 Dohyup Kim
Dohyup Kim Margaret W. Thairu
Margaret W. Thairu Allison K. Hansen
Allison K. Hansen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci., 04 August 2016
Sec. Plant Pathogen Interactions
Volume 7 - 2016 | https://doi.org/10.3389/fpls.2016.01164
This article is part of the Research TopicAdvances in Plant-Hemipteran InteractionsView all 18 articles
It has become increasingly clear that microbes form close associations with the vast majority of animal species, especially insects. In fact, an array of diverse microbes is known to form shared metabolic pathways with their insect hosts. A growing area of research in insect-microbe interactions, notably for hemipteran insects and their mutualistic symbionts, is to elucidate the regulation of this inter-domain metabolism. This review examines two new emerging mechanisms of gene regulation and their importance in host-microbe interactions. Specifically, we highlight how the incipient areas of research on regulatory “dark matter” such as epigenomics and small RNAs, can play a pivotal role in the evolution of both insect and microbe gene regulation. We then propose specific models of how these dynamic forms of gene regulation can influence insect-symbiont-plant interactions. Future studies in this area of research will give us a systematic understanding of how these symbiotic microbes and animals reciprocally respond to and regulate their shared metabolic processes.
Microbial associates that interact with insects can produce a wide array of metabolic products that complement the metabolic needs of their herbivorous hosts (Hansen and Moran, 2014). Consequently, microbes that form persistent but noninvasive associations with their hosts have the potential to provide their hosts with useful novel gene products in a short evolutionary timespan. How these symbiotic microbes and animals reciprocally respond to and regulate shared metabolic processes is a nascent but emerging area of research.
Animals, including insects, can biosynthesize some but not all of the amino acids that are required for building proteins. Food sources that are deficient in those essential amino acids (EAA), such as plant sap, present a nutritional challenge to consumers. Most insect herbivores that feed on a phloem or xylem sap diet are able to feed on this essential amino acid-deficient niche because they harbor one or more nutritional symbionts (Hansen and Moran, 2014). One model system that has been productive for teasing apart the regulatory mechanisms of shared animal-microbe metabolic processes is the pea aphid, Acyrthosiphon pisum, a sap-feeding insect in the order Hemiptera, and the bacterium Buchnera aphidicola, the mutualistic endosymbiont found in most aphids. Aphid and Buchnera physiologies are integrated for the production of amino acids and this occurs within specialized aphid cells called bacteriocytes. Specifically, Buchnera relies on the aphid for the biosynthesis of aphid-encoded non-essential amino acid pathways, and the aphid relies on Buchnera for the biosynthesis of Buchnera-encoded essential amino acid pathways. Previous work on this system supports the prevailing hypothesis that this integrated metabolism is regulated primarily by the aphid host via aphid-encoded transporters (Price et al., 2014) and aphid genes that complement Buchnera's EAA pathways (Hansen and Moran, 2011; Poliakov et al., 2011). Moreover, an aphid-encoded protein of bacterial origin can be transported into Buchnera cells and therefore a cross-domain protein translocation system exists for this intimate symbiosis (Nakabachi et al., 2005, 2014).
Gene expression of aphid bacteriocytes has been characterized at the transcriptome and proteome level (Hansen and Moran, 2011; Poliakov et al., 2011). As expected pathways involved in the amino acid metabolism are especially enriched in bacteriocytes compared to other aphid body cells (Hansen and Moran, 2011; Poliakov et al., 2011). However, the regulatory factors that lead to the development of these tissues and their signature expression profiles are not well-understood. In other animal systems, the primary regulatory factors that determine a eukaryotic cell's fate and its potential reprograming include histones, DNA methylation, noncoding RNAs, and transcription factors (Peter and Davidson, 2015). Work by Braendle et al. (2003) did identify three transcription factors that are expressed in temporal order, Dll, En, and Ubx or Abd-A, during bacteriocyte development in aphid embryos. The timing and expression of this subset of transcription factors is unique compared to any other cell type in insect embryos (Braendle et al., 2003). Currently it is unclear how these transcription factors may regulate metabolic processes or if other unknown co-factors are involved during embryonic stages or later during maternal bacteriocyte development. Moreover, it is unknown if chemical marks on histones and/or DNA (i.e., epigenetic mechanisms) (Hansen and Moran, 2014) are important in the regulation and metabolic reprogramming of these symbiotic cells, especially in response to environmental signals such as host plant nutrients or secondary plant compounds. Therefore, further understanding of how different subsets of host genes turn on and off in bacteriocyte development in response to environmental stimuli is required in order to fully understand how these intimate symbioses evolved, how they are maintained, and how they may ultimately influence host-plant-interactions.
Although evidence from the aphid and Buchnera model system suggests that the host exerts the majority of the regulatory control on this symbiosis, the role of the symbiont in gene regulation is not well-defined (Hansen and Moran, 2014). Given that Buchnera has lost the majority of the canonical mechanisms for gene regulation its capacity to be a participant in gene regulation has long seemed compromised (Shigenobu et al., 2000). Nevertheless, it was recently suggested that Buchnera regulates its own protein expression via putative post-transcriptional mechanisms (Hansen and Degnan, 2014). This recent study presents somewhat of a paradigm shift in understanding the regulation of these intracellular host-microbe mutualisms. It is now clear that symbionts with reduced genomes can potentially regulate their genomes through post-transcriptional processes, such as through regulatory small RNAs (sRNAs; Hansen and Degnan, 2014). Consequently, the potential role of microbe-mediated gene regulation cannot be ignored in these shared insect-microbe regulatory networks.
Recent advancements in the field of molecular biology and genome sequencing technologies allow for the first time the ability to predict how epigenetic mechanisms and regulatory sRNAs may impact the regulation and evolution of animal and microbial genomes. This review draws upon these emerging fields of gene regulation to investigate the potential role of epigenetics and sRNAs in both insect and microbe-mediated regulation of shared metabolisms. By understanding the dynamics and the intersection of these regulatory mechanisms, we can begin to disentangle the evolution of these shared herbivore-microbe metabolisms. Ultimately, if we can identify key molecular mechanisms that are responsible for regulating shared amino-acid metabolisms, which are widespread in symbiont-enabled herbivory, we can determine if the evolution of these mechanisms impacts insect-plant interactions.
For the first part of this review we will discuss previous literature on epigenetic mechanisms in eukaryotic gene regulation, and how epigenetics are potentially involved in herbivore-microbe interactions. For the second part of this review we will discuss previous literature on regulatory RNAs and how they are potentially involved in the regulation of these insect-microbe interactions.
Under Darwinian natural selection random genetic mutations in the DNA molecule are inherited from parent to offspring, and increase in frequency within a population if a given mutation contributes to differential reproductive success (Dobzhansky, 1937). Alternative mechanisms of adaptation, such as Lamarkian inheritance, where an individual can pass down acquired traits that are obtained during its lifetime, have been hotly debated (Pilpel and Rechavi, 2015). Evolutionary theory that involves different variations of Lamarkian inheritance has been resurrected multiple times through history (Burkhardt, 2013). One controversial variation of Lamarkian inheritance, neo-Lamarkism (Skinner, 2015), has been proposed to explain epigenetic inheritance (Jablonka and Lamb, 2015) and CRISPR-cas immunity in Bacteria and Archaea (Koonin and Wolf, 2009), because these acquired traits are not random but are induced by the environment in a predictable fashion and are inherited through generations.
Epigenetic marks referred to here as chemical marks on DNA and histones are responsible for tissue-specific gene expression in eukaryotes and thus can lead to a change in an organism's phenotype (Gama-Sosa et al., 1983). If the environment induces epigenetic marks in a repeatable way and these marks are inherited across generations then there is potential for epigenetics to play important roles in organismal adaptation in natural populations (Gadjev, 2015), which can then affect the organism's interaction with other organisms. Therefore, we propose that epigenetic mechanisms may be important for the evolution of both insect-plant and insect-microbe interactions.
One type of epigenetic mark that is widespread and generally occurs across all domains of life is DNA methylation. DNA methylation involves the enzymatic addition of a methyl group to individual nucleotide bases of DNA in chromosomes by DNA methyltransferases (DMNTs). Now more than ever, DNA methylation has become more tractable to study due to the recent advancement in sequencing technology and bioinformatics. In turn the field of epigenomics is more accessible to researchers for both model and non-model organisms. As such the number of research articles on DNA methylation has been steadily increasing (Romanoski et al., 2015). For example, according to the Web of Science (2016), the number of epigenetic research articles has doubled every 5 years since 1971, reaching just over 110,000 articles between 2011 and 2015.
The role and patterns of DNA methylation vary widely among the three domains of life (Jeltsch, 2010). Several independent losses of DNA methylation have occurred in various eukaryotic taxa, suggesting that the role of DNA methylation may not be essential for all eukaryotic species (Field et al., 2004; Wion and Casadesús, 2006). For example, in the model systems Caenorhabditis elegans (roundworm) and Drosophila melanogaster (fruit fly) DNA methylation is not functional, because of lineage specific losses of DMNTs (Goll and Bestor, 2005). Nevertheless, DMNTs are present and DNA methylation is prevalent and functional in a wide diversity of other eukaryotic taxa including plants, vertebrates, and invertebrates (Jeltsch, 2010).
In vertebrates, including humans, cytosine methylation is widespread in the genome, specifically at cytosine-phosphate-guanine (CpG) dinucleotide sites (Bird, 1986). However, CpG-rich regions called CpG islands, which are typically 300- to 3000- base pairs long and are located primarily in the promoter regions of vertebrate genes, are largely un-methylated (Bird, 1986). Methylation of even a single CpG site in a promoter region can significantly inhibit transcription of the downstream genes (Robertson et al., 1995). Such transcriptional inhibition or gene silencing is an important role for DNA methylation as it also helps maintain the integrity of the genome by silencing transposable elements (Zamudio et al., 2015). DNA methylation also has a well-established role in imprinting, such as mammalian X-chromosome inactivation (Augui et al., 2011; Balaton et al., 2015), and differential expression of parental-specific alleles (Reik et al., 1987; Li et al., 1993; Razin and Cedar, 1994). Furthermore, DNA methylation in gene-body regions (e.g., un-translated regions, exons, and introns) can also affect the activity of genes in vertebrate genomes. For example, in human cell lines the inhibition of DNA methylation in gene-body regions resulted in the alternative splicing of exons (Maunakea et al., 2013).
In invertebrates, cytosine methylation also plays an important role in gene regulation. Epigenomic research in invertebrates initially lagged behind, because the two main invertebrate model species in genetics, C. elegans and D. melanogaster, do not have active copies of DNMTs (detailed above). Nevertheless, DNA methylation has been observed in a diversity of other invertebrate species. For example, in the Pacific oyster Crassostrea gigas, different levels of CpG methylation have been observed that correlate with gene functions (Gavery and Roberts, 2010). In the Chinese white shrimp, Fenneropenaeus chinensis, tissue-specific DNA methylation was observed (He et al., 2015). Furthermore, DNA methylation has been reported in many insect species of various orders including Diptera, Hemiptera, Hymenoptera, Lepidoptera, Coleoptera, Odonata, and Orthoptera (Field et al., 2004; Richards et al., 2008; Walsh et al., 2010; Xiang et al., 2010; Zhang J. et al., 2015; Zhang M. et al., 2015). In general, methylation levels of invertebrate CpG sites are relatively low, ranging from 0.36–20% (Regev et al., 1998), compared to mammalian systems where 60–90% of all CpG dinucleotides are subject to methylation (Suzuki and Bird, 2008).
Methylome studies across different invertebrate taxa have revealed that DNA methylation is often confined to genic regions (promoters, exons, and introns) of the genome, whereas intergenic regions remain largely unmethylated (Suzuki and Bird, 2008). In hymenopteran genomes, such as parasitoid wasps, ants, and bees, low levels of DNA methylation occur within transposable elements (TEs) compared to vertebrate genomes. These results suggest that DNA methylation has no or very little association with the repression of TEs as shown in vertebrates (Yan et al., 2015). DNA methylation within invertebrate genes has been associated with gene activation and alternative splicing. For example, loss of DNA methylation from multiple CpG sites within the insecticide-detoxifying esterase gene E4 of the green peach aphid Myzus persicae was associated with a reduction of transcription of the esterase gene E4, and thus increased sensitivity to pesticides (Field et al., 1989; Field, 2000). Also, several studies have proposed that DNA methylation is associated with alternative splicing of mRNA transcripts, which leads to behavioral regulation and caste specificity in eusocial insects including bees (Foret et al., 2012; Li-Byarlay et al., 2013), ants (Bonasio et al., 2012), and termites (Terrapon et al., 2014).
Mounting evidence from the handful of non-model animal systems that have been studied suggests that environmental cues can trigger the reprogramming of cells through DNA methylation, resulting in the regulation of adaptive traits (Kucharski et al., 2008; Moczek and Snell-Rood, 2008; Alvarado et al., 2015; Table 1). As such, differential methylation patterns have the potential to produce an adaptive regulatory response to current environmental conditions. Environmental signals such as diet, stress, and anxiety have been shown to alter DNA methylation patterns during an organism's lifetime (Weaver et al., 2004; Jankard and Herman, 2008; Schwenk et al., 2013). For example, in the honey bee nutritional cues from royal jelly regulate queen determination via epigenetic mechanisms. Specifically, the gene, dynactin p62 is differentially methylated in queens compared to workers, and it is hypothesized to be a key gene in regulating different developmental pathways (Kucharski et al., 2008). In this study, when DNA methyltransferase 3 (Dnmt3) is silenced in larvae that feed on protein-rich royal jelly these larvae develop into fertile queens with fully developed ovaries (Kucharski et al., 2008). This result indicates that a nutritional signal can alter epigenetic patterns resulting in caste determination. In another honeybee study, social stimuli of bees were highly correlated with changes in DNA methylation patterns between worker and nurse bees of the same age (Lockett et al., 2012). One particular CpG site in the gene Protein kinase C-binding protein 1 (PKCbp1) with variable levels of methylation between worker and nurse bees was strongly correlated with the alternative splicing of its gene product. The direct consequences of this alternative splicing however are unclear (Lockett et al., 2012). DNA methylation also plays a role in caste determination of another social hymenopteran, the carpenter ant Camponotus floridanus, by modifying ant body size, a key trait associated with the division of labor (Alvarado et al., 2015).
