 Bruno Contreras-Moreira1,2*
Bruno Contreras-Moreira1,2* Carlos P. Cantalapiedra1
Carlos P. Cantalapiedra1 María J. García-Pereira1
María J. García-Pereira1 Sean P. Gordon3John P. Vogel3
Sean P. Gordon3John P. Vogel3 Ernesto Igartua1
Ernesto Igartua1 Ana M. Casas1
Ana M. Casas1 Pablo Vinuesa4
Pablo Vinuesa4- 1Estación Experimental de Aula Dei – Consejo Superior de Investigaciones Científicas, Zaragoza, Spain
- 2Fundación ARAID, Zaragoza, Spain
- 3DOE Joint Genome Institute, Walnut Creek, CA, USA
- 4Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
The pan-genome of a species is defined as the union of all the genes and non-coding sequences found in all its individuals. However, constructing a pan-genome for plants with large genomes is daunting both in sequencing cost and the scale of the required computational analysis. A more affordable alternative is to focus on the genic repertoire by using transcriptomic data. Here, the software GET_HOMOLOGUES-EST was benchmarked with genomic and RNA-seq data of 19 Arabidopsis thaliana ecotypes and then applied to the analysis of transcripts from 16 Hordeum vulgare genotypes. The goal was to sample their pan-genomes and classify sequences as core, if detected in all accessions, or accessory, when absent in some of them. The resulting sequence clusters were used to simulate pan-genome growth, and to compile Average Nucleotide Identity matrices that summarize intra-species variation. Although transcripts were found to under-estimate pan-genome size by at least 10%, we concluded that clusters of expressed sequences can recapitulate phylogeny and reproduce two properties observed in A. thaliana gene models: accessory loci show lower expression and higher non-synonymous substitution rates than core genes. Finally, accessory sequences were observed to preferentially encode transposon components in both species, plus disease resistance genes in cultivated barleys, and a variety of protein domains from other families that appear frequently associated with presence/absence variation in the literature. These results demonstrate that pan-genome analyses are useful to explore germplasm diversity.
Introduction
High-throughput sequencing has made it possible to assemble whole genomes and transcriptomes at an unprecedented rate, leading to the comparison of individuals of the same species. In prokaryotes, where most of these studies have been carried out, the concept of pan-genome has emerged as the sum of a core genome, shared by all isolates, plus a dispensable genome consisting of partially shared and strain-specific genes (Tettelin et al., 2005). Similar analyses are now being performed in humans and other eukaryotes with large genomes (Li et al., 2010). Among plants, some studies compared ecotypes of model species Arabidopsis thaliana and accessions of crops such as maize, barley, soybean, or rice, revealing that dispensable genes play important roles in evolution, and in the complex interplay between plants and the environment (Cao et al., 2011; Hansey et al., 2012; Dai et al., 2014; Hirsch et al., 2014; Li et al., 2014; Yao et al., 2015). In fact, it has been questioned whether these loci are really dispensable, and for this reason we call them “accessory,” another term borrowed from microbiology (Laing et al., 2010; Marroni et al., 2014). Exploration of presence/absence variation (PAV) of accessory loci increases our capacity to link genotypes to phenotypes, and indeed PAV has been found to explain phenotypic differences among cultivars beyond those revealed by standard SNP-based genotyping methods (Yano et al., 2016).
Current re-sequencing approaches usually involve Whole Genome Sequencing (WGS) of different genotypes, comparing them against a reference genome to identify SNPs and small indels (Lai et al., 2010; Li et al., 2014; Yao et al., 2015; The 1001 Genomes Consortium, 2016). In cases where there is no reference genome available, or where cost or genome complexity preclude the creation of bona fide assemblies or sequencing multiple accessions with enough coverage, it is possible to draft a pan-genome using alternative methods as proxies, such as hybridization arrays (Druka et al., 2006; Springer et al., 2009; Muñoz-Amatriaín et al., 2013), low-coverage sequencing (Yao et al., 2015) or Genotyping-By-Sequencing (Lu et al., 2015). Transcriptome sequencing (RNA-seq) has also been conducted to sample pan-genomes (Hansey et al., 2012; Hirsch et al., 2014; Gusev et al., 2016), in which case the observation of PAV is linked to differential expression, and ultimately function, and is not necessarily bound to differential gene content. Indeed, transcriptome data model the expressed pan-genome, or pan-transcriptome, as an atlas of expressed genes of the species. If a variety of tissues are sequenced with similar depth across genotypes, core and accessory transcripts should be reasonable estimates of the pan-genome. In practice, all these experimental approaches usually demand mixing datasets that vary in quality to compare sequences of related accessions.
Some state-of-the art approaches model the pan-genome as a complex data structure which can be interrogated with standard operations such as read mapping or variant calling. Several representations have been developed, going from unaligned sequences, to blocks of multiple alignments or to k-mer collections (Computational Pan-Genomics Consortium, 2016). An example of software able to represent an arbitrary pan-genome as a graph database would be PanTools (Sheikhizadeh et al., 2016). These algorithms ideally take complete chromosome arms as input and therefore can model the pan-genome at the base-pair level. However, when this kind of data is not available it is still possible to explore a pan-genome by clustering raw reads or assembled contigs. This would be the case of Cnidaria, which uses k-mer counting algorithms to efficiently compute genetic distances and classify samples (Aflitos et al., 2015). GET_HOMOLOGUES-EST, described in this paper, aims at reconstructing a gene-based pan-genome from related genotypes. By performing local alignments, it can analyze sequence features annotated in WGS genomes and assembled expressed sequences.
This software builds on previous methods which targeted bacterial datasets (Contreras-Moreira and Vinuesa, 2013; Vinuesa and Contreras-Moreira, 2015), and takes BLASTN alignments (Altschul et al., 1997) to drive Markov (OMCL) sequence clustering (van Dongen, 2000; Li et al., 2003). GET_HOMOLOGUES-EST has been adapted to the large size of plant genomic data sets, and adds new features to adequately handle redundant and fragmented transcript sequences, as those usually obtained from state-of-the-art technologies like RNA-seq, as well as incomplete/fragmented gene models from WGS assemblies. By parsing sequence clusters, it can construct pan-genome matrices (PMs) that drive the annotation of core and accessory sequences and simulations of gene pool growth. Besides, it allows the calculation of Average Nucleotide Identity (ANI) matrices that summarize intra-specific genetic diversity. These outputs can be exported to high quality plots (R Development Core Team, 2008), as requested by users and reviewers of related software (Golicz et al., 2015; Xiao et al., 2015).
The software is first benchmarked with WGS assemblies of 19 ecotypes of the model plant A. thaliana, and then with de novo assembled transcriptomes of both A. thaliana and 16 cultivated (Hordeum vulgare subsp. vulgare) and wild (H. vulgare subsp. spontaneum) barley accessions. Besides publicly available data, in-house produced transcriptomes of barley elite cultivar Scarlett (2-row) and Spanish landrace SBCC073 (6-row) are surveyed in this work. We chose these species for several reasons: (i) For their contrasting genomic features and resources; while A. thaliana is a model dicot species with a compact genome and plenty of available genomic resources (The 1001 Genomes Consortium, 2016), barley is a grass with a large and repetitive genome, with only two annotated references currently available: Morex (International Barley Genome Sequencing Consortium et al., 2012) and Haruna Nijo (Sato et al., 2015). (ii) For economic relevance, as barley is an important crop worldwide, and closely related to wheat, with germplasm diversity potentially useful for breeding programs. (iii) To add evidence to other studies which claim that plant genomes are mosaics including genes shared by all individuals and also genes found only in some genotypes, challenging the dominant model of reference-based genomic studies.
The results illustrate how the software can be used to analyze plant pan-genomes, and provide insights into their evolution. While core genes are conserved and highly expressed, accessory loci are variably expressed and accumulate non-synonymous substitutions at a higher rate and frequently belong to protein families associated to PAV. The benchmarks also test to what extent transcripts can be used to sample a pan-genome and to infer molecular phylogenies. Overall, our findings indicate that accessory loci comprise a significant portion of the genetic diversity of plants and should be explicitly annotated in genome sequences. Software tools such as GET_HOMOLOGUES-EST can be valuable for this task.
Materials and Methods
Arabidopsis thaliana Sequence Sets
Nucleotide sequences corresponding to CDS annotated in 19 A. thaliana ecotypes were retrieved from http://mtweb.cs.ucl.ac.uk/mus/www/19genomes/sequences/CDS/ (Gan et al., 2011). Seedling, root, and floral bud RNA-seq reads of 17 of those accessions were obtained from http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53197, as well as Fragments Per Kb of exon per Million fragments mapped (FPKM) values produced by mapping them to TAIR10 reference1 with TopHat v2.0.9 and Cufflinks v2.1.1 (Trapnell et al., 2009, 2010). De novo transcript assemblies were produced automatically with Trinity v2.0.2 (Haas et al., 2013) (–seqType fq –trimmomatic –normalize_reads –normalize_max_read_cov 50 –min_contig_length 200), and CDS sequences [both nucleotide open reading frames (ORFs) and peptides] inferred with script transcripts2cds.pl (see below). On average, protein-coding transcripts amounted to 74.5% of all assembled sequences. Expression counts (in Transcripts Per Million, TPM) of deduced CDS sequences allocated to occupancy classes were obtained by quasi-mapping the RNA-seq read libraries to de novo assemblies with Sailfish-0.10.0 and default parameters (Srivastava et al., 2016). Occupancy is defined as the number of ecotypes present in a sequence cluster and in this experiment takes values from 1 to 19. Clusters of genome-annotated CDS sequences were compared to de novo transcripts of increasing occupancy with script make_nr_pangenome_matrix.pl (see below, parameters -S 100 -e -l 200 -s 95), requiring % sequence identity ≥ 95 and default % alignment coverage ≥ 50, to account for truncated transcripts. These results were then used to compute the ratio of transcripts matching the same CDS.
SBCC073 and Scarlett Transcriptomes
One-week old seedlings of barley cultivar Scarlett and landrace-derived inbred line SBCC073 were vernalized for four weeks (3–8°C 8 h light) and then transferred into a growth chamber with 16 h light (intensity 250 μmol m-2 s-1)/8 h dark photoperiod, and controlled temperature (21°C during the day/18°C at night). Plants were irrigated, fertilized, and treated with fungicide, using standard procedures. Last expanded leaves were sampled after main tiller spike emergence. Samples were frozen in liquid nitrogen until they were processed to obtain RNA using the NucleoSpin RNA Plant kit (Macherey-Nagel, Düren, Germany). Quality of RNA was assessed with a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and with Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Illumina TruSeq standard RNAseq libraries were prepared at CNAG (Barcelona, Spain) and sequenced in an Illumina HiSeq2000 instrument. In total 79.8M and 156.1M paired-end 101 bp reads were obtained for SBCC073 and Scarlett, respectively. Pre-processing of raw reads was done with FASTQC v0.102 and Trimmomatic v0.22 (Bolger et al., 2014), requiring a minimum mean quality Phred score of 28 every 15 adjacent nucleotides, cropping the first nucleotides to obtain 0.25 frequency of each base per position, and keeping reads longer than 80 bp. Pass-filter reads were corrected for single base mismatches with Musket v1.0.6 (Liu et al., 2013). The filtered, corrected, and normalized reads were assembled into final contigs with Trinity. Quality was estimated by mapping back the raw reads to the assemblies with Trinity scripts bowtie_PE_separate_then_join.pl and SAM_nameSorted_to_uniq_count_stats.pl, obtaining 71.73 and 68.05% proper pairs for SBCC073 and Scarlett, respectively, which are typical values of Trinity assemblies.
