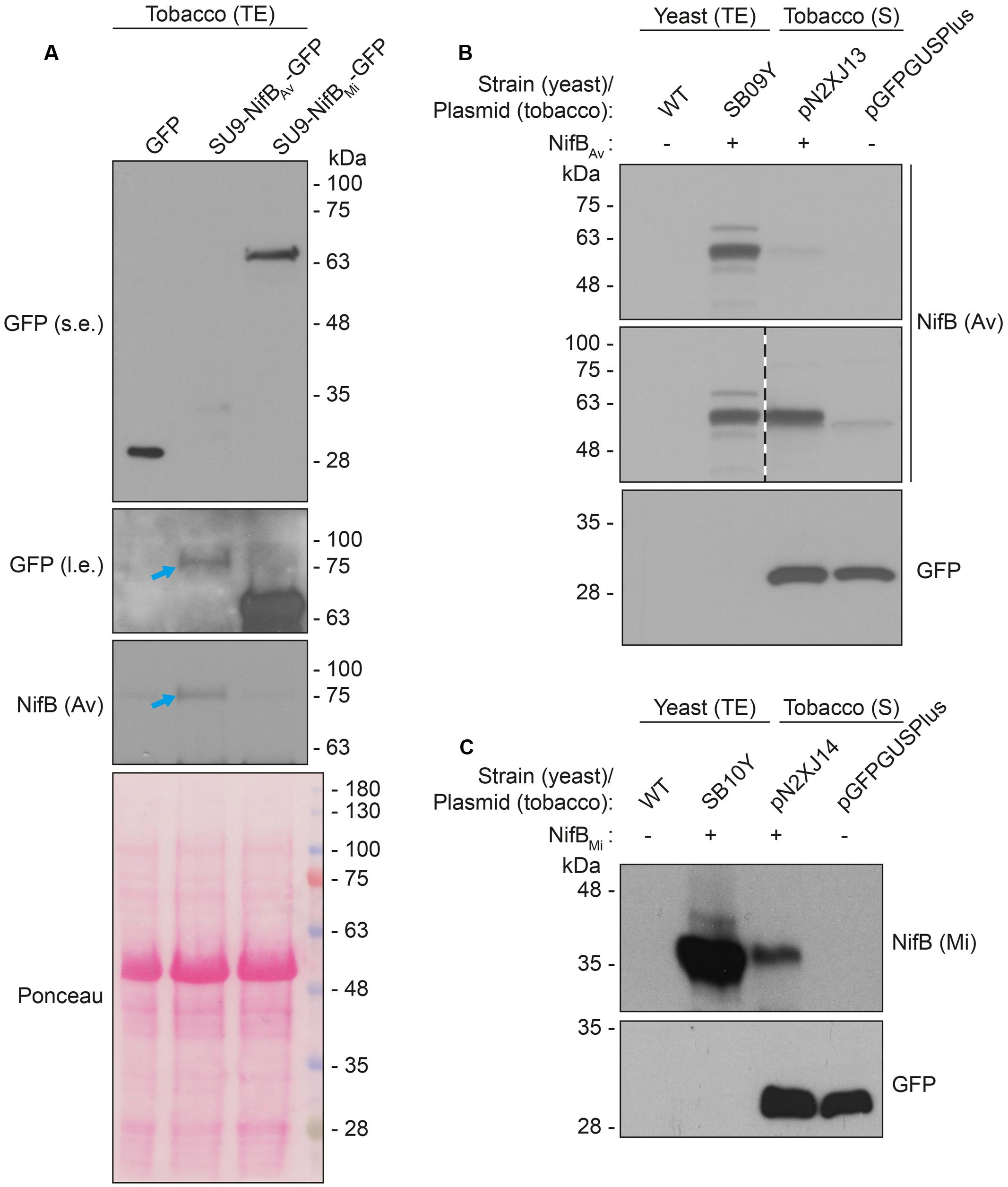
FIGURE 6. Expression and solubility of mitochondria targeted (SU9) NifBAv and NifBMi in N. benthamiana leaves. (A) Western blot analysis of total protein extracts (TE) prepared from infiltrated N. benthamiana leaves expressing GFP, SU9-NifBAv-GFP or SU9-NifBMi-GFP. Blue arrows indicate the polypeptide recognized both by GFP and NifBAv specific antibodies. Short (s.e.) and long (l.e.) film exposures of the GFP antibody probed membrane are shown. (B) Migration of SU9-NifBAv-His10 when expressed in S. cerevisiae and N. benthamiana. Migration in SDS-PAGE was determined after Western blot analysis using NifBAv specific antibodies. Total protein extracts (TE) from W303-1a S. cerevisiae cells (WT) or cells expressing SU9-NifBAv-His10 (SB09Y) were prepared. Soluble protein extracts (S) from N. benthamiana leaf cells infiltrated with A. tumefaciens containing control vector (pGFPGUSPlus) or vector for expression of SU9-NifBAv-His10 (pN2XJ13). Dotted line indicate different exposures of the right part of the membrane. See Supplementary Figure S9 for entire gel of the cropped exposure. (C) Migration of SU9-NifBMi-His10 when expressed in S. cerevisiae and N. benthamiana. Migration in SDS-PAGE was determined after Western blot analysis using NifBMi specific antibodies. Total protein extracts (TE) from W303-1a S. cerevisiae cells (WT) or cells expressing SU9-NifBMi-His10 (SB10Y) were prepared. Soluble protein extracts (S) from N. benthamiana leaf cells infiltrated with A. tumefaciens containing control vector (pGFPGUSPlus) or vector for expression of SU9-NifBMi-His10 (pN2XJ14). As control of N. benthamiana leaf infiltration, GFP expressed from the pGFPGUSPlus vector backbone was detected (B,C).
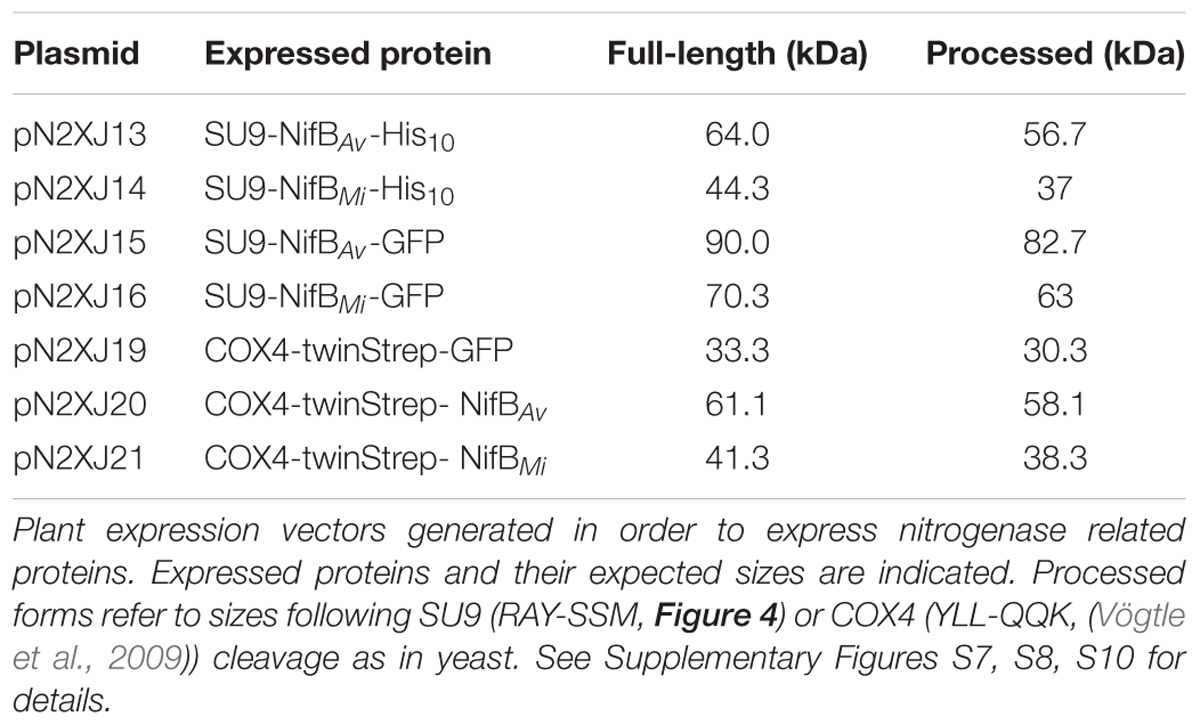
TABLE 3. Tobacco expressed nitrogenase related proteins and their expected sizes.