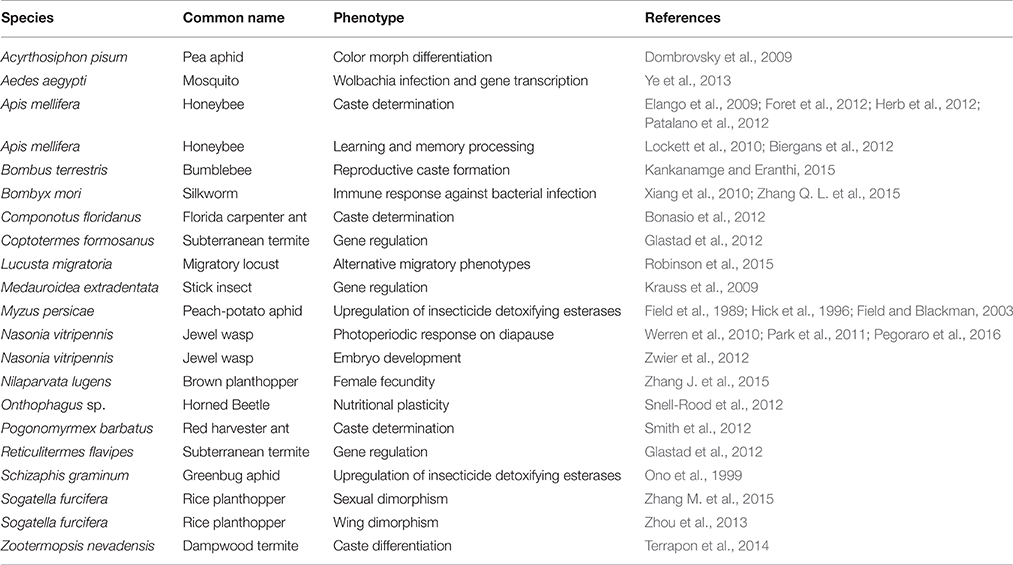
Table 1. DNA methylation in various insects and its associated phenotypic effects.
In the parasitoid wasp Nasonia vitripennis changes in photoperiod are hypothesized to induce genome-wide DNA methylation changes (Pegoraro et al., 2016). When day length is decreased female N. vitripennis wasps induce developmental arrest of their progeny (diapause). This photoperiodic response allows the larvae to survive throughout winter. Knock-down of either DNMT1a or DNMT3 in N. vitripennis parents disrupted the photoperiod-induced developmental arrest of their larvae. Although the exact mechanisms are yet to be elucidated, these results suggest that environmentally induced diapause in N. vitripennis are linked to DNA methylation.
In addition to hymenopterans, methylation contributes to adaptive regulatory responses to environmental conditions in aphids. In the green peach aphid, M. persicae, individuals resistant to an organophosphate pesticide encode a differentially methylated esterase gene that confers the resistant phenotype (Field et al., 1989, 1996; Hick et al., 1996). Biotypes of the Russian wheat aphid, Diuraphis noxia, that differ in virulence toward their host plant display differential methylation patterns for key genes that are expressed in their salivary glands, suggesting that methylation may play an important role in this insect's ability to feed on different host plant cultivars (Gong et al., 2012). In A. pisum, it has been shown that extreme temperatures may result in variation in DNA methylation patterns, which are correlated to different color phenotypes within genetic clones (Dombrovsky et al., 2009). This study revealed that the intensity of methylation in CpG-islands within aphid cuticular genes varied dramatically between three different A. pisum color morphs (white, pink, and green). Furthermore, the authors identified correlations between CpG island methylation and growth rate, morph development, and pigmentation of the aphid population by pharmacologically inhibiting the DNA methyltransferases (Dombrovsky et al., 2009). In sum, DNA methylation may help drive rapid and precise gene regulation to variable environmental conditions.
In the sections above we detailed several examples of how environmental cues can induce specific DNA methylation patterns in insects, which can result in adaptive gene expression profiles. Although microbial associations are ubiquitous in many insect systems, as of yet, there has not been extensive research on how microbes affect insect host epigenomics. Nevertheless, we predict that epigenomics may play major roles broadly in insect-plant and insect-microbe ecology and evolution. For example, insect microbial associations have facilitated numerous host plant niche expansions, and the diversification of insect lineages (Hansen and Moran, 2014). Moreover, insect symbionts can contribute to a variety of extended insect host phenotypes, which include: defense against viral pathogens, fungal pathogens, and parasitoids (Kaltenpoth et al., 2005; Oliver et al., 2005; Scarborough et al., 2005; Scott et al., 2008; Teixeira et al., 2008; Vorburger et al., 2009), conferring thermal tolerance (Dunbar et al., 2007; Brumin et al., 2011), facilitating food digestion (Brownlie et al., 2009; Salem et al., 2012), and manipulating sexual reproduction (Stouthamer and Werren, 1993). Currently, we are still in the discovery phase in identifying specific genetic mechanisms that facilitate these important microbe-induced, insect-extended phenotypes.
To the best of our knowledge the only studies that demonstrate an effect of microbes on insect epigenomics are of Wolbachia and mosquitos. Wolbachia is an intracellular bacterial symbiont that is both vertically and horizontally inherited in numerous insect species and commonly enhances its transmission through reproductive manipulations (Stouthamer and Werren, 1993). Nevertheless, mosquitos like Drosophila, do not have functional DNA methylation, because they do not encode DMNT1 and DMNT3 (Holt et al., 2002; Nene et al., 2007). However, they do encode the methyltransferase, DMNT2, which has substrate specificity for tRNAs (Goll et al., 2006), contributes to antiviral defense in Drosophila (Durdevic et al., 2013), and is involved in random genome methylation patterns (Kunert et al., 2003). In one study, when the pathogenic strain of Wolbachia (wMelPop) infects the mosquito, Aedes aegypti, the mosquito is hypomethylated because Wolbachia suppresses DNMT2 (Zhang G. et al., 2013). When DMNT2 is overexpressed in mosquito cell lines Wolbachia replication is inhibited, suggesting that suppression of DNMT2 is beneficial for Wolbachia survival. Conversely, in A. aegypti DNMT2 is induced by the Dengue virus, and this induction promotes Dengue virus replication. In vivo this antagonistic interaction ultimately results in Wolbachia suppressing the Dengue virus via DNMT2 suppression (Zhang G. et al., 2013). In another study, the Wolbachia strain wMelPop results in both the methylation and de-methylation of A. aegypti's genome (Ye et al., 2013). For the most part these changes in methylation primarily appeared to be random (Ye et al., 2013). In this study, the direct effect of differential methylation on transcription in wMelPop-infected compared to uninfected mosquitos remains unclear.
Given the paucity of evidence of symbionts affecting invertebrate host epigenetics and studies of insect hosts with functional DNA methylation systems investigating the effect of symbionts on vertebrate hosts may provide insights into possible ways symbionts may impact DNA methylation in insect genomes. In general, DNA methylation in vertebrate studies of gut-associated microbes has revealed that host immune responses and microbially derived metabolites affect host DNA methylation. For example, in mice Takahashi et al. (2011) found that the methylation level of the Toll-like receptor 4 gene in intestinal epithelial cells is significantly lower in germ-free mice compared to conventional mice. Moreover, results from this study suggests that this epigenetic modification is elicited by and important for the maintenance of commensal microbes in the gut. In another study, when the human pathogen Helicobacer pylori infects the human gut DNA methylation increases in the promoter regions of the human genes filamin C and thrombomodulin. This results in the silencing of these genes and a concomitant increase in the risk of gastric cancer (Nakajima et al., 2009). In another human microbiome study, an increase in abundance of two members of the human oral microbiome that belong to Enterobacteriaceae and Tenericutes is associated with the hypermethylation of the promoter regions of the human host gene MDR1. Hypermethylation of MDR1 can result in head and neck squamous cell carcinoma (Bebek et al., 2012). In another study on host pathogens, pathogenic viruses including human adenovirus, hepatitis B virus and HIV are known to increase genome-wide levels of methylation of their host by up-regulating DNMT1 (Fang et al., 2001; Burgers et al., 2007; Jung et al., 2007).
In addition to pathogenic and non-pathogenic gut microbes mediating human immune responses through DNA methylation, microbe-derived metabolites can also influence DNA methylation in humans and ultimately impact expressed phenotypes. For example, nutritional uptake in early postnatal humans modifies the infant's gut microbiome, which in turn affects the epigenetic patterns of the individual (Mischke and Plosch, 2013). This study proposes that changes in the composition of the gut microbiome results in altered profiles of microbe-produced metabolites such as folate and short-chain fatty acids. The same study proposed that an increase in such metabolites may influence the DNA methylation patterns of adjacent intestinal cells, which in turn results in the predisposition to obesity.
Microbial symbionts and pathogens of humans and some insects have been demonstrated to alter patterns of DNA methylation. As such microbial symbionts have the capacity to (radically) alter host phenotypes. We hypothesize that this ability is widespread in insect symbionts, particularly among co-evolved insect symbionts. These intimate partners may influence methylation of their insect hosts with functional DNA methylation systems by modulating their host's immune responses to microbes. For example, attenuating immune responses so as to permit their intracellular persistence. In addition, these symbionts can encode novel biosynthetic pathways, which may contribute microbially derived metabolites, such as folate; folate is a key source of the one carbon group used to methylate DNA (Table 2). Moreover, in co-evolved insect symbioses tissue- specific, DNA-methylation patterns in specialized insect cells that harbor obligate symbionts may facilitate the development and regulation of this long-term symbiotic relationship. Nevertheless, our understanding of the development and regulation of these symbiotic cells in insects is still nascent (Braendle et al., 2003; Hosokawa et al., 2016). Therefore, by investigating if and how epigenetic modifications affect the regulation of insect-microbe interactions, we will gain a better understanding of key biological mechanisms in symbiosis and evolution in general.
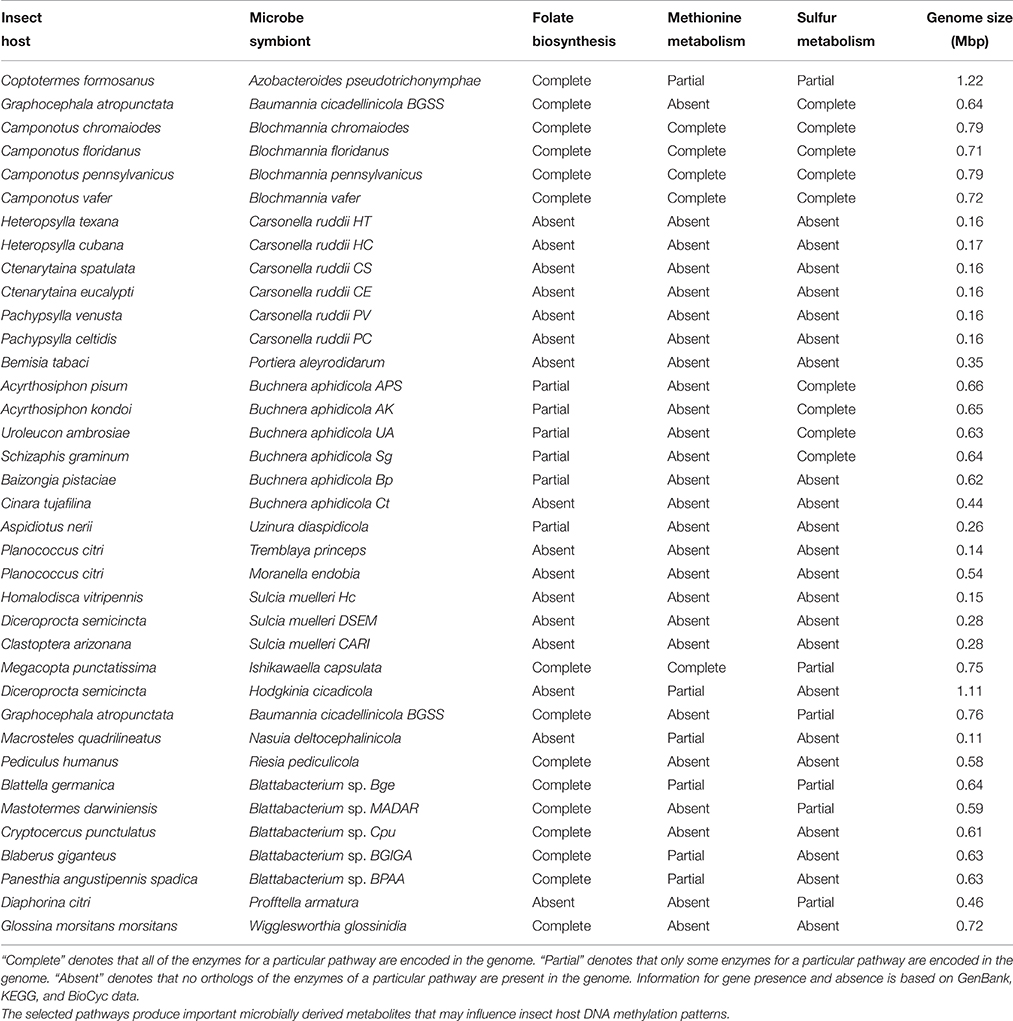
Table 2. Presence and absence of pathways from obligate symbionts of insects with fully sequenced genomes.
In recent years, there has been a rapid increase in the identification of non-coding, regulatory RNAs in Bacteria, Archaea, and Eukaryotes (Babski et al., 2014). With the improvement of sequencing technology it has been found that the non-coding RNAs expressed within in a cell include more than just the rRNAs, tRNAs, small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs). What has become clear is that these non-coding RNAs of varying sizes are important in a myriad of biological functions, which includes gene regulation. One large class of non-coding RNAs is regulatory small RNAs (sRNAs). sRNAs can be categorized depending on their function, structure, conservation among taxa, and/or size range (Kim et al., 2009; Waters and Storz, 2009). In general, the term sRNAs encompasses a large diversity of expressed RNAs that have various size cut offs depending on what domain you are studying (Babski et al., 2014). This review will broadly define sRNAs as regulatory elements that are transcribed from coding and/or non-coding regions, which have both perfect and/or imperfect base-pairing interactions with their target RNA, and are <300 nt in length.
