
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 28 August 2017
Sec. Plant Breeding
Volume 8 - 2017 | https://doi.org/10.3389/fpls.2017.01448
 Zhen Huang1,2
Zhen Huang1,2 Gary Peng1
Gary Peng1 Xunjia Liu1
Xunjia Liu1 Abhinandan Deora3Kevin C. Falk1
Abhinandan Deora3Kevin C. Falk1 Bruce D. Gossen1
Bruce D. Gossen1 Mary R. McDonald3
Mary R. McDonald3 Fengqun Yu1*
Fengqun Yu1*Clubroot, caused by Plasmodiophora brassicae, is an important disease of canola (Brassica napus) in western Canada and worldwide. In this study, a clubroot resistance gene (Rcr2) was identified and fine mapped in Chinese cabbage cv. “Jazz” using single-nucleotide polymorphisms (SNP) markers identified from bulked segregant RNA sequencing (BSR-Seq) and molecular markers were developed for use in marker assisted selection. In total, 203.9 million raw reads were generated from one pooled resistant (R) and one pooled susceptible (S) sample, and >173,000 polymorphic SNP sites were identified between the R and S samples. One significant peak was observed between 22 and 26 Mb of chromosome A03, which had been predicted by BSR-Seq to contain the causal gene Rcr2. There were 490 polymorphic SNP sites identified in the region. A segregating population consisting of 675 plants was analyzed with 15 SNP sites in the region using the Kompetitive Allele Specific PCR method, and Rcr2 was fine mapped between two SNP markers, SNP_A03_32 and SNP_A03_67 with 0.1 and 0.3 cM from Rcr2, respectively. Five SNP markers co-segregated with Rcr2 in this region. Variants were identified in 14 of 36 genes annotated in the Rcr2 target region. The numbers of poly variants differed among the genes. Four genes encode TIR-NBS-LRR proteins and two of them Bra019410 and Bra019413, had high numbers of polymorphic variants and so are the most likely candidates of Rcr2.
Plasmodiophora brassicae Woronin is a biotrophic soil-borne pathogen in the Infrakingdom Rhizaria (Nikolaev et al., 2004) that causes clubroot disease in Brassica oil and vegetable crops. It produces long-lived resting spores that are difficult to control using standard strategies such as anti-microbial compounds or crop rotations (Voorrips, 1995; Tsushima, 2000). Therefore, genetic resistance is the most effective approach to manage this disease.
Clubroot is an increasing problem on canola (Brassica napus) in western Canada and worldwide. Sources of clubroot resistance in canola are limited, but genotypes with resistance to a broad range of pathotypes of P. brassicae have been identified in the canola progenitor species B. rapa (Hasan et al., 2012; Peng et al., 2014). These lines could be used to broaden the genetic base of clubroot resistance in canola (Yu et al., 2016). Introgression of important agronomic traits such as clubroot resistance from B. rapa into canola is possible through conventional breeding (Yu et al., 2012; Gautam et al., 2013; Li et al., 2013), but it is important to identify and map the resistance genes in B. rapa so that genes can be transferred into canola efficiently.
More than 10 genes conferring resistance to clubroot have been identified in vegetable cultivars of B. rapa (Kuginuki et al., 1997; Matsumoto et al., 1998, 2012; Suwabe et al., 2003, 2006; Hirai et al., 2004; Piao et al., 2004, 2014; Saito et al., 2006; Sakamoto et al., 2008; Cho et al., 2012; Chen et al., 2013; Kato et al., 2013; Peng et al., 2014; Zhang et al., 2014). For example, resistance genes Crr1, Crr2, and Crr4 derived from the European turnip cv. “Siloga” were mapped onto B. rapa chromosomes A08, A01, and A06, respectively (Suwabe et al., 2003, 2006). Crr3 from turnip cv. “Milan White” was mapped onto A03 (Hirai et al., 2004; Saito et al., 2006), while CRk and CRc from turnip cv. “Debra” were mapped onto A03 and A02, respectively (Sakamoto et al., 2008; Matsumoto et al., 2012). CRa from turnip line ECD02 (Matsumoto et al., 1998) and CRb from turnip line ECD01 (Piao et al., 2004) were also mapped to A03. A clubroot resistance gene, Rcr1, identified in pak choy cv. “Flower Nabana” (B. rapa) was also mapped to A03 (Chu et al., 2014; Yu et al., 2016). Three resistance genes Rcr4, Rcr8, and Rcr9 from B. rapa canola breeding line T19 were mapped to A03, A02, and A08, respectively through genotyping by sequencing (Yu et al., 2017). Two genes, Crr1 and CRa, have been cloned that encode Toll-Interleukin-1 receptor/nucleotide binding site/leucine-rich repeat (TIR-NBS-LRR, TNL) proteins (Ueno et al., 2012; Hatakeyama et al., 2013).
Genetic mapping is a powerful approach for identification of causal genes associated with economically important traits and development of molecular markers tightly linked to the traits for marker assisted selection in plant breeding. Several types of molecular markers, such as restriction fragment length polymorphism, microsatellite or simple sequence repeats, and single-nucleotide polymorphisms (SNP) have been employed in genetic mapping. However, discovery of the molecular markers through conventional approaches is very time consuming, tedious, and expensive. Next-generation sequencing (NGS) for genetic mapping has facilitated the rapid development and application of genomics tools in plant breeding (Bolger et al., 2014). NGS has been used for genetic mapping. An important mapping technique, bulked segregant analysis (BSA), consists of two bulks of individual plants with extreme phenotypes, has been utilized—for many years to identify markers linked to major genes of interest, such as those for disease resistance (Michelmore et al., 1991). BSA has also been coupled with RNA sequencing (RNA-Seq) for gene mapping (Liu et al., 2012).
Screening of Brassica cultivars for clubroot resistance to the predominant pathotypes of P. brassicae identified in Canada was performed. Several B. rapa vegetable cultivars, including pak choy cv. “Flower Nabana” and Chinese cabbage (B. rapa subsp. pekinensis) cv. “Jazz,” were resistant to clubroot (Peng et al., 2014). Also, the clubroot resistance gene Rcr1 in “Flower Nabana” was identified (Chu et al., 2014). The current study describes the identification and mapping of a clubroot resistance gene from Chinese cabbage cv. “Jazz” using SNP markers identified with bulked segregant RNA-Seq (BSR-Seq).
The objectives of the study were to: (i) determine the inheritance of clubroot resistance in cv. “Jazz”; (ii) map the resistance gene; (iii) develop SNP markers tightly linked to the resistance gene; and (iv) examine DNA variation in the target region and identify the most probable candidate(s) for the gene.