Migration of the plant expressed C-terminally His-tagged versions of the SU9-NifBAv and SU9-NifBMi proteins appeared identical to the corresponding proteins expressed in yeast, supporting that the SU9 leader sequence was processed correctly also in the plant mitochondria (Figures 6B,C and Supplementary Figure S9).
To enable simultaneous and comparative detection of the two N. benthamiana expressed NifB variants, and to exclude that solubility was affected by the C-terminal GFP moiety, new constructs were generated were the His-tag was exchanged for an N-terminal 28 amino acid Twin-Strep-tag (Schmidt et al., 2013) (Supplementary Figures S8, S10). The Twin-Strep-tag is an improved version of the eight amino acid Strep-tag II that was shown superior to His-tag for use with plant tissue extracts (Witte et al., 2004). In addition, the SU9 signal was replaced by the first 29 amino acids of the yeast cytochrome c oxidase IV (COX4) protein, which has been shown to successfully target proteins to the mitochondria in tobacco and Arabidopsis thaliana (Köhler et al., 1997; Nelson et al., 2007; Pan et al., 2014). As cleavage of COX4 in yeast has been shown to occur between amino acids 25 and 26 (Vögtle et al., 2009), similar processing in N. benthamiana would leave only four amino acids in addition to the Twin-Strep-tag. To verify functionality of the COX4 peptide, and to confirm that the Twin-Strep-tag was not interfering with targeting or solubility, a COX4-twin-Strep-GFP construct was generated (Supplementary Figures S8, S10). As expected, COX4 efficiently targeted twin-Strep-GFP to mitochondria in N. benthamiana cells (Figures 7A–C). Specific and individual detection of the fluorescent signals was verified using adjacent cells expressing only one of the constructs (Figure 7C).
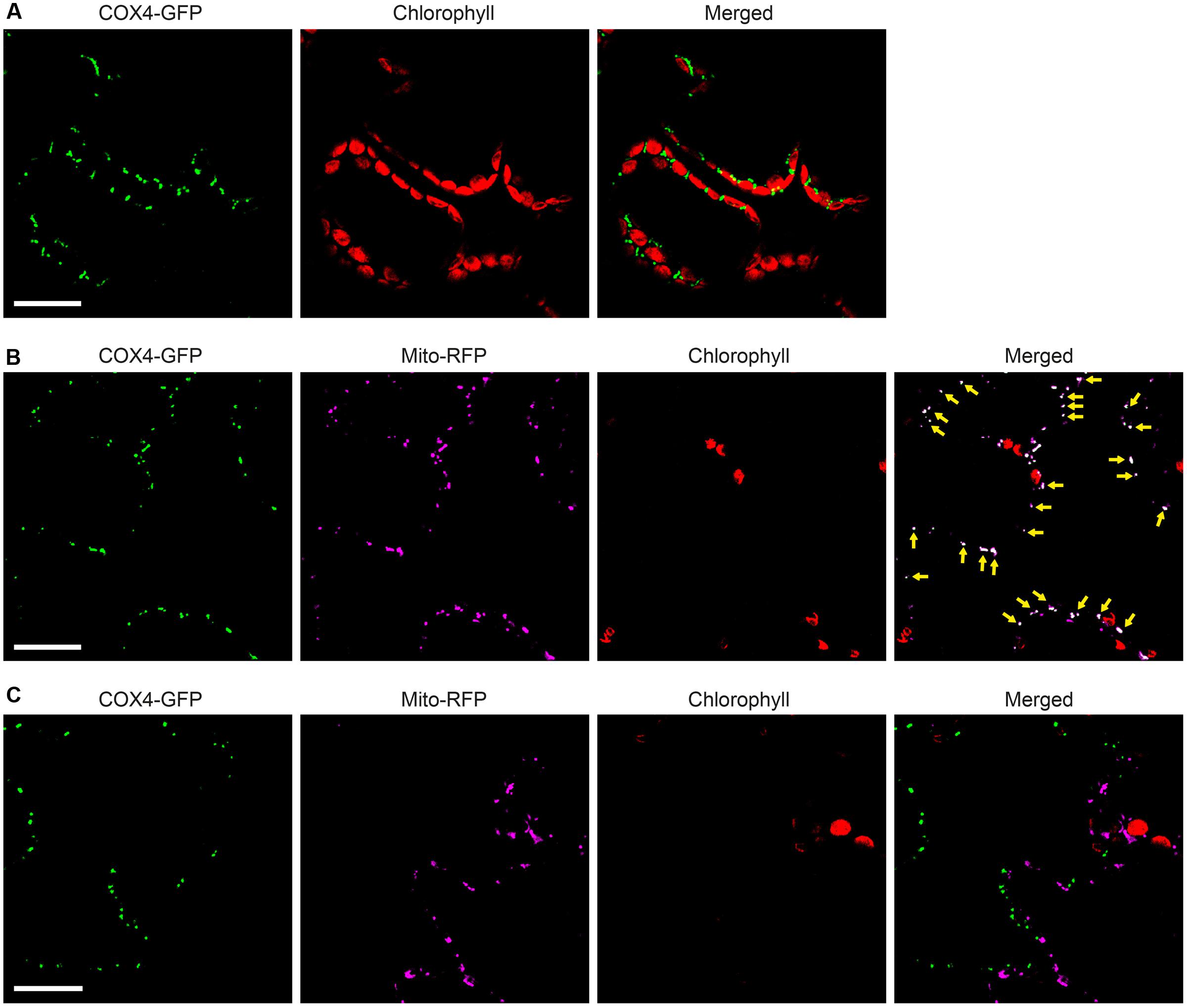
FIGURE 7. Functionality of COX4 leader sequence for mitochondria targeting of GFP in N. benthamiana leaves. (A) Mesophyll cells expressing COX4-twinStrep-GFP. GFP (green) and chlorophyll autofluorescence (red) of chloroplasts is shown. (B,C) Epidermal cells expressing COX4-twinStrep-GFP together with a fluorescent mitochondria marker (Mito-RFP). GFP (green), Mito-RFP (magenta) and chlorophyll autofluorescence (red) of chloroplasts is shown. Co-localization of COX4-twinStrep-GFP with Mito-RFP labeled structures (B) is shown as white in the merged image, and highlighted with yellow arrows. Adjacent cells expressing COX4-twinStrep-GFP or Mito-RFP (C) are shown as control to verify the specificity of the signal recorded in each channel. Scale bars show 30 μm. Confocal Microscopy conditions are specified in Materials and Methods.
Both COX4-twin-Strep-NifBAv and COX4-twin-Strep-NifBMi were readily detected in total protein extracts of A. tumefaciens infiltrated N. benthamiana leaves (Figure 8A). To test the solubility of the expressed NifB proteins, total protein extracts were separated in soluble fractions and pellet associated fractions. COX4-twin-Strep-NifBMi was detected exclusively in the soluble fraction, even upon prolonged exposure (Figure 8B). On the contrary, COX4-twin-Strep-NifBAv was more difficult to detect using the Strep-tag II antibody, and appeared to be in the non-soluble fraction. To verify the identity of the NifBAv protein detected by the Strep-tag II antibody we used NifBAv specific antibody, which confirmed that COX4-twin-Strep-NifBAv was mainly present in the pellet associated fraction (Figure 8C).