Expression counts (TPM) of SBCC073 transcripts were computed, as explained above, by quasi-mapping RNA-seq reads obtained from a mixture of tissues. This pool included seedling roots and leaves from pre-vernalization plants, post-vernalization leaves, tillers (15 days), and inflorescences (32 days). Briefly, RNA was extracted and quality-controlled as explained earlier and cDNA synthetized with the Mint-2 kit (Evrogen, Moscow, Russia). Subsequently, in vitro normalization of cDNA was performed with the kit Evrogen Trimmer, which uses a duplex-specific nuclease (DSN) to equalize the levels of cDNA molecules in the samples. Normalized samples were used to obtain sequencing libraries, following Illumina standard procedures, which were sequenced with an Illumina GAIIx instrument at Parque Científico de Madrid (Cantoblanco, Spain). This dataset has been deposited as ENA study PRJEB12639.
Barley Transcriptomes from Public Datasets
Transcript and cDNA sets of Morex and Haruna Nijo cultivars were obtained from public repositories (International Barley Genome Sequencing Consortium et al., 2012; Sato et al., 2015). The remaining transcriptomes (Dai et al., 2014) were obtained after downloading the raw sequence reads from ftp://ftp-trace.ncbi.nlm.nih.gov/sra (Short Read Archive, SRA) and automatically assembling them with Trinity, as explained for A. thaliana. CDS nucleotide and peptide sequences were inferred with script transcripts2cds.pl.
Sequences of all assembled barley transcriptomes were scanned with BLASTN against intron sequences inferred from Morex high confidence (HC) gene models (International Barley Genome Sequencing Consortium et al., 2012) and from Haruna Nijo (Sato et al., 2015) gene models, after filtering out sequence segments matching exons. Sequence alignments longer than 100 nucleotides and sequence identity ≥ 98% were annotated as introns.
Get_Homologues-EST Implementation and Novel Features
The software is built on top of BLASTN v2.2.27+ (Camacho et al., 2009) and the code base of GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013). It takes a folder containing FASTA input files, which might be gzip- or bzip2-compressed, and produces different outputs summarized on Figure 1A. It supports two sequence clustering algorithms: the fast bidirectional best hit algorithm (BDBH) and the accurate OrthoMCL algorithm (OMCL) (Li et al., 2003). Both approaches define in-paralogs as sequences with best hits in the same accession. If mandatory input nucleotide files have associated translated amino acid files, clusters of protein sequences are also produced. By default, alignment coverage is calculated with respect to the shortest sequence (see Figure 1B), since de novo assembled transcripts are frequently truncated. However, when two full-length cDNA sequences are compared, coverage is computed over the longest one. Redundant transcript isoforms or alternatively spliced genes can cause clustering problems, as they might break otherwise bidirectional best hits. Hence, the new EST protocol filters out redundant sequences with overlaps of length ≥ 40 (see Supplementary Figure S1), the same cut-off used by TGICL (Pertea et al., 2003). Another novel feature is the capability to compute ANI matrices (see below). As in the microbial release, HMMER3.1b23 is integrated to facilitate Pfam annotation of protein domains (Finn et al., 2016). However, the new EST pipeline adds the option to perform protein domain enrichment calculations with script pfam_enrich.pl (see below).
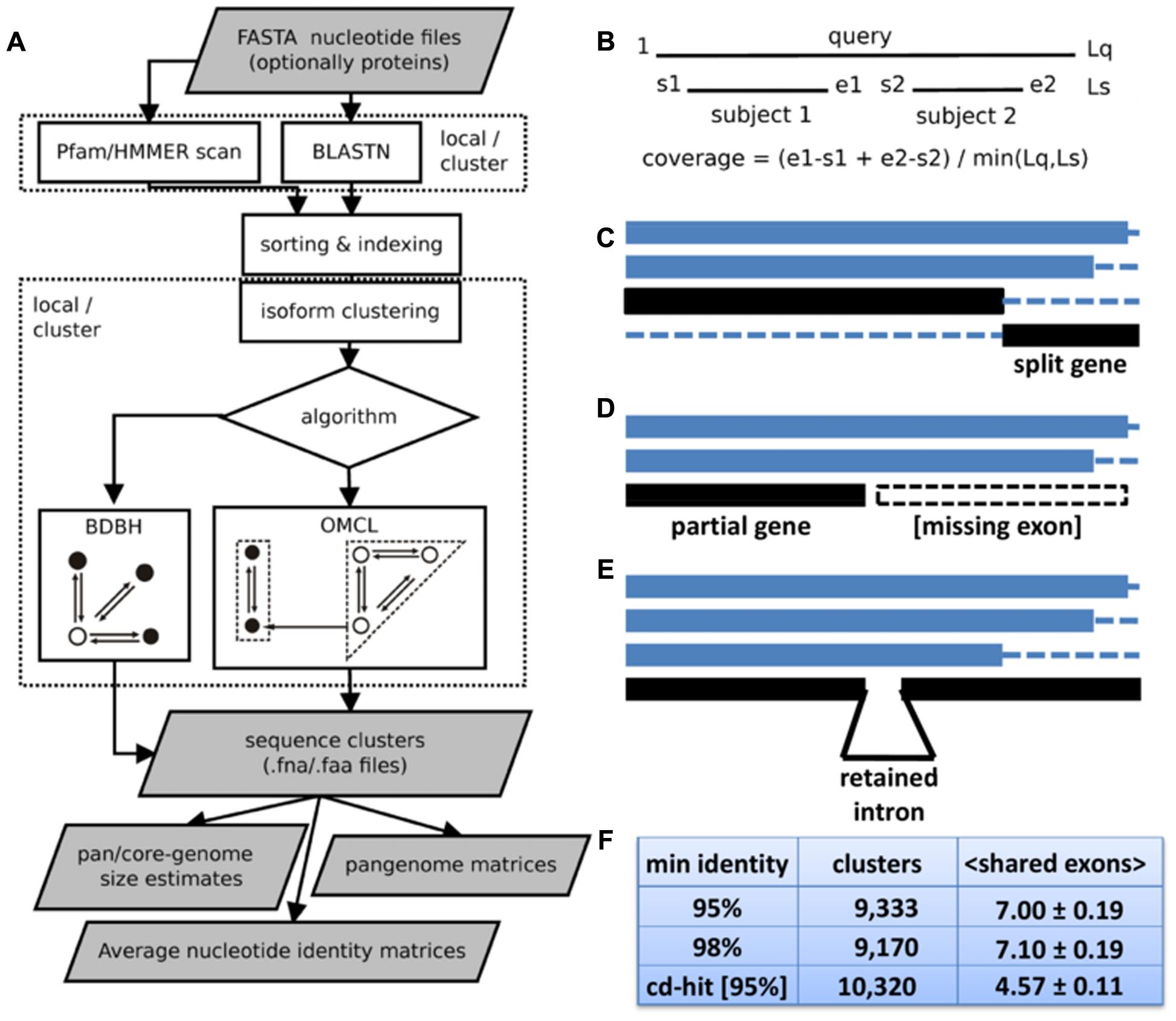
FIGURE 1. Features of GET_HOMOLOGUES-EST. (A) Flowchart of the main tasks and deliverables. BLASTN and optional Pfam scans, as well as BDBH and OMCL clustering, can be run on a local computer, preferably multi-core, or over a computer cluster. Resulting clusters are post-processed to produce pan-genome or average nucleotide identity matrices, as well as to estimate pan-, soft-core-, and core-genomes. Note that both clustering algorithms can be fine-tuned by customizing an array of parameters, of which alignment coverage is perhaps the most important. While OMCL is adequate for most applications, the niche of BDBH is the fast calculation of core sequences within large datasets. (B) Coverage calculation illustrated with the alignment of sequence ‘query’ to two aligned fragments of sequence ’subject.’ Lq and Ls are the lengths of both sequences, and s1, e1, s2, e2, and Lq are alignment coordinates. (C–E) Common problems faced when clustering RNA-seq transcripts or CDS sequences from whole-genome assemblies: split genes, partial genes and genes or transcripts with retained introns. A cluster of four sequences is shown in C, where two black fragments correspond to pieces of the same gene. Panel D shows a cluster of three sequences, where the last one corresponds to the 5′ half of a transcript, probably missing one or more exons. A cluster of four transcripts is shown in E, where the last one bears an intron not present on the others. (F) Benchmark of barley transcript clusters produced with sequence identity cut-offs of 95 and 98% and with CD-HIT-EST, requiring in all cases coverage ≥ 75%. Curated, high quality Haruna Nijo isoforms were used as a gold-standard. When the same output cluster contained several Haruna Nijo isoforms, the average number of shared exons was computed as a measure of cluster consistency. Thus, the reported mean and SEM values correspond to clusters containing > 1 isoforms of the same Haruna Nijo gene model.
Genome composition analyses can be performed to simulate pan-genome growth as more genotypes are considered. In this context, genes/transcripts from randomly permuted genomes/accessions are called novel when they fail to match previous sequences with %identity ≥ 70 and %coverage ≥ 50. These values allow sequences with retained/unprocessed introns to be matched, and correspond to the range of identities typically reported by BLASTN (megablast). The same cut-offs are in place to produce core-genome plots and, as a novel feature, soft-core plots. The script plot_pancore_matrix.pl has also been updated and can now produce intermediate snapshots of the plots.
In addition to the main script get_homologues-est.pl, this suite bundles a few other new scripts which facilitate subsequent analyses. For instance, script transcripts2cds.pl can be used to deduce CDS sequences, and their translated amino acid sequences, out of raw transcripts. This script combines ORF prediction driven by TransDecoder4 and BLASTX searches against a reference protein repository, SwissProt by default. The rules encoded in the script to produce consensus CDS sequences are listed in Supplementary Table S1. Briefly, TransDecoder and BLASTX results are compared, and those with exact overlaps are assembled into a larger CDS. In case of disagreement, BLASTX results are preferred. Performance was benchmarked by comparing deduced peptide sequences with BLASTP to the peptide sequences of gene models annotated in A. thaliana ecotype Col_0 and barley cultivar Haruna Nijo, obtaining 92.3 and 78.3% correct CDS, respectively. As overlap searching is time-consuming, a version of the script is also available that uses C++ code to compute the longest common substring efficiently. Furthermore, DIAMOND can be optionally called instead of BLASTX (Buchfink et al., 2015), reducing computing time by roughly two orders of magnitude with negligible performance differences (see documentation for details).