Chinese cabbage cv. “Jazz” (American Takii, Salinas, CA, United States) is a hybrid that is highly resistant to the five main pathotypes of P. brassicae found in Canada (Peng et al., 2014). One plant was used to pollinate a doubled-haploid canola-quality line of B. rapa, ACDC (Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada) that is self-compatible and highly susceptible to P. brassicae. Reciprocal crosses were made between “Jazz” and ACDC to produce F1 progenies. The parents and F1 plants were tested for resistance to pathotype 3 (Williams, 1966) of P. brassicae. Segregation of resistant (R) and susceptible (S) phenotypes in the F1 segregating populations was analyzed using Chi-square (χ2) tests for goodness of fit (Sokal and Rohlf, 1995).
In addition, F1 plants were self-pollinated, but only 46 of 200 plants produced seed because of self-incompatibility. Plants from the 46 self-fertile lines (F2) were used to evaluate resistance to additional pathotypes of P. brassicae. The F2 lines were equivalent to self-pollinated BC1 populations (BC1S1).
A field collection of pathotype 3 of P. brassicae was used to inoculate plants for studies on inheritance and genetic mapping. Preparation of inoculum, inoculation and plant rating followed standard protocols (Chu et al., 2014). The clubroot reaction of F2 lines to pathotypes 2, 3, 5, 6, and 8 was assessed under controlled conditions (Yu et al., 2016). Clubroot severity on 7-14 plants per line at 6 weeks after inoculation was assessed using a standard 0–3 scale. A disease severity index (DSI; Strelkov et al., 2006) was calculated using the following formula:
A highly susceptible control, Shanghai pak choy cv. “Mei Qing Choi” (Stokes Seeds, ON, Canada), was included with each group to ensure that inoculation was effective. The evaluation of clubroot reaction to pathotypes was repeated.
Correlation coefficients among the DSI values in F2 families to the five pathotypes of P. brassicae were calculated using Microsoft Excel. The significance of the correlation coefficients was determined with t-tests (Iversen and Gergen, 1997).
The F1 population from ACDC (female) × “Jazz” (male) was used for RNA-Seq. At 15 days post-inoculation, leaf tissue from 30 R plants and 30 S plants was combined to form R and S bulks, respectively; together, the two bulks comprised one biological replicate. Three replicates with a total of 90 R and 90 S plants were assessed. RNeasy Plant Mini Kit (Qiagen; Toronto, ON, Canada) with on-column deoxyribonuclease (DNase) digestion was used for RNA extraction from each sample following the manufacturer’s instructions. The RNA concentration and quality were checked using a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA, United States) and an Agilent Bioanalyzer 2100 (Agilent Technologies; Mississauga, CA, United States), to ensure that the RNA integrity number (RIN) was >8 for each sample. The preparation of cDNA libraries, RNA-Seq and DNA alignment were performed at Databio2 LLC (Ames, IA, United States). Prior to alignment, the nucleotides of each raw read were scanned for quality, and bases with PHRED quality value <15 (out of 40) (Ewing and Green, 1998; Ewing et al., 1998) were removed by the trimming pipeline at Databio2 LLC. Trimmed short reads were aligned to the reference genome (B. rapa, V1.51) using GSNAP (Wu and Nacu, 2010) as paired-end fragments with ≤2 mismatches. Short reads from the three R bulks were combined and assembled into the reference genome, as were the three S bulks, to produce separate assembly files, pooled R and pooled S.
SNP discovery and mapping of the causal gene were performed using the uniquely aligned short reads from the two pooled samples with Databio2 LLC. Polymorphisms at each potential SNP site were examined and putative SNPs were identified using the following criteria: (1) the first and last three aligned bases of each read were discarded; (2) each polymorphic base must have a PHRED base quality value of at least 15; (3) at least five unique reads must support the base-pair call. An empirical Bayesian approach was used to estimate the conditional probability of no recombination between each SNP marker and the causal gene in both the R pool and the S pool (Liu et al., 2012). The posterior probability of each SNP indicates the probability of complete linkage between the SNP and causal gene.
The same genotyping method as that described by Yu et al. (2016) was used in this study. Genomic DNA was extracted from young leaves of F1 segregating population consisting of 675 plants (including the 180 plants for RNA-Seq) using the CTAB method (Doyle and Doyle, 1990). Selected SNPs identified in the target region were confirmed using the Kompetitive Allele Specific PCR (KASP) method2 following the manufacture’s instruction. PCR reactions were performed using a StepOne Plus Real Time PCR System (Applied Biosystem, Mississauga, ON, Canada). Linkage groups were performed with JoinMap 4.1 (Van Ooijen and Voorrips, 2001).
The software SeqMan NGen 13 (DNASTAR, Madison, WI, United States) was used for short read assembly using the pooled sample assembly method (Yu et al., 2016). Standard assembling and filtering parameters were used. Discovery of SNP and InDel variants from the DNA sequences in B. rapa “Chiifu” were performed using software SeqMan Pro 13 (DNASTAR) with Q call ≥15 and depth ≥5.
Each “Jazz” plant was resistant to pathotype 3 of P. brassicae and all of the ACDC plants were susceptible, with characteristic clubroot symptoms at 5 weeks after inoculation (Figure 1). Segregation for R and S consistent with an expected ratio of 1:1 was observed in the F1 populations derived from the reciprocal crosses (Table 1). These results indicated that the resistance in “Jazz” was associated with a single dominant nuclear gene (designated as Rcr2) and that “Jazz” was heterozygous at the Rcr2 locus.

FIGURE 1. Phenotypes of the Brassica rapa parents: Chinese cabbage cv. “Jazz” (R) and susceptible canola breeding line ACDC. Plants were inoculated with pathotype 3 of Plasmodiophora brassicae.
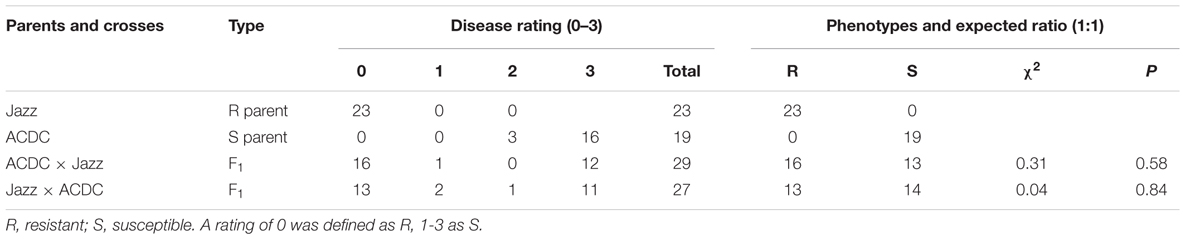
TABLE 1. Genetic analysis of clubroot resistance to pathotype 3 of Plasmodiophora brassicae in crosses of Brassica rapa cv. “Jazz” with line ACDC.
