 Ana M. Fortes
Ana M. Fortes Philippe Gallusci
Philippe Gallusci- 1Faculdade de Ciências, Instituto de Biossistemas e Ciências Integrativas, Universidade de Lisboa, Lisboa, Portugal
- 2UMR EGFV, Université de Bordeaux, Institut national de la recherche agronomique, Institut des Sciences de la Vigne et du Vin, Villenave-d’Ornon, France
Epigenetic marks include Histone Post-Translational Modifications and DNA methylation which are known to participate in the programming of gene expression in plants and animals. These epigenetic marks may be subjected to dynamic changes in response to endogenous and/or external stimuli and can have an impact on phenotypic plasticity. Studying how plant genomes can be epigenetically shaped under stressed conditions has become an essential issue in order to better understand the molecular mechanisms underlying plant stress responses and enabling epigenetic in addition to genetic factors to be considered when breeding crop plants. In this perspective, we discuss the contribution of epigenetic mechanisms to our understanding of plant responses to biotic and abiotic stresses. This regulation of gene expression in response to environment raises important biological questions for perennial species such as grapevine which is asexually propagated and grown worldwide in contrasting terroirs and environmental conditions. However, most species used for epigenomic studies are annual herbaceous plants, and epigenome dynamics has been poorly investigated in perennial woody plants, including grapevine. In this context, we propose grape as an essential model for epigenetic and epigenomic studies in perennial woody plants of agricultural importance.
Introduction
Epigenetic mechanisms regulate chromatin structure, gene expression, transposon mobility and DNA recombination (He et al., 2011; Pikaard and Scheid, 2015). They generally refer to modifications of gene expression that can be inherited through mitosis or meiosis yet without changes in the underlying DNA sequences (Eichten et al., 2014) and also include chromatin modifications that may lead to stable alteration of the transcriptional programming of non-dividing cells even after removal of the triggering signals (Avramova, 2015).
Epigenetic regulation is mediated by a complex interplay among different molecular actors. These include the DNA methylation/demethylation machinery, enzymes mediating histone post-translational modifications (PTMs), the remodeling of chromatin organization and specific classes of small RNAs and long non-coding RNAs (Lauria and Rossi, 2011; Pikaard and Scheid, 2015; Gallusci et al., 2016). Briefly, in plants 5 methyl-cytosine (m5C) is found in all sequence context, including the CG and CHG (H = A, T, or C) symmetrical motives and the non-symmetrical CHH motif (reviewed in Gehring, 2013). DNA methylation is maintained in a post-replicative way by three classes of DNA methyltransferases: DNA METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3) for CG and CHG contexts, respectively, and by the DOMAIN REARRANGED METHYLTRANSFERASE 2 (DRM2), which requires an siRNA guide and reestablishment after each cycle of DNA replication or by CMT2 for the asymmetric CHH context (Du et al., 2012; Matzke and Mosher, 2014). Finally, DNA methylation can be lost after replication when maintenance of DNA methylation is not functional or actively reversed by DNA Glycosylase-Lyases (Piccolo and Fisher, 2014).
Histone PTMs are also essential epigenetic signals that can occur at the N-terminal tail of core histones (H2A, H2B, H3, H4) through acetylation, methylation, phosphorylation and ubiquitination (Berr et al., 2011). Histone acetylation and methylation at lysine residues are established by histone acetyltransferases (HATs) and histone lysine methyltransferases (HKMTs), respectively, which are encoded by complex multigenic families. These epigenetic marks can be removed by histone deacetylases (HDACs) and histone demethylases (HDMs), respectively (Berr et al., 2011; He et al., 2011; Pikaard and Scheid, 2015).
The recent development of epigenome profiling has boosted our understanding of the dynamics and function of epigenetic marks in plants. Several approaches have been recently developed (Schmitz and Zhang, 2011; Lee and Kim, 20141). So far, histone PTM analysis relies on Chromatin Immunoprecipitation (ChIP) using specific antibodies followed by hybridization to tilling arrays (ChIP- chip, Makarevitch et al., 2015) or by high throughput sequencing (ChIP-Seq, Wang et al., 2009). DNA methylation landscape can be studied by making use of methyl sensitive restriction enzyme to enrich DNA in methylated or un-methylated sequences that are subsequently hybridized to tilling arrays or sequenced (Kim et al., 2014). Alternatively, methylated regions can be selected using m5C specific antibodies (MeDIP), and analyzed with tilling arrays (MeDip-ChIP) or by Next Generation Sequencing (Medip Seq). Both approaches were used for methylome analysis for example in Arabidopsis, or poplar (Zhang et al., 2006; Zilberman et al., 2006; Kim et al., 2014). In particular, Medip-Seq was used to analyze the changes in methylation patterns during in vitro culture of cassava (Kitimu et al., 2015). But the golden standard for methylome analysis is the combination of bisulfite conversion of DNA to high throughput sequencing that allows analyzing the methylation landscape at a single base resolution (Whole Genome Bisulfite sequencing: WGBS). The methylomes of Arabidopsis (Cokus et al., 2008; Lister et al., 2008; Stroud et al., 2013), rice (Li et al., 2012; Garg et al., 2015), maize (Eichten et al., 2013), tomato (Zhong et al., 2013), Brassica (Chalhoub et al., 2014) and many others (Niederhuth et al., 2016) have now been described using this approach.
In this perspective, we will firstly focus on the analysis of the genome wide distribution of epigenetic marks in plants under stresses. However, most species used for epigenomic studies are annual herbaceous plants and little is known about epigenomes in perennial woody plants. Indeed, omics’ approaches have been initiated in grape to understand environmental effects on plant and fruit development (Fortes et al., 2011; Agudelo-Romero et al., 2015). In addition, a few studies have indicated that epigenetic mechanisms might be involved in various aspects of grape development (Aquea et al., 2011). However, knowledge of grape epigenomes and of their variation has remained very limited until now (Niederhuth et al., 2016). Yet, grapevine presents several features that make it a relevant model for the study of epigenetic mechanisms due to the fact that is a perennial woody plant and the fruit maturation is subjected to non-climacteric molecular and hormonal regulation (Fortes et al., 2015). Grapevine varieties are preserved in their distinct genetic backgrounds through clonal propagation. However, phenotypic diversity exists within clones (Pelsy, 2010) that is unlikely to be solely driven by differences in DNA sequence. These facts contribute to the relevance of grape as a model for epigenetic and epigenomic studies in perennial woody plants of agricultural importance.