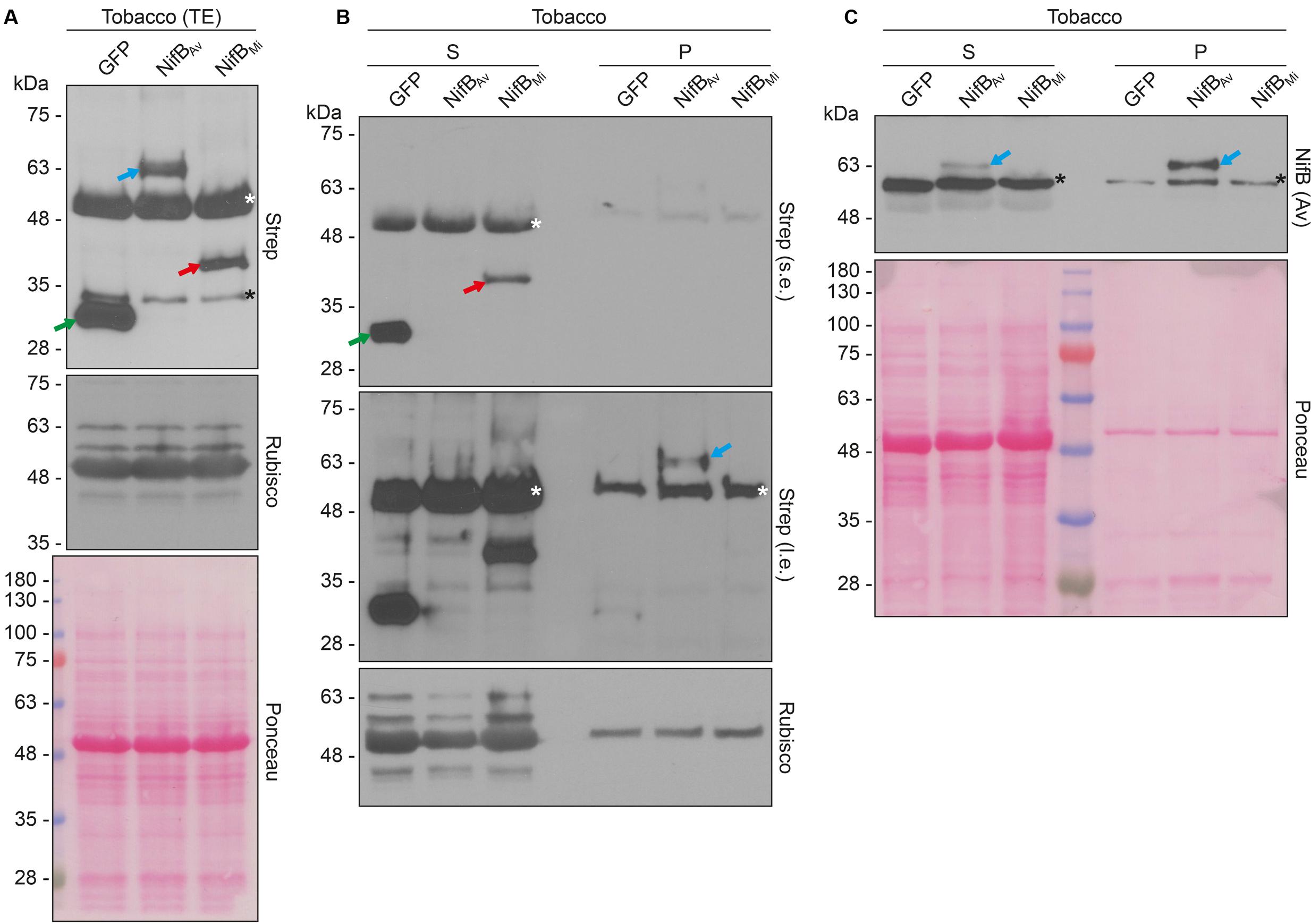
FIGURE 8. Expression and solubility of mitochondria targeted (COX4) NifBAv and NifBMi in N. benthamiana leaves. (A) Western blot analysis of total protein extracts (TE) prepared from infiltrated N. benthamiana leaves expressing COX4-twinStrep-GFP (GFP), COX4-twinStrep-NifBAv (NifBAv) or COX4-twinStrep-NifBMi (NifBMi) and separated by SDS-PAGE. The COX4-twinStrep-GFP (green arrow), COX4-twinStrep-NifBAv (blue arrow), COX4-twinStrep-NifBMi (red arrow) proteins are highlighted. A pronounced non-specific polypeptide detected using the Strep-tag antibodies (white star) co-migrated with the large subunit of Rubisco. The membrane probed with antibodies against Rubisco was also stained with Ponceau and is included as loading control. (B,C) Western blot analysis of the soluble (S) and non-soluble pellet (P) fractions of N. benthamiana leaf total extracts used in (A), using Strep-tag antibodies (B) or NifBAv antibodies (C). The COX4-twinStrep-GFP (green arrow), COX4-twinStrep-NifBAv (blue arrow), COX4-twinStrep-NifBMi (red arrow) proteins are highlighted. Non-specific bands detected using the Strep-tag antibodies (white stars) co-migrated with Rubisco (B). Non-specific bands detected with NifBAv antibodies (black stars) are also indicated (C). Short (s.e.) and long (l.e.) film exposures of the Strep-tag antibody probed membrane are shown (B). Ponceau staining of the NifBAv antibody probed membrane is shown as loading control (C).
In summary, we show that mitochondria targeting using SU9 and COX4 resulted in expression of both NifBAv and NifBMi in leaves of N. benthamiana. Leader sequence processing of all proteins appeared efficient and correct, as only one band of the expected size was detected. Similar to yeast, the NifBMi protein was more soluble than the corresponding NifBAv variant in N. benthamiana.
Discussion
Expression of functional NifB is absolutely required to engineer nitrogenase in eukaryotic organisms (e.g., plants). NifB catalyzes the formation of NifB-co, a unique [Fe–S] cluster intermediate in the biosynthesis of FeMo-co of nitrogenase (Shah et al., 1994; Curatti et al., 2006; Wiig et al., 2012; Guo et al., 2016). All diazotrophs carry at least one nifB gene (Dos Santos et al., 2012), and it is not likely that NifB-co can be produced by any other enzyme of plant origin (Vicente and Dean, 2017). As NifB-co also serves as precursor to the FeV-co of V-nitrogenase and the FeFe-co of Fe-only nitrogenase, it is required for all biological nitrogen fixation activity in nature (Bishop and Joerger, 1990; Curatti et al., 2007). Therefore, finding NifB proteins able to function in eukaryotic cells is of utmost importance.
In this work, we investigated eukaryotic expression and functionality of NifB proteins from two evolutionary distant organisms, the γ-proteobacterium A. vinelandii and the methanogen M. infernus. These NifB proteins differ in domain composition, quaternary structure, optimum temperature and stability, but both contain the same complement of [Fe–S] clusters, catalyze the SAM-radical dependent formation of NifB-co (Curatti et al., 2006; Wilcoxen et al., 2016), and have been shown to support FeMo-co biosynthesis in vivo (Arragain et al., submitted manuscript).
As most Nif proteins, NifB is extremely O2-labile. Expressed NifB variants were therefore targeted to mitochondria for respiratory protection (Lopez-Torrejon et al., 2016). Mitochondria targeting in yeast was achieved using the SU9 leader sequence, which had proved efficient for Nif protein targeting and processing in a previous study (Burén et al., 2017). Expression of the NifB variants in S. cerevisiae was coordinated with expression of A. vinelandii NifU, NifS and FdxN, as these proteins are important for the assembly of NifB [Fe–S] clusters and for NifB functionality (Jiménez-Vicente et al., 2014; Zhao et al., 2007). Both NifB variants appeared mostly insoluble and the major protein pools were pelleting together with the cellular debris of the broken cells. The accumulation insoluble NifB might be a result of overexpression or NifB hydrophobicity. It is intriguing to note that overexpression of NifB proteins often result in insoluble aggregates and that purification of NifB from different organisms, as well as purification of NifB-co itself, requires detergents (Curatti et al., 2006; Echavarri-Erasun et al., 2014; Shah et al., 1994).
The two NifB variants were also expressed and targeted to mitochondria of N. benthamiana leaf cells. Although the extraction methods and buffers used to prepare protein extracts were slightly different in yeast and tobacco, the results obtained from both systems were similar, i.e. expression levels were slightly higher and solubility was significantly better for the M. infernus NifB. Intriguingly, solubility overall appeared better in tobacco than in yeast, perhaps due to lower expression levels from the 35S promoter compared to the very strong GAL promoters. Mitochondria targeting was achieved both with SU9 and COX4 leader sequences, expanding the synthetic toolbox for Nif expression in plants. The identical migration in SDS-PAGE of each set of yeast- and tobacco-expressed NifB variants suggests that all of them underwent correct processing, which in the case of yeast SU9-NifBMi was demonstrated to occur at the predicted peptide bond between tyrosine and serine residues.