“Jazz” was identified previously as highly resistant to several pathotypes (2, 3, 5, 6, and 8) of P. brassicae (Peng et al., 2014), but Rcr2 was identified by testing with pathotype 3 only. To determine if clubroot resistance to pathotypes 2, 5, 6, and 8 in “Jazz” was associated with Rcr2, 46 F2 lines were tested for resistance to the five pathotypes. “Jazz” was highly resistant (DSI = 0), while both ACDC and the susceptible control (Shanghai pak choy cv. “Mei Qing Choi”) were highly susceptible (DSI = 100). The distributions of DSI values to the five pathotypes among the F2 lines were similar (Supplementary Figure S1). The clubroot reaction among the pathotypes was significantly correlated (Table 2).

TABLE 2. Correlation coefficients of clubroot severity among the 46 F2 lines to pathotypes 2, 3, 5, 6, and 8 of Plasmodiophora brassicae.
A total of 203.9 million raw reads were generated from the R and S pooled samples. About 98% of the reads passed the quality control standard and remained. After trimming, the average length of reads was reduced from 101 bases to 98–99 bases. A total of 86.16% of the trimmed reads from two pooled samples could be mapped to the reference genome (B. rapa_sequence_v1.5.fa), and 78.13% of the trimmed reads were unique alignments (Table 3).
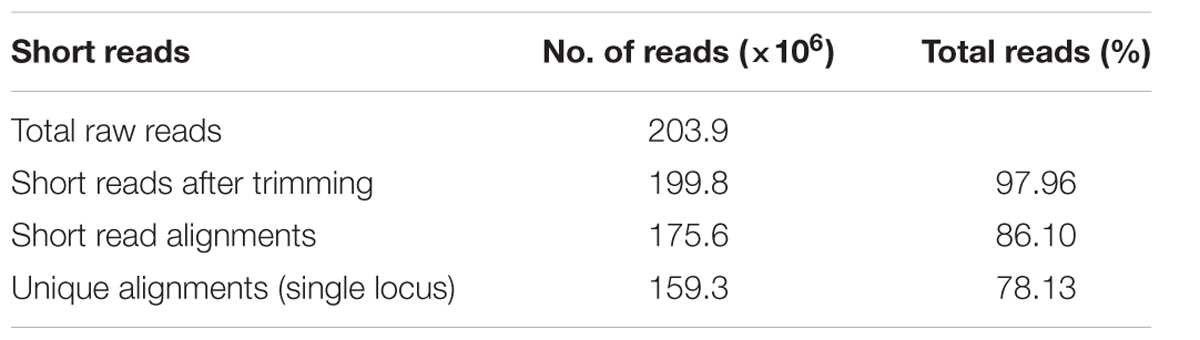
TABLE 3. Summary of RNA-Seq short reads from the R pooled sample and the S pooled sample in the F1 population from crosses of Chinese cabbage cv “Jazz” with the doubled haploid line ACDC.
Unique alignments from the pooled R and the pooled S samples were extracted separately for SNP discovery. Reads were mapped to both chromosomes and scaffolds before assigning SNPs. A total of 173,383 polymorphic SNP between the pooled R and pooled S samples were identified with 170,515 SNPs in the 10 chromosomes and 2,866 SNPs in the scaffolds (Table 4). The numbers of polymorphic SNP varied among the chromosomes, with strong positive correlation with length of chromosome (r = 0.92). This supported a previous observation (Yu et al., 2016) that there were many more polymorphic SNPs on longer chromosomes (A03, A09) than shorter chromosomes (A04, A10).

TABLE 4. Length of chromosomes and number of polymorphic SNPs identified from BSR-Seq.
Mapping of Rcr2 were carried out using the 173,383 SNPs through BSR-Seq. The linkage probability of each SNP was plotted against its physical coordinate in the B. rapa reference genome. Each of the SNP markers that had a high probability of being linked to the Rcr2 gene clustered on chromosome A03. No SNP markers with high linkage probability were observed on any other chromosomes. One significant peak was observed between 20 and 32 Mb of chromosome A03 (Figure 2A). To narrow down the interval within which Rcr2 is located, the length of chromosome A03 was scanned using a window of a fixed number of SNPs (N = 50) and a step size of five SNPs. A strong peak, indicating a high probability of complete linkage disequilibrium with Rcr2, was observed at physical position 22–26 Mb of the B. rapa reference genome (Figure 2B).
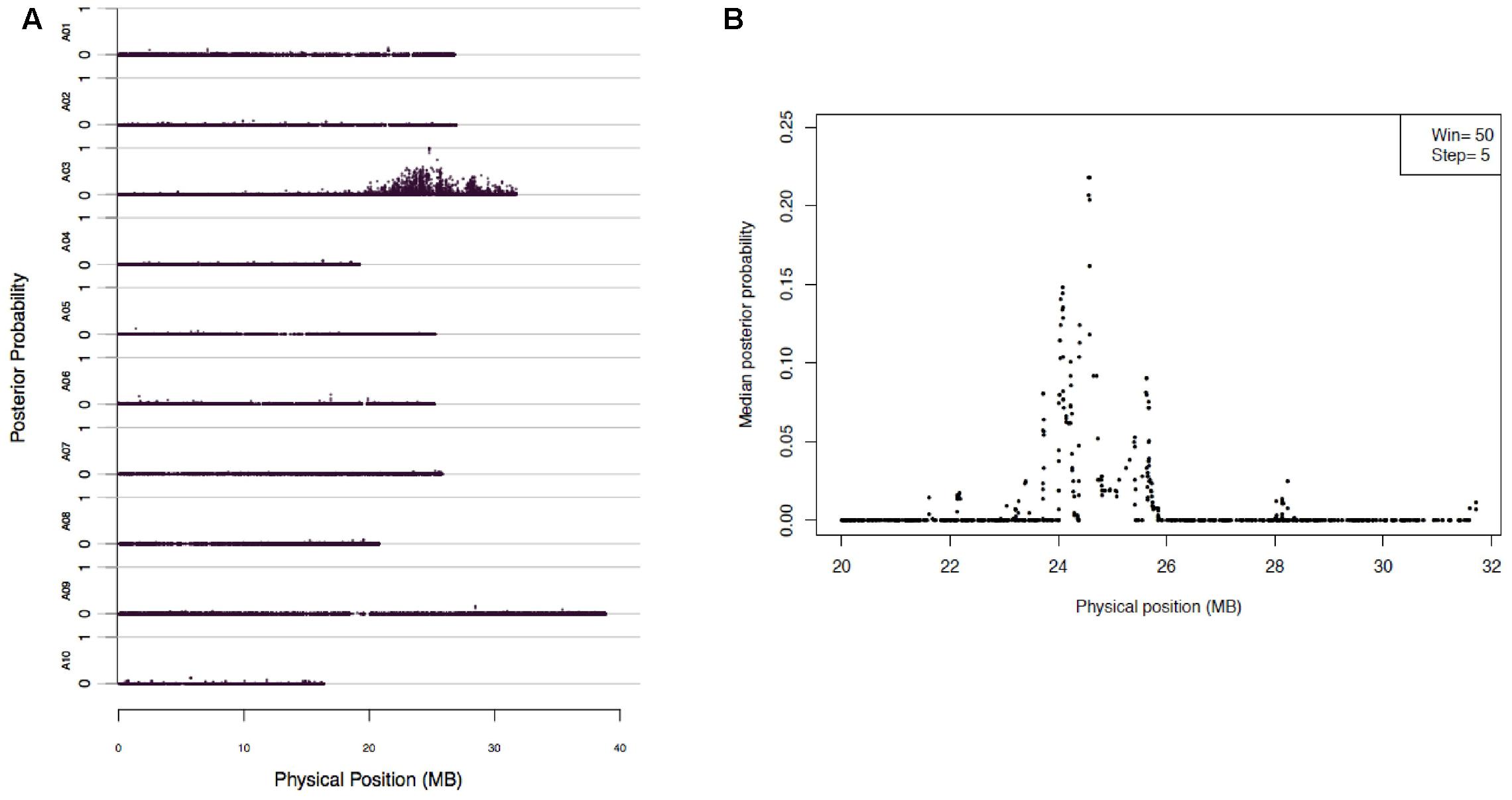
FIGURE 2. BSR-Seq to map Rcr2 according to the method described by Liu et al. (2012). (A) The physical position of each SNP marker (x-axis) was plotted versus the probability of each SNP marker being in complete linkage disequilibrium with the causal gene (y-axis). (B) Chromosome A03 was scanned by using a window containing 50 SNPs with a step size of five SNPs. Within each window, the median linkage probability obtained from a Bayesian BSA analysis across all the 50 SNPs was determined and was plotted against the middle physical position of the window.