Epigenetic Reprogramming During Abiotic Stress Responses
Recent studies have shown the differential regulation of genes encoding epigenetic regulators (Fang et al., 2014; Li et al., 2014; Su et al., 2015) as well as local chromatin and DNA methylation changes in response to a variety of abiotic stresses including cold, salinity, drought, osmolality, or mineral nutrition, thereby highlighting the relevance of epigenetic regulations in these contexts (Chen et al., 2010; Luo et al., 2012; González et al., 2013; Bocchini et al., 2015; Kim et al., 2015; Liu et al., 2015). Consistent with these results, genome wide analyses of histone PTMs and DNA methylation distribution have revealed global epigenomic reprogramming in plants under abiotic stresses. In a recent study, trimethylation at lysine 4 on histone 3 (H3K4me3), a mark normally associated with gene expression, was analyzed in Arabidopsis plants under drought stress using ChIP-seq and showed to be highly dynamic and positively correlated with the transcription level of drought induced genes in response to stress (Dijk et al., 2010). Similar results were found in rice (Zong et al., 2013) and in moss (Widiez et al., 2014). Osmotic stress also causes an increase in phosphorylated histone H3 threonine 3 (H3T3ph) located at pericentromeric regions where it is thought to help maintaining the heterochromatin structure (Wang et al., 2015). Interestingly, H3T3ph is also present in active genes where it seemed to antagonize H3K4me3, suggesting that H3T3ph may have a repressive function on gene expression during osmotic stress (Wang et al., 2015) a role also suggested for histone deacetylase HDA9 (Zheng et al., 2016). In addition, priming effects in Arabidopsis were shown to be partly mediated by remodeling of the epigenomic landscape, and involves the repressive mark H3K27me3 (Sani et al., 2013).
Recently, a specialized histone H1 variant was shown to be required for a substantial part of DNA methylation associated with environmental stress in Arabidopsis (Rutowicz et al., 2015) and two DEAD-box RNA helicases were suggested to be involved in epigenetic silencing of gene expression leading to suppression of Arabidopsis stress response (Khan et al., 2014).
In addition, DNA methylation is also critical for the responses of plant to abiotic stresses. This was initially shown by the demonstration that Arabidopsis mutants deficient in various steps of the RdDM pathway or in CHG maintenance methylation are affected in their capacity to modulate the stomatal index under low relative humidity (Tricker et al., 2012), present an hypersensitivity to heat exposure (Popova et al., 2013) or an enhanced sensitivity to phosphate starvation (Yong-Villalobos et al., 2015). These results are consistent with an important function of the DNA methylation dynamics in the regulation of abiotic stress–responsive genes. Indeed drought stress, but also nutrient deprivation cause extensive remodeling of DNA methylation patterns in Arabidopsis (Colaneri and Jones, 2013; Yong-Villalobos et al., 2015; Wibowo et al., 2016), barley (Chwialkowska et al., 2016) or Populus (Liang et al., 2014). In this latter case, modulation of DNA methylation at repetitive elements appeared essential for the control of adjacent gene expression (Liang et al., 2014) a function also suggested in maize where TEs could be used as local enhancers for stress responsive genes (Makarevitch et al., 2015). Similarly Pi deficiency in rice modulates DNA methylation at TEs located close to genes highly induced under this stress (Secco et al., 2015). In this case, however, TEs were hyper-methylated an event that occurred after gene induction most likely to prevent potentially deleterious activity of TEs located in the vicinity of highly induced stress responsive genes.
As a conclusion, the results discussed above are consistent with the idea that abiotic stresses cause significant reprogramming of chromatin not only related to gene expression, but also to the control of chromosome organization. In addition, evidence of transgenerational inheritance of plant responses to stress has been provided (Tricker et al., 2013; Migicovsky et al., 2014); although this process appears limited to and mainly mediated by the female gamete (Wibowo et al., 2016).
Epigenetic Reprogramming During Plant Biotic Stress Responses
Regarding histone Post-Translational Modifications and DNA methylation occurring upon biotic stress there is lesser information available than for abiotic stress. However, recent findings indicate that chromatin modifications contribute to plant immunity against both necrotrophic and biotrophic pathogens (reviewed by Ding and Wang, 2015). In fact, the expression of R genes which are central regulators of plant immunity was shown to be regulated by Arabidopsis E3 ubiquitin ligase genes HISTONE MONOUBIQUITINATION1 (HUB1) and HUB2 (Zou et al., 2014). Histone monoubiquitination at the R gene locus had an impact on immune responses. The loss of- function mutant bon1-1 has enhanced disease resistance to the virulent pathogen Pst DC3000 and both HUB1 and HUB2 mediate its autoimmune responses. In another case, HDA19, an Arabidopsis histone deacetylase, was shown to play a negative role in basal defense mediated by the SA-dependent signaling pathway. Loss of HDA19 causes increased expression of SA biosynthetic genes and defense genes and promotes resistance to the virulent Pst DC3000 (Choi et al., 2012). Dimethylated or trimethylated histone H3 Lys 27 (H3K27me2/3) marks silent or repressed genes involved in stress responses in plants. Li et al. (2013) showed that the rice Jumonji C protein gene JMJ705 encodes a histone Lys demethylase that specifically reverses this mark. An increase in JMJ705 expression in transgenic plants removes H3K27me3 from defense-related genes, induces their expression with involvement of jasmonic acid, and enhances plant resistance to biotic stress. Interestingly, Soyer et al. (2014) showed that chromatin-based transcriptional regulation can also act on effector gene expression in fungi during plant infection. Pathogen infection has been also reported to change histone modifications in some defense response genes (De-La-Peńa et al., 2012).
The profiling of the DNA methylomes of plants exposed to bacterial pathogen, avirulent bacteria, or salicylic acid revealed numerous stress-induced differentially methylated regions (DMRs) often coupled to differential gene expression (Dowen et al., 2012). Mutant plants globally defective in maintenance of CG methylation (met1-3) or non-CG methylation (ddc, drm1-2 drm2-2 cmt3-11) were markedly resistant to bacterial colonization.