Other new auxiliary scripts include make_nr_pangenome_matrix.pl, which produces non-redundant PMs and can compare the resulting clusters to external nucleotide or peptide sequences, plot_matrix_heatmap.sh and hcluster_matrix.sh, which can be used to produce heatmaps and distance-based dendrograms of PM or ANI matrices (see below). Finally, annotate_cluster.pl can be used to reconstruct the local alignments that support any user-selected cluster of sequences produced by get_homologues-est.pl.
The software is written in Perl and R (R Development Core Team, 2008) and is best run on a multi-core Linux/MacOSX computer or on a SGE/Open Grid computer cluster, which can be set up as explained in the manual. It offers significant performance improvements with respect to the original microbial version (Contreras-Moreira and Vinuesa, 2013), as shown in Supplementary Figure S2. Releases of the software are available with documentation, examples and an installation script that can be used to download up-to-date releases of Pfam-A and SwissProt databases. Source code and releases can be obtained from https://github.com/eead-csic-compbio/get_homologues.
Clustering Sequences and Generation of ANI and Pan-Genome Matrices
CDS and transcript sequences were grouped with get_homologues-est.pl invoking the OMCL algorithm (-M) with no cluster size restrictions (-t 0), enforcing computation of ANI matrices (-A), and otherwise default settings, including detection of redundant, overlapping isoforms (-i 40). Note that the default %identity cut-off is 95%, the same value employed by Trinity to call paralogous sequences. ANI matrices are computed by iterating across pairs (A, B) of input sequence sets, i.e., barley cultivars. For each pair, the % nucleotide identity is computed by taking the average of all the alignments of A and B sequences clustered together. CD-HIT-EST v4.5.4 (Li and Godzik, 2006) was executed with parameters -c 0.95 -G 0 -aS 0.75 to benchmark the clusters of transcripts produced by GET_HOMOLOGUES-EST. PMs matching sets of clusters were produced with script compare_clusters.pl -m.
dN/dS Estimates
Translated CDS of single-copy sequence clusters with occupancy ≥ 4 (the minimum number of sequences required for these calculations) were aligned with clustal-omega v1.2.1 (Sievers et al., 2011). The resulting alignments were translated back to codon alignments using the primers4clades suite (Contreras-Moreira et al., 2009). Each codon alignment was then passed to yn00_cds_prealigned, obtained from https://github.com/hyphaltip/subopt-kaks (Yang, 1997), to estimate ω, the ratio of non-synonymous substitutions per non-synonymous site (dN) to the number of synonymous substitutions per synonymous site (dS) of all pairs of pre-aligned sequences in a cluster. After visual inspection and statistical analyses, estimates of ω > 1.5 were used to exclude poorly aligned clusters. In this context, we define that sequence clusters with dN/dS ratio < 1 are under purifying selection and those with dN/dS > 1 are under positive selection (Yang and Bielawski, 2000).
Tree Distances and Comparisons to SNP-Based Barley Phylogenetic Tree
Average Nucleotide Identity and pan-genome presence/absence phylogenetic trees were compared among them and with a published reference tree (Dai et al., 2014) after pruning them to sets of common accessions. The symmetric and branch score distances between trees were computed with Treedist v3.695 from the Phylip package (Felsenstein, 2005), and a consensus network was computed with SplitsTree4 to visualize conserved and conflicting splits in these pairs (Huson and Bryant, 2006).
Protein Domain Annotation and Enrichment Analysis
Open reading frames contained within barley transcript sequences were inferred with script transcripts2cds.pl, producing nucleotide sequences and their corresponding peptides. Then, Pfam v28.0 (Finn et al., 2016) domains were annotated with get_homologues-est.pl –D –o, which internally calls hmmscan –acc –cut_ga. Finally, a set of Pfam-annotated CDS clusters was produced with -M -t 0 parameters, which we refer to as the “control” set. A similar “control” set of A. thaliana gene models was obtained. Enrichment of Pfam domains in sets of accessory or core clusters (“experiment” sets) was computed with script pfam_enrich.pl, which uses R function fisher.test setting p.adjust=FDR. Matched Pfam domains were counted once per cluster to avoid biases due to multiple isoforms.
Results
Algorithm Overview and Isoform Clustering
GET_HOMOLOGUES-EST takes a set of files with nucleotide sequences and runs BLASTN with all pairs of input files, as illustrated in Figure 1A. If matching files of amino acid sequences are also provided, Pfam scans can also be performed to drive domain enrichment analyses or to produce clusters with conserved domain architecture. The resulting sequence similarity tables can be optionally utilized to merge redundant isoforms (see Supplementary Figure S1), followed by a sequence clustering using the BDBH or OMCL algorithms. While BDBH seeds clusters with sequences from a selected reference genotype, and therefore skips genes absent from it (Contreras-Moreira and Vinuesa, 2013), OMCL groups nodes in a graph to build clusters which can have any composition, even without sequences from the reference (Li et al., 2003).
Among the parameters used to control these steps, alignment coverage is perhaps the most important, and it is calculated by default as depicted in Figure 1B, with respect to the shortest sequence, after adding up all non-overlapping segments reported by BLASTN. This was found to be important when handling split genes, partial genes (i.e., missing exons) or even transcripts with retained introns (Figures 1C–E, respectively). Among barley transcripts (see below), retained introns were observed in 4.4 and 2.0% transcripts, according to the Haruna Nijo and Morex reference gene models, respectively (Supplementary Tables S2 and S3). In order to measure the accuracy of GET_HOMOLOGUES-EST, clusters of barley transcripts were generated with the OMCL algorithm and compared to those produced by CD-HIT-EST (Li and Godzik, 2006), a general-purpose sequence clustering software. High-quality gene models of cultivar Haruna Nijo were taken as gold standards. Clusters obtained with GET_HOMOLOGUES-EST were found to be less affected by the presence of split genes or retained introns than CD-HIT-EST, as they grouped together more Haruna Nijo transcripts isoforms (shared exons per cluster were 7.00 ± 0.19 and 4.57 ± 0.11, respectively, see Figure 1F). An independent evaluation was carried out with 20 genes related to control of flowering in Brachypodium distachyon lines. Multiple alignment and inspection of the resulting clusters underlined the resilience of this software to broken gene models and extra/missing introns (see Supplementary Table S4).
Sequence clusters are the primary output of the software, but can be further processed to compile PM or ANI matrices. A PM counts the number of sequences from each accession/genotype present in each cluster. The total number of accessions represented in a cluster is what we call “occupancy.” Instead, ANI matrices summarize how similar input genotypes are by computing the average % nucleotide identity of clustered sequences.
Pan-Genome Analysis in Plants: The Case of Arabidopsis thaliana
The dicot species A. thaliana was the first plant to have genomic assemblies available for multiple ecotypes, and thus it was chosen as a benchmark example. A set of ecotypes, listed in Table 1, was analyzed, together with the transcriptome data available for most of them. Systematic comparison and clustering of CDS sequences of 19 A. thaliana genome assemblies support a pan-genome composed of 37,789 clusters, with a set of 26,373 genes encoded in all ecotypes (the core-genome) and a total 11,416 accessory genes. Accessory sequences can be allocated to the cloud, shell and soft-core occupancy classes (see Figure 2A), as defined for microbial genomes (Koonin and Wolf, 2008). Cloud genes (in red) have very low occupancy, as they are annotated in only one or two ecotypes, and might include artifacts or pseudogenes. In contrast, shell genes, annotated in 3 to 17 ecotypes are more likely to be biologically relevant. Soft-core genes are simply core genes which might be absent from one ecotype, perhaps due to sampling, assembly or annotation errors, and thus can be used as a robust core-genome estimate. The ANI computed with single-copy core clusters (n = 21,941) was 99.54%.
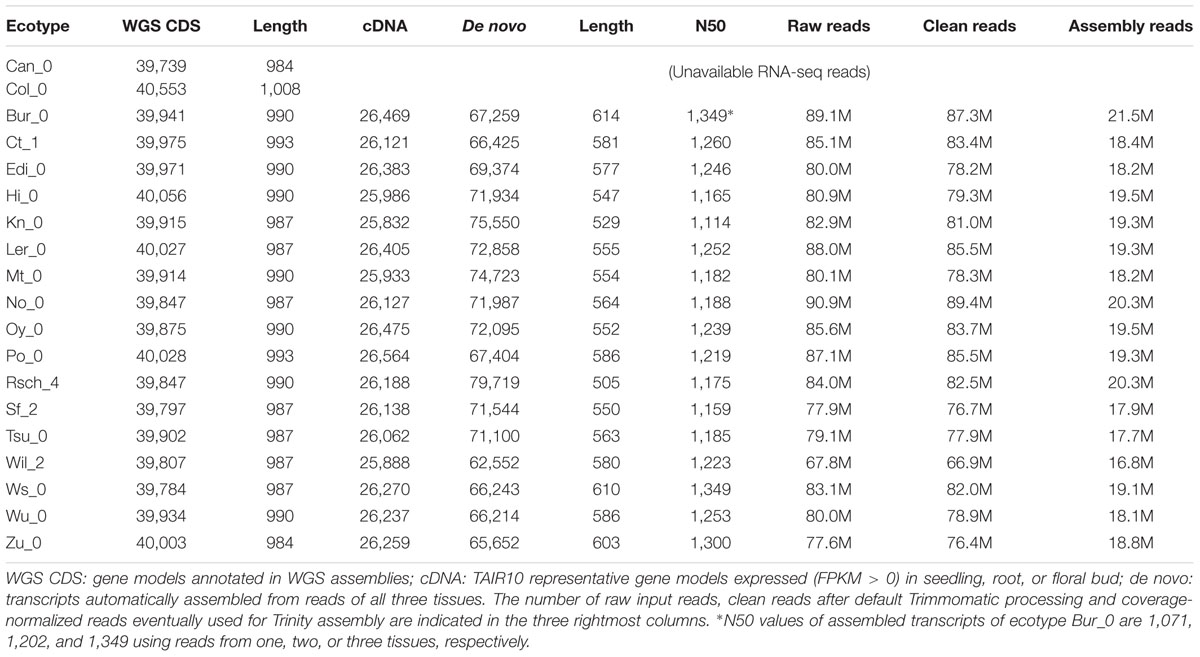
TABLE 1. Arabidopsis thaliana CDS and transcripts analyzed in this work, with median length and N50 values.