Taking advantage of the heat-resistant properties of M. infernus NifB, a protocol to enrich levels of the protein in the soluble fraction of yeast cell-free extracts was developed permitting further purification and biochemical analysis. Pure yNifBMi preparations exhibited properties characteristic of bacteria-purified NifB proteins (Curatti et al., 2006; Wilcoxen et al., 2016), including color, Fe and S content, and UV-vis spectra changes upon [Fe–S] cluster reconstitution and reduction. Importantly, yNifBMi showed activities similar to the NifBAv protein (purified from A. vinelandii cells) when used to complement ΔnifB A. vinelandii extracts in FeMo-co synthesis assays.
Whether or not NifB proteins were functional in vivo in S. cerevisiae is not known, as we have not yet been able to establish an in vivo assay for NifB activity. In this regard, we tested whether yNifBMi in the heat-treated extract was functional without reconstitution, but failed to detect activity, indicating that yNifBMi has low or no activity prior to [Fe–S] cluster reconstitution, or that some of its [Fe–S] clusters are lost during yeast cell lysis and/or heat-treatment. Importantly, addition of NifB-co to UW140 extract, in the presence or absence of yeast cell-free extract, resulted in identical activity, suggesting that the heat-treated cell-free extract did not inhibit the A. vinelandii nitrogenase activity and that lack of detectable yNifBMi activity was not due to inhibition of the yeast extract per se (data not shown).
We recently used a synthetic biology approach to assemble a yeast library for the expression of nine A. vinelandii nif genes (nifHDKUSMBEN), where the gene products were targeted to the yeast mitochondria using distinct mitochondria leader sequences (Burén et al., 2017). That work highlighted that expression levels (using distinct promoter/terminator combinations) and mitochondria signals in many cases need to be empirically tested for each gene. We also learned that two isoforms of the NifD polypeptide accumulated in the yeast mitochondria, one of which was the result of N-terminal degradation of the full-length NifD. A similar result could be observed when NifD was expressed in leaf cells of tobacco (Allen et al., 2017), where their NifD degradation isoform showed a SDS-PAGE migration very similar to ours. A plausible explanation to this NifD degradation could be instability of NifDK precursors, as stability of the NifDK tetramer is improved with protein maturation and nitrogenase co-factor (FeMo-co) insertion. In this regard, expression of functional NifB will stabilize NifDK and help increase its levels.
In summary, we have purified NifB protein expressed in a eukaryotic cell background. Following NifH and NifU (Lopez-Torrejon et al., 2016), to our knowledge this is the third Nif protein that has been purified with specific in vitro activity. Using both yeast and tobacco as expression hosts, we observed that the monomeric NifBMi (Wilcoxen et al., 2016) was expressed at higher levels, and to a higher extent accumulated as a soluble protein, than the dimeric NifBAv. This study emphasizes that simpler and more robust Nif protein variants could provide advantages in the ambitious goal of obtaining a functional plant expressed nitrogenase. Importantly, this study confirms that yeast synthetic biology provides a valuable tool in the initial designing and screening process for Nif protein expression and functionality, prior to performing more complex and time-consuming plant-based experiments.
Materials and Methods
Generation of Plasmids for Galactose-Induced Yeast Expression
Escherichia coli DH5α was used for storage and amplification of yeast expression vectors. E.coli was grown at 37°C in Luria-Bertani (LB) medium supplemented with appropriate antibiotics. Yeast optimized coding sequences for nifU, nifS, nifB (A. vinelandii and M. infernus) and fdxN with in-frame su9 leader sequences (Westermann and Neupert, 2000) were generated by GenScript, or by overlapping PCR reactions as specified below, and cloned into pESC vectors (Agilent Technologies) using standard techniques. su9-nifU and su9-nifS were cloned into pESC-URA using BamHI/HindIII and EcoRI/BglII, respectively, generating pN2GLT4. su9-fdxN and su9-His10-nifBAv were cloned into pESC-TRP using NotI/ClaI and BamHI/SalI, respectively, generating pN2GLT18. su9-nifBAv-His10 and su9-nifBMi-His10 were created using overlapping PCR, to add su9 and His10 at the 5′- and 3′-termini of nifBAv and nifBMi. Primers used for generating su9-nifBAv-His10 were 5′-ATTTTCGGTTTGTATTACTTC-3′ and 5′-CATGGAAGAGTAGGCGC-3′ (using pN2GLT18 as template), 5′-GCGCCTACTCTTCCATGGAATTGTCTGTTTTGGGT-3′ and 5′-ATGATGGTGGTGGTGATGATGATGAGCCTTAGCTTGCAAC-3′ (using pN2GLT18 as template), 5′-ATCACCACCACCATCATCACCATTAAGTCGACATGGAACA-3′ and 5′-GTACACGCGTCTGTACAGAA-3′ (using pN2GLT18 as template), to amplify su9, nifBAv and His10, respectively. 5′-ATTTTCGGTTTGTATTACTTC-3′ and 5′-GTACACGCGTCTGTACAGAA-3′ were used for the overlapping PCR reaction. Primers used for generating su9-nifBMi-His10 were 5′-ATTTTCGGTTTGTATTACTTC-3′ and 5′-CATGGAAGAGTAGGCGC-3′ (using pN2GLT18 as template), 5′-GCGCCTACTCTTCCATGGAGAAAATGTCTAAATTT-3′ and 5′-ATGATGGTGGTGGTGATGATGATGGTGTGAGAAATGCTTC-3′ (using nifBMi as template), 5′-ATCACCACCACCATCATCACCATTAAGTCGACATGGAACA-3′ and 5′-GTACACGCGTCTGTACAGAA-3′ (using pN2GLT18 as template), to amplify su9, nifBMi and His10, respectively. 5′-ATTTTCGGTTTGTATTACTTC-3′ and 5′-GTACACGCGTCTGTACAGAA-3′ were used for the overlapping PCR reaction. su9-nifBAv-His10 and su9-nifBMi-His10 were cloned into pN2GLT18, replacing su9-His10-nifBAv using BamHI/SalI, and generating pN2SB22 and pN2SB24, respectively. su9-fdxN-HA was created using overlapping PCR, to add HA at the 3′-terminus of su9-fdxN. Primers used for generating su9-fdxN-HA were 5′-GGTGGTAATGCCATGTAATATG-3′ and 5′-GCATAATCTGGAACATCATATGGATACCTTGCCTGTATTT-3′ (using pN2SB22 as template), 5′-GATGTTCCAGATTATGCTTAAGAGCTCTTAATTAACAATT-3′ and 5′-AAAGTTTAAACCGCATCAGGAAATTGTAA-3′ (using pN2SB22 as template), to amplify su9-fdxN and HA, respectively. 5′-GGTGGTAATGCCATGTAATATG-3′ and 5′-AAAGTTTAAACCGCATCAGGAAATTGTAA-3′ were used for the overlapping PCR reaction. su9-fdxN-HA was cloned into pN2SB24, replacing su9-fdxN using NotI/PacI, generating pN2SB39. To make pN2GLT18 (su9-fdxN and su9-His10-nifBAv) compatible with transformation into prototrophic S. cerevisiae CEN.PK113-7D clone DOE56, the LEU2 auxotrophic marker was replaced with the hygromycin marker hphMX4 (Burén et al., 2017), generating pN2SB15. DNA and protein sequences of all constructs are listed in Supplementary Figure S2.
Generation of Yeast Strains, Growth, and Protein Expression
Saccharomyces cerevisiae W303-1a (MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15) was the host strain for expression vectors pN2GLT4 and pN2SB22 (to generate strain SB09Y), pN2GLT4 and pN2SB24 (to generate strain SB10Y), and pN2GLT4 and pN2SB39 (to generate strain SB12Y). CEN.PK113-7D (MATa URA3 TRP1 LEU2 HIS3 MAL2-8c SUC2) strain DOE56 (having constitutive expression of mitochondria targeted NifU and NifS) (Burén et al., 2017) was the host strain for expression vector pN2SB15 (to generate strain SB03Y). Yeast transformations were carried out according to the lithium acetate method (Gietz and Schiestl, 2007).