DNA demethylation likely primes transposable elements as well as defense gene induction through the concomitant activation of their transactivators and/or the interference with other chromatin marks (Yu et al., 2013). Some immune-response genes, containing repeats in their promoter regions, are negatively regulated by DNA methylation. These defense gene loci may lose DNA methylation so that they are more easily activated at the transcriptional level (Yu et al., 2013). This is corroborated by the study of Le et al. (2014); the DNA methylases ROS1, DML2, and DML3 were shown to play a role in fungal disease resistance in Arabidopsis since a triple mutant rdd (ros1 dml2 dml3), presents down-regulation of stress response genes and increased susceptibility to a fungal pathogen. Furthermore, these authors showed that DNA demethylases target promoter transposable elements in stress responsive genes to positively regulate them.
Natural and Induced Epigenomic Variation, Phenotypic Plasticity and Breeding
Natural epigenomic variation occurs during species evolution (Hirsch et al., 2013) and together with genetic variation is likely involved in the phenotypic diversity and plasticity of plants. Epigenetic variation is sensitive to environmental inputs; epialleles induced by the environment or experimentally may be formed at a higher rate than alleles generated from genetic variation and may also be inherited leading to better adaptation to the environment (Figure 1; Hirsch et al., 2013).
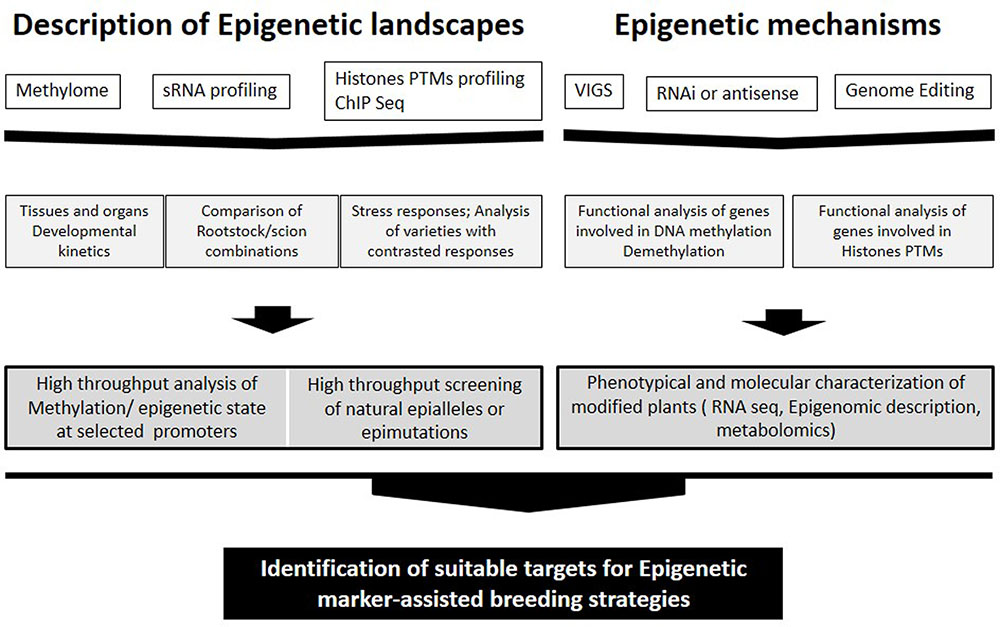
FIGURE 1. Approaches for identification of suitable targets for Epigenetic marker-assisted breeding strategies ranging from studying epigenetic landscapes to clarification of epigenetic mechanisms.
Experimentally induced epialleles have been produced in Arabidopsis by generating Epigenetic Recombinant Inbred Line (EpiRILs) populations derived from decrease in DNA methylation 1-2 (ddm1-2) or the met1 parents (Johannes et al., 2009; Reinders et al., 2009). EpiRILs were subsequently used to identify epiQTL corresponding to DMRs that determine two complex traits, flowering time and primary root length (Cortijo et al., 2014). Interestingly, these EpiRILs present variation in growth capacity (Hu et al., 2015) and are more sensitive to salinity stress than the Col0 parent line suggesting that ddm1 derived epigenotypes limit the ability to adapt to this stress (Kooke et al., 2015). As an alternative approach, a stochastically hypomethylated population was generated by selfing Brassica rapa plants previously treated with the demethylating agent 5-Azacytidine (Amoah et al., 2012). This population was used for forward screening of agronomic traits such as flowering time, seed protein content and fatty acid components. These results suggest that a portion of QTLs that have been used by breeders so far may be due to epigenetic, rather than genetic variation (Springer, 2013).
DNA methylation may also have an important role in the long term adaptation of plants (Figure 1; Garg et al., 2015). Two rice cultivars with contrasting sensitivity to drought stress and salinity showed clearly different methylation landscapes; part of the DMRs between cultivars were associated with genes involved in stress responses (Garg et al., 2015).
Indeed variation in methylation patterns have been also observed in natural populations and might be associated with specific environmental traits. In a recent study, Dubin et al. (2015) showed by analyzing Arabidopsis accessions from Northern and Southern Sweden that CHH methylation at transposons increases with temperature and this was associated with major genetic variants at the CMT2 locus. In the same study, Gene Body Methylation which was not modified by temperature was shown to be correlated with the latitude of origin; Southern accessions being less methylated than Northern one. This was associated with a lower expression of the targeted genes in Southern accessions consistent with local adaptation of the accessions.
Epialleles impacting plant traits have now been identified in many plants (Rodríguez López and Wilkinson, 2015) since the initial characterization of the cycloidea and Cnr epimutations in snapdragon and tomato, respectively (Cubas et al., 1999; Manning et al., 2006; Poole et al., 2006). For example, Vitamin E in tomato is determined by epigenetic variations linked to a SINE retrotransposon located in the promoter region of a gene involved in the vitamin synthesis. This work showed that naturally occurring epialleles may be responsible for regulation of nutritionally important metabolic QTLs and determination of agronomic traits (Quadrana et al., 2014). In another study, the complex trait of Energy use efficiency was shown to possess an epigenetic component that is stably inherited, allowing the creation of distinct isogenic sublines that can be used in breeding (Hauben et al., 2009). Thus, induced or natural epigenetic diversity may represent an unexplored resource of phenotypical variations that could be used in plant breeding programs, as recently discussed in Rodríguez López and Wilkinson (2015).