There were 490 SNP sites identified at physical position 22–26 Mb of chromosome A03 where Rcr2 was mapped using BSR-Seq (Supplementary Table S1). To fine map the gene, 675 plants were analyzed using the KASP technology, including 90 R and 90 S plants for RNA-Seq in the F1 segregating with 15 SNP markers (Supplementary Table S2). This analysis indicated that Rcr2 was flanked by SNP_A03_32 and SNP_A03_67 at 0.1 and 0.3 cM from the markers, respectively, in an interval of 0.4 cM. Five SNP markers (SNP_A03_08, SNP_A03_09, SNP_A03_11, SNP_A03_13, SNP_A03_19) co-segregated with Rcr2 (Figure 3).
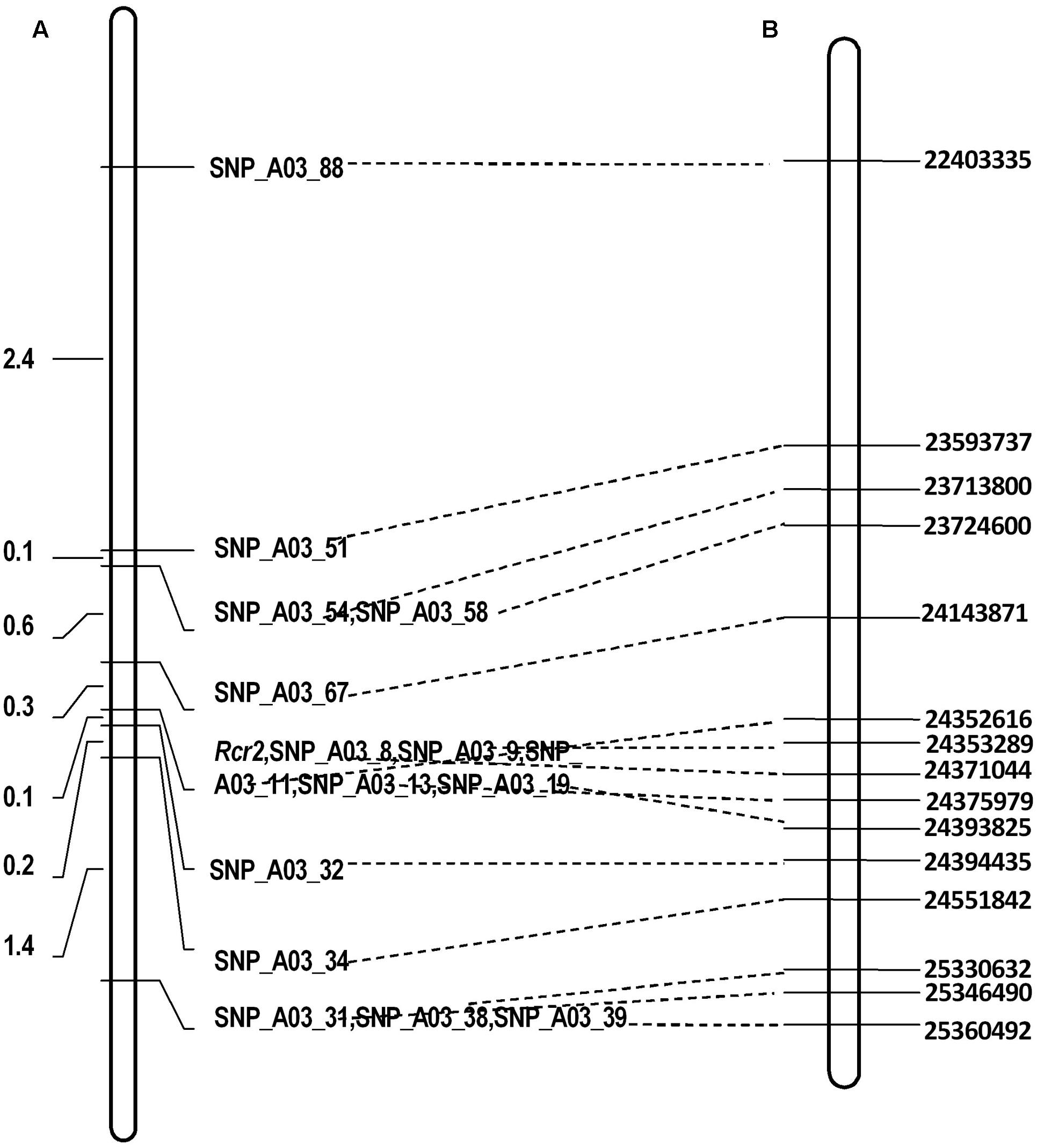
FIGURE 3. Linkage maps of the regions in which the Rcr2 gene is located. Broken lines drawn regions defined by different molecular markers on B. rapa linkage group A03. (A) Fine mapping of Rcr2 based on a F1 population consisting of 675 plants derived from ACDC × Jazz. The genetic distance is shown on the left. (B) Physical locations in bases (right) of the SNP markers.
SNP_A03_32 is located in gene Bra019406 at site 24,143,871 and SNP_A03_67 in gene Bra038775 at site 24,394,435 (Figure 3). The physical distance between these two markers was 250,565 bases. There were 36 genes annotated in this region of reference genome v1.5.