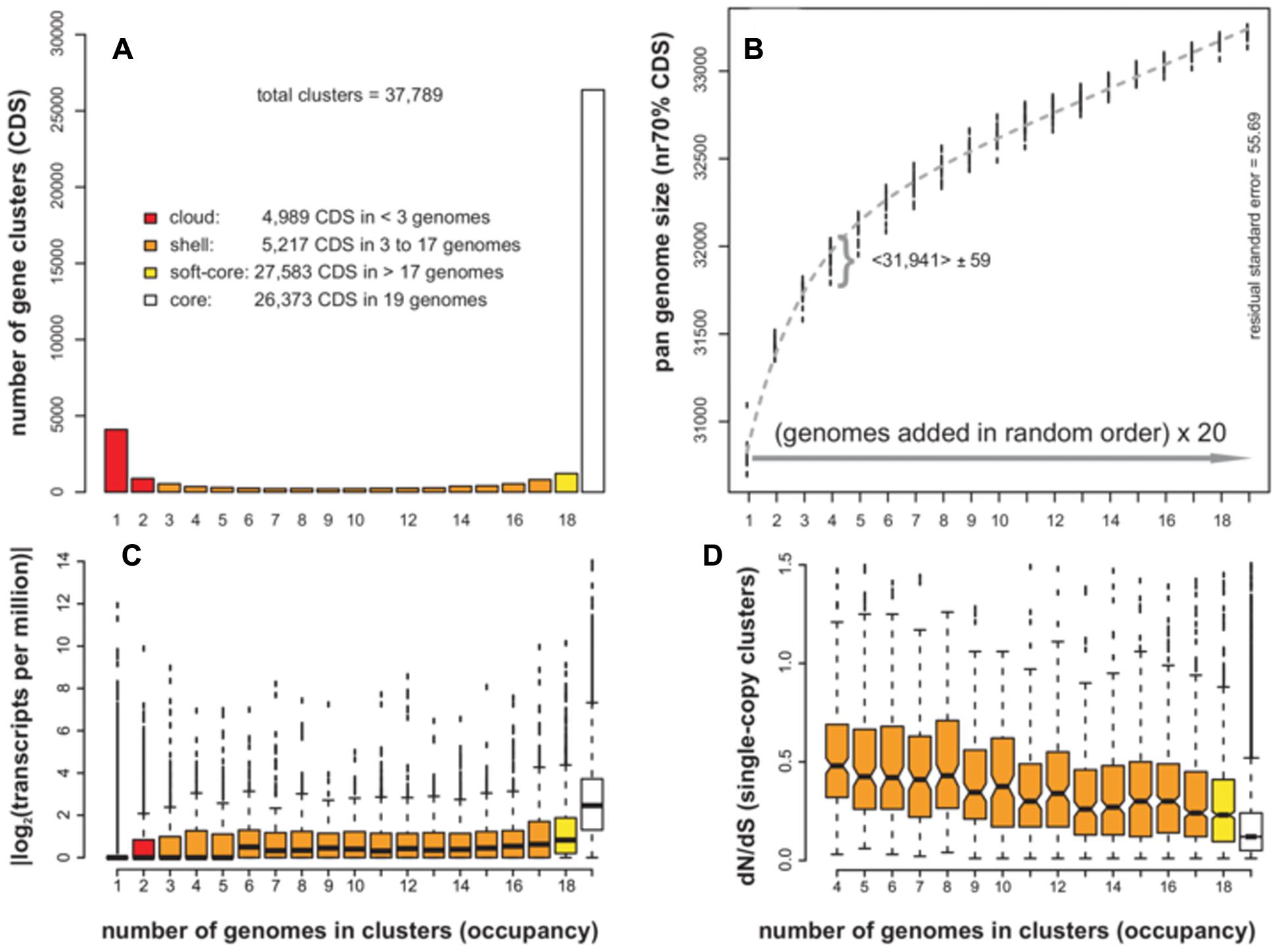
FIGURE 2. Pan-genome analyses of CDS annotated in WGS assemblies of 19 Arabidopsis thaliana ecotypes. (A) Distribution of sequence clusters as a function of their occupancy. Occupancy classes are colored as core, soft-core, shell and cloud members. (B) Pan-genome growth simulations in which accessions are added in random order, adding up 20 random permutation experiments. Novel genes contributed by the last added genome must have identity < 70% to sequences in the pool. A fitted Tettelin function is plotted in gray (Tettelin et al., 2005). A curly bracket shows the variability of size estimates after adding four genomes (mean and standard deviation are indicated). (C) Mean expression of CDS in three tissues (root, seedling, and floral bud) as a function of their occupancy. (D) dN/dS ratio of single-copy CDS clusters (n = 20,628). Notches mark 95% confidence intervals around the median. Note that clusters with occupancy <4 cannot be employed in this analysis. Top plots were produced by GET_HOMOLOGUES-EST scripts which in turn call R functions.
In order to gain a dynamic view of the A. thaliana pan-genome, the genomes can be iteratively merged in random order in an effort to identify the novel CDS sequences contributed by each ecotype, which should be sufficiently different to sequences already in the pool. Such a computational experiment, first proposed by Tettelin et al. (2005) while studying bacterial isolates of the same species, is summarized in Figure 2B. The plot shows the results of 20 sampling experiments, requiring that novel, accessory genes must not match any previous sequences with coverage ≥ 50% and sequence identity ≥ 70%. The fitted function suggests that 99% of the pan-genome is sampled after adding 15 ecotypes, with each ecotype contributing approximately 70 novel accessory genes. The mean pan-genome size after merging all 19 genomes with 20 replicates was 33,212 CDS (SD = 27). This estimate is smaller than the total size reported above due to the removal of redundant sequences.
The previous analyses showed that genes in a pan-genome can be sorted as core or accessory, and that all analyzed ecotypes contribute some accessory genes, but are they ever expressed? We tried to answer this question by taking advantage of available RNA-seq data for 17 A. thaliana ecotypes. Here, the expression of a gene cluster was computed as the average TPM of CDS contained therein. The plot in Figure 2C suggests that: (i) core genes show a higher mean expression level than accessory genes, (ii) cloud genes are hardly expressed and, (iii) shell genes have lower mean expression levels than core, but in the sampled tissues (seedling, root, and floral bud) were often observed to be highly expressed.
Finally, we look at the evolutionary conservation of pan-genome sequences as a function of their cluster occupancy. To this end, CDS clusters with one sequence per ecotype (single-copy) were selected and their average dN/dS ratio computed. A boxplot was generated with 95% confidence intervals around the median, shown as notches and computed as median ± 1.58 IQR/√n, where IQR stands for the Interquartile Range (Mcgill et al., 1978). The results in Figure 2D confirm that core genes are under significantly stronger purifying selective pressure than accessory sequences (non-overlapping notches), in which a decrease of non-synonymous substitutions is observed as their occupancies increase. Conversely, sequences under positive selection seem to be more frequent among accessory clusters (see Supplementary Table S5). Similar results were obtained when dN/dS ratios were computed on random cluster subsets (sizes n = 5 and n = 10, to exclude cluster occupancy size effects), reinforcing these observations (see Supplementary Figure S3).
Transcriptomes as Pan-Genome Proxies: A. thaliana Benchmark
While genomic sequences are the preferred raw material to model pan-genomes, the accurate assembly of large and repetitive plant genomes is challenging and expensive. In such cases, transcriptomic sequences from different ecotypes or cultivars might be a good alternative. However, transcripts can only capture a fraction of the pan-genome. Here, we exploit RNA-seq data of A. thaliana ecotypes (Table 1) in order to measure to what extent de novo transcripts can be used to sample plant pan-genomes.
Briefly, the genome-based clusters of CDS sequences presented in the previous section were compared to clusters of protein coding transcripts assembled from reads obtained from three tissues (seedling, root, and floral bud). Note that assembled transcripts are significantly shorter than annotated CDSs, with length increasing as reads from different tissues are merged (see for instance Bur_0 in Table 1). TAIR10-based cDNAs, which explicitly use gene models annotated in the underlying WGS genomes, were used as a control. The results are summarized in Table 2. First, we found that singletons, clusters of occupancy = 1, contain the shortest transcripts, matching CDS in a 2.85:1 ratio. These numbers agree with the observed low expression of cloud genes, which in consequence are poorly assembled and fragmented. In addition, 12.4% of singletons do not match the reference genome sequences, and therefore might be artifacts such as chimeras. However, clusters of occupancy ≥ 3 contain longer sequences that mostly map the genome and recall roughly two thirds of the annotated CDS in TAIR10, with specificity positively correlated with occupancy. Finally, pan-transcriptome growth simulations suggest that the number of non-redundant transcripts obtained after merging ecotypes converge to roughly thirty thousand genes if cloud clusters are disregarded (see Supplementary Figure S4). This number is ≈10% smaller than the genome-based size estimate.
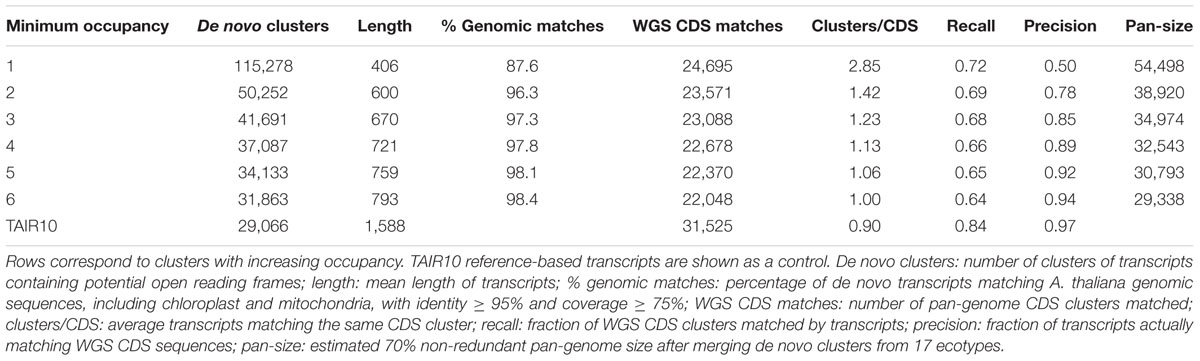
TABLE 2. Clusters of de novo assembled transcripts of 17 A. thaliana ecotypes are compared to pan-genome clusters of genome-based annotated CDS.
Drafting a Pan-Genome by Analysis of Sequences Expressed in Barley
The results of the previous section demonstrate that RNA-seq data, even without a reference genome to aid in the assembly process, might be useful to characterize the pan-genome of A. thaliana, although with some limitations. Here, we further test this approach with a collection of cultivated and wild varieties of barley, a monocot with larger (4,045 Mbp vs. 119 Mbp) and more repetitive (75.8% vs. 19.5% masked sequences) genome sequence than A. thaliana (Contreras-Moreira et al., 2016). A set of 16 barley genotypes and their transcript datasets is described in Table 3.