Grapevine Epigenomics and Epigenetics: A Model Plant for Perennial Crop Plant
Studies on Arabidopsis revealed functional aspects of epigenetic regulation of gene expression but present limitations since Arabidopsis has only 5% of methylated cytosine in the genome whereas many crops contain more than 20% (Lee and Kim, 2014). In fact, mutations in epigenetic regulators seem to have a higher impact in crops than in Arabidopsis (Mirouze and Vitte, 2014; Gallusci et al., 2016). In addition, Arabidopsis contains very few transposable elements comparing to crops (reviewed by Lee and Kim, 2014). Polymorphisms in transposon insertions and repeats can originate natural epigenetic variation. Furthermore, while the distribution of the genes along the chromosomes of Arabidopsis is fairly homogeneous, this situation may differ in crops. For example, Vitis vinifera genome is characterized by alternation of large regions with high and low gene density (Jaillon et al., 2007).
Several studies have already emerged in crops, in particular, recent analyzes carried out in tomato fruits (Zhong et al., 2013; Liu et al., 2015) constitute a relevant background for studies in grape. It is not yet known whether the epigenetic control of ripening is similar in all fleshy fruits or is limited to the tomato and related wild species (reviewed in Gallusci et al., 2016). Nevertheless, the expression patterns of several genes involved in DNA methylation and histones modifications indicate that epigenetic factors are involved in the onset of véraison in grape and a global decrease in DNA methylation may eventually occur during grape ripening (Fortes et al., 2011) as reported for tomato (Zhong et al., 2013; Liu et al., 2015). In this context, the lack of available mutants in grape constitutes a limitation comparing to tomato. However, studies addressing the methylation status of promoters of genes involved in easily identified traits can shed light on epigenetic regulation of gene expression in grape (Figure 1).
Chemical treatments that affect DNA methylation patterns could also be utilized to generate epimutations (Amoah et al., 2012) though they may not be as stable as genetic mutants. Several examples of epimutations in crops are mentioned in the review by Zhang and Hsieh (2013). Epimutagenesis may allow the opportunity to explore allelic variation and novel combinations of alleles without relying upon recombination (Springer, 2013).
Analysis of the distribution of epi-marks and DNA methylation in grape in relation with gene expression profiles and fruit quality traits would likely identify epialleles that could be used as important new targets for plant breeding (Figure 1). DNA methylation may generate multiple epialleles with various expression levels, thereby leading to continuous quantitative variation of a trait (Zeng and Cheng, 2014). On the other hand, Kitimu et al. (2015) identified candidate epimarks that distinguish between field cuttings and meristem culture cassava samples. Specific methylation signatures may be used in the future for the diagnosis of somaclonal variants and clonal stocks in grapevine.
Grape combines several specific features that could make it an appealing model to study epigenetic regulations in woody perennial plants. It is used as one of the main models for non-climacteric fruits and also flower development is programmed 1 year in advance; the impact of environmental conditions on flower and subsequently fruit development seems to be in part determined by the environmental conditions the year before. Grape also has specific requirements such as grafting, and clonal propagation. In this context, epigenetic variability could add to the genetic diversity of grape to shape the phenotypic variations observed in this plant. Consistent with this view, clonal diversity within V. vinifera varieties has been distinguished using the methylation-sensitive amplified polymorphism technique, highlighting the usefulness of using epigenetic markers in intra-varietal diversity studies (Ocaña et al., 2013). Grafting could also impact the epigenetic state of both rootstocks and shoots (scions), Figure 1. Recently, Lewsey et al. (2015) showed that mobile sRNAs regulate the DNA methylation landscape genome wide, and may be an important mechanism of genome defense in crops. They showed that site-specific transmission of epiallelic states from one accession to another can be achieved by grafting and by de novo methylation of unmethylated DNA, consistent with the idea that some effects of grafting are due to the movement of small RNAs. In grapevine, grafting with rootstocks induced the up-regulation of genes associated with DNA methylation and chromatin modification in the shoot apical meristem (Cookson and Ollat, 2013). Clarifying these mechanisms may open doors to innovative applications to enhance grapevine tolerance to stresses and grape quality.
In line with these ideas, the recent analysis of the transcriptomic changes associated with grape infection with the necrotrophic pathogen Botrytis cinerea suggested that epigenetic mechanisms are involved in the reprogramming of fruit defense (Agudelo-Romero et al., 2015). Genes coding for histones, DNA (cytosine-5)-methyltransferase, helicases, DICER and ARGONAUTE proteins were modulated during the infection, whereas those associated with TEs mobility were down-regulated (Table 1).