The numbers of polymorphic variants (SNP and InDel) between the R and S bulks was assessed in the genes in the target region. Fourteen genes (Bra038774, Bra038770, Bra038768, Bra038767, Bra038766, Bra038765, Bra038758, Bra038756, Bra038755, Bra038751, Bra038750, Bra038749, Bra019411, and Bra019408) in the region did not show any expression and no short reads were assembled into the reference genome, so no variants could be identified in the genes. Similarly, eight genes (Bra038773, Bra038772, Bra038769, Bra038763, Bra038759, Bra038748, Bra038747, and Bra019407) had short reads with sequence counts <5 in either the R or S pooled sample, and number of poly variants <1 and so variants could not be identified. The remaining 14 genes had >5 sequence counts in both the R and S pooled samples, but the numbers of polymorphic variants differed among the genes (Table 5). Four genes (Bra019409, Bra019410, Bra019412, and Bra019413) encode TNL-class disease resistance proteins, assessed using BLASTX to A. thaliana and gene ontology annotation from http://brassicadb.org/brad/index.php (Supplementary Table S3).
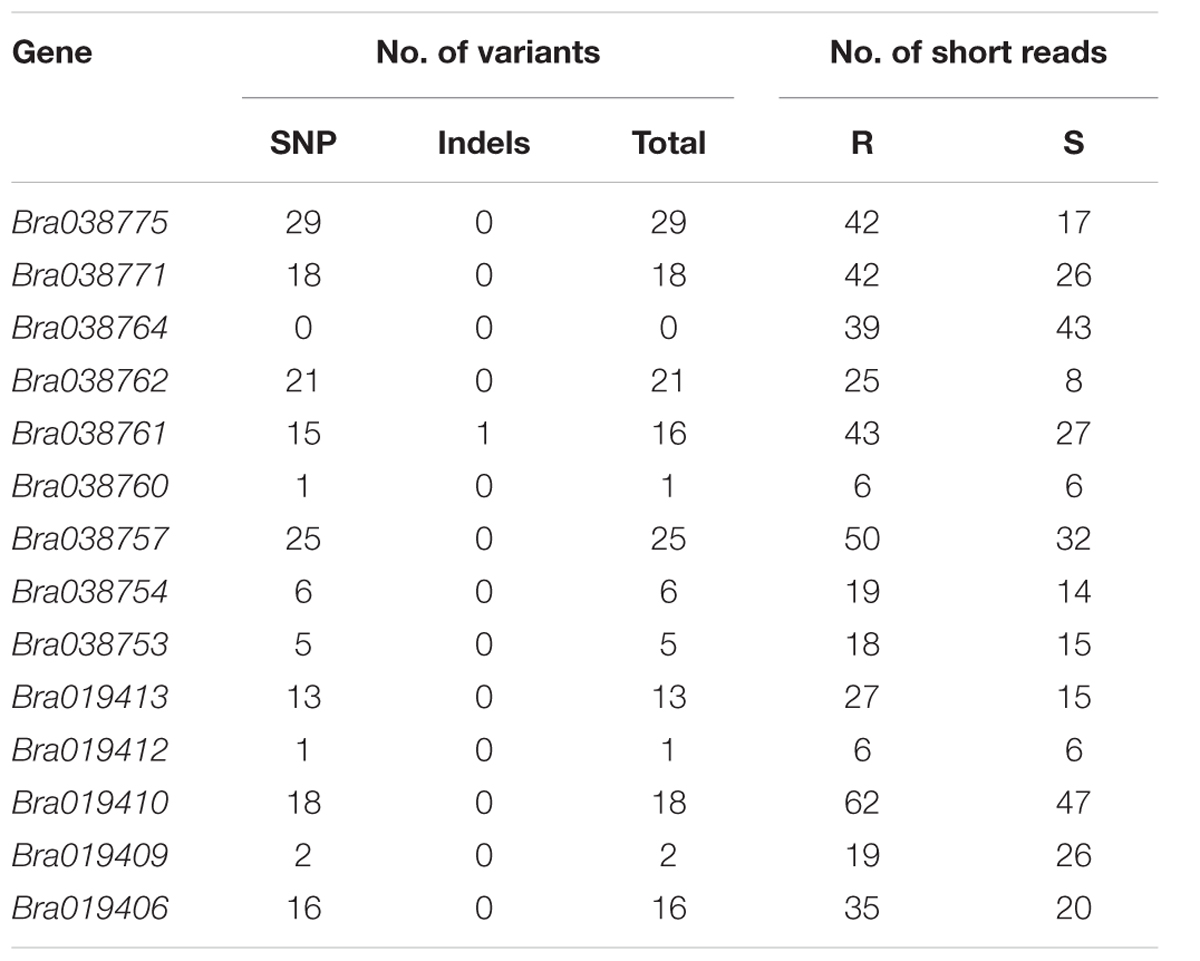
TABLE 5. Number of polymorphic variants (SNPs and Indels) in the 14 genes located in the Rcr2 interval that could be identified.
In total, 34 synonymous and non-synonymous variants were identified from the coding sequences in the four TNL genes (Supplementary Table S4). Non-synonymous variants occurred in each of the TNL genes, but Bra019410 and Bra019413 carried more non-synonymous variants that uniquely occurred in the R samples than the other two TNL genes (Figure 4). Synonymous variants also occurred in Bra019410 and Bra019413 (Figure 4). No nonsense or frameshift variants were identified.