In general, sRNAs fall into two categories: cis-encoded and trans-encoded sRNAs. Within bacteria, trans-encoded sRNAs are encoded at genomic locations that are distant from their target mRNA(s), as such they often have partial complementarity with their targets. Trans-encoded sRNAs are generally dependent on the RNA chaperone Hfq (Storz et al., 2011). Hfq proteins protect sRNAs from ribonuclease degradation and facilitate intermolecular contacts between sRNAs and their mRNA targets in the Enterobacteriaceae (reviewed in Vogel and Luisi, 2011; Sauer, 2014). Despite its important role, Hfq cannot be detected in some bacterial phyla including the Chlamydiae, Spirochaetae, Actinobacteria, Deinococcus-Thermus, Cyanobacteria, Chlorobi, and Bacteroidetes (Sun, 2002). In the Firmicutes, a gram-positive bacterial phylum, some species encode Hfq, however, unlike the Enterobacteriaceae, it does not play a major role in gene regulation (reviewed in Bouloc and Repoila, 2016). There is evidence that some of these gram-positive bacteria have analogous proteins that carry out similar roles to Hfq (reviewed in Durand et al., 2015). In many insect endosymbionts belonging to gram-negative bacteria, the homolog of Hfq has been lost (Sun, 2002). This suggests trans-encoded sRNAs are not expressed, and/or if they are expressed, other stabilizing mechanisms are employed (Hansen and Degnan, 2014).
Currently, trans-encoded sRNAs are the most extensively characterized sRNAs in bacteria. This is because of the historical difficulties that arose when trying to identify cis-encoded sRNAs (Georg and Hess, 2011). Improvements in sequencing technologies and methodologies, and bioinformatics prediction programs have led to an increase in the identification of candidate cis-encoded sRNAs, however validating the functional role of many of these accumulating candidates still lags behind (Barquist and Vogel, 2015). Cis-encoded sRNAs can be transcribed in the 5′ and/or 3′ regions of a coding sequence, within the coding sequence, or within the non-coding regions, and have perfect complementarity with their target RNAs. Many cis-encoded sRNAs in free-living bacteria have been observed to have a variety of roles, such as expression within mobile genetic elements like plasmids, transposons and phage, aiding in mRNA stability and degradation, and/or translational inhibition and attenuation (Wagner and Simons, 1994; Brantl, 2007). In addition, cis-encoded sRNAs have been identified to inhibit the synthesis of toxic proteins (Fozo et al., 2008).
Riboswitches are another common bacterial sRNA. Riboswitches are characterized by having complex folded domains—“the riboswitch,” and they encompass a non-coding portion of the mRNA that binds to various metabolites (Nahvi et al., 2002; Winkler et al., 2002; Lai, 2003; Winkler and Breaker, 2003). In the presence of specific metabolites, riboswitches undergo a conformational change which then influences transcription, translation, or other processes related to protein production (Mandal and Breaker, 2004). Early surveys of riboswitches found that within many gram-positive bacteria such as the Firmicutes, riboswitches tended to modulate transcriptional attenuation, whereas in gram-negative bacteria such as the Proteobacteria, many of the riboswitches identified regulated translational attenuation (Nudler and Mironov, 2004; Barrick and Breaker, 2007). However, as riboswitches are described in more species, the regulation of the riboswitch seems to be more closely associated to ligand/type of riboswitch (Ray and Chakdar, 2015).
In general, eukaryotic sRNAs can be divided into two families: micro RNAs (miRNAs) and small-interfering RNAs (siRNAs). Within eukaryotes, the RNAi pathway is responsible for the regulation of a diversity of endogenous genes via miRNAs, and for protecting the organism from invasive genetic material, such as viruses and transposable elements via siRNAs (Hannon, 2002). These sRNAs act as template RNA that bind to the RNA-induced silencing complex (RISC), and degrade complementary RNA sequences (Hannon, 2002). This pathway is conserved across most eukaryotes, however it has been lost in some fungal lineages and, notably, in the model system, Saccharomyces cervisiae (Billmyre et al., 2013).
miRNAs are ~21–24 nucleotides in length and are transcribed from genes or within introns and subsequently form hairpin structures that are cleaved to result in the final mature miRNA (Ha and Kim, 2014). In animals, miRNAs share partial sequence complementarity to multiple target mRNAs, much like bacterial trans-encoded sRNAs. Plant miRNAs on the other hand have high sequence complementarity (Jones-Rhoades et al., 2006). Among plants and animals, miRNAs mediate post-transcriptional gene regulation in various shared aspects of physiology and development (Flynt and Lai, 2008; de Lima et al., 2012; Vidigal and Ventura, 2015).
Unlike miRNAs, siRNAs are ~21 nt long and have endogenous or exogenous origins such as transposons and viruses. Furthermore, they are formed from double- stranded RNAs (Carthew and Sontheimer, 2009; Piatek and Werner, 2014). Also unlike miRNAs, siRNAs generally share perfect complementarity with their targets. Animal siRNAs are important in regulating transposons, heterochromatic sequences, intergenic regions, long RNA transcripts with extensive structure, and mRNAs (Ghildiyal and Zamore, 2009). It has been found that many expressed endo-siRNAs have vital roles in animal development (reviewed in Ghildiyal and Zamore, 2009). Within plants, there are three types of endo-siRNAs: cis-acting siRNAs (casiRNAs), trans-acting siRNAs (tasiRNAs), and natural antisense transcript-derived siRNAs (natsiRNAs; reviewed in Ghildiyal and Zamore, 2009; Axtell, 2013; Pattanayak et al., 2013). In general these endo-siRNAs are important in methylation (Mette et al., 2000; Llave et al., 2002; Tran et al., 2005), development (Allen and Howell, 2010), modulation of the plant's responses to stress (Jones-Rhoades and Bartel, 2004; Sunkar and Zhu, 2004; Borsani et al., 2005; Fujii et al., 2005) and plant immunity (Katiyar-Agarwal et al., 2006).
In both plants and animals, miRNAs and siRNAs share biosynthesis pathways that include RNA endonucleases Argonaute proteins and proteins Drosha and Dicer (Carthew and Sontheimer, 2009; Piatek and Werner, 2014). Though the genes involved in the biosynthesis of these sRNAs may share a common ancestor (Shabalina and Koonin, 2008), plant and animal sRNAs differ in biogenesis, function, and subsequent evolutionary trajectories (Shabalina and Koonin, 2008; Axtell et al., 2011; Mukherjee et al., 2013).
Intracellular symbionts, pathogens, and organelles are subject to repeated genetic bottlenecks, which contribute to a suite of associated genome-wide changes including genome-size reduction. This evolutionary constraint also results in biases in nucleotide composition (increased %A+T), elevated mutation rates and the fixation of deleterious and neutral mutations due to genetic drift, ultimately resulting in gene inactivation and loss (Moran et al., 2009; Moran and Bennett, 2014). Moreover, selection becomes less effective in maintaining the mechanisms of gene regulation in reduced genomes compared to free-living relatives with larger genomes and access to more diverse environments (Lambert and Moran, 1998; Hansen and Moran, 2012; Hansen and Degnan, 2014). Thus, over evolutionary time, genome sizes tend to shrink dramatically, losing numerous functional capabilities and becoming structurally rearranged compared to free-living relatives (Moran and Bennett, 2014).
Despite these dramatic changes, reduced genomes still display strong purifying selection for key genes that are essential for the host-microbe relationship, including core housekeeping genes (Moran et al., 2009; Williams and Wernegreen, 2011). Among these are genes encoding biologically active non-coding RNAs, such as tRNAs and rRNAs that display higher %G+C composition relative to coding sequences, and accumulate compensatory base substitutions to maintain important secondary structural regions (e.g., stems; Lambert and Moran, 1998; Hansen and Moran, 2012). The increased genome wide %A+T composition in reduced genomes results in numerous polyA and polyT homopolymers. Such homopolymers are readily subject to insertions or deletions (indels) that can disrupt promoters or result in non-functional proteins because of frame shifts (Dunbar et al., 2007). These indels can initiate the processes of genome erosion (Moran et al., 2009) but in several instances transcriptional slippage can occur at these polyA/T tracts, restoring the reading frame of the mRNA and yielding functional proteins (Tamas et al., 2008; Wernegreen et al., 2010). Importantly, transcriptional slippage has been: (i) observed in symbionts with reduced genomes, (ii) has been maintained over evolutionary time, and (iii) functionally has been shown to rescue the activity of important genes for the symbiont's lifestyle (Tamas et al., 2008; Wernegreen et al., 2010). Collectively these patterns indicate that even though these reduced genomes experience severe genetic drift, purifying selection is strong enough to maintain and/or co-opt compensatory mechanisms to maintain fundamental processes for the persistence of microbe lineages.
Thus, we hypothesize that bacterial symbiont genomes subject to radical gene loss and structural rearrangements over evolutionary time can compensate for the loss of essential regulatory machinery by maintaining and/or co-opting compensatory mechanisms, such as sRNAs to aid in the regulation of vital symbiotic genes and core housekeeping processes, as suggested in Hansen and Degnan (2014).
sRNAs in Bacteria and Eukaryotes have been associated with rapid diversification and adaptation of lineages to local environments (Horler and Vanderpool, 2009; Yu et al., 2010; Ames and Lovell, 2011; He et al., 2011; Raghavan et al., 2012). It has been suggested that sRNAs can rapidly evolve from non-adaptive transcripts (Raghavan et al., 2012). Non-adaptive transcripts, i.e., transcriptional noise, can result when RNA polymerase accidentally recognizes non-promoter sequences as promoter sequences, because promoters contain low information content (Mendoza-Vargas et al., 2009; Raghavan et al., 2012). In reduced genomes that experience severe genetic drift, it is unclear how novel regulatory sRNAs could evolve. However, given that purifying selection has been identified in these reduced genomes for both coding and non-coding functional RNA sequences (Lambert and Moran, 1998; Moran et al., 2009; Williams and Wernegreen, 2011; Hansen and Moran, 2012), we hypothesize that purifying selection is strong enough to maintain and/ or co-opt emergent regulatory sRNAs that are essential for the symbiont's gene regulation. The failure to maintain such essential sRNAs would be lethal for the microbe and in turn its host, if other host and/or microbe factors failed to compensate.
One example of how reduced genomes compensate to maintain essential gene expression processes comes from previous research on Buchnera from several diverse aphid species (Hansen and Moran, 2012). In this study, the authors revealed that Buchnera tRNA genes in four divergent taxa are often shorter in length than their homologs in E. coli, Buchnera's distant free-living relative. The difference in length is typically three nucleotides, and mostly reflects the loss of the encoded 3′ CCA sequence in the Buchnera tRNA genes. At the 3′ end of tRNAs, the nucleotide sequence CCA is required for amino acid activation and must either be encoded in the tRNA gene or added during tRNA maturation by the CCA-adding enzyme (cca). In Buchnera's close relatives, such as E. coli, Vibrio spp., and Pseudomonas spp., 3′ CCA is encoded in all tRNA genes that are present in Buchnera; most likely to maintain efficient translation. Nevertheless, only half of Buchnera tRNA genes encode 3′ CCA (Hansen and Moran, 2012). Importantly, using directional RNAseq, Hansen and Moran (2012) found that mature RNA transcripts of these genes possess a CCA at the 3′ end, implying CCA-addition and thus activation of amino acids. The role of the CCA-adding enzyme in E. coli and other organisms that already encode CCA in their tRNA gene sequences is to monitor the stability of tRNAs by tagging unstable tRNAs with an additional CCA for degradation (Wilusz et al., 2011; Kuhn et al., 2015). In turn, this study revealed that Buchnera is compensating for the loss of CCA in the tRNA gene sequence by co-opting the CCA-adding enzyme. If Buchnera did not co-opt this regulatory machinery for this additional role, which is not observed in Buchnera's closest free-living relatives, then the symbiont would not be able to translate proteins. Moreover, the authors observed numerous compensatory base substitutions in tRNA stems and high %GC in tRNA genes compared to coding sequences that help maintain tRNA secondary structure and function. In sum, these results provide evidence that purifying selection is strong enough in reduced genomes to (1) co-opt existing machinery to compensate for core regulatory processes and (2) maintain existing machinery to conserve essential regulatory functions.
Organelles represent the most dramatic example of tiny genomes that express regulatory sRNAs. Within eukaryotes there are two general types of organelles that have a unique evolutionary history with a microbial origin, such as mitochondria and plastids (Smith and Keeling, 2015). Mitochondria and plastids arose from bacteria that were once endosymbionts in early host eukaryotic cells (reviewed in Katz, 2012). In lineages that harbor both types of organelles, acquisition of plastids occurred after the early mitochondrial-eukaryotic lineage diversification (Gould et al., 2008). Mitochondria are believed to have evolved from a single endosymbiotic event involving an Alphaproteobacterium most likely from a Rickettsiales ancestor (Williams et al., 2007). Plastids on the other hand have a more complicated evolutionary history. All plastids evolved from the primary endosymbiosis of a cyanobacterium; however, across lineages there have also been secondary and tertiary endosymbiotic events (Gould et al., 2008). Currently, there is a growing body of research focused on organelle-associated sRNAs and their regulation (Table 3). Overall there is an emerging trend that gene regulation within the organelle is controlled by both host nuclear-encoded sRNAs and organelle-encoded sRNAs.