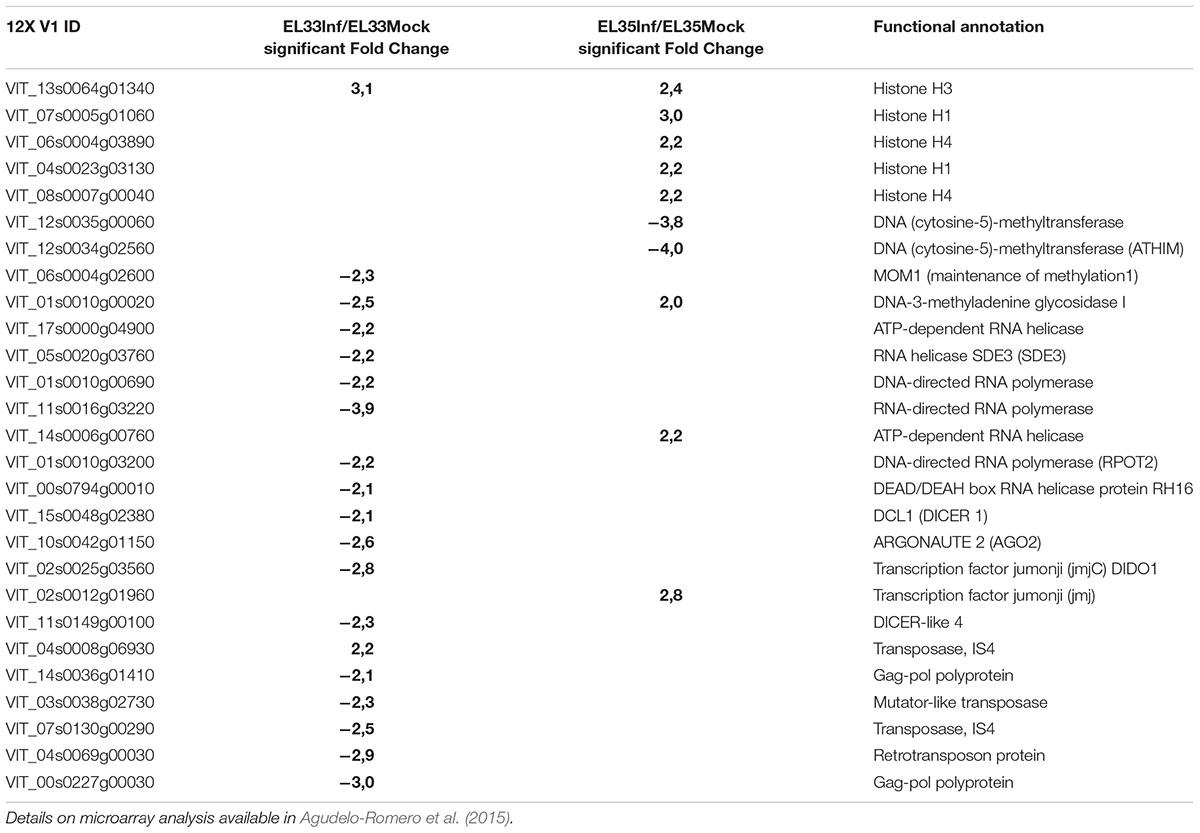
TABLE 1. Genes involved in epigenetic mechanisms differentially modulated in Trincadeira grapes infected with the fungus Botrytis cinerea at green hard stage (EL33) and véraison stage (EL35).
Base-resolution methylomes and high-throughput sRNA profilings are already available in more than 34 species (Niederhuth et al., 2016) including V. vinifera. Comparing the epigenomes of wild and cultivated Vitis species with and without biotic and non-biotic stresses will bring insights on the epigenetic basis of grapevine resistance to adverse conditions with potential impact in breeding strategies. Moreover, epigenetic marks may participate in the priming mechanisms to better withstand biotic and abiotic stresses (Crisp et al., 2016), another topic that deserves attention in order to moderate stress susceptibility and increase climate change resilience in grapevine. Interestingly, these epimarks can also be used in the future for distinguishing agronomic practices and terroir certification of wines.
Previously, transgenerational systemic acquired resistance, was demonstrated to be a prominent defense mechanism toward downy mildew pathogen and involves DNA methylation (Luna and Ton, 2012). In grapevine, a further layer of complexity can be added since memory in perennial plants is affected every year in meristems committed to flowering. Furthermore, the reason why epigenetic regulation in response to stress can be transient or transgenerational are not clear (Tricker, 2015). It is also not known the contribution of pathogen-responsive siRNAs in transgenerational immune priming and how they drive the selection of new phenotypes especially in perennial plants.
A deeper understanding of the molecular mechanisms involving tissue-specific epigenetic changes underlying genotype × environment interactions may be beneficial for long-term improvement of grapevine performance in less predictable climates with new sources of diseases.
In a near future, epigenetic marker-assisted breeding strategies will be applied to select for agronomical desirable epigenetic quantitative traits (Figure 1). Crop improvement via locus-specific epigenetic manipulation has become increasingly feasible with TALE- or CRISPR-based genome editing technologies (Mendenhall et al., 2013; Zhang and Hsieh, 2013). Such technologies can be expected to play an important role in grapevine improvement once transgenesis’ protocols are optimized for different cultivars.
Author Contributions
AF and PG designed the perspective and wrote the manuscript.
Funding
Funding was provided by the Portuguese Foundation for Science and Technology (SFRH/BPD/100928/2014, FCT Investigator FCT050, PEst-OE/BIA/UI4046/2014).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. Graham Seymour from the University of Nottingham for carefully reading the manuscript and the COST (European Cooperation in Science and Technology) Action FA1106 “Quality fruit.”