Saccharomyces cerevisiae were grown in flasks at 28°C and 200 rpm in synthetic drop-out (SD) medium (1.9 g/l yeast nitrogen base, 5 g/l ammonium sulfate, 20 g/l glucose, and Kaiser drop-out mixture (Kaiser et al., 1994) (SC -His-Leu-Trp-Ura, FORMEDIUM) supplemented with 20 mg/l adenine and 40 mg/l tryptophan, 40 mg/l histidine, 20 mg/l uracil, 60 mg/l leucine, depending on auxotrophic requirements). Plasmid for the inducible expression of SU9-FdxN and SU9-His10-NifBAv in transformed DOE56 (SB03Y) was maintained by supplementing the inoculum growth media with 300 μg/l hygromycin. Galactose induction for small-scale protein extracts was performed in the above-described SD medium in which glucose was replaced by 20 g/l galactose, and additionally supplemented with 0.1% yeast extract and 1% peptone. Total yeast protein extracts to verify protein expression were performed using mild alkali treatment (Kushnirov, 2000). Similar loading on SDS-PAGE experiments was obtained by preparing samples according to optical density, and was confirmed by using either Commassie staining of polyacrylamide gels or Ponceau staining of nitrocellulose membranes. Additionally, immunoblotting with antibodies against tubulin was used as control of gel loading and sample precipitation.
Cultures for yeast expressed NifB purifications were grown following a previously reported procedures (Lopez-Torrejon et al., 2016; Burén et al., 2017), in a 4-L fermenter (BIO-STAT). Cultures were grown at 30°C in selective SD-medium for 16 h, followed by 8 h in rich medium (0.5% yeast extract, 0.5% bactopeptone, 0.5% bactotryptone, 2.5% glucose), supplemented with 25 mg/L ammonium iron(III) citrate, 1.25 mM magnesium sulfate, 1.5 mM calcium chloride, and trace element and vitamin solutions (Lopez-Torrejon et al., 2016). Finally, protein expression was induced by addition of 2.5% galactose for 16 h. The pH was automatically maintained around 5 using 0.8 M ammonium hydroxide. Air flow was maintained at 0.75 liter air/min per liter of culture, at 300 rpm. Dissolved oxygen dropped to zero (as measured by oxygen sensor, Mettler Toledo) before addition of galactose, and remained at zero during the rest of the process.
Reverse Transcription and Quantitative Real-time Polymerase Chain Reactions
Total yeast RNA was extracted from 25 ml cultures 6 h following galactose induction. Briefly, the cells were harvested by centrifugation (10 min at 3,000 x g), washed once in Milli-Q water and then resuspended in 1 ml TRIzol reagent (ThermoFisher Scientific). Cells were broken in 2 ml screw-cap tubes using 0.5 mm glass beads (BioSpec Products) in a mixer mill (Retsch MM300) operating at 30 Hz, in 5 cycles of 1 min at 4°C. Two hundred μl chloroform was added to the lysate, vortexed for 15 sec and incubated 5 min at room temperature. Samples were then centrifuged at full speed for 5 min at 4°C. The supernatant was transferred to new tube and re-extracted with 400 μl chloroform. The supernatant containing RNA was precipitated with 1 volume isopropanol at -20°C for 20 minutes and then pelleted by centrifugation at full speed for 5 min at 4°C. The pellet was washed twice in 1 ml 70% ethanol and finally resuspended in nuclease free water. RNA concentration was measured in a nanodrop apparatus and quality was analysed by agarose gel electrophoresis. RNA was then treated with DNAse to remove eventual DNA contamination (TURBO DNA-free Kit, AM1907, Ambion). Absence of DNA was verified using polymerase chain reaction (PCR) (KAPA2G Fast HotStart ReadyMix, B4KK5609, KAPA2G). Four μg RNA was used for cDNA synthesis (High-Capacity cDNA Reverse transcription Kit, 4374966, Applied Biosystems). The presence of fdxN cDNA was verified by reverse transcription PCR (RT-PCR) using 4 distinct fdxN primer combinations (A, 5′-TGTGAACTGCTGGGCATGTG-3′ and 5′-TCTCCATCACACTCGGTGCAT-3′; B, 5′-TGCACCGAGTGTGATGGAGA-3′ and 5′-TGCCTCAGCCAATCTTTCAGGT-3′; C, 5′-ACCGAGTGTGATGGAGACTAT-3′ and 5′-GTAAGTGAACCAGGTGGGTTAG-3′; D, 5′-CCTTGGCAGGTCCTCATTT-3′ and 5′-TAGCACCTTCAACTGGACAAATA-3′). Primers targeting housekeeping genes (alg9, 5′-CACGGATAGTGGCTTTGGTGAACAATTAC-3′ and 5′-TATGATTATCTGGCAGCAGGAAAGAACTTGGG-3′; rdn18, 5′-AACTCACCAGGTCCAGACACAATAAGG-3′ and 5′-AAGGTCTCGTTCGTTATCGCAATTAAGC-3′) were selected based on Teste and colleagues (Teste et al., 2009). Quantitative real-time PCR (qPCR) (KAPA SYBR FAST Universal qPCR Kit, KK4601, KAPA2G) was performed using two fdxN primer combinations in addition to primers targeting the housekeeping genes, using an Eco Real-Time PCR System (Illumina) following user instructions.
Solubility of Yeast-Expressed NifB
Saccharomyces cerevisiae cells expressing yNifBAv and yNifBMi were resuspended in 5 volumes of lysis buffer (100 mM Tris-HCl, 400 mM NaCl, 5 mM β-mercaptoethanol (β-ME), 1 mM phenyl-methylsulfonyl fluoride (PMSF)), at pH 7 or 8 with 10% or 30% glycerol. Cells were broken in 2 ml tubes using 0.5 mm glass beads (BioSpec Products) in a mixer mill (Retsch MM300) operating at 30 Hz in 3 cycles of 1 min at 4°C. Lysates were incubated at room temperature (RT), or heated at 5°C temperature intervals from 40°C to 75°C, for 30 min. The supernatant after 20 min centrifugation at 20,000 x g and 4°C containing soluble proteins was analyzed by SDS-PAGE and immunoblot analysis.
Preparation of Yeast Anaerobic Cell-Free Extracts and NifB Purifications
Saccharomyces cerevisiae cells expressing yNifBAv and yNifBMi were resuspended in anaerobic lysis buffer (100 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% glycerol) supplemented with 2 mM dithionite (DTH), 5 mM β-ME, 1 mM PMSF, 1 μg/ml leupeptin and 5 μg/ml DNAse I. The cells were lysed in an Emulsiflex-C5 homogenizer (Avestin Inc.) at 25,000 lb per square inch. Cell-free extracts (CFE) were obtained after heat-treatment in a water bath (yNifBMi only, 65°C for 30 min), removal of cell debris and precipitated yeast proteins by centrifugation (50,000 x g for 1 h at 4°C) and filtration through a 0.2 μM pore size filter (Nalgene Rapid-Flow, Thermo Scientific). All procedures were performed under anaerobic conditions.
Saccharomyces cerevisiae cells expressing SU9-His10-NifBAv were lysed as described above, in buffer with detergents (50 mM Tris-HCl pH 8, 200 mM KCl, 10% glycerol, 5 mM β-ME, 0.05% n-dodecyl-β-D-maltopyranoside, 0.1% triton X-100 and 0.1% Tween 20) as previously described for purification of NifBMi from E. coli (Wilcoxen et al., 2016).