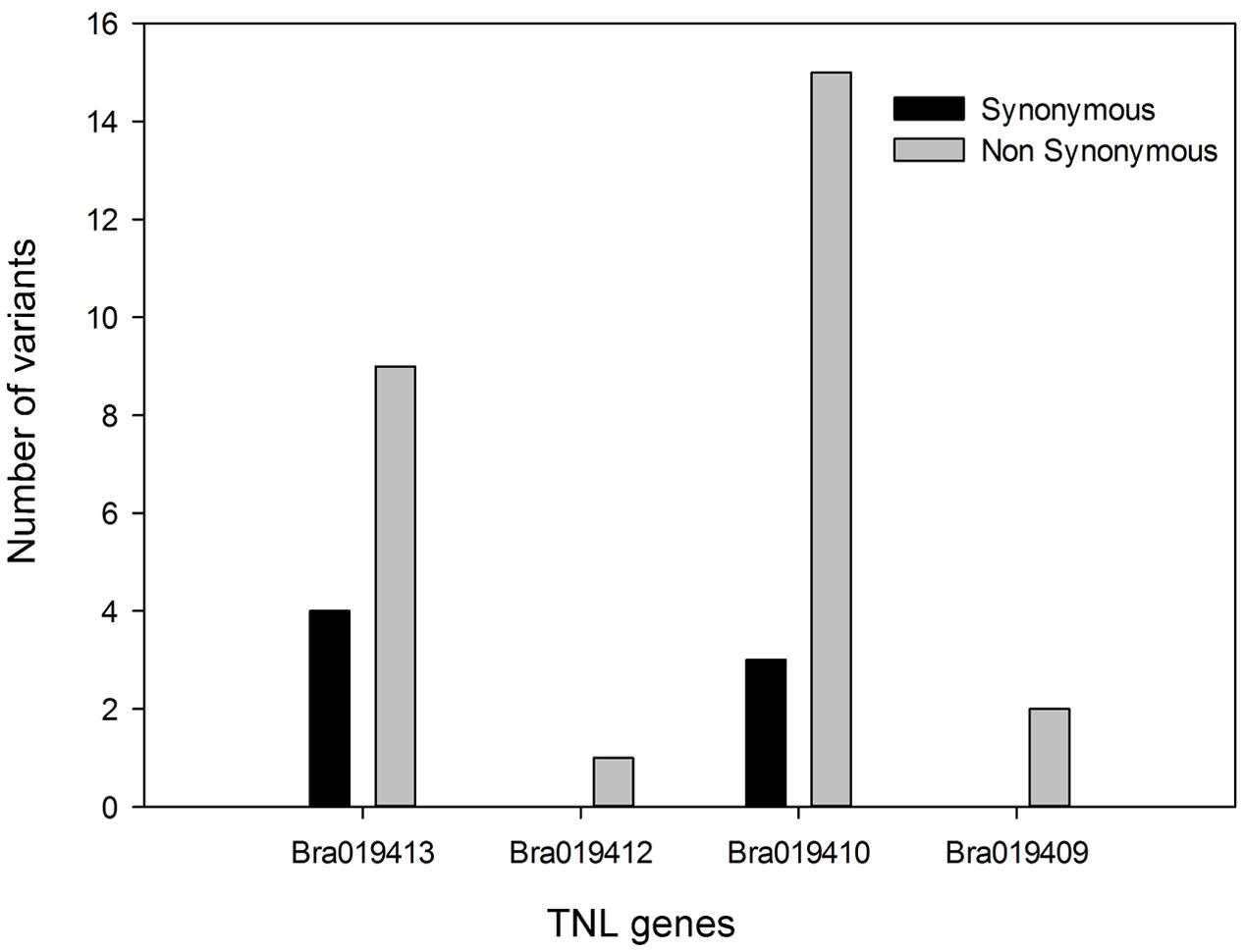
FIGURE 4. Number of unique poly variants in the four TIR-NBS-LRR genes from the pooled R samples.
RNA-Seq is a robust approach for quantifying gene expression that has been widely used for transcriptome analysis in a range of organisms (Liu et al., 2012). It also can be used for identification of DNA variants. A BSR-Seq mapping strategy that relied on RNA-Seq data was used to map the gl3 locus in maize (Liu et al., 2012). A similar strategy was used in the current study to map Rcr2 into B. rapa chromosome A03 in an interval of 4 Mb region. Almost 500 SNP sites from the BSR-Seq were identified in the Rcr2 interval, and 15 SNP markers were genotyped in the F1 segregating population (675 plants) using KASP assays. Rcr2 was further narrowed into an interval of 0.251 Mb through linkage analysis. This demonstrates that BSR-Seq in combination with KASP analysis is a powerful approach for fine mapping of causal genes.
In Canada, five pathotypes of P. brassicae (pathotypes 2, 3, 5, 6, and 8) were initially identified based on the differential system of Williams (1966), with pathotype 3 the most prevalent on canola in the prairie region (Strelkov et al., 2006, 2007). Several canola cultivars were developed that exhibited strong resistance to these pathotypes (Strelkov et al., 2007, 2016). However, this resistance has been overcome recently by new strains of the pathogen (Strelkov et al., 2016). Both Rcr2 donor (Chinese cabbage cv. “Jazz”) and Rcr1 (pak choy cv. “Flower Nabana”) donor were highly resistant to pathotypes 2, 3, 5, 6, and 8 (Peng et al., 2014). However, the Chinese cabbage cv. “Jazz” was susceptible to the new strains of the pathogen while the pak choy cv. “Flower Nabana” was resistant (F. Yu and G. Peng, unpublished data). This indicated that Rcr2 might confer a different resistance specificity from Rcr1. Another possibility was that pak choy cv. “Flower Nabana” carried additional gene(s) for resistance to the new strains. Clearly, this still needs to be determined.
Rcr2 was initially mapped to chromosome A03 based on clubroot reaction to pathotype 3. However, donor cv. “Jazz” was also resistant to each of the other four pathotypes previously identified in Canada (Peng et al., 2014). To determine if Rcr2 conferred resistance to the range of Canadian pathotypes, 46 F2 lines originating from 46 F1 plants were developed. Resistance to pathotypes 2, 3, 5, 6, and 8 was associated in each of the 46 lines (Table 2). This demonstrated that resistance to pathotypes 2, 5, 6, and 8 was significantly correlated with Rcr2 and resistance to all of the original Canadian pathotypes of P. brassicae were controlled by the same gene or linked genes. Similar results were obtained from the F2 lines containing Rcr1 (Yu et al., 2016) and BC1S1 lines containing Rcr4 (Yu et al., 2017). A resistance gene on chromosome A08 in rutabaga (B. napus subsp. napobrassica) also conferred resistance to all five pathotypes (Hasan and Rahman, 2016).
In the current study, Rcr2 was mapped in an interval of 0.4 cM, flanked by SNP markers SNP_A03_32 and SNP_A03_67 on B. rapa chromosome A03. Five markers (SNP_A03_19, SNP_A03_13, SNP_A03_09, SNP_A03_11, and SNP_A03_08) that reside in four genes (Bra019406, Bra019410, Bra019412, and Bra019413) in the target region were genotyped using the KASP method. Each of the markers co-segregated with Rcr2, indicating that they were completely linked to Rcr2. These SNP markers also co-segregated with Rcr1 (Yu et al., 2016), so it is possible that Rcr2 is co-localized with Rcr1.
Four genes in the Rcr1/Rcr2 interval belong to TNL gene families. The DNA variant profiles for the TNL genes for Rcr2 differ from those for Rcr1 (Yu et al., 2016). No nonsense or frameshift variants were found in the TNL genes (Figure 4), but all four types of variants (synonymous, non-synonymous, nonsense, and frameshift) were identified in the genes associated with Rcr1 (Yu et al., 2016). This could be associated with a lower depth of sequencing used in the current study relative to the depth used in the identification of Rcr1, such that the entire range of DNA variants in the Rcr2 interval was not fully captured.