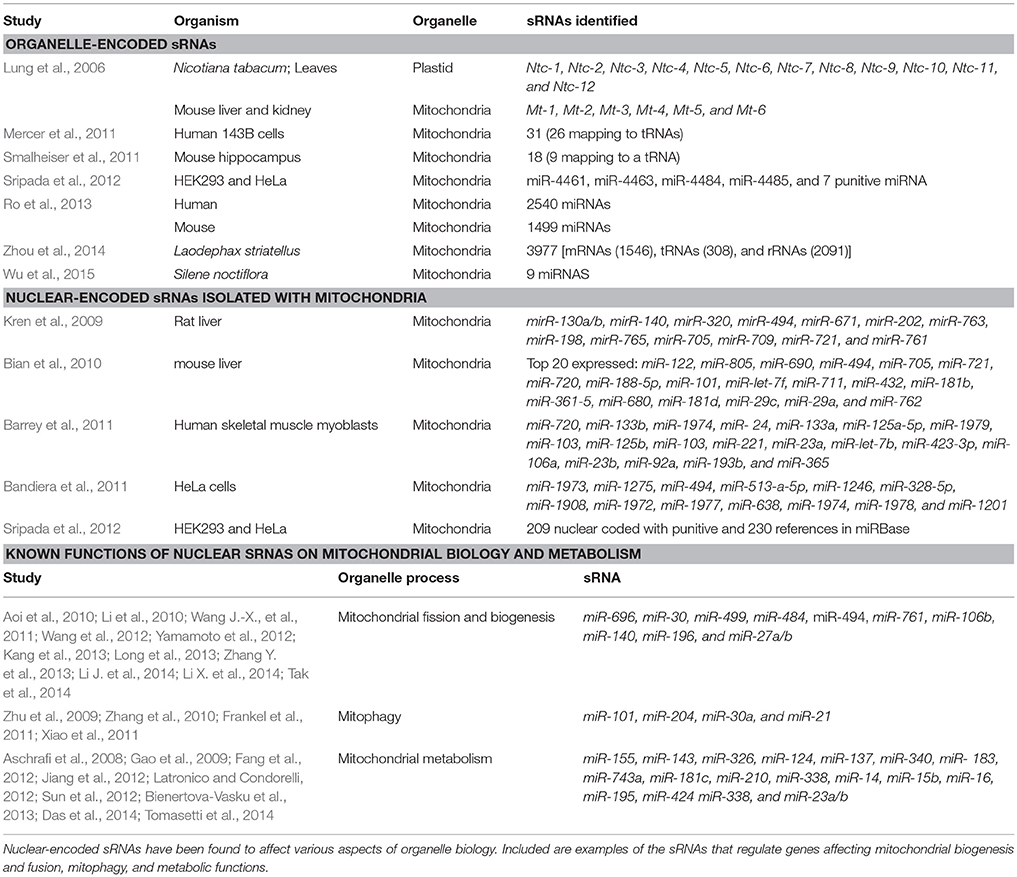
Table 3. Summary of studies isolating mitochondrial/plastid-encoded sRNAs and nuclear-encoded sRNAs found within organelles.
In the early 2000's it was thought that few if any sRNAs were expressed within organelle genomes (e.g., Lung et al., 2006). However, the identification of sRNAs has increased rapidly over the last decade for uncultivable organelles and microbes, because of lower sequencing costs and advances in sequencing technology (Hotto et al., 2011; Wang L. et al., 2011; Ro et al., 2013; Wu et al., 2015). There are two types of sRNAs identified within organelles: nuclear-encoded sRNAs, that are eukaryotic-like in origin and organelle-encoded sRNAs, that are bacterial-like in origin. Currently, the method by which nuclear-encoded sRNAs are transported into the organelle is unclear. However, there is evidence to suggest that tRNAs and 5S rRNAs can be imported into mammalian mitochondria and indirect evidence that tRNAs also can be imported into plastids (Schneider, 2011). In turn, similar pathways may be used for sRNA uptake into organelles. It is important to note that not only are nuclear-encoded sRNAs present within organelles, but argonaute (specifically, Ago2), the nuclear-encoded protein of the RISC complex, is also present within mitochondria (Bian et al., 2010; Bandiera et al., 2011). The presence of Ago2 and both nuclear-encoded and organelle-encoded sRNA within organelles supports the hypothesis that there is a complex interplay of gene regulation between the host and the organelle.
In mammals, besides providing energy for cellular functioning, mitochondria are also important in apoptosis (Lee et al., 2004; Suen et al., 2008), calcium concentration regulation (O'Rourke, 2004; Chen et al., 2005), and reactive oxygen species production (Chen et al., 2003). As such, in mammals, mitochondrial dysfunction has been linked to various diseases (Au et al., 2005; Baloyannis, 2006; Lemieux et al., 2010; Ritov et al., 2010; Gong et al., 2011). Some mitochondrial disease phenotypes are associated with the dysregulation of mitochodrial sRNAs (mitomiRs; Table 3). In general, there is less work reported in plant organelle sRNAs (Budak and Akpinar, 2015). Wang L. et al. (2011) is one of the few studies that show that chloroplast-encoded sRNAs can respond to environmental changes in the Chinese cabbage (Brassica rapa ssp. Chinensis). From these studies, we can see a potential trend emerging that organelle sRNAs expressed either from the organelle genome or the nuclear genome of their hosts are important for the regulation of these small, eroded genomes.
Overall, organelles, especially animal mitochondria, share many genomic characteristics to endosymbionts: organelles have reduced genomes when compared to free living relatives (Green, 2011; Gray, 2012), are generally low in GC content (McCutcheon and Moran, 2012; Smith, 2012), and their operons are highly fragmented (Barbrook et al., 2010; Brinza et al., 2010; Hansen and Degnan, 2014). Unlike organelles, most intracellular symbionts and pathogens with reduced genomes have not transferred regulatory genes to their host's chromosomes and still have autonomous housekeeping functions (Bennett and Moran, 2013).
Within bacterial genomes, operon structure, regulatory proteins, and sigma factors govern gene regulation. Organisms that possess reduced genomes, such as intracellular pathogens and mutualistic symbionts have lost many of these hallmark regulatory features (e.g., Mycoplasma sp. and Buchnera; Dandekar et al., 2000; Shigenobu et al., 2000). Within the human pathogen, Mycoplasma pneumoniae, it has been revealed that ~13% of coding genes have a corresponding antisense RNA (Güell et al., 2009). The function of a few of these antisense sRNA candidates has been determined and have been shown to down-regulate their target gene (Güell et al., 2009). Two of these sRNAs, NEW87 and NEW8, potentially have roles in metabolism and DNA repair and replication, respectively (Güell et al., 2009). Several of these antisense sRNAs are conserved in the closely related species Mycoplasma genitalium, however, the functionality of these sRNA transcripts including NEW87 and NEW8 have not yet been validated (Lluch-Senar et al., 2007). There is evidence that suggests that many of the transcripts that are found within reduced bacterial genomes are simply the by-product of transcriptional noise (Raghavan et al., 2012; Llorens-Rico et al., 2016), highlighting the importance of validating the function of identified sRNAs.
Another type of intracellular parasite, Rickettsia, is an intracellular alphaproteobacterial endosymbiont that is found widely within arthropod lineages. The most widely studied Rickettsia species cause vertebrate diseases such as Typhus and Rocky Mountain spotted fever (Weinert et al., 2009). These pathogenic species also spend at least part of their lifecycles in arthropod vectors. A recent study observed sRNA expression within 13 Rickettsia species and identified 1785 novel sRNAs (Schroeder et al., 2015). The number of sRNAs identified within each species varied from 15–191 and there was no correlation with genome size and number of sRNAs identified (Schroeder et al., 2015).
Wolbachia, another intracellular alphaproteobacterium, which is closely related to Rickettsia, is found within many insect taxa and other invertebrates (Hilgenboecker et al., 2008). It has varied effects on its host, such as reproductive manipulation (Werren et al., 2008), inhibition of vector-bone pathogen transmission (Moreira et al., 2009; Bian et al., 2013), and as a symbiont providing vitamins to its host (Hosokawa et al., 2010). When Wolbachia parasitically infects mosquito (A. aegypti) cell lines, it has been found to express sRNAs that potentially increase bacterial fitness by upregulating host genes that help facilitate its own replication (Mayoral et al., 2014a). It has been found that Wolbachia infection of A. aegypti results in differential expression of host miRNAs compared to uninfected control mosquitos (Hussain et al., 2011; Mayoral et al., 2014b). Hussain et al. (2011) identified that the host sRNA, aae-miR-2940, which upregulates a metalloprotease, is necessary for successful Wolbachia infection.
Until recently, there was little evidence that gene regulation occurred within unculturable mutualistic endosymbionts. It has been hypothesized that symbionts like Buchnera, display minimal gene regulation because they live in a stable intracellular environment (Shigenobu et al., 2000). Buchnera microarray experiments have provided limited evidence that this endosymbiont is able to regulate the transcription of genes underlying essential amino-acid (EEA) pathways in response to aphid nutritional demand (Moran et al., 2003, 2005; Reymond et al., 2006; Viñuelas et al., 2011). In turn, Buchnera's gene regulation at the RNA level is generally assumed to be non-existent (Hansen and Moran, 2014). However, a recent study showed that Buchnera exhibits differential expression of 80 proteins between two different Buchnera life-stages, including proteins that are involved in essential amino acid biosynthesis with no concomitant changes in mRNA expression suggesting that post-transcriptional regulation was occurring (Hansen and Degnan, 2014). To support this hypothesis, Hansen and Degnan (2014), also found that across highly divergent Buchnera lineages, there is widespread conservation of cis- and/or trans-encoded sRNAs. They also revealed that of the 80 differentially expressed proteins identified between the two Buchnera life stages, 86% have evidence of possible cis-acting regulatory sRNAs (Hansen and Degnan, 2014). Many of the differentially expressed proteins in EEA pathways also had cis sRNAs identified within their transcriptional unit or flanking them (Hansen and Degnan, 2014). These observations led the authors to hypothesize that these patterns in differential protein expression may result from post-transcriptional regulation, such as conserved sRNAs (Hansen and Degnan, 2014). Based on this study's results, we predict that these novel sRNAs potentially have important regulatory roles, especially for Buchnera's EAA pathways.
In summary, there is growing evidence that both organelles and bacteria with highly reduced genomes, have maintained key sRNAs to aid in the regulation of genes that are vital to their core biological functioning. Organelles have not only evolved mechanisms to uptake vital eukaryotic nuclear encoded sRNAs but also the necessary machinery (Ago2) to process these sRNAs. There is evidence that shows that bacteria can produce sRNAs that target eukaryotic genes, suggesting that these sRNAs either mimic eukaryotic sRNAs or can be co-opted into the RNAi pathway. This type of trans-kingdom communication via sRNAs has also been observed in fungal plant pathogens (Weiberg et al., 2013) and the malarial parasite (LaMonte et al., 2012). There is also evidence demonstrating that organelles have maintained sRNAs within their highly eroded bacterial genome. Within endosymbionts, there is evidence that sRNAs are important for regulating functions important in maintaining the symbiosis with their host. Overall, these examples of sRNA utilization supports the hypothesis that within organisms that have undergone radical gene loss, sRNAs may aid in the regulation of vital symbiotic genes and core housekeeping processes.
Recent studies on epigenomic and small-RNA regulation in eukaryotes and bacteria have challenged historical preconceptions of how these genomes are regulated and evolve. The implications of these new studies on the understanding of the regulation of shared animal-microbe metabolic processes warrant further investigation in future studies. Such studies are critical because of the importance these regulatory mechanisms have in the evolution and maintenance of these widespread herbivore symbioses. To this end, we conclude this review by proposing future directions that will aid in teasing apart the regulatory underpinnings and dynamics of shared metabolic processes that are ubiquitous between insect herbivores and their microbial symbionts.
Elucidating the mechanisms and patterns of DNA methylation and its inheritance is important for understanding the molecular biology, ecology, and the evolution of non-model animals. For example, methylation patterns are linked not only to numerous human diseases (Robertson, 2001; Richardson, 2003; Lund et al., 2004; Stenvinkel et al., 2007; Mastroeni et al., 2010; Benton et al., 2015) but also to insect and mammalian behavior (Kucharski et al., 2008; Dias and Ressler, 2014; Alvarado et al., 2015), and pesticide resistance in insects (Field et al., 1989, 1996; Hick et al., 1996; Table 1). Some of these epigenetic-associated phenotypes are inherited from parent to offspring (Dias and Ressler, 2014), which influences current views of the mechanisms of evolution, particularly the central dogma (Szyf, 2014). CpG DNA methylation is present in divergent eukaryotic taxa such as fungi, animals, and plants (Wang et al., 2006; Feng et al., 2010; Zemach et al., 2010), and therefore its effects on gene regulation and organismal ecology and evolution are of broad interest to science in general.
Aphids are an attractive and tractable model for studying epigenetic regulation of shared herbivore-microbe metabolisms. Aphids display functional DNA methylation (Walsh et al., 2010) in contrast to many classical model organisms (e.g., D. melanogaster, S. cervisiae, C. elegans; Goll and Bestor, 2005; Feng et al., 2010). Moreover, A. pisum can be reared in culture, maintained asexually or sexually, have short generation times, have co-evolved microbial symbionts, and exhibit a wide range of host plant interactions. Thus, aphids are ideally suited for characterizing novel aspects of epigenetic regulation. Results produced from this research can thus be important for understanding insect nutrition specifically, as well as the ecology and evolution of insect-host plant interactions and symbiosis more generally. In addition, many obligate symbionts in insect systems produce folate and/or other methyl- groups, which may influence insect host DNA-methylation patterns (Table 2). In turn, more work is needed on the epigenetics of animal-microbe interactions, especially for non-classical model insect systems. Currently, many of the outstanding questions on this topic (Table 4) can now be addressed using cutting-edge molecular genetics, genomics, and bioinformatics tools.

Table 4. Outstanding questions in regard to the relative importance of regulatory mechanisms that are key to shared herbivore-microbe metabolisms.