His-tagged yNifBMi was purified by Co2+ affinity chromatography under anaerobic conditions (<0.1 ppm of O2) using an AKTA Prime FPLC system (GE Healthcare) inside a glovebox (MBraun). All buffers were previously made anaerobic by sparging with N2. Before loading the affinity column, the cell-free extract was diluted to reach 50 mM Tris-HCl, while maintaining other buffer components. Typically, anaerobic cell-free extract from 100 g of cell paste was loaded at 2 ml/min onto a column filled with 5 ml of IMAC resin (GE Healthcare) equilibrated with buffer A (50 mM Tris-HCl pH 8, 400 mM NaCl, 10% glycerol, 2 mM DTH, 5 mM β-ME) and washed with four successive washes of buffer A supplemented with 0, 10, 40 and 100 mM imidazole (10-15 column volumes per wash), respectively. Bound protein was eluted in two steps, with buffer A containing 200 and 500 mM imidazole, respectively. Eluted fractions showing the desired purity were pooled and concentrated using a 100 kDa cutoff pore centrifugal membrane device (Amicon Ultra-15, Millipore), and then desalted in PD10 columns (GE Healthcare) equilibrated with buffer A. Pure yNifBMi was frozen and stored in liquid N2.
In Vitro Reconstitution of yNifBMi Fe-S Clusters, UV-Visible Spectroscopy, N-terminal Sequencing and Protein Methods
In vitro reconstitution of purified yNifBMi was performed as previously described with modifications (Curatti et al., 2006). Pure yNifBMi stored in buffer A was buffer-exchanged to buffer B (50 mM Tris-HCl pH 8, 400 mM NaCl, 10% glycerol, 5 mM β-ME) by using a PD10 column to recover “as isolated” protein. The desalted sample (20 μM NifB monomer) was incubated with 10 mM DTT at room temperature inside a glovebox (MBraun) for 10 min. (NH4)2Fe(SO4)2 and Na2S were then added at 20-fold molar excess ratio and incubated at 35°C overnight. yNifBMi was again desalted in buffer B to recover “reconstituted” protein. As isolated and reconstituted proteins were used for colorimetric Fe (Fish, 1988) and S (Beinert, 1983) determination, in vitro FeMo-co synthesis and nitrogenase activity assays, and UV-visible spectroscopy. UV-visible absorption spectra were recorded under anaerobic conditions in septum-sealed cuvettes using a Shimadzu UV-2600 spectrophotometer. When indicated, 5 mM DTH was added to reconstituted yNifBMi. UV-visible absorption spectra were recorded against buffer B as baseline. Absorbance at 800 nm was subtracted and spectra were then normalized to 279 nm. The N-terminal amino acid sequence of purified yNifBMi was determined by Edman degradation (Proteome Factory AG). Protein concentrations were measured using the BCA protein assay (PIERCE). NifB samples were pre-treated with iodoacetamide before performing the BCA assay to eliminate the interfering effect of DTH (Hill and Straka, 1988).
In Vitro Synthesis of FeMo-co and Nitrogenase Reconstitution Assay
In vitro yNifBMi dependent FeMo-co synthesis and nitrogenase reconstitution reactions were performed in 9-ml serum vials sealed with serum stoppers (Curatti et al., 2006). Complete reactions contained 17.5 μM Na2MoO4, 175 μM homocitrate, 1.75 mM (NH4)2FeSO4, 1.75 mM Na2S, 880 μM SAM, 1.23 mM ATP, 18 mM phosphocreatine, 2.2 mM MgCl2, 3 mM DTH, 40 μg/ml creatine phosphokinase, 2.2 μM NifH (dimer), 2.9 mg/ml UW140 (A. vinelandii ΔnifB) proteins, 5 μM (or 0-10 μM titration) reconstituted yNifBMi (monomer) in 22 mM Tris-HCl (pH 7.5). The reactions (total volume of 500 μl) were incubated at 30°C for 35 min to allow for FeMo-co synthesis and insertion reactions. NifB-co-dependent in vitro FeMo-co synthesis assays were performed using 2 μM NifB-co isolated from K. oxytoca (Shah et al., 1994). Following in vitro synthesis of FeMo-co, activation of apo-MoFe nitrogenase present in UW140 extract was analyzed following addition of excess NifH and ATP-regenerating mixture (total volume 1 ml) by acetylene reduction assay at 30°C for 30 min following standard procedures (Shah and Brill, 1973). Positive control reactions for acetylene reduction were carried out with pure preparations of A. vinelandii Fe protein and MoFe protein incubated with ATP-regenerating mixture at 30°C during 30 min.
Generation of Plant Expression Vectors and Protein Expression in Leaves of N. benthamiana
Escherichia coli DH5α was used for storage and amplification of plant expression vectors. E. coli was grown at 37°C in LB medium supplemented with appropriate antibiotics. su9-nifBAv-His10 and su9-nifBMi-His10 were PCR amplified using primers 5′-AAAAGGATCCAATGGCCTCCACTCGTGTCCTCG-3′ and 5′-TTTTCACGTGTTAATGGTGATGATGGTGGTG-3′, with pN2SB22 and pN2SB24 as templates, respectively. su9-nifBAv-His10 and su9-nifBMi-His10 were digested with BamHI and PmlI, and inserted into pGFPGUSPlus vector (Vickers et al., 2007) (Addgene plasmid #64401) digested with BglII and PmlI, replacing GUS and generating pN2XJ13 (su9-nifBAv-His10) and pN2XJ14 (su9-nifBMi-His10), respectively. su9-nifBAv was PCR amplified using primers 5′-AAAAGCTAGCATGGCCTCCACTCGTGTCCTCG-3′ and 5′-TTTTGCTAGCGCCTTAGCTTGCAACAAAGC-3′, with pN2SB22 as template. su9-nifBAv was digested with NheI and inserted into pGFPGUSPlus vector digested with XbaI, generating pN2XJ15 for expression of su9-nifBAv-gfp. su9-nifBMi was PCR amplified using primers 5′-AAAAGCTAGCATGGCCTCCACTCGTGTCCTCG-3′ and 5′-TTTTGCTAGCGCGTGTGAGAAATGCTTCAAGTCG-3′, with pN2SB24 as template. su9-nifBMi was digested with NheI and inserted into pGFPGUSPlus vector digested with XbaI, generating pN2XJ16 for expression of su9-nifBMi-gfp. DNA sequence encoding the enhanced 35S promoter and an in-frame fusion of the cox4 mitochondria leader sequence (Köhler et al., 1997) with the 28 amino acid Twin-Strep-tag was generated by ThermoFisher. The E35S-cox4-twinStrep DNA sequence was flanked by HindIII and BglII, with a BamHI site additionally added 5′ of the BglII site. E35S-cox4-twinStrep was digested with HindIII and BglII, and inserted into pGFPGUSPlus vector also digested with HindIII and BglII, to generate pN2SB41. DNA sequence encoding egfp was PCR amplified using primers 5′-AAAAAGGATCCATGGTGAGCAAGGGCGA-3′ and 5′-AAAAAGGTCACCTTACTTGTACAGCTCGTCCATG-3′, and pGFPGUSPlus as template. egfp was digested with BamHI and BstEII, and inserted into pN2SB41 also digested with BamHI and BstEII, creating pN2XJ17. pN2XJ17 was digested with PstI to remove the non-targeted EGFP, to generate pN2XJ19 (cox4-twinStrep-gfp). DNA sequences encoding nifBAv and nifBMi, flanked by BamHI and BstEII, were generated by ThermoFisher. nifBAv and nifBMi were digested with BamHI and BstEII, and inserted into pN2SB41 also digested with BamHI and BstEII, to generate pN2XJ20 (cox4-twinStrep-nifBAv) and pN2XJ21 (cox4-twinStrep-nifBMi). DNA and protein sequences of all constructs are listed in Supplementary Figure S8.
Agrobacterium tumefaciens strain GV3101(pMP90) was transformed with plasmids pN2XJ13, pN2XJ14, pN2XJ15, pN2XJ16, pN2XJ19, pN2XJ20, pN2XJ21 and the silencing suppressor p19 (Huang et al., 2009). The pDCL-mito-mRFP1 mitochondria marker (Mito-RFP) in A. tumefaciens strain C58 (Candat et al., 2014) was kindly provided by Prof. Macherel and Prof. Logan at the Angers University (France). A. tumefaciens mediated infiltration of N. benthamiana leaves was essentially performed as described by Leuzinger and colleagues (Leuzinger et al., 2013). Three to four days post infiltration, plant tissue was used for protein extraction or confocal microscopy.