Footnotes
References
Agudelo-Romero, P., Erban, A., Rego, C., Carbonell-Bejerano, P., Nascimento, T., Sousa, L., et al. (2015). Transcriptome and metabolome reprogramming in Vitis vinifera cv. Trincadeira berries upon infection with Botrytis cinerea. J. Exp. Bot. 66, 1769–1785. doi: 10.1093/jxb/eru517
Amoah, S., Kurup, S., Rodriguez Lopez, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A Hypomethylated population of Brassica rapa for forward and reverse Epi-genetics. BMC Plant Biol. 12:193. doi: 10.1186/1471-2229-12-193
Aquea, F., Vega, A., Timmermann, T., Poupin, M. J., and Arce-Johnson, P. (2011). Genome-wide analysis of the SET DOMAIN GROUP family in Grapevine. Plant Cell Rep. 30, 1087–1097. doi: 10.1007/s00299-011-1015-0
Avramova, Z. (2015). Transcriptional “memory” of a stress: transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 83, 149–159. doi: 10.1111/tpj.12832
Berr, A., Sha, S., and Shen, W. (2011). Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta 1809, 567–576. doi: 10.1016/j.bbagrm.2011.07.001
Bocchini, M., Bartucca, M. L., Ciancaleoni, S., Mimmo, T., Cesco, S., Pii, Y., et al. (2015). Iron deficiency in barley plants: phytosiderophore release, iron translocation, and DNA methylation. Front. Plant Sci. 6:514. doi: 10.3389/fpls.2015.00514
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, L., Luo, M., Wang, Y., and Wu, K. (2010). Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J. Exp. Bot. 61, 3345–3353. doi: 10.1093/jxb/erq154
Choi, S. M., Song, H. R., Han, S. K., Han, M., Kim, C. Y., Park, J., et al. (2012). HDA19 is required for the repression of salicylic acid biosynthesis and salicylic acid-mediated defense responses in Arabidopsis. Plant J. 71, 135–146. doi: 10.1111/j.1365-313X.2012.04977.x
Chwialkowska, K., Nowakowska, U., Mroziewicz, A., Szarejko, I., and Kwasniewski, M. (2016). Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (Hordeum vulgare L.). J. Exp. Bot. 67, 1109–1121. doi: 10.1093/jxb/erv552
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Colaneri, A. C., and Jones, A. M. (2013). Genome-wide quantitative identification of DNA differentially methylated sites in Arabidopsis seedlings growing at different water potential. PLoS ONE 8:e59878. doi: 10.1371/journal.pone.0059878
Cookson, S. J., and Ollat, N. (2013). Grafting with rootstocks induces extensive transcriptional re-programming in the shoot apical meristem of grapevine. BMC Plant Biol. 13:147. doi: 10.1186/1471-2229-13-147
Cortijo, S., Wardenaar, R., Colomé-Tatché, M., Gilly, A., Etcheverry, M., Labadie, K., et al. (2014). Mapping the epigenetic basis of complex traits. Science 343, 1145–1148. doi: 10.1126/science.1248127
Crisp, P. A., Ganguly, D., Eichten, S. R., Borevitz, J. O., and Pogson, B. J. (2016). Reconsidering plant memory: intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2:e1501340. doi: 10.1126/sciadv.1501340
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
De-La-Peña, C., Rangel-Cano, A., and Alvarez-Venegas, R. (2012). Regulation of disease-responsive genes mediated by epigenetic factors: interaction of Arabidopsis–Pseudomonas. Mol. Plant Pathol. 13, 388–398. doi: 10.1111/J.1364-3703.2011.00757.X
Dijk, K., Van Ding, Y., Malkaram, S., Riethoven, J. M., Liu, R., Yang, J., et al. (2010). Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 10:238. doi: 10.1186/1471-2229-10-238
Ding, B., and Wang, G.-L. (2015). Chromatin versus pathogens: the function of epigenetics in plant immunity. Front. Plant Sci. 6:675. doi: 10.3389/fpls.2015.00675
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. U.S.A. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Du, J., Zhong, X., Bernatavichute, Y. V., Stroud, H., Feng, S., Caro, E., et al. (2012). Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151, 167–180. doi: 10.1016/j.cell.2012.07.034
Dubin, M. J., Zhang, P., Meng, D., Remigereau, M., Osborne, E. J., Casale, F. P., et al. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4:e05255. doi: 10.7554/eLife.05255
Eichten, S. R., Briskine, R., Song, J., Li, Q., Swanson-wagner, R., Hermanson, P. J., et al. (2013). Epigenetic and genetic In fl uences on DNA methylation variation in maize populations. Plant Cell 25, 2783–2797. doi: 10.1105/tpc.113.114793
Eichten, S. R., Schmitz, R. J., and Springer, N. M. (2014). Epigenetics: beyond chromatin modifications and complex genetic regulation. Plant Physiol. 165, 933–947. doi: 10.1104/pp.113.234211
Fang, H., Liu, X., Thorn, G., Duan, J., and Tian, L. (2014). Biochemical and biophysical research communications expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun. 443, 400–405. doi: 10.1016/j.bbrc.2013.11.102
Fortes, A. M., Agudelo-Romero, P., Silva, M. S., Ali, K., Sousa, L., Maltese, F., et al. (2011). Transcript and metabolite analysis in Trincadeira cultivar reveals novel information regarding the dynamics of grape ripening. BMC Plant Biol. 11:149. doi: 10.1186/1471-2229-11-149
Fortes, A. M., Teixeira, R. T., and Agudelo-Romero, P. (2015). Complex interplay of hormonal signals during grape berry ripening. Molecules 20, 9326–9343. doi: 10.3390/molecules20059326
Gallusci, P., Hodgman, C., Teyssier, E., and Seymour, G. B. (2016). DNA methylation and chromatin regulation during fleshy fruit development and ripening. Front. Plant Sci. 7:807. doi: 10.3389/fpls.2016.00807
Garg, R., Narayana Chevala, V., Shankar, R., and Jain, M. (2015). Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 5:14922. doi: 10.1038/srep14922
Gehring, M. (2013). Genomic imprinting: insights from plants. Annu. Rev. Genet. 47, 187–208. doi: 10.1146/annurev-genet-110711-155527
González, R. M., Ricardi, M. M., and Iusem, N. D. (2013). Epigenetic marks in an adaptive water stress-responsive gene in tomato roots under normal and drought conditions. Epigenetics 8, 864–872. doi: 10.4161/epi.25524
Hauben, M., Haesendonckx, B., Standaert, E., Kelen, K., Van Der Azmi, A., Breusegem, F., et al. (2009). Energy use efficiency is characterized by an epigenetic component that can be directed. Proc. Natl. Acad. Sci. U.S.A. 106, 20109–20114. doi: 10.1073/pnas.0908755106