Insect taxa in several of the most diverse insect orders are known to harbor obligate symbionts with reduced genomes. Most of these symbionts have been characterized in blood and sap-feeding insects; the smallest known bacterial genome is a symbiont from a sap-feeding insect (Bennett and Moran, 2013). Similar to organelles, such as mitochondria, these obligate symbiont genomes are greatly reduced due to genetic drift, primarily encode genes essential for their host, are unculturable, and have lost most genes for transcriptional regulation. Nevertheless, unlike organelles these symbionts encode core housekeeping genes, and in general none of its genes were transferred to the host's chromosome (Bennett and Moran, 2013). To this end obligate symbionts like the model Buchnera are still autonomous bacterial cells, however some of these minimal genomes can regulate gene expression at the post-transcriptional level (Hansen and Degnan, 2014). Potentially in these small genomes the loss of canonical regulatory proteins has resulted in the evolution of compensatory regulatory mechanisms, such as small regulatory RNAs (Hansen and Degnan, 2014). In sum, if these genomes rely primarily on small RNAs for gene regulation, instead of proteins, this could be a prime example of how genomes revert to the “RNA world” for gene regulation. More functional and comparative genomic studies using novel manipulative techniques on unculturable microbes are required to further understand the putative regulatory role of sRNAs in symbionts with reduced genomes. Ultimately, these future studies will determine the relative importance of these microbial regulatory mechanisms in these intimate symbioses. Outstanding questions on this topic are presented in Table 4 and can now be addressed using classical microbiology techniques, cutting-edge genomics, and bioinformatics tools.
DK, MT, and AH all substantially wrote and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Patrick Degnan, Amanda Adams, Katie Frye, and two reviewers for their helpful comments and suggestions on earlier drafts and the University of Illinois at Urbana-Champaign for funding.
Allen, E., and Howell, M. D. (2010). miRNAs in the biogenesis of trans-acting siRNAs in higher plants. Semin. Cell Dev. Biol. 21, 798–804. doi: 10.1016/j.semcdb.2010.03.008
Alvarado, S., Rajakumar, R., Abouheif, E., and Szyf, M. (2015). Epigenetic variation in the Egfr gene generates quantitative variation in a complex trait in ants. Nat. Commun. 6:6513. doi: 10.1038/ncomms7513
Ames, R. M., and Lovell, S. C. (2011). Diversification at transcription factor binding sites within a species and the implications for environmental adaptation. Mol. Biol. Evol. 28, 3331–3344. doi: 10.1093/molbev/msr167
Aoi, W., Naito, Y., Mizushima, K., Takanami, Y., Kawai, Y., Ichikawa, H., et al. (2010). The microRNA miR-696 regulates PGC-1α in mouse skeletal muscle in response to physical activity. Am. J. Physiol. Endocrinol. Metab. 298, E799–E806. doi: 10.1152/ajpendo.00448.2009
Aschrafi, A., Schwechter, A. D., Mameza, M. G., Natera-Naranjo, O., Gioio, A. E., and Kaplan, B. B. (2008). MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J. Neurosci. 28, 12581–12590. doi: 10.1523/JNEUROSCI.3338-08.2008
Au, H.-K., Yeh, T.-S., Kao, S.-H., Tzeng, C.-R., and Hsieh, R.-H. (2005). Abnormal mitochondrial structure in human unfertilized oocytes and arrested embryos. Ann. N.Y. Acad. Sci. 1042, 177–185. doi: 10.1196/annals.1338.020
Augui, S., Nora, E. P., and Heard, E. (2011). Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 12, 429–442. doi: 10.1038/nrg2987
Axtell, M. J. (2013). Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 64, 137–159. doi: 10.1146/annurev-arplant-050312-120043
Axtell, M. J., Westholm, J. O., and Lai, E. C. (2011). Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 12:221. doi: 10.1186/gb-2011-12-4-221
Babski, J., Maier, L.-K., Heyer, R., Jaschinski, K., Prasse, D., Jäger, D., et al. (2014). Small regulatory RNAs in Archaea. RNA Biol. 11, 484–493. doi: 10.4161/rna.28452
Balaton, B. P., Cotton, A. M., and Brown, C. J. (2015). Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 6, 1–11. doi: 10.1186/s13293-015-0053-7
Baloyannis, S. J. (2006). Mitochondrial alterations in Alzheimer's disease. J. Alzheimers Dis. 9, 119–126. doi: 10.1177/153331750401900205
Bandiera, S., Rüberg, S., Girard, M., Cagnard, N., Hanein, S., Chrétien, D., et al. (2011). Nuclear outsourcing of RNA interference components to human mitochondria. PLoS ONE 6:e20746. doi: 10.1371/journal.pone.0020746
Barbrook, A. C., Howe, C. J., Kurniawan, D. P., and Tarr, S. J. (2010). Organization and expression of organellar genomes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 785–797. doi: 10.1098/rstb.2009.0250
Barquist, L., and Vogel, J. (2015). Accelerating discovery and functional analysis of small RNAs with new technologies. Annu. Rev. Genet. 49, 367–394. doi: 10.1146/annurev-genet-112414-054804
Barrey, E., Saint-Auret, G., Bonnamy, B., Damas, D., Boyer, O., and Gidrol, X. (2011). Pre-microRNA and mature microRNA in human mitochondria. PLoS ONE 6:e20220. doi: 10.1371/journal.pone.0020220
Barrick, J. E., and Breaker, R. R. (2007). The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 8:R239. doi: 10.1186/gb-2007-8-11-r239
Bebek, G., Bennett, K. L., Funchain, P., Campbell, R., Seth, R., Scharpf, J., et al. (2012). Microbiomic subprofiles and MDR1 promoter methylation in head and neck squamous cell carcinoma. Hum. Mol. Gen. 21, 1557–1565. doi: 10.1093/hmg/ddr593
Bennett, G. M., and Moran, N. A. (2013). Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 5, 1675–1688. doi: 10.1093/gbe/evt118
Benton, M. C., Johnstone, A., Eccles, D., Harmon, B., Hayes, M. T., Lea, R. A., et al. (2015). An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 16, 8. doi: 10.1186/s13059-014-0569-x
Bian, G., Joshi, D., Dong, Y., Lu, P., Zhou, G., Pan, X., et al. (2013). Wolbachia invades Anopheles stephensi populations and induces refractoriness to plasmodium infection. Science 340, 748–751. doi: 10.1126/science.1236192
Bian, Z., Li, L.-M., Tang, R., Hou, D.-X., Chen, X., Zhang, C.-Y., et al. (2010). Identification of mouse liver mitochondria-associated miRNAs and their potential biological functions. Cell Res. 20, 1076–1078. doi: 10.1038/cr.2010.119
Bienertova-Vasku, J., Sana, J., and Slaby, O. (2013). The role of microRNAs in mitochondria in cancer. Cancer Lett. 336, 1–7. doi: 10.1016/j.canlet.2013.05.001
Biergans, S. D., Jones, J. C., Treiber, N., Galizia, C. G., and Szyszka, P. (2012). DNA methylation mediates the discriminatory power of associative long-term memory in honeybees. PLoS ONE 7:e39349. doi: 10.1371/journal.pone.0039349
Billmyre, R. B., Calo, S., Feretzaki, M., Wang, X., and Heitman, J. (2013). RNAi function, diversity, and loss in the fungal kingdom. Chromosome Res. 21, 561–572. doi: 10.1007/s10577-013-9388-2
Bird, A. P. (1986). CpG-rich islands and the function of DNA methylation. Nature 321, 209–213. doi: 10.1038/321209a0
Bonasio, R., Li, Q., Lian, J., Mutti, N. S., Jin, L., Zhao, H., et al. (2012). Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 22, 1755–1764. doi: 10.1016/j.cub.2012.07.042
Borsani, O., Zhu, J., Verslues, P. E., Sunkar, R., and Zhu, J.-K. (2005). Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 123, 1279–1291. doi: 10.1016/j.cell.2005.11.035
Bouloc, P., and Repoila, F. (2016). Fresh layers of RNA-mediated regulation in Gram-positive bacteria. Curr. Opin. Microbiol. 30, 30–35. doi: 10.1016/j.mib.2015.12.008
Braendle, C., Miura, T., Bickel, R., Shingleton, A. W., Kambhampati, S., and Stern, D. L. (2003). Developmental origin and evolution of bacteriocytes in the aphid–Buchnera symbiosis. PLoS Biol. 1:e21. doi: 10.1371/journal.pbio.0000021
Brantl, S. (2007). Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Opin. Microbiol. 10, 102–109. doi: 10.1016/j.mib.2007.03.012
Brinza, L., Calevro, F., Duport, G., Gaget, K., Gautier, C., and Charles, H. (2010). Structure and dynamics of the operon map of Buchnera aphidicola sp. strain APS. BMC Genomics 11:666. doi: 10.1186/1471-2164-11-666
Brownlie, J. C., Cass, B. N., Riegler, M., Witsenburg, J. J., Iturbe-Ormaetxe, I., McGraw, E. A., et al. (2009). Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathog. 5:e1000368. doi: 10.1371/journal.ppat.1000368
Brumin, M., Kontsedalov, S., and Ghanim, M. (2011). Rickettsia influences thermotolerance in the whitefly Bemisia tabaci B biotype. Insect Sci. 18, 57–66. doi: 10.1111/j.1744-7917.2010.01396.x
Budak, H., and Akpinar, B. A. (2015). Plant miRNAs: biogenesis, organization and origins. Funct. Integr. Genomics 15, 523–531. doi: 10.1007/s10142-015-0451-2
Burgers, W. A., Blanchon, L., Pradhan, S., De Launoit, Y., Kouzarides, T., and Fuks, F. (2007). Viral oncoproteins target the DNA methyltransferases. Oncogene 26, 1650–1655. doi: 10.1038/sj.onc.1209950
Burkhardt, R. W. (2013). Lamarck, evolution, and the inheritance of acquired characters. Genetics 194, 793–805. doi: 10.1534/genetics.113.151852
Carthew, R. W., and Sontheimer, E. J. (2009). Origins and Mechanisms of miRNAs and siRNAs. Cell 136, 642–655. doi: 10.1016/j.cell.2009.01.035
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2003). Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278, 36027–36031. doi: 10.1074/jbc.M304854200
Chen, X., Zhang, X., Kubo, H., Harris, D. M., Mills, G. D., Moyer, J., et al. (2005). Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 97, 1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1
Dandekar, T., Huynen, M., Regula, J. T., Ueberle, B., Zimmermann, C. U., Andrade, M. A., et al. (2000). Re-annotating the Mycoplasma pneumoniae genome sequence: adding value, function and reading frames. Nucleic Acids Res. 28, 3278–3288. doi: 10.1093/nar/28.17.3278
Das, S., Bedja, D., Campbell, N., Dunkerly, B., Chenna, V., Maitra, A., et al. (2014). miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 9:e96820. doi: 10.1371/journal.pone.0096820
de Lima, J. C., Loss-Morais, G., and Margis, R. (2012). MicroRNAs play critical roles during plant development and in response to abiotic stresses. Genet. Mol. Biol. 35, 1069–1077. doi: 10.1590/S1415-47572012000600023
Dias, B. G., and Ressler, K. J. (2014). Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 17, 89–96. doi: 10.1038/nn.3594
Dobzhansky, T. (1937). Genetics and the Origin of Species, 2nd Edn, 1941; 3rd Edn, 1951. New York, NY: Columbia University Press.