Protein extracts were prepared from infiltrated N. benthamiana leaf tissue in lysis buffer (100 mM Tris-HCl pH 8, 150 mM NaCl, 10 mM MgCl2, 0.2% NP-40, 5% glycerol, 5 mM β-ME and 5 mM ethylenediaminetetraacetic acid (EDTA)). Two hours before use, 5% polyvinylpolypyrrolidone (PVPP) was added to lysis buffer and, just before use, 1 mM PMSF, 1 μg/ml leupeptin and 1x protease inhibitor cocktail (P8215, Sigma) were added. Extraction was performed at a 2:1 ratio of buffer to tissue. Ten leaf discs of 5 mm diameter each (approximate weight of 200 mg) were added to a 2-ml Eppendorf tube containing a 7-mm diameter steel ball. Tubes were kept in liquid N2 until use. Leaf tissue was broken using mixer mill (Retsch MM300) operating at 30 Hz for 1 min at 4°C. The dry tissue powder was supplemented with 400 μl lysis buffer and mixed for another 1 min at 30 Hz and 4°C. The broken tissue in lysis buffer was further incubated on an orbital shaker for 30 min at 4°C. One hundred μl extract were added to 100 μl 2x Laemmli buffer (2xLB) and heated for 10 min at 95°C to obtain the “total extract”. The rest of the extract was centrifuged at 20,000 x g for 30 min at 4°C to separate pellet from supernatant. The supernatant “soluble extract (S)” was mixed with 2xLB and heated for 10 min at 95°C. The pellet (P) was resuspended in 1 ml lysis buffer (no additional PVPP added) and centrifuged at 20,000 x g for 10 min at 4°C. Finally, the pellet was resuspended in 800 μl 2xLB and heated for 10 min at 95°C. Ten μl of each fraction were used for SDS-PAGE and immunoblot analysis. Similar sample loading on SDS-PAGE lanes was assessed either by Commassie staining of polyacrylamide gels, by Ponceau staining of transferred nitrocellulose membranes, or by immunoblotting with antibodies against Rubisco.
Confocal Microscopy of N. benthamiana Leaf Tissue
Subcellular localization of fluorescent protein tagged proteins was examined in leaves of A. tumefaciens infiltrated N. benthamiana using a Leica TCS SP8 laser scanning confocal microscope with a 40x/1.10 water immersion objective equipped with LAS X software (Leica). EGFP, RFP, and chlorophyll were excited with 488-, 561-, or 638-nm laser lines, respectively, with an emission band of 500 to 537 nm for EGFP detection, 585 to 620 nm for RFP detection, and 652 to 727 nm for chlorophyll autofluorescence. EGFP and chlorophyll was recorded simultaneously, while RFP was detected in a separate scan. Laser intensity and gain was maintained during each experiment. For each experiment, specificity of the recorded signals was verified using single transformed cells.
Antibodies
Antibodies used for immunoblotting in this study were as follows: polyclonal antibodies detecting NifUAv, NifSAv, NifBAv and NifBMi were raised against purified preparations of the corresponding A. vinelandii or M. infernus proteins. Rubisco specific antibodies were a kind gift from Prof. Göran Samuelsson, Umeå University. His-tag (H-3, sc-8036, Santa Cruz), HA-tag (3F10, 12013819001, Roche), GFP (B-2, sc-9996, Santa Cruz), Strep-tag II (StrepMAB-Classic, 2-1507-001, IBA Lifesciences) specific antibodies are commercially available.
Author Contributions
SB, XJ, GL-T, and CE-E carried out the experimental work. SB, XJ, GL-T, CE-E and LR contributed to experimental design and data analysis. SB and LR wrote the paper.
Funding
Funding for this research was provided by the Bill & Melinda Gates Foundation Grant OPP1143172 (LR).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jose María Buesa for yeast fermentations and Marcel Veldhuizen for help with cloning. We thank Emilio Jimeìnez for fruitful discussions about NifB purifications and activity assays. We thank David Macherel and David Logan for providing the RFP mitochondria marker, and Göran Samuelsson for the anti-Rubisco specific antibodies.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01567/full#supplementary-material
References
Allen, R. S., Tilbrook, K., Warden, A. C., Campbell, P. C., Rolland, V., Singh, S. P., et al. (2017). Expression of 16 nitrogenase proteins within the plant mitochondrial matrix. Front. Plant Sci. 8:287. doi: 10.3389/fpls.2017.00287
Balk, J., and Pilon, M. (2011). Ancient and essential: the assembly of iron-sulfur clusters in plants. Trends Plant Sci. 16, 218–226. doi: 10.1016/j.tplants.2010.12.006
Beinert, H. (1983). Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131, 373–378. doi: 10.1016/0003-2697(83)90186-0
Bishop, P. E., and Joerger, R. D. (1990). Genetics and molecular biology of alternative nitrogen fixation systems. Annu. Rev. Plant Physiol. Plant Mol. Biol. 41, 109–125. doi: 10.1146/annurev.pp.41.060190.000545
Borlaug, N. E. (2002). Feeding a world of 10 billion people: the miracle ahead. Vitr. Cell. Dev. Biol. Plant 38, 221–228. doi: 10.1079/IVP2001279
Bulen, W. A., and LeComte, J. R. (1966). The nitrogenase system from Azotobacter: two-enzyme requirement for N2 reduction, ATP-dependent H2 evolution, and ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 56, 979–986.
Burén, S., Young, E. M., Sweeny, E. A., López-Torrejón, G., Veldhuizen, M., Voigt, C. A., et al. (2017). Formation of nitrogenase NifDK tetramers in the mitochondria of Saccharomyces cerevisiae. ACS Synth. Biol. 6, 1043–1055. doi: 10.1021/acssynbio.6b00371
Candat, A., Paszkiewicz, G., Neveu, M., Gautier, R., Logan, D. C., Avelange-Macherel, M.-H., et al. (2014). The ubiquitous distribution of late embryogenesis abundant proteins across cell compartments in Arabidopsis offers tailored protection against abiotic stress. Plant Cell 26, 3148–3166. doi: 10.1105/tpc.114.127316
Curatti, L., Hernandez, J. A., Igarashi, R. Y., Soboh, B., Zhao, D., and Rubio, L. M. (2007). In vitro synthesis of the iron-molybdenum cofactor of nitrogenase from iron, sulfur, molybdenum, and homocitrate using purified proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 17626–17631. doi: 10.1073/pnas.0703050104
Curatti, L., Ludden, P. W., and Rubio, L. M. (2006). NifB-dependent in vitro synthesis of the iron – molybdenum cofactor of nitrogenase. Proc. Natl. Acad. Sci. U.S.A. 103, 5297–5301. doi: 10.1073/pnas.0601115103
Curatti, L., and Rubio, L. M. (2014). Challenges to develop nitrogen-fixing cereals by direct nif-gene transfer. Plant Sci. 225, 130–137. doi: 10.1016/j.plantsci.2014.06.003
Dos Santos, P. C., Fang, Z., Mason, S. W., Setubal, J. C., and Dixon, R. (2012). Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13:162. doi: 10.1186/1471-2164-13-162
Echavarri-Erasun, C., Arragain, S., Scandurra, A. A., and Rubio, L. M. (2014). “Expression and purification of NifB proteins from aerobic and anaerobic sources,” in Metalloproteins: Methods and Protocols, eds J. C. Fontecilla-Camps and Y. Nicolet (Totowa, NJ: Humana Press), 19–31. doi: 10.1007/978-1-62703-794-5_3
Emerich, D. W., and Burris, R. H. (1976). Interactions of heterologous nitrogenase components that generate catalytically inactive complexes. Proc. Natl. Acad. Sci. U.S.A. 73, 4369–4373.
Emerich, D. W., and Burris, R. H. (1978). Complementary functioning of the component proteins of nitrogenase from several bacteria. J. Bacteriol. 134, 936–943.
Fish, W. W. (1988). Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364.