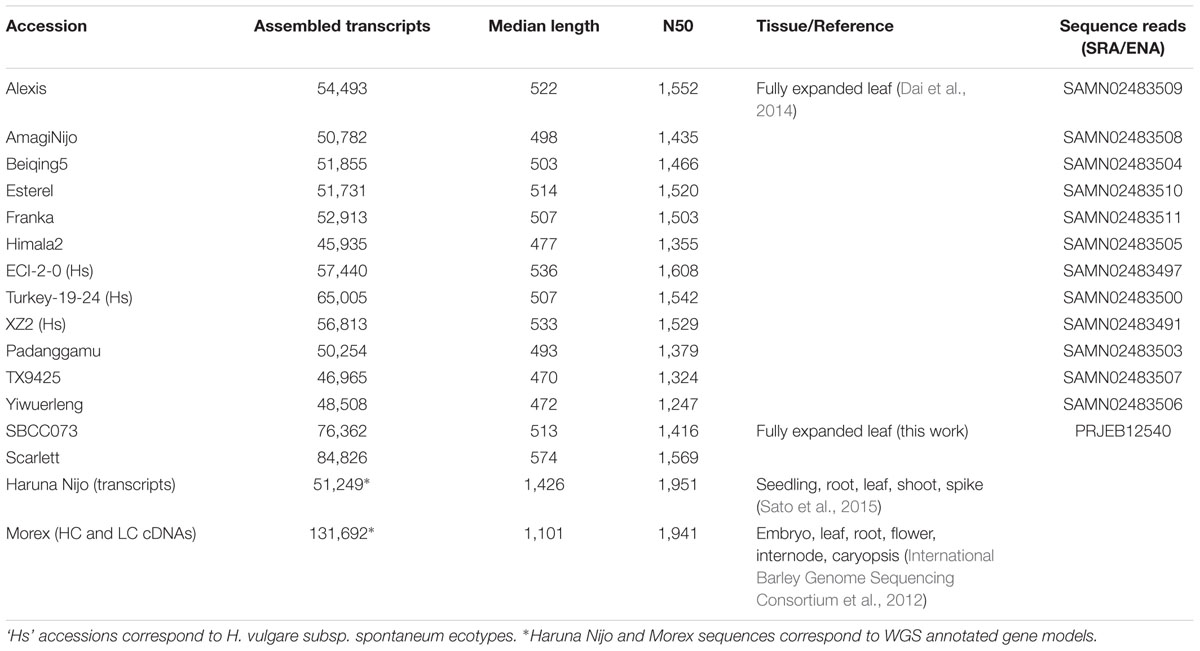
TABLE 3. Barley transcriptomes analyzed on this work.
Clusters of barley transcripts were produced with the OMCL algorithm (see Supplementary Figure S5). These were then taken to compute ANI matrices as explained in Section “Materials and Methods.” These matrices report the average percent identity between pairs of cultivars/accessions computed across all clusters in which they are found together, thus summarizing the overall similarity among them. The subset of core clusters (n = 10,922), those containing sequences from all 16 input accessions, frequently with several isoforms or fragments per accession, was analyzed first. The mean identity observed within these clusters was 98.95%. The highest value (99.22%) was found among Japanese cultivars Haruna Nijo and Amagi Nijo and the lowest value (98.65%) between ECI-2-0 and Beiqing5. The core ANI matrix was converted to a distance matrix and used to build the dendrogram shown in Figure 3. This analysis places landrace line SBCC073 next to Morex, a 6-row cultivar, and Scarlett next to 2-row cultivar Alexis. The resulting tree is largely congruent with a recently reported neighbor-joining phylogeny based on a distance matrix computed from single nucleotide exon variants identified by mapping RNA-seq transcripts against Morex WGS contigs (Dai et al., 2014). In fact, both trees display a minimal symmetric difference distance of 2 and a branch score distance of 1.419. This topological similarity is graphically depicted as a consensus network presented in Supplementary Figure S6A, which shows only one reticulation.
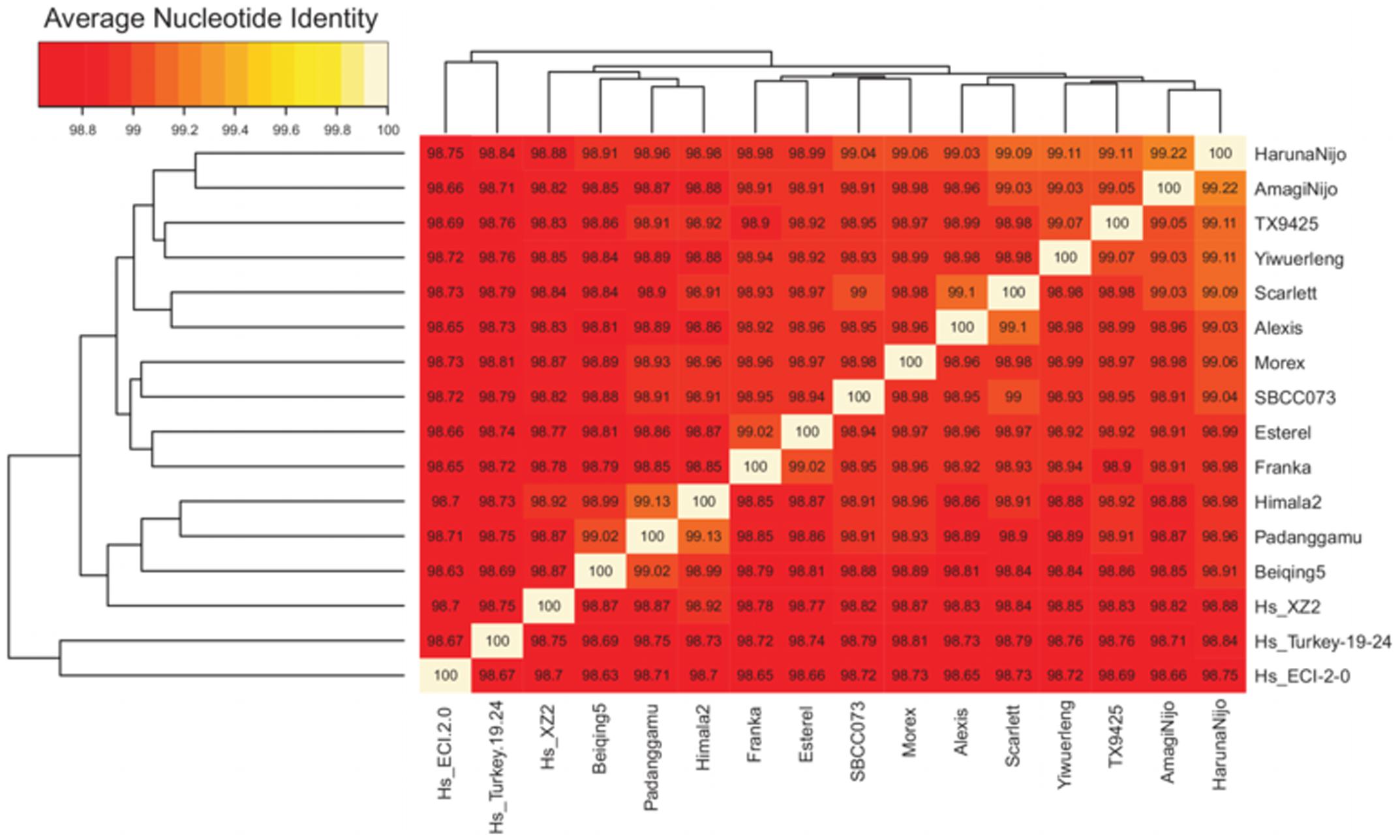
FIGURE 3. Average nucleotide % identity matrix and ordered heat map of 10,922 core barley transcripts. This figure was produced with script plot_matrix_heatmap.sh, which calls heatmap.2 function from the gplots R package. The dendrograms were computed by complete linkage clustering and Euclidean distances computed among ANI columns.
Clusters with exactly one sequence per genotype (single-copy) are usually preferred for phylogenetic inference. As there are only 166 core single-copy clusters in this collection (287 when computed with algorithm BDBH), a more convenient, larger set can be obtained by reducing the required occupancy. For instance, there were 1,775 single-copy clusters with occupancy ≥ 10. Within these, ANI among Morex and Haruna Nijo transcripts was found to be 99.54%, which is compatible with a recently published estimate of exon similarity of 99.71% (Sato et al., 2015). The resulting ANI matrix can be used to build a tree with the same topology of that in Supplementary Figure S6A, although with a branch score distance of 0.447, confirming that redundant sequences within clusters do affect identity calculations (Supplementary Figure S6B).
These cluster sets can also be taken to make PMs, as shown in the flowchart in Figure 1. In this example, PMs have 16 rows, one per barley entry, and usually many columns, corresponding to the total number of sequence clusters. Even if cloud clusters are not taken into account, as recommended by the A. thaliana benchmark, the resulting PM spans 51,245 columns, with occupancies ranging from 3 to 16. Each matrix cell takes integer values, if one or more isoforms/transcripts of that gene are captured in a given cultivar, or zero if expression was not detected. Moreover, if RNA was sampled from cultivars under different experimental conditions, that would surely be reflected in the PM. For these reasons, PAV patterns in transcript-based PMs might be poor guides for phylogeny inference. Here, we report results from analyses performed using either all samples, or only leaf sequences, avoiding mixing data from different tissues. In either case, PAV-derived trees failed to reconstruct the reference phylogeny (Dai et al., 2014), with divergent topologies and symmetric difference distances of 10 (see Supplementary Figures S6C,D). Moreover, these analyses indicate that SBCC073 and Scarlett share more expressed sequences than other cultivars, despite belonging to different lineages (Figure 3; Supplementary Figure S7). This is likely a consequence of them being processed side-by-side for this work, with plants undergoing the same treatments and tissues sampled at the same time.
Finally, barley clusters can also be used to model pan-transcriptome growth. As in the A. thaliana exercise, cloud clusters (with occupancy ≤ 2) were left out. The comparison of leaf samples produced the plots in Supplementary Figure S8 and the fitted functions estimate a core set of 9,843–10,603 transcripts encoding proteins, and a leaf pan-transcriptome of 28,762 70% non-redundant CDS after all 14 sequence sets have been merged. This simulation predicts that nine accessions are enough to sample 99% of leaf-expressed protein encoding transcripts.
Expression and Conservation of Barley Transcripts
Barley transcript clusters were further evaluated by estimating their expression in several tissues of genotype SBCC073, and also their evolutionary conservation as a function of occupancy. The top plot in Figure 4 indicates that core sequences are significantly more expressed than accessory transcripts, as computed with confidence intervals (notches) around the median, with cloud transcripts (occupancy ≤ 2) being the least expressed. These results match our observations in A. thaliana (see Figure 2C).