Dombrovsky, A., Arthaud, L., Ledger, T. N., Tares, S., and Robichon, A. (2009). Profiling the repertoire of phenotypes influenced by environmental cues that occur during asexual reproduction. Genome Res. 19, 2052–2063. doi: 10.1101/gr.091611.109
Dunbar, H. E., Wilson, A. C., Ferguson, N. R., and Moran, N. A. (2007). Aphid thermal tolerance is governed by a point mutation in bacterial symbionts. PLoS Biol. 5:e96. doi: 10.1371/journal.pbio.0050096
Durand, S., Tomasini, A., Braun, F., Condon, C., and Romby, P. (2015). sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol. Rev. 39, 316–330. doi: 10.1093/femsre/fuv007
Durdevic, Z., Hanna, K., Gold, B., Pollex, T., Cherry, S., Lyko, F., et al. (2013). Efficient RNA virus control in Drosophila requires the RNA methyltransferase Dnmt2. EMBO Rep. 14, 269–275. doi: 10.1038/embor.2013.3
Elango, N., Hunt, B. G., Goodisman, M. A. D., and Yi, S. V. (2009). DNA methylation is widespread and associated with differential gene explression in castes of the honey bee, Apis melifera. Proc. Natl. Acad. Sci. U.S.A. 106, 11206–11211. doi: 10.1073/pnas.0900301106
Fang, J. Y., Mikovits, J. A., Bagni, R., Petrow-Sadowski, C. L., and Ruscetti, F. W. (2001). Infection of lymphoid cells by integration-defective human immunodeficiency virus type 1 increases de novo methylation. J. Virol. 75, 9753–9761. doi: 10.1128/JVI.75.20.9753-9761.2001
Fang, R., Xiao, T., Fang, Z., Sun, Y., Li, F., Gao, Y., et al. (2012). MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J. Biol. Chem. 287, 23227–23235. doi: 10.1074/jbc.M112.373084
Feng, S., Cokus, S. J., Zhang, X., Chen, P. Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and Divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 107, 8689–8694. doi: 10.1073/pnas.1002720107
Field, L. M. (2000). Methylation and expression of amplified esterase genes in the aphid Myzus persicae (Sulzer). Biochem. J. 349, 863–868. doi: 10.1042/bj3490863
Field, L. M., and Blackman, R. L. (2003). Insecticide resistance in the aphid Myzus persicae (Sulzer): chromosome location and epigenetic effects on esterase gene expression in clonal lineages. Biol. J. Linn. Soc. 79, 107–113. doi: 10.1046/j.1095-8312.2003.00178.x
Field, L. M., Crick, S. E., and Devonshire, A. L. (1996). Polymerase chain reaction-based identification of insecticide resistance genes and DNA methylation in the aphid Myzus persicae (Sulzer). Insect Mol. Biol. 5, 197–202. doi: 10.1111/j.1365-2583.1996.tb00054.x
Field, L. M., Devonshire, A. L., Ffrench-Constant, R. H., and Forde, B. G. (1989). Changes in DNA methylation are associated with loss of insecticide resistance in the peach-potato aphid Myzus persicae (Sulz.). FEBS Lett. 243, 323–327. doi: 10.1016/0014-5793(89)80154-1
Field, L. M., Lyko, F., Mandrioli, M., and Prantera, G. (2004). DNA methylation in insects. Insect Mol. Biol. 13, 109–115. doi: 10.1111/j.0962-1075.2004.00470.x
Flynt, A. S., and Lai, E. C. (2008). Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat. Rev. Genet. 9, 831–842. doi: 10.1038/nrg2455
Foret, S., Kucharski, R., Pellegrini, M., Feng, S., Jacobsen, S. E., Robinson, G. E., et al. (2012). DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl. Acad. Sci. U.S.A. 109, 4968–4973. doi: 10.1073/pnas.1202392109
Fozo, E. M., Hemm, M. R., and Storz, G. (2008). Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72, 579–589. doi: 10.1128/mmbr.00025-08
Frankel, L. B., Wen, J., Lees, M., Høyer-Hansen, M., Farkas, T., Krogh, A., et al. (2011). microRNA-101 is a potent inhibitor of autophagy. EMBO J. 30, 4628–4641. doi: 10.1038/emboj.2011.331
Fujii, H., Chiou, T.-J., Lin, S.-I., Aung, K., and Zhu, J.-K. (2005). A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 15, 2038–2043. doi: 10.1016/j.cub.2005.10.016
Gadjev, I. (2015). Nature and nuture: lamarck's legacy. Biol. J. Linn. Soc. 114, 242–247. doi: 10.1111/bij.12439
Gama-Sosa, M. A., Midgett, R. M., Slagel, V. A., Githens, S., Kuo, K. C., Gehrke, C. W., et al. (1983). Tissue-specific differences in DNA methylation in various mammals. Biochim. Biophys. Acta 740, 212–219. doi: 10.1016/0167-4781(83)90079-9
Gao, P., Tchernyshyov, I., Chang, T. C., Lee, Y. S., Kita, K., Ochi, T., et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase and glutamine metabolism. Nature 458, 762–765. doi: 10.1038/nature07823
Gavery, M. R., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 11:483. doi: 10.1186/1471-2164-11-483
Georg, J., and Hess, W. R. (2011). cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 75, 286–300. doi: 10.1128/MMBR.00032-10
Ghildiyal, M., and Zamore, P. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108. doi: 10.1038/nrg2504
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. D. (2012). Evidence of a conserved functional role for DNA methylation in termites. Insect Mol. Biol. 22, 143–154. doi: 10.1111/imb.12010
Goll, M. G., and Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514. doi: 10.1146/annurev.biochem.74.010904.153721
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C. L., Zhang, X., et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Gong, L., Cui, F., Sheng, C., Lin, Z., Reeck, G., Xu, J., et al. (2012). Polymorphism and methylation of four genes expressed in salivary glands of Russian Wheat Aphid (Homoptera: Aphididae). J. Econ. Entomol. 105, 232–241. doi: 10.1603/EC11289
Gong, Y., Chai, Y., Ding, J.-H., Sun, X.-L., and Hu, G. (2011). Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 488, 76–80. doi: 10.1016/j.neulet.2010.11.006
Gould, S. B., Waller, R. F., and McFadden, G. I. (2008). Plastid evolution. Annu. Rev. Plant Biol. 59, 491–517. doi: 10.1146/annurev.arplant.59.032607.092915
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4:a011403. doi: 10.1101/cshperspect.a011403
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Güell, M., van Noort, V., Yus, E., Chen, W.-H., Leigh-Bell, J., Michalodimitrakis, K., et al. (2009). Transcriptome complexity in a genome-reduced bacterium. Science 326, 1268–1271. doi: 10.1126/science.1176951
Ha, M., and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524. doi: 10.1038/nrm3838
Hansen, A. K., and Degnan, P. H. (2014). Widespread expression of conserved small RNAs in small symbiont genomes. ISME J. 8, 2490–2502. doi: 10.1038/ismej.2014.121
Hansen, A. K., and Moran, N. A. (2011). Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. U.S.A. 108, 2849–2854. doi: 10.1073/pnas.1013465108
Hansen, A. K., and Moran, N. A. (2012). Altered tRNA characteristics and 3′ maturation in bacterial symbionts with reduced genomes. Nucleic Acids Res. 40, 7870–7884. doi: 10.1093/nar/gks503
Hansen, A. K., and Moran, N. A. (2014). The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol. Ecol. 23, 1473–1496. doi: 10.1111/mec.12421
He, B. Z., Holloway, A. K., Maerkl, S. J., and Kreitman, M. (2011). Does positive selection drive transcription factor binding site turnover? A test with Drosophila cis-regulatory modules. PLoS Genet. 7:e1002053. doi: 10.1371/journal.pgen.1002053
He, Y., Du, Y., Li, J., Liu, P., Wang, Q., and Li, Z. (2015). Analysis of DNA methylation in different tissues of Fenneropenaeus chinensis from the wild population and Huanghai No. 1. Acta Oceanol. Sin. 34, 175–180. doi: 10.1007/s13131-015-0765-x
Herb, B. R., Wolschin, F., Hansen, K. D., Aryee, M. J., Langmead, B., Irizarry, R., et al. (2012). Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat. Neurosci. 15, 1371–1373. doi: 10.1038/nn.3218
Hick, C. A., Field, L. M., and Devonshire, A. L. (1996). Changes in the methylation of amplified esterase DNA during loss and reselection of insecticide resistance in peach-potato aphids, Myzus persicae. Insect Biochem. Mol. Biol. 26, 41–47. doi: 10.1016/0965-1748(95)00059-3
Hilgenboecker, K., Hammerstein, P., Schlattmann, P., Telschow, A., and Werren, J. H. (2008). How many species are infected with Wolbachia?–A statistical analysis of current data. FEMS Microbiol. Lett. 281, 215–220. doi: 10.1111/j.1574-6968.2008.01110.x
Holt, R. A., Subramanian, G. M., Halpern, A., Sutton, G. G., Charlab, R., Nusskern, D. R., et al. (2002). The genome sequence of the malaria mosquito Anopheles gambiae. Science 298, 129–149. doi: 10.1126/science.1076181
Horler, R. S., and Vanderpool, C. K. (2009). Homologs of the small RNA SgrS are broadly distributed in enteric bacteria but have diverged in size and sequence. Nucleic Acids Res. 37, 5465–5476. doi: 10.1093/nar/gkp501
Hosokawa, T., Ishii, Y., Nikoh, N., Fujie, M., Satoh, N., and Fukatsu, T. (2016). Obligate bacterial mutualists evolving from environmental bacteria in natural insect populations. Nat. Microbiol. 1:15011. doi: 10.1038/nmicrobiol.2015.11
Hosokawa, T., Koga, R., Kikuchi, Y., Meng, X.-Y., and Fukatsu, T. (2010). Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. U.S.A. 107, 769–774. doi: 10.1073/pnas.0911476107
Hotto, A. M., Schmitz, R. J., Fei, Z., Ecker, J. R., and Stern, D. B. (2011). Unexpected diversity of chloroplast noncoding RNAs as revealed by deep sequencing of the Arabidopsis transcriptome. G3 (Bethesda) 1, 559–570. doi: 10.1534/g3.111.000752
Hussain, M., Frentiu, F. D., Moreira, L. A., O'Neill, S. L., and Asgari, S. (2011). Wolbachia uses host microRNAs to manipulate host gene expression and facilitate colonization of the dengue vector Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A. 108, 9250–9255. doi: 10.1073/pnas.1105469108
Jablonka, E., and Lamb, M. J. (2015). The inheritance of acquired epigenetic variations. Int. J. Epidemiol. 44, 1094–1103. doi: 10.1093/ije/dyv020
Jankard, R., and Herman, J. P. (2008). Limbic regulation of hypohalamo-pituitary-adrenocortical function during acute and chronic stress. Ann. N.Y. Acad. Sci. 1148, 64–73. doi: 10.1196/annals.1410.012
Jiang, S., Zhang, L.-F., Zhang, H.-W., Hu, S., Lu, M.-H., Liang, S., et al. (2012). A novel miR-155/miR-143c ascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO 31, 1985–1998. doi: 10.1038/emboj.2012.45
Jones-Rhoades, M. W., and Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14, 787–799. doi: 10.1016/j.molcel.2004.05.027
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Jung, J. K., Arora, P., Pagano, J. S., and Jang, K. L. (2007). Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res. 67, 5771–5778. doi: 10.1158/0008-5472.CAN-07-0529
Kaltenpoth, M., Gottler, W., Herzner, G., and Strohm, E. (2005). Sytmbiotic vacteria protect wasp larvae from funcgal infestation. Curr. Biol. 15, 475–479. doi: 10.1016/j.cub.2004.12.084
Kang, T., Lu, W., Xu, W., Anderson, L., Bacanamwo, M., Thompson, W., et al. (2013). MicroRNA-27 (miR- 27) targets prohibitin and impairs adipocyte differentiation and mitochondrial function in human adipose-derived stem cells. J. Biol. Chem. 288, 34394–3402. doi: 10.1074/jbc.M113.514372
Kankanamge, A., and Eranthi, H. (2015). DNA Methylation, Genomic Imprinting and Polyphenism in the Bumblebee, Bombus terrestris. Doctoral dissertation, University of Leicester, Leicester, UK.
Katiyar-Agarwal, S., Morgan, R., Dahlbeck, D., Borsani, O., Villegas, A., Zhu, J.-K., et al. (2006). A pathogen-inducible endogenous siRNA in plant immunity. Proc. Natl. Acad. Sci. U.S.A. 103, 18002–18007. doi: 10.1073/pnas.0608258103
Katz, L. A. (2012). Origin and diversification of eukaryotes. Annu. Rev. Microbiol. 66, 411–427. doi: 10.1146/annurev-micro-090110-102808
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139. doi: 10.1038/nrm2632
Koonin, E. V., and Wolf, Y. I. (2009). Is evolution Darwinian or/and Lamarckian? Biol. Direct 4:42. doi: 10.1186/1745-6150-4-42
Krauss, V., Eisenhardt, C., and Unger, T. (2009). The genome of the stick insect Medauroidea extradentata is strongly methylated within genes and repetitive DNA. PLoS ONE. 4:e7223. doi: 10.1371/journal.pone.0007223
Kren, B. T., Wong, P., Sarver, A., Zhang, X., Zeng, Y., and Steer, C. J. (2009). MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 6, 65–72. doi: 10.4161/rna.6.1.7534
Kucharski, R., Maleszka, J., Foret, S., and Maleszka, R. (2008). Nutritional control of reproductive status in honeybees via DNA methylation. Science 319, 1827–1830. doi: 10.1126/science.1153069
Kuhn, C., Wilusz, J. E., Zheng, Y., Beal, P. A., and Joshua-Tor, L. (2015). On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 160, 644–658. doi: 10.1016/j.cell.2015.01.005
Kunert, N., Marhold, J., Stanke, J., Stach, D., and Lyko, F. (2003). A Dnmt2-like protein mediates DNA methylation in Drosophila. Development 130, 5083–5090. doi: 10.1242/dev.00716
Lai, E. C. (2003). RNA sensors and riboswitches: self-regulating messages. Curr. Biol. 13, R285–R291. doi: 10.1016/S0960-9822(03)00203-3
Lambert, J. D., and Moran, N. A. (1998). Deleterious mutations destabilize ribosomal RNA in endosymbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 95, 4458–4462. doi: 10.1073/pnas.95.8.4458
LaMonte, G., Philip, N., Reardon, J., Lacsina, J. R., Majoros, W., Chapman, L., et al. (2012). Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe 12, 187–199. doi: 10.1016/j.chom.2012.06.007
Latronico, M. V. G., and Condorelli, G. (2012). The might of microRNA in mitochondria. Circ. Res. 110, 1540–1542. doi: 10.1161/CIRCRESAHA.112.271312