Galloway, J. N., Townsend, A. R., Erisman, J. W., Bekunda, M., Cai, Z., Freney, J. R., et al. (2008). Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320, 889–892. doi: 10.1126/science.1136674
Gietz, R. D., and Schiestl, R. H. (2007). Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 35–37. doi: 10.1038/nprot.2007.14
Guo, Y., Echavarri-Erasun, C., Demuez, M., Jimenez-Vicente, E., Bominaar, E. L., and Rubio, L. M. (2016). The nitrogenase FeMo-cofactor precursor formed by NifB protein: a diamagnetic cluster containing eight iron atoms. Angew. Chem. Int. Ed. Engl. 55, 12764–12767. doi: 10.1002/anie.201606447
Hill, H. D., and Straka, J. G. (1988). Protein determination using bicinchoninic acid in the presence of sulfhydryl reagents. Anal. Biochem. 170, 203–208. doi: 10.1016/0003-2697(88)90109-1
Huang, Z., Chen, Q., Hjelm, B., Arntzen, C., and Mason, H. (2009). A DNA replicon system for rapid high-level production of virus-like particles in plants. Biotechnol. Bioeng. 103, 706–714. doi: 10.1002/bit.22299
Ivleva, N. B., Groat, J., Staub, J. M., and Stephens, M. (2016). Expression of active subunit of nitrogenase via integration into plant organelle genome. PLOS ONE 11:e0160951. doi: 10.1371/journal.pone.0160951
Jeanthon, C., L’Haridon, S., Reysenbach, A. L., Vernet, M., Messner, P., Sleytr, U. B., et al. (1998). Methanococcus infernus sp. nov., a novel hyperthermophilic lithotrophic methanogen isolated from a deep-sea hydrothermal vent. Int. J. Syst. Bacteriol. 48(Pt 3), 913–919. doi: 10.1099/00207713-48-3-913
Jiménez-Vicente, E., Navarro-Rodríguez, M., Poza-Carrión, C., and Rubio, L. M. (2014). Role of Azotobacter vinelandii FdxN in FeMo-co biosynthesis. FEBS Lett. 588, 512–516. doi: 10.1016/j.febslet.2013.12.018
Johnson, D. C., Dean, D. R., Smith, A. D., and Johnson, M. K. (2005). Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 74, 247–281. doi: 10.1146/annurev.biochem.74.082803.133518
Kaiser, C., Michaelis, S., and Mitchell, A. (1994). Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Köhler, R. H., Zipfel, W. R., Webb, W. W., and Hanson, M. R. (1997). The green fluorescent protein as a marker to visualize plant mitochondria in vivo. Plant J. 11, 613–621. doi: 10.1046/j.1365-313X.1997.11030613.x
Leuzinger, K., Dent, M., Hurtado, J., Stahnke, J., Lai, H., Zhou, X., et al. (2013). Efficient agroinfiltration of plants for high-level transient expression of recombinant proteins. J. Vis. Exp. 77:e50521. doi: 10.3791/50521
Lill, R., and Muhlenhoff, U. (2008). Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700. doi: 10.1146/annurev.biochem.76.052705.162653
Lopez-Torrejon, G., Jimenez-Vicente, E., Buesa, J. M., Hernandez, J. A., Verma, H. K., Rubio, L. M., et al. (2016). Expression of a functional oxygen-labile nitrogenase component in the mitochondrial matrix of aerobically grown yeast. Nat. Commun. 7:11426. doi: 10.1038/ncomms11426
Nelson, B. K., Cai, X., and Nebenführ, A. (2007). A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136. doi: 10.1111/j.1365-313X.2007.03212.x
Oldroyd, G. E. D., and Dixon, R. (2014). Biotechnological solutions to the nitrogen problem. Curr. Opin. Biotechnol. 26, 19–24. doi: 10.1016/j.copbio.2013.08.006
Pan, R., Kaur, N., and Hu, J. (2014). The Arabidopsis mitochondrial membrane-bound ubiquitin protease UBP27 contributes to mitochondrial morphogenesis. Plant J. 78, 1047–1059. doi: 10.1111/tpj.12532
Rubio, L. M., and Ludden, P. W. (2008). Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 62, 93–111. doi: 10.1146/annurev.micro.62.081307.162737
Sanchez, P. A., and Swaminathan, M. S. (2005). Hunger in Africa: the link between unhealthy people and unhealthy soils. Lancet 365, 442–444. doi: 10.1016/S0140-6736(05)17834-9
Schmidt, T. G. M., Batz, L., Bonet, L., Carl, U., Holzapfel, G., Kiem, K., et al. (2013). Development of the Twin-Strep-tag and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr. Purif. 92, 54–61. doi: 10.1016/j.pep.2013.08.021
Shah, V. K., Allen, J. R., Spangler, N. J., and Ludden, P. W. (1994). In vitro synthesis of the iron-molybdenum cofactor of nitrogenase. Purification and characterization of NifB cofactor, the product of NIFB protein. J. Biol. Chem. 269, 1154–1158.
Shah, V. K., and Brill, W. J. (1973). Nitrogenase. IV. Simple method of purification to homogeneity of nitrogenase components from Azotobacter vinelandii. Biochim. Biophys. Acta 305, 445–454.
Smil, V. (2000). Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. Cambridge, MA: MIT Press.
Teste, M.-A., Duquenne, M., François, J. M., and Parrou, J.-L. (2009). Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol. Biol. 10:99. doi: 10.1186/1471-2199-10-99
Vicente, E. J., and Dean, D. R. (2017). Keeping the nitrogen-fixation dream alive. Proc. Natl. Acad. Sci. U.S.A. 114, 3009–3011. doi: 10.1073/pnas.1701560114
Vickers, C. E., Schenk, P. M., Li, D., Mullineaux, P. M., and Gresshoff, P. M. (2007). pGFPGUSPlus, a new binary vector for gene expression studies and optimising transformation systems in plants. Biotechnol. Lett. 29, 1793–1796. doi: 10.1007/s10529-007-9467-6
Vitousek, P. M., Aber, J. D., Howarth, R. W., Likens, G. E., Matson, P. A., Schindler, D. W., et al. (1997). Human alteration of the global nitrogen cycle: sources and consequences. Ecol. Appl. 7, 737–750.
Vögtle, F. N., Wortelkamp, S., Zahedi, R. P., Becker, D., Leidhold, C., Gevaert, K., et al. (2009). Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439. doi: 10.1016/j.cell.2009.07.045
Westermann, B., and Neupert, W. (2000). Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 16, 1421–1427.
Wiig, J. A., Hu, Y., Lee, C. C., and Ribbe, M. W. (2012). Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 337, 1672–1675. doi: 10.1126/science.1224603
Wilcoxen, J., Arragain, S., Scandurra, A. A., Jimenez-Vicente, E., Echavarri-Erasun, C., Pollmann, S., et al. (2016). Electron paramagnetic resonance characterization of three iron–sulfur clusters present in the nitrogenase cofactor maturase NifB from Methanocaldococcus infernus. J. Am. Chem. Soc. 138, 7468–7471. doi: 10.1021/jacs.6b03329
Witte, C. P., Noël, L. D., Gielbert, J., Parker, J. E., Romeis, T., Noe, L. D., et al. (2004). Rapid one-step protein purification from plant material using the eight-amino acid StrepII epitope. Plant Mol. Biol. 55, 135–147. doi: 10.1007/s11103-004-0501-y
Yuvaniyama, P., Agar, J. N., Cash, V. L., Johnson, M. K., and Dean, D. R. (2000). NifS-directed assembly of a transient [2Fe-2S] cluster within the NifU protein. Proc. Natl. Acad. Sci. U.S.A. 97, 599–604. doi: 10.1073/pnas.97.2.599
Keywords: nitrogen fixing plants, yeast, mitochondria, SAM-radical, iron-molybdenum cofactor
Citation: Burén S, Jiang X, López-Torrejón G, Echavarri-Erasun C and Rubio LM (2017) Purification and In Vitro Activity of Mitochondria Targeted Nitrogenase Cofactor Maturase NifB. Front. Plant Sci. 8:1567. doi: 10.3389/fpls.2017.01567
Received: 31 May 2017; Accepted: 28 August 2017;
Published: 12 September 2017.
Edited by:
Nikolai Provorov, All-Russian Research Institute of Agricultural Microbiology of the Russian Academy of Agricultural Sciences, RussiaReviewed by:
Oswaldo Valdes-Lopez, National Autonomous University of Mexico, MexicoZonghua Wang, Fujian Agriculture and Forestry University, China
Copyright © 2017 Burén, Jiang, López-Torrejón, Echavarri-Erasun and Rubio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis M. Rubio, bG0ucnViaW9AdXBtLmVz