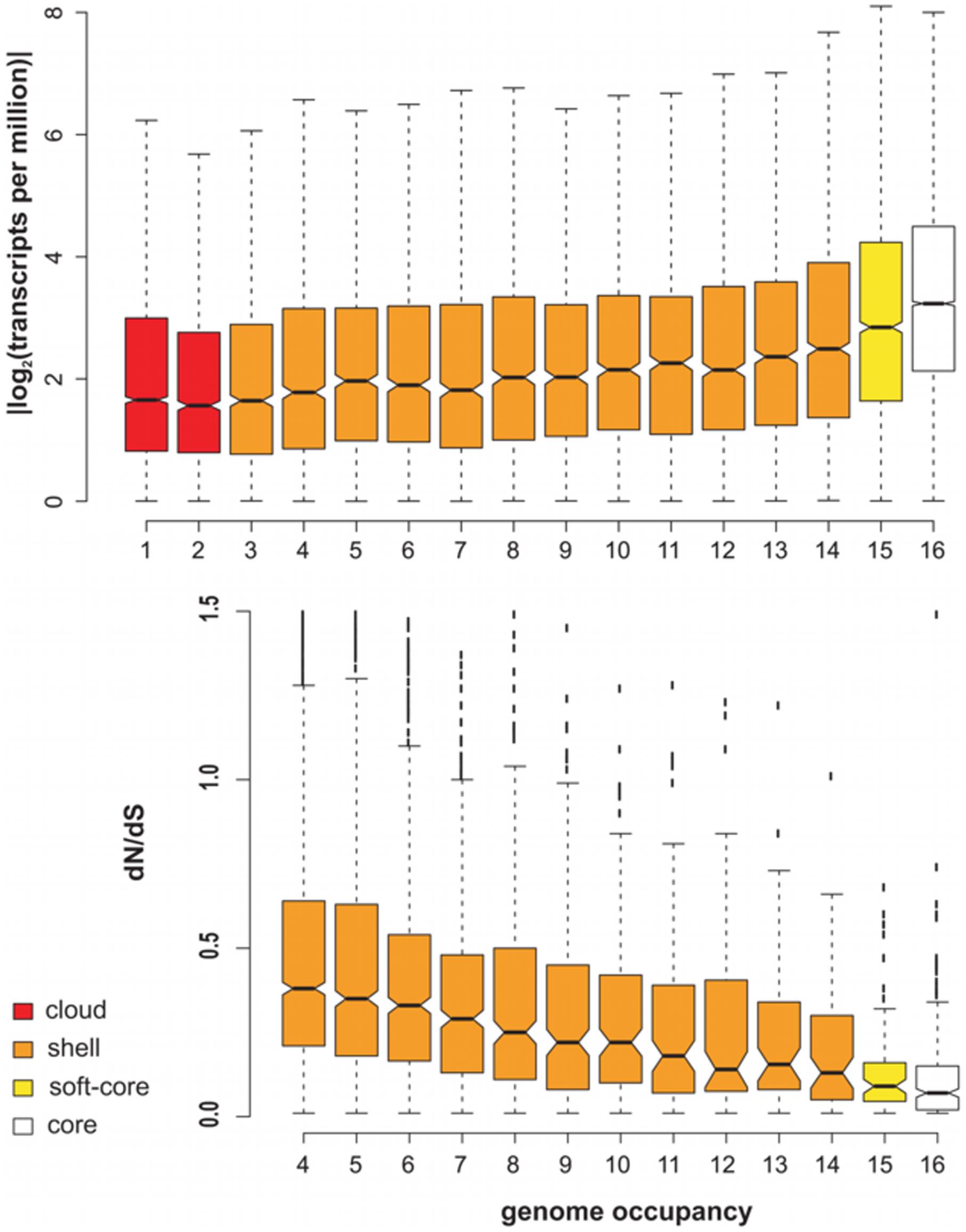
FIGURE 4. Expression and evolutionary conservation of transcripts after clustering 16 barley transcriptomes. (Top) Expression levels inferred from mapped reads of a mixture of tissues of SBCC073 as a function of occupancy. (Bottom) dN/dS ratio of sequences within single-copy deduced CDS clusters (n = 4,692). Notches mark 95% confidence intervals around the median. Note that clusters with occupancy <4 cannot be employed in this analysis. Occupancy classes are colored as core, soft-core, shell and cloud members.
CDS sequences encoded in barley transcripts were extracted, obtaining 665,694 nucleotide sequences and their corresponding peptides. Single-copy clusters of CDS sequences were then analyzed to evaluate their intra-species conservation. The bottom boxplot in Figure 4 shows that median values of dN/dS ratios (ω) and their interquartile ranges (IQRs) are inversely proportional to cluster occupancy, with shell sequences displaying the broadest IQRs and largest ω values. While these ω estimates are more skewed than those observed in A. thaliana, the confidence intervals confirm that leaf core protein-coding genes are under significantly stronger purifying selective pressure than accessory transcripts with occupancies < 10, as shown by Tukey’s honestly significant difference testing (see Supplementary Figure S9). These results resemble those of A. thaliana (Figure 2D; Supplementary Table S5).
Accessory Genes in Barley and Arabidopsis thaliana: Functional Annotation
The primary application of a PM is the identification of subsets of accessory genes which might be encoded or expressed in some genotypes but not in others, under the hypothesis that differentially represented genes might explain part of their phenotypic differences. While such questions can be readily answered with high quality WGS assemblies, with transcriptome data one must take care that the same tissues and developmental stages are being compared, and that unreliable transcripts are left out, as discussed in previous sections. With these caveats in mind, we set out to quantify how many novel leaf genes are being expressed in different barley accessions which are not currently annotated in the reference genomes, defined here as the union of Morex and Haruna Nijo gene models. Three sets of transcripts containing CDS sequences, produced with script parse_pangenome_matrix.pl and listed in Table 4, were analyzed, all of them with occupancies ≥ 3. The first two correspond to individual leaf transcriptomes assembled for this work, SBCC073 and Scarlett, while the third example illustrates the analysis of transcripts expressed in the leaves of all three wild barley genotypes. In all cases accessory sequences could be detected, with individual genotypes contributing more, but shorter, sequences than the group of wild barleys. Despite their length, a quarter of these sequences matched at least one Pfam protein domain, and thus could be functionally annotated. Three sets of accessory genes encoded in A. thaliana ecotypes, missing from the WGS genome sequence of reference accession Col_0, were also analyzed in parallel (bottom rows in Table 4). In order to compute enrichment, Pfam domains were also annotated in all barley and A. thaliana pan-genome clusters. These “control” sets contained 113,222 and 37,789 clusters, respectively.

TABLE 4. Accessory transcripts (barley) and genes (A. thaliana) not found in reference genomes (Morex ∪ Haruna Nijo and Col_0, respectively).
The most frequent protein domains encoded in accessory sequences are listed in Figure 5, with some found to be over-represented with respect to their respective “control” sets (marked with asterisks, see full list in Supplementary Data Sheet 3). Most enriched protein domains correspond to components of transposon-like sequences (reverse transcriptases, integrases, GAG domains, or Zn knuckles), which have been proposed to drive the evolution of the pan-genome (Morgante et al., 2007). Domains from NBS-LRR proteins (NB-ARC and Leucine rich repeats) appear among accessory genes of both species, but are enriched only in cultivated barleys. Conversely, while transposase components are depleted in the A. thaliana core-genome and in the set of core leaf transcripts in barley, resistance domains are significantly under-represented only in the latter (Supplementary Table S6). Other abundant domains recognized within barley accessory transcripts include kinases (including Tyr-kinases), transcription factors (Myb, FAR1), membrane transporters (ABC, Major Intrinsic Proteins) or even a domain of unknown function found mostly in grasses (Finn et al., 2016) and absent from the A. thaliana pan-genome (DUF3615, see Supplementary Table S7). Similarly, frequent domains among A. thaliana accessory genes include several examples of unknown functions, specific transposases or proteins such as prolamins, self-incompatibility S1 or C1 proteins.
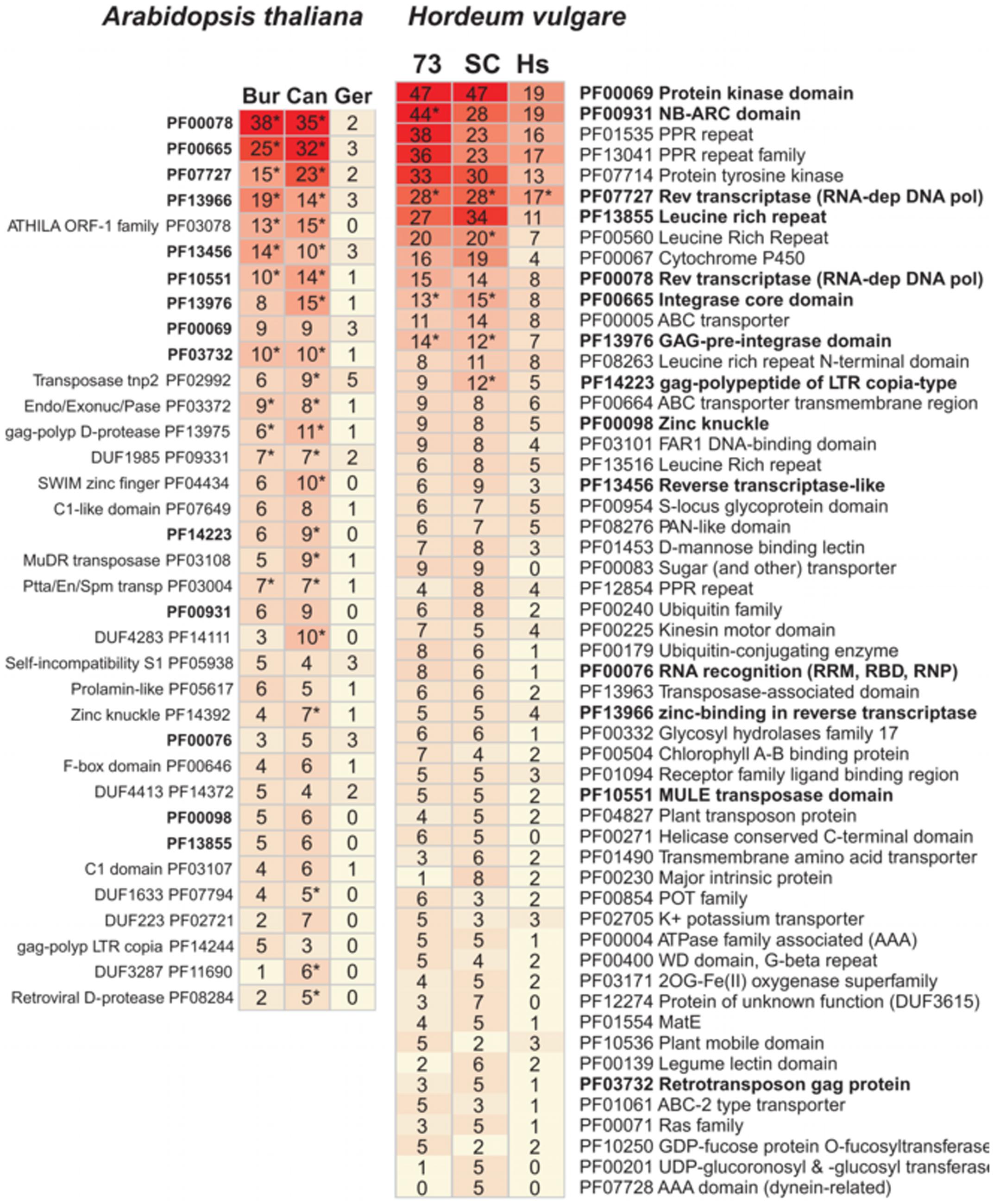
FIGURE 5. Most frequent Pfam protein domains annotated in accessory transcripts (barley, Right) and genes (A. thaliana, Left). Abbreviation “73” stands for accession SBCC073, “SC” for Scarlett and “Hs” for H. vulgare subsp. spontaneum ecotypes ECI-2-0, XZ2, and Turkey-19-24. Abbreviation “Bur” stands for ecotypes Bur_0, “Can” for Can_0 and “Ger” for German ecotypes No_0, Po_0, Wu_0 and Zu_0. Domains shown have at least five occurrences in one of the three datasets (see Supplementary Data Sheet 3 for the complete set). Domains in bold were found in both species. ∗Domains enriched compared to the “control” set of CDS-based clusters (FDR-adjusted P-value < 0.05).