Based on the analysis of polymorphic variants in the genes in the current study, it is unlikely that the genes Bra019409 and Bra019412 are candidates for Rcr2 because few polymorphic variants were identified in these genes. Bra019410 and Bra019413 carried higher number of variants, especially those that result in changes in amino acid sequence. They also carried higher number of synonymous variants, which can affect protein conformation and function (Sauna and Kimchi-Sarfaty, 2011). However, there were more polymorphic variants identified in Bra019409 and Bra019410 than in Bra019412 and Bra019413 in the Rcr1 interval (Yu et al., 2016). Rcr1/Rcr2 were mapped in the same interval as a known clubroot resistance gene, CRa. However, previous characterization of CRa (Ueno et al., 2012) did not determine how it was related to the TNL genes in the target region. The complete CRa coding sequence (GenBank: AB751517.1) consists of 4223 bp (Yu et al., 2016). A Blast search3 indicated that CRa is homologous to the TNL genes identified (especially Bra019410), but distinctly different to these genes (Yu et al., 2016). In addition, a QTL for clubroot resistance, Rcr4, has also been mapped to the Rcr1/Rcr2/CRa region (Yu et al., 2017). The TNL gene(s) that correspond with Rcr2, and the relationship of Rcr2 with Rcr1/Rcr4 or CRa, can only be identified conclusively after all of the genes have been cloned. Cloning could also be used to resolve the uncertainty of whether Rcr2 confers resistance against several pathotypes or represents several tightly linked genes.
In total, 15 robust SNP markers were associated with Rcr2. These markers could provide an effective and robust basis for introgression of Rcr2 into canola using MAS.
FY and GP conceived of the study; FY designed the experiments, analyzed data, designed KASP primers, and found SNP flanking sequences; ZH performed gene mapping and analysis of KASP markers; GP and KF provided parental lines. ZH, XL, AD, BG, and MM tested plants for resistance to clubroot; ZH and FY drafted the manuscript. All authors reviewed the manuscript and approved the final draft.
This work was funded by A-base fund from Agriculture and Agri-Food Canada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Mr. Adrian Chang for assistance on developing the F2 population and Ms. Linda McGregor for assistance on phenotyping.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01448/full#supplementary-material
FIGURE S1 | The distribution of DSI values to five pathotypes in the F2 population consisting of 46 lines derived from ACDC × Jazz.
Bolger, M. E., Weisshaar, B., Scholz, U., Stein, N., Usadel, B., and Mayer, K. F. X. (2014). Plant genome sequencing-applications for crop improvement. Curr. Opin. Biotechnol. 26, 31–37. doi: 10.1016/j.copbio.2013.08.019
Chen, J., Jing, J., Zhan, Z., Zhang, T., Zhang, C., and Piao, Z. (2013). Identification of novel QTLs for isolate-specific partial resistance to Plasmodiophora brassicae in Brassica rapa. PLoS ONE 8:e85307. doi: 10.1371/journal.pone.0085307
Cho, K. H., Park, S. H., Kim, K. T., Kim, S., Kim, J. S., and Park, B. S. (2012). Mapping quantitative trait loci (QTL) for clubroot resistance in Brassica rapa L. J. Hortic. Sci. Biotechnol. 87, 325–333. doi: 10.1080/14620316.2012.11512872
Chu, M., Song, T., Falk, K. C., Zhang, X., Liu, X., Chang, A., et al. (2014). Fine mapping of Rcr1 and analyses of its effect on transcriptome patterns during infection by Plasmodiophora brassicae. BMC Genomics 15:1166. doi: 10.1186/1471-2164-15-1166
Ewing, B., and Green, P. (1998). Base-calling of automated sequencer traces using phred. II. error probabilities. Genome Res. 8, 186–194. doi: 10.1101/gr.8.3.186
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998). Base-calling of automated sequencer traces using phred. I. accuracy assessment. Genome Res. 8, 175–185. doi: 10.1101/gr.8.3.175
Gautam, M., Ge, X., and Li, Z. (2013). Brassica (book chapter). Alien Gene Transf. Crop Plants 2, 207–229.
Hasan, M. J., and Rahman, H. (2016). Genetics and molecular mapping of resistance to Plasmodiophora brassicae pathotypes 2, 3, 5, 6, and 8 in rutabaga (Brassica napus var. napobrassica). Genome 59, 805–815. doi: 10.1139/gen-2016-0034
Hasan, M. J., Strelkov, S. E., Howard, R. J., and Rahman, H. (2012). Screening of Brassica germplasm for resistance to Plasmodiophora brassicae pathotypes prevalent in Canada for broadening diversity in clubroot resistance. Can. J. Plant Sci. 92, 501–515. doi: 10.4141/CJPS2010-006
Hatakeyama, K., Suwabe, K., Tomita, R. N., Kato, T., Nunome, T., Fukuoka, H., et al. (2013). Identification and characterization of Crr1a, a gene for resistance to Clubroot Disease (Plasmodiophora brassicae Woronin) in Brassica rapa L. PLoS ONE 8:e54745. doi: 10.1371/journal.pone.0054745
Hirai, M., Harada, T., Kubo, N., Tsukada, M., Suwabe, K., and Matsumoto, S. (2004). A novel locus for clubroot resistance in Brassica rapa and its linkage markers. Theor. Appl. Genet. 108, 639–643. doi: 10.1007/s00122-003-1475-x
Kato, T., Hatakeyama, K., Fukino, N., and Matsumoto, S. (2013). Fine mapping of the clubroot resistance gene CRb and development of a useful selectable marker in Brassica rapa. Breed. Sci. 63, 116–124. doi: 10.1270/jsbbs.63.116
Kuginuki, Y., Ajisaka, H., Yui, M., Yoshikawa, H., and Hida, K. (1997). RAPD markers linked to a clubroot-resistance locus in Brassica rapa L. Euphytica 98, 149–154. doi: 10.1023/A:1003147815692
Li, Q., Mei, J., Zhang, Y., and Li, J. (2013). A large-scale introgression of genomic components of Brassica rapa into B. napus by the bridge of hexaploid derived from hybridization between B. napus and B. oleracea. Theor. Appl. Genet. 126, 2073–2080. doi: 10.1007/s00122-013-2119-4
Liu, S., Yeh, C., Tang, H. M., Nettleton, D., and Schnable, P. S. (2012). Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 7:e36406. doi: 10.1371/journal.pone.0036406
Matsumoto, E., Ueno, H., Aruga, D., Sakamoto, K., and Hayashida, N. (2012). Accumulation of three clubroot resistance genes through marker-assisted selection in Chinese Cabbage (Brassica rapa ssp. pekinensis). J. Jpn. Soc. Hortic. Sci. 81, 184–190. doi: 10.2503/jjshs1.81.184