Lee, Y., Jeong, S.-Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. doi: 10.1091/mbc.E04-04-0294
Lemieux, H., Vazquez, E. J., Fujioka, H., and Hoppel, C. L. (2010). Decrease in mitochondrial function in rat cardiac permeabilized fibers correlates with the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1157–1164. doi: 10.1093/gerona/glq141
Li, E., Beard, C., and Jaenisch, R. (1993). Role for DNA methylation in genomic imprinting. Nature. 366, 362–365. doi: 10.1038/366362a0
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., and Li, P. (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6:e1000795. doi: 10.1371/journal.pgen.1000795
Li, J., Li, Y., Jiao, J., Wang, J., Li, Y., Qin, D., et al. (2014). Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 34, 1788–99. doi: 10.1128/MCB.00774-13
Li, X., Wang, F. S., Wu, Z. Y., Lin, J. L., Lan, W. B., and Lin, J. H. (2014). MicroRNA-19b targets Mfn1 to inhibit Mfn1-induced apoptosis in osteosarcoma cells. Neoplasma 61, 265–273. doi: 10.4149/neo_2014_034
Li-Byarlay, H., Li, Y., Stround, H., Feng, S., Newman, T. C., Kaneda, M., et al. (2013). RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc. Natl. Acad. Sci. U.S.A. 110, 12750–12755. doi: 10.1073/pnas.1310735110
Llave, C., Kasschau, K. D., Rector, M. A., and Carrington, J. C. (2002). Endogenous and silencing-associated small RNAs in plants. Plant Cell 14, 1605–1619. doi: 10.1105/tpc.003210
Llorens-Rico, V., Cano, J., Kamminga, T., Gil, R., Latorre, A., Chen, W.-H., et al. (2016). Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2:e1501363. doi: 10.1126/sciadv.1501363
Lluch-Senar, M., Vallmitjana, M., Querol, E., and Piñol, J. (2007). A new promoterless reporter vector reveals antisense transcription in Mycoplasma genitalium. Microbiology 153, 2743–2752. doi: 10.1099/mic.0.2006/007559-0
Lockett, G. A., Helliwell, P., and Maleszka, R. (2010). Involvement of DNA methylation in memory processing in the honey bee. Neuroreport 21, 812–816. doi: 10.1097/WNR.0b013e32833ce5be
Lockett, G. A., Kucharski, R., and Maleszka, R. (2012). DNA methylation changes elicited by social stimuli in the brains of worker honey bees. Genes Brain Behav. 11, 235–242. doi: 10.1111/j.1601-183X.2011.00751.x
Long, B., Wang, K., Li, N., Murtaza, I., Xiao, J.-Y., Fan, Y.-Y., et al. (2013). miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med. 65C, 371–379. doi: 10.1016/j.freeradbiomed.2013.07.009
Lund, G., Andersson, L., Lauria, M., Lindholm, M., Fraga, M. F., Villar-Garea, A., et al. (2004). DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J. Biol. Chem. 279, 29147–29154. doi: 10.1074/jbc.M403618200
Lung, B., Zemann, A., Madej, M. J., Schuelke, M., Techritz, S., Ruf, S., et al. (2006). Identification of small non-coding RNAs from mitochondria and chloroplasts. Nucleic Acids Res. 34, 3842–3852. doi: 10.1093/nar/gkl448
Mandal, M., and Breaker, R. R. (2004). Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol. 5, 451–463. doi: 10.1038/nrm1403
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., and Rogers, J. (2010). Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol. Aging 31, 2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Mayoral, J. G., Etebari, K., Hussain, M., Khromykh, A. A., and Asgari, S. (2014b). Wolbachia infection modifies the profile, shuttling and structure of microRNAs in a mosquito cell line. PLoS ONE 9:e96107. doi: 10.1371/journal.pone.0096107
Mayoral, J. G., Hussain, M., Joubert, D. A., Iturbe-Ormaetxe, I., O'Neill, S. L., and Asgari, S. (2014a). Wolbachia small noncoding RNAs and their role in cross-kingdom communications. Proc. Natl. Acad. Sci. U.S.A. 111, 18721–18726. doi: 10.1073/pnas.1420131112
McCutcheon, J. P., and Moran, N. A. (2012). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. doi: 10.1038/nrmicro2670
Mendoza-Vargas, A., Olvera, L., Olvera, M., Grande, R., Vega-Alvarado, L., Taboada, B., et al. (2009). Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS ONE 4:e7526. doi: 10.1371/journal.pone.0007526
Mercer, T. R., Neph, S., Dinger, M. E., Crawford, J., Smith, M. A., Shearwood, A. M., et al. (2011). The human mitochondrial transcriptome. Cell 146, 645–658. doi: 10.1016/j.cell.2011.06.051
Mette, M. F., Aufsatz, W., van der Winden, J., Matzke, M. A., and Matzke, A. J. (2000). Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 19, 5194–5201. doi: 10.1093/emboj/19.19.5194
Mischke, M., and Plosch, T. (2013). More than just a gut instinct-the potential interplay between a baby's nutrition, its gut microbiome, and the epigenome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R1065–R1069. doi: 10.1152/ajpregu.00551.2012
Moczek, A. P., and Snell-Rood, E. C. (2008). The basis of bee-ing different: the role of gene silencing in plasticity. Evol. Dev. 10, 511–513. doi: 10.1111/j.1525-142X.2008.00264.x
Moran, N. A., and Bennett, G. M. (2014). The tiniest tiny genomes. Annu. Rev. Microbiol. 68, 195–215. doi: 10.1146/annurev-micro-091213-112901
Moran, N. A., McLaughlin, H. J., and Sorek, R. (2009). The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science 323, 379–382. doi: 10.1126/science.1167140
Moran, N. A., Plague, G. R., Sandström, J. P., and Wilcox, J. L. (2003). A genomic perspective on nutrient provisioning by bacterial symbionts of insects. Proc. Natl. Acad. Sci. U.S.A. 100(Suppl. 2), 14543–14548. doi: 10.1073/pnas.2135345100
Moran, N. A., Tran, P., and Gerardo, N. M. (2005). Symbiosis and insect diversification: an ancient symbiont of sap-feeding insects from the bacterial phylum Bacteroidetes. Appl. Environ. Microbiol. 71, 8802–8810. doi: 10.1128/AEM.71.12.8802-8810.2005
Moreira, L. A., Iturbe-Ormaetxe, I., Jeffery, J. A., Lu, G., Pyke, A. T., Hedges, L. M., et al. (2009). A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 139, 1268–1278. doi: 10.1016/j.cell.2009.11.042
Mukherjee, K., Campos, H., and Kolaczkowski, B. (2013). Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol. Biol. Evol. 30, 627–641. doi: 10.1093/molbev/mss263
Nahvi, A., Sudarsan, N., Ebert, M. S., Zou, X., Brown, K. L., and Breaker, R. R. (2002). Genetic Control by a Metabolite Binding mRNA. Chem. Biol. 9, 1043–1049. doi: 10.1016/S1074-5521(02)00224-7
Nakabachi, A., Ishida, K., Hongoh, Y., Ohkuma, M., and Miyagishima, S.-Y. (2014). Aphid gene of bacterial origin encodes a protein transported to an obligate endosymbiont. Curr. Biol. 24, R640–R641. doi: 10.1016/j.cub.2014.06.038
Nakabachi, A., Shigenobu, S., Sakazume, N., Shiraki, T., Hayashizaki, Y., Carninci, P., et al. (2005). Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc. Natl. Acad. Sci. U.S.A. 102, 5477–5482. doi: 10.1073/pnas.0409034102
Nakajima, T., Yamashita, S., Maekita, T., Niwa, T., Nakazawa, K., and Ushijima, T. (2009). The presence of a methylation fingerprint of Helicobacter pylori infection in human gastric mucosae. Int. J. Cancer 124, 905–910. doi: 10.1002/ijc.24018
Nene, V., Wortman, J. R., Lawson, D., Haas, B., Kodira, C., Tu, Z. J., Ren, Q., et al. (2007). Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316, 1718–1723. doi: 10.1126/science.1138878
Nudler, E., and Mironov, A. S. (2004). The riboswitch control of bacterial metabolism. Trends Biochem. Sci. 29, 11–17. doi: 10.1016/j.tibs.2003.11.004
O'Rourke, B. (2004). Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ. Res. 94, 420–432. doi: 10.1161/01.RES.0000117583.66950.43
Oliver, K. M., Moran, N. A., and Hunter, M. S. (2005). Variation in resistance to parasitism in aphids is due to symbionts not host genotype. Proc. Natl. Acad. Sci. U.S.A. 102, 12795–12800. doi: 10.1073/pnas.0506131102
Ono, M., Swanson, J. J., Field, L. M., Devonshire, A. L., and Siegfried, B. D. (1999). Amplification and methylation of an esterase gene associated with insecticide-resistance in greenbugs, Schizaphis graminum (Rondani) (Homoptera: Aphididae). Insect Biochem. Mol. Biol. 29, 1065–1073. doi: 10.1016/S0965-1748(99)00082-X
Park, J., Peng, Z., Zeng, J., Elango, N., Park, T., Wheeler, D., et al. (2011). Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol. Biol. Evol. 28, 3345–3354. doi: 10.1093/molbev/msr168
Patalano, S., Hore, T. A., Reik, W., and Sumner, S. (2012). Shifting behavior: epigenetic reprogramming in eusocial insects. Curr. Opin. Cell Biol. 24, 367–373. doi: 10.1016/j.ceb.2012.02.005
Pattanayak, D., Solanke, A. U., and Kumar, P. A. (2013). Plant RNA interference pathways: diversity in function, similarity in action. Plant Mol. Biol. Rep. 31, 493–506. doi: 10.1007/s11105-012-0520-9
Pegoraro, M., Bafna, A., Davies, N. J., Shuker, D. M., and Tauber, E. (2016). DNA methylation changes induced by long and short photoperiods in Nasonia. Genome Res. 26, 203–210. doi: 10.1101/gr.196204.115
Peter, I. S., and Davidson, E. H. (2015). Genomic Control Process: Development and Evolution. New York, NY: Elsevier.
Piatek, M. J., and Werner, A. (2014). Endogenous siRNAs: regulators of internal affairs. Biochem. Soc. Trans. 42, 1174–1179. doi: 10.1042/BST20140068
Pilpel, Y., and Rechavi, O. (2015). The Lamarckian chicken and the Darwinian egg. Biol. Direct 10, 1–5. doi: 10.1186/s13062-015-0062-9
Poliakov, A., Russell, C. W., Ponnala, L., Hoops, H. J., Sun, Q., Douglas, A. E., et al. (2011). Large-scale label-free quantitative proteomics of the pea aphid-buchnera symbiosis. Mol. Cell. Proteomics 10:M110.007039. doi: 10.1074/mcp.m110.007039
Price, D. R., Feng, H., Baker, J. D., Bavan, S., Luetje, C. W., and Wilson, A. C. (2014). Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc. Natl. Acad. Sci. U.S.A. 111, 320–325. doi: 10.1073/pnas.1306068111
Raghavan, R., Sloan, D. B., and Ochman, H. (2012). Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 3:e00156–12. doi: 10.1128/mBio.00156-12
Ray, S., and Chakdar, H. (2015). Riboswitch: ancient living switch for gene regulation. Mol. Genet. Microbiol. Virol. 29, 227–239. doi: 10.3103/S0891416814040090
Razin, A., and Cedar, H. (1994). DNA methylation and genomic imprinting. Cell. 77, 473–476. doi: 10.1016/0092-8674(94)90208-9
Regev, A., Lamb, M. J., and Jablonka, E. (1998). The rold of DNA methylation in invertebrates: developmental regulation or genome defense?. Mol. Biol. Evol. 15, 880–891. doi: 10.1093/oxfordjournals.molbev.a025992
Reik, W., Collick, A., Norris, M. L., Barton, S. C., and Surani, M. A. (1987). Genomic imprinting determines methylation of parental alleles in transgenic mice. Nature 328, 248–251. doi: 10.1038/328248a0
Reymond, N., Calevro, F., Viñuelas, J., Morin, N., Rahbé, Y., Febvay, G., et al. (2006). Different levels of transcriptional regulation due to trophic constraints in the reduced genome of Buchnera aphidicola APS. Appl. Environ. Microbiol. 72, 7760–7766. doi: 10.1128/AEM.01118-06
Richards, S., Gibbs, R. A., Weinstock, G. M., Brown, S. J., Denell, R., Beeman, R. W., et al. (2008). The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955. doi: 10.1038/nature06784
Richardson, B. (2003). DNA methylation and autoimmune disease. Clin. Immunol. 109, 72–79. doi: 10.1016/S1521-6616(03)00206-7
Ritov, V. B., Menshikova, E. V., Azuma, K., Wood, R., Toledo, F. G. S., Goodpaster, B. H., et al. (2010). Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 298, E49–E58. doi: 10.1152/ajpendo.00317.2009
Ro, S., Ma, H.-Y., Park, C., Ortogero, N., Song, R., Hennig, G. W., et al. (2013). The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 23, 759–774. doi: 10.1038/cr.2013.37
Robertson, K. D. (2001). DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155. doi: 10.1038/sj.onc.1204341
Robertson, K. D., Hayward, S. D., Ling, P. D., Samid, D., and Ambinder, R. F. (1995). Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol. Cell. Biol. 15, 6150–6159. doi: 10.1128/MCB.15.11.6150
Robinson, K. L., Tohidi-Esfahani, D., Ponton, F., Simpson, S. J., Sowrd, G. A., and Lo, N. (2015). Alternative migratory locust phenotypes are associated with differences in the expression of genes encoding the methylation machinery. Insect Mol. Biol. 25, 105–115. doi: 10.1111/imb.12203
Romanoski, C. E., Glass, C. K., Stunnenberg, H. G., Wilson, L., and Almouzni, G. (2015). Epigenomics: roadmap for regulation. Nature 518, 314–316. doi: 10.1038/518314a
Salem, H., Kreutzer, E., Sudakaran, S., and Kaltenpoth, M. (2012). Actinobacteria as essential symbionts in firebugs and cotton strainers (Hemiptera, Pyrrhocoridae). Environ. Microbiol. 15, 1956–1968. doi: 10.1111/1462-2920.12001
Sauer, E. (2014). Structure and RNA-binding properties of the bacterial LSm protein Hfq. RNA Biol. 10, 610–618. doi: 10.4161/rna.24201
Scarborough, C. L., Ferrari, J., and Godfray, H. C. J. (2005). Aphid protected from pathogen by endosymbiont. Science 310, 1781–1781. doi: 10.1126/science.1120180
Schneider, A. (2011). Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu. Rev. Biochem. 80, 1033–1053. doi: 10.1146/annurev-biochem-060109-092838
Schroeder, C. L. C., Narra, H. P., Rojas, M., Sahni, A., Patel, J., Khanipov, K., et al. (2015). Bacterial small RNAs in the Genus Rickettsia. BMC Genomics 16:1075. doi: 10.1186/s12864-015-2293-7
Schwenk, R. W., Vogel, H., and Schürmann, A. (2013). Genetic and epigenetic control of metabolic health. Mol. Metab. 2, 337–347. doi: 10.1016/j.molmet.2013.09.002