Discussion
The results presented in this paper illustrate how GET_HOMOLOGUES-EST can be used to analyze plant pan-genomes from either genomic sequences or expressed tags. Our approach overcomes some intrinsic caveats, such as incomplete sequences or transcripts with retained introns, by combining local BLASTN alignments in order to calculate coverage and by using the OMCL algorithm to cluster sequences. The analyses carried out here support this software as a useful resource for these tasks, as it yields clusters which tend to group together more barley transcript isoforms than CD-HIT-EST.
Taking advantage of a collection of 19 A. thaliana ecotypes we demonstrate how plant pan-genomes can be scrutinized, and sequences allocated to core and accessory clusters that differ in their occupancy. These clusters were then used to simulate pan-genome growth by permuting genomes randomly and setting a redundancy cut-off. Mathematical functions borrowed from microbiology fit well the A. thaliana data, and can be used to estimate how many ecotypes are required to recover a percentage of the non-redundant pan-genome (Tettelin et al., 2005). As more WGS assemblies become available in the future, the predictive ability of these models could be further evaluated.
Public RNA-seq data for 17 of those A. thaliana ecotypes were gathered to check the expression of pan-genome genes and three classes of expression levels were observed, with core sequences being the most expressed, cloud the least, and shell genes in-between. Furthermore, analysis of non-synonymous substitutions across pan-genome CDS sequences reveals a trend in which core genes are under significantly stronger purifying selective pressure than accessory genes, in agreement with related reports (Bush et al., 2014). Altogether, these results indicate that the occupancy of pan-genome clusters can be used as a predictor of mutation rates and, more generally, as a proxy of their evolutionary roles.
De novo assembled transcriptomes of 17 A. thaliana ecotypes were used to explore the species pan-genome and to benchmark to what extent transcripts can be used for this job instead of genomic gene models. First, we observed that expression-based sequences are shorter on average than the homologous genome-based CDS. Second, we found that cloud transcripts contribute most low-confidence sequences, and therefore conclude that accessory genes should be identified in at least three accessions to be considered bona fide genes. Furthermore, the benchmarks show that pan-genome growth experiments performed with transcripts from three tissues under-estimate the non-redundant pool of CDS sequences by at least 10%. It is unlikely that these rules of thumb could be transferred with no modification to all plants, and they will certainly change if only one tissue is sampled, but they probably reflect to what extent RNAseq data can be used for this purpose. In addition, the terms “core” and “accessory” change their meaning when applied to transcripts, as in this context PAV is tied to differential expression. A core transcript must be encoded and expressed in all sampled genotypes. An accessory transcript makes sense only in the context of a tissue of interest, or when it is not encoded in a reference WGS genome. Despite these limitations, we anticipate that transcriptome-based analyses might potentially overcome a caveat of WGS pan-genome approaches when translating PAV to function: that of distinguishing low occupancy accessory genes from unexpressed pseudogenes.
De novo assembled transcriptomes and genome-based cDNA sequences from 16 cultivated and wild barleys were investigated with the aim of confirming some of the lessons learned with Arabidopsis. This exercise demonstrated that transcripts can be effectively clustered with this software and then used to compute ANI matrices. While these are currently computed to define bacterial species (Kim et al., 2014), here we show that barley ANI matrices are consistent with molecular phylogenies inferred from SNP data. On the contrary, our simulations suggest that PM summarizing patterns of presence/absence of transcripts fail to reliably reconstruct patterns of shared ancestry between genotypes. Instead, dendrograms derived from transcriptome-based PM likely reflect differential gene expression between accessions, that is, a functional relationship which should be assessed under controlled experimental designs to avoid batch effects, sampling bias, or other noise sources. Pan-transcriptome growth simulations suggest that nine barley genotypes are enough to retrieve 99% of the non-redundant pool of sequences expressed in the leaf, which was estimated to contain 28,762 sequences, more than twice the number reported in Morex leaves using the Barley1 GeneChip (Druka et al., 2006) and significantly more than the 19,081 non-redundant HC genes reported in by the International Barley Genome Sequencing Consortium et al. (2012).
Mapping RNA-seq reads from a mixture of tissues from barley landrace SBCC073 allowed to independently estimate the expression patterns of barley pan-transcriptome clusters, and largely confirmed the trend observed in A. thaliana. Moreover, dN/dS calculations with barley transcripts encoding ORFs reproduced to some extent the previous observation that core genes are under stronger purifying selection than accessory loci. It can therefore be concluded that the expression and conservation properties of pan-genomes can be approximated from RNA-seq experimental data.
The construction of a transcript-based PM allowed identifying accessory leaf CDS sequences not annotated in the current barley references. The average size of these de novo assembled protein-coding sequences was rather small, as expected for transcripts of low expression and occupancy, although long enough to allow recognition of protein domains. Among the annotated protein families, a few Pfam domains related to transposons were found enriched with respect to the complete transcriptome in different tests (SBCC073, Scarlett, wild barleys and several A. thaliana ecotypes). We speculate that these observations might hint at some connection between these mobile genetic elements and the generation of accessory genes. Actually, recent studies are increasingly highlighting the role of transposons in the evolution of structural and functional features of plant genomes (Lisch, 2013). Of most interest to crop scientists is the fact that loci controlling adaptive responses to the environment are frequent transposition targets (Quadrana et al., 2016). The description in barley of a cluster of accessory resistance genes in a transposon-rich region subscribes this observation (Cantalapiedra et al., 2016). Unfortunately there are no physical maps of these genotypes to systematically test this hypothesis, for instance by checking for the co-occurrence of accessory loci and transposons.
Apart from transposons domains, other abundant domains annotated in accessory sequence tags closely match protein families associated to copy number variation in barley, such as Ser/Thr and Tyr-protein kinases, RNA recognition domains or resistance NBS-LRR genes (Muñoz-Amatriaín et al., 2013). As these observations were made after hybridization experiments, they can be taken as confirmatory of our own observations, in which variants were actually sequenced and assembled. Moreover, some of these protein families have been reported to harbor most large-effect SNPs in maize inbred lines (Lai et al., 2010) and among presence/absence variants in A. thaliana (Bush et al., 2014).
Components of NBS-LRRs were found among accessory sequences in both species. However, they were significantly enriched only in cultivars. Such enrichment in the accessory part of the genome has been reported in other crops, such as Brassica oleracea (Golicz et al., 2016) or soybean (Mchale et al., 2012). This can be related to the pattern of evolution of this protein family, presenting specific mechanisms to generate diversity and elevated rates of non-synonymous substitutions (Leister et al., 1998; Michelmore and Meyers, 1998), as NBS-LRR genes are an important part of the immune system of plants (Jones and Dangl, 2006). In wheat, the domestication process caused loss of NBS-LRR ancestor genes, ensued by gene gain events through gene duplication and diversification, to keep up with pathogen evolution (Gu et al., 2015), resulting in large differences in NBS-LRR content between accessions.
Overall, it seems that some multi-genic families grow by gaining accessory genes both in dicot and monocot plants. For such sequences to be broadly used in plant breeding it will be necessary to catalog them and assign them relative positions within physical or genetic maps (Jupe et al., 2013). Some strategies based on association mapping of PMs are already being proposed in other plants (Lu et al., 2015; Gusev et al., 2016; Jin et al., 2016).
Conclusion
Our results describe a scenario in which central, conserved genes are kept on a core set, are highly expressed and found on most genotypes, while accessory genes, with low mean expression, accumulate amino acid substitutions at a higher rate. The analyses with both transcripts and CDS of A. thaliana and barley, illustrate how tools such as GET_HOMOLOGUES-EST can help upgrading plant genomics to pan-genomics.
Author Contributions
BC-M, EI, AC, and PV designed the study. BC-M, CC, and PV produced source code and documentation. BC-M, CC, MG-P, and AC carried out research. BC-M, CC, SG, JV, AC, EI, and PV analyzed the data. BC-M, CC, SG, JV, EI, AC, and PV wrote the manuscript.