Matsumoto, E., Yasui, C., Ohi, M., and Tsukada, M. (1998). Linkage analysis of RFLP markers for clubroot resistance and pigmentation in Chinese cabbage (Brassica rapa ssp. pekinensis). Euphytica 104, 79–86. doi: 10.1023/A:1018370418201
Michelmore, R. W., Paran, I., and Kesseli, R. V. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. U.S.A. 88, 9828–9832. doi: 10.1073/pnas.88.21.9828
Nikolaev, S. I., Berney, C., Fahrni, J. F., Bolivar, I., Polet, S., Mylnikov, A. P., et al. (2004). The twilight of heliozoa and rise of Rhizaria, an emerging supergroup of amoeboid eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 101, 8066–8071. doi: 10.1073/pnas.0308602101
Peng, G., Falk, K. C., Gugel, R. K., Franke, C., Yu, F., James, B., et al. (2014). Sources of resistance to Plasmodiophora brassicae (clubroot) pathotypes virulent on canola. Can. J. Plant Pathol. 36, 89–99. doi: 10.1080/07060661.2013.863805
Piao, W., Liang, S., Li, X., Li, P., Yu, S., Lim, Y. P., et al. (2014). Genetic detection of clubroot resistance loci in a new population of Brassica rapa. Hortic. Environ. Biotechnol. 55, 540–547. doi: 10.1007/s13580-014-0079-5
Piao, Z. Y., Deng, Y. Q., Choi, S. R., Park, Y. J., and Lim, Y. P. (2004). SCAR and CAPS mapping of CRb, a gene conferring resistance to Plasmodiophora brassicae in Chinese cabbage (Brassica rapa ssp. pekinensis). Theor. Appl. Genet. 108, 1458–1465. doi: 10.1007/s00122-003-1577-5
Saito, M., Kubo, N., Matsumoto, S., Suwabe, K., Tsukada, M., and Hirai, M. (2006). Fine mapping of the clubroot resistance gene, Crr3, in Brassica rapa. Theor. Appl. Genet. 114, 81–91. doi: 10.1007/s00122-006-0412-1
Sakamoto, K., Saito, A., Hayashida, N., Taguchi, G., and Matsumoto, E. (2008). Mapping of isolate-specific QTLs for clubroot resistance in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Theor. Appl. Genet. 117, 759–767. doi: 10.1007/s00122-008-0817-0
Sauna, Z. E., and Kimchi-Sarfaty, C. (2011). Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691. doi: 10.1038/nrg3051
Sokal, R., and Rohlf, F. J. (1995). Biometry: The Principles and Practice of Statistics in Biological Research. San Francisco, CA: W. H. Freeman.
Strelkov, S. E., Hwang, S. F., Manolii, V. P., Cao, T., and Feindel, D. (2016). Emergence of new virulence phenotypes of Plasmodiophora brassicae on canola (Brassica napus) in Alberta, Canada. Eur. J. Plant Pathol. 145, 517–529. doi: 10.1007/s10658-016-0888-8
Strelkov, S. E., Manolii, V. P., Cao, T., Xue, S., and Hwang, S. F. (2007). Pathotype classification of Plasmodiophora brassicae and its occurrence in Brassica napus in Alberta, Canada. J. Phytopathol. 155, 706–712. doi: 10.1111/j.1439-0434.2007.01303.x
Strelkov, S. E., Tewari, J. P., and Smith-Degenhardt, E. (2006). Characterization of Plasmodiophora brassicae populations from Alberta, Canada. Can. J. Plant Pathol. 28, 467–474. doi: 10.1080/07060660609507321
Suwabe, K., Tsukazaki, H., Iketani, H., Hatakeyama, K., Fujimura, M., Nunome, T., et al. (2003). Identification of two loci for resistance to clubroot (Plasmodiophora brassicae Woronin) in Brassica rapa L. Theor. Appl. Genet. 107, 997–1002. doi: 10.1007/s00122-003-1309-x
Suwabe, K., Tsukazaki, H., Iketani, H., Hatakeyama, K., Fujimura, M., Nunome, T., et al. (2006). Simple sequence repeat-based comparative genomics between Brassica rapa and Arabidopsis thaliana? the genetic origin of clubroot resistance. Genetics 173, 309–319. doi: 10.1534/genetics.104.038968
Tsushima, S. (2000). Perspective of integrated pest management-a case study: clubroot disease of crucifers. J. Pestic. Sci. 25, 296–299. doi: 10.1584/jpestics.25.296
Ueno, H., Matsumoto, E., Aruga, D., Kitagawa, S., Matsumura, H., and Hayashida, N. (2012). Molecular characterization of the CRa gene conferring clubroot resistance in Brassica rapa. Plant Mol. Biol. 80, 621–629. doi: 10.1007/s11103-012-9971-5
Van Ooijen, J. V., and Voorrips, R. E. (2001). JoinMap Version 3.0 Software for the Calculation of Genetic Linkage Maps. Wageningen: Kyazma B.V.
Voorrips, R. E. (1995). Plasmodiophora brassicae? aspects of pathogenesis and resistance in Brassica oleracea. Euphytica 83, 139–146. doi: 10.1007/BF01678041
Williams, P. H. (1966). A system for the determination of races of Plasmodiophora brassicae that infect cabbage and rutabaga. Phytopathology 56, 624–626.
Wu, T. D., and Nacu, S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. doi: 10.1093/bioinformatics/btq057
Yu, F., Lydiate, D. J., Guge, R., Sharpe, A., and Rimmer, S. R. (2012). Introgression of Brassica rapa subsp. sylvestris blackleg resistance into B. napus. Mol. Breed. 30, 1495–1506. doi: 10.1007/s11032-012-9735-6
Yu, F., Zhang, X., Huang, Z., Chu, M., Song, T., Falk, K. C., et al. (2016). Identification of genome-wide variants and discovery of variants associated with Brassica rapa clubroot resistance gene Rcr1 through bulked segregant RNA sequencing. PLoS ONE 11:e0153218. doi: 10.1371/journal.pone.0153218
Yu, F., Zhang, X., Peng, G., Falk, K. C., Strelkov, S. E., and Gossen, B. D. (2017). Genotyping-by-sequencing reveals three QTL for clubroot resistance to six pathotypes of Plasmodiophora brassicae in Brassica rapa. Sci. Rep. 7:4516. doi: 10.1038/s41598-017-04903-2
Keywords: clubroot, RNA-Seq, SNPs, fine mapping, Brassica rapa, Plasmodiophora brassicae
Citation: Huang Z, Peng G, Liu X, Deora A, Falk KC, Gossen BD, McDonald MR and Yu F (2017) Fine Mapping of a Clubroot Resistance Gene in Chinese Cabbage Using SNP Markers Identified from Bulked Segregant RNA Sequencing. Front. Plant Sci. 8:1448. doi: 10.3389/fpls.2017.01448
Received: 31 May 2017; Accepted: 04 August 2017;
Published: 28 August 2017.
Edited by:
Jacqueline Batley, University of Western Australia, AustraliaReviewed by:
Chunyu Zhang, Huazhong Agricultural University, ChinaCopyright © 2017 Huang, Peng, Liu, Deora, Falk, Gossen, McDonald and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengqun Yu, ZmVuZ3F1bi55dUBhZ3IuZ2MuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.