He, G., Elling, A. A., and Deng, X. W. (2011). The epigenome and plant development. Annu. Rev. Plant Biol 62, 411–435. doi: 10.1146/annurev-arplant-042110-103806
Hu, Y., Morota, G., Rosa, G. J. M., and Gianola, D. (2015). Prediction of plant height in Arabidopsis thaliana using DNA methylation data. Genetics 201, 779–793. doi: 10.1534/genetics.115.177204
Hirsch, S., Baumberger, R., and Grossniklaus, U. (2013). Epigenetic variation, inheritance, and selection in plant populations. Cold Spring Harb. Symp. Quant. Biol. 77, 97–104. doi: 10.1101/sqb.2013.77.014605
Jaillon, O., Aury, J.-M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Johannes, F., Porcher, E., Teixeira, F. K., Saliba-colombani, V., Albuisson, J., Heredia, F., et al. (2009). Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 5:e1000530. doi: 10.1371/journal.pgen.1000530
Khan, A., Garbelli, A., Grossi, S., Florentin, A., Batelli, G., Acuna, T., et al. (2014). The Arabidopsis STRESS RESPONSE SUPPRESSOR DEAD-box RNA helicases are nucleolar- and chromocenter-localized proteins that undergo stress-mediated relocalization and are involved in epigenetic gene silencing. Plant J. 79, 28–43. doi: 10.1111/tpj.12533
Kim, J.-M., Sasaki, T., Ueda, M., Sako, K., and Seki, M. (2015). Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 6:114. doi: 10.3389/fpls.2015.00114
Kim, K., Do Baidouri, M. E. l., and Jackson, S. A. (2014). Accessing epigenetic variation in the plant methylome. Brief. Funct. Genomics 13, 318–327. doi: 10.1093/bfgp/elu003
Kitimu, S. R., Taylor, J., March, T. J., Tairo, F., Wilkinson, M. J., and Lopez, C. M. R. (2015). Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 6:590. doi: 10.3389/fpls.2015.00590
Kooke, R., Johannes, F., Wardenaar, R., Becker, F., Etcheverry, M., Colot, V., et al. (2015). Epigenetic basis of morphological variation and phenotypic plasticity in Arabidopsis thaliana. Plant Cell 27, 337–348. doi: 10.1105/tpc.114.133025
Lauria, M., and Rossi, V. (2011). Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta 1809, 369–378. doi: 10.1016/j.bbagrm.2011.03.002
Le, T.-N., Schumann, U., Smith, N. A., Tiwari, S., Au, P., Zhu, Q.-H., et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 15, 458. doi: 10.1186/s13059-014-0458-3
Lee, S., and Kim, N. (2014). Transposable elements and genome size variations in plants. Genomics Inform. 12, 87–97. doi: 10.5808/GI.2014.12.3.87
Lewsey, M. G., Hardcastle, T. J., Melnyk, C. W., Molnar, A., Valli, A., and Urich, M. A. (2015). Mobile small RNAs regulate genome-wide DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 113, 1–10. doi: 10.1073/pnas.1515072113
Li, H., Yan, S., Zhao, L., Tan, J., Zhang, Q., Gao, F., et al. (2014). Histone acetylation associated up-regulation of the cell wall related genes is involved in salt stress induced maize root swelling. BMC Plant Biol. 14:105. doi: 10.1186/1471-2229-14-105
Li, T., Chen, X., Zhong, X., Zhao, Y., Liu, X., Zhou, S., et al. (2013). Jumonji C domain protein JMJ705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 25, 4725–4736. doi: 10.1105/tpc.113.118802
Li, X., Zhu, J., Hu, F., Ge, S., Ye, M., Xiang, H., et al. (2012). Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics 13:300. doi: 10.1186/1471-2164-13-300
Liang, D., Zhang, Z., Wu, H., Huang, C., Shuai, P., Ye, C., et al. (2014). Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 15(Suppl. 1):S9. doi: 10.1186/1471-2156-15-S1-S9
Lister, R., O’Malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Liu, R., How-Kit, A., Stammitti, L., Teyssier, E., Rolin, D., Mortain-Bertrand, A., et al. (2015). A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. U.S.A. 112, 10804–10809. doi: 10.1073/pnas.1503362112
Luna, E., and Ton, J. (2012). The epigenetic machinery controlling transgenerational systemic acquired resistance. Plant Signal. Behav. 7, 615–618. doi: 10.4161/psb.20155
Luo, M., Liu, X., Singh, P., Cui, Y., Zimmerli, L., and Wu, K. (2012). Chromatin modifications and remodeling in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 129–136. doi: 10.1016/j.bbagrm.2011.06.008
Makarevitch, I., Waters, A. J., West, P. T., Stitzer, M., Hirsch, C. N., Ross-ibarra, J., et al. (2015). Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 11:e1004915. doi: 10.1371/journal.pgen.1004915
Manning, K., Tor, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Mendenhall, E. M., Williamson, K. E., Reyon, D., Zou, J. Y., Ram, O., Joung, J. K., et al. (2013). Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 31, 1133–1136. doi: 10.1038/nbt.2701
Migicovsky, Z., Yao, Y., and Kovalchuk, I. (2014). Transgenerational phenotypic and epigenetic changes in response to heat stress in Arabidopsis thaliana. Plant Signal. Behav. 9, e27971. doi: 10.4161/psb.27971
Mirouze, M., and Vitte, C. (2014). Transposable elements, a treasure trove to decipher epigenetic variation: insights from Arabidopsis and crop epigenomes. J. Exp. Bot. 65, 2801–2812. doi: 10.1093/jxb/eru120
Niederhuth, C. E., Bewick, A. J., Ji, L., Alabady, M. S., Kim, K., Do Li, Q., et al. (2016). Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 17, 194. doi: 10.1186/s13059-016-1059-0
Ocaña, J., Walter, B., and Schellenbaum, P. (2013). Stable MSAP markers for the distinction of Vitis vinifera cv pinot noir clones. Mol. Biotechnol. 55, 236–248. doi: 10.1007/s12033-013-9675-3
Pelsy, F. (2010). Molecular and cellular mechanisms of diversity within grapevine varieties. Heredity (Edinb). 104, 331–340. doi: 10.1038/hdy.2009.161
Piccolo, F. M., and Fisher, A. G. (2014). Getting rid of DNA methylation. Trends Cell Biol. 24, 136–143. doi: 10.1016/j.tcb.2013.09.001
Pikaard, C. S., and Scheid, O. M. (2015). Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 6:a019315. doi: 10.1101/cshperspect.a019315
Poole, M., Hong, Y., Thompson, A. J., King, G. J., Manning, K., To, M., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Popova, O. V., Dinh, H. Q., Aufsatz, W., and Jonak, C. (2013). The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol. Plant 6, 396–410. doi: 10.1093/mp/sst023