Funding
This work was supported by DGA – Obra Social La Caixa (grant number GA-LC-059-2011), Spanish MINECO (AGL2013-48756-R and CSIC13-4E-2490) and CONACyT-México (grant 179133) and DGAPA-PAPIIT/UNAM IN211814. CC was funded by Spanish MINECO grant BES-2011-045905 linked to AGL2010-21929. BC-M was funded by Fundación ARAID. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David Des Marais and Jason Stajich for discussions, advice and the opportunity to test this software on Brachypodium transcriptomes. We also acknowledge comments from referees which helped improve the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00184/full#supplementary-material
Footnotes
- ^ ftp://ftp.arabidopsis.org/home/tair/Sequences/
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- ^ http://hmmer.org
- ^ https://transdecoder.github.io/
References
Aflitos, S. A., Severing, E., Sanchez-Perez, G., Peters, S., De Jong, H., and De Ridder, D. (2015). Cnidaria: fast, reference-free clustering of raw and assembled genome and transcriptome NGS data. BMC Bioinformatics 16:352. doi: 10.1186/s12859-015-0806-7
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bush, S. J., Castillo-Morales, A., Tovar-Corona, J. M., Chen, L., Kover, P. X., and Urrutia, A. O. (2014). Presence-absence variation in A. thaliana is primarily associated with genomic signatures consistent with relaxed selective constraints. Mol. Biol. Evol. 31, 59–69. doi: 10.1093/molbev/mst166
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cantalapiedra, C. P., Contreras-Moreira, B., Silvar, C., Perovic, D., Ordon, F., Gracia, M. P., et al. (2016). A Cluster of nucleotide-binding site–leucine-rich repeat genes resides in a barley powdery mildew resistance quantitative trait loci on 7HL. Plant Genome 9. doi: 10.3835/plantgenome2015.10.0101
Cao, J., Schneeberger, K., Ossowski, S., Gunther, T., Bender, S., Fitz, J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. doi: 10.1038/ng.911
Computational Pan-Genomics Consortium (2016). Computational pan-genomics: status, promises and challenges. Brief. Bioinform. doi: 10.1093/bib/bbw089 [Epub ahead of print].
Contreras-Moreira, B., Castro-Mondragon, J. A., Rioualen, C., Cantalapiedra, C. P., and Van Helden, J. (2016). RSAT::Plants: motif discovery within clusters of upstream sequences in plant genomes. Methods Mol. Biol. 1482, 279–295. doi: 10.1007/978-1-4939-6396-6_18
Contreras-Moreira, B., Sachman-Ruiz, B., Figueroa-Palacios, I., and Vinuesa, P. (2009). primers4clades: a web server that uses phylogenetic trees to design lineage-specific PCR primers for metagenomic and diversity studies. Nucleic Acids Res. 37, W95–W100. doi: 10.1093/nar/gkp377
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Dai, F., Chen, Z. H., Wang, X., Li, Z., Jin, G., Wu, D., et al. (2014). Transcriptome profiling reveals mosaic genomic origins of modern cultivated barley. Proc. Natl. Acad. Sci. U.S.A. 111, 13403–13408. doi: 10.1073/pnas.1414335111
Druka, A., Muehlbauer, G., Druka, I., Caldo, R., Baumann, U., Rostoks, N., et al. (2006). An atlas of gene expression from seed to seed through barley development. Funct. Integr. Genomics 6, 202–211. doi: 10.1007/s10142-006-0025-4
Felsenstein, J. (2005). ”PHYLIP (Phylogeny Inference Package) Version 3.6. Seattle, WA: Department of Genome Sciences.
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Gan, X., Stegle, O., Behr, J., Steffen, J. G., Drewe, P., Hildebrand, K. L., et al. (2011). Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423. doi: 10.1038/nature10414
Golicz, A. A., Batley, J., and Edwards, D. (2015). Towards plant pangenomics. Plant Biotechnol. J. 14, 1099–1105. doi: 10.1111/pbi.12499
Golicz, A. A., Bayer, P. E., Barker, G. C., Edger, P. P., Kim, H., Martinez, P. A., et al. (2016). The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7, 13390. doi: 10.1038/ncomms13390
Gu, L., Si, W., Zhao, L., Yang, S., and Zhang, X. (2015). Dynamic evolution of NBS-LRR genes in bread wheat and its progenitors. Mol. Genet. Genomics 290, 727–738. doi: 10.1007/s00438-014-0948-8
Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B. W., et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252. doi: 10.1038/ng.3506
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hansey, C. N., Vaillancourt, B., Sekhon, R. S., De Leon, N., Kaeppler, S. M., and Buell, C. R. (2012). Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PLoS ONE 7:e33071. doi: 10.1371/journal.pone.0033071
Hirsch, C. N., Foerster, J. M., Johnson, J. M., Sekhon, R. S., Muttoni, G., Vaillancourt, B., et al. (2014). Insights into the maize pan-genome and pan-transcriptome. Plant Cell 26, 121–135. doi: 10.1105/tpc.113.119982
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
International Barley Genome Sequencing Consortium, Mayer, K. F., Waugh, R., Brown, J. W., Schulman, A., Langridge, P., et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716. doi: 10.1038/nature11543
Jin, M., Liu, H., He, C., Fu, J., Xiao, Y., Wang, Y., et al. (2016). Maize pan-transcriptome provides novel insights into genome complexity and quantitative trait variation. Sci. Rep. 6, 18936. doi: 10.1038/srep18936
Jones, J. D., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Jupe, F., Witek, K., Verweij, W., Sliwka, J., Pritchard, L., Etherington, G. J., et al. (2013). Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 76, 530–544. doi: 10.1111/tpj.12307
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. doi: 10.1093/nar/gkn668
Lai, J., Li, R., Xu, X., Jin, W., Xu, M., Zhao, H., et al. (2010). Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030. doi: 10.1038/ng.684
Laing, C., Buchanan, C., Taboada, E. N., Zhang, Y., Kropinski, A., Villegas, A., et al. (2010). Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 11:461. doi: 10.1186/1471-2105-11-461
Leister, D., Kurth, J., Laurie, D. A., Yano, M., Sasaki, T., Devos, K., et al. (1998). Rapid reorganization of resistance gene homologues in cereal genomes. Proc. Natl. Acad. Sci. U.S.A. 95, 370–375. doi: 10.1073/pnas.95.1.370
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, R., Li, Y., Zheng, H., Luo, R., Zhu, H., Li, Q., et al. (2010). Building the sequence map of the human pan-genome. Nat. Biotechnol. 28, 57–63. doi: 10.1038/nbt.1596
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, Y. H., Zhou, G., Ma, J., Jiang, W., Jin, L. G., Zhang, Z., et al. (2014). De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052. doi: 10.1038/nbt.2979
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Liu, Y., Schroder, J., and Schmidt, B. (2013). Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 29, 308–315. doi: 10.1093/bioinformatics/bts690
Lu, F., Romay, M. C., Glaubitz, J. C., Bradbury, P. J., Elshire, R. J., Wang, T., et al. (2015). High-resolution genetic mapping of maize pan-genome sequence anchors. Nat. Commun. 6, 6914. doi: 10.1038/ncomms7914
Marroni, F., Pinosio, S., and Morgante, M. (2014). Structural variation and genome complexity: is dispensable really dispensable? Curr. Opin. Plant Biol. 18, 31–36. doi: 10.1016/j.pbi.2014.01.003
Mcgill, R., Tukey, J., and Larse, W. (1978). Variations of box plots. Am. Stat. 32, 12–16. doi: 10.2307/2683468
Mchale, L. K., Haun, W. J., Xu, W. W., Bhaskar, P. B., Anderson, J. E., Hyten, D. L., et al. (2012). Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 159, 1295–1308. doi: 10.1104/pp.112.194605
Michelmore, R. W., and Meyers, B. C. (1998). Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 8, 1113–1130.
Morgante, M., De Paoli, E., and Radovic, S. (2007). Transposable elements and the plant pan-genomes. Curr. Opin. Plant Biol. 10, 149–155. doi: 10.1016/j.pbi.2007.02.001
Muñoz-Amatriaín, M., Eichten, S. R., Wicker, T., Richmond, T. A., Mascher, M., Steuernagel, B., et al. (2013). Distribution, functional impact, and origin mechanisms of copy number variation in the barley genome. Genome Biol. 14, R58. doi: 10.1186/gb-2013-14-6-r58
Pertea, G., Huang, X., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., et al. (2003). TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652. doi: 10.1093/bioinformatics/btg034
Quadrana, L., Bortolini Silveira, A., Mayhew, G. F., Leblanc, C., Martienssen, R. A., Jeddeloh, J. A., et al. (2016). The Arabidopsis thaliana mobilome and its impact at the species level. Elife 5:e15716. doi: 10.7554/eLife.15716
R Development Core Team (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Sato, K., Tanaka, T., Shigenobu, S., Motoi, Y., Wu, J., and Itoh, T. (2015). Improvement of barley genome annotations by deciphering the Haruna Nijo genome. DNA Res. 23, 21–28. doi: 10.1093/dnares/dsv033
Sheikhizadeh, S., Schranz, M. E., Akdel, M., De Ridder, D., and Smit, S. (2016). PanTools: representation, storage and exploration of pan-genomic data. Bioinformatics 32, i487–i493. doi: 10.1093/bioinformatics/btw455
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Springer, N. M., Ying, K., Fu, Y., Ji, T., Yeh, C. T., Jia, Y., et al. (2009). Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 5:e1000734. doi: 10.1371/journal.pgen.1000734
Srivastava, A., Sarkar, H., Gupta, N., and Patro, R. (2016). RapMap: a rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes. Bioinformatics 32, i192–i200. doi: 10.1093/bioinformatics/btw277
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ”pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
The 1001 Genomes Consortium (2016). 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166, 481–491. doi: 10.1016/j.cell.2016.05.063
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
van Dongen, S. (2000). A Cluster Algorithm for Graphs. Amsterdam: National Research Institute for Mathematics and Computer Science in the Netherlands.
Vinuesa, P., and Contreras-Moreira, B. (2015). Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: a case study of pIncA/C plasmids. Methods Mol. Biol. 1231, 203–232. doi: 10.1007/978-1-4939-1720-4_14
Xiao, J., Zhang, Z., Wu, J., and Yu, J. (2015). A brief review of software tools for pangenomics. Genomics Proteomics Bioinformatics 13, 73–76. doi: 10.1016/j.gpb.2015.01.007
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556.
Yang, Z., and Bielawski, J. P. (2000). Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503. doi: 10.1016/S0169-5347(00)01994-7
Yano, K., Yamamoto, E., Aya, K., Takeuchi, H., Lo, P. C., Hu, L., et al. (2016). Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 48, 927–934. doi: 10.1038/ng.3596
Keywords: comparative genomics, pan-genome, RNA-seq, core-genome, accessory genome, Arabidopsis thaliana, barley
Citation: Contreras-Moreira B, Cantalapiedra CP, García-Pereira MJ, Gordon SP, Vogel JP, Igartua E, Casas AM and Vinuesa P (2017) Analysis of Plant Pan-Genomes and Transcriptomes with GET_HOMOLOGUES-EST, a Clustering Solution for Sequences of the Same Species. Front. Plant Sci. 8:184. doi: 10.3389/fpls.2017.00184
Received: 24 October 2016; Accepted: 30 January 2017;
Published: 14 February 2017.
Edited by:
Michael Eric Schranz, Wageningen University and Research Centre, NetherlandsReviewed by:
Ying Zhang, University of Rhode Island, USASumit Kumar Bag, CSIR-National Botanical Research Institute, India
Copyright © 2017 Contreras-Moreira, Cantalapiedra, García-Pereira, Gordon, Vogel, Igartua, Casas and Vinuesa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Contreras-Moreira, YmNvbnRyZXJhc0BlZWFkLmNzaWMuZXM=