Scott, J. J., Oh, D. C., Yuceer, M. C., Klepzig, K. D., Clardy, J., and Currie, C. R. (2008). Bacterial protection of beetle-fungus mutualism. Science 322, 63. doi: 10.1126/science.1160423
Shabalina, S. A., and Koonin, E. V (2008). Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. (Amst). 23, 578–587. doi: 10.1016/j.tree.2008.06.005
Shigenobu, S., Watanabe, H., Hattori, M., Sakaki, Y., and Ishikawa, H. (2000). Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407, 81–86. doi: 10.1038/35024074
Skinner, M. K. (2015). Environmental epigenetics and a unified theory of the molecular aspects of evolution: a neo-Lamarckian concept that facilitates neo-Darwinian evolution. Genome Biol. Evol. 7, 1296–1302. doi: 10.1093/gbe/evv073
Smalheiser, N. R., Lugli, G., Thimmapuram, J., Cook, E. H., and Larson, J. (2011). Mitochondrial small RNAs that are up-regulated in hippocampus during olfactory discrimination training in mice. Mitochondrion 11, 994–995. doi: 10.1016/j.mito.2011.08.014
Smith, C. R., Mutti, N. S., Jasper, W. C., Naidu, A., Smith, C. D., and Gadau, J. (2012). Patterns of DNA methylation in development, division of labor and hybridization in an ant with genetic caste determination. PLoS ONE 7:e42433. doi: 10.1371/journal.pone.0042433
Smith, D. R. (2012). Updating our view of organelle genome nucleotide landscape. Front. Genet. 3:175. doi: 10.3389/fgene.2012.00175
Smith, D. R., and Keeling, P. J. (2015). Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. U.S.A. 112, 10177–10184. doi: 10.1073/pnas.1422049112
Snell-Rood, E. C., Troth, A., and Moczek, A. P. (2012). DNA methylation as a mechanism of nutritional plasticity: limited support from horned beetles. J. Exp. Zool. Part B Mol. Dev. Evol. 320, 22–34. doi: 10.1002/jez.b.22479
Sripada, L., Tomar, D., Prajapati, P., Singh, R., Singh, A. K., and Singh, R. (2012). Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS ONE 7:e44873. doi: 10.1371/journal.pone.0044873
Stenvinkel, P., Karimi, M., Jahansson, S., Axelsson, J., Suliman, M., Lindholm, B., et al. (2007). Impact of inflammation on epigenetic DNA methylation – a novel risk factor for cardiovascular disease? J. Intern. Med. 261, 488–499. doi: 10.1111/j.1365-2796.2007.01777.x
Storz, G., Vogel, J., and Wassarman, K. M. (2011). Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43, 880–891. doi: 10.1016/j.molcel.2011.08.022
Stouthamer, R., and Werren, J. H. (1993). Microbes associated with parthenogenesis in wasps of the genus Trichogramma. J. Invertebr. Pathol. 61, 6–9. doi: 10.1006/jipa.1993.1002
Suen, D.-F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Sun, X. (2002). Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res. 30, 3662–3671. doi: 10.1093/nar/gkf508
Sun, Y., Zhao, X., Zhou, Y., and Hu, Y. (2012). miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 28, 1346–1352. doi: 10.3892/or.2012.1958
Sunkar, R., and Zhu, J.-K. (2004). Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16, 2001–2019. doi: 10.1105/tpc.104.022830
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. doi: 10.1038/nrg2341
Szyf, M. (2014). Lamarck revisited: epigenetic inheritance of ancestral odor fear conditioning. Nat. Neurosci. 17, 2–4. doi: 10.1038/nn.3603
Tak, H., Kim, J., Jayabalan, A. K., Lee, H., Kang, H., Cho, D.-H., et al. (2014). miR-27 regulates mitochondrial networks by directly targeting the mitochondrial fission factor. Exp. Mol. Med. 46:e123. doi: 10.1038/emm.2014.73
Takahashi, K., Sugi, Y., Nakano, K., Tsuda, M., Kurihara, K., Hosono, A., et al. (2011). Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J. Biol. Chem. 286, 35755–35762. doi: 10.1074/jbc.M111.271007
Tamas, I., Wernegreen, J., Nystedt, B., Kauppinen, S. N., Darby, A. C., Gomez-Valero, L., et al. (2008). Endosymbiont gene functions impaired and rescued by polymerase infidelity at poly(A) tracts. Proc. Natl. Acad. Sci. U.S.A. 105, 14934–14939. doi: 10.1073/pnas.0806554105
Teixeira, L., Ferreira, A., and Ashburner, M. (2008). The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 6:e2. doi: 10.1371/journal.pbio.1000002
Terrapon, N., Cai, L., Robertson, H., Ji, L., Meng, X., Booth, W., et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat. Commun. 5:3636. doi: 10.1038/ncomms4636
Tomasetti, M., Neuzil, J., and Dong, L. (2014). MicroRNAs as regulators of mitochondrial function: role in cancer suppression. Biochim. Biophys. Acta 1840, 1441–1453. doi: 10.1016/j.bbagen.2013.09.002
Tran, R. K., Henikoff, J. G., Zilberman, D., Ditt, R. F., Jacobsen, S. E., and Henikoff, S. (2005). DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr. Biol. 15, 154–159. doi: 10.1016/j.cub.2005.01.008
Vidigal, J. A., and Ventura, A. (2015). The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol. 25, 137–147. doi: 10.1016/j.tcb.2014.11.004
Viñuelas, J., Febvay, G., Duport, G., Colella, S., Fayard, J.-M., Charles, H., et al. (2011). Multimodal dynamic response of the Buchnera aphidicola pLeu plasmid to variations in leucine demand of its host, the pea aphid Acyrthosiphon pisum. Mol. Microbiol. 81, 1271–1285. doi: 10.1111/j.1365-2958.2011.07760.x
Vogel, J., and Luisi, B. F. (2011). Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9, 578–589. doi: 10.1038/nrmicro2615
Vorburger, C., Sandrock, C., Gouskov, A., Castaneda, L. E., and Ferrari, J. (2009). Genotypic variation and the role of defensive endosymbionts in an all- parthenogenetic host-parasitoid interaction. Evolution 63, 1439–1450. doi: 10.1111/j.1558-5646.2009.00660.x
Wagner, E. G., and Simons, R. W. (1994). Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 48, 713–742. doi: 10.1146/annurev.mi.48.100194.003433
Walsh, T. K., Brisson, J. A., Robertson, H. M., Gordon, K., Jaubert-Possamai, S., Tagu, D., et al. (2010). A functional DNA methylation system in the pea aphid, Acyrthosiphone pisum. Insect Mol. Biol. 19, 215–228. doi: 10.1111/j.1365-2583.2009.00974.x
Wang, J.-X., Jiao, J.-Q., Li, Q., Long, B., Wang, K., Liu, J.-P., et al. (2011). miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 17, 71–78. doi: 10.1038/nm.2282
Wang, K., Long, B., Jiao, J.-Q., Wang, J.-X., Liu, J.-P., Li, Q., et al. (2012). miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 3, 781. doi: 10.1038/ncomms1770
Wang, L., Yu, X., Wang, H., Lu, Y.-Z., de Ruiter, M., Prins, M., et al. (2011). A novel class of heat-responsive small RNAs derived from the chloroplast genome of Chinese cabbage (Brassica rapa). BMC Genomics 12:289. doi: 10.1186/1471-2164-12-289
Wang, Y., Jorda, M., Jones, P. J., Maleszka, R., Ling, X., Robertson, H. M., et al. (2006). Functional CpG methylation system in a social insect. Science 314, 645–664. doi: 10.1126/science.1135213
Waters, L. S., and Storz, G. (2009). Regulatory RNAs in bacteria. Cell 136, 615–628. doi: 10.1016/j.cell.2009.01.043
Weaver, I. C. G., Cervoni, N., Champagne, F. A., D'Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. doi: 10.1038/nn1276
Web of Science (2016). Thomson Reuters. Available online at: https://webofknowledge.com (Accessed April 25, 2016).
Weiberg, A., Wang, M., Lin, F.-M., Zhao, H., Zhang, Z., Kaloshian, I., et al. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123. doi: 10.1126/science.1239705
Weinert, L. A., Werren, J. H., Aebi, A., Stone, G. N., and Jiggins, F. M. (2009). Evolution and diversity of Rickettsia bacteria. BMC Biol. 7:6. doi: 10.1186/1741-7007-7-6
Wernegreen, J. J., Kauppinen, S. N., and Degnan, P. H. (2010). Slip into something more functional: selection maintains ancient frameshifts in homopolymeric sequences. Mol. Biol. Evol. 27, 833–839. doi: 10.1093/molbev/msp290
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Werren, J. H., Richards, S., Desjardins, C. A., Niehuis, O., Gadau, J., Colbourne, J. K., et al. (2010). Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348. doi: 10.1126/science.1178028
Williams, K. P., Sobral, B. W., and Dickerman, A. W. (2007). A robust species tree for the alphaproteobacteria. J. Bacteriol. 189, 4578–4586. doi: 10.1128/JB.00269-07
Williams, L. E., and Wernegreen, J. J. (2011). Purifying selection, sequence composition, and context-specific indel mutations shape intraspecific variation in a bacterial endosymbiont. Genome Biol. Evol. 4, 44–51. doi: 10.1093/gbe/evr128
Wilusz, J. E., Whipple, J. M., Phizicky, E. M., and Sharp, P. A. (2011). tRNAs marked with CCACCA are targeted for degradation. Science 334, 817–821. doi: 10.1126/science.1213671
Winkler, W. C., and Breaker, R. R. (2003). Genetic control by metabolite-binding riboswitches. Chembiochem 4, 1024–1032. doi: 10.1002/cbic.200300685
Winkler, W., Nahvi, A., and Breaker, R. R. (2002). Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 419, 952–956. doi: 10.1038/nature01145
Wion, D., and Casadesús, J. (2006). N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
Wu, Z., Stone, J. D., Štorchová, H., and Sloan, D. B. (2015). High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genomics 16:938. doi: 10.1186/s12864-015-2155-3
Xiang, H., Zhu, J., Chen, Q., Dai, F., Li, X., Li, M., et al. (2010). Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 28, 516–520. doi: 10.1038/nbt.1626
Xiao, J., Zhu, X., Bin, H., Zhang, Y., Kang, B., Wang, Z., et al. (2011). MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J. Biomed. Sci. 18:35. doi: 10.1186/1423-0127-18-35
Yamamoto, H., Morino, K., Nishio, Y., Ugi, S., Yoshizaki, T., Kashiwagi, A., et al. (2012). MicroRNA-494 regulates mitochondrial biogenesis in skeletal muscle through mitochondrial transcription factor A and Forkhead box j3. Am. J. Physiol. Endocrinol. Metab. 303, E1419–E1427. doi: 10.1152/ajpendo.00097.2012
Yan, H., Bonasio, R., Simola, D. F., Liebig, J., Berger, S. L., and Reinberg, D. (2015). DNA methylation in social insects: how epigenetics can control behavior and longevity. Annu. Rev. Entomol. 60, 435–452. doi: 10.1146/annurev-ento-010814-020803
Ye, Y. H., Wollfit, M., Huttley, G. A., Rancès, E., Caragata, E. P., Popovici, J., et al. (2013). Infection with a virulent strain of Wolbachia disrupts genome wide-patterns of cytosine methylation in the mosquito Aedes aegypti. PLoS ONE 8:e66482. doi: 10.1371/journal.pone.0066482
Yu, Y. T., Yuan, X., and Velicer, G. J. (2010). Adaptive evolution of an sRNA that controls Myxococcus development. Science 328, 993. doi: 10.1126/science.1187200
Zamudio, N., Barau, J., Teissandier, A., Walter, M., Borsos, M., Servant, N., et al. (2015). DNA methylation restrains transposons from adopting a chromatin signature permissive for meiotic recombination. Genes Dev. 29, 1256–1270. doi: 10.1101/gad.257840.114
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zhang, G., Hussain, M., O'Neill, S. L., and Asgari, S. (2013). Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A. 110, 10276–10281. doi: 10.1073/pnas.1303603110
Zhang, J.-G., Wang, J.-J., Zhao, F., Liu, Q., Jiang, K., and Yang, G.-H. (2010). MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta. 411, 846–852. doi: 10.1016/j.cca.2010.02.074
Zhang, J., Xing, Y., Li, Y., Yin, C., Ge, C., and Li, F. (2015). DNA methyltransferases have an essential role in female fecundity in brown planthopper, Nilaparvata lugens. Biochem. Biophys. Res. Commun. 464, 83–88. doi: 10.1016/j.bbrc.2015.05.114
Zhang, M., Chen, J., Liang, S., Li, G., Wang, F., and Ahmad, I. (2015). Differentially methylated genomic fragments related with sexual dimorphism of rice pests, Sogatella furcifera. Insect Sci. 22, 731–738. doi: 10.1111/1744-7917.12179
Zhang, Q. L., Su, L. E., and Lu, Z. Q. (2015). Expression of DNA methylation and histone acetylation related genes in response to bacterial infection in the silkworm, Bombyx mori. Acta Entomol. Sin. 58, 941–949.
Zhang, Y., Yang, L., Gao, Y.-F., Fan, Z.-M., Cai, X.-Y., Liu, M.-Y., et al. (2013). MicroRNA-106b induces mitochondrial dysfunction and insulin resistance in C2C12 myotubes by targeting mitofusin- 2. Mol. Cell. Endocrinol. 381, 230–240. doi: 10.1016/j.mce.2013.08.004
Zhou, X., Chen, J., Shike Liang, M., and Wang, F. (2013). Differential DNA methylation between two wing phenotypes adults of Sogatella furcifera. Genesis 51, 819–826. doi: 10.1002/dvg.22722
Zhou, G., Wang, T., Lou, Y., Cheng, J., Zhang, H., and Xu, J. H. (2014). Identification and characterization of microRNAs in small brown planthopper (Laodephax striatellus) by next-generation sequencing. PLoS ONE 9:e103041. doi: 10.1371/journal.pone.0103041
Zhu, H., Wu, H., Liu, X., Li, B., Chen, Y., Ren, X., et al. (2009). Regulation of autophagy by a beclin1- targeted microRNA, miR-30a, in cancer cells. Autophagy 5, 816–823. doi: 10.4161/auto.9064
Keywords: symbiosis, epigenomics, DNA methylation, small RNAs, insect-plant interactions
Citation: Kim D, Thairu MW and Hansen AK (2016) Novel Insights into Insect-Microbe Interactions—Role of Epigenomics and Small RNAs. Front. Plant Sci. 7:1164. doi: 10.3389/fpls.2016.01164
Received: 26 April 2016; Accepted: 20 July 2016;
Published: 04 August 2016.
Edited by:
Linda Walling, University of California, Riverside, USAReviewed by:
Igor Kovalchuk, University of Lethbridge, CanadaCopyright © 2016 Kim, Thairu and Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Allison K. Hansen, YWtoQGlsbGlub2lzLmVkdQ==
†First co-authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.