Quadrana, L., Almeida, J., Asís, R., Duffy, T., Dominguez, P. G., Bermúdez, L., et al. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5, 3027. doi: 10.1038/ncomms5027
Reinders, J., Wulff, B. B. H., Mirouze, M., Marí-Ordóñez, A., Dapp, M., Rozhon, W., et al. (2009). Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 23, 939–950. doi: 10.1101/gad.524609
Rodríguez López, C. M., and Wilkinson, M. J. (2015). Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 6:397. doi: 10.3389/fpls.2015.00397
Rutowicz, K., Puzio, M., Halibart-Puzio, J., Lirski, M., Kotliñski, M., Kroteñ, M. A., et al. (2015). A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol. 169, 2080–2101. doi: 10.1104/pp.15.00493
Sani, E., Herzyk, P., Perrella, G., Colot, V., and Amtmann, A. (2013). Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 14:R59. doi: 10.1186/gb-2013-14-6-r59
Schmitz, R. J., and Zhang, X. (2011). High-throughput approaches for plant epigenomic studies. Curr. Opin. Plant Biol. 14, 130–136. doi: 10.1016/j.pbi.2011.03.010
Secco, D., Wang, C., Shou, H., Schultz, M. D., Chiarenza, S., Nussaume, L., et al. (2015). Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. Elife 4:e09343. doi: 10.7554/eLife.09343
Soyer, J. L., El Ghalid, M., Glaser, N., Ollivier, B., Linglin, J., Grandaubert, J., et al. (2014). Epigenetic control of effector gene expression in the plant pathogenic fungus Leptosphaeria maculans. PLoS Genet. 10:e1004227. doi: 10.1371/journal.pgen.1004227
Springer, N. M. (2013). Epigenetics and crop improvement. Trends Genet. 29, 241–247. doi: 10.1016/j.tig.2012.10.009
Stroud, H., Greenberg, M. V. C., Feng, S., Bernatavichute, Y. V., and Jacobsen, S. E. (2013). Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364. doi: 10.1016/j.cell.2012.10.054
Su, L.-C., Deng, B., Liu, S., Li, L.-M., Hu, B., Zhong, Y.-T., et al. (2015). Isolation and characterization of an osmotic stress and ABA induced histone deacetylase in Arachis hypogaea. Front. Plant Sci. 6:512. doi: 10.3389/fpls.2015.00512
Tricker, P. J. (2015). Transgenerational inheritance or resetting of stress-induced epigenetic modifications: two sides of the same coin. Front. Plant Sci. 6:699. doi: 10.3389/fpls.2015.00699
Tricker, P. J., George Gibbings, J., Rodríguez López, C. M., Hadley, P., and Wilkinson, M. J. (2012). Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 63, 3799–3814. doi: 10.1093/jxb/ers076
Tricker, P. J., López, C. M. R., Gibbings, G., Hadley, P., and Wilkinson, M. J. (2013). Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int. J. Mol. Sci. 14, 6674–6689. doi: 10.3390/ijms14046674
Wang, X., Elling, A. A., Li, X., Li, N., Peng, Z., He, G., et al. (2009). Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 21, 1053–1069. doi: 10.1105/tpc.109.065714
Wang, Z., Casas-mollano, J. A., Xu, J., Riethoven, J. M., and Zhang, C. (2015). Osmotic stress induces phosphorylation of histone H3 at threonine 3 in pericentromeric regions of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 112, 8487–8492. doi: 10.1073/pnas.1423325112
Wibowo, A., Becker, C., Marconi, G., Durr, J., Price, J., Hagmann, J., et al. (2016). Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by dna glycosylase activity. Elife 5, 1–27. doi: 10.7554/eLife.13546
Widiez, T., Symeonidi, A., Luo, C., Lam, E., Lawton, M., and Rensing, S. A. (2014). The chromatin landscape of the moss Physcomitrella patens and its dynamics during development and drought stress. Plant J. 79, 67–81. doi: 10.1111/tpj.12542
Yong-Villalobos, L., González-Morales, S. I., Wrobel, K., Gutiérrez-Alanis, D., Cervantes-Peréz, S. A., Hayano-Kanashiro, C., et al. (2015). Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc. Natl. Acad. Sci. U.S.A. 112, E7293–E7302. doi: 10.1073/pnas.1522301112
Yu, A., Lepère, G., Jay, F., Wang, J., Bapaume, L., Wang, Y., et al. (2013). Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 110, 2389–2394. doi: 10.1073/pnas.1211757110
Zeng, F., and Cheng, B. (2014). Transposable element insertion and epigenetic modification cause the multiallelic variation in the expression of FAE1 in Sinapis alba. Plant Cell 26, 2648–2659. doi: 10.1105/tpc.114.126631
Zhang, C., and Hsieh, T. (2013). Heritable epigenetic variation and its potential applications for crop improvement. Plant Breed. Biotechnol. 1, 307–319. doi: 10.9787/PBB.2013.1.4.307
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006). Resource Mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201. doi: 10.1016/j.cell.2006.08.003
Zheng, Y., Ding, Y., Sun, X., Xie, S., Wang, D., Liu, X., et al. (2016). Histone deacetylase HDA9 negatively regulates salt and drought stress responsiveness in Arabidopsis. J. Exp. Bot. 67, 1703–1713. doi: 10.1093/jxb/erv562
Zhong, S., Fei, Z., Chen, Y.-R., Zheng, Y., Huang, M., Vrebalov, J., et al. (2013). Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159. doi: 10.1038/nbt.2462
Zilberman, D., Gehring, M., Tran, R. K., Ballinger, T., and Henikoff, S. (2006). Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39, 61–69. doi: 10.1038/ng1929
Zong, W., Zhong, X., You, J., and Xiong, L. (2013). Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol. 81, 175–188. doi: 10.1007/s11103-012-9990-2
Keywords: DNA methylation, epigenomics, grape, Histone Post-Translational Modifications, small RNAs, Vitis vinifera
Citation: Fortes AM and Gallusci P (2017) Plant Stress Responses and Phenotypic Plasticity in the Epigenomics Era: Perspectives on the Grapevine Scenario, a Model for Perennial Crop Plants. Front. Plant Sci. 8:82. doi: 10.3389/fpls.2017.00082
Received: 02 November 2016; Accepted: 16 January 2017;
Published: 06 February 2017.
Edited by:
José Tomás Matus, Centre for Research in Agricultural Genomics, SpainReviewed by:
Carlos Marcelino Rodriguez Lopez, University of Adelaide, AustraliaPatricio Arce, Pontifical Catholic University of Chile, Chile
Copyright © 2017 Fortes and Gallusci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana M. Fortes, YW1mb3J0ZXNAZmMudWwucHQ= Philippe Gallusci, cGhpbGlwcGUuZ2FsbHVzY2lAaW5yYS5mcg==